

Abstract
BACKGROUND:
The majority of non-small cell lung cancer (NSCLC) patients harboring activating EGFR mutations respond well to osimertinib (AZD9291), a third generation mutation-selective epidermal growth factor receptor (EGFR) inhibitor. The current study focuses on determining whether targeting MEK/ERK signaling prevents or delays the development of acquired resistance to osimertinib.
Methods:
Drug effects on cell survival were determined by measuring cell number alterations. Apoptosis was assessed using flow cytometry for detection of annexin V-positive cells and Western blotting for protein cleavage. Alterations of proteins in cells were detected with Western blotting. Drug effects on delaying the emergence of osimertinib resistance were evaluated using colony formation in vitro and xenografts in nude mice in vivo, respectively.
RESULTS:
Osimertinib combined with a MEK or ERK inhibitor synergistically decreased cell survival with enhanced induction of apoptosis in EGFR-mutant, but not EGFR wild-type NSCLC cells. These combinations were also very effective in killing cell clones with primary intrinsic resistance to osimertinib. Continuous and intermittent pharmacologic inhibition of MEK/ERK signaling delayed the emergence of osimertinib resistance both in vitro and in vivo.
CONCLUSIONS:
Our results provide strong preclinical evidence in support of targeting MEK/ERK signaling as a strategy to delay or prevent acquired resistance to osimertinib in the clinic to improve long-term therapeutic efficacy of osimertinib. From a clinical standpoint, our data support the evaluation of an intermittent treatment schedule of osimertnib combining with a MEK or ERK inhibitor in patients with EGFR-mutated NSCLC.
Keywords: MEK/ERK, EGFR, osimertinib, resistance, lung cancer
Precis:
The key finding in this study is that inclusion of MEK/ERK inhibition in the treatment of EGFR-mutant NSCLC with osimertinib using either a concurrent or an intermittent schedule substantially delayed the emergence of acquired resistance to osimertinib without increasing toxicity. This finding highlights the importance of MEK/ERK inhibition in maintaining the long-term benefit of osimertinib through delaying the emergence of acquired resistance to osimertinib.
INTRODUCTION
Despite advances in early diagnosis and innovative treatments, lung cancer, which presents in 80% of cases as non-small cell lung cancer (NSCLC), is still the leading cause of cancer death worldwide. The sub-group of NSCLC patients harboring epidermal growth factor receptor (EGFR) activating mutations such as exon 19 deletion (19del) and exon 21 point mutation (L858R), make up ~10% and ~40% of Western and Asian NSCLC populations, respectively1. EGFR tyrosine kinase inhibitors (EGFR-TKIs), particularly the third generation osimertinib (TAGRISSO™ or AZD9291), have achieved great success in terms of improving progression-free survival and overall survival of patients. A recently completed phase 3 trial has shown that patients receiving osimertinib had a median overall survival of 38.6 months2. Different from other first and second-generation EGFR-TKIs, osimertinib selectively and irreversibly inhibits EGFR activating mutations such as 19del and L858R as well as the resistant T790M mutation without any meaningful effect on the wild-type (WT) EGFR3. Osimertinib is now an FDA-approved drug for the frontline treatment of this subgroup of NSCLC patients with activating EGFR mutations (first-line) as well as salvage therapy of patients with acquired resistance to 1st generation EGFR-TKIs due to T790M mutation (second-line). Unfortunately, like other EGFR-TKIs, patients eventually relapse from osimertinib treatment due to the development of acquired resistance to osimertinib, limiting the long-term benefit for patients. Hence, managing the emergence of osimertinib-acquired resistance has become a significant challenge, and an important issue to address for long-term control of this disease in the clinic3–5.
It is well-known that MEK/ERK signaling is a key pathway downstream of EGFR that mediates EGFR-dependent regulation of cancer cell growth and survival6–8. Our studies and others have demonstrated that targeting MEK/ERK signaling through either MEK or ERK inhibition can achieve impressive effects in overcoming acquired resistance to osimertinib in different preclinical models8–12. A clinical case study reported that a osimertinib-resistant NSCLC patient with B-Raf mutation responded to the treatment with osimertinib and trametinib combination13. Moreover, MEK inhibition using trametinib combined with WZ4002, another third generation EGFR-TKI, was shown to delay the emergence of acquired resistance to WZ4002 in WZ4002-sensitive models14. The current study focused on determining whether targeting MEK/ERK signaling with either MEK or ERK inhibition impacts the emergence of acquired resistance to osimertinib. We were particularly interested in the effects of different combinatorial schedules on delaying emergence of acquired resistance to osimertinib and the long-term safety or toxicity of the combinations in vivo in order to provide preclinical data to inform clinical trial designs for testing this strategy.
MATERIALS AND METHODS
Reagents
Osimertinib, trametinib (GSK1120212), and VRT752271 (ulikertinib or BVD-523) were the same as described previously8, 10, 15. Other reagents and antibodies used in this study were the same as described in our previous studies8, 10, 15.
Cell lines and cell culture
NSCLC cell lines used in this study and culture conditions were the same as described previously8, 15. These cell lines, except for A549, have not been genetically authenticated recently.
Cell viability assay
Cell numbers in 96-well cell culture plates were estimated by sulforhodamine B (SRB) assay as previously described16. Combination index (CI) for drug interaction (e.g., synergy) was calculated using CompuSyn software (ComboSyn, Inc.).
Detection of apoptosis
Apoptosis was detected with Annexin V/7-AAD apoptosis detection kit (BD Biosciences; San Jose, CA) according to the manufacturer’s instructions. Caspase and PARP cleavage were detected by Western blot analysis as additional indicators of apoptosis.
Western blot analysis
The procedures for preparation of whole-cell protein lysates and subsequent Western blotting were the same as we described previously8, 15.
Assays for delaying emergence of osimertinib resistance in vitro
Cells seeded in 24-well plates were exposed to different treatments and refed with fresh medium containing the same treatments every four days until the end of the study. The remaining cells were stained with crystal violet. Delaying assays in 96-well plates were conducted as reported previously14. Cell treatments were the same as in 24-well plates.
Evaluation of effects of treatments on delaying emergence of acquired resistance to osimertinib in vivo using mouse xenograft model
Animal experiments were approved by the Institutional Animal Care and Use Committee (IACUC) of Emory University. Treatments included vehicle control, osimertinib (15 mg/kg/day, daily, og), trametinib (1 mg/kg, once daily or once every two days, og) and their combination. Tumor volumes were measured using caliper measurements and calculated with the formula V = (length × width2)/2. Mice were sacrificed when tumor sizes on average were over 800 mm3.
RESULTS
Osimertinib exerts transient inhibitory effects on MEK/ERK signaling in comparison with a sustained suppression on Akt
WZ4002 was reported to induce rebound activation of MEK/ERK signaling in NSCLC lines with EGFR activating mutations after a sustained treatment14. Therefore, we determined whether osimertinib exerts similar effects on reactivating MEK/ERK signaling in different EGFR-mutant NSCLC cell lines. Time-course analysis showed that osimertinib treatment decreased p-ERK levels at early time points up to 24 h. With longer treatment times, such as 48 and 72 h, osimertinib exhibited reduced activity in decreasing p-ERK levels (e.g., HCC827 and H1975) and even increased the levels of p-ERK (e.g., PC-9). Interestingly, osimertinib decreased p-ERK levels through the period of testing in H1650 cells, although this cell line is relatively insensitive to osimertinib and other EGFR-TKIs. In contrast, we found that osimertinib maintained its effects on decreasing p-Akt levels, even at 72 h treatment, in every tested cell line (Fig. 1A). Therefore, osimertinib apparently exerts a transient suppressive effect on MEK/ERK signaling, but a sustained inhibition on Akt.
