

Abstract
Crohn’s disease (CD), one of the major forms of inflammatory bowel disease (IBD), is characterized by chronic inflammation of the gastrointestinal tract and associated with aberrant CD4+ Th1 and Th17 responses. Protein kinase 2 (CK2) is a conserved serine-threonine kinase involved in signal transduction pathways which regulate immune responses. CK2 promotes Th17 cell differentiation and suppresses the generation of Foxp3+ regulatory T cells. The function of CK2 in CD4+ T-cells during the pathogenesis of CD is unknown. We utilized the T-cell induced colitis model, transferring CD45RBhi naïve CD4+ T-cells from CK2αfl/fl controls and CK2αfl/fldLck-Cre mice into Rag 1−/− mice. CD4+ T-cells from CK2αfl/fldLck-Cre mice failed to induce wasting disease and significant intestinal inflammation, which was associated with decreased IL-17A+, IFN-γ+ and double positive IL-17A+ IFN-γ+ CD4+ T-cells in the spleen and colon. We determined that CK2α regulates CD4+ T-cell proliferation through a cell-intrinsic manner. CK2α is also important in controlling CD4+ T-cell responses by regulating NFAT2, which is vital for T-cell activation and proliferation. Our findings indicate that CK2α contributes to the pathogenesis of colitis by promoting CD4+ T-cell proliferation and Th1 and Th17 responses, and that targeting CK2 may be a novel therapeutic treatment for patients with CD.
INTRODUCTION
Protein Kinase CK2 is a highly conserved serine-threonine kinase that is expressed in all eukaryotic organisms1. CK2 is responsible for the phosphorylation of serine and threonine residues specified by acidic side chains in many proteins, including growth factor receptors, transcription factors, and cytoskeletal proteins 2, 3. Aberrant expression and high CK2 kinase activity are characteristic of many cancers, promoting tumor survival and growth, and CK2 is a promising therapeutic target for malignant diseases 4. CK2 exists in tetrameric complexes consisting of two catalytic subunits (CK2α and/or CK2α’) and two regulatory subunits (CK2β), The regulatory subunit is not essential for activity, but it confers specificity and can affect the ability of the catalytic subunits to phosphorylate certain substrates 5. CK2 enhances the activity of several signaling pathways that are essential for cell proliferation and differentiation, including the NF-κB, PI3K/AKT/mTOR and JAK/STAT pathways 1, 6, 7. CK2 directly phosphorylates NF-κB p65 and IκB to enhance NF-κB signaling, and phosphorylates AKT to activate the mTOR pathway 8. Our previous studies provided the first evidence that CK2 is critical for activation of the JAK/STAT signaling pathway in tumor cells and T-cells 7–10.
Inflammatory Bowel Diseases (IBDs) are chronic relapsing inflammatory disorders of the gastrointestinal tract and can be classified into two major subtypes, Crohn’s disease (CD) and ulcerative colitis (UC) 11, 12. It is widely accepted that IBDs are triggered by an inappropriate immune response, primarily by CD4+ T-cells to antigens of commensal gut bacteria in genetically susceptible cohorts 13. In CD, there is a bias toward the production of proinflammatory cytokines associated with T helper (Th) 1 (IFN-γ) and Th17 (IL-17) cells 12, 14, whereas UC is thought to be associated with Th2 cells producing IL-5 and IL-13 15, 16. Accordingly, one of the main therapeutic strategies for IBDs is to target CD4+ T-cells.
Growing evidence suggests that CK2 can modulate the function of immune cells, including CD4+ T-cells 9, 10, 17–19. Historically, CK2 was thought to be constitutively expressed and active 1, however, we recently demonstrated that CK2 protein and kinase activity are induced in CD4+ T-cells upon T cell receptor (TCR) stimulation 9. Interestingly, Ulges et al., and our group described the function of CK2 in regulating the Th17/T regulatory cell (Treg) axis 9, 19. Utilizing CX-4945, a CK2α and CK2α’ specific small molecule inhibitor, siRNA knockdown of CK2α, as well as genetic deletion of CK2α in CD4+ T-cells, our group demonstrated that CK2 activity promotes Th17 cell differentiation and inhibits generation of Foxp3+ Treg cells 9, 10. Mechanistically, we determined that CK2α promotes Th17 cell differentiation and suppresses Tregs through the negative regulation of the transcription factor FoxO1 10. Ulges et al., demonstrated that T-cell specific deletion of CK2β also results in defective Th17 development and enhanced Treg generation 19. Taken together, these results suggest that both the catalytic activity conferred by CK2α and CK2α’ and CK2β-mediated regulatory mechanisms are important for Th17-promoting signaling pathways during CD4+ T-cell activation and lineage commitment. Importantly, targeting of CK2 systemically with pharmacological inhibition or by CD4+ T-cell specific deletion of either CK2α or CK2β resulted in significant protection in a preclinical model of Multiple Sclerosis, Experimental Autoimmune Encephalomyelitis (EAE), which was associated with decreased Th17 cells and increased Tregs 9, 10, 19. CK2β is also involved in the suppressive function of CD4+ Foxp3+ Tregs against allergy-promoting Th2 cells 18. In addition, CK2 is critical for monocyte-derived dendritic cells to mature and produce cytokines to polarize effector T-cells in response to chemicals related to allergic contact dermatitis 17. Thus, CK2 appears to have important roles in regulating both innate and adaptive immune responses 20. CK2 expression and activity is enhanced in epithelial cells during murine and human intestinal inflammation, and it was suggested that CK2 promotes mucosal homeostasis in colitis 21. However, the exact function of CK2 in T-cells during colitis is unknown and is the focus of this study.
We investigated the role of CK2α in CD4+ T-cells during chronic intestinal inflammation. We demonstrate that CK2α expression in CD4+ T-cells is critical for the induction of colitis. Genetic abrogation of CK2α in CD4+ T-cells led to diminished Th1 and Th17 cell differentiation in both the spleen and colon in the T-cell transfer colitis model. We also demonstrated that CK2α is essential for normal CD4+ T-cell proliferation and controls CD4+ T-cell accumulation and responses in the spleen and colon through a cell-intrinsic manner. Mechanistically, CK2α regulates IL-12/STAT4 pathway activation by controlling IL-12 receptor (IL-12R) expression, which then promotes Th1 cell differentiation. In addition, CK2α controls expression of the transcription factor BATF and costimulatory molecules CD40L and ICOS by regulating expression of NFAT2, a transcription factor vital for T-cell activation and proliferation. Thus, our data demonstrate that CK2α contributes to the pathogenesis of colitis by promoting CD4+ T-cell proliferation and Th1 and Th17 responses.
RESULTS
CK2α Expression and Kinase Activity in CD4+ T-cells is Required for Colitis.
To assess the role of CK2α in T-cells in the development of colitis, CK2αfl/fldLck-Cre mice, in which CK2α is specifically deleted in T-cells, were used 10. We previously determined that CD4+ T-cells lacking CK2α, the major catalytic subunit of CK2, exhibit a significant decrease in overall CK2 kinase activity 10. We transferred CD45RBhi CD4+ T-cells isolated from CK2αfl/fl or CK2αfl/fldLck-Cre mice into lymphopenic Rag1−/− mice with T-cell and B-cell deficiency to induce colitis 22. CD4+ T-cells from CK2αfl/fl mice induced severe colitis, as assessed by body weight loss, colon length and weight and inflammation score (Figs. 1A–1E). In contrast, CD4+ T-cells from CK2αfl/fldLck-Cre mice induced only mild intestinal inflammation in Rag1−/− mice, as documented by lack of body weight loss (Fig. 1A), longer colon length (Fig. 1B) and lighter colon weight (Fig. 1C). Consistent with these findings, histopathological analysis showed less severe inflammation in the colon of Rag1−/− mice that received CD4+ T-cells from CK2αfl/fldLck-Cre mice compared to CD4+ T-cells from CK2αfl/fl mice (Figs. 1D and 1E). Serum concentrations of proinflammatory cytokines and chemokines such as IFN-γ, TNF-α, IL-12p40, CCL5, CXCL9 and CXCL10 were analyzed, and there were significantly lower concentrations of proinflammatory cytokines and chemokines in Rag1−/− mice that received CD4+ T-cells from CK2αfl/fl dLck-Cre mice compared to CK2αfl/fl mice (Fig. 1F). To exclude the possibility of genotoxicity of dLck-Cre, we transferred naïve CD4+ T-cells from dLck-Cre mice into Rag−/− mice. CD4+ T-cells from dLck-Cre mice induced severe body weight loss compared to CD4+ T-cells from CK2αfl/fldLck-Cre mice, and were comparable to CD4+ T-cells from CK2αfl/fl mice (Fig. 1A). These results indicate that CK2α expression in CD4+ T-cells is required for development of colitis in this T-cell adoptive transfer model.
Figure 1. CK2α Expression in CD4+ T-cells is Required for Colitis.
