

Abstract
Objective –
Our recent work demonstrates that PTEN is an important regulator of SMC phenotype. SMC-specific PTEN deletion promotes spontaneous vascular remodeling and PTEN loss correlates with increased atherosclerotic lesion severity in human coronary arteries. In mice, PTEN overexpression reduces plaque area and preserves SMC contractile protein expression in atherosclerosis and blunts AngII-induced pathological vascular remodeling, suggesting that pharmacologic PTEN upregulation could be a novel therapeutic approach to treat vascular disease.
Approach and Results –
To identify novel PTEN activators, we conducted a high-throughput screen using a fluorescence based PTEN promoter-reporter assay. After screening ~3400 compounds, 11 hit compounds were chosen based on level of activity and mechanism of action. Following in vitro confirmation, we focused on 5-azacytidine, a DNMT1 inhibitor, for further analysis. In addition to PTEN upregulation, 5-azacytidine treatment increased expression of genes associated with a differentiated SMC phenotype. 5-azacytidine treatment also maintained contractile gene expression and reduced inflammatory cytokine expression after PDGF stimulation, suggesting 5-azacytidine blocks PDGF-induced SMC de-differentiation. However, these protective effects were lost in PTEN-deficient SMCs. These findings were confirmed in vivo using carotid ligation in SMC-specific PTEN knockout mice treated with 5-azacytidine. In WT controls, 5-azacytidine reduced neointimal formation and inflammation while maintaining contractile protein expression. In contrast, 5-azacytidine was ineffective in PTEN knockout mice, indicating that the protective effects of 5-azacytidine are mediated through SMC PTEN upregulation.
Conclusions –
Our data indicates 5-azacytidine upregulates PTEN expression in SMCs, promoting maintenance of SMC differentiation and reducing pathological vascular remodeling in a PTEN-dependent manner.
Keywords: Vascular Smooth Muscle Cell; PTEN, Drug Screen; Vascular Remodeling; DNA Methylation
Subject Terms: Vascular Disease, Vascular Biology, Smooth Muscle Proliferation and Differentiation
Graphical Abstract
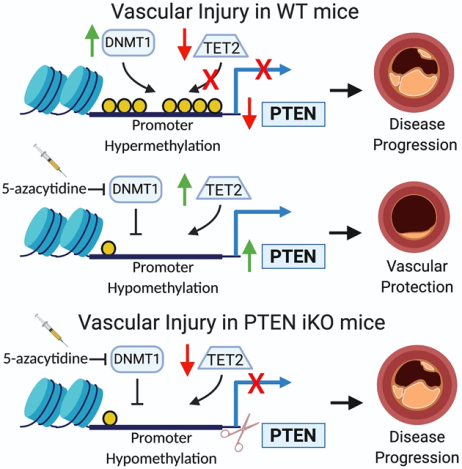
Introduction
Cardiovascular diseases remain the leading cause of death in the United States and globally, posing by a huge financial burden due to healthcare-associated costs. In the United States, coronary artery disease is the leading cause of cardiovascular related death and is estimated to underlie one third of all deaths in those over 35 years of age. Atherosclerosis and restenosis are chronic and acute inflammatory vascular diseases, respectively, characterized by significant vascular remodeling1,2. Activation of resident vascular smooth muscle cells (SMCs) plays a critical role in remodeling and is a key event promoting vascular disease progression. Under physiologic conditions, vascular smooth muscle cells are maintained in a differentiated, quiescent phenotype characterized by the expression of SMC-specific contractile proteins such as smooth muscle myosin heavy chain (Myh11/SMMHC) and alpha-smooth muscle actin (Acta2/αSMA)3,4. However SMCs are not terminally differentiated, and in response to injury or pathogenic stimuli, they can undergo phenotypic transition (de-differentiation) and adopt a more proliferative, pro-inflammatory, pro-fibrotic phenotype. The concept of SMC de-differentiation is well established, and phenotypically modulated SMCs play an important role contributing to the development of pathological vascular remodeling. However, the mechanisms regulating SMC de-differentiation are not fully understood, and there are currently no therapeutic agents aimed specifically towards maintaining a differentiated SMC phenotype.
Previously, we have shown that inactivation of the tumor suppressor phosphatase and tensin homolog (PTEN), a lipid and protein phosphatase known to antagonize the PI3K/Akt signaling pathway, is associated with SMC activation and vascular disease progression through both phosphatase-dependent and -independent effects5–8. Our recent data demonstrate a novel phosphatase-independent function for nuclear PTEN in SMC transcriptional control through direct association with serum response factor (SRF) and its muscle-specific co-factor myocardin6. Formation of this complex facilitates selective SRF binding to CArG boxes in the promoter regions of SM-specific contractile genes to maintain SMC differentiation, suggesting that PTEN plays a central role in maintenance of SMC differentiation6. Highlighting the clinical relevance, we showed that reduced PTEN expression is directly correlated with SMC de-differentiation and increased atherosclerotic lesion complexity in human coronary arteries9. Recent studies using systemic PTEN overexpressing mice10 indicate that PTEN upregulation is able to preserve the differentiated SMC phenotype, block inflammation, and prevent pathologic vascular remodeling in the setting of Angiotensin II-mediated hypertension11 and atherosclerosis (our unpublished findings). Thus, we sought to identify small molecules capable of upregulating PTEN expression, and, to add translational significance, determine if pharmacologic PTEN upregulation would be a viable approach to reduce injury-induced vascular remodeling.
There are few small molecules known to induce PTEN expression, leaving the therapeutic potential of PTEN relatively untapped. To address this, we developed a robust high-throughput compound screening (HTS) promoter-reporter assay in SMCs, which we used to identify small molecules with the ability to upregulate PTEN promoter activity to induce PTEN expression. Here, we describe the results of a 3,406 compound screen using our PTEN promoter-reporter assay to identify upregulators of PTEN expression. Following the screen, and subsequent in vitro confirmatory testing, we identified 5-azacytidine, a DNA methyltransferase-1 (DNMT1) inhibitor, as a hit compound with the potential to upregulate PTEN expression in SMCs. PTEN promoter hypermethylation has been identified as a mechanism repressing PTEN expression in other cell systems, and in homocysteine-stimulated SMCs12–21. Additionally, altered DNA methylation in SMCs has been identified in atherosclerotic vessels, and treatment with 5-azacytidine has been reported to be protective in mouse models of vascular disease22,23. However, in cardiovascular disease, the functional and biological consequences of PTEN hypermethylation and the genes that mediate the protective effects of 5-azacytidine treatment have not yet been defined. Our results indicate that 5-azacytidine treatment causes PTEN upregulation, resulting in inhibition of SMC de-differentiation and resistance to injury-mediated pathological vascular remodeling.
Complete Materials and Methods
The authors declare that all supporting data are available within the article [and its online supplementary files].
SMC Promoter-Reporter Cell Line Generation
Plasmid constructs were generated using a pCDH-CMV-MCS-EF1α-copGFP lentivector (System Biosciences Cat.# CD511B-1). To create a positive control reporter plasmid, the mCherry ORF was inserted at the multi cloning site using restriction enzymes BamHI and NotI (New England BioLabs) to create a pCDH-CMV-mCherry-EF1α-copGFP vector. Genomic DNA from HEK293 cells was extracted and a 4.3 kbp region of the human PTEN promoter region (−90 to −4412 from translation start site) was amplified by PCR using Platinum SuperFi DNA Polymerase (Thermo Fisher Scientific) and specific primers containing restriction sites (forward primer with ClaI site: 5’-GACTATCGAT TGCTTGGAGACCAGTGTATTGT-3’ and reverse primer with NheI site: 5’-TGTGCTAGC TGGACTTGGCGGTAGCTGAT-3’). The human PTEN promoter sequence was retrieved from GRCh38 human assembly through UCSC genome browser (http://genome.ucsc.edu/). 4000 bases plus 5’ UTR exons were obtained upstream of the PTEN gene (RefSeq: NM_000314) for cloning primer design. The final cloned sequence encompasses 4323 base pairs ranging from 87860058–87864380 on chromosome 10. The resulting PCR product was run on a 1% agarose gel and the cloned PTEN promoter fragment was extracted using a QIAquick gel extraction kit (Qiagen 28704). Using the pCDH-CMV-mCherry-EF1α-copGFP positive control vector, the CMV promoter was excised through restriction digestion with ClaI and NheI and the cloned PTEN promoter fragment inserted using DNA ligation to create a pCDH-PTEN-mCherry-EF1α-copGFP vector. NEB 5-alpha Competent E. coli were transformed with either the pCDH-CMV-mCherry-EF1α-copGFP or pCDH-PTEN-mCherry-EF1α-copGFP constructs using a High Efficiency Transformation Protocol (New England Biolabs C2987H). Transformed E. coli were grown overnight on selection plates containing Luria Bertani broth (LB) supplemented with 100 μg/mL carbenicillin. Individual colonies from the selection plates were expanded by overnight culture in LB broth supplemented with 100 μg/mL carbenicillin. After overnight culture, E. coli cells were pelleted at 10,000xg for 20 min and plasmids were purified using an EndoFree Plasmid Maxi Kit (Qiagen 12362). DNA sequencing was performed to validate insertion of the full sequence of the cloned promoter into the pCDH-PTEN-mCherry-EF1α-copGFP vector. HEK293T cells were plated at 7×105 cells/60 mm dish in Dulbecco’s Modified Eagle’s Medium (DMEM) containing 10% FCS, 4.5 g/L glucose, L-glutamine (2 mM), and penicillin-streptomycin, then transfected for 24 hr with 1 μg purified plasmid using standard transfection protocol, previously described8. Virus containing media was harvested at 48 and 72 hr post transfection. Primary rat aortic SMCs were cultured in Minimum Essential Medium (MEM) containing 10% FCS, L-glutamine (2 mM), and penicillin-streptomycin and plated at 1×106 cells/100 mm dish prior for transduction with virus-containing media. SMCs were transduced with viral media for 48 hr, then expanded for 7 days prior to harvesting for FACS. For FACS analysis, single cell suspensions of transduced SMCs expressing the pCDH-CMV-MCS-EF1α-copGFP, pCDH-CMV-mCherry-EF1α-copGFP or pCDH-PTEN-mCherry-EF1α-copGFP vectors were analyzed for endogenous GFP and mCherry expression. Flow cytometry was performed on a Galios cytometer (Becton Dickenson). Cells were sorted based on high GFP expression to identify construct-positive SMCs. GFP-positive SMCs were then maintained and expanded for use in high-throughput compound screen.
