

Abstract
The emergence of resistance to available antileishmanial drugs advocates identification of new drug targets and their inhibitors for visceral leishmaniasis. Here, we identified Leishmania donovani heat shock protein 78 (LdHSP78), a putative caseinolytic protease, as important for parasite infection of host macrophages and a potential therapeutic target. Enrichment of LdHSP78 in infected humans, hamsters, and parasite amastigotes suggested its importance for disease persistence. Heterozygous knockouts of L. donovani HSP78 (LdHSP78+/−) and Leishmania mexicana HSP78 (LmxHSP78+/−) were generated using a flanking UTR-based multifragment ligation strategy and the CRISPR-Cas9 technique, respectively to investigate the significance of HSP78 for disease manifestation. The LdHSP78+/− parasite burden was dramatically reduced in both murine bone marrow-derived macrophages and hamsters, in association with enrichment of proinflammatory cytokines and NO. This finding implies that LdHSP78+/− parasites cannot suppress immune activation and escape NO-mediated toxicity in macrophages. Furthermore, phosphorylation of the mitogen-activated protein kinase p38 was enhanced and phosphorylation of extracellular signal-regulated kinase 1/2 was decreased in cells infected with LdHSP78+/− parasites, compared with WT parasites. Virulence of the LdHSP78+/− strain was restored by episomal addition of the LdHSP78 gene. Finally, using high-throughput virtual screening, we identified P1,P5-di(adenosine-5′)-pentaphosphate (Ap5A) ammonium salt as an LdHSP78 inhibitor. It selectively induced amastigote death at doses similar to amphotericin B doses, while exhibiting much less cytotoxicity to healthy macrophages than amphotericin B. In summary, using both a genetic knockout approach and pharmacological inhibition, we establish LdHSP78 as a drug target and Ap5A as a potential lead for improved antileishmanial agents.
Keywords: Leishmania, transgenic parasite, molecular modeling, high-throughput virtual screening, heat shock protein 78 (HSP78), Ap5A, caseinolytic protease, kinase signaling, macrophage, innate immunity, drug screening
Under general as well as inducible environmental stresses, organisms respond by synthesizing a group of chaperons called heat shock proteins (HSPs) (1). HSPs facilitate survivability of organisms by assisting in maturation, activation, translocation, and degradation of misfolded nascent proteins (1). Interestingly, for some pathogens like Leishmania donovani, which have a digenetic life cycle, inducible heat stress is a vital part of their biology. They are transmitted from the poikilothermic sand fly gut to the homeothermic mammalian macrophage, which involves a shift in temperature of almost 10 °C. This temperature upshift is a key trigger for the differentiation of Leishmania donovani promastigotes (sand fly stage) to amastigotes (mammalian stage) (2). Heat stress unleashed by the macrophage leads to enrichment of various Leishmania HSPs, which determine the pathology of the disease. In cutaneous leishmaniasis, Leishmania major can tolerate heat shock up to 35 °C, which results in tropical skin lesions; in visceral leishmaniasis (VL), L. donovani can sustain temperatures as high as 37–40 °C, which correlates with visceralization of the parasites (3). Besides management of heat stress, protozoan HSPs play a substantial role in virulence. A recent transcriptomic analysis revealed a positive correlation between the Plasmodium HSPs and regulatory interleukin levels in patients with malignant malaria (4).
In other organisms, HSPs belonging to the caseinolytic protease B (ClpB) group, like bacterial ClpB (Escherichia coli), yeast (Saccharomyces cerevisiae) HSP104, and plant HSP101, have extraordinary capacity to rescue disaggregated proteins during severe heat stress, which is otherwise lethal for the organisms (5, 6). To date, functional study of only one ClpB protein, HSP100 (LmxHSP100), has been reported in Leishmania biology (6). It has been shown that heat-stressed parasites are strongly enriched with LmxHSP100, and LmxHSP100−/− null mutants of L. major are unable to establish infection within murine peritoneal macrophages, as well as BALB/c mice (6). LdHSP100−/− null mutants of L. donovani showed impaired amastigote development, and LdHSP100 is also found to be involved in the transport of exosome-mediated virulence factors during infection (6).
A major global challenge for antileishmanial therapy is overcoming chemoresistant and treatment failure cases arising from treatment with the classical antileishmanial agents available on the market, such as antimonial salts, amphotericin B (AmB), miltefosine, and paromomycin (7, 8). Most of these drugs are repurposed and non-target based. Recent studies have highlighted protozoan HSPs, such as HSP90 in Plasmodium falciparum and Trypanosoma evansi, as potential drug targets. These factors prompted us to explore the role of LdHSP78 (UniProt code LdBPK_020680), a putative member of the ClpB protease family, as a potential drug target in L. donovani infection.
A BLAST search revealed that LdHSP78 expresses a 78-kDa protein orthologous to LmjF27.2630 of L. major and Tb927.2.3030 of Trypanosoma brucei (6, 9). Significant sequence structural similarity has been observed between LdHSP78 and a ClpB protein from Thermus thermophilus that is composed of an N-terminal domain, two nucleotide-binding domains (NBDs) (NBD1 and NBD2), and a middle coiled coil domain (MD region) (10, 11). Here, we propose the probable prominence of LdHSP78 for VL progression. The importance of HSP78 for survival of the parasites was assessed by HSP78 knockout generation in L. donovani (LdHSP78+/−) and Leishmania mexicana (LmxHSP78+/−), with the virulence phenotype of LdHSP78+/− parasites being rescued by episomal overexpression of LdHSP78 (resKO). However, the lethality of null mutants (Ld/LmxHSP78−/−) and successful conditional knockout of LdHSP78 generation suggested probable essentiality of the gene for Leishmania, but more experimental validations are awaited. Infectivity and the capacity to suppress the antileishmanial immune responses activated by host macrophages were determined by infecting murine bone marrow-derived macrophages (BMDMs) and Syrian golden hamsters with WT, LdHSP78+/−, and resKO parasites. Furthermore, P1,P5-di(adenosine-5′)-pentaphosphate (Ap5A) ammonium salt, a synthetic ATP analog, was identified as a pharmacological inhibitor of LdHSP78 protein. The antileishmanial efficacy of Ap5A was evaluated and compared with that of the generic antileishmanial drug AmB in healthy BMDMs, promastigotes, and amastigotes. Notably, Ap5A induced much less collateral damage to host cells, unlike AmB. Hence, our study reveals LdHSP78 as a potential antileishmanial therapeutic target while identifying its inhibitor, Ap5A, as a promising lead molecule against VL.
Results
LdHSP78 is a stress-regulating protein that is enriched during VL
LdHSP78 is a putative ATP-dependent Clp subunit-containing protein (UniProt code LdBPK_020680). It is encoded by a single exon located at L. donovani chromosome 2 and is highly conserved throughout the Leishmania spp. while being nonorthologous to human proteins (no human orthologs with >20% sequence identity are available). Involvement of HSP78 in VL was initially perceived from semiquantitative PCR of LdHSP78 transcripts from L. donovani-infected BMDMs (Fig. 1A). Moreover, enrichment of the same was detected in the peripheral blood, spleen, and liver of 3-month-infected golden Syrian hamsters (Fig. 1B). A study conducted with a small cohort of VL patient blood samples correlated with the mRNA expression profile of animals (Fig. 1B). To establish chaperone-like properties of LdHSP78, we mimicked the host phagosomal microenvironment by growing L. donovani promastigotes in M199 (pH 4.0 and pH 5.5) at 37 °C under axenic conditions. Almost 40–55-fold higher expression of LdHSP78 mRNA was obtained at pH 5.5 and 37 °C, compared with that obtained at pH 7.0 and 22 °C (Fig. 1C). These data collectively indicated that HSP78 levels were higher not only during physiological stress of the parasites but also under VL conditions.
Figure 1.
