

Abstract
Tumor cells and the tumor ecosystem rapidly evolve in response to therapy. This tumor evolution results in the rapid emergence of drug resistance that limits the magnitude and duration of response to therapy including chemotherapy, targeted therapy and immunotherapy. Thus, there is an urgent need to understand and interdict tumor evolution to improve patient benefit to therapy. Reverse phase protein arrays (RPPA) provides a powerful tool to evaluate and develop approaches to target the processes underlying one form of tumor evolution: adaptive evolution. Tumor cells and the tumor microenvironment rapidly evolve through rewiring of protein networks to bypass the effects of therapy. In this review, we present the concepts underlying adaptive resistance and use of RPPA in understanding resistance mechanisms and identification of effective drug combinations. We further demonstrate that this novel information is resulting in biomarker driven trials aimed at targeting adaptive resistance and improving patient outcomes.
Keywords: Reverse phase protein arrays, adaptive resistance, combination therapy, tumor evolution, Targeted therapy
Introduction
We have entered an era where we can characterize the tumor and its microenvironment with an unprecedented breadth and depth. The ability to deeply evaluate the genomic events driving tumor initiation and progression has ushered in personalized targeted and immune therapies aimed at selecting the optimal therapy for each individual: precision oncology. The original enthusiasm for the modern era of precision oncology was driven by remarkable responses to imatinib mesylate in chronic myelogenous leukemia (CML) to the extent that patients with CML now live a lifespan that is not shortened by their disease (Hochhaus et al. 2017). However, despite major progress and remarkable responses in a subset of patients, responses to single agent targeted therapy are transient due to the rapid emergence of resistance. This stresses the need to develop and implement rational combination therapies that will interdict adaptive resistance, and convert the high response rates observed with targeted therapies into durable responses that approximate cures. The implementation of effective combination therapies has its own attendant problems. These include challenges in identification of effective combinations, imperfect prediction which patients will benefit, and management of potential overlapping toxicities of combination therapies that limit their utility. An ability to select effective combinations for individual patients will require that we move beyond the current focus on DNA mutations, fusions and copy number aberrations (CNA) as the dominant method for personalized therapy selection. We have demonstrated that changes at the DNA and RNA level are poorly reflected at the protein level which represents the functional unit in the cell and the most common therapeutic target (Akbani et al. 2014; Johansson et al. 2019; Lu et al. 2016; Zhao et al. 2018). As DNA and RNA changes poorly reflect post translational modifications and the location and function of protein complexes, proteomic data includes important information content not available by nucleotide analysis. Further, even using systems biology approaches, integrating the complexity of the genomic changes at the DNA level into functional events that can be effectively targeted has remained elusive.
Detail protein analysis by RPPA affords an ability to understand the balance between efficacy and toxicity that drives therapeutic index. The deep mechanistic understanding arising from proteomic analysis can provide insights not only into rational drug combinations and biomarkers to select patients who will benefit but as importantly into dose and schedules that alleviate toxicity that cannot be predicted by other approaches.
As noted above, emergence of resistance represents the key challenge that needs to be overcome to improve patient outcomes in response to therapy. Resistance occurs in three major forms: 1) Innate resistance where the tumor and the ecosystem are intrinsically resistant to the selected therapy. This requires a rapid change in therapeutic approach so as not to deliver inactive therapy; 2) Adaptive resistance where the tumor and the microenvironment rapidly evolve in response to therapeutic stress resulting in treatment resistance. Adaptive resistance is generally epigenetic with rewiring of signaling networks as a consequence of therapeutic stress; and 3) Acquired resistance where selection of resistant subclones or generation of new genomic alterations results in genomic resistance. Once it emerges, genomic resistance has proven difficult to reverse. Intratumoral heterogeneity is a major contributor to acquired resistance as drug resistant subclones present prior to therapy initiation can become dominant and result in tumor progression despite ongoing therapy.
This review will focus on adaptive resistance as a mechanism of therapeutic resistance as well as a therapeutic opportunity. Thus, the emphasis will be on model systems wherein longitudinal data are available. Studies based on functional proteomics data obtained by RPPA from both the basal state and drug perturbed state, and conclusions arising from the data will be presented. While systems biology theory indicates that the information content available from a perturbed system is much greater than that which can be obtained from an unperturbed system, there remains significant information that can be gleaned from a single static time point in model systems and from a basal patient biopsy. Indeed, obtaining serial biopsies from patients remains a major challenge in terms of cost, morbidity, and patient compliance. Thus, the ability to determine which drug combinations would benefit a specific patient based on a basal biopsy would be extremely beneficial. Both approaches will be presented throughout this review.
Systems are robust and designed to adapt to stress primarily as a consequence of environmental stresses. These include dietary and inhaled toxins, injuries, and infections. The “system” must change rapidly in response to stress, either to wait for the stress to pass or to directly deal with it. These responses to a major degree are mediated by network rewiring. The stresses may also represent selection of cells with particular “bistable” states. A bistable state represents a cell population that, due to relatively stable rewiring of networks, enters more than one “state”. This state can be “remembered” after cell division or alternatively stochastic events in the cell will result in a stable network for each “state”. Regardless of the underlying mechanism, the network rewiring can render a tumor cell resistant to therapeutic stress in much the same way as normal cells become resistant to environmental stresses. This represents co-option of a normal cellular process by tumor cells and the tumor microenvironment. Cancer cells appear to have a limited number of “rewiring” options to bypass therapeutic stress. Thus, if bypass mechanisms are discoverable, they could provide an effective approach to select effective combination therapies. Importantly, however, since adaptive resistance occurs primarily through network rewiring, it is necessary to implement effective approaches to identify the protein changes that mediate adaptive resistances. Our operating precept is thus that adaptive responses to therapy generate multiple drug escape routes through the rewiring of cellular signaling mechanisms and modulation of the immune and cellular microenvironment in a patient-specific manner.
RPPA platform
RPPA, as implemented by our team based on a technology develop by Petricoin and Liotta (see chapter xx in this book), provides a high throughput, cost effective approach to determine the effects of therapeutic challenge on protein networks. We have validated 460 antibodies as applicable to the RPPA platform with 25% of the antibodies interacting with post translationally modified proteins including phosphorylation, acetylation, methylation and cleavage. Direct comparisons have demonstrated that high quality and high affinity antibodies are more sensitive than mass spectrometry, particularly for post-translational modifications (Mertins et al. 2014). Further, RPPA requires much less material than conventional mass spectrometry approaches, although multiple reaction monitoring (MRM) and related technologies including immunoMRM have the potential to increase sensitivity and decrease input. Importantly, cold ischemia following sample collection appears to have only modest effects on protein amounts with the most significant increases observed being in phosphorylation of stress proteins (Hennessy et al. 2010; Mertins et al. 2014). However, sample collection approaches such as needle aspirates, core biopsies and surgical excision as well as warm ischemia during surgery can markedly affect phosphoprotein levels. Perhaps more critically in terms of longitudinal analysis of patient samples, heterogeneity between lesions samples and in particular changes in tumor and stroma content are critical confounding factors (Labrie et al. 2019; Zhao et al. 2018).
