

Abstract
The majority of studies of genetic association with disease have been performed in Europeans. This European bias has important implications for risk prediction of diseases across global populations. In this commentary, we justify the need to study more diverse populations using both empirical examples and theoretical reasoning.
It has been well documented that genetic studies of human disease, especially large-scale ones, have not captured the level of diversity that exists globally, as they are predominantly based on populations of European ancestry (Popejoy and Fullerton, 2016). The under-representation of ethnically diverse populations impedes our ability to fully understand the genetic architecture of human disease and exacerbates health inequalities. Further, the lack of ethnic diversity in human genomic studies means that our ability to translate genetic research into clinical practice or public health policy may be dangerously incomplete, or worse, mistaken. For example, attempts to use estimates of genetic risk from European-based studies in non-Europeans may result in inaccurate assessment of risk and lack of interventions in under-studied populations. In this commentary, we discuss examples that illustrate why inclusion of ethnically diverse populations in human genetic studies facilitates identification of genetic risk factors for Mendelian and complex diseases. Additionally, we discuss why lack of replication across populations of genetic associations with complex traits, including disease risk, is expected based on the evolutionary history of populations across the globe. Lastly, we discuss challenges and future directions for promoting equity in human genomic studies.
The Impact of Genetic Diversity on Mendelian Disease
For Mendelian diseases, a pathogenic variant usually causes disease regardless of the population in which it occurs (Figure 1A). However, in some cases, such as X-linked G6PD deficiency and favism, a condition will only present upon specific environmental exposure (i.e., fava bean consumption causing hemolytic anemia). Given diverse evolutionary histories, different mutations in the same gene may account for a given disease in diverse populations (allelic heterogeneity). Variation in causative mutations may confound diagnoses or treatments.
Figure 1. Factors Affecting Ability to Replicate Genotype-Phenotype Associations across Populations (Transferability).
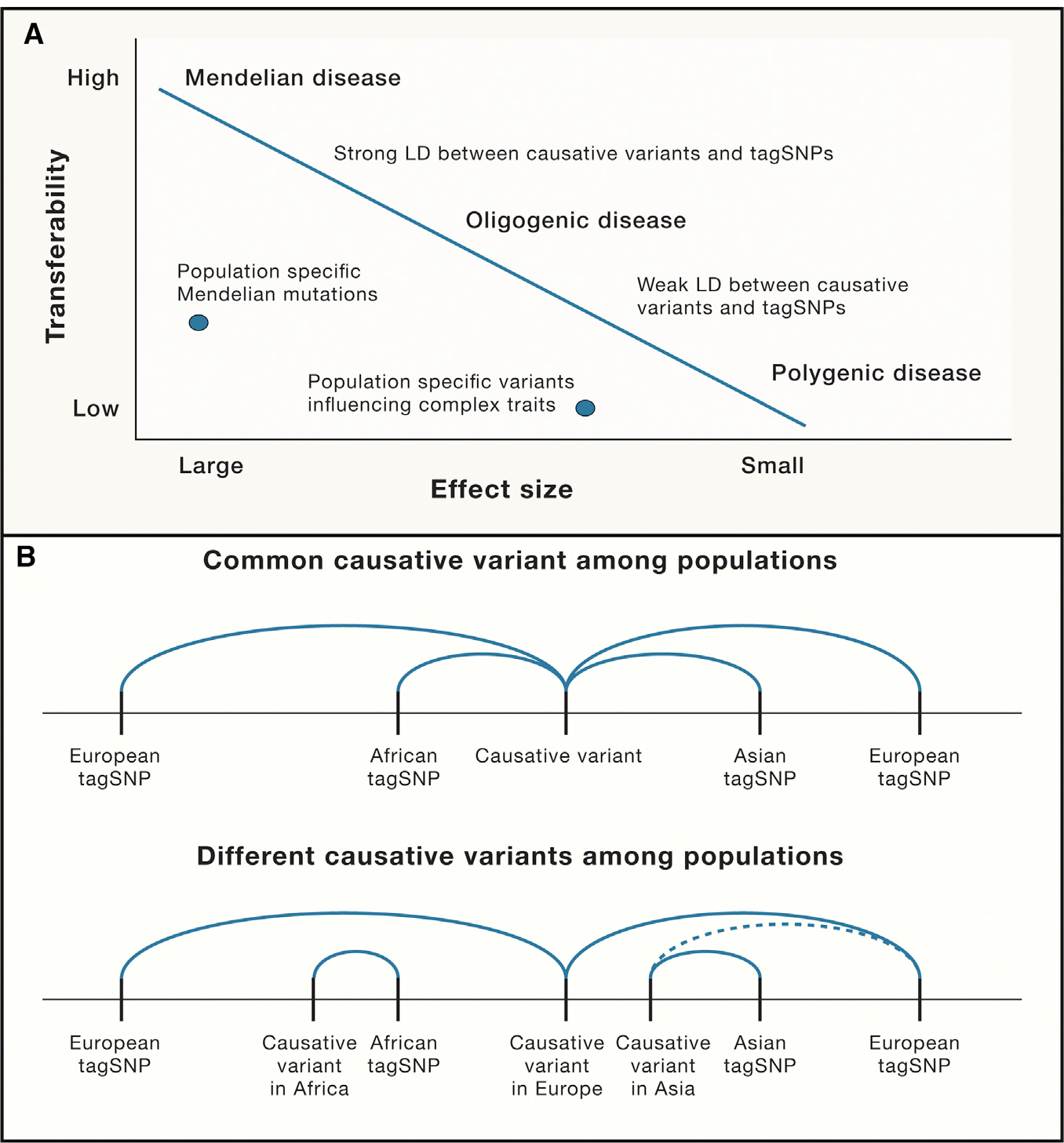
(A) The role of effect size on transferability—monogenic (Mendelian) diseases with large effect sizes will cause disease wherever they occur. In contrast, genetic associations with complex traits affected by a few genes with moderate effect sizes (oligogenic) are less likely to transfer across populations. Lastly, for conditions affected by many genes (polygenic) with small effect sizes, transferability may be limited. For all cases, if risk is assessed by tagSNPs, the degree of LD will affect transferability.