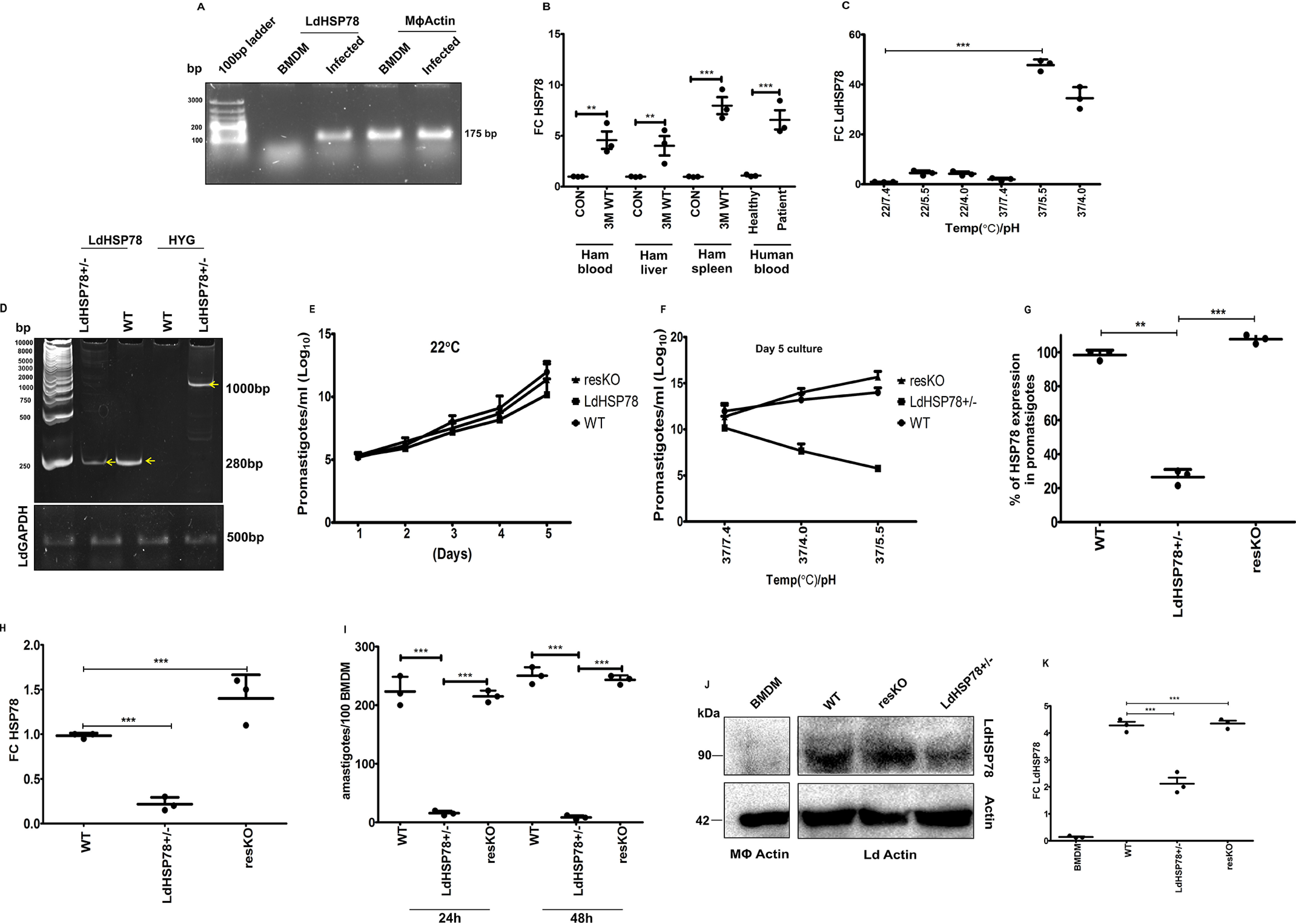
LdHSP78 is an essential chaperone for infection. A, semiquantitative PCR of LdHSP78 mRNA in L. donovani-infected BMDMs. B, RT-PCR of LdHSP78 mRNA from 3-month-infected hamster (Ham) spleen, liver, and blood and VL patient peripheral blood. The Ct value of LdHSP78 was normalized against that of L. donovani GAPDH and plotted as fold change (FC) (2−ΔΔCt). C, RT-PCR of LdHSP78 mRNA from promastigotes cultured at different temperature and pH values. The Ct value of LdHSP78 was normalized against that of L. donovani GAPDH and plotted as fold change (FC) (2−ΔΔCt). D, partial deletion of LdHSP78 validation by urea-PAGE-based separation of PCR products, amplified from WT (control transfected) and LdHSP78+/− parasites (L. donovani actin is the loading control). E and F, Determination of LdHSP78+/− promastigote fitness under (E) normal conditions (22 °C and pH 7.4) and (F) amastigote-like conditions (37 °C and pH 4.0, 5.5, and 7.4). Growth kinetics of promastigotes were determined from 5-day cultures (to obtain maximum growth). G and H, RT-PCR-based determination of LdHSP78 mRNA loads of WT, LdHSP78+/−, and resKO promastigotes (G) and amastigotes (H) in infected BMDMs. I, Giemsa staining of BMDMs infected with WT, LdHSP78+/−, and resKO parasites for 24 and 48 h at a 1:10 ratio. J and K, Western blotting of lysates from uninfected BMDMs (negative control) and WT, LdHSP78+/−, and resKO promastigotes (loading controls were macrophage actin and L. donovani actin). Fold changes of LdHSP78 were calculated from densitometric values of the bands. Each group is represented by three samples, and each experiment was performed in triplicate. Statistical significance (**, p < 0.01; ***, p < 0.001) was calculated using one-way analysis of variance followed by Dunnett's multiple-comparison test and unpaired two-tailed t test followed by Welch correction (each dot is representative of a single sample; n = 3 samples per group). Data represent mean ± S.D. (GraphPad Prism v5.0).
Generation of HSP78 transgenic cell lines
LdHSP78 knockout parasite cell lines were generated using both a flanking UTR-based multifragment ligation strategy (Fig. S1A) and the CRISPR-Cas9 technique (Fig. S1N). For UTR-mediated knockout generation, 5′ (997 bp) and 3′ (946 bp) flanking regions (UTRs) of LdHSP78 were directionally cloned (Fig. S1, A and B) at both sides of the hygromycin (HYG) (3245 bp) and puromycin (PUR) (3005 bp) cassettes, using specialized primers that incorporated overhangs with unique SfiI digestion sites at each fragment end (Fig. S1, D and E). The PacI-digested linearized 5397-bp HYG construct was transfected in log-phase parasites for generation of heterozygous knockouts (Fig. S1E). A separate population of WT parasites was maintained as mock-transfected cells (transfected with empty vector). LdHSP78-HYG (LdHSP78+/−) cells were cultured in 50 μg/ml HYG-supplemented M199. Next, pure LdHSP78+/− clones were collected by isolating single colonies from the serially diluted LdHSP78+/− cells grown on HYG-supplemented M199 agar.
Genomic DNA was isolated from log-phase WT and LdHSP78+/− promastigotes, and PCR was performed using primers spanning the flanking region to the ORFs of HSP78 and HYG (Table S1, primers 5–8). The presence of both HSP78 and HYG in genomic DNA of LdHSP78+/− parasites suggested successful partial replacement of one LdHSP78 allele with the HYG cassette (Fig. 1D). The 5′ UTR (997 bp) and 3′ UTR (946 bp) flanking regions, which had been chosen for knockout generation, were scanned against all 36 chromosomes of L. donovani. Sequence identity and coverage match of 100% were obtained only at chromosome 2 loci flanking the HSP78 gene (Fig. S1H), and no sequence overlap with other chromosomes was observed, confirming the least off-target effect and high efficacy of the flanking region-mediated deletion technique.
LdHSP78 was overexpressed in LdHSP78+/− cell lines (resKO cell lines) by cloning the episomal vector pXG-GFP(+) (generously provided by Stephen Beverly, Washington University) at the BamHI and EcoRV sites (Fig. S1, I–L). L. donovani is a diploid organism; therefore, to completely deplete LdHSP78, two drug cassettes (HYG and PUR) were transfected successively. PacI-digested linearized 5397-bp HYG (Fig. S1E) and 4974-bp PAC (Fig. S1F) cassettes were serially transfected in log-phase parasites for generation of null mutants. Attempts were made to culture LdHSP78-HYG/PAC (LdHSP78−/−) cells in 50 μg/ml HYG- and 20 μg/ml PUR-supplemented medium. However, live parasite counts confirmed that null mutants started dying with days of culturing. Our multiple attempts to obtain live parasites from HYG- and PUR-supplemented M199 medium failed probably due to the essential nature of the LdHSP78 gene for the promastigotes.
Parasites bearing an extrachromosomal copy of genes generally allow deletion of the chromosomal copies of essential genes. Therefore, promastigotes were transfected with the pXG-HSP78 episome (pXG-GFP-HSP78-Neo+) to maintain an extra copy of genes, followed by double deletion of the chromosomal copies using HYG and PUR cassettes. The conditional knockout (cond KO- LdHSP78−/−epiHSP78) cells were maintained in the presence of HYG, neomycin (NEO), and PUR drug selection medium for 15 days, followed by depletion of NEO from the medium. Interestingly, we found that, despite NEO selection withdrawal (required for pXG-HSP78 maintenance), cells still grew well and, following 7 days of culture, we isolated genomic DNA. LdHSP78, HYG, NEO, and PUR drug cassettes were PCR amplified. For amplification of HSP78 and NEO genes, primers were designed against their respective ORF regions; for HYG and PUR cassettes, amplification primers spanning the 5′ flanking region of the HSP78 gene and the respective drug ORF regions were designed (Table S1, primers 43–50). PCR data indicated that, in the absence of episomal NEO, LdHSP78−/− cells are holding back the extrachromosomal copy of LdHSP78 for their survival (Fig. S1M).
We also attempted to design null mutants of LmxHSP78 using the CRISPR-Cas9 system (in silico analysis showed >98% sequence homology of LdHSP78 to LmHSP78) in Cas9-positive L. mexicana cell lines (generously provided by Jeremy Mottram's laboratory) (Fig. S1N), which indicated that a HSP78 null mutation is probably lethal in L. mexicana. In this model also, only heterozygous parasites survived in the presence of blasticidin (50 μg/ml) (Fig. S1O). Genomic DNA PCR showed successful replacement of one LmHSP78 allele with the blasticidin cassette (Fig. S1P). Western blot analysis depicted significant reduction of LmHSP78 in heterozygous cell lines, compared with WT cell lines (Fig. S1Q). These findings indicated that HSP78 might be essential for survival of the parasite, although more experimental validations are required to confirm this.