Fig. 1. Osimertinib inhibits ERK and Akt with different dynamics (A) and, when combined with MEK or ERK inhibition, synergistically decreases cell survival in NSCLC cell lines harboring mutant EGFR (B and C), but not in those with WT EGFR (D and E).
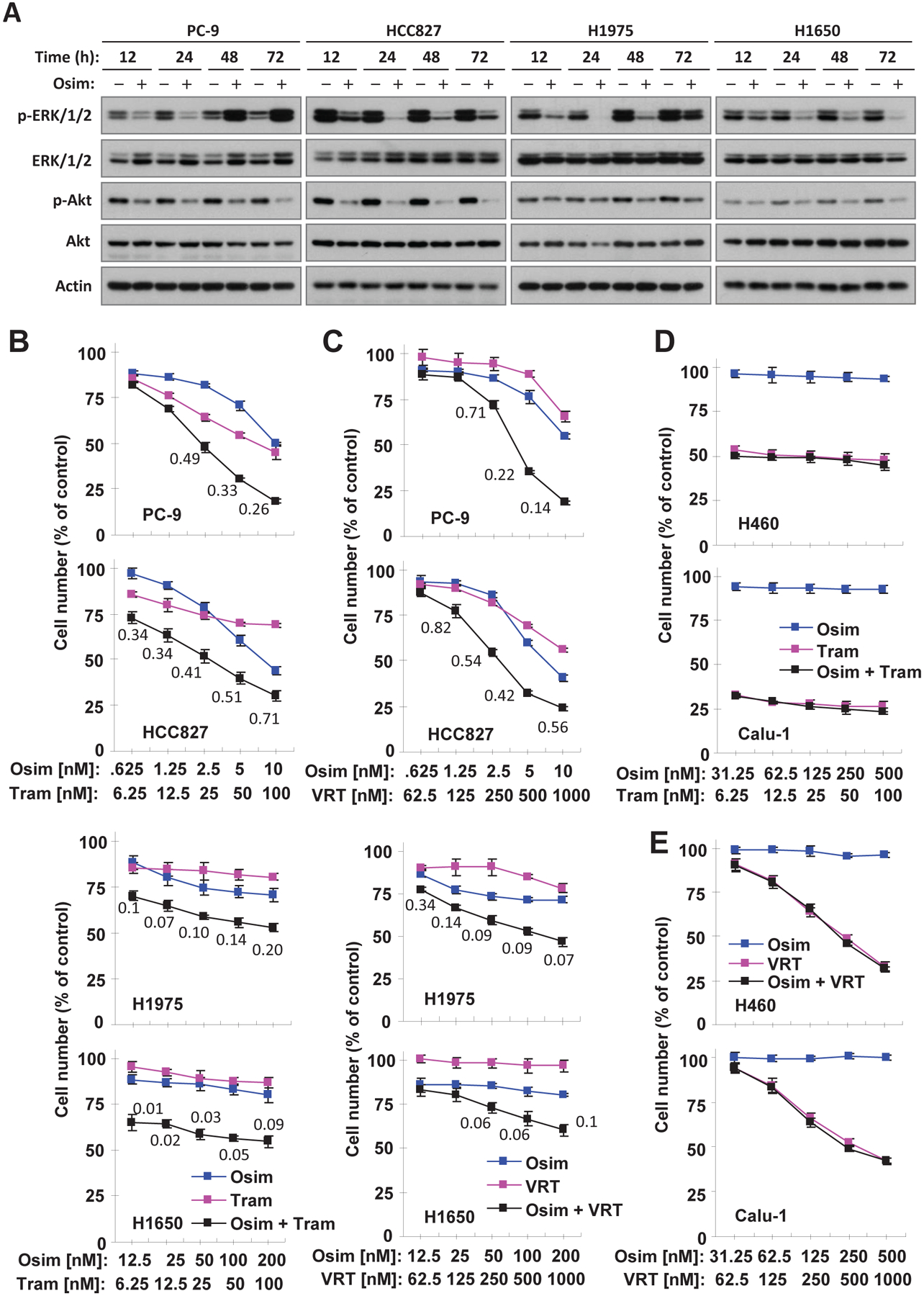
A, The given cell lines were exposed to DMSO and 50 nM (PC-9), 100 nM (HCC827) or 250 nM (H1975 and H1650) osimertinib (Osim), respectively, for the different times as indicated. Western blotting was used to detect different proteins. B-E, The indicated cell lines plated in 96-well plates were exposed to varied concentrations of osimertinib alone, trametinib (Tram) or VRT52271 (VRT) alone or their respective combinations. After 3 days, cell numbers were estimated using the SRB assay. The data are means ± SDs of four replicate determinations. The numbers inside the graphs are CIs for the given combinations.
Osimertinib combined with a MEK or ERK inhibitor synergistically decreases the survival of EGFR-mutant, but not EGFR WT NSCLC cell lines
We next determined the effects of osimertinib combined with the MEK inhibitor, trametinib, or the ERK inhibitor, VRT752271, on the growth of these EGFR-mutant NSCLC cell lines. We found that the combination of osimertinib with trametinib was much more potent than either agent alone in decreasing the survival of the tested cell lines, with CIs < 1 (Fig. 1B) indicating synergistic effects on decreasing the survival of EGFR-mutant cell lines. Similar results were also generated with the combination of osimertinib and VRT752771 (Fig. 1C). Both combinations had no effect on the growth of two NSCLC cell lines with WT EGFR (Calu-1 and H460; Figs. 1D and E). In addition, the combination of osimertinib and trametinib did not augment the growth suppression of three other EGFR WT NSCLC cell lines (A549, H596 and H226) (Fig. S1). Therefore, the combination of osimertinib with MEK or ERK inhibition selectively and synergistically suppresses the growth of EGFR-mutant NSCLC cells only.
Targeting MEK/ERK combined with osimertinib enhances Bim levels and induction of apoptosis in EGFR-mutant NSCLC cells
We next assessed whether osimertinib combined with MEK or ERK inhibition augments induction of apoptosis in EGFR-mutant NSCLC cells. As presented in Fig. 2A, the combination of osimertinib with trametinib or VRT752271 was significantly more active than either agent alone in increasing the proportion of annexin V-positive (apoptotic) cells in all three tested cell lines (PC-9, HCC827 and H1650). Consistently, these combinations also enhanced cleavage of cacase-3 and PARP in comparison with each single agent, evidenced by detection of the highest levels of cleaved caspase-3 and PARP in both PC-9 and HCC827 cells exposed to the combinations (Fig. 2B). Collectively, these data indicate that osimertinib combined with MEK or ERK inhibition enhances induction of apoptosis in EGFR-mutant NSCLC cells.
Fig. 2. The combination of osimertinib with either trametinib or VRT752271 augments induction of apoptosis including increases in annexin V-positive cells (A) and in protein cleavage (B) accompanied with enhanced Bim elevation (B) in EGFR-mutant NSCLC cells.