(B) The role of differential linkage disequilibrium (LD) on transferability. (Top) A single common causative variant may be tagged by different SNPs in different populations; (bottom) causative variants differ by population (allelic heterogeneity) that are tagged by different SNPs (solid lines) or tagged by the same SNP but with weaker LD (dotted line). In either case, tagSNPs derived from a single population (e.g., European) may be inadequate to allow replication of associations across study populations.
Cystic fibrosis (CF) is a Mendelian disease common in Europe (1 in 2000–3000 births) and rarer in African Americans (1 in 17,000 births). A consequence of low CF prevalence in African-descent individuals is that CF is often underdiagnosed. In Europeans, the most common causative allele is ΔF508 in the CFTR gene, accounting for more than 70% of cases. However, ΔF508 accounts for only about 29% of CF cases in people of the African diaspora (Stewart and Pepper, 2017). In contrast, a different mutation 3120+1G→A accounts for between 15% and 65% of CF patients in South Africans with African ancestry (Padoa et al., 1999). Indeed, more than 2,000 rare mutations in CFTR underlie considerable clinical heterogeneity and guide different treatment modalities. Such is the case for the CF medication ivacaftor that selectively targets mutations affecting the receptor gating capacity of the CFTR protein. Knowing and testing for specific pathogenic variants that vary in frequency across populations is crucial for appropriate clinical intervention.
There may also be population-specific mutations in understudied populaions that cause health disparities. For example, transthyretin (TTR) amyloid car-diomyopathy (ATTR-CM), due to a mutant transthyretin protein producing accumulation of amyloid fibrils, is an important and underdiagnosed cause of heart failure (HF) in African Americans. A TTR pathogenic missense mutation (V122I) is almost exclusively found in African descent subjects, with a carrier frequency in African Americans of 3%–4%. V122I acts in a dominant manner and accounts for as much as 10% of all HF cases in African Americans (Buxbaum et al., 2006). As new treatments targeting TTR gene expression or stabilizing the abnormal transthyretin are becoming available, genetic screening for this prevalent mutation in people of African descent can provide critical information with respect to both diagnostic accuracy and therapy, a case in point for precision medicine.
Identifying pathogenic variants causing Mendelian disease is more complicated when one considers locus heterogeneity. For example, there are more than 300 genes involved in retinal disease; over 3000 mutations in 65 genes cause retinitis pigmentosa (RP) with different modes of inheritance (https://sph.uth.edu/retnet/). As many of these mutations have only been characterized in Europeans, we know little about the genetic causes of retinal disease across ethnically diverse populations.