LdHSP78 is an essential chaperon for infection persistence in BMDMs
Chaperone-like activity of HSP78 was already indicated from its enrichment during pH and heat stress in L. donovani promastigotes (Fig. 1C). Physical fitness of WT, LdHSP78+/−, and resKO promastigotes was studied initially at 22 °C, which indicated equivalent growth rates for all three cell lines at day 5 (Fig. 1E). Subsequently, it was found that, at pH 5.5 and 37 °C, the growth rate of LdHSP78+/− parasites was reduced significantly, denoting the role of HSP78 in pH and heat stress management (Fig. 1F). Quantitative expression of LdHSP78 mRNA obtained from promastigotes indicated ∼80% reduction of the mRNA in LdHSP78+/− promastigotes and almost 95% restoration of mRNA in resKO promastigotes (Fig. 1G). Analysis of LdHSP78 mRNA levels from BMDMs infected with WT, LdHSP78+/−, and resKO parasites also depicted a similar profile of LdHSP78 transcripts in LdHSP78+/− amastigotes (Fig. 1H). This was accompanied by a significant drop in the parasite burden at 24 h and 48 h postinfection in LdHSP78+/− parasite-infected BMDMs (Fig. 1I). We successfully reestablished the virulence of LdHSP78+/− parasites by episomal overexpression of LdHSP78. Complete restoration of LdHSP78 mRNA was found in resKO parasite-infected BMDMs (Fig. 1H). Investigation of the infection index of resKO parasites in BMDMs also corroborated with mRNA data, establishing the significance of LdHSP78 for infection in vitro (Fig. 1I).
Protein level validation showed depletion of LdHSP78 in heterozygous cell lines
The most immunodominant peptide (127 amino acids; indicated in purple in Fig. S2A) of LdHSP78 was codon optimized and subcloned from pGEX-6P.1-Amp+ to the pET41a-Kan+ vector (Fig. S2, B–D). The hydrophobicity of the peptide was analyzed using a hydropathy plot, which indicated that the peptide sequence chosen is enriched with hydrophilic amino acids and ideal as a B-cell epitope (Fig. S2E). The epitope was then overexpressed and purified in the native form using nickel-nitrilotriacetic acid resin, and BALB/c mice were immunized with it (Fig. S2, F–H). Sera collected after three boosters were used for immunoblotting of the purified epitope, to analyze the quality of the generated antibody (Fig. S2I). Thereafter, partial depletion of LdHSP78 and LmHSP78 (previously mentioned) was validated by Western blotting from parasite lysates obtained from promastigote cell lines generated using flanking UTR-mediated and CRISPR-Cas9–mediated knockout technologies, respectively. Almost 50% or more depletion of HSP78 protein was obtained in LmHSP78 (Fig. S2Q) and LdHSP78+/−, while complete restoration of LdHSP78 protein was achieved in resKO parasites (Fig. 1, J and K). The specificity of anti-HSP78 antibody was cross-validated using BMDM lysate, and no immunoreactive band was obtained (Fig. 1, J and K).
Down-regulation of proinflammatory cytokine synthesis during LdHSP78+/− infection in BMDMs
One of the major issues with antileishmanial therapy is overcoming immunosuppression induced by parasites. Our next question was whether partial depletion of LdHSP78 would result in inability of parasites to overcome host immune activation. Significant down-regulation of proinflammatory cytokines and up-regulation of anti-inflammatory cytokines were observed in WT parasite-infected BMDMs. In contrast, in LdHSP78+/− parasite-infected macrophages, mRNA and protein levels of anti-inflammatory cytokines, such as transforming growth factor β (TGFβ) (Fig. 2, A and E), IL10 (Fig. 2, B and F), and IL27A (Fig. 2, C and G), were significantly depleted, whereas IL12A (Fig. 2, D and H) was significantly enriched. Thus, cytokine profiling revealed that proinflammatory cytokines were significantly inhibited during LdHSP78+/− parasite infection.
Figure 2.

Cytokine profiling of BMDMs infected with LdHSP78-depleted and -overexpressing parasites indicated sustained proinflammatory cytokine responses. RT-PCR (A–D) and sandwich ELISAs (E–H) of proinflammatory (IL12A) and anti-inflammatory (IL10, IL27A, and TGFβ) cytokines from WT, LdHSP78+/−, and resKO parasite-infected BMDMs. Eight hours after infection, total RNA was isolated from BMDMs and subjected to RT-PCR. Data were normalized using mouse GAPDH as a control gene. For TGFβ and IL10 ELISAs, culture supernatants were collected 12 h after infection; for IL12A and IL27A ELISAs, 48-h postinfection supernatants were collected. FC, fold change. Each experimental set is representative of three samples, and error bars are mean ± S.D. Statistical significance was analyzed using one-way analysis of variance followed by Dunnett's multiple comparison test. **, p < 0.01; ***, p < 0.001.
MAPK activation and NO generation instigated LdHSP78+/− parasite clearance from BMDMs
Extracellular signal-regulated kinase 1/2 (ERK1/2) and p38 mitogen-activated protein kinase (MAPK) are the two major MAPKs that are involved in the regulation of cytokine profiles and NO synthesis (12). These MAPKs require phosphorylation at their specific amino acids for their activation. Infection with WT parasites induced significant up-regulation of phospho-ERK1/2 (p-p44/p42, Thr202/Tyr204) (Fig. 3, A and E) and decreased levels of phospho-p38 (Thr180/Tyr182) (Fig. 3, B and F), while total MAPK expression levels remain constant. In contrast, reverse activation profiles of these proteins were found in LdHSP78+/− parasite-infected macrophages (Fig. 3).
Figure 3.

Expression of phospho-p38, iNOS, and NO increased during LdHSP78+/− parasite infection in BMDMs. A–H, lysates were collected from BMDMs infected for 24 h with WT, LdHSP78+/−, and resKO parasites and Western blotted for phospho-ERK1/2 and total ERK1/2 (phospho-ERK1/2 bands were normalized to ERK1/2 bands) (A and E), phospho-p38 and total p38 (phospho-p38 bands were normalized to p38 bands) (B and F), arginase 1 (Arg1) (C and G), and iNOS (D and H) (arginase 1 and iNOS bands were normalized to β-actin bands). Lipopolysaccharide (LPS) (10 μg/ml) was the positive control and mouse β-actin was the loading control. Scattered plots adjacent to each blot represented mean densitometric values of three blots performed for each protein. I, the Griess assay was performed with culture supernatants of WT, LdHSP78+/−, and resKO parasite-infected BMDMs collected 24 and 48 h postinfection. Each experimental set is representative of three samples, and error bars are mean ± S.D. Statistical significance was analyzed using one-way analysis of variance followed by Dunnett's multiple-comparison test. *, p < 0.05; **, p < 0.01; ***, p < 0.001.
Phosphorylation of ERK1/2 leads to enrichment of arginase 1 levels, which helps in parasite survival, whereas phospho-p38 MAPK leads to enrichment of inducible NOS (iNOS) expression, which mediates clearance of parasites by generation of NO (13). Therefore, we examined the levels of iNOS and arginase 1 expression, which suggested that LdHSP78+/− parasites were unable to exhaust iNOS and to enhance arginase 1 expression (Fig. 3, C, D, G, and H). Significant amounts of NO generation were obtained from BMDMs infected with the mutant parasites, compared with the WT ones (Fig. 3I). After overexpression of LdHSP78 in resKO parasites, the immunosuppressive nature of parasites was significantly restored, with concomitant suppression of phospho-p38, iNOS and NO generation.
Partial loss of HSP78 affects the infectivity index of parasites in an in vivo experimental infection model
Golden Syrian hamsters are a well-established model that successfully mimics the pathophysiology of human VL infection (14, 15). Before infecting animals, the percentages of infective metacyclic promastigotes were determined in all of the parasite cultures by lipophosphoglycan-independent Ficoll density gradient analysis using 10–40% Ficoll gradients (Fig. S3A). Phase-contrast microscopy showed that >60% of metacyclics were present in 6–9-day stationary-phase promastigotes of WT, LdHSP78+/−, and resKO parasites (Fig. S3B). RT-PCR expression of the L. donovani SHERPIN gene revealed comparable amounts of successful metacyclic formation (Fig. S3C). Thus, we ensured that all of the following infections were carried out with stationary-phase metacyclic parasites only.
To understand the role of LdHSP78 in animal infection, we first assessed the pathophysiological symptoms of the hamsters infected with three different groups of parasites, up to 3 months postinfection. Similar to infected humans, VL, WT, and resKO parasite-infected hamsters exhibited progressive splenomegaly and hepatomegaly, with high amastigote burdens in organ smears (Fig. 4A and Table S2). In LdHSP78+/− parasite-infected hamsters, splenic and hepatic lesion formation was delayed significantly, with lower amastigote burdens in spleen and liver stamp smears (Fig. 4A and Table S2). Leishman Donovan unit data indicated that infection with WT and resKO parasites resulted in almost 20-fold greater splenic macrophage parasite loads and 25-fold greater hepatic macrophage parasite loads, compared with LdHSP78+/− parasite infection (Fig. 4B). Since detection of live parasites in the organs provides a better depiction of disease severity and the virulence of parasites, limiting dilution assays (LDAs) with 1 mg/ml minced spleen and liver tissues were performed for animals infected with three different cell lines. LDA data indicated severe declines in parasite burdens, from 1010 WT parasites to 103 LdHSP78+/− parasites in spleen and from 1015 WT parasites to 104 LdHSP78+/− parasites in liver 12 weeks postinfection (Fig. 4C). Interestingly, in resKO parasite-infected animals, there was almost complete restoration of LDA results in both spleen and liver, indicating that LdHSP78 overexpression restored virulence of the LdHSP78+/− parasites significantly (Fig. 4C).
Figure 4.