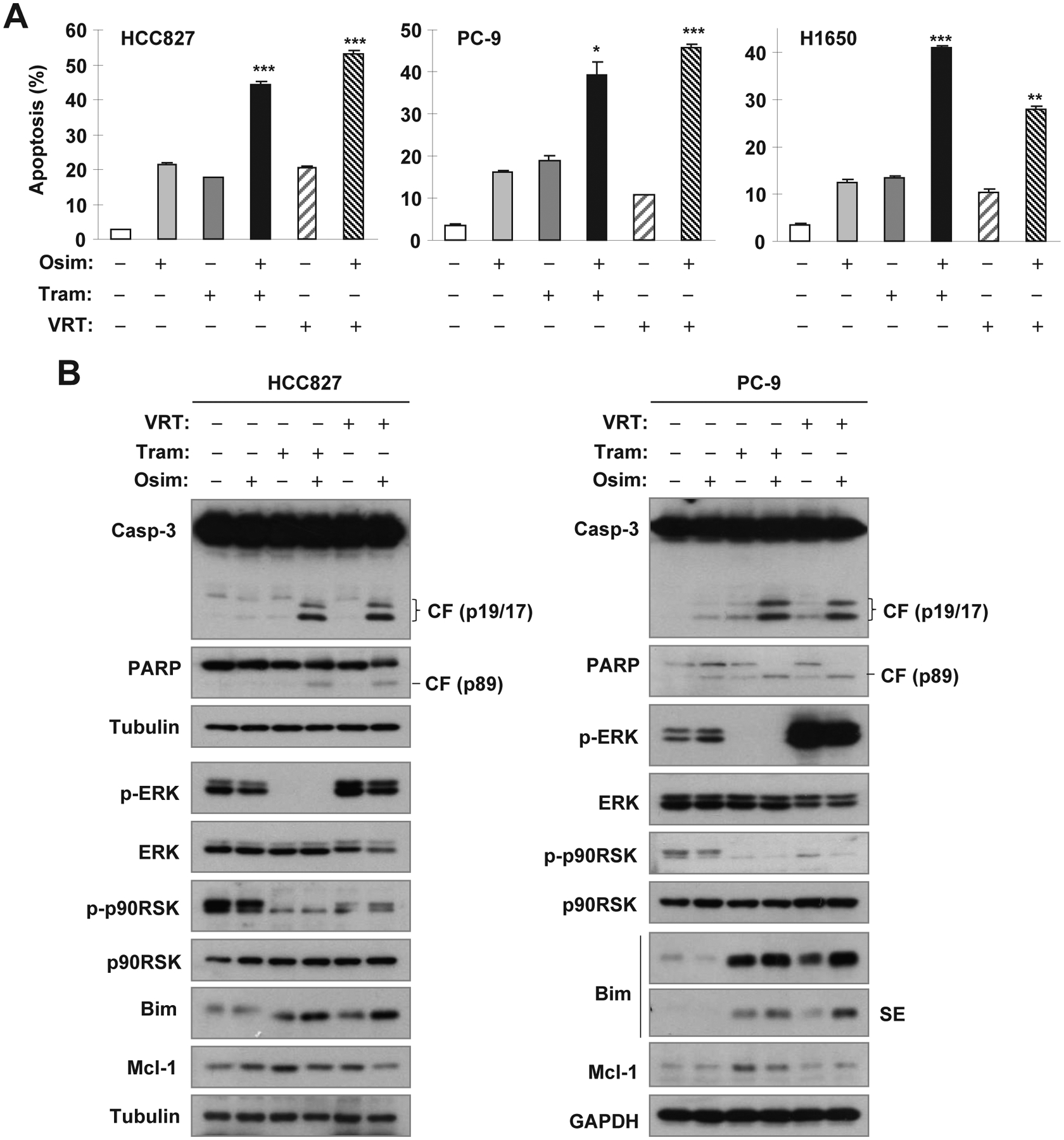
The indicated cell lines were treated with DMSO, 5 nM osimertinib (Osim) alone, 25 nM trametinib (Tram) alone, 0.5 μM VRT52271 (VRT) alone or osimertinib combined with Tram or VRT for 48 h. Apoptosis was evaluated by annexin V flow cytometry (A) and by detection of caspase and PARP cleavage with Western blotting (B). The data in A are means ± SDs of duplicate determinations. * P < 0.05, ** P < 0.01 or *** P < 0.001 at least compared with effect caused by either single agent alone.
Under the tested conditions, treatment with both trametinib and VRT752271 effectively suppressed phosphorylation of ERK and p90RSK (ERK substrate), respectively (Fig. 2B), confirming their effects on the suppression of MER/ERK signaling. Moreover, we detected the highest levels of Bim in cells exposed to these combinations although increased levels of Bim were also detected in cells treated with trametinib or VRT752271 alone (Fig. 2B). Therefore, the combination of osimertinib with MEK or ERK inhibition enhances Bim levels, a key event known to mediate osimertinib-induced apoptosis8, in EGFR-mutant NSCLC cells.
Intrinsically osimertinib-resistant cells are present within sensitive EGFR-mutant NSCLC cell populations
Give the heterogeneous nature of cancer cells, we questioned whether there are pre-existing clones that are intrinsically resistant to osimertinib. To address this question, we treated PC-9 cells once with a very high concentration of osimertinib (2 μM), which eliminated the majority of sensitive cells. We then expanded individual clones of surviving cells and assessed their resistance to osimertinib. Among 16 surviving clones, three (#4, #8 and #12) were fully resistant to osimertinib while the remainder were partially resistant (Fig. 3A). We named the three resistant clones PC-9/intrinsic osimertinib-resistant clone #4 (PC-9/IORC#4), PC-9/IORC#8 and PC-9/IORC#12, respectively. The same strategy was also applied to HCC827 cells. We found that 11 surviving clones were partially resistant to osimertinib although we did not identify fully resistant clones from this cell line (Fig. S2). Therefore, we suggest that intrinsically osimertinib-resistant cells exist within EGFR-mutant NSCLC cell populations that are otherwise sensitive
Fig. 3. Surviving clones from PC-9 cells treated with a one-time high concentration of osimertinib display resistance to osimertinib (A) and are sensitive to the combination of osimertinib with MEK or ERK inhibition (B-E).
