

Abstract
Clinical guidance is often sought when prescribing drugs for patients with primary mitochondrial disease. Theoretical considerations concerning drug safety in patients with mitochondrial disease may lead to unnecessary withholding of a drug in a situation of clinical need. The aim of this study was to develop consensus on safe medication use in patients with a primary mitochondrial disease. A panel of 16 experts in mitochondrial medicine, pharmacology, and basic science from six different countries was established. A modified Delphi technique was used to allow the panellists to consider draft recommendations anonymously in two Delphi rounds with predetermined levels of agreement. This process was supported by a review of the available literature and a consensus conference that included the panellists and representatives of patient advocacy groups. A high level of consensus was reached regarding the safety of all 46 reviewed drugs, with the knowledge that the risk of adverse events is influenced both by individual patient risk factors and choice of drug or drug class. This paper details the consensus guidelines of an expert panel and provides an important update of previously established guidelines in safe medication use in patients with primary mitochondrial disease. Specific drugs, drug groups, and clinical or genetic conditions are described separately as they require special attention. It is important to emphasise that consensus‐based information is useful to provide guidance, but that decisions related to drug prescribing should always be tailored to the specific needs and risks of each individual patient. We aim to present what is current knowledge and plan to update this regularly both to include new drugs and to review those currently included.
Keywords: drugs, in vitro studies, in vivo studies, mitochondrial diseases, mitochondrial toxicity, safety
1. INTRODUCTION
Mitochondrial diseases are a group of inherited metabolic disorders that can present at any age and often exhibit multisystem involvement and high morbidity and mortality.1 The prevalence is conservatively estimated at 1 in 4300 live births.2 The natural course of these diseases is progressive and currently no disease‐modifying therapies are available for the vast majority. Important considerations in the management of patients with mitochondrial diseases include early treatment of organ‐specific complications and avoidance of potential triggers of decompensation including catabolic stressors (eg, fasting, intercurrent illness, pyrexia, trauma, or surgery) or medications that are toxic to mitochondrial function.3
For pharmacological treatment of patients risk‐benefit considerations and a search for safer, better tolerated or more effective alternatives are part of normal clinical practice. Prescribing drugs to patients with mitochondrial disease is associated with the additional consideration of the drug's potential to negatively influence mitochondrial function.4 Due to the heterogeneity in manifestations of mitochondrial disease, reported effects of a pharmacologic agent in an individual patient do not automatically account for all mitochondrial patients.
Mechanisms of drug‐induced mitochondrial toxicity observed in pre‐clinical models include: (a) inhibition of one or more of the electron transport chain complexes (ETCs); (b) uncoupling of mitochondrial oxidative phosphorylation (OXPHOS) by dissipation of the membrane potential and thus disconnecting the ETC from ATP synthase; (c) inhibition of mitochondrial OXPHOS by binding to ATP synthase; (d) inhibition of mitochondrial protein synthesis and biogenesis by affecting mitochondrial DNA (mtDNA) replication and/or fusion of mitochondria; and (e) formation of reactive oxygen species (ROS) as a consequence of any or all of the four above‐mentioned mechanisms.5
Clinical guidelines on the safe use of medications in patients with mitochondrial disease are available but substantial practice variation is a potential source of outcome disparity. Reasons for this have included the paucity of high‐quality evidence and the historical lack of screening for mitochondrial toxicity in drug development. For the vast majority of existing licensed drugs, mitochondrial toxicity is unknown. Thus, when evaluating drug safety in patients with a mitochondrial disease, we must rely on information from in vitro and in vivo pre‐clinical studies and published case reports. Many studies have investigated drug side effects by analysing mitochondrial function in healthy cell lines exposed to high doses of these compounds. It is very difficult to extrapolate these results to what would happen in cells from patients, or what the clinical consequences might be in vivo for patients with mitochondrial disease.
Conscious of the lack of available evidence, we convened a workshop aimed at developing a consensus about safe medication use in patients with a primary mitochondrial disease. Consensus was based on a review of the literature and the clinical experience of paediatricians, internists and neurologists experienced in treating affected patients. The results of this Delphi workshop were used to develop guidance for physicians prescribing drugs for patients with mitochondrial diseases. This article does not contain any studies with human or animal subjects performed by any of the authors.
2. METHODS
A modified Delphi‐based technique was used to develop a consensus view of drug safety in patients with a mitochondrial disease by a group of internationally acknowledged experts. The Delphi technique was developed by the RAND (research and development) Corporation in 1953 to elicit and process value judgments.6 The technique consists of a structured and repetitive survey of at least two rounds, which continues until consensus is reached among panellists. Between each round, feedback is provided to the panellists.