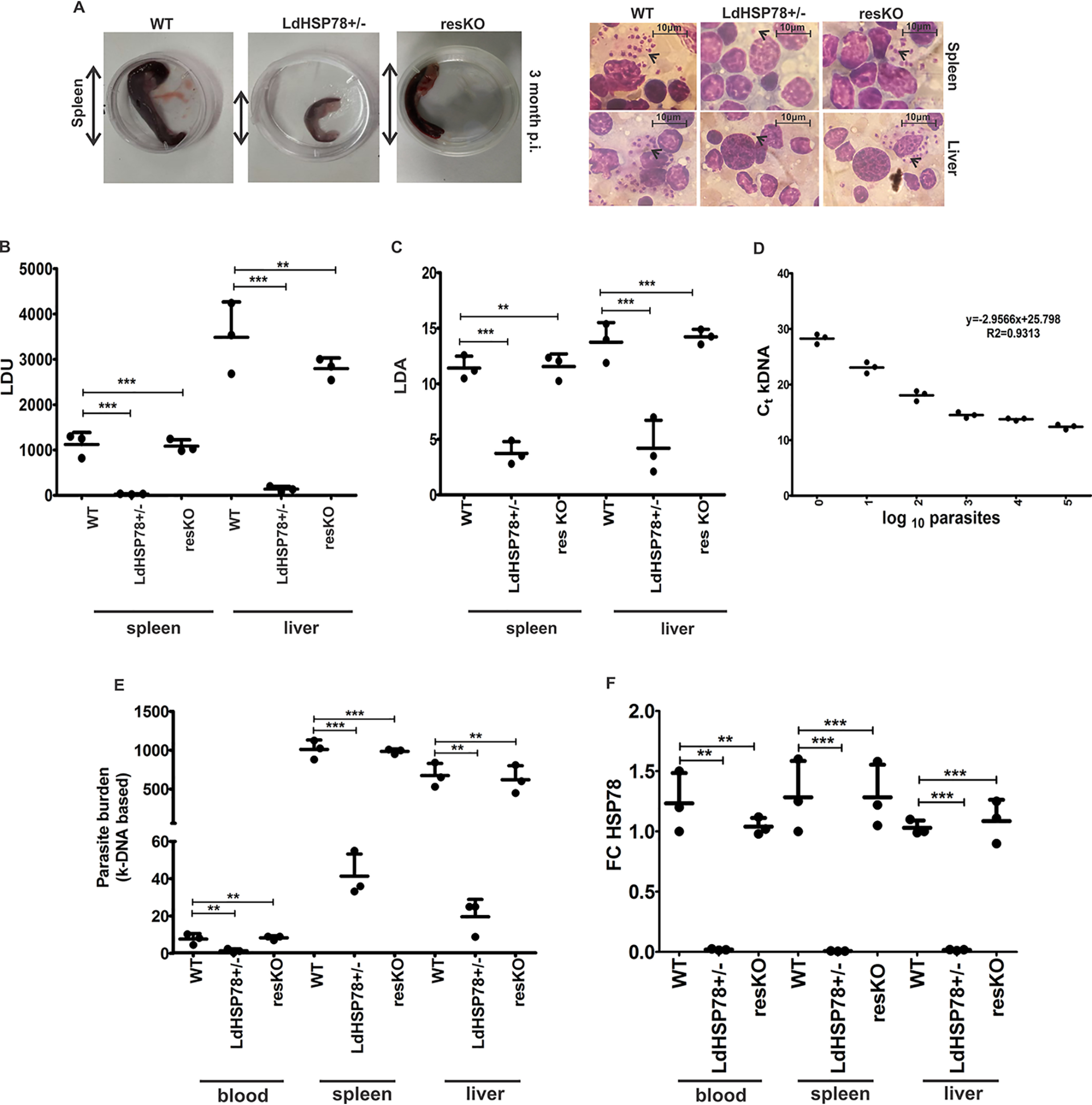
LdHSP78+/− parasites showed compromised infectivity in hamsters. A, comparative pathophysiology of WT, LdHSP78+/−, and resKO parasite-infected spleens procured from hamsters 3 months postinfection (p.i.), showing splenomegaly during WT and resKO parasite infection. B, Leishman-Donovan units (LDU) from phase-contrast microscopy of Giemsa-stained images (scale bar, 10 μm). C, LDA results (log10 parasite numbers/ml) for WT, LdHSP78+/−, and resKO parasite-infected spleen and liver. D, genomic DNA isolated from WT parasites was serially diluted, and RT-PCR for kDNA and L. donovani GAPDH was performed. Ct values obtained were used to prepare the standard curve of kDNA versus parasite count in log10 count/ml. E, Ct values of kDNA from hamster whole blood, spleen, and liver were obtained and plotted in the standard curve of kDNA to calculate the parasite burden of each organ, and results were plotted in this scatter graph. F, RT-PCR of LdHSP78 mRNA from infected hamster organs and whole blood was performed. FC, fold change. Each experimental set is representative of three samples, and error bars are mean ± S.D. Statistical significance was analyzed using one-way analysis of variance followed by Dunnett's multiple-comparison test. **, p < 0.01; ***, p < 0.001.
Decreased fitness of LdHSP78+/− parasites within hamsters could be confirmed by highly sensitive kinetoplastidial DNA (kDNA)-dependent determination of parasite counts in peripheral blood, spleen, and liver. Parasites were serially diluted from 105 parasites/ml to 1 parasite/ml, and a kDNA standard curve was prepared following a method mentioned elsewhere (Fig. 4D) (16). Genomic DNA isolated from hamster peripheral blood, spleen, and liver were then subjected to RT-PCR for kDNA amplification. Based on kDNA Ct values obtained from organs, almost 10 ± 2 parasites/ml WT parasites, 2 ± 1 parasites/ml LdHSP78+/− parasites, and 15 ± 2 parasites/ml resKO parasites in animal blood were obtained (Fig. 4E). Moreover, parasite loads in LdHSP78+/− parasite-infected organs were much lower than those in WT and resKO parasite-infected animals (Fig. 4E and Table S3). Subsequently, RT-PCR data for LdHSP78 mRNA from blood and organs of WT, LdHSP78+/−, and resKO parasite-infected animals depicted almost 5-fold (in blood), 10-fold (in spleen), and 4.5-fold (in liver) reductions during LdHSP78+/− parasite infection, compared with WT and resKO parasite infections (Fig. 4F). The fold changes indicated that the proliferation rate of parasites was significantly compromised in hamsters after partial deletion of LdHSP78.
Protective cytokines were enriched during LdHSP78+/− parasite infections
Establishment of VL in hamsters largely depends on the fine balance between Th1 and Th2 responses in spleen (17). L. donovani parasites cause enrichment of the immunosuppressive cytokines like IL10 and TGFβ, which supports parasite persistence, whereas parasite clearance is triggered by enrichment of IL12 and IFNγ (17, 18). Findings shown in Fig. 4 suggested that partial deletion of LdHSP78 triggered rapid clearance of mutant parasites in hamsters. Therefore, fold changes in regulatory cytokine mRNA expression in infected hamster blood, spleen, and liver were compared. As indicated by RT-PCR, IL10 and TGFβ were depleted 12 weeks postinfection (Fig. 5, A and B), whereas IFNγ was significantly enriched (Fig. 5C), in blood, liver, and spleen of LdHSP78+/− parasite-infected animals. Cytokine ELISAs also indicated that IL10 titers were exhausted and IL12 was enriched in LdHSP78+/− parasite-infected hamsters, which was completely reversed with resKO parasite infection (Fig. 5, D and E). These findings indicated that, due to enrichment of protective cytokines, LdHSP78+/− parasites were unable to establish infections in hamster organs.
Figure 5.

Protective cytokine responses and NO generation were augmented in LdHSP78+/− parasite-infected hamsters. A–C, IL10 (A), TGFβ (B), and IFNγ (C) levels in whole blood, spleen, and liver of WT, LdHSP78+/−, and resKO parasite-infected hamsters from three groups were measured by RT-PCR. D and E, sandwich ELISAs for IL10 (D) and IL12 (E) were performed with blood samples collected at regular time intervals up to 3 months. F and G, enrichment of iNOS (F) and NO (G) in whole blood, liver, and spleen of hamsters was determined by RT-PCR and Griess assays, respectively. Hamster GAPDH was used as the control for all RT-PCR assays. FC, fold change. Each experimental set is representative of three animals, and error bars are mean ± S.D. Statistical significance was analyzed using one-way analysis of variance followed by Dunnett's multiple-comparison test. *, p < 0.05, **, p < 0.01; ***, p < 0.001.
NO triggered LdHSP78+/− parasites clearance from hamsters
Earlier, we demonstrated that, after genetic depletion of LdHSP78, clearance of L. donovani amastigotes was instigated by macrophage NO (Fig. 3I). Therefore, fold changes in iNOS mRNA in blood, spleen, and liver of infected hamsters were studied, which suggested that iNOS expression was higher in LdHSP78+/− parasite-infected hamsters, compared with WT and resKO parasite-infected hamsters (Fig. 5F). Subsequently, the Griess assay demonstrated time-dependent enrichment of NO in sera of LdHSP78+/− parasite-infected animals, compared with their virulent and rescued parasite-infected counterparts (Fig. 5G). These studies indicated that, in in vivo systems, greater NO enrichment perhaps leads to abrogation of infection in LdHSP78+/− parasite-infected hamsters, compared with WT and resKO parasite infections.