High-throughput screening protocol
High-throughput screening (HTS) was performed at the High-throughput and High Content Screening Core Facility in the Skaggs School of Pharmacy and Pharmaceutical Science at the University of Colorado Anschutz Medical Campus24. PTEN promoter-reporter and positive control SMCs were plated onto 384-well black plates (Corning Inc.) at a concentration of 2,000 cells in 40 μL of phenol red free MEM containing 10% FCS, L-glutamine (2 mM), and penicillin-streptomycin and allowed to adhere for 24 hrs. SMCs were then washed with 1x PBS and media changed to phenol red free MEM containing 1% FCS, L-glutamine (2 mM), and penicillin-streptomycin. To validate the assay prior to beginning the screen, positive control SMCs and PTEN promoter-reporter SMCs were plated on two separate days and treated with 0%, 0.5% or 1% DMSO, then Z’ scores calculated25. After assay validation, screening began by treating PTEN promoter-reporter SMCs with library compounds at 10 μM, with Z’ scores calculated for each plate. Cells for the assay were plated using a Janus Liquid Handling Robot (PerkinElmer). The compound library consisted of NIH Clinical Collection 1 (450 compounds) and 2 (281 compounds), the Spectrum Collection (2320 compounds), and a Selleckchem Kinase Inhibitor Library (355 compounds). SMCs were imaged 72 hr following treatment with library compounds to assess the level of mCherry expression; DRAQ5 (nuclei identification) and SYTOXBlue (dead cell identification) were added immediately prior to imaging. Imaging was performed using the Operetta high content imaging system (PerkinElmer) and results were analyzed using Harmony Software (PerkinElmer). DMSO stimulated PTEN promoter-reporter SMCs were used as a negative control for the assay. mCherry signal intensity was quantified as the average signal intensity per cell. Using the Harmony Software, non-GFP positive cells were gated out of the analysis by setting a lower limit for GFP expression. This removed construct-negative cells from mCherry signal quantification. A threshold of 2.0-fold induction of mCherry intensity relative to basal mCherry expression in DMSO-stimulated PTEN promoter-reporter cells was used to identify positive hits. Compounds causing severe growth restriction (<40% of negative control nuclei count) or excessive cytotoxicity (greater than 1:1 ratio of dead:live cells) were excluded from further analysis. Well images from all compounds causing >2.0 fold mCherry induction were analyzed to exclude auto-fluorescent compounds or compounds that precipitated out of solution to remove false positives. Following the initial screen, a confirmatory screen of the preliminary hit compounds was performed in duplicate at 10 μM (see Supplemental Table I for list of compounds). Following the confirmatory screen at 10 μM, we identified confirmed hit compounds that met the >2.0-fold mCherry induction threshold in both the preliminary and confirmatory screen. These confirmed hits were then screened in duplicate in a dose response format at 5.0 μM, 1.0 μM, and 0.2 μM. A dose-responsive threshold of mCherry induction above negative control was used to classify a hit compound at the 5.0 μM, 1.0 μM, and 0.2 μM doses. Hit compounds chosen for further in vitro testing were purchased from commercial vendors and suspended in 100% DMSO (see Supplemental Table II for list of compounds).
Cell Culture, In vitro hit validation and RT-qPCR
Primary rat aortic smooth muscle cells (SMCs) were isolated and cultured as previously described26. For in vitro PTEN depletion studies, stable PTEN-deficient SMC clones expressing a lentiviral short hairpin RNA (shRNA) sequence based on the mouse PTEN gene (NCBI accession NM008960) were utilized. The generation of these PTEN-depleted SMC clones was described previously8. In all cell culture experiments, SMCs were maintained in MEM containing 10% FCS, L-glutamine (2 mM), and penicillin-streptomycin and were used as primary cultured cells until passage 15 unless otherwise specified. For RT-qPCR experiments, SMCs were maintained in phenol red free MEM containing 10% FCS, L-glutamine (2 mM), and penicillin-streptomycin then plated at 3×105 cells/well into 6-well plates (Corning Inc.), allowed to adhere for 24 hr, then media changed to phenol red free MEM containing 1% FCS, L-glutamine (2 mM), and penicillin-streptomycin with compounds dosed in triplicate at a final compound concentration of 10 μM, resulting in final 0.1% DMSO concentration. SMCs treated with 0.1% DMSO were used as a vehicle control. SMCs were stimulated with compounds for 24, 48, or 72 hrs in the presence or absence of PDGF-BB (20 ng/mL Millipore GF149), then total RNA was isolated using RLT lysis buffer (Qiagen) supplemented with 0.1% β-mercaptoethanol. Samples were processed with QIAshredder and RNeasy Plus kits (Qiagen) to isolate total RNA. Following RNA isolation, cDNA was made using the qScript XLT cDNA SuperMix (Quantabio). Sequence-specific primers were designed for the following genes Pten (Phosphatase and tensin homolog deleted on chromosome 10), Myh11 (Smooth muscle myosin heavy chain), Acta2 (α-smooth muscle actin), Cnn1 (Calponin), Ccl2 (C-C motif chemokine ligand 2), Ccl7 (C-C motif chemokine ligand 7), Tet2 (Tet methylcytosine dioxygenase 2), Gapdh (Glyceraldehyde-3-phosphate dehydrogenase). Primer sequences are available in Supplemental Table III. RT-qPCR was performed as previously described11 and Gapdh was used for normalization. Data were normalized to SMCs stimulated with 0.1% DMSO. mRNA data presented is the mean expression from three independent experimental replicates.
Animals
Inducible SMC-specific PTEN iKO mice and 5-azacytidine injections –
PTENfl/fl (Tak Mak, Ontario Cancer Institute, University of Toronto, Toronto, Ontario, Canada), SMMHC (Myh11)-CreERT2 transgenic mice (Stephen Offermanns, University of Heidelberg, Heidelberg, Germany), and Rosa26-YFP mice (#006148; B6;129S6-Gt(ROSA)26Sortm1(EYFP)Cos/J; The Jackson Laboratory) were bred to generate tamoxifen-inducible SMC-specific PTEN-knockout mice (PTENflox/flox-Myh11-CreERT2;Rosa26-YFP) (PTEN iKO) fully backcrossed on the C57BL6/J genetic background, as described previously5. Control wild type (WT) mice expressed Myh11-CreERT2;Rosa26-YFP, but were WT for PTEN. All mice carried an inducible SMC-specific YFP reporter in order to fate-map SMCs in the setting of injury even if they lose all characteristics of SMCs (i.e. loss of SMC-specific contractile marker expression). Mice were previously backcrossed more than 10 generations to the C57BL6/J background. Only male mice were used for in vivo experiments, as the Myh11-CreERT2 BAC transgene inserted onto the Y chromosome. Mice (8–10 weeks old) received 1.5 μg tamoxifen injections i.p. for 7 consecutive days to induce PTEN knockout and YFP knock-in. Tamoxifen was allowed to wash out for 7 days. After tamoxifen washout, carotid artery ligation injury was performed as described previously7. Briefly, left carotid arteries were completely ligated just proximal to the carotid bifurcation. 5-azacytidine (Sigma A2385) was prepared at 0.5 mg/mL in sterile saline immediately prior to injection, then injected i.p. at 2 mg/kg 3x weekly for 3 weeks prior to tissue harvest. As a control, an equivalent volume of sterile saline was injected i.p. on the same schedule. 5-azacytidine injections began at the time of carotid ligation injury. 3 weeks post carotid ligation injury, uninjured and injured carotid arteries were harvested, fixed in 4% buffered PFA for 24 hr, soaked in 30% sucrose for 24 hr for cryoprotection, and embedded in OCT for immunofluorescent staining. Mice were maintained in the Center for Comparative Medicine and all procedures were performed under a protocol approved by the Institutional Animal Care and Use Committee at the University of Colorado Anschutz Medical Campus.
The 5-azacytidine dose used for in vivo studies was chosen based on very careful pharmacokinetic and pharmacodynamic studies conducted previously. 5-azacytidine is an FDA approved compound for use in humans. Pharmacokinetic and pharmacodynamic information related to 5-azacytidine was published by the FDA and EMA. The pharmacokinetic properties of 5-azacytidine in mice have been previously published, while more recent papers have described the pharmacokinetic properties of 5-azacytidine concentrations in human plasma samples. 5-azacytidine is rapidly absorbed, with plasma concentrations peaking around 15–30 minutes post dosing, and then is rapidly cleared from blood. The 2 mg/kg dosage used in the present study was chosen based on the non-clinical toxicology information provided by the FDA. 2.0 mg/kg was the lowest dose where there was a significant increase in tumor development in mice after 50–52 weeks of dosing. For a short-term experiment as our study design, 2 mg/kg was reported to be well tolerated and result in few adverse side effects as a 2.2 mg/kg dose 3x weekly was tolerated for 52 weeks in previous studies. Please see the following links for information regarding 5-azacytidine PK/PD information and dosages:
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3662854/
https://media.celgene.com/content/uploads/sites/11/2-VID-EPAR-WC500050242.pdf
https://ascopubs.org/doi/full/10.1200/JCO.2005.07.450?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%3dpubmedhttps://www.accessdata.fda.gov/drugsatfda_docs/nda/2004/50-794_Vidaza_Pharmr_P1.pdf
https://www.accessdata.fda.gov/drugsatfda_docs/label/2008/050794s011lbl.pdf
Morphology, Tissue Staining and Microscopy
Morphological Analysis –
While intima-media (I/M) ratio is recognized as the gold standard measurement for neointima formation, percent stenosis was used in this study as injury-induced medial changes varied from animal to animal and within groups, thus skewing I/M ratio analysis. Arterial specimens were sectioned from the site of ligation to identify the region with maximal luminal narrowing. These sections were selected for morphometric measurements of percent stenotic area and adventitial area, as described previously7. To measure percent stenosis, hematoxylin stained arterial sections were visualized using an Olympus light microscope and measurements made using SPOT software. Adventitial remodeling was measured as the ratio of adventitial area over outer medial circumference.
Immunofluorescence CD68-YFP-αSMA-SMMHC –
OCT-embedded tissues were sectioned at 5.0 μM. Slides were brought to RT for 15 minutes and prepared for staining by rehydrating in diH2O for 10 minutes and then washed once with 1x PBS. Slides were permeabilized for 10 minutes in 100% MeOH and then washed with 0.05% Tween 20 for 5 minutes, followed by an additional wash for 5 minutes with 1x PBS. Slides were blocked with 3% horse serum in PBS for 60 minutes at RT prior to the addition of primary antibodies. Rat derived anti-mouse CD68 (BioRad; MCA1957) was used at 1:100 dilution, while anti-αSMA and anti-YFP-FITC were used as described above. Anti-CD68, anti-αSMA, rabbit anti-SMMHC (1:500 Abcam ab125884), and anti-GFP-FITC primary antibodies were diluted in 1x PBS + 3% horse serum, then applied overnight at 4° C. Following incubation, slides were washed with 1x PBS, then incubated with Alexa Fluor 647-conjugated goat-anti-rabbit secondary antibody (1:500 Invitrogen A21244) and AlexaFluor 594-conjugated donkey-anti-rat secondary antibody (1:500 Invitrogen A21209) for 1 hr at RT. Slides were washed with 1x PBS and mounted with Vectashield Antifade Mounting Medium with DAPI (Vector H-1200). Negative controls included incubating arterial sections with comparable IgGs followed by secondary antibodies. Images were visualized using a Keyence BZ-X710 microscope and BZ-X Viewer Software. Cell counting for CD68+ macrophages was performed with ImageJ with the Cell Counter plugin.
Movat’s pentachrome stain –
Russel-Movat pentachrome staining was performed with modifications to the manufacturer’s instructions (American MasterTech) for OCT-embedded tissue. Slides were brought to RT for 15 minutes and prepared for staining by rehydrating in diH2O for 10 minutes. Slides were immersed in Verhoeff’s elastic stain for 4 minutes and then rinsed in running diH2O for 4 minutes. Slides were immersed in 1% Alcian Blue Solution for 10 minutes and then rinsed in running diH2O for 2 minutes. All other steps were followed per the manufacturer’s protocol. Tissues were stained with Verhoeff’s elastic stain, Alcian blue for mucin, crocein scarlet-acid fuchsin for muscle, and saffron solution for collagen. The slides were dehydrated and mounted with VectaMount Permanent Mounting Medium (Vector). Images were visualized using a Keyence BZ-X710 microscope and BZ-X Viewer Software.
Masson’s Trichrome stain –
Masson’s Trichrome stain was performed with modifications to the manufacturer’s instructions (American MasterTech) for OCT-embedded tissue. Slides were brought to RT for 15 minutes and prepared for staining by rehydrating in diH2O for 10 minutes. Slides were then immersed in Bouin’s Fluid at RT overnight, then rinsed in running diH2O for 10 minutes. The following protocol was then performed with all slides being immersed in solution and then rinsed in running diH2O unless otherwise noted: working Weigert’s Hematoxylin for 4 minutes and rinse for 3 minutes, Beibrich Scarlet-Acid Fuchsin for 10 minutes and rinse for 2 minutes, Phosphomolybdic/ Phosphotungstic Acid for 10 minutes with no rinse, Aniline Blue Solution for 90 seconds and rinse for 2 minutes, and 1% Acetic Acid for 3 minutes with no following rinse. The slides were then dehydrated and mounted with VectaMount Permanent Mounting Medium (Vector). Images were visualized using a Keyence BZ-X710 microscope and BZ-X Viewer Software.
