

Abstract
Due to their relatively large molecular sizes and delicate nature, biologic drugs such as peptides, proteins, and antibodies often require high and repeated dosing, which can cause undesired side effects and physical discomfort in patients and render many therapies inordinately expensive. To enhance the efficacy of biologic drugs, they could be encapsulated into polymeric hydrogel formulations to preserve their stability and help tune their release in the body to their most favorable profile of action for a given therapy. In this study, a series of injectable, thermoresponsive hydrogel formulations were evaluated as controlled delivery systems for various peptides and proteins, including insulin, Merck proprietary peptides (glucagon-like peptide analogue and modified insulin analogue), bovine serum albumin, and immunoglobulin G. These hydrogels were prepared using concentrated solutions of poly(lactide-co-glycolide)–block-poly(ethylene glycol)–block-poly(lactide-co-glycolide) (PLGA–PEG–PLGA), which can undergo temperature-induced sol–gel transitions and spontaneously solidify into hydrogels near the body temperature, serving as an in situ depot for sustained drug release. The thermoresponsiveness and gelation properties of these triblock copolymers were characterized by dynamic light scattering (DLS) and oscillatory rheology, respectively. The impact of different hydrogel-forming polymers on release kinetics was systematically investigated based on their hydrophobicity (LA/GA ratios), polymer concentrations (20, 25, and 30%), and phase stability. These hydrogels were able to release active peptides and proteins in a controlled manner from 4 to 35 days, depending on the polymer concentration, solubility nature, and molecular sizes of the cargoes. Biophysical studies via size exclusion chromatography (SEC) and circular dichroism (CD) indicated that the encapsulation and release did not adversely affect the protein conformation and stability. Finally, a selected PLGA–PEG–PLGA hydrogel system was further investigated by the encapsulation of a therapeutic glucagon-like peptide analogue and a modified insulin peptide analogue in diabetic mouse and minipig models for studies of glucose-lowering efficacy and pharmacokinetics, where superior sustained peptide release profiles and long-lasting glucose-lowering effects were observed in vivo without any significant tolerability issues compared to peptide solution controls. These results suggest the promise of developing injectable thermoresponsive hydrogel formulations for the tunable release of protein therapeutics to improve patient’s comfort, convenience, and compliance.
1. Introduction
Peptides and proteins now constitute a significant portion of therapeutics for various disease indications, owing to their favorable safety profile, target specificity, and pharmacokinetics (PK) compared to small-molecule drugs.1−5 However, the stability and conformational integrity of these molecules still remain a significant challenge for the discovery and development of peptide/protein therapeutics.6,7 Due to their complex, dynamic, and fragile three-dimensional structures, proteins are often susceptible to aggregation and denaturation in the presence of harsh proteolytic and chemical environment in the human body, which might require repetitive dosing for many biologics, potentially compromising the patient’s comfort and compliance. To this end, hydrogels have emerged as a promising class of drug delivery vehicles owing to their abilities to meet the critical delivery challenges:8−11 (a) protecting the delicate cargo over the extended period of therapy; (b) enabling the release of cargo in a tunable manner to maintain drug concentration within the therapeutic window; (c) serving as a drug depot that significantly minimizes the number/frequency of administration with lower side effects; (d) offering on-demand drug release in response to specific stimuli through highly tunable structures; and (e) enhancing biocompatibility with a suitable choice of materials.
In particular, temperature-sensitive hydrogels have attracted considerable interest for several therapeutic applications.12−15 The thermogelling polymers based on poly(N-isopropylacrylamide) and polyester block copolymers can remain in solution at low or room temperature but rapidly convert to solidifying gels upon injection at body temperature (37 °C).9,13,16−20 These temperature-sensitive materials would be amenable to incorporate biomacromolecules such as proteins via simple mixing and protect the cargoes from potential enzymatic degradation. Specifically, the solution of the polymer–protein mixture allows for easy injection via a syringe, and the rapid gelation upon injection at a targeted location would serve as a drug depot for long-term sustained delivery. The drug release kinetics can be efficiently controlled by the choice of a gel-forming polymer backbone with suitable amphiphilicity and biodegradability. Particularly, temperature-sensitive hydrogels based on poly(lactide-co-glycolide)-block-poly(ethylene glycol)-block-poly(lactide-co-glycolide) (PLGA–PEG–PLGA) have attracted considerable interest as a sustained drug delivery depot owing to their biocompatibility and promising safety profile.21−23 For example, ReGel is a thermogel formulation developed based on PLGA–PEG–PLGA systems and has been evaluated in clinical trials for the delivery of a poorly soluble drug, paclitaxel (OncoGel), as a local chemotherapy.15 In addition to the delivery of hydrophobic small-molecule drugs, peptides and proteins can also be entrapped in the hydrophilic domain of the hydrogels and release through diffusion from the network, through which the release of protein therapeutics, such as insulin,24 exenatide,25,26 liraglutide,13 and interleukin-2 (IL-2),27 has been evaluated previously. Despite the promise of these studies, there have been limited understandings in the interplay of these protein cargoes with the hydrogel systems during encapsulation and release, especially given the well-known acidic degradation byproducts of PLGA and the delicate nature of various proteins. Therefore, a fundamental study correlating the molecular and physicochemical characteristics of PLGA–PEG–PLGA hydrogels (hydrophobicity/solubility, gelation time, and mechanical strength) with the release kinetics and biophysical/conformational stability of protein cargoes in various sizes would provide valuable insight and understanding in the potential translation of such hydrogel systems into real clinical products of long-acting protein therapeutics.
In this work, PLGA–PEG–PLGA hydrogels were investigated for the sustained release of various biologic cargoes, including insulin, Merck proprietary peptides (glucagon-like peptide analogue and modified insulin analogue), bovine serum albumin (BSA), and immunoglobulin G (IgG) (Scheme 1). The effects of polymer concentrations, the hydrophobicity of two LA/GA block ratios, and the impact of polysaccharide-based excipients on the release kinetic of insulin were studied. Additionally, the impact of molecular weight and solubility of encapsulated macromolecular cargoes (insulin, BSA, and IgG) on the release rate from hydrogels was also assessed. The biophysical stability and conformational properties of these released proteins were confirmed via size exclusion chromatography (SEC) and circular dichroism (CD). Finally, the glucose-lowering efficacy and pharmacokinetics performance of selected PLGA–PEG–PLGA hydrogel formulations that contain Merck proprietary peptides (glucagon-like peptide analogue and modified insulin analogue) were evaluated in diabetic mouse and minipig models.
Scheme 1. (a) Structure of PLGA–PEG–PLGA Triblock Copolymer Used In This Study; (b) Thermogelation Mechanism of the Polymer Solution at an Elevated Temperature; (c) In Vivo Efficacy and Pharmacokinetics Studies of Therapeutic Peptides in Diabetic Animal Models.

2. Results
PLGA–PEG–PLGA triblock copolymers show a low critical gelation concentration (ca. 12–30 wt %) and have a low critical gelation temperature (ca. 25–37 °C), which render them promising materials for drug delivery applications.12,28 Moreover, the aqueous solution of these polymers shows a reversible temperature-sensitive hydrogel formation. At low temperature (ca. 4 °C), the polymer forms micelles in solution, where end PLGA blocks form the core and PEG blocks are exposed toward the aqueous phase (Scheme 1). At an elevated temperature (ca. 37 °C), the outer PEG blocks start dehydrating, leading to increased interactions between hydrophobic chains. At this stage, micellar particles interact and form aggregates leading to the formation of a percolated hydrogel network structure.12,29,30 In this study, the PLGA–PEG–PLGA polymers tested exhibited a low critical gelation temperature of approximately 34 °C, providing a flexible transition window for in vivo applications.
2.1. Thermoresponsive Properties of PLGA–PEG–PLGA Triblock Copolymers
To understand the changes in the co-assembly behavior of the polymers with temperature, we studied the sizes of micelles formed in diluted polymer solutions (0.1%) at different temperatures (Figure 1a,b). For 94/6–25 and 3/1–25% polymers, the initially formed micelle sizes were 24 and 43 nm, respectively, at 4 °C. No appreciable changes were observed in assembly sizes in both cases at temperatures up to 30 °C. However, when the temperature was increased to 37 °C, larger particle sizes were observed for both polymer systems (42 and 73 nm for 94/6 and 3/1 polymers, respectively; Figure 1a,b and Figure S1). As the gelation process is expected to generate networks of polymer threads, the size values at this step suggest a qualitative measure of the changes in the assembly properties at the diluted concentration to help understand the transformation from the micelle to gel network structure of concentrated polymer solutions.
Figure 1.

DLS measurements of 94/6 (a) and 3/1 (b) PLGA-PEG-PLGA (LA/GA ratio) polymer solutions (0.1%) showing the changes in particle sizes with temperature.