Our current antibody list is available at https://www.mdanderson.org/research/researchresources/core-facilities/functional-proteomics-rppa-core.html. The antibodies have been selected with a major emphasis on cancer initiation and progression pathways, as well as pathways relating to approved and emerging therapeutic agents. Further, the antibodies have been credentialed for use in RPPA by positive correlations with mass spectrometry data, RNA transcriptional profiling, and western blotting across a wide range of cell lines and cell conditions. The pathways interrogated by the platform include differentiation, proliferation, cell cycle progression, apoptosis, autophagy, metabolism, epithelial mesenchymal transition (EMT), DNA damage repair, replication stress, cell surface tyrosine kinase receptors, proliferation, as well as signaling pathways mediated by integrins, TGFα/β, transmembrane receptors, AMPK, TSC/mTOR, PI3K/AKT, RAS/MAPK, Hippo, and Wnt/beta-catenin. The RPPA platform encompasses tumor cell intrinsic mediators as well as extracellular matrix, stroma, growth factors and immune system components. Indeed, as immunotherapy has demonstrated utility in the therapy for a number of tumor lineages, we have greatly extended our antibody repertoire to cover this area. We have analyzed thousands of patient samples including approximately 8000 encompassing the majority of The Cancer Genome Atlas (TCGA) sample set (see TCGA papers) with extensive DNA and RNA data as well as over 1200 cell lines. Various aspects of the data are available from our The Cancer Proteome Atlas website: TCPAportal.org (Li et al. 2013; Li et al. 2017; Chen et al. 2019), as well as for the CCLE cell line dataset (Ghandi et al. 2019) from depmap.org and the TCGA data from cbioportal.org. Our TCPA portal not only provides a downloadable data resource but also tools for exploration and visualization of the data (Chen et al. 2019; Li et al. 2013). Further, we are pleased to host RPPA data from any organization at the site. We continue to make data derived from our RPPA platform as well as our list of validated antibodies available to the community to facilitate research and to provide improved outcomes for our patients.
Using RPPA to understand adaptive responses
Analysis of adaptive responses to therapeutic agents requires an analysis of changes over time. Indeed, we have now analyzed a sparse matrix consisting of over 12,000 longitudinal analyses of protein changes in approximately 150 cell lines across multiple lineages in response to therapeutic agents that is scheduled for release through the Cancer Proteome Atlas website (TCPAportal.org).
We have formulated our adaptive resistance approach as a platform to identify and implement combination therapies for specific patients in a manner that is designed to deliver on the promise of personalized medicine. The approaches are designed to identify combinations that cross the current silos of surgery, targeted, immune, chemo, and radiation therapies to develop optimal approaches that encompass all therapeutic options available to patients. Importantly, we have been highly successful in translating these studies to novel patient trials. To fulfill these goals, we thus developed the Combinatorial Adaptive Response Therapy (CART) platform to use RPPA to characterize and target adaptive responses to therapeutic stresses, thus inducing cell death and preventing acquisition of drug resistance (Budina-Kolomets et al. 2016; Echevarria-Vargas et al. 2018; Fang et al. 2019; Krepler et al. 2017; Krepler et al. 2016; Kwong et al. 2015; Labrie et al. 2019; Lu et al. 2017; Lu et al. 2016; Muranen et al. 2016; Muranen et al. 2012; Sun et al. 2017; Sun et al. 2018; Zervantonakis et al. 2017; Zhang et al. 2016). Using this platform, we have shown that rapid changes in the tumor and tumor ecosystem reflecting adaptive resistance mediates resistance to targeted therapies, but importantly can also constitute therapeutically tractable vulnerabilities to identify rational combinations and overcome resistance. In CART, highly-characterized cancer cell lines, genomically-manipulated cell lines, cell line-derived xenografts, PDX, genetically-manipulated mice (GEMM) or patient samples exposed to a therapeutic agent are systematically analyzed for adaptive responses to the drug. Identification of proteins, signaling pathways and functional events that are altered as a compensatory response to survive the therapeutic stress provides a guideline and filter for targets that can be tested to determine whether inhibition of the altered signaling pathway(s) will interdict the survival effects mediated by adaptive responses. Strikingly, targeting proteins that are induced using drugs, siRNAs, or shRNAs induced synergistic responses for almost 80% of predicted targets, an unprecedented efficacy rate.
CART is applicable to transcriptional changes measured by RNASeq, but the analysis is complicated by the number of RNA candidates assessed and in particular in determining causal events from transcriptional profiles. In contrast, the targets on the RPPA platform have been selected to inform on pathways of importance in cancer and, in particular, drug resistance pathways, thus making deconvoluting data from the RPPA platform much simpler. Further, the smaller number of targets assessed by RPPA decreases challenges with the multiple comparisons problem. To further increase the power of the approach, adaptive responses can be assessed as pathways rather than as individual molecules (Labrie et al. 2019; Lu et al. 2016). Indeed, in a number of systems, RPPA has proven to have marked predictive value for response to therapeutic agents, evaluation of signaling networks, to predict mechanisms of resistance and patient outcomes, in some cases, exceeding that of transcriptional profiling (Cancer Genome Atlas 2015; Hill et al. 2016; Kwong et al. 2015; Yuan et al. 2014; Zhao et al. 2018). However, the information content that can be obtained is enhanced by integration of data from multiple levels.
We have applied CART to a broad set of therapeutic agents including inhibitors of BRAF, MEK, PI3K pathway, BRD4, DNA damage, checkpoint, and polyADP ribose polymerase inhibitors (PARPi). In melanoma patients, longitudinal analysis of samples from patients resistant to BRAF inhibitors have demonstrated, for example, that there are multiple pathways that can bypass the action of BRAF inhibitors including reactivation of the RAS/MAPK pathway as well as RAS/MAPK independent pathways that are not predicted by mutational status (Kwong et al. 2015). These studies have also identified candidate biomarkers of response and resistance to BRAF inhibitors (Kwong et al. 2015). The results have subsequently been confirmed by demonstrating that reactivation of phospho-MAPK signaling as well as activation of the PI3K pathway can mediate BRAF and MEK inhibitor resistance in PDX models (Krepler et al. 2016). The latter observation was confirmed in co-clinical trials by demonstrating re-sensitization of BRAF/MEK resistant melanoma PDXs by introduction of inhibitors of the PI3K pathway (Krepler et al. 2016). Studies of resistance to BRAF and MEK inhibitors in melanoma have also implicated mitochondrial biogenesis, telomere dysfunction, PAK and HSP70 as potential targetable mechanisms of resistance (Budina-Kolomets et al. 2016; Lu et al. 2017; Zhang et al. 2016; Zhang et al. 2018). Extensive characterization of a large collection of melanoma PDX including those sensitive and resistant to targeted therapy and immune checkpoint inhibitors demonstrated tyrosine kinase receptor upregulation including IGF1R that is a potent activator of the PI3K pathway and identified multiple potential therapy combinations as well as biomarkers of response and resistance (Krepler et al. 2017).