Western Blot Analysis
SMCs were maintained in MEM containing 10% FCS, L-glutamine (2 mM), and penicillin-streptomycin then plated at 6×105 cells/well into 60 mm dishes (Corning Inc.) and allowed to adhere for 24 hrs, then media changed to MEM containing 0.1% FCS, L-glutamine (2 mM), and penicillin-streptomycin for 24 hrs. Cells were washed with 1x Hank’s Balanced Salt Solution, and media changed to MEM containing 1% FCS, L-glutamine (2 mM), and penicillin-streptomycin and 10 μM 5-azacytidine resulting in a final 0.1% DMSO concentration. SMCs treated with 0.1% DMSO were used as a vehicle control. SMCs were stimulated with 5-azacytidine for 72 hrs in the presence or absence of PDGF-BB, then total protein was harvested. SMCs were lysed using ice-cold M-PER Mammalian Protein Extraction Reagent (ThermoScientific 78501) supplemented with 10 μL/mL protease inhibitor cocktail (Sigma P8340). Cell lysates were sonicated for 10 seconds on ice, then spun at 14,000xg for 10 min at 4° C and supernatant harvested. Solubilized proteins were loaded in equal amounts, then separated by SDS-PAGE and transferred for Western blotting, as described previously6. Antibodies to total PTEN (Cell Signaling 9559L) or phosphoSer473-Akt (Cell Signaling 4060S) were used at 1:1000 dilution, while β-actin (Sigma A2547) was used at a 1:60,000 dilution and served as a loading control. Band intensity was quantified using ImageJ, and normalized to β-actin expression.
Statistics
Data were analyzed using PRISM 8 (GraphPad Software, Inc.). Column statistics and D’Agostino and Pearson omnibus normality tests were performed to determine the mean, standard deviation, and normality of the data. For data that passed normality test, a 1-way ANOVA was used to calculate if the overall P value was significant, followed by Bonferroni’s multiple comparison to determine differences between groups. For comparison between two groups (i.e. control versus 5-azacytidine treatment), unpaired, two-tailed Student’s t tests for normal data were used and reported with exact P values. A P value of 0.05 or less was considered significant.
Results
PTEN promoter-reporter plasmid generation and high-throughput screen assay validation
In order to identify compounds that upregulate PTEN promoter activity, we developed a lentiviral, fluorescent based promoter-reporter assay in SMCs using a pCDH-CMV-MCS-EF1α-copGFP lentivector as a foundation for plasmid development (See Methods) (Supplemental Figure IA). Using this assay, we performed a high-throughput compound screen (HTS) in collaboration with the High Throughput Screening and Chemical Biology Core Facility at the University of Colorado Anschutz Medical Campus24,27. In all lentivectors, GFP expression was driven by the constitutive EF1α promoter, which facilitated flow sorting for GFP-positive cells to obtain a pure population of lentivector-positive SMCs (Supplemental Figure IB) It is important to note that there is mCherry expression in the PTEN promoter-reporter SMCs due to basal PTEN promoter activity and expression in these cells (Supplemental Figure IB right panel). Induction of mCherry expression was used as a readout for PTEN promoter activity.
We confirmed detection of both GFP and mCherry in the PTEN promoter-reporter and positive control SMCs by taking live cell images using the Operetta high content imaging system (PerkinElmer). Basal mCherry expression in unstimulated PTEN promoter-reporter SMCs was readily detectable above background (Supplemental Figure IC, top right panel), but was well below mCherry expression in positive control SMCs (Supplemental Figure IC bottom right panel). The mCherry signal intensity from the PTEN promoter-reporter and positive control cells was quantified using Harmony Software (PerkinElmer), and a Z’ factor was calculated to ensure a robust, reproducible assay prior to screening any compounds (Supplemental Figure ID). Compounds in the screening library were suspended in 100% DMSO. PTEN promoter-reporter and positive control SMCs were treated with various concentrations of DMSO to ensure DMSO treatment alone did not impair assay viability or alter fluorescent intensity as assessed by Z’ factor calculation (Supplemental Figure ID).
Compound screening identifies 5-azacytidine as an inducer of PTEN expression
After the development and validation of our promoter-reporter assay, we screened a 3,406 compound library to identify compounds that induced mCherry expression. mCherry expression was quantified following 72 hr treatment with library compounds (Supplemental Figure IIA) and >2.0 fold induction of mCherry signal above basal mCherry expression in the negative controls was used as a threshold to identify hit compounds. DMSO-stimulated PTEN promoter-reporter cells were used as a negative control. After excluding false positives, cytotoxic, and growth restrictive compounds, 151 hit compounds were identified. Representative images of mCherry induction caused by treatment with two compounds that were identified as hits are shown in Supplemental Figure IIB. The 151 preliminary hit compounds (Supplemental Table I) were then rescreened at a 10 μM concentration followed by dose-response screening, confirming 57 compounds as hits (Supplemental Figure IIC). 11 of the 57 compounds (Supplemental Table II) were identified for further in vitro testing based on their level of activity or known mechanism of action. Stimulation with the 11 final hit compounds induced mCherry expression in the PTEN promoter-reporter SMCs in a dose-responsive manner (Supplemental Figure IID).
Each of the 11 final hit compounds were tested for their ability to induce PTEN mRNA expression by treating WT SMCs with each compound at 10 μM. Only 5 of the 11 hit compounds significantly upregulated PTEN mRNA levels (Figure 1A). As there may be epigenetic or other regulatory differences between the endogenous PTEN promoter and the 4.3 kb fragment in our promoter-reporter system, it is likely that this discrepancy is due to moving from a semi-artificial promoter-reporter system to directly measuring PTEN mRNA expression in SMCs. The time-course of PTEN mRNA expression after treatment with the hit compounds revealed two distinct patterns. KU55933, R406 and 5-azacytidine treatment caused PTEN mRNA levels to peak at 48 hrs, then fall back to near baseline after 72 hr treatment (Figure 1B–C), suggesting that treatment with these compounds leads to direct PTEN promoter activation. In contrast, Deguelin and Itraconazole treatment resulted in PTEN mRNA levels to rise gradually and peak after 72 hr treatment (Figure 1B), suggesting that treatment with these compounds activates secondary pathways leading to downstream PTEN promoter activation. 5-azacytidine, a DNMT1 inhibitor, was chosen for further investigation as it caused the highest level of PTEN mRNA induction at both the 24 and 48 hr timepoints, and significantly upregulated PTEN protein expression after 72 hr treatment (Figure 1D). In addition, treatment with another DNMT1 inhibitor, 5-aza-2’-deoxycytidine resulted in a similar induction of PTEN (Supplemental Figure III).
Figure 1. Compound validation in vitro using PTEN mRNA and protein expression identifies the DNA methyltransferase-1 inhibitor, 5-azacytidine.
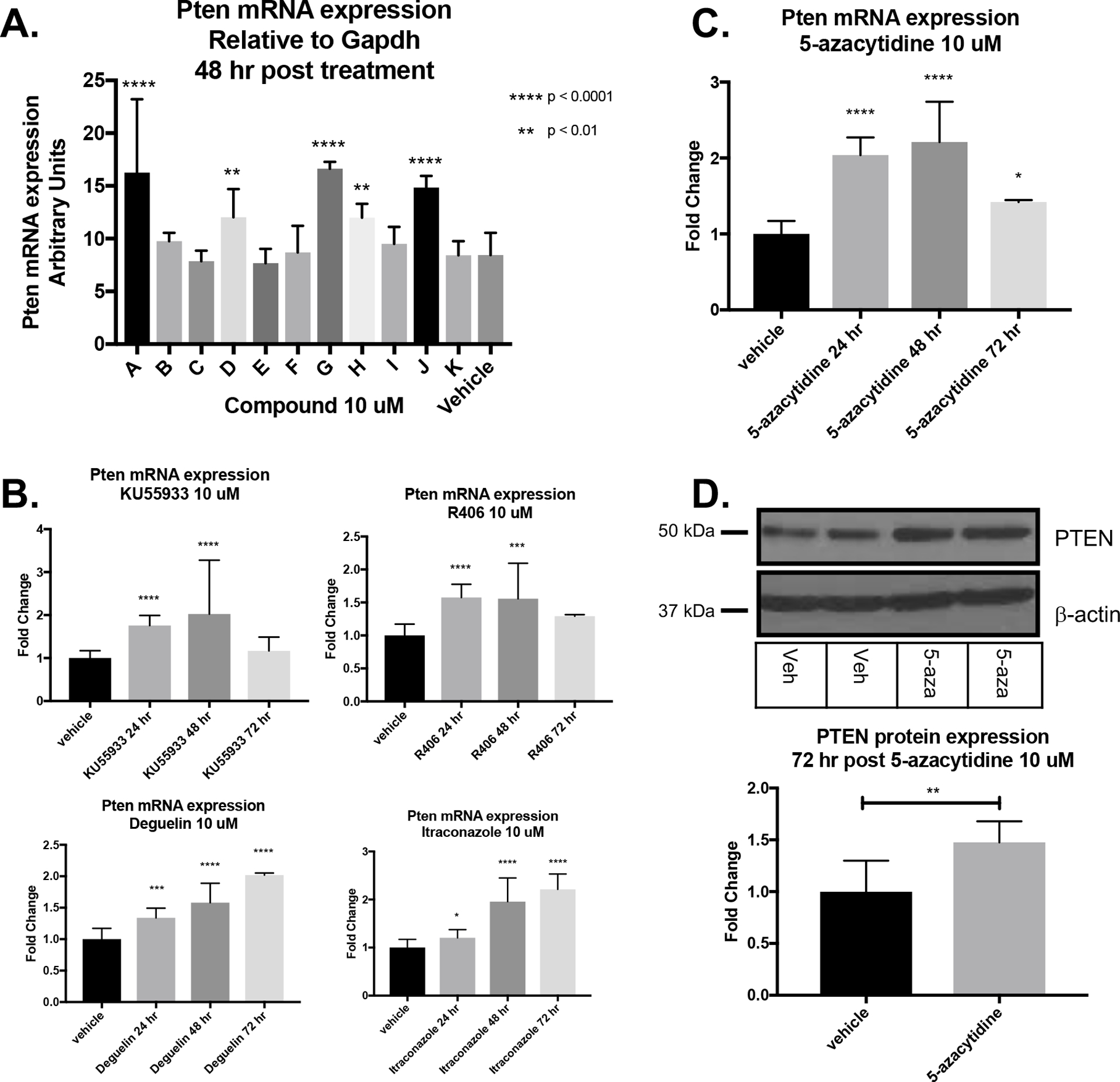
(A) WT SMCs were plated and treated in duplicate with hit compounds at 10 μM for 48 hr prior to harvest. PTEN mRNA expression was assessed by RT-qPCR. Data represent three independent experiments using replicate samples from each experiment for each hit compound that exhibited in vitro activity from above. 5 compounds resulted in PTEN mRNA upregulation. Paired T tests comparing each hit compound separately against DMSO control; **p<0.01; ****p<0.0001. (B) Timecourse of PTEN mRNA induction by indicated hit compounds. (C) Timecourse of PTEN mRNA induction after treatment with 10 μM 5-azacytidine. (D) Representative Western blot image of PTEN protein (top) and quantification (bottom graph) indicating PTEN upregulation in SMCs after 72 hr 5-azacytidine treatment. Data from three independent experimental replicates. Paired T test; p<0.01.
5-azacytidine treatment promotes a differentiated SMC phenotype
Previously, we have shown that PTEN plays an essential role in the maintenance of a differentiated SMC phenotype through both phosphatase-dependent and -independent effects6,7. In order to examine whether 5-azacytidine-mediated increased PTEN expression affected SMC phenotype, we measured basal expression of SMC contractile genes and Tet2, a TET methylcytosine dioxygenase family member that catalyzes the first step in DNA demethylation and is a recognized master regulator of SMC differentiation28. Treatment with 10 μM 5-azacytidine in vitro increased expression of the contractile genes Acta2, Myh11 and Cnn1 as well as Tet2 (Figure 2A). PDGF-BB is known to induce SMC de-differentiation, characterized by a loss of contractile gene expression and increased inflammatory cytokine expression7,29. Previously we demonstrated that PTEN loss is associated with SMC de-differentiation in both rodents and humans8,9. Here, we found that SMC treatment with 20 ng/mL PDGF-BB resulted in loss of Pten and Myh11 expression, and increased expression of the pro-inflammatory cytokine Ccl2, consistent with SMC de-differentiation (Figure2B). Additionally, 5-azacytidine treatment not only upregulated Pten and Myh11 expression, but blocked PDGF-induced downregulation of Pten and Myh11 and PDGF-BB-mediated induction of Ccl2 (Figure 2B). These data suggest that 5-azacytidine treatment promotes a differentiated SMC phenotype and actively blocks PDGF-BB-mediated SMC dedifferentiation.