2.2. Rheological Behaviors of Hydrogels at Different Polymer Concentrations
Polymer solutions with varying concentrations and LA/GA ratios were subsequently studied using oscillatory rheology to understand their suitability as in situ drug depots. To this end, gelation time/kinetics and changes in viscoelastic properties as a function of time at 37 °C (Figure 2 and Supporting Information Figure S2) were studied. Specifically, the storage modulus (G′) and loss modulus (G″) were monitored, where the storage modulus (G′) represents the elastic/solid-like property and the loss modulus indicates (G″) the viscous/liquid-like property of polymeric materials.31 At low temperature, polymers in the buffer solution behaved more liquid-like as evidenced by the G″ > G′ values at the beginning of the experiment in all cases (Figure 2). However, G′ of the solutions rapidly increased when subjected to 37 °C incubation. This was attributed to the formation of hydrogels from the polymer solutions, and the kinetics could be quantified from the gelation time or gel point (tgel, defined as the temperature, where G′ = G″ and measured at the cross-over point; Figure 2).32
Figure 2.

(a–d) Moduli of PLGA–PEG–PLGA hydrogels at 37 °C for different polymer concentrations and LA/GA contents (abbreviated as LA/GA–polymer concentration); (e) summary of the storage, loss moduli, and gelation time for the experimented hydrogel systems at 37 °C. *Gelation time of 3/1–25% hydrogel is less than 2 s at 37 °C as gelation occurred during the temperature ramp step.
To explore the optimal polymer concentration, we studied the dynamic viscoelastic properties of hydrogels through observing tgel and changes in the G′ and G″ values. The tgel was expected to be lowered with increasing polymer concentration and hydrophobicity. It was expected that a higher polymer concentration would lower the gelation time. Indeed, the measured values of tgel were found to be 150, 90, and 80 s for polymer concentrations of 20, 25, and 30%, respectively (for LA/GA ratio 94/6). The moduli of the systems, G′ and G″, also followed a similar trend. At 37 °C, the G′ values sharply increased for all hydrogel precursor solutions, indicating rapid gelation. The storage moduli near the cross-over point (tgel) were 20, 95, and 164 Pa for 94/6–20, 94/6–25, and 94/6–30% polymer solutions, respectively. However, the 3/1–25% polymer system behaved differently with faster gelation (instantaneously formed hydrogel upon exposure to 37 °C; tgel could not be measured as the cross-over point was achieved very fast) and a higher G′ value (276 Pa at 90 s), which was counterintuitive as hydrogels formed by copolymers with lower LA/GA ratios are typically less stable due to weaker hydrophobic interactions.33 This could be potentially attributed to the local aggregation of the polymer that increased the viscosity and eventually phase-separated after 3 days of exposure at 37 °C (see Figure S3 in the Supporting Information). Nevertheless, the concentration-dependent increase in the mechanical strength of LA/GA 96/4 hydrogels is consistent with the existing reports in similar thermogel systems,14,34 suggesting this to be a facile approach in tailoring the mechanical properties and stability of the hydrogels.
2.3. Release Kinetics of Insulin from Temperature-Sensitive Hydrogels with Different Polymer Concentrations and LA/GA Contents
The release characteristics of various thermoresponsive gel systems were evaluated using insulin as a model molecule. Figure 3 illustrates the cumulative release of insulin over a period of 35 days. Initially, ∼20% of insulin was released within 2 days from all hydrogels irrespective of the polymer concentration and LA/GA content. This uncontrolled burst release behavior can be attributed to the loosely bound cargo molecules located at the vicinity of the hydrogel–buffer interface. In the second phase, the release of insulin became more gradual and showed concentration dependence for 94/6 hydrogels, with >90% of insulin being released from 20, 25, and 30% hydrogels within 14, 21, and 28 days, respectively. However, the 3/1–25% system showed a rather slow and sustained release behavior with only additional ∼25% of cargo release over a period of 17 days (from 38% at day 4 to 63% at day 21). For this system, insulin release was completed after 35 days. Interestingly, these release profiles can be explained based on the rheological properties of the hydrogels (Figure 2e). As the storage modulus gradually increases with the polymer concentration for 94/6 systems, the release kinetics becomes slower. However, the 3/1 system showed a significantly higher G′ value that directly influenced the release profile providing rather slower and sustained kinetics. It is important to note that the release mechanism of cargo molecules from the hydrogel systems was dictated by both diffusion and degradation kinetics of the polyester backbone of the polymers. While diffusion and slow hydrolytic degradation are the major mechanisms of insulin release for 94/6 systems, the anomalous slow release from the 3/1 system could be due to aggregation and local phase separation/precipitation that imparted greater hydrolytic resistance to the hydrogel. Comparable insulin release data was reported by Payyappilly et al. for thermoresponsive poly(ethylene glycol)–poly(e-caprolactone)–poly(ethylene glycol) (PEG–PCL–PEG) hydrogels,34 in which such dependence of insulin release kinetics on the polymer concentration and gel strength (i.e., storage modulus) was also observed. These results confirm that protein release kinetics from these hydrogels can be easily tuned, which provides insights into the design of hydrogels that meet the targeted therapeutic profile of a specific disease indication.
Figure 3.

Cumulative release kinetics of insulin from different PLGA–PEG–PLGA hydrogels (n = 3).
2.4. Stability and Biophysical Characterization of Insulin Released from Hydrogels
The released insulin was evaluated for structural stability with SEC and CD spectroscopy over the course of the release. Figure 4 demonstrates the SEC profiles of the released insulin, which are indicative of the possibility of any aggregation during the encapsulation and release process. Although there was some minor shift in retention time, which could be due to the drastic differences in insulin concentration, as well as the possible polymer residues present in the release samples, no significant aggregation was observed throughout the entire release period. The secondary structural property of the released insulin was further tested with CD spectroscopy (Figure 5). Insulin shows the CD spectra that are typical of an α-helical protein with negative bands occurring at 208 and 222 nm regions.35,36 As illustrated in Figure 5, no significant change of protein conformation was observed even after 35 days of release study. It was also confirmed in previous studies with other systems that the secondary conformation of insulin was maintained in comparison to that of native insulin (ca. up to 7 days) after releasing from the hydrogels.34,37 These results indicate that the majority of the encapsulated insulin retained the structural integrity and conformation through the entire course of release from the hydrogels, which could likely be attributed to the limited molecular motions/interactions and hydrophilic microenvironment within the hydrogels. Owing to the reasonable release duration and well-defined gelation kinetics of 94/6 hydrogels at a 25% concentration, this composition was employed in all further studies of the hydrogels.
Figure 4.
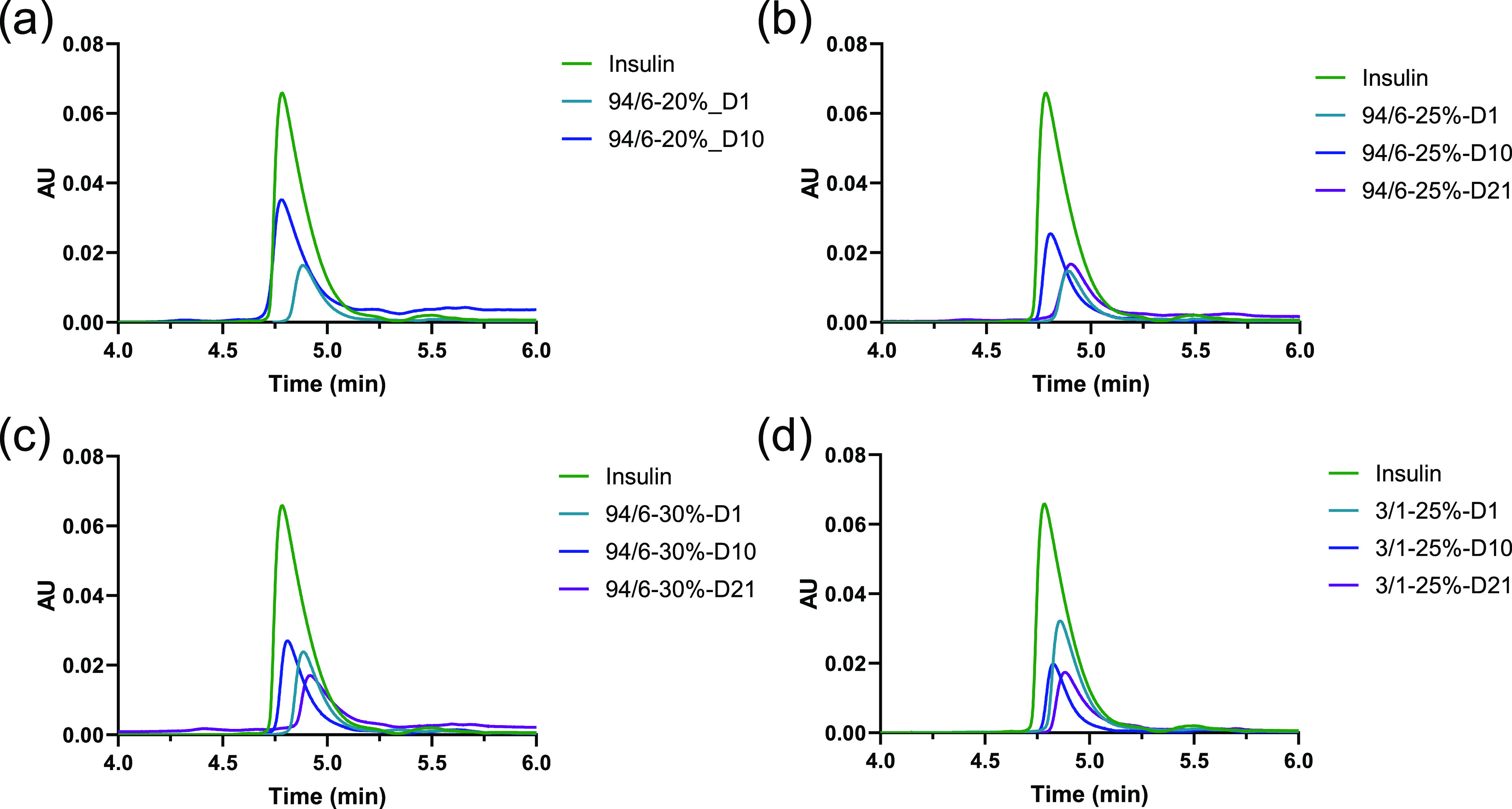
SEC of the released insulin at different time intervals from hydrogels: (a) 94/6–20%, (b) 94/6–25%, (c) 94/6–25%, and (d) 3/1–25%.