2.1. Formation of panellists
Clinicians and researchers with expertise in mitochondrial medicine and pharmacology were invited. Potential participants were selected based on their known experience in the field of mitochondrial medicine and pharmacology. To broaden the panel of experts, we also asked invitees to provide names of other potential participants. Since drug prescription could vary from country to country and from continent to continent, we endeavoured to achieve geographical balance in the selection of panellists. Candidate panellists were invited by e‐mail outlining the study aims and the Delphi process. Patient representatives from the International Mito Patients advocacy group IMP (https://www.mitopatients.org/) were also invited. The consensus panel finally consisted of 16 participants and two patient representatives from IMP. All the participants are experts in mitochondrial medicine, including 13 clinicians, two pharmacologists and a basic scientist. Participants were from five different European countries and from the USA.
2.2. Selection of drugs
Due to the limited duration of the workshop (2 days), only a finite number of drugs could be considered and drugs to be studied were, therefore, selected by using a previously published list on the website of the patient advocacy group IMP (https://www.mitopatients.org/mitodisease/potentially-harmful-drugs) supplemented with a few drugs commonly prescribed to patients affected by mitochondrial disease (eg, anaesthetic agents, analgesics, antibiotics, antiepileptic drugs). Two facilitators (MM and MdV) formulated a list of 32 drugs and drug classes and circulated this to the panellists. Subsequently, panellists had the opportunity to add drugs which were in their view most needed a consensus opinion. Due the limited time, the total number of drugs/drug classes was limited to 46. The decision to study some drugs as a group was based on the fact that the class of drugs have a common pharmacological mode of action or similar off‐target side effects.
2.3. Delphi‐based process
The process was performed in three stages (Figure 1).
Preparatory phase: Potential panellists were invited and the list of drugs and drug classes to be studied was compiled. Each panellist was assigned 2 to 4 drugs or drug classes and was asked to perform a thorough literature search from recognised medical databases. Participants were requested to send a one page summary of their results, including the following items: mode of action; theoretical effect on mitochondrial function; known effects on mitochondrial function; effects observed in patients with mitochondrial disease, cell lines from patients with mitochondrial disease or control cell lines; what type of study (eg, case report, in vitro) and a comprehensive list of references consulted during the review process. The panellists were also asked to submit one or two statements about the drugs they had investigated, including a statement about the safety of the drug in patients with mitochondrial disease.
First Delphi round: The literature reviews of the drugs were summarised. All panellists were provided with this bundle of drug summaries, as background information, before they were requested to give their opinion in the first voting round. The statements were rewritten by the facilitators and sent to all panellists by an online survey (ie, Google Form), who had the opportunity to indicate their agreement anonymously on a linear five‐point scale anchored at each end by ‘Score 1: Absolutely disagree’ and ‘Score 5: Absolutely agree’. Opportunity was provided for participants to add comments in order to correct areas of ambiguity or suggestions for improvement. Strong consensus agreement was predefined as the mean result ≥4 and ≥70% agreement among the panellists (scores of 4 or 5 on the linear scale). If only one of these two criteria was met, good consensus was considered. If neither consensus criteria was met, then the statement was considered to lack consensus agreement. Statements that did not achieve consensus in the initial round were scheduled for discussion at the workshop as pre‐agreed.
Second Delphi round: This phase took place during a two‐day workshop. During the workshop, panellists were provided with feedback regarding the drugs for which the statements had not reached consensus. The feedback consisted of presentations of the literature search. After each presentation, there was an opportunity to ask questions, to comment and to exchange personal experiences in prescribing the specific drug in question. At the end of each session during which seven to nine drugs had been discussed, the panellists were asked to re‐rate the statements concerning these drugs by the online survey, that is, the second voting round. Results were analysed to evaluate the level of agreement for each statement. Consensus was pre‐defined as in the first Delphi round.
Figure 1.