Figure 2. 5-azacytidine upregulates basal SMC pro-differentiation gene expression and blocks PDGF-induced SMC de-differentiation.
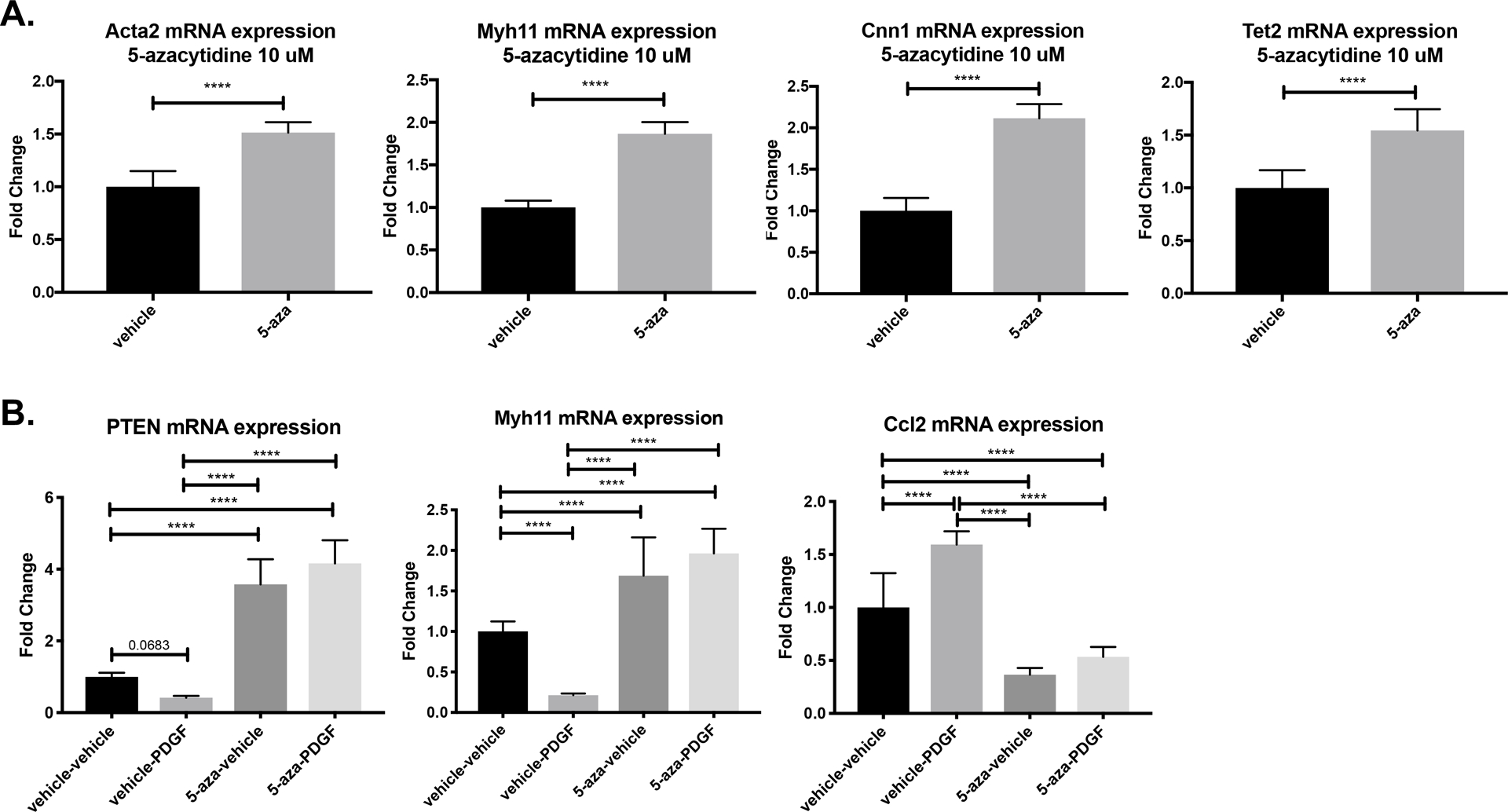
(A) WT SMCs were cultured in 0.1% CS EMEM and treated with 10 μM 5-azacytidine or DMSO vehicle control for 48 hrs prior to harvest. RNA was harvested and RT-qPCR was used to detect the indicated genes. Paired T test; ****p<0.0001. (B) WT SMCs were cultured in 0.1% CS EMEM and pre-treated with 10 μM 5-azacytidine or DMSO vehicle control for 24 hrs, then simultaneously stimulated with 20 ng/mL PDGF-BB and a second dose of 10 μM 5-azacytidine. RNA harvested 24 hr post PDGF-BB stimulation and RT-qPCR used to detect the indicated genes. ANOVA with Bonferroni post hoc; ***p<0.0001. All data are from triplicate technical replicates within each experiment and three independent experimental replicates.
Pro-differentiation effects of 5-azacytidine are mediated via PTEN upregulation
In order to examine whether the pro-differentiation effects of 5-azacytidine are mediated specifically through PTEN upregulation, we utilized PTEN knockdown (PTEN KD) SMCs, which stably express a lentiviral PTEN shRNA7,8. SMCs expressing a lentiviral non-targeting shRNA (control SMCs) were used as a control. Compared to control SMCs, PTEN KD SMCs exhibit a functional depletion of PTEN, as measured by increased phosphorylated Akt (Supplemental Figure IV). We found that 5-azacytidine upregulated PTEN mRNA expression in control SMCs, but PTEN induction was blocked in PTEN KD SMCs, indicating that the PTEN shRNA was sufficient to block the PTEN upregulation mediated by 5-azacytidine (Figure 3A). Importantly, compared to control SMCs, 5-azacytidine-mediated upregulation of pro-differentiation genes, such as Tet2 and Acta2, was lost in PTEN KD SMCs (Figure 3B). We have previously shown PTEN loss in SMCs has significant pro-inflammatory effects5. Compared to control SMCs and consistent with our previous findings5,7, PTEN KD SMCs exhibited higher basal inflammatory cytokine expression (Figure 3B). Whereas 5-azacytidine treatment in control SMCs almost completely blocked basal expression of the inflammatory cytokines Ccl2 and Ccl7, 5-azacytidine was unable to downregulate Ccl2 expression in PTEN KD SMCs, and caused only a minor reduction in Ccl7 expression (Figure 3B). These data support the conclusion that, under basal conditions, 5-azacytidine exerts its SMC pro-differentiation effects through PTEN upregulation. To examine the effects of 5-azacytidine on SMC differentiation in the context of combined loss of PTEN and PDGF treatment, control and PTEN KD SMCs were stimulated with PDGF-BB. In control SMCs, PDGF-BB stimulation resulted in reduced PTEN protein expression (Figure 3C). However, 5-azacytidine rescued PTEN inhibition in control SMCs, which was not observed in PTEN KD SMCs (Figure 3C). Quantification of PTEN protein expression in response to PDGF-BB and 5-azacytidine treatment is shown in Figure 3C. These data indicate that 5-azacytidine is able to block the de-differentiating effects of PDGF-BB in control SMCs, but the protective effects of 5-azacytidine are lost in PTEN KD SMCs. Collectively, these results support that 5-azacytidine treatment blunts the de-differentiating effects of PDGF-BB specifically through maintenance of PTEN expression.
Figure 3. Pro-differentiation effects of 5-azacytidine in vitro are mediated through PTEN induction.
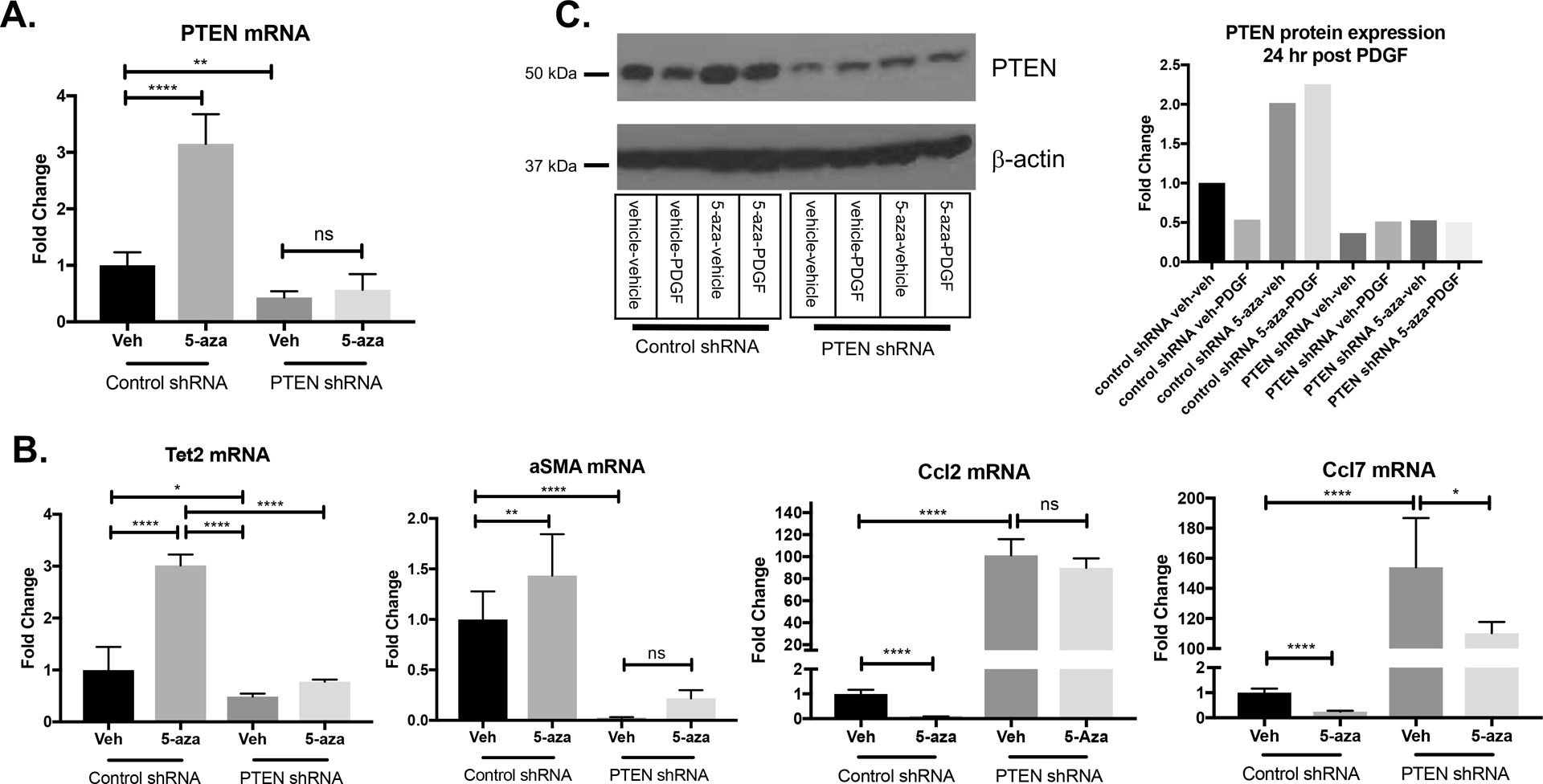
SMCs stably expressing control non-targeting shRNA or PTEN targeting shRNA were cultured in 0.1% CS EMEM. (A) SMCs were treated with 5-azacytadine for 24 hrs and RNA was harvested for RT-qPCR analysis of PTEN mRNA. 10 μM 5-azacytidine induced PTEN upregulation in control shRNA SMCs, but induction is blocked in SMCs stably expressing PTEN shRNA. ANOVA with Bonferroni post hoc; **p<0.01; ****p<0.0001. (B) SMCs were treated as in (A) and RT-qPCR used to analyze the indicated genes. Compared to controls, 5-azacytidine was unable to upregulate the differentiation-associated genes, Acta2 and Tet2, in PTEN depleted SMCs. In contrast, while 5-azacytidine blocked basal expression of the inflammatory genes Ccl2 and Ccl7 in control shRNA SMCs, the increased basal expression in PTEN depleted SMCs was not reversed. (C) SMCs were treated with 5-azacytidine and PDGF-BB for 24 hrs and protein was harvested for Western analysis of PTEN. Western blots (left) showing PTEN protein expression in control shRNA SMCs or PTEN KD SMCs following treatment with 5-azacytidine and PDGF-BB for 24 hrs. β-actin is shown as a loading control. (Right) Quantification of PTEN band intensity, normalized to β-actin expression. Fold change is expressed relative to control shRNA SMCs stimulated with vehicle.