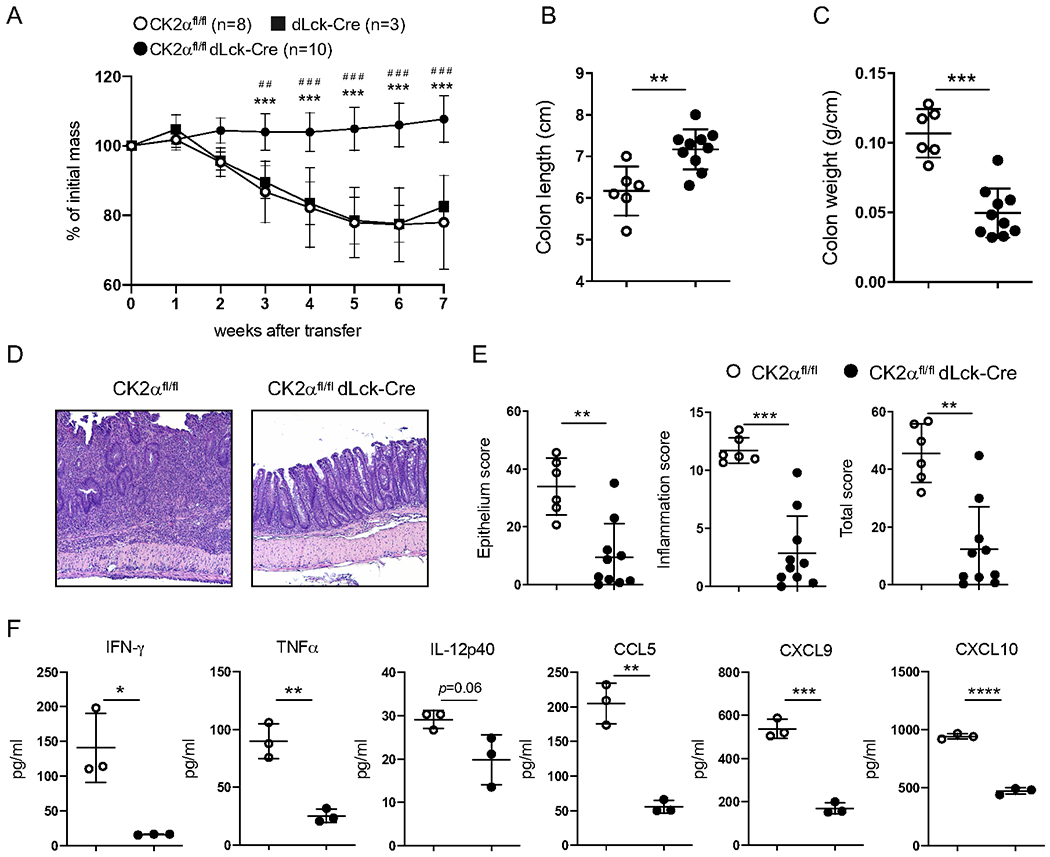
C57BL/6 Rag1−/− mice (male or female) were injected i.v. with 1 x 106 naïve CD4+ T-cells from sex and age-matched CK2αfl/fl, dLck-Cre or CK2αfl/fldLck-Cre mice at the age of 8-10 weeks. Body weight of recipients was monitored weekly. Seven weeks after transfer, mice were sacrificed and assessed for intestinal inflammation. (A) Weight change of recipient mice. CK2αfl/fl, n=8; dLck-Cre, n=3; CK2αfl/fldLck-Cre, n=10. * CK2αfl/fl compared to CK2αfl/fldLck-Cre; # dLck-Cre compared to CK2αfl/fldLck-Cre. (B) Colon length and (C) colon weight. Each dot represents one mouse. (D) Representative pictures of recipient colon sections stained with H&E. (E) Pathological scores of colons in recipient mice. (F) Cytokines and chemokines in serum were detected by Multiplex ELISA. Data represent pooled results from three independent experiments. CK2αfl/fl, n=6; CK2αfl/fldLck-Cre, n=10. Bars represent the mean ± SD. * p<0.05, ** p<0.01, *** p<0.001, ****p<0.0001, ## p<0.01, ### p<0.001.
CK2α Controls T-cell Accumulation and Cytokine Responses in the Spleen and Colon.
The T-cell adoptive transfer colitis model is associated with the accumulation of CD4+ Th1 and Th17 cells in the intestine 22. To analyze the direct effects of CK2α on CD4+ T-cell responses, we isolated cells from the spleen and colon of Rag1−/−mice that received CD4+ T-cells from CK2αfl/fl or CK2αfl/fldLck-Cre mice and analyzed CD4+ T-cell accumulation and T helper cell responses at 7 weeks after transfer. There were significantly fewer total mononuclear cells (Fig. 2A) and CD4+ T-cells (Fig. 2B) in both the spleen and colon from Rag1−/− recipients of CK2α-deficient CD4+ T-cells compared to the control group. Furthermore, we performed intracellular flow cytometric analysis of IL-17A and IFN-γ expression in CD4+ T-cells. Three distinct populations were detected in Rag1−/− recipients of CD4+ T-cells: IFN-γ+, IL-17A+ and double positive IFN-γ+IL-17A+ cells (Fig. 2C). We observed significant reductions in the percentage of IFN-γ+ and IFN-γ+IL-17A+ CD4+ T-cells in the spleen and colon from Rag1−/− recipients of CK2α-deficient CD4+ T-cells (Fig. 2D). There were significant reductions in the absolute cell numbers of IFN-γ+ CD4+ T-cells in the spleen and colon, and a significant reduction in the absolute cell numbers of IL-17A+ and IFN-γ+IL-17A+CD4+ T-cells in the colon (Fig. 2E). These findings indicate that CK2α controls both Th1 and Th17 cell differentiation in vivo. Collectively, these data suggest that CK2α regulates the accumulation, differentiation and colitogenic properties of CD4+ T-cells in the T-cell adoptive transfer colitis model.
Figure 2. CK2α Controls T-cell Accumulation in the Spleen and Colon.
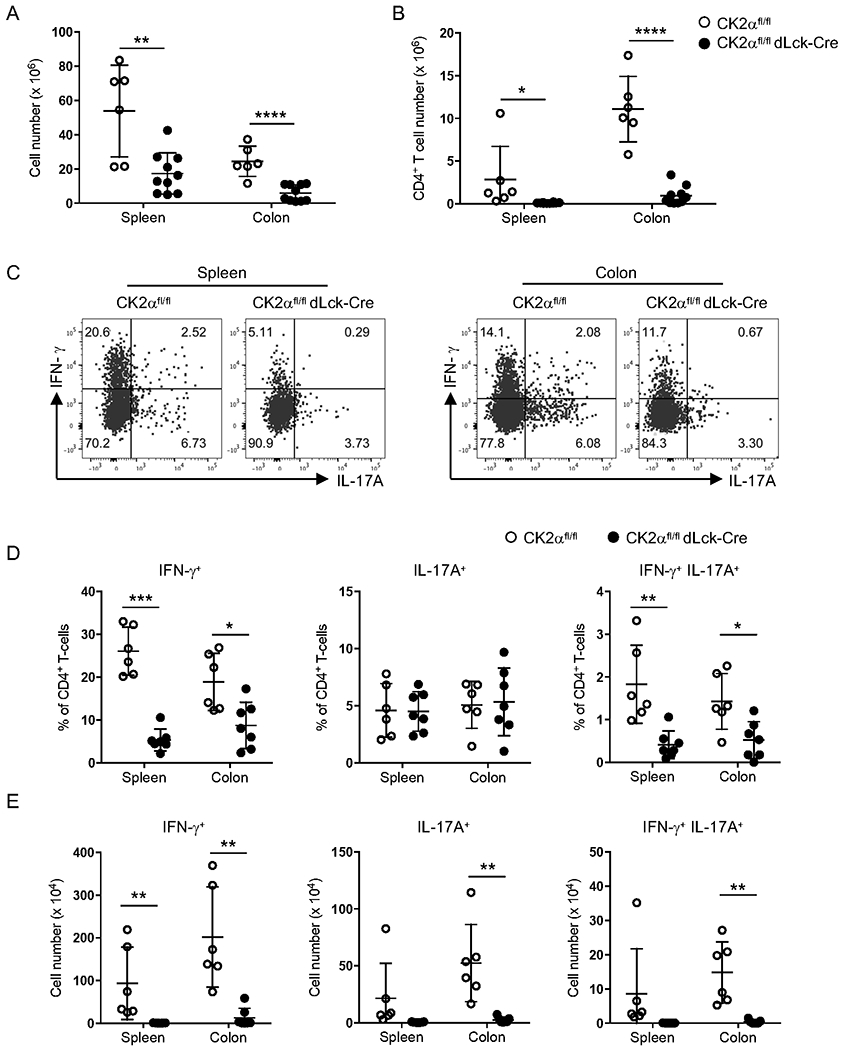
Absolute number of mononuclear cells (A) and CD4+ T-cells (B) in spleen and colon were detected at 7 weeks after transfer. (C) Representative flow cytometry profile of IL-17A and IFN-γ production by CD4+ T-cells in the spleen and colon. (D) Percentages and (E) absolute cell numbers of IFN-γ+, IL-17A+ and IFN-γ+ IL-17A+ CD4+ T-cells in spleen and colon is shown. Data represent pooled results from three independent experiments. CK2αfl/fl, n=6; CK2αfl/fldLck-Cre, n=7. Bars represent the mean ± SD. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
CK2α Controls Th1 Differentiation through the IL-12/STAT4 Pathway.