Genetic modifiers can also complicate the understanding of the biology underlying differences in disease presentation. Sickle cell disease (SCD), which is caused by homozygosity for a missense mutation (Glu6Val) in the b-globin gene (HBB), is an example. Every year about 300,000 newborns are diagnosed with SCD, and the SCD mortality in Africa among children less than 5 years old can reach 90%. Despite the high mortality of homozygotes, this mutation is maintained at high frequency in malaria endemic regions of Africa because heterozygous individuals are protected from malaria, resulting in balancing selection. Modifiers of SCD severity include maintenance of high fetal hemoglobin (HbF) expression, normally completely lost by 12 months of age, which results in fewer sickling crises and less severe disease. In Saudi patients with the Benin haplotype, HbF expression persists in adults, reaching twice the levels observed in African patients with the same haplotype (Piel et al., 2017), and other genes can also modify the expression of HbF. Further, distinct traits can modify SCD presentation, including alpha-thalassemia, the prevalence and allelic spectrum of which varies across populations (Piel et al., 2017). Hence, presentation of SCD may vary among populations due to gene-gene interactions (epistasis). The reason for phenotypic differences among populations with respect to SCD disease modifiers remains largely unexplained, despite clear clinical relevance. Larger studies across diverse populations of genetic factors influencing SCD presentation are needed.
As whole-genome sequencing (WGS) is increasingly used to infer the causes of rare undiagnosed diseases, reference genomes from more ethnically diverse populations are particularly important. This is because one criterion for identifying putative causal variants is by confirming rarity across populations. Therefore, if databases do not include sufficient data from ethnically diverse populations we may mistakenly infer that a benign variant is pathogenic. For example, the gnomAD exome and genome database includes ~60% European sequences and less than 10% sequences from individuals of African ancestry at present (https://macarthurlab.org/2017/02/27/the-genome-aggregation-database-gnomad/).
The Impact of Genetic Diversity on Complex Traits
As of 2018, the majority of genome-wide association studies (GWAS), which aim to identify genetic variants associated with complex traits including disease risk, have been conducted in European (52%) or Asian (21%) populations (Figure 2, left). When we consider the number of individuals included in GWAS based on ethnicity, 78% are European, 10% are Asian, 2% are African, 1% are Hispanic, and all other ethnicities represent < 1% of GWAS (Figure 2, right). These disparities are unacceptable, particularly since GWAS findings may not replicate across ethnic groups.
Figure 2. Summary of GWAS Studies by Ancestry for Studies in the GWAS Catalog through January 2019.
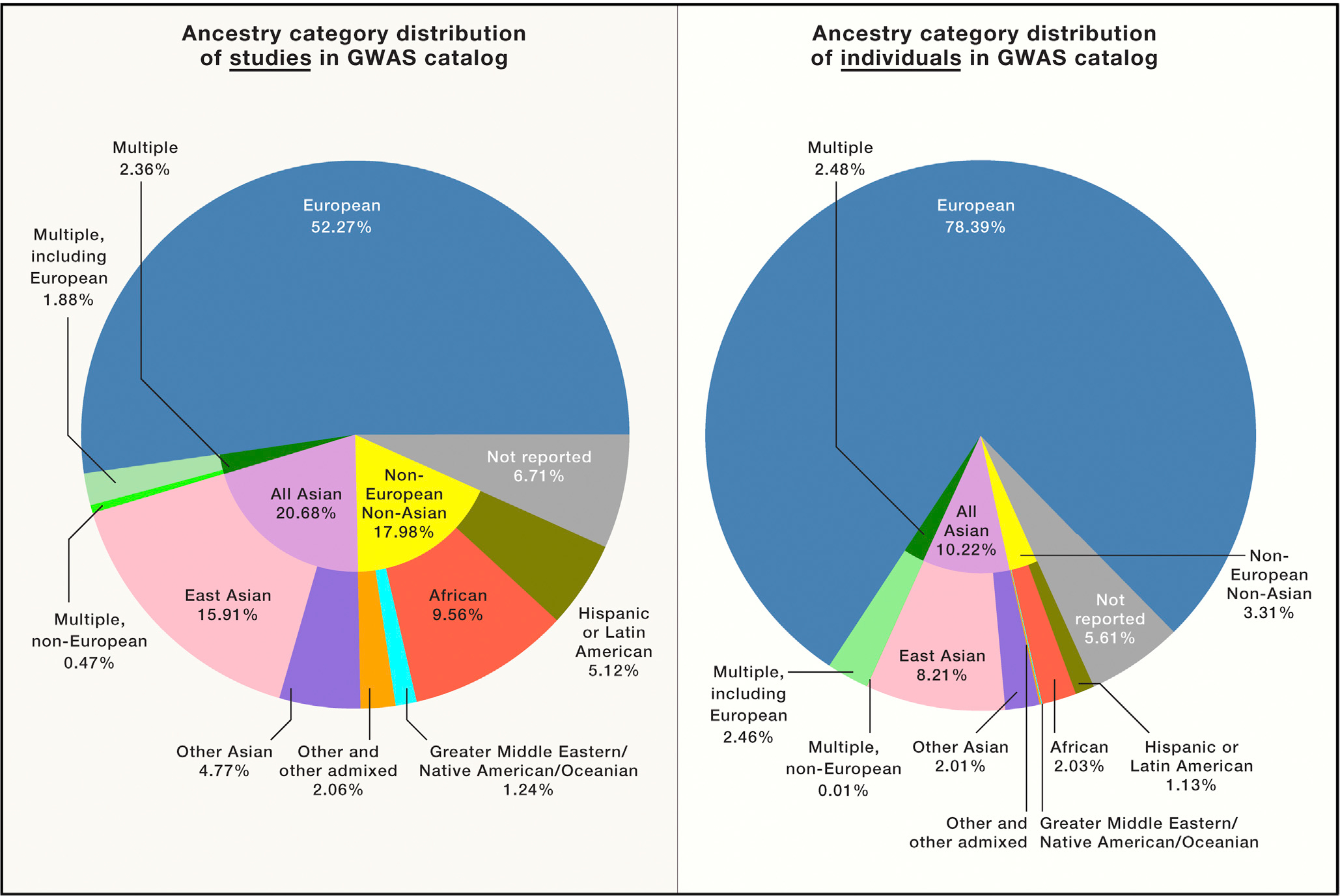
We show the distribution of ancestry categories in percentages included in GWAS (https://www.ebi.ac.uk/gwas/home) based on the study (left) and based on the total number of individuals (right).