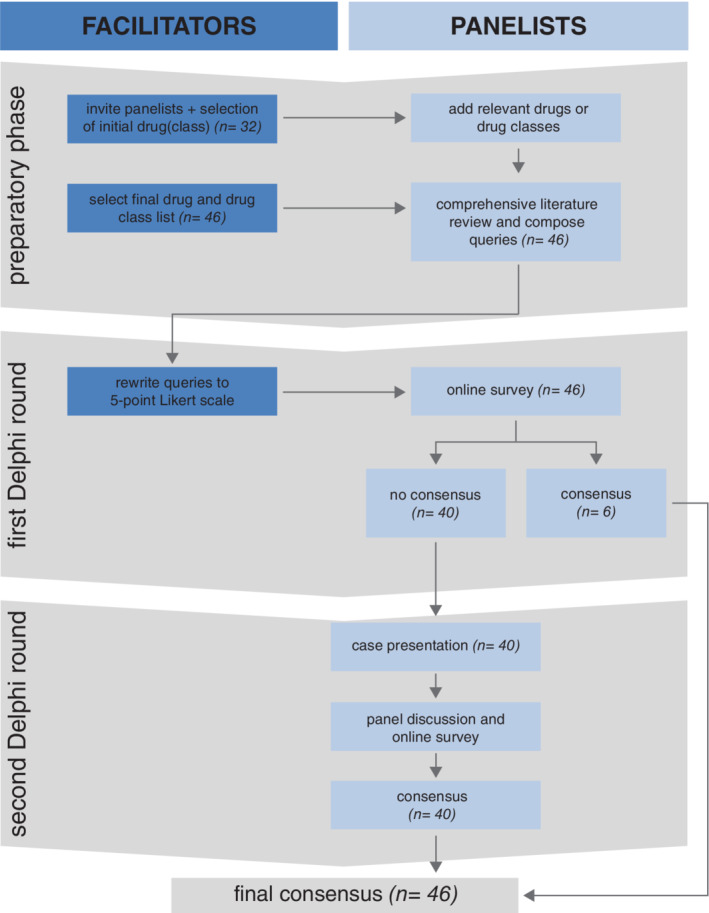
Flowchart of the Delphi‐based process showing the activities and results of the three stages. n = number of drugs/drug groups
3. RESULTS
3.1. Preparatory phase
In the preparatory phase the participants performed and summarised a literature review regarding the drugs or drug groups they were asked to study (see Supplementary data for the summaries of these literature reviews). Furthermore, a total of 55 statements for the voting rounds were composed by the participants to cover the selected 46 drugs/drug groups.
3.2. Delphi rounds
All the participants responded in the first Delphi round, all of them participated in the workshop meeting and all of them responded in the second voting round. During the first voting round, prior to the workshop meeting, consensus was not achieved for 40 drugs owing to a low level of agreement (<70% and/or <4). Good or strong consensus was reached for six drugs or drug groups; enalapril, paracetamol, midazolam, carbamazepine, oxcarbazepine, and haloperidol.
According to our pre‐agreed Delphi process, the levels of evidence in the available literature pertaining to drugs that did not reach consensus were discussed during the meeting. The main point of discussion for each drug was the knowledge gap between pre‐clinical drug studies and how these relate to the patient with a mitochondrial disease. Drug effects measured in vitro or in vivo were either not, or not fully, translatable to clinical consequences for the patient for several reasons. Most studies were designed to investigate toxicity of the drug, and consequently examined the effects of extremely high drug doses (ie, to certify the occurrence of a toxic response and associated off‐target mechanisms). Therapeutic drug levels would plausibly show less or no negative effects on mitochondrial function. Moreover, cell lines used in the majority of studies did not have a primary mitochondrial defect and thus none of the compensatory processes associated with mitochondrial dysfunction. Investigating the effects of drugs in cell lines from patients with primary mitochondrial disease was therefore considered worthwhile. In several studies, the formation of ROS was used as an outcome parameter; however, in many cases, the source of the increased ROS concentration, mitochondrial or extra‐mitochondrial, was not demonstrated. After discussing and weighing the pre‐clinical data, the expert team discussed their own clinical experiences of observed safety or side effects of the drug(s) in question in patients with mitochondrial disease.
This process was followed by the second voting round. Good or strong consensus was achieved for all the drugs or drug groups. The consensus results for all the statements are shown in Table 1 and Figure 2. Immediately following the workshop, it was discussed that one of the statements could have been (for the panellists) or could be (future use of the information by colleagues) phrased misleadingly. It concerned the statement about the use of steroids in patients with primary mitochondrial disease. Therefore, a more clearly defined statement was formulated and the panellists were asked to vote on this statement via an online survey. The result of this voting showed strong consensus.
Table 1.