Figure 5.
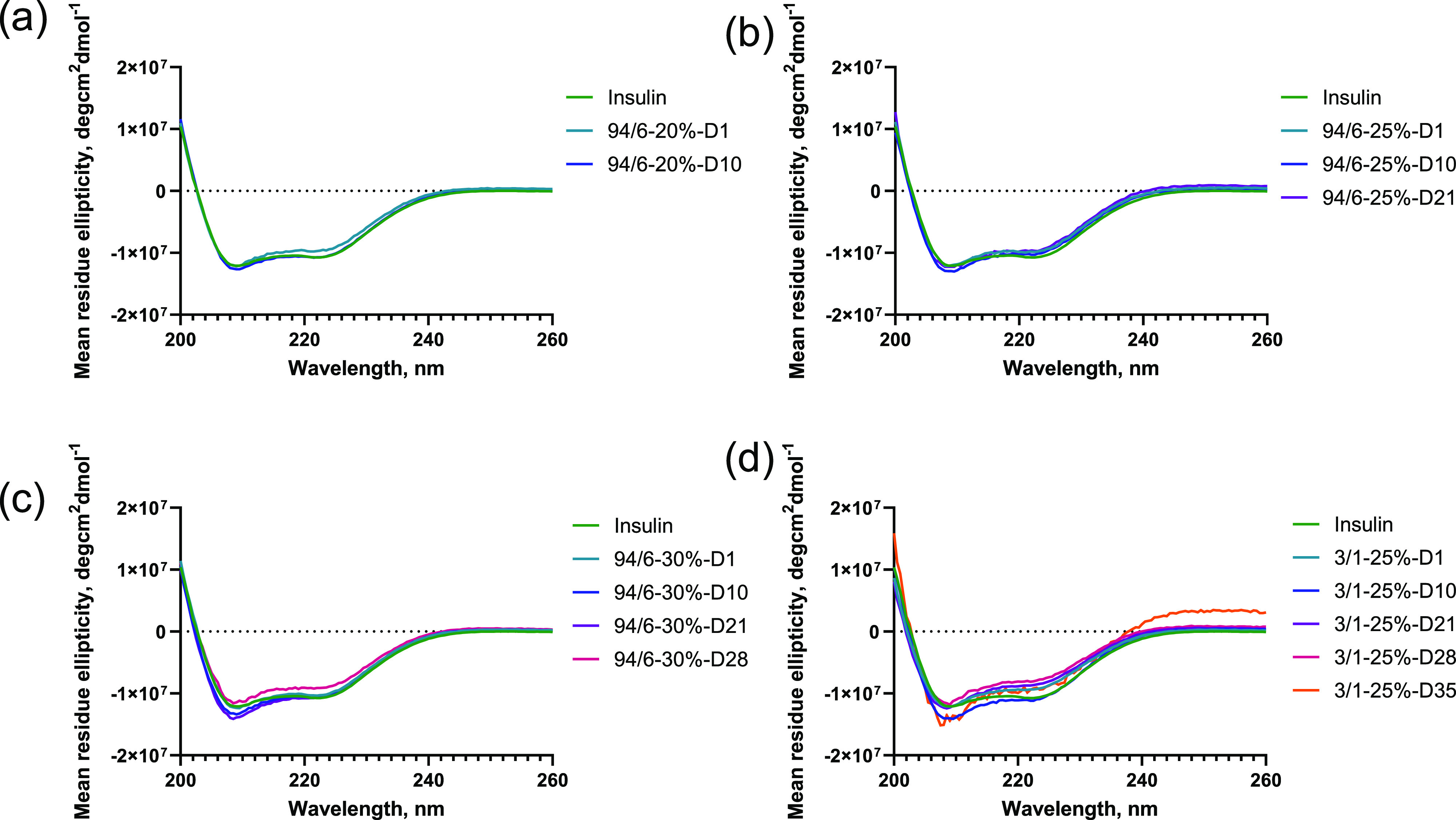
CD spectroscopy of the released insulin at different time intervals from hydrogels: (a) 94/6–20%, (b) 94/6–25%, (c) 94/6–25%, and (d) 3/1–25% (n = 3).
2.5. Effect of Excipients on Rheological Behaviors of Hydrogels
Sodium alginate (ALG), hyaluronic acid (HA), and hydroxypropyl methyl cellulose (HPMC) have been utilized in pharmaceutical formulations as thickeners, binders, and stabilizers for emulsions.38−43 It was reported that the addition of these polysaccharide excipients to poloxamer-based hydrogels can improve the mechanical strength and gel stability through potential hydrogen-bonding and hydrophobic interactions.41−43 To understand the applicability of these polysaccharide excipients to the PLGA–PEG–PLG systems, different 94/6–25% hydrogels were prepared with final concentrations of 0.75, 1, and 1% of ALG, HA, and HPMC, respectively. For ALG, the lower amount (0.75%) was found to be the maximum acceptable concentration for obtaining a stable hydrogel. Afterward, the polymer solutions were mixed with different excipients for rheological measurements. No significant change in gelation time was observed for ALG and HPMC, whereas tgel for HA was found to be slightly lower, 77 s (Figure 6a–c and Supporting Information Figure S4). Interestingly, the moduli values for the different excipient-containing hydrogels were significantly different. Although it was expected that the utilization of excipients would render thicker hydrogels with even higher storage moduli compared to the base 94/6–25% hydrogel, only ALG-incorporated hydrogel showed a higher G′ value in comparison. For HPMC, we did not observe any change in the G′ value. Although HA imparted the fastest gelation, the G′ value had decreased. These observations could be explained based on the stability of the excipient-loaded hydrogels at 37 °C, where precipitation occurred during incubation. The differential dehydration of the PEG block in the PLGA–PEG–PLGA copolymer in the presence of different excipients could be a major reason for such changes in storage modulus.
Figure 6.