Although genomic aberrations in ovarian cancer in the PI3K and RAS/MAPK pathway are rare in high grade serous ovarian cancers, activation of the pathways is relatively common. We thus explored adaptive responses to pathway inhibition in relevant ovarian cancer models using CART. Studies of PI3K pathway inhibitors in ovarian cancer cell models demonstrated that matrix-attached cells are resistant to this therapy due to drug-induced transcriptional upregulation of cellular survival programs mediated by Bcl-2, EGFR, or IGF1R (Muranen et al. 2012). Further studies indicated that tumor upregulation of the proto-oncogene transcriptional regulators c-MYC and YAP1 can mediate resistance to PI3K pathway inhibitors through inhibition of the p38 stress kinase. Furthermore, they suggest that therapies that inactivate YAP or MYC or augment p38 activity could enhance the efficacy of PI3K pathway inhibitors (Muranen et al. 2016). Subsequent analysis of responses to PI3K pathway inhibitors in fourteen highly characterized ovarian cancer PDXs, reflecting the characteristics of human cancer (Liu et al. 2017), demonstrated a key role for apoptotic balance in mediating resistance to pathway inhibition. A subset of apoptotic mediators were predictive of PI3K pathway inhibitor sensitivity (BIM, caspase-3, BCL-XL), whereas others predicted resistance (MCL-1, XIAP) (Zervantonakis et al. 2017). Importantly, inhibition of these adaptive responses abrogates resistance to PI3K pathway inhibitors suggesting combinations that warrant exploration in clinical trials (Muranen et al. 2012; Zervantonakis et al. 2017; Muranen et al. 2016). Although most ovarian cancer models are resistant to monotherapy treatment with MEK inhibitors, CART analysis demonstrated a consistent upregulation of BIM suggesting that the cells would be primed for responses to anti-apoptotic mediators, a hypothesis that was confirmed experimentally (Iavarone et al. 2019). Further, BIM and pERK levels provide biomarkers for response to combinations of MEK and BCL2/XL inhibitors (Iavarone et al. 2019). Once again, CART analysis was predictive of effective drug combinations that warrant exploration in clinical trials.
Inhibition of downstream signaling pathways including the PI3K and MAPK pathways leads to compensatory feedback up regulation of upstream pathways and in particular cell surface tyrosine kinase receptors (O’Reilly et al. 2006; Simpkins et al. 2012). The effects of MEK inhibitors in highly characterized ovarian cancer models (Ince et al. 2015) can be reversed by interdicting src signaling resulting in synergistic responses in vitro and in vivo (Simpkins et al. 2012). Response to MEK inhibitors in ovarian cancer can be predicted by a MAPK pathway signature derived from RPPA as well as by the ability of MEK inhibitors to downregulate the pathway (Hew et al. 2016). The effects of MEK inhibitors are synergistic with inhibition of estrogen signaling in a subset of ovarian cancer models. Based on RPPA analysis, the combination of MEK inhibitors and fulvestrant induce changes in mediators of cell cycle regulators, DNA damage repair, and lysosomal function providing opportunities for additional drug combinations (Hew et al. 2016).
PARP, a critical component of the single strand break (SSB) repair (SSBR) pathway, came into focus as a target when single-strand break repair (SSBR) was identified as a synthetic lethal partner with defects in the homologous repair (HR) pathway induced by BRCA1/2 mutations (Pilie et al. 2019). The original excitement was based on the concept that compromise in base excision repair induced by blocking PARP1 enzyme activity would result in conversion of SSB to double strand breaks (DSB) during DNA replication, thus inducing synthetic lethality in cancer cells with homologous recombination (HR) defects that repair DSB while normal cells retain the ability to repair DSB through HR. Since PARP1 participates in additional DNA repair processes including inhibition of nonhomologous end joining (NHEJ), in alt-NHEJ, and in recruitment of DNA repair proteins consistent with HR-independent PARPi activity, patients without HR-deficient tumors may also benefit from PARP inhibitors (PARPi). Furthermore, several PARPi “trap” PARP proteins at sites of DNA damage, with trapped PARP being more toxic than SSB or DSB. More recently, it has become clear that PARP plays a major role in replication fork protection (Pilie et al. 2019; Sun et al. 2018). Thus, PARPi compromise replication forks resulting in DNA damage and replication stress independent of its role in SSBR. Replication stress as well as HR result in long tracts of single stranded DNA that is protected by RPA32. Increased replication stress and HR can result in RPA32 depletion and replication catastrophe. Thus PARPi have multiple effects on both the S phase and G2 DNA damage checkpoints that present potential therapeutic opportunities (Pilie et al. 2019; Sun et al. 2018).
PARPi monotherapy has demonstrated activity in multiple tumor lineages in association with aberrations in HR repair (Pilie et al. 2019). However only a subset of patients whose umors which have BRCA1/2 mutations have demonstrated benefit from PARPi monotherapy. Further, while there may be activity of PARPi monotherapy in tumors that are HR competent, it is modest. Unfortunately as with most targeted therapies, the responses to PARPi monotherapy are transient due to the rapid emergence of resistance (Pilie et al. 2019). The responses to PARPi monotherapy however, suggest that PARPi could represent a platform on with to build rational drug combinations. Thus, we have implemented a program based on RPPA analysis of adaptive responses and mechanisms of resistance to extend the population of patients who benefit from PARPi beyond those with HR defects, including mutations in BRCA1/2 and to generate durable responses with acceptable toxicity. We have sought approaches: 1) that will induce HR defects in tumors that are HR intact, 2) identify and reverse both intrinsic and adaptive mechanisms of resistance of PARPi, 3) capitalize on the replication stress and DNA damage induced by PARPi, and 4) synergize with the activation of the immune system and particularly STING responses induced by PARPi. The overarching hypothesis is that by targeting the adaptive responses to PARPi, we will be able to attain our goals of prolonged responses with acceptable toxicity across a broad population of patients. This hypothesis is strongly supported by our previous studies, wherein targeting adaptive responses to PARPi with inhibitors of S phase and G2 DNA damage checkpoints (Fang et al. 2019; George et al. 2017), immune checkpoints (Labrie et al. 2019; Shen et al. 2019; Wang et al. 2019), PI3K (Konstantinopoulos et al. 2019; Matulonis et al. 2017), MEK (Sun et al. 2017), or BRD4 (Sun et al. 2018) pathways has induced synergistic responses in both in vitro and in vivo models leading to clinical trials (NCT03162627, NCT02208375, NCT02659241, NCT03586661, NCT02316834, NCT03544125, NCT03801369, NCT03637491) where improved patient outcomes were observed (Konstantinopoulos et al. 2019; Matulonis et al. 2017). Some examples are described in the next paragraphs.