We previously determined that CK2α is critical for Th17 cell differentiation in the EAE model, but that Th1 cell differentiation was not affected 9, 10. However, a decreased frequency of Th1 IFN-γ+ CD4+ T-cells was found in the spleen and colon from Rag1−/− recipients of CK2α-deficient CD4+ T cells (Fig. 2D), suggesting that in the colitis model, CK2α may influence Th1 cell differentiation. As such, we next examined how CK2α may regulate the differentiation of Th1 cells. CK2α-deficient CD4+ T-cells exhibited a defect in polarization to the Th1 phenotype in vitro, as IFN-γ production by CK2α-deficient CD4+ T-cells was significantly reduced (Figs. 3A and 3B). IL-12 signaling through STAT4 is essential for induction of IFN-γ production and commitment of Th1 cells 23, 24. To determine the effect of CK2α on the activation of STAT4, naïve CD4+ T-cells were activated for 24 h, then stimulated with IL-12 for the indicated time points. Robust STAT4 phosphorylation in response to IL-12 was detected in CD4+ T-cells from CK2αfl/fl mice, which was diminished in CK2α-deficient CD4+ T-cells (Figs. 3C and 3D). IL-12 signals through IL-12R to induce STAT4 activation 25. We next examined expression levels of both IL-12Rβ1 and IL-12RP2 in CD4+ T-cells. CK2α-deficient CD4+ T-cells expressed significantly lower levels of IL-12Rβ1 and IL-12Rβ2 than CK2α-sufficient CD4+ T-cells upon TCR activation (Figs. 3E and 3F). Thus, in the absence of CK2α, decreased IL-12R expression results in reduced IL-12-induced STAT4 phosphorylation, thereby resulting in impaired Th1 cell differentiation.
Figure 3. CK2α Controls Th1 Differentiation through the IL-12/STAT4 Pathway.
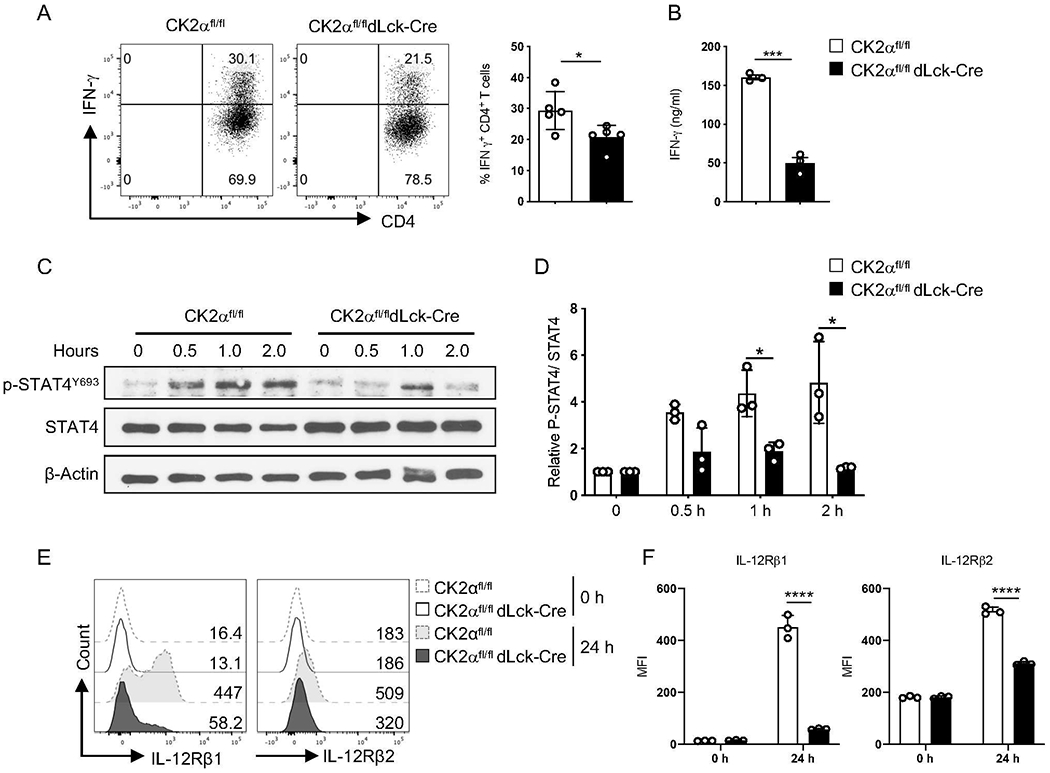
(A) Naïve CD4+ T-cells from CK2αfl/fl or CK2αfl/fl dLck-Cre mice were polarized under Th1 conditions [anti-CD3 (1 μg/ml), anti-CD28 (1 μg/ml), IL-12 (10 ng/ml) and anti-IL-4 (10 μg/ml)] for 3 days. IFN-γ production by CD4+ T-cells was detected by intracellular staining. Representative flow cytometry profiles of IFN-γ production by CD4+ T-cells is shown (n = 5). (B) IFN-γ in the supernatant was measured by ELISA (n=3). (C) Naïve CD4+ T-cells from CK2αfl/fl mice or CK2αfl/fl dLck-Cre mice were activated with anti-CD3 (1 μg/ml) and anti-CD28 (1 μg/ml) for 24 h, stimulated with IL-12 (10 ng/ml) for the indicated time points, then phosphorylated P-STAT4 Y693 in CD4+ T-cells was detected by immunoblotting. (D) Relative ratio of phosphorylated P-STAT4 Y693 to total STAT4 at the indicated time points is shown (n=3). (E) Naïve CD4+ T-cells from CK2αfl/fl or CK2αfl/fl dLck-Cre mice were activated with anti-CD3 (1 μg/ml) and anti-CD28 (1 μg/ml) for 24 h, and expression levels of IL-12Rβ1 and IL-12Rβ2 detected by flow cytometry. Representative line graphs are shown. (F) Quantitation of MFI of IL-12Rβ1 and IL-12Rβ2 expression is shown. n=3 in each group. Bars represent the mean ± SD. * p<0.05, *** p< 0.001, **** p< 0.0001.
CK2α is Essential for CD4+ T-cell Proliferation but Not Survival In Vitro and In Vivo.
The failure of CK2α-deficient CD4+ T-cells to accumulate in the spleen and colon (Figs. 2A and 2B) may reflect a failure to proliferate and/or survive in these organs. To address these possibilities, we assessed the proliferation capacity of CK2α-sufficient and -deficient CD4+ T-cells both in vitro and in vivo. CFSE labeled CD4+ T-cells were stimulated with graded concentrations of anti-CD3 and anti-CD28 Abs, and proliferation of CD4+ T-cells was assessed by CFSE dilution. CK2α-deficient T-cells exhibited reduced proliferation compared to CD4+ T-cells from CK2αfl/fl mice (Figs. 4A and 4B). We also analyzed the survival of CK2α-sufficient and -deficient CD4+ T-cells by Annexin V staining, and no differences were observed (Sup. Figs. 1A and 1B). We next transferred CFSE labeled CK2α-sufficient or -deficient CD4+ T-cells into Rag1−/− mice to analyze their proliferative ability in vivo. There was significantly less proliferation of CK2α-deficient CD4+ T-cells compared with CD4+ T-cells from CK2αfl/fl mice in the spleen (Figs. 4C and 4D), but no differences in Annexin V+ CD4+ T-cells between the two groups were observed (Sup. Figs. 1C and 1D). Altogether, these findings indicate that CK2α controls T-cell accumulation by affecting cell proliferation but not survival.
Figure 4. CK2α is Essential for CD4+ T-cell Proliferation In Vitro and In Vivo.
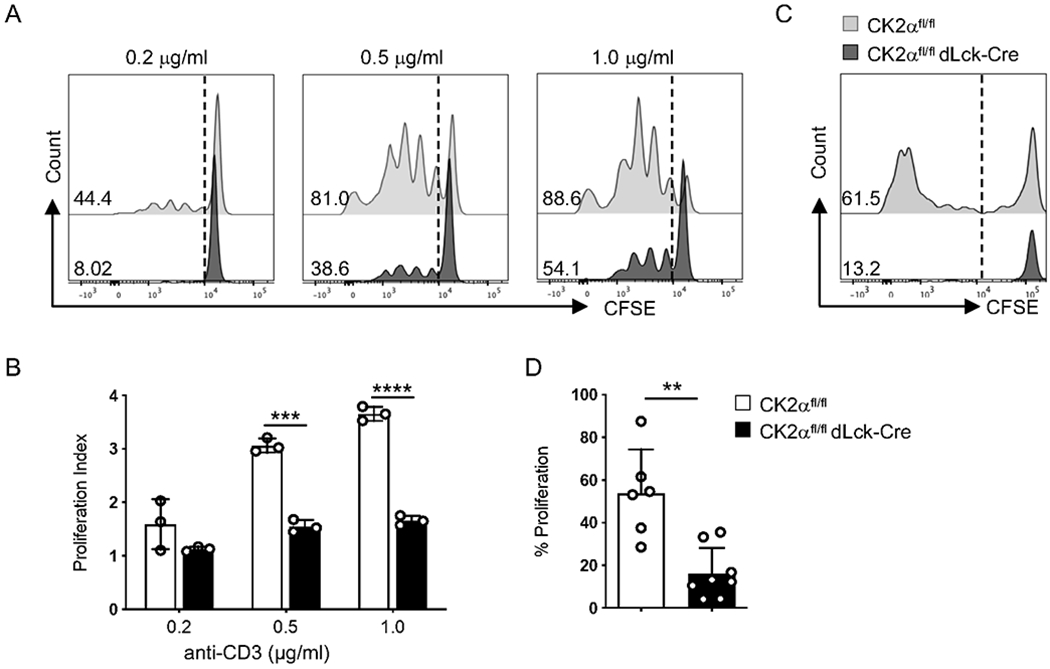
(A) CFSE labeled naïve CD4+ T-cells were activated with the indicated concentrations of anti-CD3 (0.2 μg/ml, 0.5 μg/ml, 1.0 μg/ml) and 1.0 μg/ml of anti-CD28 Abs for 72 h, and proliferation was assessed by CFSE dilution. Representative line graphs are shown. (B) Proliferation index of CD4+ T-cells is shown (n = 3). (C) 1 x 106 CFSE labeled naïve CD4+ T-cells from CK2αfl/fl or CK2αfl/fl dLck-Cre mice were transferred into Rag1−/− mice by i.v. injection. Mice were sacrificed 5 days later, and proliferation of CD4+ T-cells from the spleen was assessed by CFSE dilution. Representative line graphs are shown. (D) Frequencies of cells undergoing proliferation is shown. Data represent pooled results from three independent experiments. CK2αfl/fl, n=6; CK2αfl/fldLck-Cre, n=8. Bars represent the mean ± SD. ** p<0.01, *** p<0.001, **** p<0.0001.