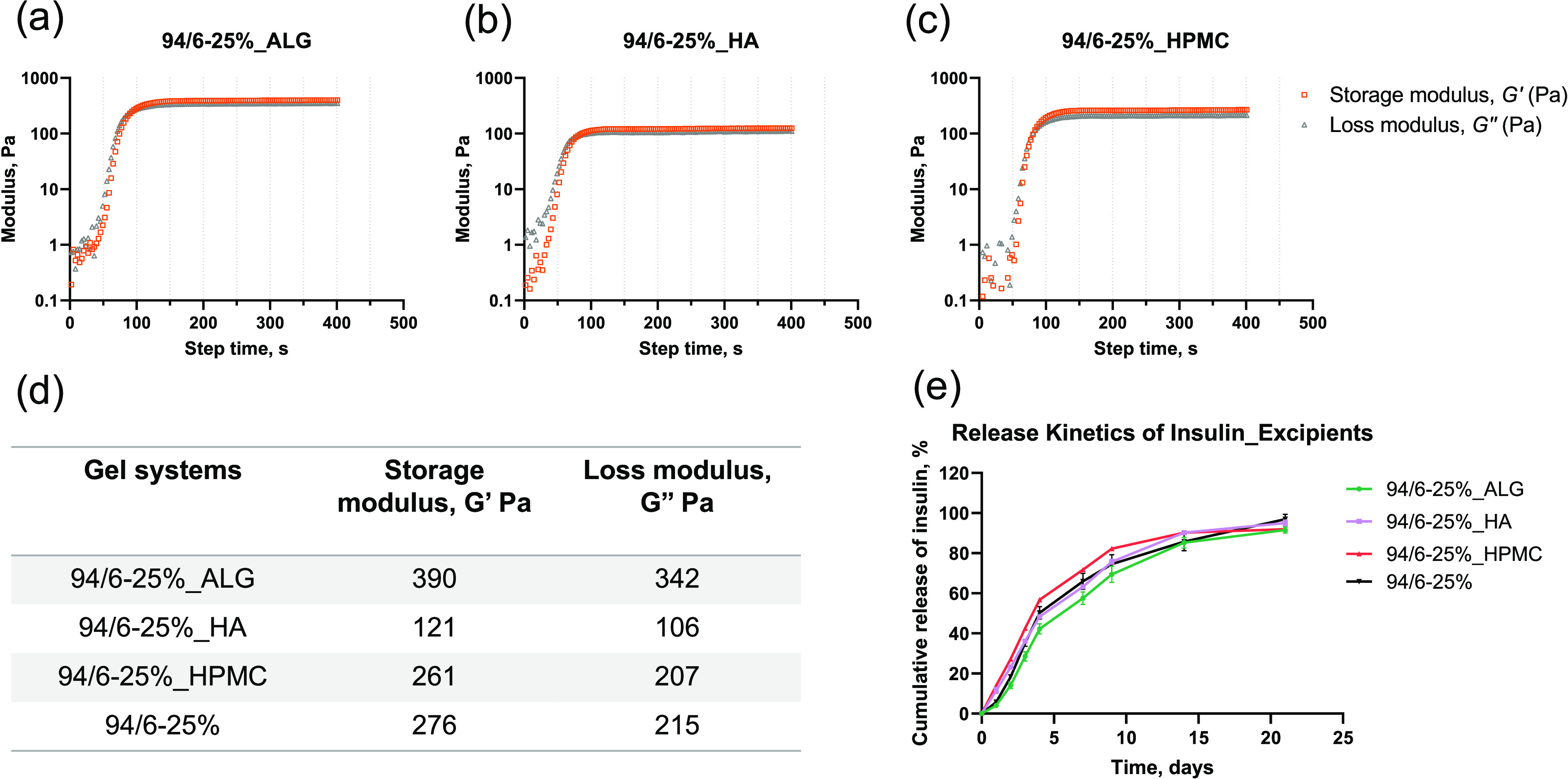
(a–c) Moduli of 94/6–25% PLGA–PEG–PLGA hydrogels in the presence of different excipients (abbreviated as LA–GA_polymer concentration_excipients); (d) summary of storage and loss moduli and gelation time for the experimented hydrogel systems; and (e) cumulative release kinetics for 94/6–25% PLGA–PEG–PLGA hydrogels with excipients (n = 3).
2.6. Effect of Excipients on Release Kinetics of Insulin
Next, the release kinetics of insulin was evaluated from different excipient-loaded hydrogels (Figure 6e). It is hypothesized that the ALG-loaded hydrogel should show the slowest insulin release kinetics due to its highest G′ value. Indeed, it was found that the hydrogel incorporated with ALG showed the slowest release kinetics (58% compared to 66, 63, and 72% for no excipient-, HA-, and HPMC-loaded hydrogels, respectively, at day 7), in agreement with the rheology data (Figure 6d). However, the release of insulin from HA (63% at day 7) and HPMC (72% at day 7) hydrogels was comparable with or slightly higher than that of the parent 94/6–25% hydrogel (66% at day 7), respectively. All hydrogels released the insulin cargo within 21 days, which was similar to the unmodified hydrogel. From the release data and rheological measurements, it was clear that the alginate-loaded hydrogel could be marginally beneficial in slowing down the release further, which is consistent with the previous reports in poloxamer-based systems where alginate addition was employed to improve gel strength for ophthalmic delivery.41 Similarly, all excipient-loaded hydrogels were found to stabilize the secondary structure of insulin without significant aggregation (see the CD spectra and SEC profile in Supporting Information Figures S5 and S6). These results demonstrate that the interactions of the polysaccharide excipients with the PLGA–PEG–PLGA hydrogel could be further leveraged to fine-tune the mechanical properties and modulate the release kinetics of encapsulated cargoes.
2.7. Effect of Protein Sizes on Release Kinetics and Stability Studies
Finally, to further explore the release duration and compatibility of these hydrogels with proteins of different sizes and structural complexity in addition to insulin (5.8 kDa), the 94/6–25% hydrogel formulations were incorporated with bovine serum albumin (BSA, 66.5 kDa) and immunoglobulin G antibody (IgG, 150 kDa) to study the release kinetics and postrelease protein stability. Based on the diffusion rate of proteins with varied MWs, it is expected that the release kinetics from the hydrogel should be fastest for the smallest MW protein insulin and would gradually slow down for BSA and IgG. Interestingly, BSA showed the fastest release that was completed within 4 days, despite having significantly higher MW compared to insulin (80 and 35% releases for BSA and insulin by day 3, respectively; Figure 7). In contrast, IgG followed the expected trend and provided a gradual steady release compared to all other proteins (only 18% release by day 3; Figure 7). This rationale behind this specific release kinetics could be based on three parameters: (a) diffusion, (b) solubility, and (c) hydrolytic degradation of the hydrogels. For BSA, the release profile was only controlled via diffusion as gel matrix degradation would be minimal within a couple of days. Similarly, the physical diffusion of IgG would be the major release mechanism at the initial stage, which could be subsequently coupled with gel matrix degradation as time increases. However, the unique release profile of insulin could be attributed to the differences in solubility in the hydrogel matrix. Although insulin was solubilized while loading into the hydrogel, an increase in turbidity was observed in over a few hours, which indicated partial precipitation of insulin inside the hydrogel matrix. Thus, the release of insulin from hydrogel was initially dictated by both solubility equilibria and protein diffusion. At the later stage, gel matrix degradation became faster and contributed to the overall release kinetics. Thus, the solubility of the cargo would also be another contributing factor to control drug release kinetics. These data are consistent with the previous reports in which protein precipitation has been reported to contribute significantly to the prolonged release profiles of small proteins and antibodies from the hydrogels.44,45 In this study, 20 mg/mL was selected as the protein-loading concentration for comparison, but it is expected that the protein-loading capacity of these hydrogels would be even higher. Ultimately, the maximum protein loading would be dependent on the overall viscosity profiles of the formulation that could still enable syringe injection (i.e., protein size, protein viscosity, polymer concentration, etc.).
Figure 7.

Cumulative release kinetics for 94/6–25% PLGA–PEG–PLGA hydrogels incorporated with cargoes of different MWs (n = 3).
Similarly, the structural stability of the released BSA and IgG was evaluated with SEC and CD spectroscopy (Figure S7). CD spectra of BSA (Figure S7a) showed the predominant α-helical conformation with no deterioration of the secondary structure confirmed by bands at 208 and 220 nm.46 IgG, on the other hand, majorly consisted of β-sheets (a band at 218 nm).46,47 No distinguishable conformational change was observed in this case as well (Figure S7b). Although it is known that BSA is acid-labile and IgG antibody has complex higher-order structures, both proteins were found to be stable over the course of the encapsulation and release processes and did not show any significant aggregation in the SEC profiles (Figure S7c,d). Taken together, these results indicate the versatility and compatibility of the hydrogel systems in preserving protein stability and releasing protein cargoes across a wide range of molecular sizes over an extended period of time.
2.8. In Vivo Peptide Delivery from Hydrogels
To study the in vivo performance of these hydrogels, the glucose-lowering efficacy of glucagon-like peptide (peptide A, 3.5 kDa)-loaded hydrogels was evaluated in nonfasted diabetic mice (shown in Figure 8a). The mice receiving a single injection of blank hydrogel without drug maintained a high blood glucose level (ca. 80% at 72 h) during the entire course of the experiment. As expected, the blood glucose levels of mice were effectively lowered and maintained below 50–65% compared to the starting glucose values over the course of 48–72 h after a single subcutaneous administration of the peptide A-loaded gel formulation, whereas the glucose levels quickly recovered to baseline level (ca. 73%) at approximately 48 h after solution injection of peptide A. These results confirm that the peptide was released from the hydrogel formulations in a controlled manner with detectable and comparable bioactivity, demonstrating the potential of these formulations for repeated administration of peptide therapeutics in long-term glycemic control.
Figure 8.

(a) In vivo release of a glucagon-like peptide (peptide A, 3.5 kDa) from the 25% PLGA–PEG–PLGA (LA/GA = 94/6) formulation in diabetic mice (n = 5) after a single subcutaneous administration. Glucose levels were monitored as a function of time. Glucose values were normalized against the starting level prior to injection. Statistical analysis was performed by a two-way ANOVA Dunnett’s multiple comparison test (**P < 0.01 and ***P < 0.001) vs blank hydrogel control; (b) in vivo release of a modified insulin peptide analogue (peptide B, 12 kDa) from the 25% PLGA–PEG–PLGA (LA/GA = 94/6) formulation in diabetic minipigs (n = 6) after a single subcutaneous administration. Plasma concentration of the modified insulin was measured as a function of time; statistical analysis was not applied to the PK results due to the drastic difference in doses between the solution and hydrogel groups and that a direct dose comparison is not possible without inducing hypoglycemia.