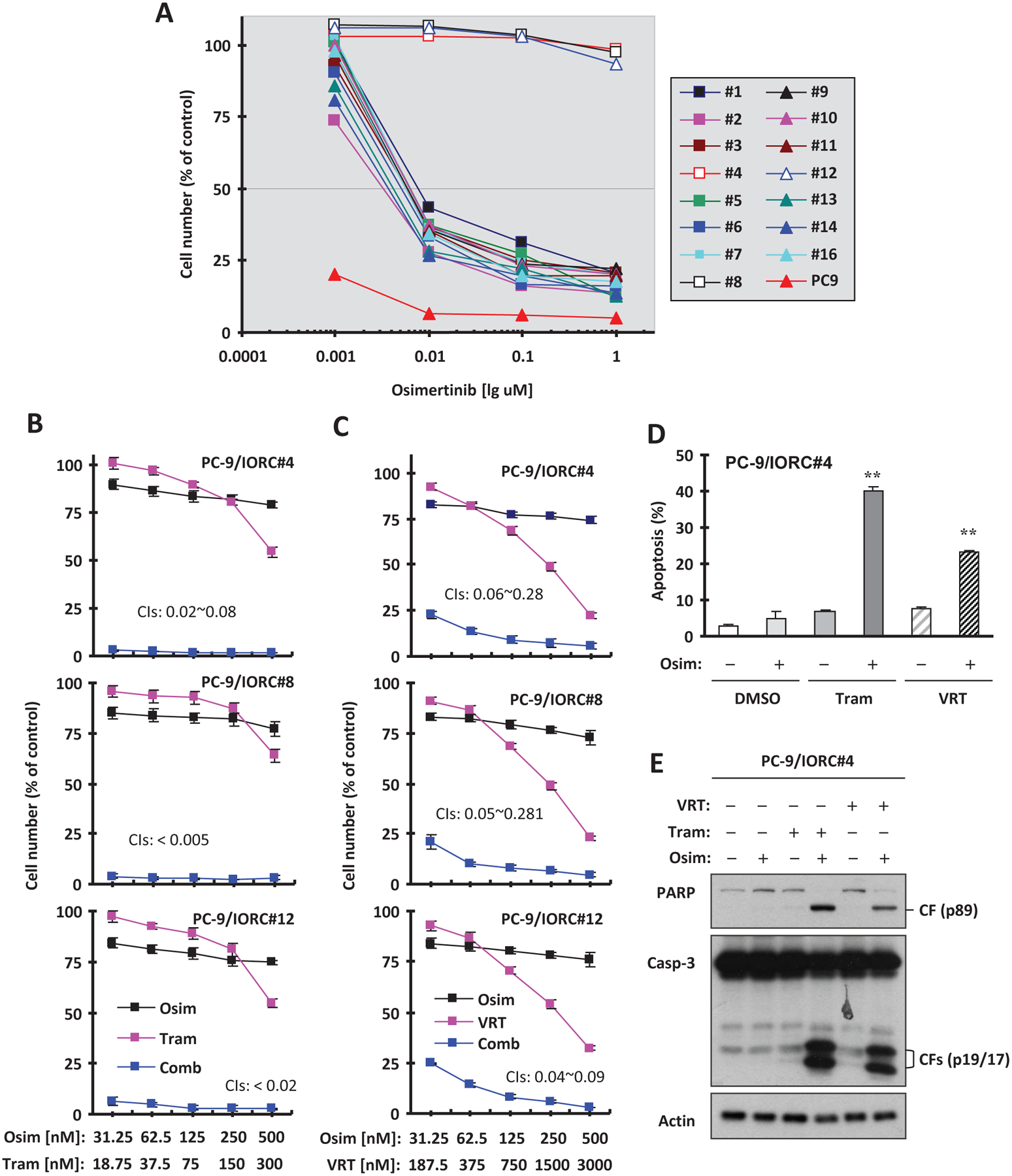
A, PC-9 cells were exposed to 2 μM osimertinib for 3 days. The surviving cells were re-plated after 1:10 dilution with medium. Single clones were picked up after approximately 10 days and expanded. PC-9 and its derived clones as indicated were seeded in 96-well plates and treated with different concentrations of osimertinib for 3 days. Cell numbers were estimated with the SRB assay. B and C, The three most resistant clones as indicated were plated in 96-well plates and on the second day exposed to the varied concentrations of osimertinib (Osim) alone, trametinib (Tram) or VRT52271 (VRT) alone or their respective combinations. After 3 days, cell numbers were estimated using the SRB assay. The data are means ± SDs of four replicate determinations. The numbers inside the graphs are CIs for the given combinations. D and E, PC-9/IOR#4 cells were treated with 200 nM osimertinib, 100 nM trametinib, 0.5 μM VRT752271 or osimertinib combined with trametinib or VRT752271 for 24 h. Apoptosis was evaluated by annexin V flow cytometry (D) and by detection of caspase and PARP cleavage with Western blotting (E). The data in D are means ± SDs of duplicate determinations. ** P < 0.01 at least compared with effect caused by either single agent alone.
Osimertinib combined with MEK or ERK inhibition is highly active in killing intrinsic osimertinib-resistant clones through induction of apoptosis
Given the high activity of osimertinib combined with MEK inhibition against EGFR-mutant NSCLC cells with acquired resistance to osimertinib as we previously reported8, we next examined the effects of osimertinib combined with trametinib or VRT752271 on the growth of these intrinsically osimertinib-resistant clones. Both combinations were highly effective in decreasing the survival of these clones, whereas each single agent in the combinations had limited effects on decreasing the survival of these cell lines (Figs. 3B and C). The ICs for these combinations were far smaller than 1, indicating super-synergistic effects in killing these primarily resistant clones. We also looked at the effects of these combinations on induction of apoptosis in the representative PC-9/IORC#4 cells and found that osimertinib combined with either trametinib or VRT752271 enhanced induction of apoptosis while either single agent had minimal effect on inducing apoptosis as evidenced by enhancing annexin V-positive cells (Fig. 3D) and cleavage of caspase-3 and PARP (Fig. 3E). Therefore, it is clear that the combination of osimertinib with either MEK or ERK inhibition is highly active in killing intrinsic osimertinib-resistant clones through the induction of apoptosis.
The combination of osimertinib with MEK inhibition prevents the emergence of osimertinib resistance in vitro
Following the above promising results, we determined whether targeting MER/ERK signaling indeed prevents or delays the development of acquired resistance to osimertinib. For this purpose, we first used in vitro models to test the emergence of osimertinib resistance and the impact of combination treatment with osimertinib and trametinib on this process. We employed an osimertinib dose of 500 nM, a concentration close to clinically achievable steady-state plasma level of osimertinib in NSCLC patients receiving a 80 mg/day dosage17. Treatment of PC-9 cells in 24-well cell culture plates with 500 nM osimertinib alone effectively eliminated cells within 4 days. However, resistant clones emerged on day 30 and became dominant by day 40 (Fig. 4A), indicating the appearance of resistance. Treatment with trametinib at 20 nM alone did not apparently affect the growth of PC-9 cells; but concurrent treatment with the combination of osimertinib and trametinib eliminated cells and plates remained free of resistant clones during the treatment course up to 64 days (Fig. 4A), indicating effective prevention of the emergence of acquired resistance to osimertinib. To validate this finding, we further conducted a similar experiment in 96-well plates with 60 replicate wells for each treatment. Here we defined positive wells (i.e., with growth of resistant cells) as over 50% cell confluence. We did not detect positive wells until week 5 post treatment with osimertinib alone. After 10 weeks, 80% of wells exposed to osimertinib alone were positive, but none exposed to concurrent treatment with trametinib and osimertinib were positive (Fig. 4B). Similarly, trametinib alone at the tested conditions did not affect the growth of PC-9 cells.
Fig. 4. The combination of osimertinib and trametinib with different schedules delays the emergence of osimertinib resistance in vitro.