CK2α Controls T-cell Accumulation in the Spleen and Colon through a Cell-intrinsic Manner.
To further address whether CK2α affects CD4+ T-cell function through cell-intrinsic or cell-extrinsic mechanisms, we took advantage of a co-transfer model 26. We administered equal numbers of congenic CD45.1 C57BL/6 and CD45.2 CK2αfl/fldLck-Cre naïve CD4+ T-cells into Rag1−/− recipients. As a control, we transferred a 1:1 ratio of CD45.1 C57BL/6 and CD45.2 CK2αfl/fl naïve CD4+ T-cells into Rag1−/− recipients. Two weeks after transfer, the ratio between CK2α-sufficient CD4+ T-cells to C57BL/6 CD4+ T-cells in spleen, mesenteric lymph nodes (MLNs), and colon lamina propria was slightly lower compared to the ratio right before cell transfer (Figs. 5A and 5C). However, the relative ratio was significantly higher than the ratio between CD4+ T-cells from CK2α-deficient mice to congenic C57BL/6 CD4+ T-cells (Figs. 5B and 5C). The relative ratio was consistent in spleen, MLNs and colon lamina propria in both groups (Figs. 5A, 5B and 5C), and remained constant 4 weeks after transfer in both groups, indicating that there is no proliferation defect of CK2α-sufficient CD4+ T-cells compared to C57BL/6 CD4+ T-cells (Sup. Fig. 2). CD4+ T-cell responses in the spleen and colon in the co-transfer system were analyzed. We observed that the percentages of IFN-γ+ and IFN-γ+IL-17A+ producing cells in CK2α-deficient CD4+ T cells were significantly lower compared to their C57BL/6 counterparts in the spleen and colon (Figs. 5D and 5E). There were no differences in CD4+ T-cell responses between C57BL/6 and CK2αfl/fl T-cells, as shown by comparable percentages of IFN-γ+ and IFN-γ+IL-17A+ CD4+ T-cells between C57BL/6 and CK2αfl/fl T-cells (Sup. Fig. 3). These results demonstrate that the requirement of cell-intrinsic CK2α signaling for T-cell accumulation in the intestine cannot be rescued by the presence of CK2α-sufficient T-cells and their subsequent downstream inflammatory signals. Therefore, CK2α activity in CD4+ T-cells is required for their effective accumulation through a cell-intrinsic manner.
Figure 5. CK2α Controls T-cell Accumulation in the Spleen and Colon through a Cell-intrinsic Manner.
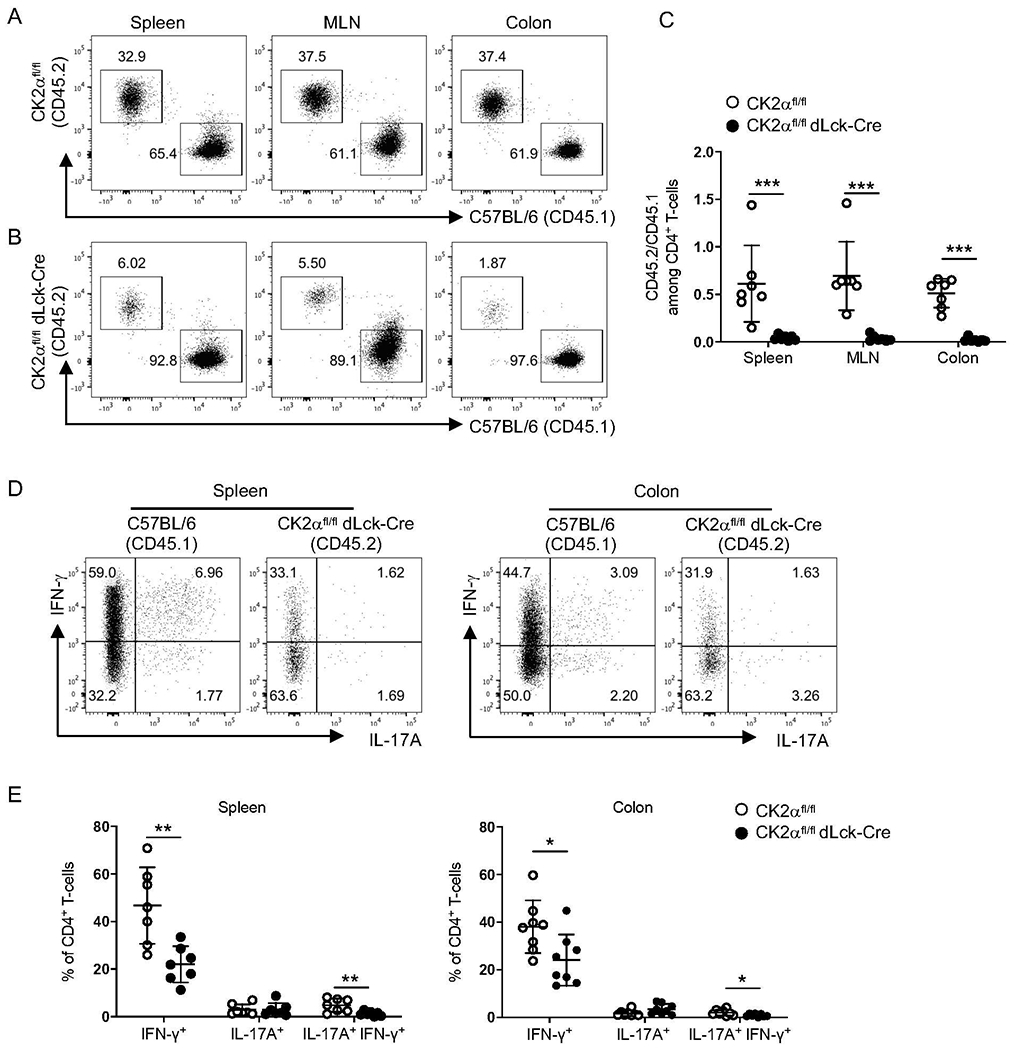
Naïve CD4+ T-cells from CK2αfl/fl mice (CD45.2+) or CK2αfl/fl dLck-Cre mice (CD45.2+) were co-transferred with naïve CD4+ T-cells from C57BL/6 mice (CD45.1+) at a ratio of 1:1 (5 x 105: 5 x 105) into Rag1−/− mice by i.v. injection. Recipients were sacrificed 2 weeks after transfer, and the percentage of C57BL/6 CD4+ T-cells (CD45.1+) and CK2α-sufficient or -deficient CD4+ T-cells (CD45.2+) in the spleen, mesenteric lymph node (MLN) and colon was detected by flow cytometry. (A and B) Representative flow cytometry profile of CD4+ T-cells recovered from the indicated sites. (C) Ratio of CD45.2/CD45.1 CD4+ T-cells in spleen, MLN and colon. (D) Representative flow cytometry profiles of IFN-γ and IL-17A production by CD4+ T-cells derived from C57BL/6 and CK2αfl/fl dLck-Cre mice in the spleen and colon. (E) Frequencies of IFN-γ+, IL-17A+ and IL-17A+ IFN-γ+ CD4+ T-cells derived from C57BL/6 and CK2αfl/fl dLck-Cre mice in the spleen and colon is shown. Data represent pooled results from three independent experiments. CK2αfl/fl, n=8; CK2αfl/fldLck-Cre, n=8. Bars represent the mean ± SD. * p<0.05, *** p<0.001.
CK2α Controls CD4+ T-cell Responses, In Part, by Regulating Expression of NFAT2.
To understand the molecular mechanisms that confer CK2α function in CD4+ T-cells in the development of colitis, CD4+ T-cells were isolated from the colon 6 weeks after transfer and subjected to RNA sequencing. A total of 122 genes were significantly increased and 132 genes were significantly decreased in CK2α-deficient CD4+ T-cells compared to CK2α-sufficient CD4+ T-cells (Fig. 6A). GSEA of the data revealed that differentially expressed genes were associated with the T cell receptor (TCR) signaling pathway (Fig. 6B). CK2α-deficient CD4+ T-cells express lower levels of genes associated with TCR and co-stimulatory signaling pathways, such as Nfatc1, Batf, Icos and Cd40lg (Fig. 6C), which are important for T-cell activation and proliferation 27–30. We validated RNA-sequencing data by analyzing gene expression levels in CD4+ T-cells from Rag1−/− mice with colitis by RT-PCR, and demonstrated significantly lower expression levels of Nfatc1, Icos, Cd40lg and Batf in CD4+ T-cells from CK2αfl/fldLck-Cre mice compared to CK2αfl/fl mice (Fig. 6D). Studies have shown that CD40L and ICOS are NFAT-dependent molecules and the transcription factor BATF is also regulated by NFAT in T follicular helper (Tfh) cells 31–34. To determine a possible role of NFAT2 in gene expression in CD4+ T-cells in vitro, CD4+ T-cells from CK2αfl/fl and CK2αfl/fldLck-Cre mice were stimulated with anti-CD3 and anti-CD28 Abs for 24 h, and expression levels of NFAT2 were analyzed by flow cytometry. CD4+ T-cells from CK2αfl/fldLck-Cre mice expressed significantly lower levels of NFAT2 after TCR stimulation compared to CD4+ T-cells from CK2αfl/flmice (Fig. 6E). Next, we sought to determine whether the defect in CD4+ T-cell function in CK2αfl/fldLck-Cre mice was due to decreased NFAT2 expression. To do this, a constitutively-active form of NFAT2 (caNFAT2) 31 was overexpressed in CK2αfl/fldLck-Cre CD4+ T-cells in vitro. CK2α-deficient CD4+ T-cells expressed lower levels of ICOS, CD40L and BATF compared to CK2α-sufficient CD4+ T-cells, and caNFAT2 rescued expression levels of ICOS, and partially rescued CD40L and BATF levels (Figs. 6F and 6G). Importantly, caNFAT2 also partially rescued the defect of CD4+ T-cell proliferation in CK2αfl/fldLck-Cre mice, as assessed by Ki-67 staining (Figs. 6H and 6I). Altogether, these data demonstrate that CK2α controls CD4+ T-cell function, in part, by regulating NFAT2 expression.