Voting results of the final Delphi round per statement
| Question | Mean | % of people voting 4 or 5 | Consensus |
|---|---|---|---|
| General | |||
| We need to update the IMP table of potentially harmful drugs for mitochondrial patients version 3 (https://www.mitopatients.org/mitodisease/potentially‐harmful‐drugs) | 4.87 | 100 | Strong |
| Good clinical practice including general indications, contraindications, clinical monitoring and side effects for all drugs must always kept in mind (with or without mitochondrial genetic defect). They will not be discussed in this consensus | 4.94 | 100 | Strong |
| For all drugs where clear evidence in vivo of mitochondrial toxicity is absent or poor, they can be used with careful monitoring in the first few days of treatment for potential side effects and measurement of blood lactate | 4.19 | 93.7 | Strong |
| There is a great need for further studies to determine a) the criteria for drug mitochondrial toxicity in humans, and b) which specific drugs are toxic for mitochondria and must be avoided | 4.25 | 75 | Strong |
| Analgesics‐Antipyretics‐NSAIDs‐Corticosteroids | |||
| Paracetamol is not contraindicated in primary mitochondrial disease (PMD) | 4.56 | 87.5 | Strong |
| Do you consider that steroids are safe to use in acutely ill patients with PMD? | 4.46 | 100 | Strong |
| It is safe to use steroids in patients with Kearns‐Sayre syndrome | 4.13 | 86.6 | Strong |
| NSAIDs can be safely used in PMD | 4.31 | 87.5 | Strong |
| It is reasonable to avoid NSAIDs for long periods in PMD with renal or hepatic or gastrointestinal involvement | 4.31 | 93.75 | Strong |
| Use of aspirin is safe in PMD | 4.56 | 93.75 | Strong |
| Alcohol | |||
| Alcohol in large amounts (above recommended daily intake) is generally toxic and should be avoided | 4.37 | 93.75 | Strong |
| Alcohol consumption within the limits recommended by national guidelines appears non‐toxic in PMD | 5 | 100 | Strong |
| Anaesthetics | |||
| It is safe to use articaine in PMD | 4.75 | 100 | Strong |
| It is safe to use bupivacaine in PMD | 4.81 | 100 | Strong |
| It is safe to use lidocaine in PMD | 5 | 100 | Strong |
| It is safe to use volatile anaesthetics in PMD | 4.62 | 100 | Strong |
| It is safe to use fentanyl in PMD | 4.75 | 100 | Strong |
| Ketamine is safe in general anaesthesia for patients with PMD | 4.75 | 100 | Strong |
| Barbiturates are safe in general anaesthesia for patients with PMD | 4.56 | 93.75 | Strong |
| Propofol is safe in induction anaesthesia in PMD | 3.81 | 81.25 | Consensus |
| Extra caution and monitoring should be considered for patients with PMD manifesting predominantly with myopathic phenotype when neuromuscular blockade is required for general anaesthesia and surgery | 4.25 | 87.5 | Strong |
| Non depolarizing neuromuscular blocking agents are safe for general anaesthesia in patients with PMD | 4.56 | 100 | Strong |
| Antibiotics | |||
| As a general approach, short term (< 7 days) antibiotic treatment is unlikely to be a problem in PMD. Infection is a much greater risk than short term antibiotics | 4.75 | 100 | Strong |
| If indicated, linezolid could be used in mitochondrial disease, with careful lactate monitoring, particularly in children and other patients with pre‐existent lactic acidaemia | 4.56 | 100 | Strong |
| It is safe to use quinolones in PMD | 4.44 | 100 | Strong |
| Aminoglycosides should be avoided in patients with predisposing mitochondrial DNA mutations (eg, m.1555A > G and m.1494C > T) for ototoxicity | 4.81 | 100 | Strong |
| Topical chloramphenicol use is safe in PMD | 4.62 | 100 | Strong |
| It is safe to use tetracyclines in PMD | 4.75 | 100 | Strong |
| It is safe to use ceftriaxone in PMD | 4.87 | 100 | Strong |
| Antidepressant‐Neuroleptic drugs | |||
| The use of antipsychotics medications when they are clinically indicated is not contraindicated in PMD | 4.19 | 87.5 | Strong |
| Quetiapine can be safely used in PMD despite some studies in rodents or cell lines indicate potential mitochondrial toxicity | 4.31 | 93.75 | Strong |
| Fluphenazine could be safely used in PMD | 4 | 75 | Strong |
| Haloperidol can be safely used in PMD despite some studies in rodents or cell lines indicate potential mitochondrial toxicity | 3.81 | 75 | Consensus |
| It is safe to use tricyclic antidepressants in PMD | 4.87 | 100 | Strong |