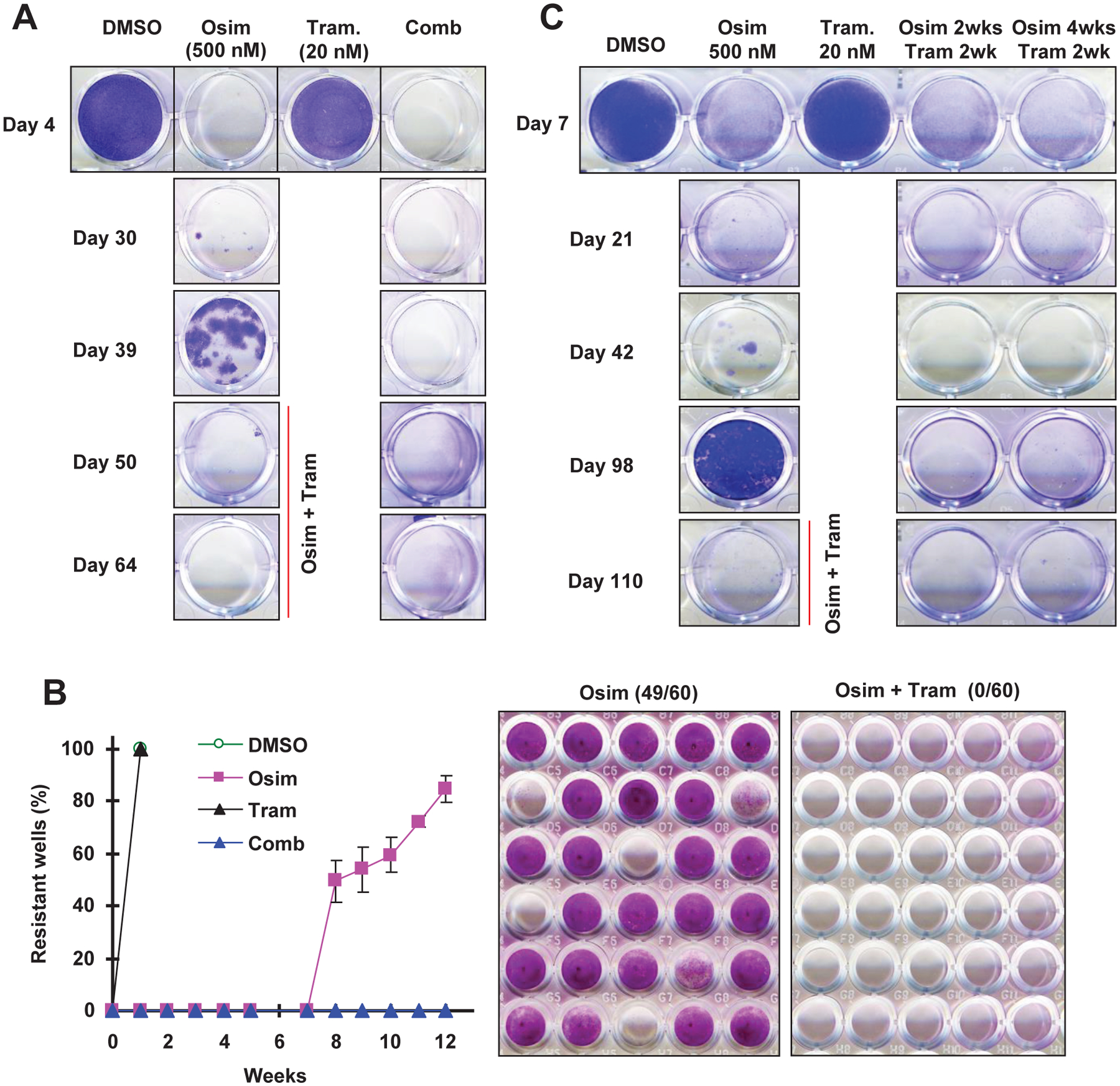
A, PC-9 cells seeded in 24-well plates were exposed to DMSO, 500 nM osimertinib (Osim), 20 trametinib (Tram) and osimertinib plus trametinib, respectively. The same treatments were repeated every 4 days. The cells were stained and pictured on the indicated days. After 39 days, the cells treated with osimertinib alone were switched to treatment with the combination of osimertinib and trametinib. B, PC-9 cells seeded in 96-well plates were exposed to the treatments as described in A. Cell densities were scored weekly for 10 weeks. At the end of the experiment, the cells were fixed and stained with SRB (right panel). The data (left panel) are the means ± SDs of 3 plates, each of which had 60 replicate wells. C, The experimental procedure was basically the same as described in A except for the combination treatments as follows: the combination of osimertinib and trametinib was included for 2 weeks after every 2-weeks or 4-weeks osimertinib treatment. After 98 days, resistant cells treated with osimertinib alone were switched to treatment with the combination of osimertinib and trametinib.
We also tested the effects of different intermittent combination schedules on preventing the emergence of acquired resistance to osimertinib. In this experiment, PC-9 resistant clones in dishes exposed to osimertinib alone were detected after 40 days and almost reached confluence after 98 days. However, visible PC-9 resistant clones were not detected even after 110 days in dishes exposed to a repeating treatment cycle of two different intermittent combination schedules: 1) osimertinib alone for 2 weeks followed by concurrent treatment with osimertinib and trametinib for 2 weeks; and 2) osimertinib alone for 4 weeks followed by concurrent treatment with osimertinib and trametinib for 2 weeks (Fig. 4C). These results indicate that intermittent treatment schedules with osimertinib and trametinib work as effectively as the concurrent treatment schedule in preventing osimertinib resistance.
Since the combination of osimertinib and a MEK inhibitor (e.g., trametinib) is very effective against cells and tumors with osimertinib acquired resistance, as we recently reported8, we then exposed dishes with the appearance of osimertinib-resistant clones in the above described experiments to the combination of osimertinib and trametinib. Consistently, these resistant cells were effectively eliminated (Figs. 4A and C), indicating their responsiveness to this combinatorial treatment.
The combination of osimertinib with MEK inhibition either concurrently or intermittently prevents or delays emergence of acquired resistance to osimertinib in vivo
Finally, we determined whether targeting MEK/ERK signaling is able to prevent or delay the development of acquired resistance to osimertinib in vivo with a tolerable safety profile. To this end, we treated PC-9 xenografts with osimertinib in the absence and presence of trametinib for a sustained period and compared the time to emergence of osimertinib resistance and alterations in body weight. Osimertinib was given to mice at a dose of 15 mg/kg, which represents an equivalent dose of close to 80 mg daily in humans. Trametinib was given once every two days while osimertinib was administered to mice once daily (Fig. 5A). As observed in vitro, the growth of PC-9 xenografts in mice receiving osimertinib treatment alone was effectively inhibited for the first 30 days. After that, these tumors started to grow back and rapidly reached a peak size of around 500 mm3 after 50 days, consistent with the development of treatment resistance. Interestingly, these tumors maintained a relatively slow rate of growth over the next 30 days of treatment. Trametinib alone at the tested condition had no effect on suppressing the growth of PC-9 xenografts. However, the growth of PC-9 xenografts in mice receiving co-treatment with osimertinib and trametinib was effectively suppressed through the end of the treatment period (i.e., 83 days). There was clear regression with the combination after about 60 days, when the tumor sizes dropped below their initial tumor sizes. One mouse even showed undetectable tumor, indicating cure (Fig. 5B). After the termination of treatment, we observed the mice for an additional 31 days and found that tumors in the remaining 5 mice receiving osimertinib and trametinib grew larger (Fig. 5B), suggesting the need for continuous treatment. With this concurrent treatment schedule, no differences were seen in body weights between mice receiving osimertinib and co-treatment with osimertinib and trametinib (Fig. 5C), suggesting that the combination of osimertinib and trametinib does not cause an additive increase in toxicity.
Fig. 5. Concurrent treatment with osimertinib and trametinib combination (A) delays the emergence of osimertinib resistance in vivo (B) without enhancing toxicity (C).