Figure 6. CK2α Controls CD4+ T-cell Responses by Regulating Expression of NFAT2.
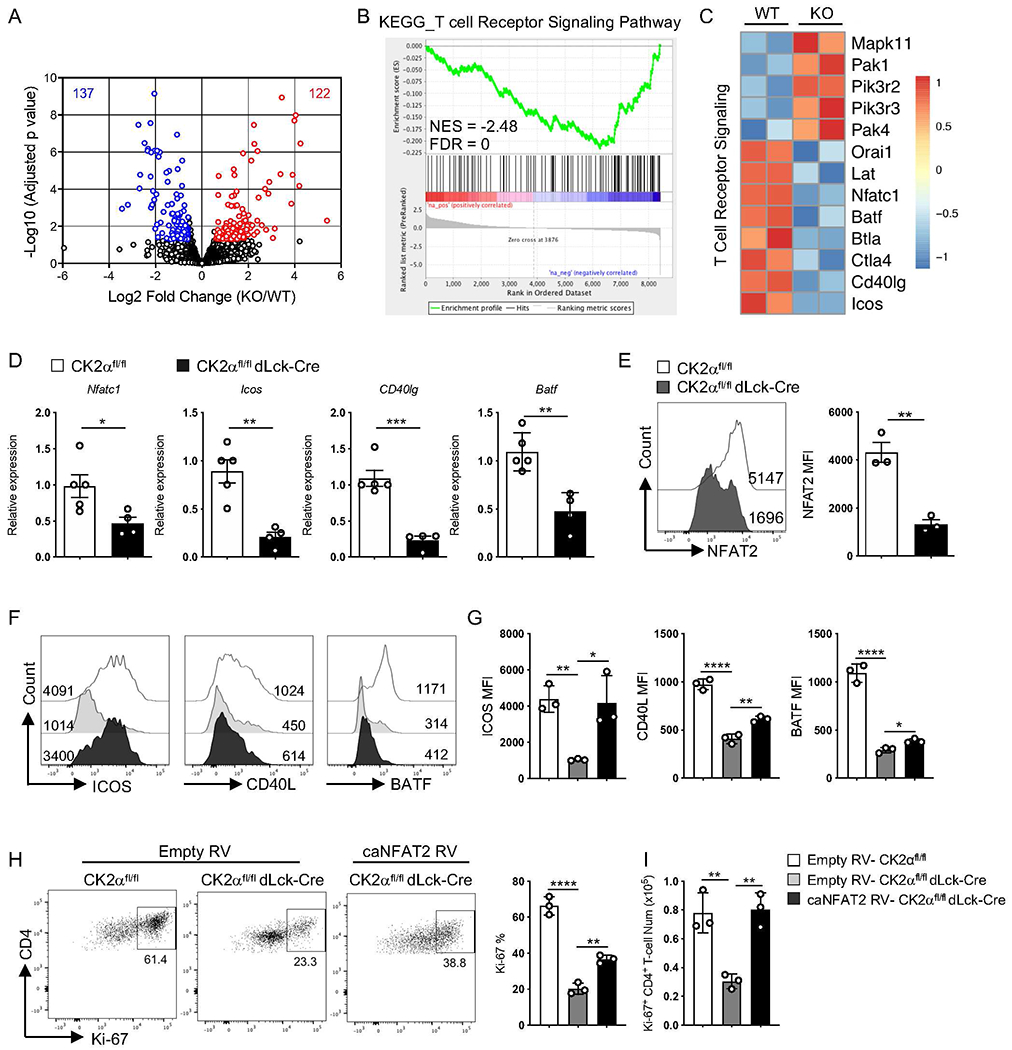
RNA sequencing of CK2αfl/fl CD4+ T-cells (WT) and CK2αfl/fl dLck-Cre CD4+ T-cells (KO) from the colon at 6 weeks after colitis induction was performed (three to five mice were combined per experiment, two experiments in each group). (A) Summary of genes differentially regulated by CK2α using the following cutoffs are shown: p < 0.05, fold change >1.5. (B) GSEA plot shows enrichment of RNA sequencing data compared with KEGG pathway dataset. (C) Heat map shows TCR signaling relative gene expression. (D) Selected gene expression was validated in CD4+ T-cells from colitis Rag1−/− mice by qRT-PCR. CK2αfl/fl, n=5; CK2αfl/fldLck-Cre, n=4. (E) Naive CD4+ T-cells from CK2αfl/fl and CK2αfl/fl dLck-Cre mice were activated with 0.5 μg/ml of anti-CD3 and 1.0 μg/ml of anti-CD28 Abs for 24 h, and expression of NFAT2 was detected by flow cytometry. Representative line graphs and quantitation are shown. n=3 in each group. (F) Naïve CD4+ T-cells from CK2αfl/fl and CK2αfl/fl dLck-Cre mice were activated and transduced with caNFAT2 or empty control vector. GFP+ transduced cells were sorted and stimulated with 0.1 μg/ml of anti-CD3 and 1.0 μg/ml of anti-CD28 Abs for 48 h, and expression of ICOS, CD40L and BATF was detected by flow cytometry. Representative line graphs are shown. (G) Quantitation of relative MFI of ICOS, CD40L and BATF is shown. n=3 in each group. (H) GFP+ cells were sorted and stimulated with 0.1 μg/ml of anti-CD3 and 1.0 μg/ml of anti-CD28 Abs for 48 h, and proliferating cells detected by Ki-67 staining. Representative line graphs and quantitation are shown. (I) Quantitation of absolute number of Ki-67+ CD4+ T-cells is shown. n=3 in each group. Bars represent the mean ± SD. * p<0.05, ** p<0.01, *** p<0.001, **** p<0.0001.
DISCUSSION
The majority of our current knowledge on CK2 comes from extensive studies in the context of cancer 35. However, growing evidence suggests the crucial involvement of CK2 in both innate and adaptive immune responses 20. In this study, we demonstrate that CK2α expression and kinase activity is required for CD4+ T-cells to induce colitis. We found that CK2α controls CD4+ T-cell proliferation and differentiation into Th1 and Th17 cells, which then consequently affects the pathogenesis of T-cell mediated colitis.
Studies show that there is a bias toward the production of proinflammatory cytokines associated with Th1 and Th17 cells in patients with CD 12, 14. We took advantage of the T-cell adoptive transfer colitis model, which is associated with the accumulation of CD4+ Th1 and Th17 cells in the intestine 22, to elucidate the function of CK2α in CD4+ T-cells. Strikingly, we found that CK2α-deficient CD4+ T-cells failed to induce severe inflammation in the colon (Fig. 1), which was associated with decreased numbers of CD4+ T-cells and IFN-γ- and IL-17A- producing CD4+ T-cells both in the spleen and colon. These data indicate that CK2α controls Th1 and Th17 differentiation in the context of colitis. This is consistent with our previous studies which showed that CK2α regulates Th17 differentiation in the EAE model 9, 10. However, there was no significant change in Th1 cell responses in the EAE model. Furthermore, in the EAE model, CK2α deficiency in CD4+ T-cells promoted the differentiation of Foxp3+ T regulatory (Treg) cells, which was not observed in the colitis model. Thus, the regulatory functions of CK2α in CD4+ T-cell differentiation may be different in distinct inflammatory disorders or target organs (CNS versus colon) due to microenvironmental differences. Secondly, the differential response of Th1 cells in vitro may be caused by the strength of TCR stimulation. In this study, we use lower concentrations of anti-CD3 (0.2-1.0 μg/ml) compared to our previous study (10 μg/ml) 10, to activate CD4+ T-cells in vitro to mimic the in vivo colitis condition, and found that CK2α-deficient CD4+ T-cells showed impaired Th1 differentiation (Fig. 3A). Collectively, in the T-cell adoptive transfer colitis model, CK2α regulates the differentiation of both Th1 and Th17 cells.
Importantly, STAT4 is essential for Th1 differentiation and function, and is predominantly activated by IL-12 and to a lesser extent by IFNα and IL-23 36, 37. Genome wide association studies (GWAS) have shown that the STAT4 gene is significantly associated with both CD and UC 38, 39, and the IL-12-IL-23 signaling pathway is an attractive therapeutic target in these diseases 40. In this study, we demonstrate that CK2 is critical for IL-12-induced phosphorylation of STAT4 (Fig. 3C) by directly controlling IL-12 receptor expression (Figs. 3E and 3F), thereby linking CK2α to the signaling pathway critical for Th1 cell differentiation. Our previous study showed that CK2 was a novel interaction partner of JAK1/2 and potentiated downstream STAT3 activation and gene expression 7. Thus, there is the possibility that CK2α may directly affect JAK2/STAT4 activation through TCR-induced CK2α expression, and this will be addressed in future studies.