| It is safe to use chlorpromazine in PMD | 4.75 | 100 | Strong |
| It is safe to use clozapine in PMD | 4.56 | 100 | Strong |
| It is safe to use risperidone in PMD | 4.56 | 100 | Strong |
| Antidiabetic drugs | |||
| It is safe to use metformin in PMD | 4.56 | 100 | Strong |
| It is safe to use glitazone in PMD | 4.37 | 100 | Strong |
| Antiepileptic drugs | |||
| Since there are no descriptions of toxicity of midazolam or other benzodiazepines (BDZ) in PMD, it is correct to assume that midazolam or other BDZ could be used in acute seizure in PMD, or be used as anaesthetic | 4.56 | 100 | Strong |
| Valproic acid should be avoided only in POLG patients | 4.25 | 81.25 | Strong |
| In non‐POLG patients with mitochondrial disease, without liver disease, valproic acid could be used to manage refractory epilepsy and refractory mood disorders | 4.4 | 100 | Strong |
| Carbamazepine is safe in PMD | 4.12 | 75 | Strong |
| Oxcarbazepine is not contraindicated in PMD | 4.37 | 93.75 | Strong |
| Oral phenobarbital is safe in patients with PMD | 4.6 | 100 | Strong |
| In refractory mitochondrial status epilepticus, barbiturates in appropriate settings could be used for long duration infusion | 4.53 | 100 | Strong |
| It is safe to use gabapentin in PMD | 4.86 | 100 | Strong |
| It is safe to use phenytoin in PMD | 4.33 | 86.66 | Strong |
| It is safe to use levetiracetam in PMD | 4.86 | 100 | Strong |
| It is safe to use perampanel in PMD | 4.13 | 80 | Strong |
| It is safe to use topiramate in PMD | 4.46 | 100 | Strong |
| In refractory mitochondrial status epilepticus, propofol is safe for long duration infusion (up to 48 hours) | 4.47 | 100 | Strong |
| Ketamine is safe for long duration infusion (eg, refractory status epilepticus) in PMD | 4.31 | 93.75 | Strong |
| Bisphosphonates | |||
| It is safe to use bisphosphonates in PMD | 4.25 | 100 | Strong |
| Cardiovascular drugs | |||
| It is safe to use amiodarone in PMD | 4.06 | 93.3 | Strong |
| It is safe to use beta‐blockers in PMD | 4.46 | 100 | Strong |
| Enalapril is safe in PMD | 4.06 | 81.25 | Strong |
| Fibrate drugs‐Statins | |||
| It is safe to use fibrate in PMD | 4.62 | 100 | Strong |
| It is safe to use statins in PMD as long as guidelines concerning monitoring of CK and symptoms are followed | 4.5 | 100 | Strong |
Abbreviation: PMD, primary mitochondrial disease.
Figure 2.
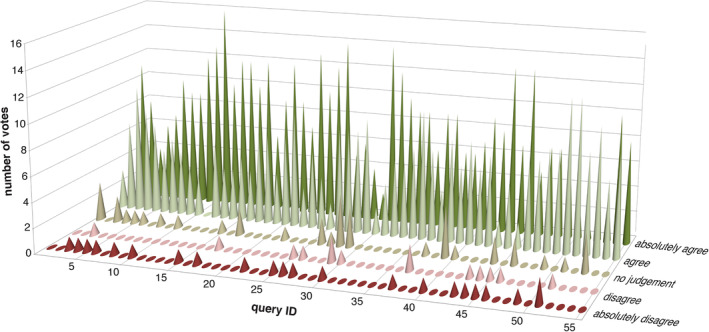
Voting results of the final Delphi round, showing the distribution of votes per statement
The general conclusion of the Delphi process, all based on consensus (level 4 evidence), was that all the 46 drugs or drug groups studied are considered to be generally safe for patients with mitochondrial disease although some specific restrictions were considered for certain molecular defects and particular clinical situations. These specific conditions will be reviewed in the Discussion.
4. DISCUSSION
In this study, 16 experts in mitochondrial medicine, pharmacology and basic science provided their professional opinion concerning the safe use of medications in patients with primary mitochondrial disease, to assist clinician and patient decision‐making. During the Delphi‐based process, it became apparent that for many drugs clinical experience conferred most weight, since these observations reflected the effect of the drug in actual patients with mitochondrial diseases. It is important to emphasise that consensus‐based information is useful to provide guidance, but that decisions related to drug prescribing should always be tailored to the specific needs and risks of each individual patient. Recognising the need for a personalised drug prescription is essential to avoid the pitfalls of one‐size‐fits‐all clinical standards, especially in patients with a mitochondrial disease, a group characterised by enormous clinical heterogeneity. Moreover, good clinical practice, including general indications, contraindications, clinical monitoring and side effects for all drugs must always be considered, whether it is a patient with mitochondrial disease or not.
Some of the statements devised were genotype‐specific. However, the majority could be applied generically to the disease group (ie, mitochondrial disease) as a whole. Caveats for certain drugs or class of drugs were noted, as follows (Table 2).