Sequence and structural analysis revealed highly conserved NBDs and Clp domain in LdHSP78
Phylogenetic analysis shows wide representation of the HSP78 protein across various phyla (Fig. S4). The 3D model of the L. donovani HSP78 protein was generated via MODELLER v9.836 (19), with the coordinates of the ClpB protein from Thermus thermophilus (PDB code 1QVR) being used as a potential template. The 3D model was validated using PROCHECK (20) and Verify 3D (21) structure validation tools (Fig. S5). L. donovani HSP78 protein can be distinguished into five domains, e.g. N-terminal domain (positions 1–136), D1 large domain or NBD1 (positions 151–331), D1 small domain (positions 332–535) containing the ClpB linker, D2 large domain or NBD2 (positions 545–756), and D2 small domain (positions 757–817). NBD1 and NBD2 are relatively more conserved than other domains (Fig. 6A) across all phyla. However, ranges of conservation for all five domains are similar in all phyla, as well as in Euglenozoa (contains the Kinetoplastida class) and Ascomycota, the other largest phylum where HSP78 is present (Fig. 6A). The N-terminal domain of HSP78 is found to be mostly α-helical, with parallel β-strands inserted near its N terminus. The N-terminal domain is connected to NBD1 through a flexible linker (positions 137–150). The D1 domain is composed of a set of α-helices along with short β-strands and a long coiled coil structure. The coiled coil structure is known as the ClpB linker, containing two short coiled coil motifs. The C-terminal D2 small domain is a mixed α/β structure and lacks the long coiled coil insertion (Fig. 6A).
Figure 6.
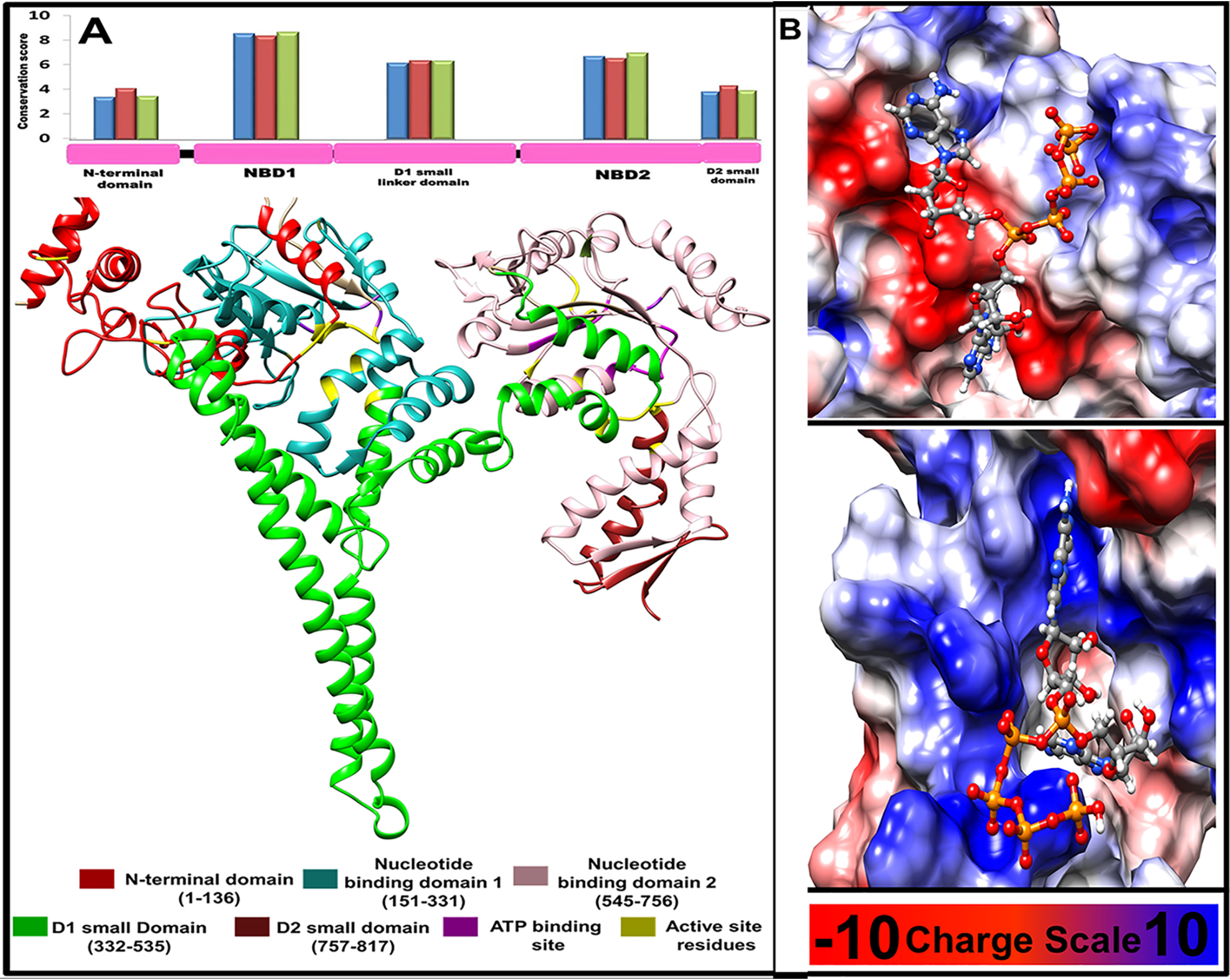
Structural features of L. donovani HSP78 protein. A, 2D and 3D domain organizations of L. donovani HSP78 protein and their relative conservation. Blue, red, and green bars show conservation of individual domain calculated using the representative sequences from all phyla, Euglenozoa, and Ascomycota, respectively. B, binding mode and probable interactions of the selected inhibitor Ap5A, which shows significant binding potential for NBD1 and NBD2 shown in upper and lower panel, respectively.
High-throughput virtual screening identified the ATP analog Ap5A as a probable LdHSP78 chemical inhibitor
The high-throughput virtual screening (HTVS) technique was implemented based on two aspects, de novo and substrate analog-based screening. Both de novo screening and substrate (ATP) analog-based HTVS techniques were applied using the first conformer of 2,838,166 compounds available in the Phase database (22) and 9998 compounds in the PubChem database (23), respectively. Compounds were docked at the nucleotide-binding sites using virtual screening techniques incorporated in the GLIDE package from the Schrodinger suite (24). The molecules that passed the filtration criteria (1460 and 1409 among Phase molecules and 9480 and 9480 among PubChem molecules binding at NBD1 and NBD2, respectively) were further considered for docking with the GOLD program (25), using GOLD fitness scores. Docking complexes possessing similar modes of binding (root mean square deviation of ≤5Å) among the top 30% solutions were considered probable inhibitors. This screening yielded 33 and 14 probable molecules binding at NBD1 and NBD2, respectively, whereas substrate analog-based screening suggested 3 and 45 molecules as NBD1 and NBD2 binders, respectively (Fig. S6). Furthermore, a third set of probable inhibitors that are likely to bind both of the NBDs were identified for both de novo and analog-based screening methods after comparison of the top 100 GLIDE docking solutions.
Inhibitors bound at NBD1 and NBD2 primarily interacted with 44 and 19 unique HSP78 residues via 66.4% and 60.4% hydrogen bonding, 12.1% and 16.5% electrostatic bonding, 11.2% and 5.2% salt bridge interactions, and 10.2% and 17.9% hydrophobic interactions, respectively (Fig. S7). Ap5A displayed the best docking properties and capability of binding to both of the NBD pockets. Fig. 6B shows the mode of interaction of Ap5A binding to NBD1 and NBD2.
Chemical inhibition of LdHSP78 by Ap5A resulted in reduced infectivity index of L. donovani in BMDMs
The cytotoxicity of Ap5A ammonium salt was assessed in WT, LdHSP78+/−, and resKO promastigote cell lines using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction assay. It was observed that the IC50 of Ap5A was 14.5 ± 1.92 μm for WT parasites. Ap5A is a structural analog of ATP. Parasites are enriched with multiple ATP-dependent proteins that are as crucial as LdHSP78 for survival; therefore, Ap5A might exert nonspecific effects on the promastigotes. Interestingly, LdHSP78+/− parasites exhibited higher sensitivity to Ap5A treatment (IC50 = 0.25 ± 0.077 μm), and resKO cell lines (with overexpression of LdHSP78) regained resistance against Ap5A, with an IC50 equivalent to that of WT parasites (IC50 = 15.0 ± 0.90 μm) (Fig. 7A). To further emphasize the specificity of Ap5A for LdHSP78, we compared the growth responses of heterozygous knockouts (LdHSP78+/−) and Ap5A-treated WT promastigotes under pH- and temperature-stressed conditions. Ap5A treatment closely mimicked the growth pattern of the promastigotes with partial depletion of LdHSP78. Inhibitor treatment enhanced the sensitivity of WT promastigotes to physiological stresses, similar to that of the LdHSP78+/− phenotype (Fig. 7B). These findings cumulatively indicated that Ap5A is probably specific for LdHSP78 in the promastigotes.
Figure 7.

Ap5A showed comparable antileishmanial efficacy with AmB. A, E, F, and H, the viability of axenic promastigotes (A and F) and healthy BMDMs (E and H) treated with Ap5A and AmB was evaluated using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide reduction assay. B, pH sensitivity and temperature sensitivity of Ap5A (15 μm)-treated WT and LdHSP78+/−promastigotes were compared by determination of promastigote growth. C and G, Giemsa staining was performed to calculate the amastigote burden of BMDMs, infected with L. donovani for 24 h and treated with either Ap5A or AmB. A and D, the comparative sensitivity of WT, LdHSP78+/−, and resKO promastigotes and amastigotes for Ap5A treatment was analyzed to understand the specificity of Ap5A for LdHSP78. Each experimental set is representative of three samples, and error bars are mean ± S.D. Statistical significance was analyzed using one-way analysis of variance followed by Dunnett's multiple-comparison test.