CK2 participates in diverse biological processes, including transcription, translation, cell growth and proliferation in normal and disease states, particularly in cancers 4. CK2 promotes cancer cell growth and proliferation as well as suppresses cell apoptosis 41, which are associated with worse prognosis. Here, we demonstrate that CK2α is critical for CD4+ T-cell proliferation but has no effect on CD4+ T-cell survival, especially under lower doses of TCR stimulation (Fig. 4). This indicates that CK2 has cell-specific functions in cancer cells and immune cells. Considering that aberrant aggregated CD4+ T-cells in the intestine is one symptom in patients with IBD 11, 42, targeting CK2 in CD4+ T-cells to limit cell proliferation might be a therapeutic strategy in IBD.
Another striking finding from the RNAseq data was that NFAT2, ICOS, CD40L and BATF expression was significantly decreased in CK2α-deficient CD4+ T-cells isolated from the colon (Fig. 6). These genes are associated with the TCR signaling pathway. NFAT transcription factors are key regulators of T-cell activation and exhaustion, and consist of five members, NFAT1, NFAT2, NFAT3, NFAT4 and NFAT5 27. Three members of the NFAT family including NFAT1, NFAT2 and NFAT4, are present in T-cells, and play vital roles in T-cell development, activation, differentiation and function 27, 43. During T-cell activation, NFAT proteins are dephosphorylated by activated calcineurin, which leads to their nuclear translocation and induction of NFAT-mediated gene transcription 44. NFAT1 binds to IFN-γ promoter regions to regulate Th1 cell responses 45. NFAT1 and NFAT2 also directly bind to the IL-17 promoter region to regulate cytokine production 46, 47. Moreover, NFAT2 cooperates with NFAT1 to control the expression levels of ICOS, CXCR5 and CD40L, as well as control the expression of IRF4 and BATF in the differentiation of Tfh cells 31–34. Interestingly, GWAS studies have shown that NFAT2 and ICOS are newly identified candidate genes in IBD susceptibility loci associated with modulation of T-cell responses 48. In addition, CD40L+ T-cells are increased in patients with IBD 49. ICOS-ICOSL and CD40-CD40L are important costimulatory pathways to amplify immune responses and promote inflammation 50. One study demonstrated that CK2 binds the N-terminus of NFAT2 and phosphorylates important amino acid residues to regulate NFAT2 activity 51. Our data show that CK2α can also regulate NFAT2 expression both at the mRNA and protein levels in CD4+ T-cells (Figs. 6D and E). More importantly, we demonstrated that overexpression of caNFAT2 in CK2α-deficient CD4+ T-cells partially rescued the defect in T-cell proliferation and also partially rescued expression of the NFAT2 dependent molecules ICOS, CD40L and BATF (Figs. 6F, 6G, 6H and 6I). These findings indicate that the CK2-NFAT2 axis is vital for regulating T-cell activation, proliferation and differentiation, and thereafter leads to aberrant CD4+ T-cell immune responses in T-cell induced colitis. The exact manner by which CK2α regulates NFAT2 expression is still unclear and under study. Important to note is that caNFAT2 did not completely rescue the CD4+ T-cell phenotype caused by CK2α deficiency. This raises the possibility that CK2α has other targets, not only NFAT2, which are essential for CD4+ T-cell responses. These are important questions to be addressed in future studies.
Taken together, our data reinforce the critical function of CK2α in CD4+ T-cells. We demonstrated that CK2α is vital for CD4+ T-cell colitogenic properties, regarding aspects of regulating Th1 and Th17 cell differentiation and cell proliferation. Mechanistically, CK2α controls STAT4 activation by controlling IL-12R expression in CD4+ T-cells to regulate Th1 cell differentiation, and regulates NFAT2 expression to influence CD4+ T-cell function upon T-cell activation. These findings suggest that CK2α may be a therapeutic target in IBD and potentially in other T-cell mediated diseases.
METHODS
Mice.
CK2αfl/fldLck-Cre mice 10, CK2αfl/fl mice 10, Rag1−/− mice and CD45.1 C57BL/6 mice were bred and maintained under specific pathogen free conditions in the animal facility at the University of Alabama at Birmingham (UAB). Male and female mice between 8- and 12-week old were used for all experiments. All experimental procedures involving animals were reviewed and approved by the Institutional Animal Care and Use Committee of UAB.
CD4+ T-cell Purification.
For in vivo transfer, total CD4+ T-cells were first purified from the spleen and peripheral lymph nodes using the Dynabeads™ CD4 Positive Isolation Kit (Invitrogen, Carlsbad, CA), then naïve CD4+ T-cells were sorted as CD4+CD25−CD45RBhi on an Aria II (BD Bioscience, San Jose, CA), routinely to 98% purity. For in vitro activation and polarization, naïve CD4+ T-cells were enriched from the spleen and peripheral lymph nodes by the EasySep™ Mouse Naïve CD4+ T Cell Kit (STEMCELL Technologies Inc, Vancouver, BC), routinely to 90- 95% purity.
T-cell Transfer Model of Colitis.
Naïve CD4+CD25−CD45RBhi T-cells from CK2αfl/fl, dLck-Cre or CK2α fl/fldLck-Cre mice were transferred via intravenous (i.v.) tail injection to sex and age-matched Rag1−/− mice (1 x 106 cells per mouse) 22. Weight loss of recipient Rag1−/− mice was monitored weekly, and mice were sacrificed 6-7 weeks after transfer. Samples of proximal, middle, and distal colon were fixed in 10% formalin. Paraffin embedded samples were cut into 6 μm sections followed by hematoxylin and eosin staining, and histopathology was evaluated in a double blinded manner by a veterinary pathologist. Epithelium damage and inflammation severity were scored separately and a total score was given accordingly. Briefly, epithelium damage was evaluated in all sections by hyperplasia, goblet cell loss, degeneration and necrosis, ulceration and dysplasia. Inflammation severity was evaluated by crypt exudate, lamina propria and submucosal inflammatory cell accumulation intensity, submucosal edema distribution and transmural inflammation. Severity of lesions were graded as follows: 0 = normal, 1 = mild, 2 = moderate, 3 = severe 52, 53.
In co-transfer experiments, naïve CD4+CD25−CD45RBhi T-cells from CK2αfl/fl or CK2αfl/fldLck-Cre mice were co-transferred with naïve CD4+CD25−CD45RBhi T-cells from CD45.1 C57BL/6 mice at the ratio of 1:1 (5 x 105: 5 x 105) to sex and age-matched Rag1−/− mice. Recipient Rag1−/− mice were sacrificed 2 and 4 weeks after transfer to evaluate CD4+ T-cell accumulation and responses in the spleen and colon.
Lymphocyte Preparation.
Single-cell suspensions of spleen and lymph nodes were prepared as previously described 9, 10 and resuspended in R10 medium (RPMI 1640 with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 10 mM HEPES, 1 mM sodium pyruvate, and 50 μM β-mercaptoethanol). Colon lamina propria lymphocytes were prepared using the Lamina Propria Dissociation Kit (Miltenyi Biotec, Auburn, CA) according to the manufacturer’s instructions. Cells were then isolated using a 40/75% discontinuous Percoll gradient, washed, and resuspended in R10 medium.
Flow Cytometry.
Cell surface staining and intracellular staining was performed as previously described 10. For cytokine production analysis, cells were stimulated with 50 ng/ml of PMA (Sigma-Aldrich, St. Louis, MO) and 750 μg/ml of ionomycin (Sigma-Aldrich, St. Louis, MO) in the presence of GolgiStop (BD Biosciences, San Jose, CA) for 4 h. After staining of cell surface molecules, cells were fixed and permeabilized using the Foxp3 Staining Buffer Set (eBioscience, Grand Island, NY)9. Stained cells were run on an LSRII flow cytometer (BD Biosciences, San Jose, CA), and data were analyzed using FlowJo software (Tree Star, Inc, Ashland, OR). The following antibodies were used in this study (all Biolegend except where noted otherwise): anti-CD3ε PerCP-Cy5.5/PE-Cy7/APC (clone 145-2C11); anti-CD4 Pacific Blue/FITC /Alexa Fluor 647/APC-Cy7/ PE (clone RM4-5); anti-CD11b FITC (clone M1-70); anti-CD11b BUV395 (clone M1-70, BD Bioscience), anti-CD45 APC-Cy7 (clone 30-F11); anti-CD45.1 Alexa Fluor 488/PerCP-Cy5.5 (clone A20); anti-CD45.2 Alexa 647/APC-Cy7 (clone 104); anti-CD45RB FITC (clone C363.16A), anti-CD25 Alexa Fluor 647/PE-Cy7 (clone PC61.5); anti-IFN-γ Pacific Blue/PE-Cy7 (clone XMG1.2); anti-IL-17A APC (clone TC11-18H10); anti-Ki-67 Brilliant Violet 421 (clone 16A8); anti-CD40L PE-Cy7 (clone 24-31); anti-ICOS APC (clone 15F9); anti-BATF PE (clone S39-1060, BD Bioscience); NFAT2 (Cell Signaling Technology); Alexa Fluor 647 Anti-Rabbit IgG (H+L) (Jackson Immuno Research Labs); anti-CD212 (IL-12Rβ1) PE (Clone 114, BD Bioscience); and anti-IL-12Rβ2 Alexa Fluor 488 (R&D System).
Naïve CD4+ T-cell Activation and Polarization.