5-azacytadine prevents vascular remodeling after carotid artery injury
To determine if systemic delivery of 5-azacytidine in vivo induces PTEN, WT mice were subjected to carotid artery ligation injury and treated with 5-azacytidine. As shown in Supplemental Figure V, compared to vehicle control, 5-azacytidine treatment resulted in increased PTEN mRNA expression in whole lung tissue and in injured carotid arteries. To determine whether 5-azacytidine treatment was able to reduce pathological vascular remodeling after injury, we subjected WT mice (Myh11-CreERT2;Rosa26-YFP) to carotid ligation injury, which results in pronounced neointima formation, and both medial and adventitial remodeling. Since de-differentiated SMCs are known to contribute to the pathological remodeling processes that occur in this model5,30, all mice carried an inducible SMC-specific YFP marker in order to fate-map SMCs in response to injury even if they lose all characteristics of SMCs (i.e. loss of SMC-specific contractile markers). Prior to injury, mice were injected with tamoxifen for 7 consecutive days to induce reporter knockin followed by a washout period. Following injury, mice were injected with 5-azacytidine i.p. at 2 mg/kg 3x weekly for 3 weeks; 5-azacytidine treatment began immediately following injury (5-azacytidine dose and route of administration was determined based on previous studies as outlined in detail in the “Complete Materials and Methods” in the Supplemental Materials). Compared to saline controls, hematoxylin and Movat’s pentachrome staining demonstrated that 5-azacytidine treatment promoted maintenance of vascular architecture and reduced neointima formation and adventitial remodeling after injury (Figure 4A, top and top middle left and middle panels). This was associated with reduced collagen deposition and perivascular fibrosis, as assessed by Masson’s staining (Figure 4A, top and top middle right panels). Quantification of percent stenosis and adventitial remodeling are shown in Figure 4B&C. These in vivo findings are consistent with our in vitro findings and support that 5-azacytidine reduces injury-mediated SMC de-differentiation to maintain vascular integrity.
Figure 4. 5-azacytidine blocks injury-induced pathological vascular remodeling and neointima formation in a PTEN-dependent manner.
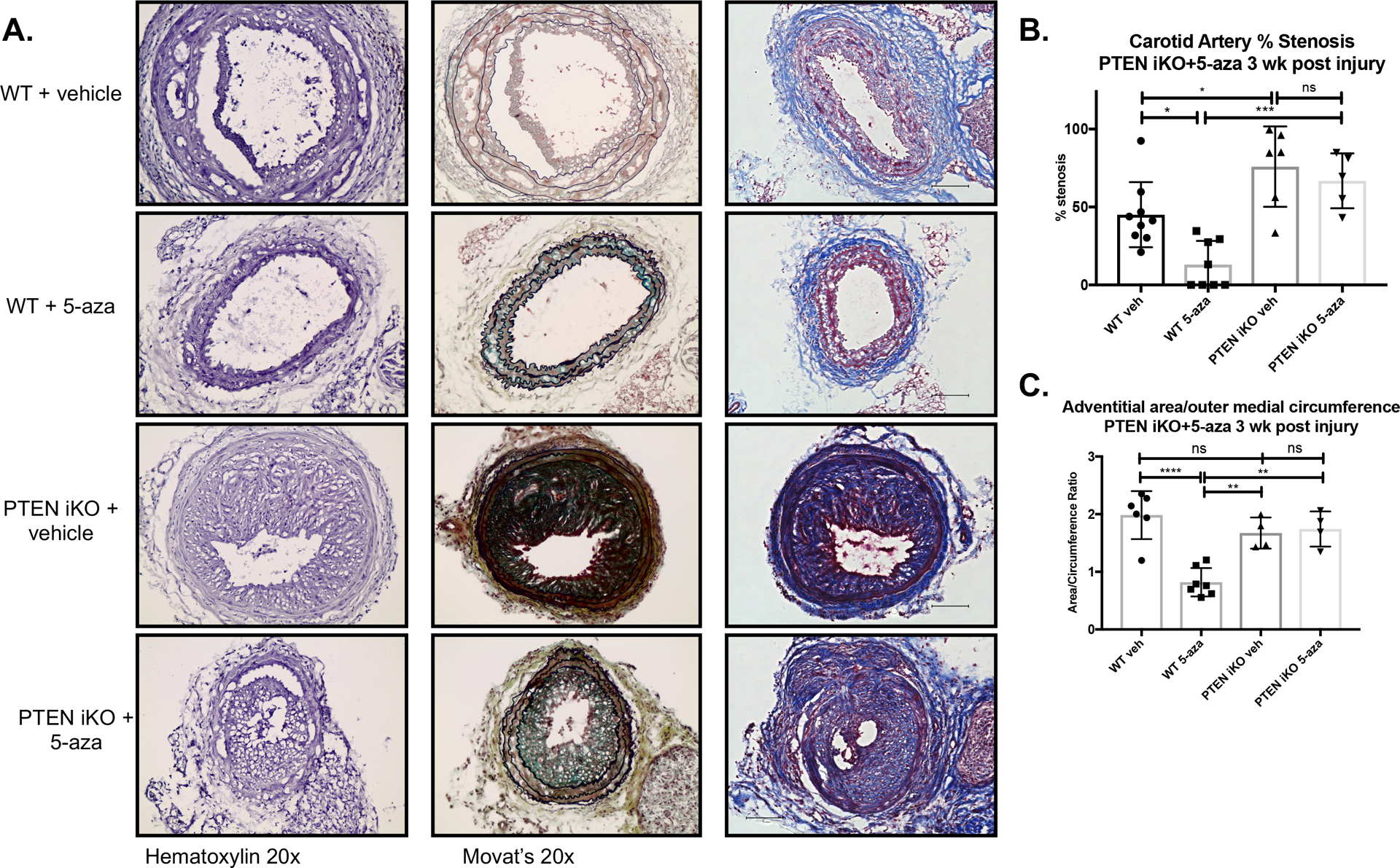
Male WT (Myh11-CreERT2;Rosa26-YFP) and SMC-specific PTEN iKO (PTENflox/flox-Myh11-CreERT2;Rosa26-YFP) mice received 1.5 μg tamoxifen injections i.p. for 7 consecutive days to induce PTEN knockout in PTEN iKO mice and YFP knock-in in both WT and PTEN iKO mice. The left carotid artery was ligated to induce vascular injury and neointima formation as described in Materials and Methods. Mice received 2 mg/kg 5-azacytidine or vehicle control i.p. injections 3x weekly for 3 weeks prior to tissue harvest. The right uninjured control and left injured carotid arteries were harvested three weeks post-injury, fixed in 4% PFA, and embedded in OCT. (A) Arterial sections were stained for hematoxylin (left panels), Movat’s pentachrome stain (middle panels), or Masson’s trichrome stain (right panels). Shown are representative stained images of injured left carotid arteries. (B&C) Percent stenosis (B) and adventitial remodeling (C) were measured using Image J, as described in Materials and Methods. (B) N=9 (WT vehicle), N=8 (WT 5-azacytidine), N=6 PTEN iKO vehicle), N=5 (PTEN iKO 5-azacytidine). (C) N=6 (WT vehicle), N=7 (WT 5-azacytidine), N=4 PTEN iKO vehicle), N=4 (PTEN iKO 5-azacytidine). ANOVA with Bonferroni post hoc. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001.
SMC-specific PTEN KO blocks protective effects of 5-azacytidine after carotid injury
In order to investigate whether the in vivo protective effects of 5-azacytidine were mediated through PTEN induction in SMCs, we utilized SMC-specific PTEN knockout (PTEN iKO) mice (PTENfl/fl-Myh11-CreERT2;Rosa26-YFP) compared to wild-type mice (PTEN WT) as controls. PTEN knockout and SMC YFP reporter knockin were induced as described above prior to performing carotid ligation injury. 5-azacytidine was injected at 2 mg/kg i.p. as described above. Consistent with our previous findings5, injury-mediated neointima formation was exacerbated in PTEN iKO mice compared to PTEN WT mice, indicating PTEN loss in SMCs is a significant contributor to pathological vascular remodeling (Figure 4A&B). While 5-azacytidine reduced vascular remodeling in PTEN WT mice, as measured by percent stenosis and total adventitial area, the anti-remodeling effects were blunted in PTEN iKO mice, which is consistent with our in vitro data and supports the conclusion that 5-azacytidine mediates pro-differentiation protective effects through its ability to induce PTEN expression (Figure 4A–C). Additionally, compared to uninjured carotid arteries (Supplemental Figure VI), 5-azacytidine treatment attenuated CD68+ macrophage accumulation in PTEN WT mice but it had no effect on macrophage accumulation in PTEN iKO mice (Figure 5). Negative control IgG stained images are shown in Supplemental Figure VIII. Injury-mediated de-differentiation of SMCs was analyzed by tracking YFP+ mature SMC-derived cells and coexpression of the SMC-specific contractile proteins, αSMA and SMMHC, markers of mature, differentiated SMCs. As we reported previously5,31, mature SMCs contribute to the majority of neointimal cells, as assessed by the large numbers of YFP+ intimal cells (Figures 5&6). However, compared to uninjured vessels (Supplemental Figures VI&VII), many YFP+ cells in both the media and neointima exhibited reduced levels of the contractile proteins αSMA and SMMHC. This was particularly apparent in injured arteries from PTEN iKO mice. Compared to saline controls, in PTEN WT mice treated with 5-azacytidine, expression of αSMA and SMMHC was maintained in YFP+ SMC-derived cells (Figures 5&6). In contrast, αSMA and SMMHC expression was significantly reduced in YFP+ cells that comprise a majority of the neointima in PTEN iKO mice, even after 5-azacytidine treatment (Figures 5&6). Collectively, these data support that 5-azacytidine-mediated PTEN induction maintains SMCs in a quiescent, anti-inflammatory, differentiated state, and that 5-azacytidine-mediated PTEN induction is critical to the protective, anti-remodeling effects of 5-azacytidine treatment.
Figure 5. 5-azacytidine reduces injury-mediated macrophage accumulation and enhances SMC contractile protein expression in a PTEN-dependent manner.