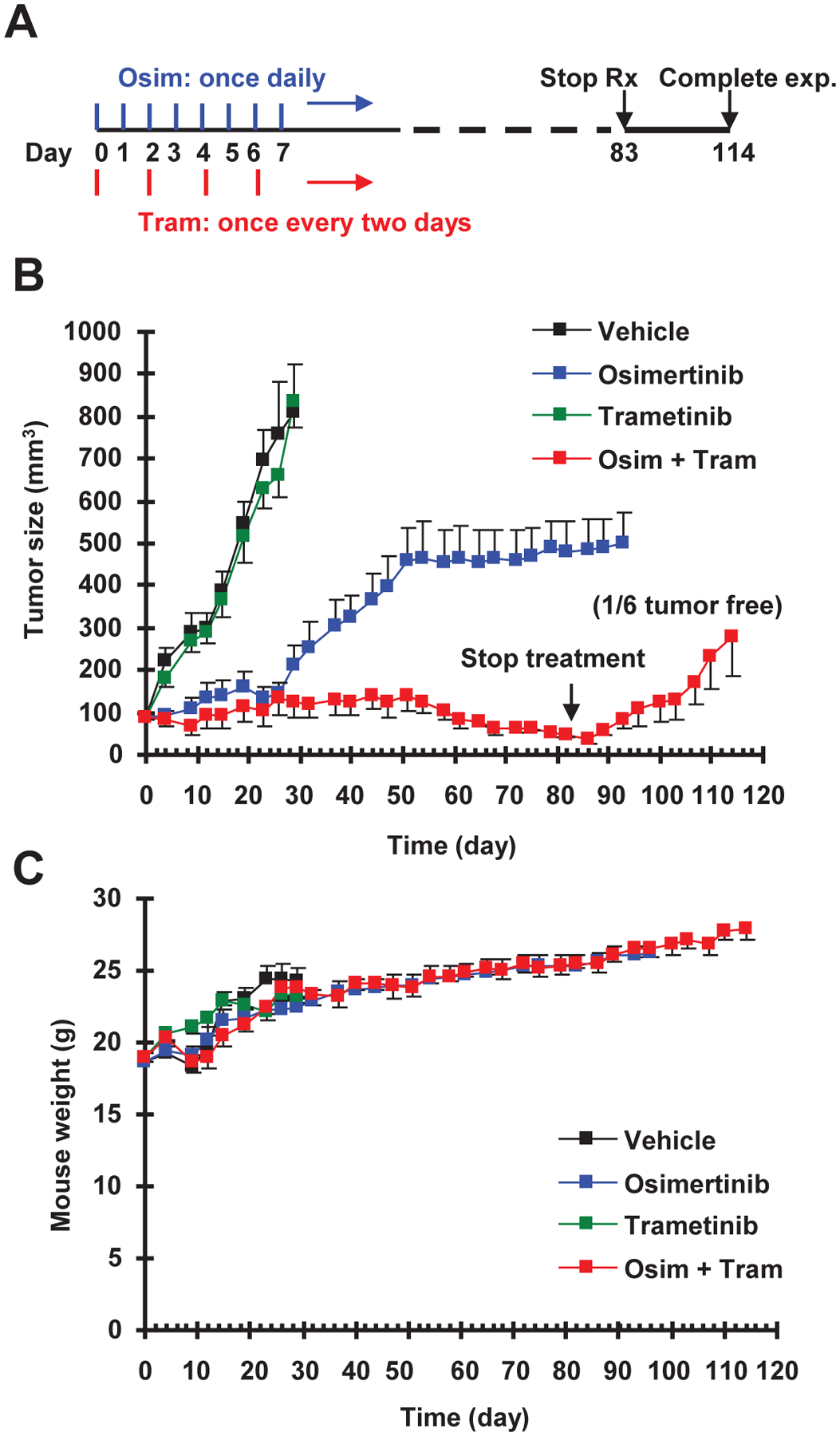
A, A schema for the concurrent treatment schedule. B and C, PC-9 xenografts in nude mice (6 mice/group) were treated with vehicle, osimertinib (15 mg/kg, og, once/day), trametinib (1 mg/kg, og, once/two days) or the combination of osimertinib and trametinib. Tumor sizes and mouse body weights were measured every three days and presented as means ± SEMs.
We next investigated whether intermittent inclusion of MEK inhibition in the osimertinib treatment course achieves a similar effect as concurrent treatment with osimertinib and trametinib in preventing or delaying the emergence of acquired resistance to osimertinib. Hence, we conducted another experiment to test the effects of two different intermittent treatment schedules on delaying the development of acquired osimertinib resistance. In the first schedule, mice with PC-9 xenografts were treated with osimertinib alone for 2 consecutive weeks followed by concurrent treatment with osimertinib and trametinib for another 2 weeks. The second schedule involved 4-weeks treatment with osimertinib alone followed by 2-weeks combined treatment with osimertinib plus trametinib. Both treatment schedules were repeated until the end of the experiments (Fig. 6A). As seen in Fig. 6B, treatment with osimertinib alone was effective at suppressing the growth of PC-9 xenografts for the first 30 days and then lost its efficacy after prolonged treatment. Tumors reached a peak at around 600 mm3 within the next 30 days but showed no further growth thereafter, as we observed in the first experiment (Fig. 5) described above. For these mice with resistant PC-9 xenografts, we then switched treatment with osimertinib alone to the combination of osimertinib and trametinib after 94 days. These large tumors (> 600 mm3) responded quite well to the combination and shrank substantially (< 200 mm3 in size) within just 16 days. Importantly, the growth of PC-9 xenografts in mice receiving either schedule #1 or schedule #2 intermittent treatment with combined osimertinib and trametinib was sustainably suppressed over the 3-month treatment period. Growth suppression of these tumors by both intermittent treatments was maintained for 16 days after treatment was stopped. Importantly, there were two mice in each of the treatment groups with undetectable tumors, indicating their tumor-free or cured status. Again, there was no significant difference in body weights of mice in the intermittent combination groups compared to the osimertinib alone group (Fig. 6C). These results again suggest the safety of the combined treatment.
Fig. 6. Intermittent treatment schedules with osimertinib and trametinib combination (A) delay the emergence of osimertinib resistance in vivo (B) without enhancing toxicity (C).