Screening for mtDNA mutations associated with predisposition to aminoglycoside susceptibility should be considered before treatment with aminoglycosides.7 It is strongly recommended to screen for these mtDNA mutations before elective long‐term treatment with aminoglycosides is planned. In some emergency situations aminoglycosides provide very effective broad‐spectrum antibiotic treatment and should be prescribed until immediate danger has passed or microbial sensitivities have been established and a suitable alternative antibiotic can be administered. The benefits of the drug outweigh the risks in these situations. The mitochondrial 12S rRNA is a hot spot for mutations associated with both aminoglycoside‐induced and non‐syndromic hearing loss. Of those, the homoplasmic m.1555A > G and m.1494C > T mutations in the highly conserved coding region of the 12S rRNA have been associated with hearing loss worldwide.8
Valproic acid should be used only in exceptional circumstances. In most national guidelines for the treatment of status epilepticus, valproic acid is not the first choice drug. Furthermore, the drug is absolutely contraindicated in patients with mitochondrial disease due to POLG mutations. Additionally, valproic acid should not be used in patients with known liver disease and/or clinical signs suspicious for POLG disease, such as epilepsia partialis continua, explosive onset of focal epilepsy or rhythmic high amplitude delta with superimposed spikes (RHADS) on EEG.9
The last drug group that deserves particular attention is the group of neuromuscular blocking agents. Extra caution and monitoring should be performed for patients manifesting a predominantly myopathic phenotype.
Although historically there have been largely theoretical concerns around general anaesthetic use in patients with mitochondrial disease, adverse events are exceptionally rare. Consensus was unanimous that these drugs and drug classes were deemed safe. General surgery is a potentially risky procedure for any patient with mitochondrial disease. Catabolism should be prevented by minimising preoperative fasting and administering intravenous glucose perioperatively during prolonged anaesthesia, unless the patient is on a ketogenic diet.3
The duration of drug administration may also play a role in whether not side effects develop. Short term use of midazolam is considered safe, for example, in the acute management of epileptic seizures or for a short anaesthetic procedure. In specific clinical situations, a longer duration of administration of a certain drug can be justified, despite an increased risk of side effects or disease progression. This accounts for situations in which no alternative treatment options are available and the absence of treatment could have a more detrimental effect of disease progression. Examples include the use of propofol or barbiturate infusions in the management of refractory status epilepticus. Duration of treatment should be guided by individual patient needs and their response to specific treatments.
Many patients with a mitochondrial disease have renal impairment, including patients with the m.3243A > G mtDNA mutation or genetic defects in RMND1.10, 11, 12 Drug dose adjustment should be considered particularly when active drug moieties are renally cleared, for example, levetiracetam.
Metabolic acidosis (lactic acidosis) may occur in patients with mitochondrial disease; therefore, drugs that can cause acidosis should be prescribed with caution, with advice to report symptoms of metabolic acidosis and regular clinical review and monitoring of acid‐base status in blood.
Table 2.
Points of attention regarding drug prescription in patients with a mitochondrial disease (detailed description in Section 4)
| Specific drug/drug group/clinical condition/genotype | Points of attention |
|---|---|
| Specific drug/drug group/genotype | |
| Aminoglycosides | The mitochondrial 12S rRNA is a hot spot for mutations associated with both aminoglycoside‐induced and non‐syndromic hearing loss. Screening for these mtDNA mutations is strongly recommended before elective long‐term treatment is planned. The benefits of the drug in emergency treatment, as a very effective broad‐spectrum antibiotic, outweigh the risks in these situations. |
| Valproic acid | Should be used only in exceptional circumstances. The drug is absolutely contraindicated in patients with mitochondrial disease due to POLG mutations. Valproic acid should not be used in patients with known liver disease and/or clinical signs suspicious for POLG disease. |
| Neuromuscular blocking agents | Extra caution and monitoring should be performed for patients manifesting a predominantly myopathic phenotype. |
| Specific clinical condition | |
| General anaesthesia and surgery | Catabolism should be prevented by minimising preoperative fasting and administering intravenous glucose perioperatively during prolonged anaesthesia, unless the patient is on a ketogenic diet. |
| Duration of treatment | The duration of drug administration may play a role in whether or not side effects develop. Duration of treatment should be guided by individual patient needs and their response to specific treatments. |
| Renal impairment | Many patients with a mitochondrial disease have renal impairment; drug dose adjustment should be considered particularly when active drug moieties are renally cleared. |
| Metabolic acidosis (lactic acidosis) | Metabolic acidosis (lactic acidosis) may occur in patients with mitochondrial disease, therefore drugs that can cause acidosis should be prescribed with caution. Regular clinical review and monitoring of acid‐base status in blood is recommended. |
Abbreviation: PMD, primary mitochondrial disease.