Using RPPA analysis, we demonstrated that PARPi markedly altered the apoptotic pathway and in particular decreased BIM levels (Sun et al. 2017). BIM is regulated through both the PI3K and MAPK pathways, however, our subsequent studies demonstrated that the effects of PARPi on BIM were primarily due to activation of the MAPK pathway. This was supported by the observation that cell lines with RAS mutations were markedly resistant to PARPi and further that cells rendered resistant to PARPi gained RAS mutations or MAPK pathway activation (Sun et al. 2017). The resistance to PARPi was reversed by both MEK and erk inhibitors leading to a clinical trial with PARP and MEK inhibitors (NCT03162627). Initial results from the dose escalation phase of the trial were presented recently demonstrating marked benefit for patients with KRAS mutant tumors, that are normally resistant to therapy. Using a robust approach to identify cells with HR defects combined with mechanistic studies based on RPPA, we demonstrated that BRD4 inhibitors decreased CTIP, which is a critical component of the HR repair pathway (Sun et al. 2018). The combination of BRD4 and PARP inhibitors was synergistic particularly in RAS mutant model systems that were resistant to PARPi. As indicated above, PARPi induce replication stress and increase double strand breaks leading to activation of the S and G2 damage checkpoints as indicated by RPPA analysis amongst other approaches. We thus assessed combinations of PARPi and inhibitors of the S and G2 checkpoints (ATR and Wee1, (Fang et al. 2019)). Remarkably the combinations were synergistic resulting in tumor regressions in a number of model systems (Fang et al. 2019). However, the combinations were poorly tolerated in mice similar to observations in early human trials (Pilie et al 2019). Using RPPA, we demonstrated that the responses, replication stress and DNA damage induced by PARPi and Wee1 inhibitors (Wee1i) persisted for several days after drug removal (Fang et al. 2019). This suggested that sequential therapy could maintain efficacy while ameliorating toxicity, which we subsequently confirmed in multiple relevant systems. We then demonstrated that the differential effect of sequential therapy on tumor cells and normal cells was due to ongoing replication stress in cancer but not normal cells. This has led to the development and approval of the STAR trial to test the hypothesis that sequential therapy with PARP and Wee1 inhibitors will maintain efficacy while ameliorating toxicity in human cancer patients.
A number of criteria need to be fulfilled for targeting adaptive therapy to be effective in patients including: adaptive responses must be patient-specific with different responses in different patients, adaptive responses must be consistent in different tumor lesions within a single patient, and adaptive responses must be targetable. To determine whether these criteria are met, we performed a series of window of opportunity (WOO) ovarian cancer trials (NCT02659241, NCT02316834) using PARP, and S and G2 DNA damage checkpoint (WEE1) inhibitors leveraging the ability to obtain biopsies from multiple sites in ovarian cancer. In the PARPi WOO trials, multiple sites in the peritoneal cavity were biopsied during an initial laparoscopy, patients received 7–14 days of therapy and then multiple biopsies from different lesions were collected at the time of definitive surgery and analyzed by RPPA (Labrie et al. 2019). The goal was to focus on adaptive responses. To achieve this, results from the on-treatment RPPA analysis is divided by the pretreatment data. Importantly, patient specific effects that dominate the RPPA data are removed by dividing treated by untreated samples. In order to visualize the data, we “rank order” changes in proteins by summing median centered protein levels normalized to control. Proteins are represented on the x axis and patient samples on the y axis with consistent decreases at the left end (blue) and increases at the right end (red) of the x axis of the heat map (Figure 1A). Analysis of 9 lesions across 3 patients treated with the PARPi, talazoparib, demonstrated that each of the three lesions in each patient demonstrated a similar response except for 1 sample from patient 2 that likely had limited tumor content. As indicated at the bottom of figure 1A and in colors for the arrows to the heat map, to a different degree in each patient, PARPi upregulated the apoptotic, PI3K and MAPK pathways, EMT, immune activity, and the G2M checkpoint, all of which are targetable. Notably all of the adaptive responses had been observed in our preclinical studies with the exception of induction of immune responses and the marked increase in PDGFRβ in patient #2 (dark red band in all samples) which is also targetable. Figure 1B presents a more detailed analysis of the spectrum of changes in each patient with associated potential therapeutic opportunities. For example, patient 1 demonstrates induction of the G2-M checkpoint, immune engagement, and PI3K pathway. In contrast, patient 2 demonstrates engagement of the immune system and induction of PDGFRβ without activation of the other pathways. Patient 3 is markedly different from patient 1 and 2 with ongoing DNA damage, modest immune induction and engagement of the MAPK pathway. Thus, each patient has a spectrum of combinations that are expected to be active. Perhaps most importantly none of the combinations could be predicted from the untreated biopsy but only from the biopsy on therapy in response to therapeutic stresses. Taken together, the data indicate that there are: 1) a limited spectrum of adaptive responses; 2) adaptive responses are patient-specific; 3) multiple lesions in the same patient engage a consistent suite of adaptive responses, and 4) all patients demonstrate a targetable adaptive response. Based on this data, we have implemented breast cancer trials wherein patients have an initial biopsy receive PARPi monotherapy for one cycle followed by a biopsy and then addition of a targeted therapy to interdict or capitalize on the adaptive responses (NCT03544125, NCT03801369).
Figure 1A. Adaptive responses to PARP inhibitors predict rational combinations Individual patients have different adaptive responses Interlesional proteomic heterogeneity is markedly decreased following therapy.
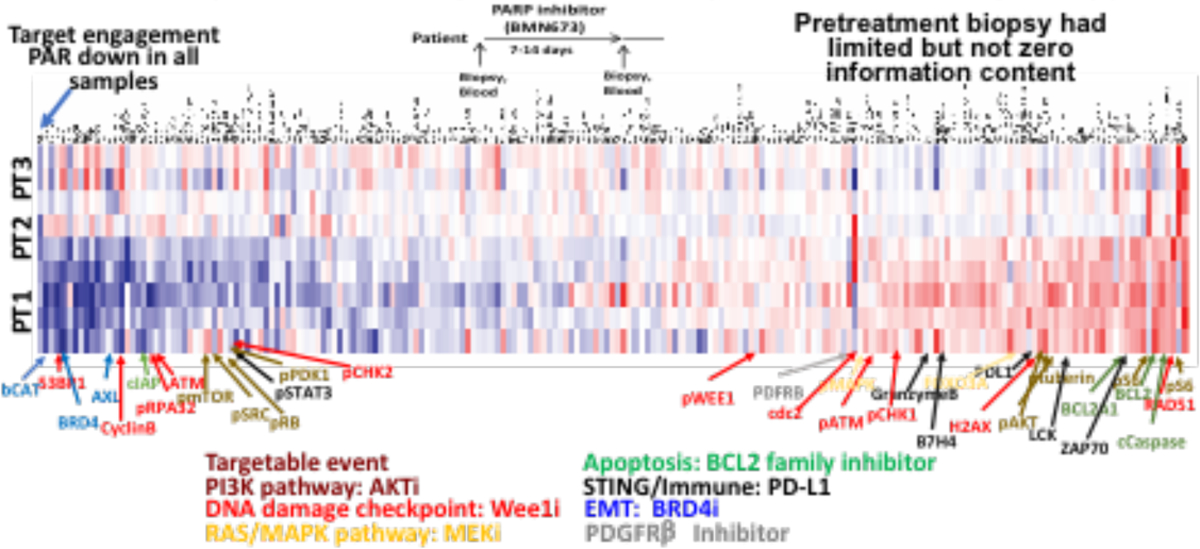
1A Adaptive responses across 3 separate lesions in 3 ovarian cancer patients treated with monotherapy PARPi in a window of opportunity trial (design is at the top). Samples were assessed by RPPA with 300 antibodies to key proteins of interest. Data is presented as the ratio of treated to untreated biopsy to remove patient specific effects and to allow focus on adaptive responses. Antibodies are plotted on the x axis organized from high (red) to low (green) based on the average of all samples and samples are plotted on the y axis.