Infected macrophages were treated for 1 h with vehicle control (20 mm PBS) and a range of Ap5A doses from 1 μm to 15 μm, and cells were kept for 24 h after drug removal in order to understand the leishmanicidal efficacy of Ap5A on amastigotes in BMDMs. The infectivity index calculated by Giemsa staining suggested that the IC50 dose of Ap5A for amastigotes was 0.8 ± 0.18 μm (Fig. 7C). Furthermore, the sensitivity of WT, LdHSP78+/−, and resKO amastigotes to Ap5A was determined to nullify the nonspecific effect of Ap5A on the amastigotes. The IC50 of Ap5A for WT amastigotes was 1.00 ± 0.24 μm and that for LdHSP78+/− amastigotes was 0.7 ± 0.15 μm. Similar to resKO promastigotes, overexpression of LdHSP78 led to regained resistance of resKO amastigotes to Ap5A (IC50 = 1.2 ± 0.14 μm) (Fig. 7D). Thus, the sensitivity of amastigotes to Ap5A also suggested that Ap5A might have specific effects on LdHSP78. However, the IC50 of Ap5A for healthy BMDMs was 90 ± 1.55 μm, which was indicative of its poor toxicity against host cells (Fig. 7E).
IC50 and selective inhibitory (SI) values of Ap5A were compared with those of AmB to understand the efficacy of Ap5A as a potent antileishmanial agent. Therefore, similar experiments were carried out using AmB, which showed that all three promastigote cell lines have almost equivalent sensitivities, i.e. the IC50 for WT promastigotes is 0.5 ± 0.12 μm, that for LdHSP78+/− promastigotes is 0.45 ± 0.13 μm, and that for resKO promastigotes is 0.56 ± 0.07 μm (Fig. 7F). Although AmB has a much lower IC50 for promastigotes (0.5 ± 0.12 μm), the IC50 for amastigotes (1.25 ± 0.59 μm) is very similar to or slightly higher than that of Ap5A (Fig. 7G). Nevertheless, the cytotoxicity of AmB for healthy BMDMs is much higher, and the corresponding IC50 (0.4 ± 0.19 μm) overlaps with its promastigote and amastigote IC50 values (Fig. 7H), which further indicated that AmB harms the host cells at a much lower concentration than the concentration at which it kills the parasites. Comparison of SI values for two different inhibitors were undertaken using the World Health Organization guidelines (SI = IC50 for BMDMs/IC50 for amastigotes). The SI value of Ap5A was 112.5, and the SI value for AmB was 0.8. These data suggested that Ap5A might have promising target-specific antileishmanial efficacy, compared with AmB.
Discussion
Our study identified LdHSP78, a putative AAA+ domain-containing ClpB family member, as a potential virulence factor of the parasite and explored its role as a therapeutic target. We recognized that LdHSP78 is enriched in VL patient blood and infected hamster organs. To confirm the essentiality of HSP78 in virulence, heterozygous HSP78 knockout parasites of two different species of Leishmania were generated by employing conventional homologous recombination-mediated and advanced CRISPR-Cas9-mediated genetic manipulation tools. Null mutation of LmHSP78/LdHSP78 was found to be lethal, and heterozygous mutants had significantly compromised virulence in both in vitro and in vivo infection models. Interestingly, LdHSP78+/− parasites regained their virulence phenotype upon successful add-back of the deleted HSP78 allele in the form of episome. Partial deletion of LdHSP78 culminated in the inability of the parasites to bypass most of the activated immune responses of the host. Mechanistic understanding of LdHSP78+/− parasite clearance suggested that enrichment of phospho-p38 and proinflammatory cytokines preceded activation of NO, which possibly led to LdHSP78+/− clearance from macrophages. Thus, after ensuring the essential role of LdHSP78 during infection, we identified Ap5A, an ATP analog, as a potential inhibitor of LdHSP78 and studied its in vitro antileishmanial efficacy. We found that Ap5A could successfully clear the parasite load at almost the same dose as AmB in BMDMs but showed greatly reduced toxicity to healthy host macrophages, which is a major hurdle in using AmB.
VL is one of the major global threats next to malaria, causing an estimated 0.5 million new cases annually (WHO reports, 2018) (42, 43). Under such circumstances, VL therapeutics call for urgent identification of novel parasite target proteins that are amenable to high-throughput screening and design of druggable chemical products. In this postgenomic era, integrative computational approaches facilitate the identification and prioritization of candidate targets (26). LdHSP78 is highly conserved in trypanosomes, with no human orthologs, and belongs to the AAA+ superfamily of ATP-dependent HSPs, which efficiently relieves microorganisms from apoptotic shock generated by disaggregated proteins (27). L. donovani promastigotes initiate infection by transdifferentiating into amastigotes in the presence of high temperatures and extremely acidic pH (phagosomal pH) inside mammalian macrophages, which is a rate-limiting step for infection initiation. Parasite HSPs play very crucial roles for overriding the stresses induced during this stage (28, 29). Previously, functional significance of HSP83, HSP90, and HSP23 in stress regulation of Leishmania was reported (28, 30, 31). However, the prominence of an HSP belonging to the ClpB group as a potential drug target in L. donovani has not been explored. Enrichment of LdHSP78 in L. donovani-infected BMDMs, hamster organs, and VL patients reinforced its association with active VL.
Clp proteins are reportedly essential for virulence and the persistence of intracellular pathogens such as Listeria monocytogenes, Francisella tularensis, and Toxoplasma gondii inside host macrophages. Therefore, we verified the essentiality of LdHSP78 for the persistence of L. donovani inside macrophages by generating HSP78 knockout parasites and then rescued the phenotype using episomal overexpression of HSP78. Significant reduction in LdHSP78+/− parasite burdens in BMDMs, paralleled by reduced abundance of LdHSP78 mRNA expression in amastigotes, indicated LdHSP78 as one of the factors for survival of amastigotes in macrophages.
However, all of our attempts to generate chromosomal null mutants of HSP78 in both L. donovani and L. mexicana cells, using different genetic manipulation tools, failed. Null mutant parasites survived only in the presence of the episomal vector pXG-GFP expressing HSP78, which was stably maintained even after removal of selection. These findings indicated probable essentiality of the HSP78 gene for promastigote survival, which needs more experimental investigation for confirmation.
Hubel et al. reported that HSP100 depletion greatly affects the amastigote stage of development of L. major within macrophages (6). We obtained profound LdHSP78 enrichment especially at pH 5.5 and 37 °C. It was interesting that only pH or temperature stress was unable to induce the enrichment but dual stress (both high temperature and low pH), which mimics the macrophage microenvironment, induced LdHSP78 enrichment. Moreover, LdHSP78+/− parasites showed impaired growth rates under similar conditions, which indicates that LdHSP78 plays a crucial role as a chaperone during the amastigote stage of development.
To confirm a parasite protein as a potential drug target, it is essential to understand whether knockout parasites are able to survive inside the host and to bypass host defense machineries. Establishment of infection in macrophages is reflected by the cytokine profile. L. donovani induces anti-inflammatory cytokines and exhausts proinflammatory cytokine levels in a MAPK-dependent manner, which finally leads to transcription of arginase 1 and suppression of iNOS, aiding survival of the parasites in macrophages (32–36). Proinflammatory cytokines like IL12A were enriched during LdHSP78+/− parasite infection. In addition, enhanced phosphorylation of p38 MAPK and up-regulation of iNOS clearly indicated that macrophages are capable of showing active immune responses against LdHSP78+/− parasites. For persistence within macrophages, virulent L. donovani inhibits iNOS synthesis to be shielded from NO-mediated toxicity (33, 37). In agreement with enriched iNOS expression, NO synthesis was augmented during LdHSP78+/− parasite infection, compared with WT parasite infection. Down-regulation of NO was regained after introduction of the LdHSP78 allele in LdHSP78+/− parasites. These studies cumulatively indicated that HSP78 depletion facilitates nonviability of parasites in macrophages in a NO-dependent manner.
Before identification of a potential inhibitor, the role of LdHSP78 in hamster VL was also studied. Comparative infectivity analysis of WT, LdHSP78+/−, and resKO parasites in hamsters indicated that heterozygous knockout of LdHSP78 greatly reduced the capacity of the parasites to proliferate within the infection foci of the animals, such as spleen, liver, and peripheral blood. Besides the decrease in parasite burden and recovery in pathological symptoms of infection, proinflammatory cytokines were enriched and NO titers were increased in LdHSP78+/− parasite-infected animals, indicating that the partial depletion of HSP78 disables the parasites from establishing full-grown infections in hamsters. This motivated us to screen for potential HSP78 inhibitor molecules using the HTVS method and to test their antileishmanial efficacies.
It is worth mentioning that ATP analogs represent a novel as well as wide repertoire of pharmacological inhibitors for different diseases, such as cancer, pathological calcification, and multiple myeloma (38–41). In silico analysis revealed HSP78 as a putative ATP-dependent HSP; thereafter, by HTVS we screened Ap5A ammonium salt, which is an ATP analog. In this study, the inhibitory role of Ap5A has been substantiated in L. donovani infection. However, detailed structural, functional, and pharmacological aspects of Ap5A and its derivatives will be deciphered in the future. A comparative study of the leishmanicidal efficacy of Ap5A ammonium salt with that of AmB revealed that Ap5A had an IC50 for amastigotes of 0.8 μm with an SI value of 112.5, whereas AmB had an IC50 for amastigotes of 1.25 μm with an SI value 0.8. This indicates that Ap5A can be a potential alternative for AmB, which shows profound toxic effects on the host. However, in order to find better derivatives of the inhibitor, having cytotoxicity for amastigotes in the nanomolar range, a library of Ap5A derivatives was already generated computationally. From them, 23 novel molecules possessing better docking scores and other relevant druggability parameters were selected to initiate a future novel drug development scheme against LdHSP78 protein (Fig. S8).