To further understand the in vivo release profiles and the translation to these technologies in large animal models, a modified insulin peptide analogue (peptide B, 12 kDa) was selected as a model molecule and encapsulated in 94/6–25% PLGA–PEG–PLGA formulation. The peptide was first dissolved in the polymer solution and injected subcutaneously in (T1DM) Yucatan minipigs. As shown in Figure 8b, the plasma concentration of insulin released from the hydrogel formulation was shown to maintain at above/close to 0.1 nM over the course of approximately 100 h, indicating a prolonged release of the payload. In contrast, the peptide released from the insulin solution formulation (control) was undetectable within 24 h. Due to the strong potency of insulin, it presents a persistent risk for overdosing that can result in life-threatening hypoglycemia events, which has been a constant challenge/limitation for many existing insulin therapies.48 Owing to its controlled release characteristics, the hydrogel formulation was able to accommodate a significantly high dose (ca. ∼16-fold higher) of the modified insulin peptide without causing any hypoglycemia effects (ca. 15 nmol in hydrogel vs 0.9 nmol in solution), which could potentially improve the therapeutic index of insulin.
3. Discussion
It has long been recognized that the degradation of PLGA-based polymers generates acidic byproducts, which can lead to a low pH microenvironment within PLGA-based delivery systems during incubation.49 For example, reports from Langer and co-workers indicated that the acidic microclimate pH within PLGA microspheres was in the range of 1.5–3.5, which could be potential stress for the instability of encapsulated proteins.50 Uchida et al. also reported on the acid-induced degradation of insulin from PLGA microspheres albeit a very slow release rate.51 Although the release of insulin and other peptides/proteins has been previously studied in PLGA–PEG–PLGA systems, only a few studies have explored the structural integrity and conformation properties of the encapsulated proteins during the incubation and release processes. Interestingly, it has been reported that significant conformational changes of liraglutide (amphiphilic polypeptide with a hydrophobic 16-carbon side chain) were observed when incubated with PLGA–PEG–PLGA systems, indicating that the hydrophilicity/hydrophobicity of the encapsulated proteins might have an impact on their interactions with the matrix and subsequently their conformational stabilities.13 The biophysical studies of the released proteins as characterized by SEC and CD in this report demonstrated that the PLGA–PEG–PLGA hydrogels do not adversely affect the stability of encapsulated hydrophilic proteins investigated, including small peptides like insulins that are prone to aggregation, acid-labile proteins like BSA, and complex proteins with higher-order structures such as IgG antibody. This could be likely attributed to the highly hydrophilic environment within the hydrogel structures, where most of the acidic degradation byproducts could easily diffuse out of the matrix and quickly be diluted in the surrounding release media without interacting with the encapsulated proteins extensively (as opposed to the potential accumulation of acidic monomers in the relatively more hydrophobic environment within a microsphere).
One potential area of improvement for the PLGA–PEG–PLGA hydrogel systems is their relatively short release duration for proteins (i.e., typically a few weeks in vitro), as these molecules can easily diffuse through the hydrophilic chains as the polymers dissolve during incubation. Given the extensive efforts in chemical/hydrophobic modifications of these polymers (e.g., PLGA/PEG length, end capping, LA/GA ratios),12 we have also explored approaches based on physical entanglements of polysaccharide-based excipients as an alternative to diversify the toolbox of modulating protein release kinetics and extending release duration. Consistent with the previous reports in poloxamer-based systems,41 it is demonstrated in this study that alginate can improve the mechanical strength of the hydrogels and prolong protein release from the matrix, likely due to the hydrogen bonding between the two polymers, which may lead to a stronger gel network. More detailed investigations of the PLGA–PEG–PLGA matrix interactions with polysaccharide-based excipients and the feasibility of cross-linking these polysaccharides into the network for mechanical reinforcement are underway and will be the subject of future reports.
Additionally, the hydrogels tested in this study were well tolerated in both animal models without any significant injection site reactions (albeit some minor swelling issues in few animals), suggesting the biocompatibility and biodegradability of these formulations. More importantly, results in this report suggest the translation of the controlled release potential of PLGA–PEG–PLGA hydrogel systems in large animal models (i.e., minipigs), which is a valuable complement to many existing reports in rodent models and of particular interest to the field of diabetes treatment considering the many physiological similarities between human and pig.52 It is anticipated that the translation of these technologies into the pharmaceutical industries would be extremely valuable in disease areas where repetitive injections may be required (e.g., diabetic therapies, cancer immunotherapy, and ophthalmic delivery). Other critical considerations are the feasibility for scalable industrial production and cost of goods, especially for therapeutic areas like diabetes where the pricing and manufacturing costs are most important given the large patient population. With the advances in polymer chemistry and green synthesis, it is envisioned that large-scale GMP manufacturing of these thermosensitive polymers would be possible in an economically and environmentally friendly fashion in the near future.30
4. Conclusions
In summary, we have investigated different PLGA–PEG–PLGA hydrogels to modulate the release kinetics of several biomacromolecules. The hydrogels with two different LA/GA rations were studied at different polymer concentrations. The particle size measurements revealed the thermoresponsive behavior at different temperatures. The results from these experiments were correlated with the rheological properties by studying the gelation kinetics and gel strength. The 94/6 LA/GA system with a 25% polymer concentration was found to be superior in terms of optimum stability, gelation properties, and desired release kinetics with minimal material content. Interestingly, we observed that the release of biomacromolecular cargoes from hydrogels is not only dependent on molecular diffusion but also significantly impacted by the cargo solubility inside the gel matrix. Further characterization of released biologics with CD spectra and SEC measurements showed that the hydrogels can serve as a useful reservoir to protect the biologics from denaturation or aggregation over the entire period of release. Finally, the delivery efficacy of the selected hydrogel was evaluated under two different in vivo systems to understand the pharmacodynamics and pharmacokinetics of two therapeutically relevant peptides. This study systematically investigates different PLGA–PEG–PLGA hydrogel systems and correlate different structural and molecular features of both hydrogel material and biologics cargoes. These results offer insight into the rational design of developing long-acting hydrogel formulations of protein therapeutics and suggest the potential of these injectable thermoresponsive hydrogel systems to improve patient’s comfort, convenience, and compliance with reduced therapeutic dose and minimized dosing frequency.
5. Experimental Section
5.1. Materials
Poly(lactic-co-glycolic acid)–b-poly(ethylene glycol)–b-poly(lactic-co-glycolic acid) copolymers (PLGA–PEG–PLGA; LA/GA ratios 94:6 and 3:1; MW 1700–1500–1700 Da, research grade) were procured from Polyscitech (Akina Incorporated). Sodium alginate (ALG, low viscosity), hyaluronic acid (HA, low molecular weight), and hydroxypropyl methyl cellulose (HPMC) were purchased from Sigma-Aldrich and Fisher Scientific. Insulin and bovine serum albumin (BSA) were obtained from Sigma-Aldrich. Immunoglobulin G (IgG, Human Plasma, MyBioSource) was purchased from Fisher Scientific. Merck proprietary peptides A and B (glucagon-like peptide analogue and modified insulin analogue) were synthesized internally. All chemicals were used without any further purification unless otherwise mentioned.
5.2. General Procedure for Preparation of PLGA–PEG–PLGA Hydrogels, Encapsulation, and Release of Protein Cargoes
Hydrogels with different concentrations of PLGA–PEG–PLGA triblock copolymers were prepared with the following procedure. All concentrations were reported in wt/vol percentages. Approximately 3 g of each polymer was weighed in 4 mL glass vials, and 10 mL of phosphate-buffered saline (PBS) buffer (pH 7.4) was added to make 30% solutions. Next, the mixtures were stirred overnight at 4 °C to ensure complete solubilization of the polymers. These stock solutions were further diluted with an appropriate volume of PBS buffer (at 4 °C) to make 20 and 25% solutions. These solutions were referred to as 94/6–20, 94/6–25, 94/6–30, and 3/1–25%, respectively (abbreviated as LA/GA ratio–polymer concentration).
For the preparation of hydrogel, 0.25 mL of the polymer solution was taken in a glass vial. Subsequently, the vial was incubated in a 37 °C incubator (with orbital shaking at 35 rpm) to trigger the gelation process.