Figure 1B. Pathway analysis provides patient specific targets.
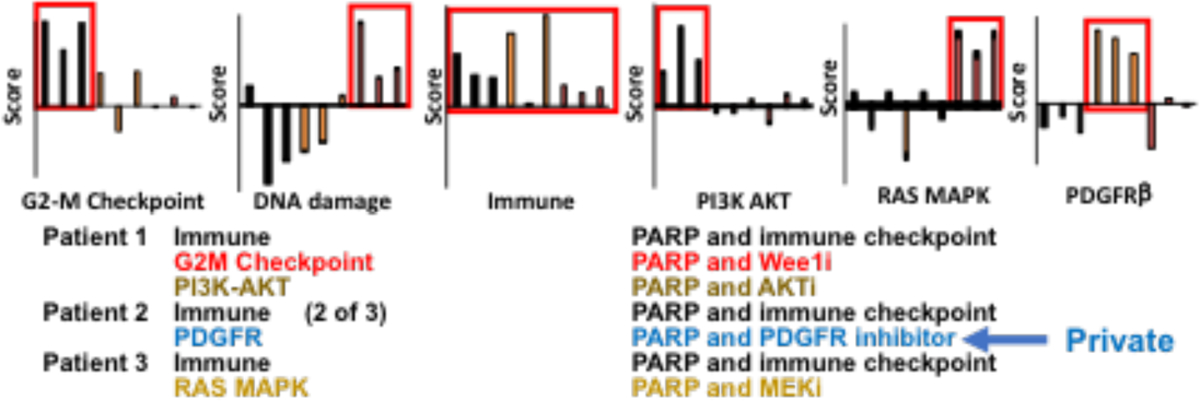
1B Pathway analysis and a novel therapeutic opportunity (PDGFR)Responses are remarkably consistent across lesions in each patient with therapeutic opportunities being different between patients.
The Future
RPPA is a powerful tool to identify adaptive responses to therapy. Indeed, using RPPA as a tool to characterize model systems, basal protein levels in patient samples and importantly adaptive responses to therapy, we have identified and validated a number of combination therapies predicted to abrogate the adaptive responses. We have explored underlying mechanisms and validated the efficacy of the combinations, importantly leading to clinical trials that are demonstrating patient benefit. Further an understand underlying mechanisms arising from RPPA has led to approaches to bypass toxicity including novel dosing and scheduling approaches. RPPA is being used as a tool in clinical trials as indicated in chapter xx (Petricoin). There are a number of limitations to the approach including dependence on fresh frozen samples. We have, however, moved multiplex functional proteomics analysis to the CLIA laboratory to facilitate our clinical trials using a barcoded antibody approach in collaboration with Nanostring (Lee et al. 2018; Merritt et al. 2019). We use RPPA to identify key biomarkers and then transition a small set of targets encompassing key pathways that would be used to select drug combinations for individual patients to the CLIA laboratory. Thus, RPPA serves as our key discovery tool that is used to drive the development of rational combination trials. In the future, the sensitivity, cost effectiveness, and breadth of analysis afforded by RPPA analysis offers the potential to drive mechanistic analysis of tumors that inform on development of rational drug combinations and biomarkers. As the quality control and suite of validated antibodies increases, the power of RPPA will drive studies from the bench to bedside and back provided deep mechanistic understanding and improved patient outcomes.
References
- Akbani R, Ng PK, Werner HM, Shahmoradgoli M, Zhang F, Ju Z, Liu W, Yang JY, Yoshihara K, Li J, Ling S, Seviour EG, Ram PT, Minna JD, Diao L, Tong P, Heymach JV, Hill SM, Dondelinger F, Stadler N, Byers LA, Meric-Bernstam F, Weinstein JN, Broom BM, Verhaak RG, Liang H, Mukherjee S, Lu Y, Mills GB (2014) A pan-cancer proteomic perspective on The Cancer Genome Atlas. Nat Commun 5:3887. doi: 10.1038/ncomms4887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budina-Kolomets A, Webster MR, Leu JI, Jennis M, Krepler C, Guerrini A, Kossenkov AV, Xu W, Karakousis G, Schuchter L, Amaravadi RK, Wu H, Yin X, Liu Q, Lu Y, Mills GB, Xu X, George DL, Weeraratna AT, Murphy ME (2016) HSP70 Inhibition Limits FAK-Dependent Invasion and Enhances the Response to Melanoma Treatment with BRAF Inhibitors. Cancer Res 76 (9):2720–2730. doi: 10.1158/0008-5472.CAN-15-2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N (2015) Genomic Classification of Cutaneous Melanoma. Cell 161 (7):1681–1696. doi: 10.1016/j.cell.2015.05.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M-J, Li J, Akbani R, Wang Y, Lu Y, Mills GB, Liang H (2019) TCPA v6.0: An Integrative Platform for Pan-cancer Analysis of Functional Proteomic Data Mol Cell Proteomics In press [DOI] [PMC free article] [PubMed]
- Echevarria-Vargas IM, Reyes-Uribe PI, Guterres AN, Yin X, Kossenkov AV, Liu Q, Zhang G, Krepler C, Cheng C, Wei Z, Somasundaram R, Karakousis G, Xu W, Morrissette JJ, Lu Y, Mills GB, Sullivan RJ, Benchun M, Frederick DT, Boland G, Flaherty KT, Weeraratna AT, Herlyn M, Amaravadi R, Schuchter LM, Burd CE, Aplin AE, Xu X, Villanueva J (2018) Co-targeting BET and MEK as salvage therapy for MAPK and checkpoint inhibitor-resistant melanoma. EMBO Mol Med 10 (5). doi: 10.15252/emmm.201708446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang Y, McGrail DJ, Sun C, Labrie M, Chen X, Zhang D, Ju Z, Vellano CP, Lu Y, Li Y, Jeong KJ, Ding Z, Liang J, Wang SW, Dai H, Lee S, Sahni N, Mercado-Uribe I, Kim TB, Chen K, Lin SY, Peng G, Westin SN, Liu J, O’Connor MJ, Yap TA, Mills GB (2019) Sequential Therapy with PARP and WEE1 Inhibitors Minimizes Toxicity while Maintaining Efficacy. Cancer Cell 35 (6):851–867 e857. doi: 10.1016/j.ccell.2019.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George E, Kim H, Krepler C, Wenz B, Makvandi M, Tanyi JL, Brown E, Zhang R, Brafford P, Jean S, Mach RH, Lu Y, Mills GB, Herlyn M, Morgan M, Zhang X, Soslow R, Drapkin R, Johnson N, Zheng Y, Cotsarelis G, Nathanson KL, Simpkins F (2017) A patient-derived-xenograft platform to study BRCA-deficient ovarian cancers. JCI Insight 2 (1):e89760. doi: 10.1172/jci.insight.89760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghandi M, Huang FW, Jane-Valbuena J, Kryukov GV, Lo CC, McDonald ER 3rd, Barretina J, Gelfand ET, Bielski CM, Li H, Hu K, Andreev-Drakhlin AY, Kim J, Hess JM, Haas BJ, Aguet F, Weir BA, Rothberg MV, Paolella BR, Lawrence MS, Akbani R, Lu Y, Tiv HL, Gokhale PC, de Weck A, Mansour AA, Oh C, Shih J, Hadi K, Rosen Y, Bistline J, Venkatesan K, Reddy A, Sonkin D, Liu M, Lehar J, Korn JM, Porter DA, Jones MD, Golji J, Caponigro G, Taylor JE, Dunning CM, Creech AL, Warren