The impact of this study could be multifaceted, starting from revealing for the first time the importance of the highly conserved ClpB parasite chaperon as an antileishmanial drug target, followed by screening of potential structural analogs (using the HTVS method) against the protein along with its functional validation. In summary, we established that targeting ATP-dependent LdHSP78 with a pharmacological inhibitor can open a new avenue for therapeutic interventions against VL.
Materials and methods
Ethics statement
All animal experiments were approved by the Animal Ethical Committee (Protocol 147/1999/CPSCEA) of the Indian Institute of Chemical Biology, according to the National Regulatory Guidelines issued by the Committee for the Purpose of Control and Supervision on Experimental Animals, under the Division of Animal Welfare, Ministry of Environment and Forest, Government of India.
For human studies, 4–5-ml venous blood samples were collected in individual heparinized vials from three anonymous patients who were initially diagnosed with VL symptoms, such as prolonged fever, hepatosplenomegaly, and pancytopenia, from Rajendra Memorial Research Institute of Medical Science (Patna, Bihar, India). Moreover, samples were tested to be VL positive by the rK39 strip test. Healthy individual blood samples (similar quantities) were collected from both male and female subjects 23–30 years of age. Written approval was obtained from all participants in their local languages. Human sample-related experiments were approved by the Human Ethics Committee of CSIR-Indian Institute of Chemical Biology, abiding by the Declaration of Helsinki principles.
Heterozygous knockout using the UTR-mediated technique
In order to replace LdHSP78, UTR sequences flanking the HYG gene were cloned; for genomic add-back of the deleted cassette and generation of resKO parasites, the 2454-bp-long HSP78 gene was cloned by using appropriate primers (Table S3). Details are given in the supporting information.
CRISPR-Cas9-mediated heterozygous knockout generation
Using CRISPR-Cas9 technology, LmHSP78 deletion was attempted in axenic T7- and Cas9-positive Leishmania mexicana cell lines (designed and generously provided by Prof. Jeremy Mottram, University of York, UK). The detailed protocol is provided in the supporting information. Detailed description of the materials and methods is provided in the supporting information.
Data availability
All data are contained within the manuscript and Supporting material. Additional raw data will be shared on request from the following contact persons, a. Dr. Saikat Chakrabarti (saikat@iicb.res.in) b. Dr. Nahid Ali (nail@iicb.res.in).
Supplementary Material
Acknowledgments
We thank Oishee Chakrabarti (Saha Institute of Nuclear Physics, Kolkata, India) for her valuable advice at different stages of the study. We thank Stephen Beverley (Washington University School of Medicine), Philip Yates (Oregon Health & Science University), and Jeremy Mottram (University of York) for providing the constructs, T7/Cas9-positive L. mexicana, and E. coli ccdB Survival-T1R cells. We acknowledge Janmenjoy Midya for assistance in animal maintenance, rearing, and handling. S.D. thanks Sohitri Mukherjee (St. Xavier's College, Kolkata, India) for her contributions in cloning and protein-related experiments. We also acknowledge the central instrumentation facility, CSIR-IICB, and Uday Bandyopadhyay and his laboratory (CSIR-IICB) for cloning-related advice.
This article contains supporting information.
Author contributions—S. D., N. A., and S. C. conceptualization; S. D., A. B., M. K., N. A., and S. C. resources; S. D., A. B., M. K., and S. C. data curation; S. D., A. B., M. K., N. A., and S. C. formal analysis; S. D., N. A., and S. C. supervision; S. D., N. A., and S. C. funding acquisition; S. D., A. B., M. K., N. A., and S. C. validation; S. D., A. B., and M. K. investigation; S. D., A. B., M. K., S. A. E., M. A., and N. A. methodology; S. D., A. B., and S. C. writing-original draft; S. D., A. B., M. K., and S. C. writing-review and editing; A. B. and M. K. software; N. A. and S. C. visualization; N. A. and S. C. project administration.
Funding and additional information—S. C. acknowledges CSIR-IICB for infrastructure support, CSIR Network Project BSC-0114, Systems Medicine Cluster Grant GAP357 from the Department of Biotechnology, and High-Risk High-Reward Grant HRR/2016/000093 from the Department of Science and Technology for financial support. N. A. acknowledges the J. C. Bose National Fellowship and UK Research and Innovation via the Global Challenges Research Fund (Grant MR/P027989/1) for financial support.
Conflict of interest—The authors declare that they have no conflicts of interest with the contents of this article.
- HSP
- heat shock protein
- VL
- visceral leishmaniasis
- NBD
- nucleotide-binding domain
- ClpB
- caseinolytic protease B
- Ap5A
- P1,P5-di(adenosine-5′)-pentaphosphate
- TGFβ
- transforming growth factor β
- MAPK
- mitogen-activated protein kinase
- ERK1/2
- extracellular signal-regulated kinase 1/2
- kDNA
- kinetoplastidial DNA
- HTVS
- high-throughput virtual screening
- iNOS
- inducible nitric oxide synthase
- NEO
- neomycin
- PUR
- puromycin
- HYG
- hygromycin
- LDA
- limiting dilution assay
- SI
- selective inhibitory
- BMDM
- bone marrow-derived macrophage
- AmB
- amphotericin B
References
- 1. Li Z., and Srivastava P. (2004) Heat-shock proteins. Curr. Protoc. Immunol. Appendix 1 10.1002/0471142735.ima01ts58 [DOI] [PubMed] [Google Scholar]
- 2. Zilberstein D., and Shapira M. (1994) The role of pH and temperature in the development of Leishmania parasites. Annu. Rev. Microbiol. 48, 449–470 10.1146/annurev.mi.48.100194.002313 [DOI] [PubMed] [Google Scholar]
- 3. Callahan H. L., Portal I. F., Bensinger S. J., and Grogl M. (1996) Leishmania spp: temperature sensitivity of promastigotes in vitro as a model for tropism in vivo. Exp. Parasitol. 84, 400–409 10.1006/expr.1996.0128 [DOI] [PubMed] [Google Scholar]
- 4. Pallavi R., Roy N., Nageshan R. K., Talukdar P., Pavithra S. R., Reddy R., Venketesh S., Kumar R., Gupta A. K., Singh R. K., Yadav S. C., and Tatu U. (2010) Heat shock protein 90 as a drug target against protozoan infections: biochemical characterization of HSP90 from Plasmodium falciparum and Trypanosoma evansi and evaluation of its inhibitor as a candidate drug. J. Biol. Chem. 285, 37964–37975 10.1074/jbc.M110.155317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lee S., Sowa M. E., Choi J. M., and Tsai F. T. (2004) The ClpB/Hsp104 molecular chaperone: a protein disaggregating machine. J. Struct. Biol. 146, 99–105 10.1016/j.jsb.2003.11.016 [DOI] [PubMed] [Google Scholar]
- 6. Hubel A., Krobitsch S., Horauf A., and Clos J. (1997) Leishmania major Hsp100 is required chiefly in the mammalian stage of the parasite. Mol. Cell Biol. 17, 5987–5995 10.1128/mcb.17.10.5987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ponte-Sucre A., Gamarro F., Dujardin J. C., Barrett M. P., Lopez-Velez R., Garcia-Hernandez R., Pountain A. W., Mwenechanya R., and Papadopoulou B. (2017) Drug resistance and treatment failure in leishmaniasis: a 21st century challenge. PLoS Negl. Trop. Dis. 11, e0006052 10.1371/journal.pntd.0006052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Castro M. D. M., Cossio A., Velasco C., and Osorio L. (2017) Risk factors for therapeutic failure to meglumine antimoniate and miltefosine in adults and children with cutaneous leishmaniasis in Colombia: a cohort study. PLoS Negl. Trop. Dis. 11, e0005515 10.1371/journal.pntd.0005515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leonhardt S. A., Fearson K., Danese P. N., and Mason T. L. (1993) HSP78 encodes a yeast mitochondrial heat shock protein in the Clp family of ATP-dependent proteases. Mol. Cell Biol. 13, 6304–6313 10.1128/mcb.13.10.6304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Weibezahn J., Schlieker C., Bukau B., and Mogk A. (2003) Characterization of a trap mutant of the AAA+ chaperone ClpB. J. Biol. Chem. 278, 32608–32617 10.1074/jbc.M303653200 [DOI] [PubMed] [Google Scholar]
- 11. Schlieker C., Weibezahn J., Patzelt H., Tessarz P., Strub C., Zeth K., Erbse A., Schneider-Mergener J., Chin J. W., Schultz P. G., Bukau B., and Mogk A. (2004) Substrate recognition by the AAA+ chaperone ClpB. Nat. Struct. Mol. Biol. 11, 607–615 10.1038/nsmb787 [DOI] [PubMed] [Google Scholar]
- 12. Chio C. C., Chang Y. H., Hsu Y. W., Chi K. H., and Lin W. W. (2004) PKA-dependent activation of PKC, p38 MAPK and IKK in macrophage: implication in the induction of inducible nitric oxide synthase and interleukin-6 by dibutyryl cAMP. Cell. Signal. 16, 565–575 10.1016/j.cellsig.2003.10.003 [DOI] [PubMed] [Google Scholar]