For encapsulation of protein cargoes in hydrogels, 5 mg of each cargo molecule (insulin, bovine serum albumin, immunoglobulin G) was weighed in a glass vial and added with 0.25 mL of polymer solution (with varying concentrations) to obtain a drug-loading concentration of 20 mg/mL. The mixture was stirred in ice for 1 h to ensure complete mixing. Subsequently, it was incubated at 37 °C to trigger the gelation. After 1 h of incubation at 37 °C (to ensure complete gelation for all samples), 2.5 mL of PBS buffer (at 37 °C) was slowly added as release media on top of the freshly formed hydrogel, and this was further incubated at 37 °C (with previous conditions). The hydrogel samples in vials were incubated in a 37 °C incubator (with orbital shaking at 35 rpm). At a specific time interval, the buffer with the released cargo molecule was completely withdrawn and replenished with an equal amount of fresh PBS buffer.11,13,25,26
5.3. Preparation of Hydrogels in the Presence of Polysaccharide Excipients
PLGA–PEG–PLGA hydrogels containing different polysaccharide excipients (ALG, HA, and HPMC) were prepared by mixing the appropriate amount of excipients with the 25% polymer solutions (LA/GA ratio 94:6; referred to as 94/6–25% ALG, 94/6–25% HA, and 94/6–25% HPMC, respectively). The final concentrations of ALG, HA, and HPMC in the hydrogels were 0.75, 1, and 1%, respectively. For ALG, the amount is set to <1% to avoid precipitation of the mixture after the addition of alginate salt.
5.4. Dynamic Light-Scattering (DLS) Measurements
The particle size measurements were performed with a Malvern Nanozetasizer-ZS instrument. The prepared polymer solutions (94/6–25 and 3/1–25% without excipients) were diluted to a 1 mg/mL (0.1%) concentration with PBS (pH 7.4) buffer and studied with DLS measurements at different temperatures to study the gelation process from micellar solutions. The samples were incubated at the desired preset temperatures (4, 10, 20, 30, and 37 °C) in the DLS instrument and equilibrated for 1 h before subjecting to DLS measurements. The number, volume, and intensity distribution data were obtained directly from the Malvern Zetasizer software v7.13 (see the Supporting Information for details). Only volume percent data was normalized based on the highest count value and is discussed in Figure 1.
5.5. Rheological Characterization of Hydrogels
Rheological properties of the hydrogels were performed on a stress-controlled rheometer (ARES-G2, TA instruments) using a Peltier plate and a 1° steel cone and plate geometry. In all cases, 0.4 mL of polymer solutions (with and without excipients) were pipetted on top of the rheometer plate and equilibrated for 5 min at 10 °C. The samples were covered with solvent traps to minimize water evaporation during the experiment. Next, the temperature of the Peltier was increased from 10 to 37 °C to form in situ hydrogels. Dynamic oscillatory time sweeps were performed to monitor the in situ gelation and mechanical properties of different hydrogel compositions at angular frequencies of 6 rad/s and 1% strain amplitude chosen from the linear viscoelastic region. The storage or elastic modulus (G′) and loss or viscous modulus (G″) were measured as a function time at 37 °C.
5.6. Quantification of Protein Release from Hydrogels over Time
The protein release sample was collected from the 4 mL sample vials at predetermined time intervals at a 37 °C incubation and stored at −20 °C until analyzed. Ultraperformance liquid chromatography (UPLC) was employed to quantify insulin (MW 5.8 kDa, pI ∼ 5.3), and absorption studies (at 280 nm) in a microplate reader were performed for BSA (MW 66.5 kDa, pI ∼ 4.7) and IgG (MW 150 kDa, pI ∼ 5.5–8.3).
5.7. Biophysical Characterization of Released Protein
The stability of protein cargoes released from hydrogels at different time intervals was studied with size exclusion chromatography (SEC) and circular dichroism (CD) spectroscopy.
SEC was performed on an Acquity H-Class UPLC (Waters) with protein BEH SEC 125 and 200 Å columns (1.7 μm, 4.6 mm × 300 mm). In a typical experiment, 10 μL of protein sample was injected into a UPLC and eluted with a 50 mM phosphate buffer containing 0.45 M l-arginine hydrochloride (pH 7) at a flow rate of 0.3 mL/min. The detection of protein samples was performed with a UV detector (Waters UV/visible detector 2489) at 280 nm. The chromatogram was analyzed with the Empower 3 Chromatography Data Software.
CD spectroscopy of the released protein samples collected from the hydrogel matrix was recorded on a JASCO J-715 spectrophotometer. To record the spectra, 400 μL of sample solution was pipetted in a quartz cuvette of 1 mm path length and scanned from 200 to 260 nm at 25 °C (scan rate: 10 nm/min, interval: 2 nm, average of two spectra). The measurements were taken in triplicate, and the average values were plotted as mean residue ellipticity.
5.8. In Vivo Delivery of Peptides through PLGA–PEG–PLGA Hydrogels
5.8.1. Glucose-Lowering Experiments in Diabetic Mice
All animal studies were approved by the Merck Institutional Animal Use and Care Committee (IACUC). High-fat diet–streptozotocin-treated (HFD/STZ) mice were generated in-house as previously described by Mu et al.54 Briefly, 16 weeks old humanized glucagon receptor mice (hGCGR)55 from Taconic Farm (Rensselaer, NY), previously fed for 8 weeks with 60% Kcal from fat (HFD, D12492 Research Diets, New Brunswick, NJ), were dosed intraperitoneally during three consecutive days with a low dose of streptozotocin (40 mg/kg, 5 mL/kg; Sigma-Aldrich, St. Louis, MO) formulated in sodium citrate buffer at pH 4.5. To follow the induction of the disease, blood glucose and body weight were measured weekly in fed state after STZ treatment during 3 weeks before the peptide treatment. Animals with similar degrees of hyperglycemia (250–400 mg/dL) and body weight (∼40 g) were randomly divided into various vehicle or compound treatment groups. The mice were maintained under controlled conditions of lighting (12 h light/dark), temperature (23 ± 2 °C), and humidity (55 ± 15%) with access ad libitum to HFD.
To study the glucose-lowering efficacy of hydrogel formulation containing a glucagon-like peptide analogue (peptide A, 3.5 kDa, calculated pI ∼8.3), the formulation was evaluated in high-fat diet/streptozotocin-induced diabetic mice (HFD/STZ mice in the fed state). Peptide A was loaded at a concentration of 2 mg/mL in a 25% (w/v) polymer solution of PLGA–PEG–PLGA (LA/GA ratio 94:6) in PBS. The formulation was injected once subcutaneously (dose volume: 5 mL/kg, approximately 200 μL) in the mice (n = 5 per group), and the glucose level was measured with a One-Touch Ultra Glucometer (LifeScan, Milpitas CA) by tail nick at predetermined time points over the course of 72 h. Additionally, peptide A dissolved in phosphate buffer solutions was also administered subcutaneously as a control in a separate experiment with continuous glucose monitoring over 72 h. All of the mice were kept under nonfasting conditions with free access to food and water until the end of the experiment. Statistical analysis on the glucose level was performed by a two-way ANOVA Dunnett’s multiple comparison test (**P < 0.01 and ***P < 0.001) vs blank hydrogel control. Mice were kept alive for future studies following Merck IACUC guidelines for reuse of animals.
5.8.2. Pharmacokinetic Experiments in Diabetic Minipigs
All animal studies were approved by the Merck Institutional Animal Use and Care Committee (IACUC). Male Yucatan minipigs (5–7 months of age) were rendered type 1 diabetic by Alloxan injections following a proprietary protocol developed by Sinclair Research Center (Auxvasse, MO). Induction was considered successful if basal glucose levels exceeded 150 mg/dL. Minipigs used in these studies had plasma glucose levels of approximately 300–400 mg/dL and were instrumented with two jugular vein vascular access ports (VAPs). The animals were maintained at Sinclair Research Center and their glucose levels, when not in study, were controlled by the administration of insulin NPH at the time of their meals. The animals were thus kept healthy and remained in the colony for 3–5 years while being used in our studies. After each study, the animals were allowed to recover for at least 1 week prior to being re-enrolled in new studies.
To further evaluate the in vivo performance of these hydrogel formulations in large animal models, an appropriate amount of modified insulin peptide analogue (peptide B, 12 kDa, calculated pI ∼ 4.4) was dissolved at a target concentration of 9 mg/mL in 25% (w/v) polymer solution of PLGA–PEG–PLGA (LA/GA ratio 94:6) in PBS. Then, approximately 1 mL (dose volume: 0.02 mL/kg) of the polymer aqueous solution containing the peptide was subcutaneously injected in type 1 diabetes mellitus (T1DM) Yucatan minipigs (n = 6, ∼50 kg) using a 25G needle. Similarly, peptide B dissolved in phosphate buffer solutions was also administered subcutaneously as a control in a separate experiment. Blood samples were collected at predetermined time points over 24–168 h via the VAP in K3-EDTA tubes, supplemented with 100 μg/mL aprotinin, and kept on ice until processing, which occurred within 30 min of collection. After centrifugation at 3000 rpm, 4°C, for 8 min, the plasma was collected and pharmacokinetics (PK) of the peptide in the plasma was analyzed using LC–mass spectrometry (LC–MS).
Acknowledgments
The authors acknowledge the funding support and summer internship program of Merck Sharp & Dohme Corp., a subsidiary of Merck & Co., Inc., Kenilworth, NJ. The authors also thank NIGMS of the National Institute of Health (GM-136395) for partially supporting the efforts carried out at the University of Massachusetts Amherst.