The ability to replicate genetic associations across diverse populations can be affected by several factors. Differences in linkage disequilibrium (LD) across ethnicities influences how well causal variants are captured by tagging SNPs identified in a single population (Figure 1B). Markers in LD with risk variants in Europeans may not be in LD in other populations because LD patterns reflect different demographic histories that vary globally. Modern humans originated in Africa within the past 300,000 years and migrated out of Africa within the past 80,000 years. Africans have maintained larger and more sub-structured populations resulting in diverse patterns of LD across the continent (Tishkoff et al., 2009). In contrast, the migration out of Africa resulted in a population bottleneck followed by a series of founding events as modern humans spread across the globe. Thus, non-Africans have more extended regions of LD, the precise structure of which is determined by their population histories (Figure 1B). These differences in LD among populations can make transethnic mapping particularly informative for identifying risk variants.
The lack of replication of GWAS among populations could also be due to differnces in genetic architecture. Differences in genetic architecture among ethnically diverse groups could be due to population specific variation as well as changes in allele frequency that arise as a product of genetic drift, local selection, or both. For example, founder populations can have differences in prevalence of a complex disease, or related intermediate phenotypes, compared to the source population and may be particularly informative for complex disease mapping. In the Finnish population that underwent founder events, there are many low-frequency loss-of-function variants that associate with complex phenotypes, including lipid levels (Lim et al., 2014). Variation in phenotypic prevalence for complex diseases can also be found in Ashkenazim, French Canadians, Icelanders, and Sardinians—populations that experienced founder effects. However, differentiating between the effects of selection and drift on genetic diversity in these populations, not always mutually exclusive, needs to be resolved on a case by case basis.
In small, bottlenecked populations or those practicing consanguineous mating, homozygosity is enriched. Homozygosity mapping in such populations has long been a successful strategy to locate recessive disease genes. More recently, the identification of rare homozygous loss-of-function mutations due to consanguinity in apparently healthy individuals has provided important insights into gene function and has paved the way to the discovery or validation of drug targets, as in the Human Knockout Project that focuses on Pakistanis (Saleheen et al., 2017). Loss-of-function variants can also shed light on biological pathways that have relevance across populations, as has been the case with the discovery of PCSK9, an important gene for regulating LDL levels. This discovery was facilitated by studying people of African descent with PCSK9 nonsense mutations, but the knowledge has translated into a drug with global utility. Beneficial loss of function mutations are promising targets for treatment, as it is easier to develop therapeutics that turn gene products off rather than on.