For in vitro activation, naïve CD4+ T-cells were cultured in R10 medium and stimulated with plate-bound anti-CD3 (0.2 μg/ml, 0.5 μg/ml, 1.0 μg/ml) (Clone 145-2C11, BioX Cell, West Lebanon, NH) and soluble anti-CD28 Abs (1.0 μg/ml) (Clone 37.51, BioX Cell, West Lebanon, NH) for 24 and 48 h. For Th1 cell polarization, cells were stimulated with plate-bound anti-CD3 (1.0 μg/ml) and soluble anti-CD28 Abs (1.0 μg/ml) in the presence of IL-12 (10 ng/ml, Biolegend) and anti-IL-4 Ab (10 μg/ml) (Clone 11B11, BioX Cell, West Lebanon, NH) for 72 h. For phospho-STAT4 detection, cells were activated with plate-bound anti-CD3 (1.0 μg/ml) and soluble anti-CD28 (1.0 μg/ml) for 24 h, then stimulated with IL-12 (10 ng/ml) for the indicated time points.
Proliferation Assays.
For the in vitro proliferation assay, naïve CD4+ T-cells were incubated with 5 μM CFSE, washed, and activated with plate-bound anti-CD3 (0.2 μg/ml, 0.5 μg/ml, 1.0 μg/ml) and soluble CD28 Abs (1.0 μg/ml) for 72 h. CFSE dilution was detected by flow cytometry, and proliferation index was calculated by FlowJo. For the in vivo proliferation assay, naïve CD4+CD25−CD45RBhi T-cells were labeled with 5 μM CFSE, then 1 x 106 CFSE labeled CD4+ T-cells were transferred into sex and age-matched Rag1−/− mice. Rag1−/− mice were sacrificed at day 5 after transfer, and proliferation of CD4+ T-cells from the spleen was analyzed by flow cytometry.
Apoptosis Assays.
For the in vitro apoptosis assay, naïve CD4+ T-cells were activated with plate-bound anti-CD3 (0.2 μg/ml, 0.5 μg/ml, 1.0 μg/ml) and soluble CD28 Abs (1.0 μg/ml) for 72 h. Cells were stained with Annexin V APC (BioLegend, San Diego, CA) according to the manufacturer’s protocol and analyzed by flow cytometry, as previously demonstrated 8. For the in vivo apoptosis assay, naïve CD4+CD25−CD45RBhi T-cells were transferred into sex and age-matched Rag1−/− mice. Rag1−/− mice were sacrificed at day 5 after transfer, and apoptosis of CD4+ T-cells from the spleen was analyzed by Annexin V staining,
Multiplex ELISA.
Blood was collected from recipient Rag1−/− mice when sacrificed, and serum was obtained and analyzed using the Mouse Cytokine/Chemokine multiplex ELISA assay (Millipore, St. Louis, MO) according to the manufacturer’s protocol and as previously described54.
ELISA Analysis.
Naïve CD4+ T-cells (1 x 106/ml) from CK2αfl/fl or CK2α fl/fldLck-Cre mice were polarized under Th1 conditions. IFN-γ levels in the supernatant were measured using the MAX™ Standard Set Mouse IFN-γ ELISA (BioLegend, San Diego, CA).
Immunoblotting.
CD4+ T-cells were lysed in buffer containing 1% Triton X-100 (Sigma-Aldrich), protein lysates were separated by electrophoresis, transferred to a nitrocellulose membrane, and then blotted with Phospho-STAT4 (Tyr693), STAT4 (Cell Signaling Technology, Danvers, MA) and β-Actin (Sigma-Aldrich, St. Louis, MO) Abs, as previously described 9, 10. Quantification of immunoblots were performed using ImageJ.
RNA Isolation, RNA Sequencing, and Quantitative RT-PCR.
RNA sequencing was performed as previously described 9. Briefly, colonic CD4+ T-cells were sorted from recipient Rag1−/− mice at 6 weeks after transfer. Total RNA was extracted from FACS-sorted CD4+ T-cells using the miRNeasy Mini Kit (Qiagen, Venlo, Netherlands) according to the manufacturer’s protocols and submitted to GENEWIZ (South Plainfield, NJ) for RNA sequencing (RNA-seq) and bioinformatics analysis. RNA sequencing data was submitted to the Gene Expression Omnibus (GEO) Repository under accession number GSE131965. Genes with an FDR less than 0.05 with a fold change more than 1.5 or less than −1.5 were considered as differentially expressed genes. Further pathway analysis was performed by Gene Set Enrichment Analysis (GSEA) available through the Broad Institute.
For quantitative RT-PCR (qRT-PCR) analysis, 500-1000 ng of RNA was reverse transcribed into cDNA using M-MLV Reverse Transcriptase (Promega) as previously described 9. cDNA was subjected to qRT-PCR using TaqMan primers (Thermo Fisher Scientific). Relative gene expression was calculated according to the ΔΔ threshold cycle (Ct) method.
Retroviral Transduction.
Naïve CD4+ T-cells were activated under Th0 conditions [anti-CD3 (1 μg/ml), anti-CD28 (1 μg/ml), anti-IFN-γ (10 μg/ml) and anti-IL-4 (10 μg/ml)], and then CD4+ T-cells were transduced 24 h after activation by spin-infection in the presence of concentrated retroviral supernatant and 1 μg/ml polybrene (Sigma-Aldrich, St. Louis, MO). Forty-eight h after transduction, CD4+ T-cells were transferred into new plates with fresh media containing 10 ng/ml IL-2 and rested for 4 days. Transduced CD4+ T-cells were sorted as CD4+ GFP+ on an Aria II (BD Bioscience, San Jose, CA), 2 x 105 cells per well were stimulated with plate-bound anti-CD3 (0.1 μg/ml) and soluble anti-CD28 Abs (1 μg/ml) for 48 h, and then expression levels of Ki-67, BATF, CD40L and ICOS were detected by FACS staining.
Statistics.
Levels of significance for comparison between two groups was determined by Student’s t test distribution and by one-sided two-sample Mann-Whitney rank sum test for inflammation scores. Multiple comparisons were performed by ordinary one-way ANOVA. The p values are indicated as follows: *p < 0.05, **p <0.01, ***p <0.001, ****p <0.0001. All error bars represent mean ± SD. All statistical analyses (excluding RNA-seq, described above) were performed using Prism software (GraphPad).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health Grants R01NS057563 (to E.N.B.), R01CA194414 (to E.N.B.), R21AG058836 (to B. S.), National Multiple Sclerosis Society Grant RG-1606-24794 (to H.Q.) and American Diabetes Association ADA Fellowship Award #1-19-PDF-164 (to J. T.). The Comprehensive Flow Cytometry Core at the University of Alabama at Birmingham is supported by National Institutes of Health Grants P30 AR048311 and P30 AI27667. We thank Dr. Charles O. Elson III (UAB), Dr. Laurie Harrington (UAB) and their lab members for assistance with experimental design and constructive discussions. Dr. Trenton Schoeb (UAB) assisted with the histological tissue analysis.