Glossary
Abbreviations Used
- LA/GA-x%
PLGA–PEG–PLGA polymer with LA/GA = 94/6 or 3/1, and the polymer concentration is x% (w/v)
- ALG
sodium alginate
- HA
hyaluronic acid
- HPMC
hydroxypropyl methyl cellulose
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02009.
Correlation coefficients and size distribution by number; volume and intensity from DLS measurements; additional data on rheology measurements; images of hydrogels; CD spectra and SEC profiles for released insulin in the presence of different excipients and for released BSA and IgG (PDF)
Gelation video describing the rapid formation of hydrogel (MP4)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Mitragotri S.; Burke P. A.; Langer R. Overcoming the Challenges in Administering Biopharmaceuticals: Formulation and Delivery Strategies. Nat. Rev. Drug Discovery 2014, 13, 655–672. 10.1038/nrd4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dutta K.; Hu D.; Zhao B.; Ribbe A. E.; Zhuang J.; Thayumanavan S. Templated Self-Assembly of a Covalent Polymer Network for Intracellular Protein Delivery and Traceless Release. J. Am. Chem. Soc. 2017, 139, 5676–5679. 10.1021/jacs.7b01214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown L. R. Commercial Challenges of Protein Drug Delivery. Expert Opin. Drug Delivery 2005, 2, 29–42. 10.1517/17425247.2.1.29. [DOI] [PubMed] [Google Scholar]
- Genuth S.; Martin P. Control of Hyperglycemia in Adult Diabetics by Pulsed Insulin Delivery. Diabetes 1977, 26, 571–581. 10.2337/diab.26.6.571. [DOI] [PubMed] [Google Scholar]
- Leader B.; Baca Q. J.; Golan D. E. Protein Therapeutics: A Summary and Pharmacological Classification. Nat. Rev. Drug Discovery 2008, 7, 21–39. 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- Böttger R.; Hoffmann R.; Knappe D. Differential Stability of Therapeutic Peptides with Different Proteolytic Cleavage Sites in Blood, Plasma and Serum. PloS One 2017, 12, e0178943 10.1371/journal.pone.0178943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K.; Ejaz W.; Dutta K.; Thayumanavan S. Antibody Delivery for Intracellular Targets: Emergent Therapeutic Potential. Bioconjugate Chem. 2019, 30, 1028–1041. 10.1021/acs.bioconjchem.9b00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Y.; Li L.; Scott R. A.; Kiick K. L. 50th Anniversary Perspective: Polymeric Biomaterials: Diverse Functions Enabled by Advances in Macromolecular Chemistry. Macromolecules 2017, 50, 483–502. 10.1021/acs.macromol.6b02389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermonden T.; Censi R.; Hennink W. E. Hydrogels for Protein Delivery. Chem. Rev. 2012, 112, 2853–2888. 10.1021/cr200157d. [DOI] [PubMed] [Google Scholar]
- Satav S. S.; Bhat S.; Thayumanavan S. Feedback Regulated Drug Delivery Vehicles: Carbon Dioxide Responsive Cationic Hydrogels for Antidote Release. Biomacromolecules 2010, 11, 1735–1740. 10.1021/bm1005454. [DOI] [PubMed] [Google Scholar]
- Majumder P.; Baxa U.; Walsh S. T. R.; Schneider J. P. Design of a Multicompartment Hydrogel That Facilitates Time-Resolved Delivery of Combination Therapy and Synergized Killing of Glioblastoma. Angew. Chem., Int. Ed. 2018, 57, 15040–15044. 10.1002/anie.201806483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.; Chu W.; Zhuo X.; Zhang Y.; Gou J.; Ren T.; He H.; Yin T.; Tang X. Modified PLGA–PEG–PLGA Thermosensitive Hydrogels with Suitable Thermosensitivity and Properties for Use in a Drug Delivery System. J. Mater. Chem. B 2017, 5, 1551–1565. 10.1039/C6TB02158A. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Li Y.; Shen W.; Li K.; Yu L.; Chen Q.; Ding J. Controlled Release of Liraglutide Using Thermogelling Polymers in Treatment of Diabetes. Sci. Rep. 2016, 6, 31593 10.1038/srep31593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi K.; Wang Y.-L.; Qu Y.; Liao J.-F.; Chu B.-Y.; Zhang H.-P.; Luo F.; Qian Z.-Y. Synthesis, Characterization and Application of Reversible PDLLA-PEG-PDLLA Copolymer Thermogels in Vitro and in Vivo. Sci. Rep. 2016, 6, 19077 10.1038/srep19077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstad N. L.; Fowers K. D. Oncogel (Regel/Paclitaxel) - Clinical Applications for a Novel Paclitaxel Delivery System. Adv. Drug Delivery Rev. 2009, 61, 785–794. 10.1016/j.addr.2009.04.010. [DOI] [PubMed] [Google Scholar]
- Yu L.; Sheng W.; Yang D.; Ding J. Design of Molecular Parameters to Achieve Block Copolymers with a Powder Form at Dry State and a Temperature-Induced Sol-Gel Transition in Water without Unexpected Gelling Prior to Heating. Macromol. Res. 2013, 21, 207–215. 10.1007/s13233-013-1021-x. [DOI] [Google Scholar]
- Bodratti A. M.; Alexandridis P. Formulation of Poloxamers for Drug Delivery. J. Funct. Biomater. 2018, 9, 11 10.3390/jfb9010011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K.; Shi X.; Lin X.; Yao C.; Shen L.; Feng Y. Poloxamer-Based in Situ Hydrogels for Controlled Delivery of Hydrophilic Macromolecules after Intramuscular Injection in Rats. Drug Delivery 2015, 22, 375–382. 10.3109/10717544.2014.891272. [DOI] [PubMed] [Google Scholar]
- Seo B.-B.; Choi H.; Koh J.-T.; Song S.-C. Sustained Bmp-2 Delivery and Injectable Bone Regeneration Using Thermosensitive Polymeric Nanoparticle Hydrogel Bearing Dual Interactions with Bmp-2. J. Controlled Release 2015, 209, 67–76. 10.1016/j.jconrel.2015.04.023. [DOI] [PubMed] [Google Scholar]
- Park M.-R.; Chun C. J.; Ahn S. W.; Ki M.-H.; Cho C.-S.; Song S.-C. Sustained Delivery of Human Growth Hormone Using a Polyelectrolyte Complex-Loaded Thermosensitive Polyphosphazene Hydrogel. J. Controlled Release 2010, 147, 359–367. 10.1016/j.jconrel.2010.07.126. [DOI] [PubMed] [Google Scholar]
- Kapoor D. N.; Bhatia A.; Kaur R.; Sharma R.; Kaur G.; Dhawan S. Plga: A Unique Polymer for Drug Delivery. Ther. Delivery 2015, 6, 41–58. 10.4155/tde.14.91. [DOI] [PubMed] [Google Scholar]
- Makadia H. K.; Siegel S. J. Poly Lactic-Co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polymers 2011, 3, 1377–1397. 10.3390/polym3031377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Q.; Hashimoto M.; Dang T. T.; Hoare T.; Kohane D. S.; Whitesides G. M.; Langer R.; Anderson D. G. Preparation of Monodisperse Biodegradable Polymer Microparticles Using a Microfluidic Flow-Focusing Device for Controlled Drug Delivery. Small 2009, 5, 1575–1581. 10.1002/smll.200801855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi S.; Kim S. W. Controlled Release of Insulin from Injectable Biodegradable Triblock Copolymer Depot in Zdf Rats. Pharm. Res. 2003, 20, 2008–2010. 10.1023/B:PHAM.0000008050.99985.5c. [DOI] [PubMed] [Google Scholar]
- Yu L.; Li K.; Liu X. J.; Chen C.; Bao Y. C.; Ci T. Y.; Chen Q. H.; Ding J. D. In Vitro and in Vivo Evaluation of a Once-Weekly Formulation of an Antidiabetic Peptide Drug Exenatide in an Injectable Thermogel. J. Pharm. Sci. 2013, 102, 4140–4149. 10.1002/jps.23735. [DOI] [PubMed] [Google Scholar]
- Li K.; Yu L.; Liu X. J.; Chen C.; Chen Q. H.; Ding J. D. A Long-Acting Formulation of a Polypeptide Drug Exenatide in Treatment of Diabetes Using an Injectable Block Copolymer Hydrogel. Biomaterials 2013, 34, 2834–2842. 10.1016/j.biomaterials.2013.01.013. [DOI] [PubMed] [Google Scholar]