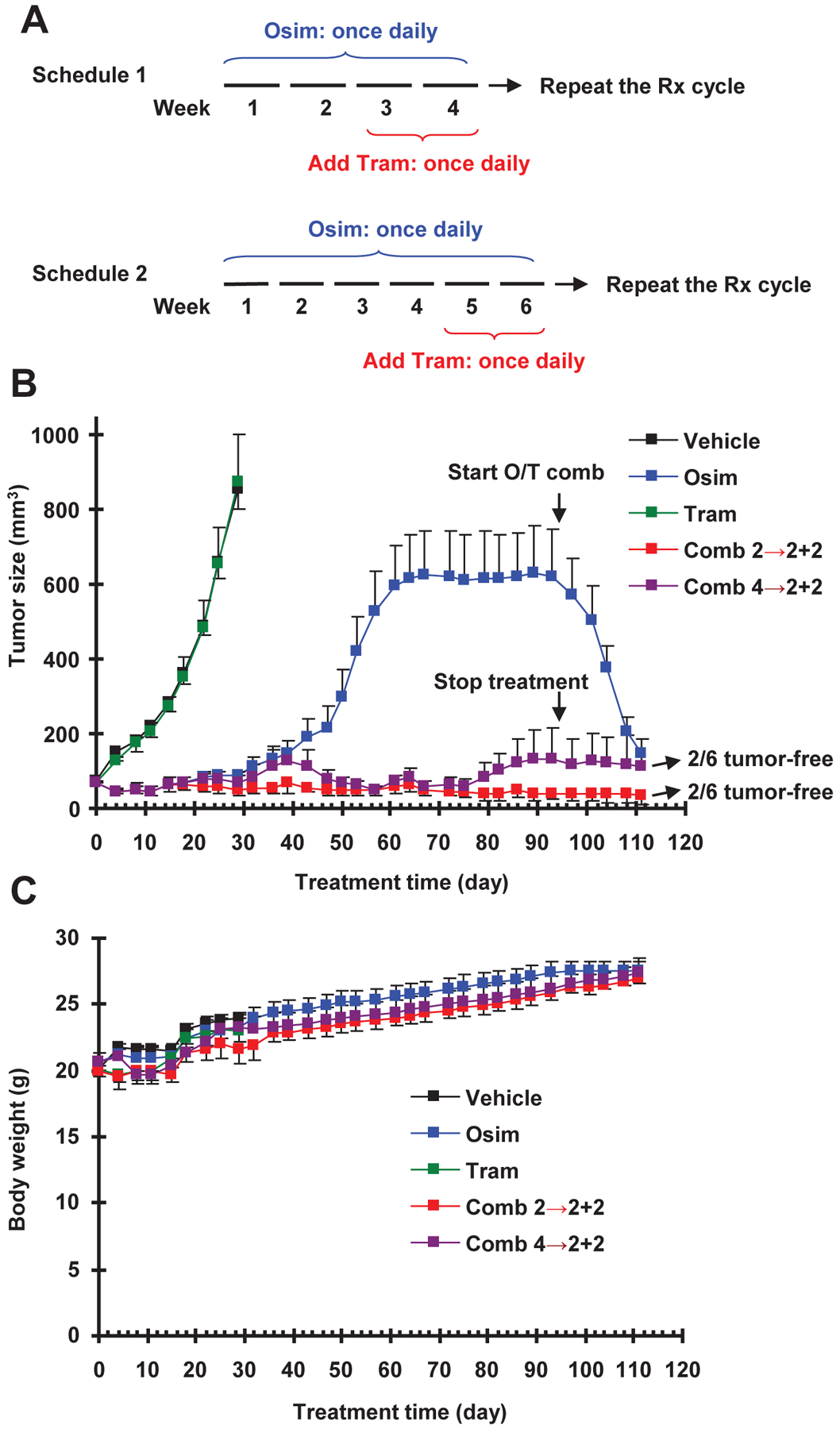
A, Schema of intermittent treatment schedules tested: Schedule #1 involved 2-weeks osimertinib treatment followed by 2-weeks osimertinib and trametinib combination and Schedule #2 used 4-weeks osimertinib treatment followed by 2-weeks osimertinib and trametinib combination. These schedules were repeated until the end of the experiments. B and C, PC-9 xenografts in nude mice (6 mice/group) were treated with vehicle, osimertinib (15 mg/kg, og, once/day), trametinib (1 mg/kg, og, once/day) or the combination of osimertinib and trametinib given according to Schedule 1 or Schedule 2. Tumor sizes and mouse body weights were measured every three days and presented as means ± SEMs.
DISCUSSION
With the approval of osimertinib as first line therapy for EGFR mutated NSCLC, there are at least two distinct strategies that can be adopted in the clinic to improve patient outcomes. The first strategy involves developing novel combination approaches to salvage patients after they develop acquired resistance. Another approach involves using novel combination approaches as part of initial therapy, in order to delay/prevent resistance and promote the efficacy of osimertinib. Previous studies including those from our own lab have demonstrated that targeting MEK/ERK signaling with either a MEK or ERK inhibitor, when combined with osimertinib, exerts impressive effects on overcoming acquired resistance to osimertinib likely through augmented induction of apoptosis8, 10, 11. In addition to this activity, the current study further shows that targeting MEK/ERK signaling is also very effective in delaying the emergence of acquired resistance to osimertinib.
We view delaying emergence of acquired resistance as a promising strategy; however, the use of combination approaches in this process for sustained periods of time also poses the risk of higher toxicity for patients. EGFR-TKIs including osimertinib effectively inhibit MEK/ERK signaling in sensitive NSCLC harboring activating EGFR mutations8, thus overlapping with the functional mechanisms of MEK or ERK inhibitors. As such, the concern is that the combination of these two groups of agents, particularly for long-term application, may accordingly increase toxicity while augmenting therapeutic efficacy. In our previous study, we did not see increased toxicity in 2- or 3-week therapeutic studies against osimertinib-resistant tumors when osimertinib was combined with a MEK or ERK inhibitor in nude mice8, 10. Similarly, the concurrent combination of osimertinib and trametinib administered to mice for over 3 months did not apparently affect mouse growth in this study, indicating its safety. The purpose of designing and testing the activity of intermittent treatment schedules in delaying osimertinib resistance was to limit the administration of trametinib in the osimertinib treatment course to avoid or minimize any potentially increased toxicity. Therefore, the evaluation and outcomes of intermittent treatment schedules described in this study are of great clinical interest to improve the efficacy of osimertinib without posing a risk of undue adverse events. It needs to be pointed out that dosage of trametinib (1 mg/kg; once daily or once every two days) used in mice is higher than that used in human for treatment of NSCLC patients, which is equivalent to around 0.41 mg/kg if calculated based on the 2 mg recommended dosage (once daily) using the guide for human to animal dose conversion18. Considering the promising results demonstrated in this study and long-time half-life of trametinib in human19, reduced dosage or prolonged time of administration for trametinib in the combination may be adjusted. Hence, future clinical application of trametinib in the combination can be further optimized.
Another important finding in this study is that the combination of osimertinib with either a MEK inhibitor or ERK inhibitor was ineffective in further decreasing the survival of NSCLC cell lines with WT EGFR while synergistically decreasing the survival of EGFR-mutant NSCLC cell lines, suggesting that this combinatorial strategy may not affect the growth of cells or tissues (e.g., normal tissues) with WT EGFR. This finding may explain why the combination of osimertinib with either MEK or ERK inhibition is a safe strategy for both delaying and overcoming acquired resistance to osimertinib.
Our overall goal is to prevent or delay the emergency of acquired resistance to osimertinib through a strategy based on the current osimertinib treatment protocol in the clinic. The schedules tested in this study in principle fulfill this requirement without changing the current osimertinib treatment procedure by simply including a MEK or ERK inhibitor either concurrently or intermittently. Our data show that the intermittent schedules, particularly osimertinib for 2 weeks followed by osimertinib plus trametinib for 2 weeks, work as well as or even better than the concurrent treatment schedule in delaying osimertinib resistance. We did not see relapse in the intermittent treatment groups, but saw relapse in the concurrent treatment group after stopping treatment, although the underlying mechanism is unclear. Regardless, we observed tumor-free mice in each combination treatment group, which collectively accounts for a cure rate of 27.8% (5/18 mice in total). This is important clinically and suggests that a proportion of patients may achieve long-term remission with these combination treatment strategies. Considering the nature of time-consuming experimental procedures, this study only used the PC-9 xenograft model, the most sensitive one to osimertinib, to test the efficacies of osimertinib and trametinib combinations in different combinational schedules on delaying or abrogating the emergence of acquired resistance to osimertinib. This is the limitation. Studies with more models including patient-derived xenografts would definitely strengthen the notions demonstrated in this study and thus are warranted.