The present study has limitations. Although consensus methods are widely used to inform clinical practice in the absence of empirical data, expert judgement ranks low in the hierarchy of evidence. However, we felt compelled to give transparent opinions based on the available preclinical studies and clinical evidence, to prevent unnecessary withholding of important drugs from patients with primary mitochondrial disease.
A recurrent issue throughout our discussions was the lack of translation between pre‐clinical studies and the clinical situation, where physicians experience that patients appear to tolerate most drugs. Multiple factors will be responsible for this lack of translation, for example, exposure time. We analysed the compounds comparing the C max values (ie, in vivo) or concentration ranges (ie, in vitro) of the pre‐clinical exposures employed alongside the C max of the clinical dose used and human toxic concentrations. The head‐to‐head comparisons can be seen in Table 3. For only 18 out of 44 drugs or drug classes was the drug concentration employed in rodent or cell models comparable with the therapeutic levels found in clinical populations. This finding demonstrates that the lack of translation to the clinical situation is due to the use of higher or toxic drug levels in the majority of pre‐clinical studies.
Table 3.
Head‐to‐head comparisons pre‐clinical and clinical drug concentrations of the evaluated drugs
| Drug (Class) | Matching preclinical and clincial levels? | References | |||||
|---|---|---|---|---|---|---|---|
| Pre‐clinical data | Clinical data | ||||||
| Pre‐clinical C max or concentration range (μg⋅mL−1) | Highest level of complexity evaluated | Model system | C max (μg⋅mL−1) | Toxicity plasma level (μg⋅mL−1) | |||
|
Aminoglycosides a Gentamycin Tobramycin Amikacin Streptomycin Neomycin |
24 |
In vitro |
Primary rat cochlear cells |
15‐20 |
12 12 30 40 |
Yes |
Schulz and Schmoldt13; Quan et al14; Hodiamont et al15 |
| Amiodaroneb | 1.0 | In vivo | Isolated liver mitochondria from treated Wistar rats | 1.0‐2.5 | 2.5 | Yes | Schulz and Schmoldt13 |
| Articaine | 28‐284 | In vitro | Human leukocytes | 0.58 | Noc | Oertel et al16; Günaydin and Demiryürek17 | |
|
Barbiturates a Amobarbital Pentobarbital Phenobarbital Secobarbital |
5.8‐232 |
In vitro |
Isolated liver Mitochondria |
2.2‐4.4 |
5.0 10 30 7.0 |
No |
Schulz and Schmoldt13; Dalmora et al18; Santos et al19 |
|
Beta blockers a Atenolol Carvedilol Metoprolol Nebivolol Propranolol |
8.2 |
In vitro |
H9C2 myocardial cells |
0.047 |
2.0 12 0.48d 1.0 |
No |
Schulz and Schmoldt13; Gehr et al, 1999; Sgobbo et al, 2007 |
|
Bisphosphonates s Alendronate
Clodronate Risedronate
Ibandronate Zoledronic acid |
12‐50
14‐57 |
In vitro
In vitro |
Gastric (RGM1) and small intestinal (IEC6) epithelial cells Gastric (RGM1) and small intestinal (IEC6) epithelial cells |
0.038
0.00097‐0.0039 |
No
No |
Mitchell et al20; Yun et al21; Nagano et al22 | |
| Bupivacaine | 2.6‐3.9 | In vitro | Primary rat cardiomyocytes | 0.49‐1.9 | 2.0 | Yes | Schulz and Schmoldt13; Li et al23; Bethea24 |
| Carbamazepine | 5.9‐236 | In vitro | Isolated rat liver mitochondria | 1.5‐6.8 | 10 | Yes | Schulz and Schmoldt13; Mahmood and Chamberlin25; Santos et al19 |
| Ceftriaxone | 555 | In vitro | Purified rat carnitine/acylcarnitine transporter | 223‐276 | No | Pochini et al26 | |
| Chloramphenicol | 100 | In vitro | Primary human fibroblasts | 4.9‐12 | 25 | No | Schulz and Schmoldt13 |