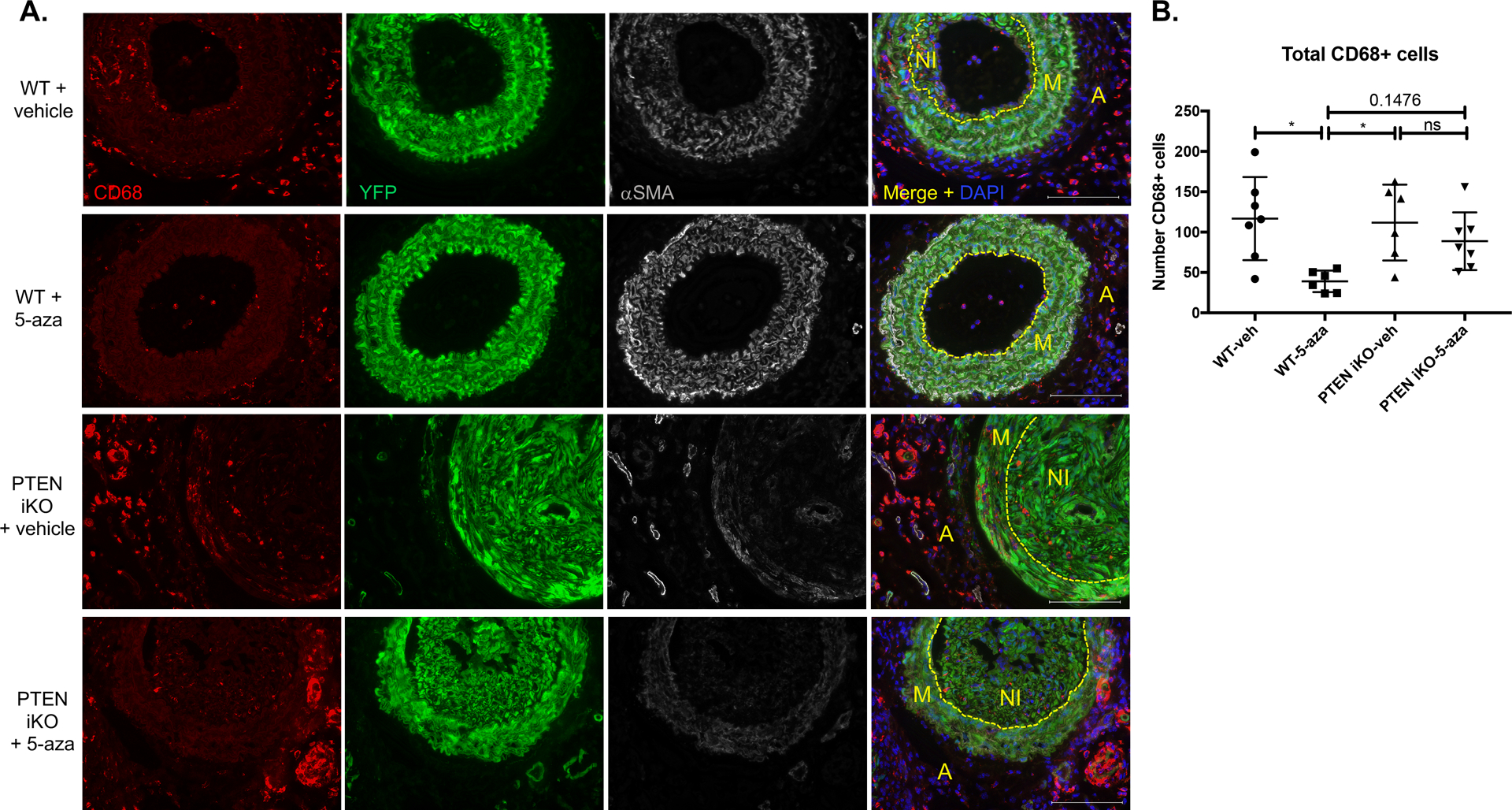
Male WT (Myh11-CreERT2;Rosa26-YFP) and SMC-specific PTEN iKO (PTENflox/flox-Myh11-CreERT2;Rosa26-YFP) mice received tamoxifen injections followed by carotid artery ligation, as described in Figure 6. Mice received 2 mg/kg 5- azacytidine or vehicle control i.p. injections 3x weekly for 3 weeks prior to tissue harvest. Left injured carotid arteries were harvested three weeks post-injury, fixed in 4% PFA, and embedded in OCT. Arterial sections were immunofluorescently stained for CD68 (red), YFP (green), and αSMA (white); nuclei were stained for DAPI (blue). Representative images from N=7 (WT vehicle), N=6 (WT 5-azacytidine), N=6 (PTEN iKO vehicle), and N=7 (PTEN iKO 5-azacytidine). Scale bars = 100μm. M = arterial media; A = arterial adventitia, NI = neointima. Dashed lines indicate the internal elastic laminae. Note decreased CD68+ macrophage accumulation and increased αSMA expression in injured arteries from WT mice treated with 5-azacytidine, but not from PTEN iKO mice treated with 5-azacytidine. (B) Total CD68+ macrophages were counted from stained slides from each group. N=7 (WT vehicle), N=6 (WT 5-azacytidine), N=6 (PTEN iKO vehicle), and N=7 (PTEN iKO 5-azacytidine). ANOVA with Bonferroni post hoc. *p<0.05.
Figure 6. 5-azacytidine enhances SMC contractile protein expression in a PTEN-dependent manner.
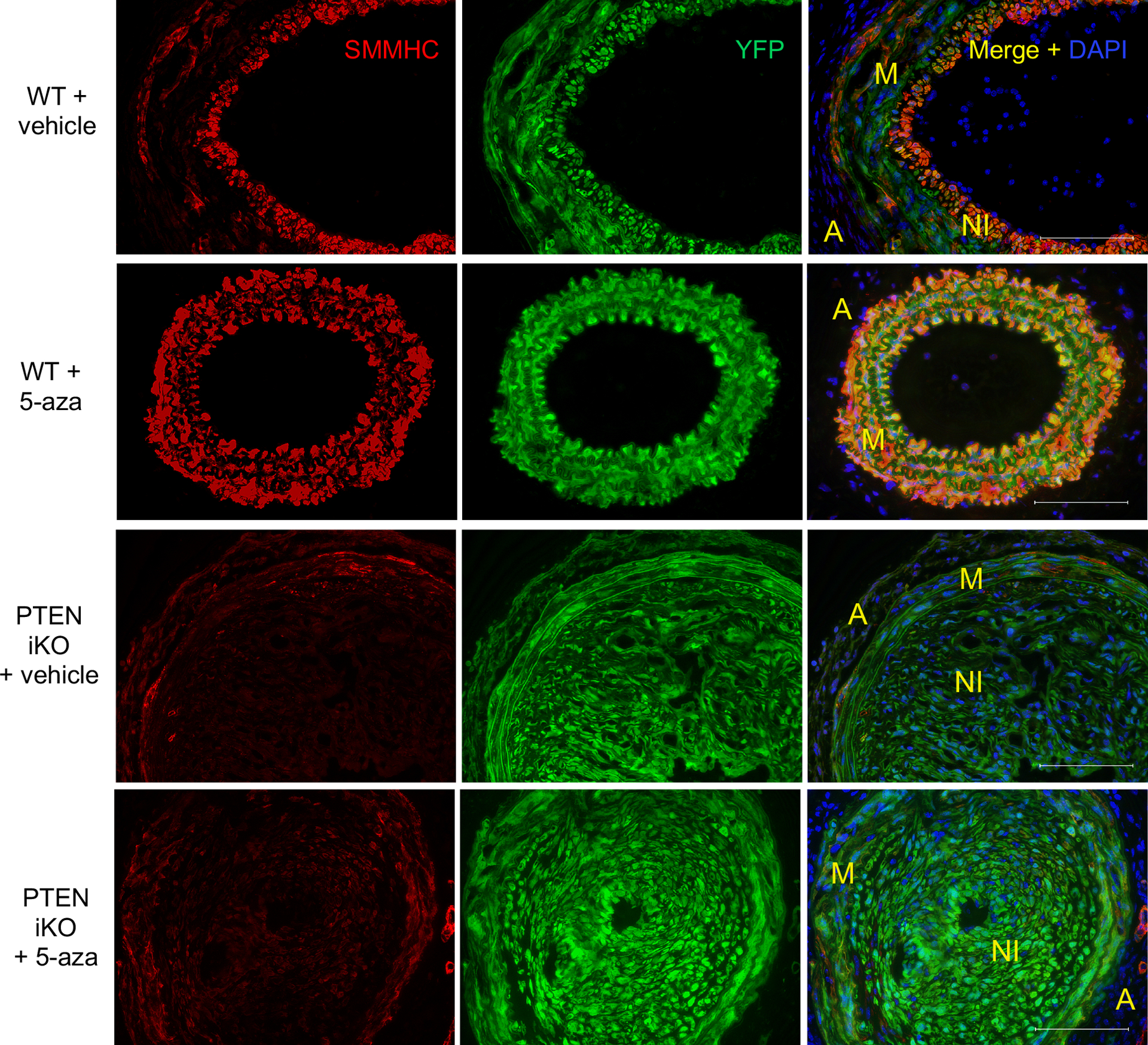
WT and PTEN iKO mice were treated as described in Figures 6&7. Injured carotid arteries were harvested 3-weeks post-injury and sections were immunofluorescently stained for YFP (green) and SMMHC (red); nuclei were stained for DAPI (blue). Representative images from N=7 (WT vehicle), N=6 (WT 5-azacytidine), N=6 (PTEN iKO vehicle), and N=7 (PTEN iKO 5-azacytidine). Scale bars = 100μm. M = arterial media; A = arterial adventitia, NI = neointima. Note increased SMMHC expression in injured arteries from WT mice treated with 5-azacytidine, but not from PTEN iKO mice treated with 5-azacytidine.
Discussion
The concept of SMC phenotypic modulation after vascular injury or during disease progression is well recognized, and SMCs are known to contribute to the pathological changes that occur by transitioning towards a proliferative, pro-inflammatory, pro-fibrotic phenotype3,30,32. Our previous research demonstrates that inactivation of PTEN is associated with SMC activation/de-differentiation and vascular disease progression through both phosphatase-dependent and -independent effects5,6,8. This suggests that PTEN inhibits SMC de-differentiation by antagonizing multiple pathological pathways, and implicates loss of PTEN as a major mechanism facilitating disease progression. Based on our previous findings9,11, in this study, we hypothesized that pharmacologic PTEN upregulation is a novel approach to inhibit pathological vascular remodeling. To address this, we performed an unbiased, high-throughput compound screen (HTS) to identify compounds with the potential to upregulate PTEN expression in SMCs. Results from the HTS and confirmatory in vitro testing identified 5-azacytidine, a potent DNMT1 inhibitor that acts as a DNA hypomethylating agent, as a compound with the potential to induce PTEN expression.
Epigenetic mechanisms, as a cause of cardiovascular disease, have received increased interest and provide an entirely novel paradigm for these diseases33–35. Three primary mechanisms govern gene expression through epigenetic mechanisms, DNA methylation, noncoding RNAs, and histone modification. Methylation of CpG islands within regulatory regions of DNA is regulated by DNA methyltransferases (DNMT1,3a,3b) and methylcytosine deoxygenases (TET1–3). These enzymes play an important role in controlling transcriptional activity by regulating chromatin accessibility to transcription factors36. While generally associated with gene repression, emerging research indicates that DNA methylation can also affect the activity of transcription factors regulating gene expression37. Epigenetic mechanisms play a critical role in SMC differentiation3,38,39, as TET2-mediated contractile gene promoter DNA hypomethylation28 as well as acetylation of histones flanking CArG elements of SMC genes promote chromatin accessibility and contractile gene expression38. Changes in overall DNA methylation patterns have been defined in the setting of atherosclerosis40–42 and 5-azacytidine treatment has been reported to be protective in mouse models of vascular disease22,43. 5-azacytidine (Vidaza) and the closely related 5-aza-2’-deoxycytidine (Decitabine) are currently approved for use in humans for treating several forms of myelodysplastic syndrome, acute myeloid leukemia, and chronic myelomonocytic leukemia44. As such, treatment with 5-azacytidine may represent a novel, clinically translatable approach to treat vascular disease. 5-azacytidine-mediated PTEN upregulation has previously been investigated in the setting of cancer, where hypermethylation of tumor suppressor gene promoters is known to be a key contributor to tumor progression13,14,45. Additionally, a recent report demonstrated that PTEN promoter methylation and subsequent reduction in PTEN expression occurs in hypoxia-induced pulmonary hypertension, and that Decitabine treatment was able to block PTEN promoter methylation and partially rescue PTEN expression23.