AC, McFarland JM, Zamanighomi M, Kauffmann A, Stransky N, Imielinski M, Maruvka YE, Cherniack AD, Tsherniak A, Vazquez F, Jaffe JD, Lane AA, Weinstock DM, Johannessen CM, Morrissey MP, Stegmeier F, Schlegel R, Hahn WC, Getz G, Mills GB, Boehm JS, Golub TR, Garraway LA, Sellers WR (2019) Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569 (7757):503–508. doi: 10.1038/s41586-019-1186-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennessy BT, Lu Y, Gonzalez-Angulo AM, Carey MS, Myhre S, Ju Z, Davies MA, Liu W, Coombes K, Meric-Bernstam F, Bedrosian I, McGahren M, Agarwal R, Zhang F, Overgaard J, Alsner J, Neve RM, Kuo WL, Gray JW, Borresen-Dale AL, Mills GB (2010) A Technical Assessment of the Utility of Reverse Phase Protein Arrays for the Study of the Functional Proteome in Non-microdissected Human Breast Cancers. Clin Proteomics 6 (4):129–151. doi: 10.1007/s12014-010-9055-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hew KE, Miller PC, El-Ashry D, Sun J, Besser AH, Ince TA, Gu M, Wei Z, Zhang G, Brafford P, Gao W, Lu Y, Mills GB, Slingerland JM, Simpkins F (2016) MAPK Activation Predicts Poor Outcome and the MEK Inhibitor, Selumetinib, Reverses Antiestrogen Resistance in ER-Positive High-Grade Serous Ovarian Cancer. Clin Cancer Res 22 (4):935–947. doi: 10.1158/1078-0432.CCR-15-0534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill SM, Heiser LM, Cokelaer T, Unger M, Nesser NK, Carlin DE, Zhang Y, Sokolov A, Paull EO, Wong CK, Graim K, Bivol A, Wang H, Zhu F, Afsari B, Danilova LV, Favorov AV, Lee WS, Taylor D, Hu CW, Long BL, Noren DP, Bisberg AJ, Consortium H-D, Mills GB, Gray JW, Kellen M, Norman T, Friend S, Qutub AA, Fertig EJ, Guan Y, Song M, Stuart JM, Spellman PT, Koeppl H, Stolovitzky G, Saez-Rodriguez J, Mukherjee S (2016) Inferring causal molecular networks: empirical assessment through a community-based effort. Nat Methods 13 (4):310–318. doi: 10.1038/nmeth.3773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, Baccarani M, Deininger MW, Cervantes F, Fujihara S, Ortmann CE, Menssen HD, Kantarjian H, O’Brien SG, Druker BJ, Investigators I (2017) Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N Engl J Med 376 (10):917–927. doi: 10.1056/NEJMoa1609324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iavarone C, Zervantonakis IK, Selfors LM, Palakurthi S, Liu JF, Drapkin R, Matulonis UA, Hallberg D, Velculescu VE, Leverson JD, Sampath D, Mills GB, Brugge JS (2019) Combined MEK and BCL-2/XL Inhibition Is Effective in High-Grade Serous Ovarian Cancer Patient-Derived Xenograft Models and BIM Levels Are Predictive of Responsiveness. Mol Cancer Ther 18 (3):642–655. doi: 10.1158/1535-7163.MCT-18-0413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince TA, Sousa AD, Jones MA, Harrell JC, Agoston ES, Krohn M, Selfors LM, Liu W, Chen K, Yong M, Buchwald P, Wang B, Hale KS, Cohick E, Sergent P, Witt A, Kozhekbaeva Z, Gao S, Agoston AT, Merritt MA, Foster R, Rueda BR, Crum CP, Brugge JS, Mills GB (2015) Characterization of twenty-five ovarian tumour cell lines that phenocopy primary tumours. Nat Commun 6:7419. doi: 10.1038/ncomms8419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson HJ, Socciarelli F, Vacanti NM, Haugen MH, Zhu Y, Siavelis I, Fernandez-Woodbridge A, Aure MR, Sennblad B, Vesterlund M, Branca RM, Orre LM, Huss M, Fredlund E, Beraki E, Garred O, Boekel J, Sauer T, Zhao W, Nord S, Hoglander EK, Jans DC, Brismar H, Haukaas TH, Bathen TF, Schlichting E, Naume B, Consortia Oslo Breast Cancer Research C, Luders T, Borgen E, Kristensen VN, Russnes HG, Lingjaerde OC, Mills GB, Sahlberg KK, Borresen-Dale AL, Lehtio J (2019) Breast cancer quantitative proteome and proteogenomic landscape. Nat Commun 10 (1):1600. doi: 10.1038/s41467-019-09018-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konstantinopoulos PA, Barry WT, Birrer M, Westin SN, Cadoo KA, Shapiro GI, Mayer EL, O’Cearbhaill RE, Coleman RL, Kochupurakkal B, Whalen C, Curtis J, Farooq S, Luo W, Eismann J, Buss MK, Aghajanian C, Mills GB, Palakurthi S, Kirschmeier P, Liu J, Cantley LC, Kaufmann SH, Swisher EM, D’Andrea AD, Winer E, Wulf GM, Matulonis UA (2019) Olaparib and alpha-specific PI3K inhibitor alpelisib for patients with epithelial ovarian cancer: a dose-escalation and dose-expansion phase 1b trial. Lancet Oncol 20 (4):570–580. doi: 10.1016/S1470-2045(18)30905-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krepler C, Sproesser K, Brafford P, Beqiri M, Garman B, Xiao M, Shannan B, Watters A, Perego M, Zhang G, Vultur A, Yin X, Liu Q, Anastopoulos IN, Wubbenhorst B, Wilson MA, Xu W, Karakousis G, Feldman M, Xu X, Amaravadi R, Gangadhar TC, Elder DE, Haydu LE, Wargo JA, Davies MA, Lu Y, Mills GB, Frederick DT, Barzily-Rokni M, Flaherty KT, Hoon DS, Guarino M, Bennett JJ, Ryan RW, Petrelli NJ, Shields CL, Terai M, Sato T, Aplin AE, Roesch A, Darr D, Angus S, Kumar R, Halilovic E, Caponigro G, Jeay S, Wuerthner J, Walter A, Ocker M, Boxer MB, Schuchter L, Nathanson KL, Herlyn M (2017) A Comprehensive Patient-Derived Xenograft Collection Representing the Heterogeneity of Melanoma. Cell Rep 21 (7):1953–1967. doi: 10.1016/j.celrep.2017.10.