Local adaptation can also influence the genetic architecture of complex traits, which may not necessarily be related to the initial selection event(s), as many genes have pleiotropic effects. For example, in African Americans with nondiabetic progressive chronic and end stage kidney disease, two African-specific risk variants (G1 and G2) in the apolipoprotein L1 gene (APOL1) are strongly associated with these conditions. These variants confer an increased risk of approximately 7- to 10-fold for kidney disease, and, together, partially explain the higher incidence of end stage renal disease in African Americans as compared to European Americans (Freedman et al., 2018; Genovese et al., 2010). It has been argued that these variants are at high frequency in populations of West African descent because they are protective against sleeping sickness caused by Trypanosoma brucei protozoa. These same variants at APOL1 are also associated with a broad range of nondiabetic kidney diseases, including severe lupus nephritis and sickle cell nephropathy (Freedman et al., 2018).
Another potential complication causing lack of replication of GWAS across ethnic groups could be epistasis due to differences in genetic backgrounds (G × G) as well as gene-environment (G × E) interactions that vary among populations. A recent multi-ethnic, genome-wide study identified genetic loci interacting with physical activity to affect blood lipid levels, illustrating G × E interactions and the value of including different ancestries in studies of complex traits. Variants in four loci were found to interact with physical activity to influence lipid levels. These interactions were discovered in a transethnic mapping study that included European, African, Asian, and Hispanic populations. Two out of the four loci (SNTA1 and CNTNAP2) were identified because Africans and Hispanics showed a relatively high frequency of the variants associated with lipid levels compared to other populations (Kilpeläinen et al., 2019). Had the study only been performed in Europeans, these effects would not have been discovered.
Even when diverse populations are studied, specific gene effects in these populations may not be evident, as often diverse populations are studied only as part of large meta-analyses that estimate associations from combined data. The result of this analytical strategy is to identify variants that have mostly similar effects across populations, but it can reduce the ability to detect population-specific genetic risk factors. Although studies that perform meta-analyses clearly discover true risk variants, they often fail to identify those variants that differ in frequency among populations, thereby missing an unknown proportion of the genetic risk.
Polygenic risk scores (PRS) are obtained by computing the effect size of thousands of genetic variants from a discovery sample, then combining and applying them to the genetic profiles from other individuals to predict risk of complex disease. It has been recently claimed that PRS can attain cumulative effect sizes comparable to monogenic diseases. PRS have been used to assess European individuals at high genetic risk (odds ratio > 3) for polygenic diseases such as coronary artery disease (8% of the population) and Type 2 diabetes (3.4% of the population) (Khera et al., 2018). For the reasons discussed above for complex disease (e.g., differences in LD and heterogeneity), PRS may not be transferable across diverse populations. Indeed, inconsistencies in the directions of effect of risk variants have been observed across ethnic groups (Martin et al., 2017). In general, these limitations on transferability of PRS will underestimate or overestimate disease risk in understudied populations. For instance, a UK study of PRS for schizophrenia showed a 10-fold higher score in Africans and African-Americans than in Europeans, but this does not reflect true disease risk, indicating that the PRS was not informative across populations (Curtis, 2018). Thus, there is an urgent need to determine the accuracy of PRS replicability before this approach is implemented in the clinic.
The Significance of Diversity for Pharmacogenetics and Pharmacogenomics
Genetic variation among populations can affect how efficacious a drug is or how likely it is to cause adverse events. For example, warfarin, the most prescribed oral anticoagulant worldwide, has a narrow therapeutic range, and dose requirements vary considerably between patients, posing a treatment challenge. Inter-individual variation in dosage effects is influenced by single nucleotide polymorphisms (SNPs) in CYP2C9, VKORC1, CYP4F2, and another variant near the CYP2C gene cluster. Algorithms incorporating genotype information exist (although not widely implemented), to administer the most appropriate warfarin dosage. Studies have shown that in Europeans the proportion of variance in drug metabolism explained by SNPs in the CYP2C9, VKORC1, and CYP4F2 genes is 18%, 30%, and 11%, respectively (Johnson et al., 2017); however, in patients of African descent these variants explain much less of the differences in drug metabolism. Hence, the algorithms derived from Europeans do not translate into better and safer treatment across ethnic groups. Identification of genetic variants that influence drug metabolism across global populations is needed to accurately predict drug response in individuals of diverse ethnicities.