Footnotes
Conflict of Interest Statement
The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Dominguez I, Sonenshein GE, Seldin DC. Protein kinase CK2 in health and disease: CK2 and its role in Wnt and NF-kappaB signaling: linking development and cancer. Cell Mol Life Sci 2009; 66(11-12): 1850–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duncan JS, Litchfield DW. Too much of a good thing: The role of protein kinase CK2 in tumorigenesis and prospects for therapeutic inhibition of CK2. Biochim Biophys Acta 2008; 1784(1): 33–47. [DOI] [PubMed] [Google Scholar]
- 3.Bian Y, Ye M, Wang C, Cheng K, Song C, Dong M et al. Global screening of CK2 kinase substrates by an integrated phosphoproteomics workflow. Sci Rep 2013; 3: 3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rabalski AJ, Gyenis L, Litchfield DW. Molecular Pathways: Emergence of Protein Kinase CK2 (CSNK2) as a potential target to inhibit survival and DNA damage response and repair pathways in cancer cells. Clin Cancer Res 2016; 22(12): 2840–2847. [DOI] [PubMed] [Google Scholar]
- 5.Litchfield DW. Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J 2003; 369(Pt 1): 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Maira G, Salvi M, Arrigoni G, Marin O, Sarno S, Brustolon F et al. Protein kinase CK2 phosphorylates and upregulates Akt/PKB. Cell Death Differ 2005; 12(6): 668–677. [DOI] [PubMed] [Google Scholar]
- 7.Zheng Y, Qin H, Stuart F, Deng L, Litchfield DW, Terfferi A et al. A CK2-dependent mechanism for activation of the JAK-STAT signaling pathway. Blood 2011; 118(1): 156–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng Y, McFarland BC, Drygin D, Yu H, Bellis SL, Kim H et al. Targeting protein kinase CK2 suppresses prosurvival signaling pathways and growth of glioblastoma. Clin Cancer Res 2013; 19(23): 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gibson SA, Yang W, Yan Z, Liu Y, Rowse AL, Weinmann AS et al. Protein kinase CK2 controls the fate between Th17 cell and regulatory T cell differentiation. J Immunol 2017; 198: 4244–4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gibson SA, Yang W, Yan Z, Qin H, Benveniste EN. CK2 controls Th17 and regulatory T cell differentiation through inhibition of FoxO1. J Immunol 2018; 201(2): 383–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bouma G, Strober W. The immunological and genetic basis of inflammatory bowel disease. Nat Rev Immunol 2003; 3(7): 521–533. [DOI] [PubMed] [Google Scholar]
- 12.Cho JH. The genetics and immunopathogenesis of inflammatory bowel disease. Nat Rev Immunol 2008; 8(6): 458–466. [DOI] [PubMed] [Google Scholar]
- 13.Xavier RJ, Podolsky DK. Unravelling the pathogenesis of inflammatory bowel disease. Nature 2007; 448(7152): 427–434. [DOI] [PubMed] [Google Scholar]
- 14.Zenewicz LA, Antov A, Flavell RA. CD4 T-cell differentiation and inflammatory bowel disease. Trends Mol Med 2009; 15(5): 199–207. [DOI] [PubMed] [Google Scholar]
- 15.Fuss IJ, Neurath M, Boirivant M, Klein JS, de la Motte C, Strong SA et al. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J Immunol 1996; 157(3): 1261–1270. [PubMed] [Google Scholar]
- 16.Heller F, Florian P, Bojarski C, Richter J, Christ M, Hillenbrand B et al. Interleukin-13 is the key effector Th2 cytokine in ulcerative colitis that affects epithelial tight junctions, apoptosis, and cell restitution. Gastroenterology 2005; 129(2): 550–564. [DOI] [PubMed] [Google Scholar]
- 17.de Bourayne M, Gallais Y, El Ali Z, Rousseau P, Damiens MH, Cochet C et al. Protein kinase CK2 controls T-cell polarization through dendritic cell activation in response to contact sensitizers. J Leukoc Biol 2017; 101(3):703–715. [DOI] [PubMed] [Google Scholar]
- 18.Ulges A, Klein M, Reuter S, Gerlitzki B, Hoffmann M, Grebe N et al. Protein kinase CK2 enables regulatory T cells to suppress excessive T2 responses in vivo. Nat Immunol 2015; 16(3):267–75. [DOI] [PubMed] [Google Scholar]
- 19.Ulges A, Witsch EJ, Pramanik G, Klein M, Birkner K, Buhler U et al. Protein kinase CK2 governs the molecular decision between encephalitogenic TH17 cell and Treg cell development. Proc Natl Acad Sci U S A 2016; 113(36): 10145–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gibson SA, Benveniste EN. Protein Kinase CK2: An emerging regulator of immunity. Trends Immunol 2018; 39(2): 82–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Koch S, Capaldo CT, Hilgarth RS, Fournier B, Parkos CA, Nusrat A. Protein kinase CK2 is a critical regulator of epithelial homeostasis in chronic intestinal inflammation. Mucosal Immunol 2013; 6(1): 136–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ostanin DV, Bao J, Koboziev I, Gray L, Robinson-Jackson SA, Kosloski-Davidson M et al. T cell transfer model of chronic colitis: concepts, considerations, and tricks of the trade. Am J Physiol Gastrointest Liver Physiol 2009; 296(2): G135–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT et al. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature 1996; 382: 171–174. [DOI] [PubMed] [Google Scholar]
- 24.Kaplan MH, Sun Y-L, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature 1996; 382: 174–177. [DOI] [PubMed] [Google Scholar]
- 25.Watford WT, Hissong BD, Bream JH, Kanno Y, Muul L, O’Shea JJ. Signaling by IL-12 and IL-23 and the immunoregulatory roles of STAT4. Immunol Rev 2004; 202: 139–156. [DOI] [PubMed] [Google Scholar]
- 26.Ahern PP, Schiering C, Buonocore S, McGeachy MJ, Cua DJ, Maloy KJ et al. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 2010; 33(2): 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Macian F NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol 2005; 5(6): 472–484. [DOI] [PubMed] [Google Scholar]
- 28.Murphy TL, Tussiwand R, Murphy KM. Specificity through cooperation: BATF-IRF interactions control immune-regulatory networks. Nat Rev Immunol 2013; 13(7): 499–509. [DOI] [PubMed] [Google Scholar]
- 29.Dong C, Juedes AE, Temann UA, Shresta S, Allison JP, Ruddle NH et al. ICOS co-stimulatory receptor is essential for T-cell activation and function. Nature 2001; 409(6816): 97–101. [DOI] [PubMed] [Google Scholar]
- 30.Grewal IS, Xu J, Flavell RA. Impairment of antigen-specific T-cell priming in mice lacking CD40 ligand. Nature 1995; 378: 617–620. [DOI] [PubMed] [Google Scholar]
- 31.Vaeth M, Eckstein M, Shaw PJ, Kozhaya L, Yang J, Berberich-Siebelt F et al. Store-operated Ca(2+) entry in follicular T cells controls humoral immune responses and autoimmunity. Immunity 2016; 44(6): 1350–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsytsykova AV, Tsitsikov EN, Geha RS. The CD40L promoter contains nuclear factor of activated T cells-binding motifs which require AP-1 binding for activation of transcription. J Biol Chem 1996; 271(7): 3763–3770. [DOI] [PubMed] [Google Scholar]
- 33.Martinez GJ, Hu JK, Pereira RM, Crampton JS, Togher S, Bild N et al. Cutting Edge: NFAT transcription factors promote the generation of follicular helper T cells in response to acute viral infection. J Immunol 2016; 196(5): 2015–2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Koch S, Reppert S, Finotto S. NFATc1 deletion in T lymphocytes inhibits the allergic trait in a murine model of asthma. Clin Exp Allergy 2015; 45(8): 1356–1366. [DOI] [PubMed] [Google Scholar]
- 35.Chua MM, Ortega CE, Sheikh A, Lee M, Abdul-Rassoul H, Hartshorn KL et al. CK2 in cancer: cellular and biochemical mechanisms and potential therapeutic target. Pharmaceuticals (Basel) 2017; 10(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cho SS, Bacon CM, Sudarshan C, Rees RC, Finbloom D, Pine R et al. Activation of STAT4 by IL-12 and IFN-a: Evidence for the involvement of ligand-induced tyrosine and serine phosphorylation. J Immunol 1996; 157: 4781–4789. [PubMed] [Google Scholar]
- 37.Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2000; 13: 715–725. [DOI] [PubMed] [Google Scholar]
- 38.Jostins L, Ripke S, Weersma RK, Duerr RH, McGovern DP, Hui KY et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012; 491(7422): 119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Glas J, Seiderer J, Nagy M, Fries C, Beigel F, Weidinger M et al. Evidence for STAT4 as a common autoimmune gene: rs7574865 is associated with colonic Crohn’s disease and early disease onset. PLoS One 2010; 5(4): e10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Moschen AR, Tilg H, Raine T. IL-12, IL-23 and IL-17 in IBD: immunobiology and therapeutic targeting. Nat Rev Gastroenterol Hepatol 2019; 16(3): 185–196. [DOI] [PubMed] [Google Scholar]
- 41.Ahmed K, Gerber DA, Cochet C. Joining the cell survival squad: an emerging role for protein kinase CK2. Trends Cell Biol 2002; 12(5): 226–230. [DOI] [PubMed] [Google Scholar]
- 42.Smids C, Horjus Talabur Horje CS, Drylewicz J, Roosenboom B, Groenen MJM, van Koolwijk E et al. Intestinal T cell profiling in inflammatory bowel disease: Linking T cell subsets to disease activity and disease course. J Crohns Colitis 2018; 12(4): 465–475. [DOI] [PubMed] [Google Scholar]
- 43.Vaeth M, Feske S. NFAT control of immune function: New Frontiers for an Abiding Trooper. F1000Res 2018; 7: 260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hogan PG, Chen L, Nardone J, Rao A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev 2003; 17(18): 2205–2232. [DOI] [PubMed] [Google Scholar]
- 45.Lee DU, Avni O, Chen L, Rao A. A distal enhancer in the interferon-gamma (IFN-gamma) locus revealed by genome sequence comparison. J Biol Chem 2004; 279(6): 4802–4810. [DOI] [PubMed] [Google Scholar]
- 46.Gomez-Rodriguez J, Sahu N, Handon R, Davidson TS, Anderson SM, Kirby MR et al. Differential expression of interleukin-17A and −17F is coupled to T cell receptor signaling via inducible T cell kinase. Immunity 2009; 31(4): 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ghosh S, Koralov SB, Stevanovic I, Sundrud MS, Sasaki Y, Rajewsky K et al. Hyperactivation of nuclear factor of activated T cells 1 (NFAT1) in T cells attenuates severity of murine autoimmune encephalomyelitis. Proc Natl Acad Sci U S A 2010; 107(34): 15169–15174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015; 47(9): 979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liu Z, Colpaert S, D’Haens GR, Kasran A, deBoer M, Rutgeerts P et al. Hyperexpression of CD40 ligand (CD154) in inflammatory bowel disease and its contribution to pathogenic cytokine production. J Immunol 1999; 163: 4049–4057. [PubMed] [Google Scholar]
- 50.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol 2013; 13(4): 227–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Porter CM, Havens MA, Clipstone NA. Identification of amino acid residues and protein kinases involved in the regulation of NFATc subcellular localization. J Biol Chem 2000; 275(5): 3543–3551. [DOI] [PubMed] [Google Scholar]
- 52.Bleich A, Mahler M, Most C, Leiter EH, Liebler-Tenorio E, Elson CO et al. Refined histopathologic scoring system improves power to detect colitis QTL in mice. Mamm Genome 2004; 15(11): 865–871. [DOI] [PubMed] [Google Scholar]
- 53.Mahler M, Bristol IJ, Leiter EH, Workman AE, Birkenmeier EH, Elson CO et al. Differential susceptibility of inbred mouse strains to dextran sulfate sodium-induced colitis. Am J Physiol 1998; 274(3): G544–551. [DOI] [PubMed] [Google Scholar]
- 54.McFarland BC, Marks MP, Rowse AL, Fehling SC, Gerigk M, Qin H et al. Loss of SOCS3 in myeloid cells prolongs survival in a syngeneic model of glioma. Oncotarget 2016; 7(15): 20621–20635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.