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krepler C, Xiao M, Sproesser K, Brafford PA, Shannan B, Beqiri M, Liu Q, Xu W, Garman B, Nathanson KL, Xu X, Karakousis GC, Mills GB, Lu Y, Ahmed TA, Poulikakos PI, Caponigro G, Boehm M, Peters M, Schuchter LM, Weeraratna AT, Herlyn M (2016) Personalized Preclinical Trials in BRAF Inhibitor-Resistant Patient-Derived Xenograft Models Identify Second-Line Combination Therapies. Clin Cancer Res 22 (7):1592–1602. doi: 10.1158/1078-0432.CCR-15-1762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong LN, Boland GM, Frederick DT, Helms TL, Akid AT, Miller JP, Jiang S, Cooper ZA, Song X, Seth S, Kamara J, Protopopov A, Mills GB, Flaherty KT, Wargo JA, Chin L (2015) Co-clinical assessment identifies patterns of BRAF inhibitor resistance in melanoma. J Clin Invest 125 (4):1459–1470. doi: 10.1172/JCI78954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labrie M, Kim TB, Ju Z, Lee S, Zhao W, Fang Y, Lu Y, Chen K, Ramirez P, Frumovitz M, Meyer L, Fleming ND, Sood AK, Coleman RL, Mills GB, Westin SN (2019) Adaptive responses in a PARP inhibitor window of opportunity trial illustrate limited functional interlesional heterogeneity and potential combination therapy options. Oncotarget 10 (37):3533–3546. doi: 10.18632/oncotarget.26947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Geiss GK, Demirkan G, Vellano CP, Filanoski B, Lu Y, Ju Z, Yu S, Guo H, Bogatzki LY, Carter W, Meredith RK, Krishnamurthy S, Ding Z, Beechem JM, Mills GB (2018) Implementation of a Multiplex and Quantitative Proteomics Platform for Assessing Protein Lysates Using DNA-Barcoded Antibodies. Mol Cell Proteomics 17 (6):1245–1258. doi: 10.1074/mcp.RA117.000291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lu Y, Akbani R, Ju Z, Roebuck PL, Liu W, Yang JY, Broom BM, Verhaak RG, Kane DW, Wakefield C, Weinstein JN, Mills GB, Liang H (2013) TCPA: a resource for cancer functional proteomics data. Nat Methods 10 (11):1046–1047. doi: 10.1038/nmeth.2650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao W, Akbani R, Liu W, Ju Z, Ling S, Vellano CP, Roebuck P, Yu Q, Eterovic AK, Byers LA, Davies MA, Deng W, Gopal YN, Chen G, von Euw EM, Slamon D, Conklin D, Heymach JV, Gazdar AF, Minna JD, Myers JN, Lu Y, Mills GB, Liang H (2017) Characterization of Human Cancer Cell Lines by Reverse-phase Protein Arrays. Cancer Cell 31 (2):225–239. doi: 10.1016/j.ccell.2017.01.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JF, Palakurthi S, Zeng Q, Zhou S, Ivanova E, Huang W, Zervantonakis IK, Selfors LM, Shen Y, Pritchard CC, Zheng M, Adleff V, Papp E, Piao H, Novak M, Fotheringham S, Wulf GM, English J, Kirschmeier PT, Velculescu VE, Paweletz C, Mills GB, Livingston DM, Brugge JS, Matulonis UA, Drapkin R (2017) Establishment of Patient-Derived Tumor Xenograft Models of Epithelial Ovarian Cancer for Preclinical Evaluation of Novel Therapeutics. Clin Cancer Res 23 (5):1263–1273. doi: 10.1158/1078-0432.CCR-16-1237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Liu S, Zhang G, Bin W, Zhu Y, Frederick DT, Hu Y, Zhong W, Randell S, Sadek N, Zhang W, Chen G, Cheng C, Zeng J, Wu LW, Zhang J, Liu X, Xu W, Krepler C, Sproesser K, Xiao M, Miao B, Liu J, Song CD, Liu JY, Karakousis GC, Schuchter LM, Lu Y, Mills G, Cong Y, Chernoff J, Guo J, Boland GM, Sullivan RJ, Wei Z, Field J, Amaravadi RK, Flaherty KT, Herlyn M, Xu X, Guo W (2017) PAK signalling drives acquired drug resistance to MAPK inhibitors in BRAF-mutant melanomas. Nature 550 (7674):133–136. doi: 10.1038/nature24040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Ling S, Hegde AM, Byers LA, Coombes K, Mills GB, Akbani R (2016) Using reverse-phase protein arrays as pharmacodynamic assays for functional proteomics, biomarker discovery, and drug development in cancer. Semin Oncol 43 (4):476–483. doi: 10.1053/j.seminoncol.2016.06.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matulonis UA, Wulf GM, Barry WT, Birrer M, Westin SN, Farooq S, Bell-McGuinn KM, Obermayer E, Whalen C, Spagnoletti T, Luo W, Liu H, Hok RC, Aghajanian C, Solit DB, Mills GB, Taylor BS, Won H, Berger MF, Palakurthi S, Liu J, Cantley LC, Winer E (2017) Phase I dose escalation study of the PI3kinase pathway inhibitor BKM120 and the oral poly (ADP ribose) polymerase (PARP) inhibitor olaparib for the treatment of high-grade serous ovarian and breast cancer. Ann Oncol 28 (3):512–518. doi: 10.1093/annonc/mdw672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt CR, Ong GT, Church S, Barker K, Geiss G, Hoang M, Jung J, Liang Y, McKay-Fleisch J, Nguyen K, Sorg K, Sprague I, Warren C, Warren S, Zhou Z, Zollinger DR, Dunaway DL, Mills GB, Beechem JM (2019) High multiplex, digital spatial profiling of proteins and RNA in fixed tissue using genomic detection methods. BioRxiv. doi: 10.1101/559021 [DOI] [PubMed] [Google Scholar]