G6PD deficiency is another important example of how genetic variation can affect drug safety. Individuals who are enzyme deficient are at risk of hemolysis due to several drugs, including some used to treat malaria, e.g., primaquine. Neglecting genetic differences among populations has consequences, most importantly for those subjects who are at risk of serious side effects. In 2008, a failure to properly take into account deficiency-causing mutations in subSaharan Africa (where G6PD deficiency can reach a frequency of 25% due to protection against malaria infection) led to the withdrawal of an effective antimalarial drug combination (chlorproguanil-dapsone), present on the market since 2003. This happened even though the combination could have been safely used in G6PD non-enzymatically deficient individuals (Luzzatto, 2010).
A recent WGS study of bronchodilator drug response (BDR) to albuterol in 1,440 children with asthma discovered associations with variants near or within plausible candidate genes (Mak et al., 2018). Samples were selected from patients who were low and high responders to albuterol, an “extreme phenotype” design which increases power to detect association with rare variants. The subjects were Americans of Mexican, Puerto Rican, and African ancestry; the latter two populations present both the highest asthma prevalence and mortality and the lowest albuterol BDR. Mexican Americans have amongst the lowest prevalence of asthma in the United States. The study, supported by functional data, led to the identification of both population-specific and shared variants (rare and common) associated with BDR. Yet a major hurdle arose: the lack of access to comparable cohorts of similar age and ethnicity for replication. This example underscores the real need to increase the number of studies of ethnically diverse populations in bio-medical research.
Challenges and Future Directions
It is clear that patterns of genetic variation among populations can affect both disease risk and treatment efficacy and safety. Yet, a majority of studies still occur in European ancestry populations and the results can have limited utility across populations. This bias effectively translates into poorer disease prediction and treatment for individuals of under-represented ancestries. Importantly, studying diverse populations increases our ability to broadly understand genetic disease architectures that will, ultimately, lead to increased precision in medical care.
In this commentary we have focused on genetic variation, but there are other types of variation among populations that influence phenotypic diversity. For example, the transcriptome, proteome, metabolome, and microbiome, which can all influence disease risk, are affected by genomic differences, but they also capture environmental differences, all of which affect disease susceptibility and outcomes. Failure to reach beyond Europeans can severely limit our knowledge base. Unfortunately, the Genotype-Tissue Expression (GTEx) project reflects the same bias that GWAS does, with more than 85% of samples being of European descent.
In the United States there are ongoing efforts to include minority populations in biobank initiatives that have the capacity to yield extensive genetic data, including whole-exome and genome sequencing, and to connect genetic profiling to a wealth of electronic health records (Rader and Damrauer, 2016). The access to such a large amount of information opens the possibility of complementing the phenotype to genotype approach, typical of a GWAS, to its reverse: a genotype to phenotype strategy (PheWAS), where, for example, a given variant is tested for association with many recorded phenotypes (the “phenome”) due to pleiotropy. Although these resources can be useful in understanding and addressing health issues of minorities in high-income countries and thereby decrease some disparities, they do not recapitulate the genetic and environmental variation present in low- and middle-income countries (LMICs). Thus, there is a need to include the many overlooked populations that have so far been excluded from genetic research and its potential benefits.
Despite the plea to include more diverse sampling in genomic studies and the critical knowledge these studies can bring, we recognize that obstacles remain. Recruiting diverse populations can be difficult in many settings, in some cases due to a mistrust in biomedical research stemming from past experiences of exploitation. To prevent this, local ethics committees have a key role in reviewing and approving proposals, securing compliance with guidelines, and the implementation of a broad range of requirements, from appropriate treatment of study subjects to full involvement of local stakeholders in all stages of research. To generate quality genetic associations, it is essential to obtain reliable phenotype data, which in turn requires both personnel and adequate facilities. In places where diversity may be the greatest (i.e., LMICs), investment in infrastructure and professional training is a primary need.
These and other challenges require a concerted effort by both the research communities and funding agencies to include ethnically diverse populations in human genetics studies. Such access and subsequent research will improve our understanding of the genetics of disease and improve the quality of health care, for all.