Here, we demonstrate that 5-azacytidine treatment causes increased activity at the PTEN promoter, leading to increased PTEN mRNA and protein expression in basal, unstimulated SMCs and maintenance of PTEN expression in SMCs stimulated with PDGF-BB. Similarly, another DNMT1 inhibitor, 5-aza-2’-deoxycytidine, exerted similar effects on PTEN mRNA strengthening the conclusion that PTEN promoter hypomethlylation is the mechanism of action of these compounds. Additionally, we report that 5-azacytidine treatment is able to upregulate expression of genes associated with a quiescent, differentiated SMC phenotype, and block the de-differentiating, pro-inflammatory effects of PDGF-BB stimulation. Furthermore, using both in vitro PTEN knockdown and in vivo SMC-specific PTEN knockout studies, we show that the pro-differentiation, anti-remodeling effects of 5-azacytidine are significantly reduced in the setting of SMC PTEN depletion. This suggests that 5-azacytidine largely mediates its protective effects in the vasculature via induction of PTEN and maintenance of a differentiated SMC phenotype. Moving forward, investigation of sequence-specific DNA methylation in the PTEN promoter to determine regions that are hypermethylated after vascular injury/PDGF-BB stimulation and hypomethylated after treatment with 5-azacytidine may identify important regulatory sites that affect PTEN expression in SMCs.
These findings could have translational significance as loss of SMC homeostasis and SMC activation are known to contribute to a variety of vascular pathologies, such as restenosis and atherosclerosis. While 5-azacytidine is currently approved for use in humans, we appreciate that systemic delivery of a potent hypomethylating agent may cause unintended off-target effects. Localized drug delivery, perhaps in the form of a drug-eluting stent (DES), could be a viable approach to treat patients. Currently, FDA-approved DES are based on derivatives of rapamycin and paclitaxel, both general cell cycle inhibitors. 5-azacytidine may offer an advantage over such compounds because in addition to reducing SMC proliferation and inflammation, it promotes a differentiated SMC phenotype via PTEN induction and PTEN-mediated transcriptional effects on SMC contractile genes. Consequently, this would reduce the development of restenosis caused by SMC activation thus maintaining vessel wall integrity and function.
Published studies from our laboratory suggest that systemic PTEN elevation has an anti-fibrotic effect in the vasculature11. Itraconazole was identified as a hit compound in our HTS and its ability to induce PTEN expression was validated in vitro. Interestingly, itraconazole was also identified by another group in a separate high-throughput compound screen as an anti-fibrotic compound with the potential to inhibit rat-derived hepatic stellate cell differentiation towards a myofibroblast-like phenotype following TGF-β stimulation46. DNMT1-mediated hypermethylation of the PTEN promoter has been shown to contribute to hepatic stellate cell activation and liver fibrogenesis in rats18. Together, these data indicate that itraconazole or 5-azacytidine-mediated PTEN upregulation may play an anti-fibrotic role in multiple organ systems. Further investigation into the role of itraconazole-mediated PTEN upregulation on anti-fibrotic effects appears warranted based on these results.
PTEN is most commonly studied in the context of its cytosolic lipid phosphatase activity. Indeed, a recent report identified indole-3-carbinol (I3C) treatment as a pharmacological method to increase PTEN’s lipid phosphatase activity at the plasma membrane47. I3C was tested in our HTS, and while it caused a slight upregulation in mCherry expression, it did not reach our >2.0-fold induction threshold (data not shown). This is consistent with the previous report, which indicated that I3C reactivates PTEN phosphatase activity through alterations in subcellular localization rather than overall expression levels. However, recent studies have elucidated an important role for nuclear PTEN, as it has been shown to interact directly with transcriptional machinery on chromatin to alter RNA Pol II phosphorylation status and regulate gene expression48,49. Our previous report demonstrated that nuclear PTEN plays an essential role in maintainence of a differentiated SMC phenotype through interactions with SRF and myocardin6. Additionally, we reported that overall PTEN expression and levels of nuclear PTEN are depleted following vascular injury or in response to other de-differentiating stimuli6. We have chosen to focus on pharmacologic PTEN upregulation due to the fact that increasing overall PTEN expression within the cell may preserve the phosphatase-dependent cytosolic and phosphatase-independent nuclear effects of PTEN, both of which are important in inhibiting SMC phenotypic transitions. Further studies into subcellular PTEN localization following 5-azacytidine treatment are planned, with the expectation that increasing overall PTEN expression will lead to a corresponding increase in PTEN nuclear localization.
Interestingly, in contrast to WT SMCs, we found that 5-azacytidine is unable to upregulate TET2 in the setting of PTEN depletion, which may place PTEN upstream of TET2 regulation, potentially via phosphatase-dependent Akt/mTOR inhibition. Previous studies have clearly demonstrated that TET2 is a master regulator of SMC phenotype, through regulation of chromatin accessibility at CArG boxes in SMC contractile gene promoters by altering histone methylation status28. H3K4 di-methylation has been shown to be a SMC-lineage mark that is used as a docking site by myocardin to facilitate contractile gene expression38,50. Other groups have proposed that a myocardin accessory protein facilitates myocardin docking to these H3K4 di-methylated histones38. Our data suggest that PTEN may function as that myocardin accessory protein, as we have previously shown the formation of a PTEN-myocardin-SRF complex in the nucleus of differentiated SMCs6. This raises an intriguing possibility wherein PTEN and TET2 work in concert to maintain a differentiated SMC phenotype by regulating the epigenetic landscape and transcription factor interaction at contractile gene promoters. In this proposed model, PTEN facilitates expression of TET2, which consequently promotes DNA hypomethylation and alteration of CArG box histone methylation to promote chromatin accessibility. In addition, PTEN interaction with myocardin-SRF would then drive myocardin occupancy at these accessible CArG elements, resulting in contractile gene expression and maintenance of SMC-differentiation.
To summarize, using an unbiased, high-throughput compound screening approach, we identified the DNA hypomethylating agent, 5-azacytidine, as a direct inducer of PTEN expression. As DNA hypermethylation is associated with the progression of diseases such as atherosclerosis40–42, PTEN promoter hypermethylation represents a potential potent mechanism to repress PTEN expression in the setting of cardiovascular disease. Our data support the concept that pharmacologic PTEN upregulation via promoter demethylation represents a novel therapeutic approach to reduce pathological vascular remodeling.
Supplementary Material
Highlights.
Unbiased high throughput drug screen and in vitro confirmation identified 5-azacytidine as a compound that causes PTEN upregulation.
5-azacytidine treatment in vitro increases expression of SMC differentiation markers and blocks PDGF-induced SMC de-differentiation in a PTEN-dependent manner.
Treatment with 5-azacytidine in vivo blocks neointima formation, adventitial remodeling and immune cell recruitment while maintaining SMC contractile gene expression following carotid ligation.
SMC-specific PTEN knockout abolishes the protective effects of 5-azacytidine treatment in vivo, suggesting that 5-azacytidine mediates its protective effects via PTEN upregulation.
Acknowledgments
Generation of the PTEN promoter-reporter construct and the high throughput screen was conducted in collaboration with Dr. Daniel LaBarbera’s group at the School of Pharmacy High Throughput Screening and Chemical Biology core at the University of Colorado Anschutz Medical Campus (UCAMC). The cell-based screening system used for the screen was developed in collaboration with Dr. Keith Koch in the Consortium for Fibrosis Research & Translation at the UCAMC. The contents of this manuscript are solely the responsibility of the authors and do not necessarily represent the official views of the NIH.
KAS and MCMW-E designed the studies. KAS, SL, and MFM performed experiments. SL assisted with mouse surgeries and tissue harvest. LL, QZ, BE, KK, and DL assisted with the design and execution of the high throughput screen. MFM managed mouse colonies associated with this project and served as lab manager for the MCMW-E lab. KAS and MCMW-E analyzed and interpreted the experimental data. KAS and MCMW-E wrote the manuscript. AJJ, AMD, KSM, and RAN edited the manuscript.
Sources of Funding
The work was supported by grants R01 HL121877 and R01 HL123616 from the National Heart, Lung, and Blood Institute, NIH and SOMTR CFReT Pilot Award to MCMW-E, grant 1F31HL147393 from the National Heart, Lung, and Blood Institute, NIH to KAS and grant 18POST34030397 from the American Heart Association to SL.
Abbreviations:
- SMC
vascular smooth muscle cell
- αSMA/Acta2
alpha-smooth muscle actin
- SMMHC/Myh11
smooth muscle myosin heavy chain
- PTEN
phosphatase and tensin homolog
- SRF
serum response factor
- HTS
high throughput screen
- DNMT1
DNA methyltransferase-1
- FACS
fluorescence-activated cell sorting
- GFP
green fluorescent protein
- DMEM
Dulbecco’s Modified Eagle’s Medium
- MEM
Minimum Essential Medium
- TET2
Tet methylcytosine dioxygenase 2
- EdU
5-ethynyl-2’-deoxyuridine
- YFP
yellow fluorescent protein
- OCT
Optimal Cutting Temperature compound
- WT
wild type
- PTEN iKO
inducible SMC-specific PTEN knockout
- PI3K
Phosphoinositide 3-kinase
- Akt
Protein kinase B
- PDGF
Platelet derived growth factor
- HEK293
Human embryonic kidney 293 cells
- DMSO
Dimethylsulfoxide
- shRNA
Short hairpin RNA
- RT-qPCR
Quantitative reverse transcription PCR
- Ccl2
C-C motif chemokine ligand 2
- Ccl7
C-C motif chemokine ligand 7
- TGF-β
Transforming growth factor beta
- RNA Pol II
RNA polymerase II
- H3K4
Histone H3 lysine K4
Footnotes
Disclosures
None.