- Mertins P, Yang F, Liu T, Mani DR, Petyuk VA, Gillette MA, Clauser KR, Qiao JW, Gritsenko MA, Moore RJ, Levine DA, Townsend R, Erdmann-Gilmore P, Snider JE, Davies SR, Ruggles KV, Fenyo D, Kitchens RT, Li S, Olvera N, Dao F, Rodriguez H, Chan DW, Liebler D, White F, Rodland KD, Mills GB, Smith RD, Paulovich AG, Ellis M, Carr SA (2014) Ischemia in tumors induces early and sustained phosphorylation changes in stress kinase pathways but does not affect global protein levels. Mol Cell Proteomics 13 (7):1690–1704. doi: 10.1074/mcp.M113.036392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranen T, Selfors LM, Hwang J, Gallegos LL, Coloff JL, Thoreen CC, Kang SA, Sabatini DM, Mills GB, Brugge JS (2016) ERK and p38 MAPK Activities Determine Sensitivity to PI3K/mTOR Inhibition via Regulation of MYC and YAP. Cancer Res 76 (24):7168–7180. doi: 10.1158/0008-5472.CAN-16-0155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muranen T, Selfors LM, Worster DT, Iwanicki MP, Song L, Morales FC, Gao S, Mills GB, Brugge JS (2012) Inhibition of PI3K/mTOR leads to adaptive resistance in matrix-attached cancer cells. Cancer Cell 21 (2):227–239. doi: 10.1016/j.ccr.2011.12.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Reilly KE, Rojo F, She QB, Solit D, Mills GB, Smith D, Lane H, Hofmann F, Hicklin DJ, Ludwig DL, Baselga J, Rosen N (2006) mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res 66 (3):1500–1508. doi: 10.1158/0008-5472.CAN-05-2925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilie PG, Tang C, Mills GB, Yap TA (2019) State-of-the-art strategies for targeting the DNA damage response in cancer. Nat Rev Clin Oncol 16 (2):81–104. doi: 10.1038/s41571-018-0114-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen J, Zhao W, Ju Z, Wang L, Peng Y, Labrie M, Yap TA, Mills GB, Peng G (2019) PARPi Triggers the STING-Dependent Immune Response and Enhances the Therapeutic Efficacy of Immune Checkpoint Blockade Independent of BRCAness. Cancer Res 79 (2):311–319. doi: 10.1158/0008-5472.CAN-18-1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simpkins F, Hevia-Paez P, Sun J, Ullmer W, Gilbert CA, da Silva T, Pedram A, Levin ER, Reis IM, Rabinovich B, Azzam D, Xu XX, Ince TA, Yang JY, Verhaak RG, Lu Y, Mills GB, Slingerland JM (2012) Src Inhibition with saracatinib reverses fulvestrant resistance in ER-positive ovarian cancer models in vitro and in vivo. Clin Cancer Res 18 (21):5911–5923. doi: 10.1158/1078-0432.CCR-12-1257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Fang Y, Yin J, Chen J, Ju Z, Zhang D, Chen X, Vellano CP, Jeong KJ, Ng PK, Eterovic AKB, Bhola NH, Lu Y, Westin SN, Grandis JR, Lin SY, Scott KL, Peng G, Brugge J, Mills GB (2017) Rational combination therapy with PARP and MEK inhibitors capitalizes on therapeutic liabilities in RAS mutant cancers. Sci Transl Med 9 (392). doi: 10.1126/scitranslmed.aal5148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun C, Yin J, Fang Y, Chen J, Jeong KJ, Chen X, Vellano CP, Ju Z, Zhao W, Zhang D, Lu Y, Meric-Bernstam F, Yap TA, Hattersley M, O’Connor MJ, Chen H, Fawell S, Lin SY, Peng G, Mills GB (2018) BRD4 Inhibition Is Synthetic Lethal with PARP Inhibitors through the Induction of Homologous Recombination Deficiency. Cancer Cell 33 (3):401–416 e408. doi: 10.1016/j.ccell.2018.01.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Sun K, Xiao Y, Feng B, Mikule K, Ma X, Feng N, Vellano CP, Federico L, Marszalek JR, Mills GB, Hanke J, Ramaswamy S, Wang J (2019) Niraparib activates interferon signaling and potentiates anti-PD-1 antibody efficacy in tumor models. Sci Rep 9 (1):1853. doi: 10.1038/s41598-019-38534-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Van Allen EM, Omberg L, Wagle N, Amin-Mansour A, Sokolov A, Byers LA, Xu Y, Hess KR, Diao L, Han L, Huang X, Lawrence MS, Weinstein JN, Stuart JM, Mills GB, Garraway LA, Margolin AA, Getz G, Liang H (2014) Assessing the clinical utility of cancer genomic and proteomic data across tumor types. Nat Biotechnol 32 (7):644–652. doi: 10.1038/nbt.2940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zervantonakis IK, Iavarone C, Chen HY, Selfors LM, Palakurthi S, Liu JF, Drapkin R, Matulonis U, Leverson JD, Sampath D, Mills GB, Brugge JS (2017) Systems analysis of apoptotic priming in ovarian cancer identifies vulnerabilities and predictors of drug response. Nat Commun 8 (1):365. doi: 10.1038/s41467-017-00263-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Frederick DT, Wu L, Wei Z, Krepler C, Srinivasan S, Chae YC, Xu X, Choi H, Dimwamwa E, Ope O, Shannan B, Basu D, Zhang D, Guha M, Xiao M, Randell S, Sproesser K, Xu W, Liu J, Karakousis GC, Schuchter LM, Gangadhar TC, Amaravadi RK, Gu M, Xu C, Ghosh A, Xu W, Tian T, Zhang J, Zha S, Liu Q, Brafford P, Weeraratna A, Davies MA, Wargo JA, Avadhani NG, Lu Y, Mills GB, Altieri DC, Flaherty KT, Herlyn M (2016) Targeting mitochondrial biogenesis to overcome drug resistance to MAPK inhibitors. J Clin Invest 126 (5):1834–1856. doi: 10.1172/JCI82661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang G, Wu LW, Mender I, Barzily-Rokni M, Hammond MR, Ope O, Cheng C, Vasilopoulos T, Randell S, Sadek N, Beroard A, Xiao M, Tian T, Tan J, Saeed U, Sugarman E, Krepler C, Brafford P, Sproesser K, Murugan S, Somasundaram R, Garman B, Wubbenhorst B, Woo J, Yin X, Liu Q, Frederick DT, Miao B, Xu W, Karakousis GC, Xu X, Schuchter LM, Mitchell TC, Kwong LN, Amaravadi RK, Lu Y, Boland GM, Wei Z, Nathanson K, Herbig U, Mills GB, Flaherty KT, Herlyn M, Shay JW (2018) Induction of Telomere Dysfunction Prolongs Disease Control of Therapy-Resistant Melanoma. Clin Cancer Res 24 (19):4771–4784. doi: 10.1158/1078-0432.CCR-17-2773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W, Li J, Akbani R, Liang H, Mills GB (2018) Credentialing Individual Samples for Proteogenomic Analysis. Mol Cell Proteomics 17 (8):1515–1530. doi: 10.1074/mcp.RA118.000645 [DOI] [PMC free article] [PubMed] [Google Scholar]