ACKNOWLEDGMENTS
We thank Dana Crawford and Reed Pyeritz for critical feedback, and we thank Jacob Haut and Matthew Hansen for their assistance with constructing Figure 2. S.A.T. is supported by NIH grants 1R01DK104339, and 1R01GM113657 and an American Diabetes Association Pathway to Stop Diabetes Grant Pathway to Diabetes Visionary Award 1-19-VSN-02.
REFERENCES
- Buxbaum J, Jacobson DR, Tagoe C, Alexander A, Kitzman DW, Greenberg B, Thaneemit-Chen S, and Lavori P (2006). Transthyretin V122I in African Americans with congestive heart failure. J. Am. Coll. Cardiol 47, 1724–1725. [DOI] [PubMed] [Google Scholar]
- Curtis D (2018). Polygenic risk score for schizophrenia is more strongly associated with ancestry than with schizophrenia. Psychiatr. Genet 28, 85–89. [DOI] [PubMed] [Google Scholar]
- Freedman BI, Limou S, Ma L, and Kopp JB (2018). APOL1-Associated Nephropathy: A Key Contributor to Racial Disparities in CKD. Am. J. Kidney Dis 72 (5S1), S8–S16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genovese G, Friedman DJ, Ross MD, Lecordier L, Uzureau P, Freedman BI, Bowden DW, Langefeld CD, Oleksyk TK, Uscinski Knob AL, et al. (2010). Association of trypanolytic ApoL1 variants with kidney disease in African Americans. Science 329, 841–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JA, Caudle KE, Gong L, WhirlCarrillo M, Stein CM, Scott SA, Lee MT, Gage BF, Kimmel SE, Perera MA, et al. (2017). Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Pharmacogenetics-Guided Warfarin Dosing: 2017 Update. Clin. Pharmacol. Ther 102, 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, and Kathiresan S (2018). Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet 50, 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilpeläinen TO, Bentley AR, Noordam R, Sung YJ, Schwander K, Winkler T.W. c H., Chasman DI, Manning A, Ntalla I, et al. ; Lifelines Cohort Study (2019). Multiancestry study of blood lipid levels identifies four loci interacting with physical activity. Nat. Commun 10, 376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim ET, Würtz P, Havulinna AS, Palta P, Tukiainen T, Rehnström K, Esko T, Mägi R, Inouye M, Lappalainen T, et al. ; Sequencing Initiative Suomi (SISu) Project (2014). Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet. 10, e1004494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzzatto L (2010). The rise and fall of the antimalarial Lapdap: a lesson in pharmacogenetics. Lancet 376, 739–741. [DOI] [PubMed] [Google Scholar]
- Mak ACY, White MJ, Eckalbar WL, Szpiech ZA, Oh SS, Pino-Yanes M, Hu D, Goddard P, Huntsman S, Galanter J, et al. ; NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium (2018). Whole-Genome Sequencing of Pharmacogenetic Drug Response in Racially Diverse Children with Asthma. Am. J. Respir. Crit. Care Med 197, 1552–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AR, Gignoux CR, Walters RK, Wojcik GL, Neale BM, Gravel S, Daly MJ, Bustamante CD, and Kenny EE (2017). Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. Am. J. Hum. Genet 100, 635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padoa C, Goldman A, Jenkins T, and Ramsay M (1999). Cystic fibrosis carrier frequencies in populations of African origin. J. Med. Genet 36, 41–44. [PMC free article] [PubMed] [Google Scholar]
- Piel FB, Steinberg MH, and Rees DC (2017). Sickle Cell Disease. N. Engl. J. Med 376, 1561–1573. [DOI] [PubMed] [Google Scholar]
- Popejoy AB, and Fullerton SM (2016). Genomics is failing on diversity. Nature 538, 161–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rader DJ, and Damrauer SM (2016). “Pheno”menal value for human health. Science 354, 1534–1536. [DOI] [PubMed] [Google Scholar]
- Saleheen D, Natarajan P, Armean IM, Zhao W, Rasheed A, Khetarpal SA, Won HH, Karczewski KJ, O’Donnell-Luria AH, Samocha KE, et al. (2017). Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature 544, 235–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart C, and Pepper MS (2017). Cystic Fibrosis in the African Diaspora. Ann. Am. Thorac. Soc 14, 1–7. [DOI] [PubMed] [Google Scholar]
- Tishkoff SA, Reed FA, Friedlaender FR, Ehret C, Ranciaro A, Froment A, Hirbo JB, Awomoyi AA, Bodo JM, Doumbo O, et al. (2009). The genetic structure and history of Africans and African Americans. Science 324, 1035–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]