References
- 1.Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature. 2011;473:317–325. [DOI] [PubMed] [Google Scholar]
- 2.Jukema JW, Verschuren JJW, Ahmed TAN, Quax PHA. Restenosis after PCI. Part 1: pathophysiology and risk factors. Nat Rev Cardiol 2011 91. 2011;9:53–62. [DOI] [PubMed] [Google Scholar]
- 3.Alexander MR, Owens GK. Epigenetic Control of Smooth Muscle Cell Differentiation and Phenotypic Switching in Vascular Development and Disease. Annu Rev Physiol. 2012;74:13–40. [DOI] [PubMed] [Google Scholar]
- 4.Majesky MW. Vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2016;36:e82–e86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nemenoff RA, Horita H, Ostriker AC, Furgeson SB, Simpson PA, VanPutten V, Crossno J, Offermanns S, Weiser-Evans MCM. SDF-1α induction in mature smooth muscle cells by inactivation of PTEN is a critical mediator of exacerbated injury-induced neointima formation. Arterioscler Thromb Vasc Biol. 2011;31:1300–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Horita H, Wysoczynski CL, Walker LA, Moulton KS, Li M, Ostriker A, Tucker R, McKinsey TA, Churchill MEA, Nemenoff RA, Weiser-Evans MCM. Nuclear PTEN functions as an essential regulator of SRF-dependent transcription to control smooth muscle differentiation. Nat Commun. 2016;7:10830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Furgeson SB, Simpson PA, Park I, VanPutten V, Horita H, Kontos CD, Nemenoff RA, Weiser-Evans MCM. Inactivation of the tumour suppressor, PTEN, in smooth muscle promotes a pro-inflammatory phenotype and enhances neointima formation. Cardiovasc Res. 2010;86:274–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nemenoff RA, Simpson PA, Furgeson SB, Kaplan-Albuquerque N, Crossno J, Garl PJ, Cooper J, Weiser-Evans MCM. Targeted deletion of PTEN in smooth muscle cells results in vascular remodeling and recruitment of progenitor cells through induction of stromal cell-derived factor-1α. Circ Res. 2008;102:1036–1045. [DOI] [PubMed] [Google Scholar]
- 9.Moulton KS, Li M, Strand K, Burgett S, McClatchey P, Tucker R, Furgeson SB, Lu S, Kirkpatrick B, Cleveland JC, Nemenoff RA, Ambardekar AV., Weiser-Evans MCM. PTEN deficiency promotes pathological vascular remodeling of human coronary arteries. JCI Insight. 2018;3:e97228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Cao I, Song MS, Hobbs RM, Laurent G, Giorgi C, De Boer VCJ, Anastasiou D, Ito K, Sasaki AT, Rameh L, Carracedo A, Vander Heiden MG, Cantley LC, Pinton P, Haigis MC, Pandolfi PP. Systemic elevation of PTEN induces a tumor-suppressive metabolic state. Cell. 2012;149:49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lu S, Strand KA, Mutryn MF, Tucker RM, Jolly AJ, Furgeson SB, Moulton KS, Nemenoff RA, Weiser-Evans MCM. PTEN (Phosphatase and Tensin Homolog) Protects Against Ang II (Angiotensin II)-Induced Pathological Vascular Fibrosis and Remodeling—Brief Report. Arterioscler Thromb Vasc Biol. 2020;40:394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ma SC, Zhang HP, Jiao Y, Wang YH, Zhang H, Yang XL, Yang AN, Jiang YD. Homocysteine-induced proliferation of vascular smooth muscle cells occurs via PTEN hypermethylation and is mitigated by Resveratrol. Mol Med Rep. 2018;17:5312–5319. [DOI] [PubMed] [Google Scholar]
- 13.Goel A, Arnold CN, Niedzwiecki D, Carethers JM, Dowell JM, Wasserman L, Compton C, Mayer RJ, Bertagnolli MM, Boland CR. Frequent Inactivation of PTEN by Promoter Hypermethylation in Microsatellite Instability-High Sporadic Colorectal Cancers. Cancer Res. 2004;64:3014–3021. [DOI] [PubMed] [Google Scholar]
- 14.Mirmohammadsadegh A, Marini A, Nambiar S, Hassan M, Tannapfel A, Ruzicka T, Hengge UR. Epigenetic silencing of the PTEN gene in melanoma. Cancer Res. 2006;66:6546–6552. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, Hadley C, Jackson IL, Zhang Y, Zhang A, Spasojevic I, Haberle IB, Vujaskovic Z. Hypo-CpG methylation controls PTEN expression and cell apoptosis in irradiated lung. Free Radic Res. 2016;50:875–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Song D, Ni J, Xie H, Ding M, Wang J. DNA demethylation in the PTEN gene promoter induced by 5-azacytidine activates PTEN expression in the MG-63 human osteosarcoma cell line. Exp Ther Med. 2014;7:1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J, Gong P, Lyu X, Yao K, Li X, Peng H. Aberrant CpG island methylation of PTEN is an early event in nasopharyngeal carcinoma and a potential diagnostic biomarker. Oncol Rep. 2014;31:2206–2212. [DOI] [PubMed] [Google Scholar]
- 18.Bian EB, Huang C, Ma TT, Tao H, Zhang H, Cheng C, Lv XW, Li J. DNMT1-mediated PTEN hypermethylation confers hepatic stellate cell activation and liver fibrogenesis in rats. Toxicol Appl Pharmacol. 2012;264:13–22. [DOI] [PubMed] [Google Scholar]
- 19.Ning Y, Huang H, Dong Y, Sun Q, Zhang W, Xu W, Li Q. 5-Aza-2’-deoxycytidine inhibited PDGF-induced rat airway smooth muscle cell phenotypic switching. Arch Toxicol. 2013;87:871–881. [DOI] [PubMed] [Google Scholar]
- 20.Huo X, Li Z, Zhang S, Li C, Guo M, Lu J, Lv J, Du X, Chen Z. Analysis of the expression level and methylation of tumor protein p53, phosphatase and tensin homolog and muts homolog 2 in N-methyl-N-nitrosourea-induced thymic lymphoma in C57BL/6 mice. Oncol Lett. 2017;14:4339–4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma SC, Cao JC, Zhang HP, Jiao Y, Zhang H, He YY, Wang YH, Yang XL, Yang AN, Tian J, Zhang MH, Yang XM, Lu GJ, Jin SJ, Jia YX, Jiang YD. Aberrant promoter methylation of multiple genes in VSMC proliferation induced by Hcy. Mol Med Rep. 2017;16:7775–7783. [DOI] [PubMed] [Google Scholar]
- 22.Zhuang J, Luan P, Li H, Wang K, Zhang P, Xu Y, Peng W. The yin-yang dynamics of DNA methylation is the key regulator for smooth muscle cell phenotype switch and vascular remodeling. Arterioscler Thromb Vasc Biol. 2017;37:84–97. [DOI] [PubMed] [Google Scholar]
- 23.Xing XQ, Li B, Xu SL, Zhang CF, Liu J, Deng YS, Yang J. 5-Aza-2′-deoxycytidine, a DNA methylation inhibitor, attenuates hypoxic pulmonary hypertension via demethylation of the PTEN promoter. Eur J Pharmacol. 2019;855:227–234. [DOI] [PubMed] [Google Scholar]
- 24.Abraham AD, Esquer H, Zhou Q, Tomlinson N, Hamill BD, Abbott JM, Li L, Pike LA, Rinaldetti S, Ramirez DA, Lunghofer PJ, Gomez JD, Schaack J, Nemkov T, D’Alessandro A, Hansen KC, Gustafson DL, Messersmith WA, LaBarbera DV. Drug Design Targeting T-Cell Factor-Driven Epithelial–Mesenchymal Transition as a Therapeutic Strategy for Colorectal Cancer. J Med Chem. 2019;62:10182–10203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Chung, Oldenburg. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. [DOI] [PubMed] [Google Scholar]
- 26.Mourani PM, Garl PJ, Wenzlau JM, Carpenter TC, Stenmark KR, Weiser-Evans MCM. Unique, Highly Proliferative Growth Phenotype Expressed by Embryonic and Neointimal Smooth Muscle Cells is Driven by Constitutive Akt, mTOR, and p70S6K Signaling and is Actively Repressed by PTEN. Circulation. 2004;109:1299–1306. [DOI] [PubMed] [Google Scholar]
- 27.Yoo BH, Axlund SD, Kabos P, Reid BG, Schaack J, Sartorius CA, LaBarbera DV. A High-Content Assay to Identify Small-Molecule Modulators of a Cancer Stem Cell Population in Luminal Breast Cancer. J Biomol Screen. 2012;17:1211–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu R, Jin Y, Tang WH, Qin L, Zhang X, Tellides G, Hwa J, Yu J, Martin KA. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity. Circulation. 2013;128:2047–2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Majesky MW, Reidy MA, Bowen-Pope DF, Hart CE, Wilcox JN, Schwartz SM. PDGF ligand and receptor gene expression during repair of arterial injury. J Cell Biol. 1990;111:2149–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Owens GK, Kumar MS, Wamhoff BR. Molecular Regulation of Vascular Smooth Muscle Cell Differentiation in Development and Disease. Physiol Rev. 2004;84:767–801. [DOI] [PubMed] [Google Scholar]
- 31.Majesky MW, Horita H, Ostriker A, Lu S, Regan JN, Bagchi A, Dong XR, Poczobutt J, Nemenoff RA, Weiser-Evans MCM. Differentiated Smooth Muscle Cells Generate a Subpopulation of Resident Vascular Progenitor Cells in the Adventitia Regulated by Klf4 Novelty and Significance. Circ Res. 2017;120:296–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Owens GK. Molecular control of vascular smooth muscle cell differentiation and phenotypic plasticity. In: Novartis Foundation Symposium. 2007;283:174–191. [DOI] [PubMed] [Google Scholar]
- 33.Schiattarella GG, Madonna R, Van Linthout S, Thum T, Schulz R, Ferdinandy P, Perrino C. Epigenetic modulation of vascular diseases: Assessing the evidence and exploring the opportunities. Vascul Pharmacol. 2018;107:43–52. [DOI] [PubMed] [Google Scholar]
- 34.Al-Hasani K, Mathiyalagan P, El-Osta A. Epigenetics, cardiovascular disease, and cellular reprogramming. J Mol Cell Cardiol. 2019;128:129–133. [DOI] [PubMed] [Google Scholar]
- 35.Rosa-Garrido M, Chapski DJ, Vondriska TM. Epigenomes in Cardiovascular Disease. Circ Res. 2018;122:1586–1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Feinberg AP, Tycko B. The history of cancer epigenetics. Nat Rev Cancer. 2004;4:143–153. [DOI] [PubMed] [Google Scholar]
- 37.Helling BA, Gerber AN, Kadiyala V, Sasse SK, Pedersen BS, Sparks L, Nakano Y, Okamoto T, Evans CM, Yang IV., Schwartz DA. Regulation of MUC5B expression in idiopathic pulmonary fibrosis. Am J Respir Cell Mol Biol. 2017;57:91–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.McDonald OG, Wamhoff BR, Hoofnagle MH, Owens GK. Control of SRF binding to CArG box chromatin regulates smooth muscle gene expression in vivo. J Clin Invest. 2006;116:36–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gomez D, Swiatlowska P, Owens GK. Epigenetic control of SMC identity and lineage memory. Arter Thromb Vasc Biol. 2015;35:2508–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aavik E, Babu M, Ylä-Herttuala S. DNA methylation processes in atheosclerotic plaque. Atherosclerosis. 2019;281:168–179. [DOI] [PubMed] [Google Scholar]
- 41.Lacey M, Baribault C, Ehrlich KC, Ehrlich M. Atherosclerosis-associated differentially methylated regions can reflect the disease phenotype and are often at enhancers. Atherosclerosis. 2019;280:183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, Condom E, Ramírez-Ruz J, Gomez A, Gonçalves I, Moran S, Esteller M. DNA methylation map of human atherosclerosis. Circ Cardiovasc Genet. 2014;7:692–700. [DOI] [PubMed] [Google Scholar]
- 43.Zhong W, Li B, Xu Y, Yang P, Chen R, Wang Z, Shao C, Song J, Yan J. Hypermethylation of the Micro-RNA 145 Promoter Is the Key Regulator for NLRP3 Inflammasome-Induced Activation and Plaque Formation. JACC Basic to Transl Sci. 2018;3:604–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Derissen EJB, Beijnen JH, Schellens JHM. Concise Drug Review: Azacitidine and Decitabine. Oncologist. 2013;18:619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.García JM, Silva J, Peña C, Garcia V, Rodriguez R, Cruz MA, Cantos B, Provencio M, España P, Bonilla F. Promoter methylation of the PTEN gene is a common molecular change in breast cancer. Genes Chromosom Cancer. 2004;41:117–124. [DOI] [PubMed] [Google Scholar]
- 46.Bollong MJ, Yang B, Vergani N, Beyer BA, Chin EN, Zambaldo C, Wang D, Chatterjee AK, Lairson LL, Schultz PG. Small molecule-mediated inhibition of myofibroblast transdifferentiation for the treatment of fibrosis. Proc Natl Acad Sci U S A. 2017;114:4679–4684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee YR, Chen M, Lee JD, Zhang J, Lin SY, Fu TM, Chen H, Ishikawa T, Chiang SY, Katon J, Zhang Y, Shulga YV, Bester AC, Fung J, Monteleone E, Wan L, Shen C, Hsu CH, Papa A, Clohessy JG, Teruya-Feldstein J, Jain S, Wu H, Matesic L, Chen RH, Wei W, Pandolfi PP. Reactivation of PTEN tumor suppressor for cancer treatment through inhibition of a MYC-WWP1 inhibitory pathway. Science. 2019;364:eaau0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abbas A, Romigh T, Eng C. PTEN interacts with RNA polymerase II to dephosphorylate polymerase II C-terminal domain. Oncotarget. 2019;10:4951–4959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Steinbach N, Hasson D, Mathur D, Stratikopoulos EE, Sachidanandam R, Bernstein E, Parsons RE. PTEN interacts with the transcription machinery on chromatin and regulates RNA polymerase II-mediated transcription. Nucleic Acids Res. 2019;47:5573–5586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Spin JM, Maegdefessel L, Tsao PS. Vascular smooth muscle cell phenotypic plasticity: Focus on chromatin remodelling. Cardiovasc Res. 2012;95:147–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.