- 13. Stempin C. C., Dulgerian L. R., Garrido V. V., and Cerban F. M. (2010) Arginase in parasitic infections: macrophage activation, immunosuppression, and intracellular signals. J. Biomed. Biotechnol. 2010, 683485 10.1155/2010/683485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Melby P. C., Chandrasekar B., Zhao W., and Coe J. E. (2001) The hamster as a model of human visceral leishmaniasis: progressive disease and impaired generation of nitric oxide in the face of a prominent Th1-like cytokine response. J. Immunol. 166, 1912–1920 10.4049/jimmunol.166.3.1912 [DOI] [PubMed] [Google Scholar]
- 15. Loria-Cervera E. N., and Andrade-Narvaez F. J. (2014) Animal models for the study of leishmaniasis immunology. Rev. Inst. Med. Trop. Sao Paulo 56, 1–11 10.1590/S0036-46652014000100001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bhattacharya P., Ghosh S., Ejazi S. A., Rahaman M., Pandey K., Ravi Das V. N., Das P., Goswami R. P., Saha B., and Ali N. (2016) Induction of IL-10 and TGFβ from CD4+CD25+FoxP3+ T cells correlates with parasite load in Indian kala-azar patients infected with Leishmania donovani. PLoS Negl. Trop. Dis. 10, e0004422 10.1371/journal.pntd.0004422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Medina-Colorado A. A., Osorio E. Y., Saldarriaga O. A., Travi B. L., Kong F., Spratt H., Soong L., and Melby P. C. (2017) Splenic CD4+ T cells in progressive visceral leishmaniasis show a mixed effector-regulatory phenotype and impair macrophage effector function through inhibitory receptor expression. PLoS ONE 12, e0169496 10.1371/journal.pone.0169496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Asad M., Sabur A., Shadab M., Das S., Kamran M., Didwania N., and Ali N. (2019) EB1-3 chain of IL-35 along with TGF-β synergistically regulate anti-leishmanial immunity. Front. Immunol. 10, 616 10.3389/fimmu.2019.00616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Webb B., and Sali A. (2016) Comparative protein structure modeling using MODELLER. Curr. Protoc. Bioinformatics 54, 5.6.1–5.6.37. 10.1002/cpbi.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Laskowski R. A., Rullmannn J. A., MacArthur M. W., Kaptein R., and Thornton J. M. (1996) AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 8, 477–486 10.1007/BF00228148 [DOI] [PubMed] [Google Scholar]
- 21. Eisenberg D., Luthy R., and Bowie J. U. (1997) VERIFY3D: assessment of protein models with three-dimensional profiles. Methods Enzymol. 277, 396–404 10.1016/s0076-6879(97)77022-8 [DOI] [PubMed] [Google Scholar]
- 22. Dixon S. L., Smondyrev A. M., and Rao S. N. (2006) PHASE: a novel approach to pharmacophore modeling and 3D database searching. Chem. Biol. Drug Des. 67, 370–372 10.1111/j.1747-0285.2006.00384.x [DOI] [PubMed] [Google Scholar]
- 23. Kim S., Thiessen P. A., Bolton E. E., Chen J., Fu G., Gindulyte A., Han L., He J., He S., Shoemaker B. A., Wang J., Yu B., Zhang J., and Bryant S. H. (2016) PubChem Substance and Compound databases. Nucleic Acids Res. 44, D1202–D1213 10.1093/nar/gkv951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., and Shenkin P. S. (2004) Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 47, 1739–1749 10.1021/jm0306430 [DOI] [PubMed] [Google Scholar]
- 25. Jones G., Willett P., Glen R. C., Leach A. R., and Taylor R. (1997) Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 267, 727–748 10.1006/jmbi.1996.0897 [DOI] [PubMed] [Google Scholar]
- 26. Sosa E. J., Burguener G., Lanzarotti E., Defelipe L., Radusky L., Pardo A. M., Marti M., Turjanski A. G., and Fernandez Do Porto D. (2018) Target-Pathogen: a structural bioinformatic approach to prioritize drug targets in pathogens. Nucleic Acids Res. 46, D413–D418 10.1093/nar/gkx1015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neckers L., and Tatu U. (2008) Molecular chaperones in pathogen virulence: emerging new targets for therapy. Cell Host Microbe 4, 519–527 10.1016/j.chom.2008.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hombach A., Ommen G., Chrobak M., and Clos J. (2013) The Hsp90-Sti1 interaction is critical for Leishmania donovani proliferation in both life cycle stages. Cell. Microbiol. 15, 585–600 10.1111/cmi.12057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Goulhen F., Hafezi A., Uitto V. J., Hinode D., Nakamura R., Grenier D., and Mayrand D. (1998) Subcellular localization and cytotoxic activity of the GroEL-like protein isolated from Actinobacillus actinomycetemcomitans. Infect. Immun. 66, 5307–5313 10.1128/IAI.66.11.5307-5313.1998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hombach A., Ommen G., MacDonald A., and Clos J. (2014) A small heat shock protein is essential for thermotolerance and intracellular survival of Leishmania donovani. J. Cell Sci. 127, 4762–4773 10.1242/jcs.157297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Krobitsch S., and Clos J. (1999) A novel role for 100 kD heat shock proteins in the parasite Leishmania donovani. Cell Stress Chaperones 4, 191–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ansari N. A., Kumar R., Gautam S., Nylen S., Singh O. P., Sundar S., and Sacks D. (2011) IL-27 and IL-21 are associated with T cell IL-10 responses in human visceral leishmaniasis. J. Immunol. 186, 3977–3985 10.4049/jimmunol.1003588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Saha S., Mondal S., Ravindran R., Bhowmick S., Modak D., Mallick S., Rahman M., Kar S., Goswami R., Guha S. K., Pramanik N., Saha B., and Ali N. (2007) IL-10- and TGF-β-mediated susceptibility in kala-azar and post-kala-azar dermal leishmaniasis: the significance of amphotericin B in the control of Leishmania donovani infection in India. J. Immunol. 179, 5592–5603 10.4049/jimmunol.179.8.5592 [DOI] [PubMed] [Google Scholar]
- 34. Bhattacharya P., Gupta G., Majumder S., Adhikari A., Banerjee S., Halder K., Majumdar S. B., Ghosh M., Chaudhuri S., Roy S., and Majumdar S. (2011) Arabinosylated lipoarabinomannan skews Th2 phenotype towards Th1 during Leishmania infection by chromatin modification: involvement of MAPK signaling. PLoS ONE 6, e24141 10.1371/journal.pone.0024141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Junghae M., and Raynes J. G. (2002) Activation of p38 mitogen-activated protein kinase attenuates Leishmania donovani infection in macrophages. Infect. Immun. 70, 5026–5035 10.1128/iai.70.9.5026-5035.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chandra D., and Naik S. (2008) Leishmania donovani infection down-regulates TLR2-stimulated IL-12p40 and activates IL-10 in cells of macrophage/monocytic lineage by modulating MAPK pathways through a contact-dependent mechanism. Clin. Exp. Immunol. 154, 224–234 10.1111/j.1365-2249.2008.03741.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Anderson C. F., Lira R., Kamhawi S., Belkaid Y., Wynn T. A., and Sacks D. (2008) IL-10 and TGF-β control the establishment of persistent and transmissible infections produced by Leishmania tropica in C57BL/6 mice. J. Immunol. 180, 4090–4097 10.4049/jimmunol.180.6.4090 [DOI] [PubMed] [Google Scholar]
- 38. Lecka J., Ben-David G., Simhaev L., Eliahu S., Oscar J. Jr., Luyindula P., Pelletier J., Fischer B., Senderowitz H., and Sevigny J. (2013) Nonhydrolyzable ATP analogues as selective inhibitors of human NPP1: a combined computational/experimental study. J. Med. Chem. 56, 8308–8320 10.1021/jm400918s [DOI] [PubMed] [Google Scholar]
- 39. Monkkonen H., Kuokkanen J., Holen I., Evans A., Lefley D. V., Jauhiainen M., Auriola S., and Monkkonen J. (2008) Bisphosphonate-induced ATP analog formation and its effect on inhibition of cancer cell growth. Anticancer Drugs 19, 391–399 10.1097/CAD.0b013e3282f632bf [DOI] [PubMed] [Google Scholar]
- 40. Samsel M., and Dzierzbicka K. (2011) Therapeutic potential of adenosine analogues and conjugates. Pharmacol. Rep. 63, 601–617 10.1016/S1734-1140(11)70573-4 [DOI] [PubMed] [Google Scholar]
- 41. Krett N. L., Davies K. M., Ayres M., Ma C., Nabhan C., Gandhi V., and Rosen S. T. (2004) 8-Amino-adenosine is a potential therapeutic agent for multiple myeloma. Mol. Cancer Ther. 3, 1411–1420 [PubMed] [Google Scholar]
- 42. World Health Organization (2020) Leishmaniasis, Geneva, Switzerland: https://www.who.int/news-room/fact-sheets/detail/leishmaniasis. [Google Scholar]
- 43. Singh O. P., Gedda M. R., Mudavath S. L., Srivastava O. N., and Sundar S. (2019) Envisioning the Innovations in Nanomedicine to Combat Visceral Leishmaniasis: For Future Theranostic Application. Nanomedicine (Lond). 14, 1911–1927 10.2217/nnm-2018-0448 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are contained within the manuscript and Supporting material. Additional raw data will be shared on request from the following contact persons, a. Dr. Saikat Chakrabarti (saikat@iicb.res.in) b. Dr. Nahid Ali (nail@iicb.res.in).