- Samlowski W. E.; McGregor J. R.; Jurek M.; Baudys M.; Zentner G. M.; Fowers K. D. Regel (R) Polymer-Based Delivery of Interleukin-2 as a Cancer Treatment. J. Immunother. 2006, 29, 524–535. 10.1097/01.cji.0000211306.05869.25. [DOI] [PubMed] [Google Scholar]
- Gong C.; Qi T.; Wei X.; Qu Y.; Wu Q.; Luo F.; Qian Z. Thermosensitive Polymeric Hydrogels as Drug Delivery Systems. Curr. Med. Chem. 2012, 20, 79–94. 10.2174/0929867311302010009. [DOI] [PubMed] [Google Scholar]
- Shim M. S.; Lee H. T.; Shim W. S.; Park I.; Lee H.; Chang T.; Kim S. W.; Lee D. S. Poly(D,L-Lactic Acid-co-Glycolic Acid)-b-Poly(Ethylene Glycol)-b-Poly (D,L-Lactic Acid-co-Glycolic Acid) Triblock Copolymer and Thermoreversible Phase Transition in Water. J. Biomed. Mater. Res. 2002, 61, 188–196. 10.1002/jbm.10164. [DOI] [PubMed] [Google Scholar]
- Liow S. S.; Dou Q. Q.; Kai D.; Karim A. A.; Zhang K. Y.; Xu F. J.; Loh X. J. Thermogels: In Situ Gelling Biomaterial. ACS Biomater. Sci. Eng. 2016, 2, 295–316. 10.1021/acsbiomaterials.5b00515. [DOI] [PubMed] [Google Scholar]
- Aimetti A. A.; Machen A. J.; Anseth K. S. Poly(Ethylene Glycol) Hydrogels Formed by Thiol-Ene Photopolymerization for Enzyme-Responsive Protein Delivery. Biomaterials 2009, 30, 6048–6054. 10.1016/j.biomaterials.2009.07.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortimer S.; Ryan A. J.; Stanford J. L. Rheological Behavior and Gel-Point Determination for a Model Lewis Acid-Initiated Chain Growth Epoxy Resin. Macromolecules 2001, 34, 2973–2980. 10.1021/ma001835x. [DOI] [Google Scholar]
- Qiao M. X.; Chen D. W.; Ma X. C.; Liu Y. J. Injectable Biodegradable Temperature-Responsive PLGA-PEG-PLGA Copolymers: Synthesis and Effect of Copolymer Composition on the Drug Release from the Copolymer-Based Hydrogels. Int. J. Pharm. 2005, 294, 103–112. 10.1016/j.ijpharm.2005.01.017. [DOI] [PubMed] [Google Scholar]
- Payyappilly S.; Dhara S.; Chattopadhyay S. Thermoresponsive Biodegradable Peg-Pcl-Peg Based Injectable Hydrogel for Pulsatile Insulin Delivery. J. Biomed. Mater. Res., Part A 2014, 102, 1500–1509. 10.1002/jbm.a.34800. [DOI] [PubMed] [Google Scholar]
- Mishra N. K.; Joshi K. B.; Verma S. Inhibition of Human and Bovine Insulin Fibril Formation by Designed Peptide Conjugates. Mol. Pharmaceutics 2013, 10, 3903–3912. 10.1021/mp400364w. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Venkatesu P. Prevention of Insulin Self-Aggregation by a Protic Ionic Liquid. RSC Adv. 2013, 3, 362–367. 10.1039/C2RA22277A. [DOI] [Google Scholar]
- Minh K. N.; Huynh C. T.; Gao G. H.; Kim J. H.; Huynh D. P.; Chae S. Y.; Lee K. C.; Lee D. S. Biodegradable Oligo(Amidoamine/Beta-Amino Ester) Hydrogels for Controlled Insulin Delivery. Soft Matter 2011, 7, 2994–3001. 10.1039/c0sm01285h. [DOI] [Google Scholar]
- Tønnesen H. H.; Karlsen J. Alginate in Drug Delivery Systems. Drug Dev. Ind. Pharm. 2002, 28, 621–630. 10.1081/DDC-120003853. [DOI] [PubMed] [Google Scholar]
- Brown T. J.; Falzon J. L.; Pho M.; Thomas N.; Brownlee G. Evaluation of Hyaluronic Acid as a Pharmacologically Active Excipient for Gemcitabine in the Treatment of Pancreatic Cancer. Cancer Res. 2006, 66, 724. [Google Scholar]
- Ishikawa T.; Watanabe Y.; Takayama K.; Endo H.; Matsumoto M. Effect of Hydroxypropylmethylcellulose (HPMC) on the Release Profiles and Bioavailability of a Poorly Water-Soluble Drug from Tablets Prepared Using Macrogol and Hpmc. Int. J. Pharm. 2000, 202, 173–178. 10.1016/S0378-5173(00)00426-9. [DOI] [PubMed] [Google Scholar]
- Lin H.-R.; Sung K. C.; Vong W.-J. In Situ Gelling of Alginate/Pluronic Solutions for Ophthalmic Delivery of Pilocarpine. Biomacromolecules 2004, 5, 2358–2365. 10.1021/bm0496965. [DOI] [PubMed] [Google Scholar]
- Mayol L.; Biondi M.; Quaglia F.; Fusco S.; Borzacchiello A.; Ambrosio L.; La Rotonda M. I. Injectable Thermally Responsive Mucoadhesive Gel for Sustained Protein Delivery. Biomacromolecules 2011, 12, 28–33. 10.1021/bm1008958. [DOI] [PubMed] [Google Scholar]
- Wang W.; Wat E.; Hui P. C. L.; Chan B.; Ng F. S. F.; Kan C.-W.; Wang X.; Hu H.; Wong E. C. W.; Lau C. B. S.; Leung P.-C. Dual-Functional Transdermal Drug Delivery System with Controllable Drug Loading Based on Thermosensitive Poloxamer Hydrogel for Atopic Dermatitis Treatment. Sci. Rep. 2016, 6, 24112 10.1038/srep24112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Wetering P.; Metters A. T.; Schoenmakers R. G.; Hubbell J. A. Poly(Ethylene Glycol) Hydrogels Formed by Conjugate Addition with Controllable Swelling, Degradation, and Release of Pharmaceutically Active Proteins. J. Controlled Release 2005, 102, 619–627. 10.1016/j.jconrel.2004.10.029. [DOI] [PubMed] [Google Scholar]
- Liang Y.; Coffin M. V.; Manceva S. D.; Chichester J. A.; Jones R. M.; Kiick K. L. Controlled Release of an Anthrax Toxin-Neutralizing Antibody from Hydrolytically Degradable Polyethylene Glycol Hydrogels. J. Biomed. Mater. Res., Part A 2016, 104, 113–123. 10.1002/jbm.a.35545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Rose J.; Plantevin S.; Auffan M.; Bottero J.-Y.; Vidaud C. Protein Corona Formation for Nanomaterials and Proteins of a Similar Size: Hard or Soft Corona?. Nanoscale 2013, 5, 1658–1668. 10.1039/c2nr33611a. [DOI] [PubMed] [Google Scholar]
- Koutsopoulos S.; Unsworth L. D.; Nagai Y.; Zhang S. G. Controlled Release of Functional Proteins through Designer Self-Assembling Peptide Nanofiber Hydrogel Scaffold. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 4623–4628. 10.1073/pnas.0807506106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaykov A. N.; Mayer J. P.; DiMarchi R. D. Pursuit of a Perfect Insulin. Nat. Rev. Drug Discovery 2016, 15, 425–439. 10.1038/nrd.2015.36. [DOI] [PubMed] [Google Scholar]
- Ding A. G.; Schwendeman S. P. Acidic Microclimate Ph Distribution in Plga Microspheres Monitored by Confocal Laser Scanning Microscopy. Pharm. Res. 2008, 25, 2041–2052. 10.1007/s11095-008-9594-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu K.; Pack D. W.; Klibanov A. M.; Langer R. Visual Evidence of Acidic Environment within Degrading Poly(Lactic-Co-Glycolic Acid) (Plga) Microspheres. Pharm. Res. 2000, 17, 100–106. 10.1023/A:1007582911958. [DOI] [PubMed] [Google Scholar]
- Uchida T.; Yagi A.; Oda Y.; Nakada Y.; Goto S. Instability of Bovine Insulin in Poly(Lactide-co-Glycolide) (PLG) Microspheres. Chem. Pharm. Bull. 1996, 44, 235–236. 10.1248/cpb.44.235. [DOI] [PubMed] [Google Scholar]
- Larsen M. O.; Rolin B. Use of the Gottingen Minipig as a Model of Diabetes, with Special Focus on Type 1 Diabetes Research. ILAR J. 2004, 45, 303–313. 10.1093/ilar.45.3.303. [DOI] [PubMed] [Google Scholar]
- Mu J.; Qureshi S. A.; Brady E. J.; Muise E. S.; Candelore M. R.; Jiang G.; Li Z.; Wu M. S.; Yang X.; Dallas-Yang Q.; Miller C.; Xiong Y.; Langdon R. B.; Parmee E. R.; Zhang B. B. Anti-Diabetic Efficacy and Impact on Amino Acid Metabolism of Gra1, a Novel Small-Molecule Glucagon Receptor Antagonist. PloS one 2012, 7, e49572 10.1371/journal.pone.0049572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiao L.-L.; Cascieri M. A.; Trumbauer M.; Chen H.; Sullivan K. A. Generation of Mice Expressing the Human Glucagon Receptor with a Direct Replacement Vector. Transgenic Res. 1999, 8, 295–302. 10.1023/A:1008922521461. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.