In this study, we clearly identified some intrinsic osimertinib-resistant clones within the largely sensitive EGFR-mutant NSCLC cell populations. These resistant clones may contribute to the eventual emergence of relapse or resistance to osimertinib in the clinic. Therefore, it is likely that the development of resistance to osimertinib is caused by selection and expansion of intrinsically resistant clones and emergence of cells with acquired resistance. Targeting MEK/ERK signaling was very effective in killing these intrinsic osimertinib-resistant clones as demonstrated in this study. Therefore, early intervention with MEK or ERK inhibition may be helpful for eliminating or suppressing the expansion of these intrinsic osimertinib-resistant clones, allowing a longer duration of disease control in the clinic. This is likely an important mechanism accounting for the effectiveness of targeting MERK/ERK signaling in preventing or delaying the emergence of resistance to osimertinib.
Despite the promising therapeutic efficacy of osimertinib in the clinic, some patients (about 20%) carrying activating EGFR mutation do not respond to osimertinib20. This is also true of some EGFR-mutant NSCLC cell lines (e.g., H1650). Beyond the sensitive cell lines (e.g., PC-9 and HCC827), the less sensitive cell lines (e.g., H1975 and H1650) also responded well to the combination of osimertinib with either a MEK inhibitor or ERK inhibitor as evidenced by synergistic effects on inducing apoptosis and decreasing cell survival. Therefore, targeting MEK/ERK signaling may also enhance the therapeutic efficacy of osimertinib in patients with EGFR-mutant NSCLC that does not respond well to osimertinib monotherapy in the clinic.
In summary, the findings from both in vitro and in vivo preclinical models in this study have demonstrated that targeting MEK/ERK signaling effectively prevents or delays the emergence of osimertinib resistance. These findings provide strong preclinical evidence in support of further investigation of targeting MEK/ERK signaling to delay the emergence of acquired resistance to osimertinib in the clinic and improve the long-term therapeutic efficacy of osimertinib.
Supplementary Material
ACKNOWLEDGEMENT
We are grateful to Dr. Anthea Hammond in our department for editing the manuscript. TKO, SSR and SYS are Georgia Research Alliance Distinguished Cancer Scientists.
Funding support: NIH/NCI R01 CA223220 (to SYS) and UG1 CA233259 (to SSR and SYS), Emory Winship Cancer Institute lung cancer research pilot funds (to SYS) and Lee Foundation Award to the Winship Lung Cancer Program for supporting the pilot project
Footnotes
Conflicts of interest disclosures:
SSR is on consulting/advisory board for AstraZeneca, BMS, Merck, Roche, Tesaro and Amgen. TKO is on consulting/advisory board for Novartis, Celgene, Lilly, Sandoz, Abbvie, Eisai, Takeda, Bristol-Myers Squibb, MedImmune, Amgen, AstraZeneca and Boehringer Ingelheim. No potential conflicts of interest were disclosed for other authors.
REFERENCES
- 1.Tartarone A, Lerose R. Clinical approaches to treat patients with non-small cell lung cancer and epidermal growth factor receptor tyrosine kinase inhibitor acquired resistance. Ther Adv Respir Dis. 2015;9: 242–250. [DOI] [PubMed] [Google Scholar]
- 2.Ramalingam SS, Vansteenkiste J, Planchard D, et al. Overall Survival with Osimertinib in Untreated, EGFR-Mutated Advanced NSCLC. N Engl J Med. 2020;382: 41–50. [DOI] [PubMed] [Google Scholar]
- 3.Lim SM, Syn NL, Cho BC, Soo RA. Acquired resistance to EGFR targeted therapy in non-small cell lung cancer: Mechanisms and therapeutic strategies. Cancer Treat Rev. 2018;65: 1–10. [DOI] [PubMed] [Google Scholar]
- 4.Ramalingam SS, Yang JC, Lee CK, et al. Osimertinib As First-Line Treatment of EGFR Mutation-Positive Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2017: JCO2017747576. [DOI] [PubMed] [Google Scholar]
- 5.Ricordel C, Friboulet L, Facchinetti F, Soria JC. Molecular mechanisms of acquired resistance to third-generation EGFR-TKIs in EGFR T790M-mutant lung cancer. Ann Oncol. 2018;29: i28–i37. [DOI] [PubMed] [Google Scholar]
- 6.Rolfo C, Giovannetti E, Hong DS, et al. Novel therapeutic strategies for patients with NSCLC that do not respond to treatment with EGFR inhibitors. Cancer Treat Rev. 2014;40: 990–1004. [DOI] [PubMed] [Google Scholar]
- 7.Wee P, Wang Z. Epidermal Growth Factor Receptor Cell Proliferation Signaling Pathways. Cancers (Basel). 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi P, Oh YT, Deng L, et al. Overcoming Acquired Resistance to AZD9291, A Third-Generation EGFR Inhibitor, through Modulation of MEK/ERK-Dependent Bim and Mcl-1 Degradation. Clin Cancer Res. 2017;23: 6567–6579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eberlein CA, Stetson D, Markovets AA, et al. Acquired Resistance to the Mutant-Selective EGFR Inhibitor AZD9291 Is Associated with Increased Dependence on RAS Signaling in Preclinical Models. Cancer Res. 2015;75: 2489–2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li Y, Zang H, Qian G, Owonikoko TK, Ramalingam SR, Sun SY. ERK inhibition effectively overcomes acquired resistance of epidermal growth factor receptor-mutant non-small-cell lung cancer cells to osimertinib. Cancer. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Della Corte CM, Ciaramella V, Cardone C, et al. Antitumor Efficacy of Dual Blockade of EGFR Signaling by Osimertinib in Combination With Selumetinib or Cetuximab in Activated EGFR Human NCLC Tumor Models. J Thorac Oncol. 2018;13: 810–820. [DOI] [PubMed] [Google Scholar]
- 12.La Monica S, Minari R, Cretella D, et al. Acquired BRAF G469A Mutation as a Resistance Mechanism to First-Line Osimertinib Treatment in NSCLC Cell Lines Harboring an EGFR Exon 19 Deletion. Target Oncol. 2019;14: 619–626. [DOI] [PubMed] [Google Scholar]
- 13.Dagogo-Jack I, Piotrowska Z, Cobb R, et al. Response to the Combination of Osimertinib and Trametinib in a Patient With EGFR-Mutant NSCLC Harboring an Acquired BRAF Fusion. J Thorac Oncol. 2019;14: e226–e228. [DOI] [PubMed] [Google Scholar]
- 14.Tricker EM, Xu C, Uddin S, et al. Combined EGFR/MEK Inhibition Prevents the Emergence of Resistance in EGFR mutant Lung Cancer. Cancer Discov. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shi P, Oh YT, Zhang G, et al. Met gene amplification and protein hyperactivation is a mechanism of resistance to both first and third generation EGFR inhibitors in lung cancer treatment. Cancer Lett. 2016;380: 494–504. [DOI] [PubMed] [Google Scholar]
- 16.Sun SY, Yue P, Dawson MI, et al. Differential effects of synthetic nuclear retinoid receptor-selective retinoids on the growth of human non-small cell lung carcinoma cells. Cancer Res. 1997;57: 4931–4939. [PubMed] [Google Scholar]
- 17.Brown K, Comisar C, Witjes H, et al. Population pharmacokinetics and exposure-response of osimertinib in patients with non-small cell lung cancer. Br J Clin Pharmacol. 2017;83: 1216–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7: 27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Infante JR, Fecher LA, Falchook GS, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13: 773–781. [DOI] [PubMed] [Google Scholar]
- 20.Skoulidis F, Papadimitrakopoulou VA. Targeting the Gatekeeper: Osimertinib in EGFR T790M Mutation-Positive Non-Small Cell Lung Cancer. Clin Cancer Res. 2017;23: 618–622. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.