| Chlorpromazine | 0.32‐1.9 | In vitro | Rat ovarian theca cells | 1.0 | Yes | Schulz and Schmoldt13 | |
| Clozapine | 8.2‐25 | In vitro | Mouse myoblasts (C2C12), adipocytes (3 T3‐L1), hepatocytes (FL‐83B) and monocytes (RAW 264.7) | 0.10‐0.77 | 0.6 | No | Schulz and Schmoldt13 |
| Enalapril | 0.0091e | In vivo | Isolated cardiac mitochondria from spontaneously hypertensive rats | 0.023‐0.21 | No | Kelly et al27; Holenarsipur et al28; Piotrkowski et al29; Higuchi et al, 1994 | |
| Ethanol | 3680‐27 600 | In vitro | Human retinal pigment epithelial cells (ARPE‐19) | 577f | 1000 | No | Schulz and Schmoldt13; Bonet‐Ponce et al30; Klockhoff et al31 |
| Fentanyl | 0.5⋅10−3‐2⋅10−3 | In vitro | Human hepatoma HepG2 cells | 0.39⋅10−3‐23⋅10−3 | Yes | Djafarzadeh et al32 | |
|
Fibrate drugs a Bezafibrate Ciprofibrate Fenofibrate Gemfibrozil |
72‐145 |
In vitro |
Primary fibroblasts and myoblasts from MP |
10.6
8.6‐26 29.5 |
No |
Miller and Spence33; Abshagen et al34; Bastin et al35 | |
| Fluphenazine | 0.043‐44 | In vitro | Swiss albino mice brain slices | 0.056 | No | Balijepalli et al, 1999; Midha et al, 1983 | |
| Gabapentinb | 16‐33 | In vivo | Wistar rat striatum mitochondria | 4.8 | 85 | Yes | Schulz and Schmoldt13; Chen et al36; |
|
Glitazones Troglitazone Rosiglitazone pioglitazone Ciglitazone |
5.5‐22 4.5‐18 4.5‐18 |
In vitro In vitro In vitro |
Human hepatoma cells Human hepatoma cells Human hepatoma cells |
0.37‐2.2 0.12‐0.15 0.10‐3.5 |
No No No |
Eckland and Danhof37; Loi et al38; Balfour and Plosker39; Hu et al40 | |
| Haloperidolb | 0.0049 | In vivo | Brain and muscle mitochondria from treated Sprague‐Dawley rats | 0.0076 | 0.05 | Yes | Schulz and Schmoldt13; Barrientos et al41; Lei et al,42; Desai, et al43 |
| Halothane | 18‐395 | In vitro | Isolated pig heart mitochondria | 90‐225g | Yes | Hanley et al44; Atallah and Geddes45 | |
| Ketamine | 5.5‐17 | In vivo | Brain mitochondria from treated Wistar rats | 0.042 | 7.0 | No | Venâncio et al46; Wellington et al47; Moaddel et al48; Yanagihara et al49; Schulz and Schmoldt13 |
| Lidocaine | 234‐2340 | In vitro | Rat dorsal root ganglion | 0.157‐0.552 | 6.0 | No | Onizuka et al50; Schulz and Schmoldt13 |
| Linezolid | 5‐15 | In vitro | Mouse neurons | 12.5 | ND | Yes | Bobylev et al51 |
| Metformin | 50‐1292 | In vitro | Isolated mouse skeletal muscle mitochondria and various cell lines (ie, NT2196, NMuMG, MFC10A, and MCF7) | 1.3 | 5.0 | No | Schulz and Schmoldt13; Shu et al52; Andrzejewski et al53 |
| Midazolam | 33‐326 | In vitro | Isolated rat and skeletal muscle mitochondria | 0.15 | 1.0 | No | Schulz and Schmoldt13; Link et al54; Colleoni et al55 |
|
NSAIDs a Diclofenac Ibuprofen Indomethacin Naproxen Celecoxib |
18‐35 |
In vitro |
Isolated duodenum mitochondria |
4.2 96 |
50 200 4.0 200 |
No |
Schulz and Schmoldt13; Sandoval‐Acuña et al56; Caille et al57 |
| Oxarbazepine | 76 | In vitro | Rat embryo Hippocampal neurons | 1.1 | No | Araújo et al,58; Tartara et al59 | |
| Paracetamol | 756 | In vitro | Isolated mouse liver mitochondria and primary hepatocytes | 18‐21 | 100 | No | Schulz and Schmoldt13; Sevilla‐Tirado et al60; Burcham and Harman61 |
| Phenytoin | 6.3‐252 | In vitro | Isolated rat liver mitochondria | 2.0 | No | Santos et al19; Suthisisang et al62 | |
| Propofol | 4.5‐18 | In vitro | Isolated rat liver mitochondria | 2.1‐29h | Yes | Branca et al63; Khan et al64 | |
| Quetiapine | 9.6‐77 | In vitro | Isolated rat liver mitochondria | 53‐117 | 1.8d | Yes | Schulz and Schmoldt13; Modica‐Napolitano et al65; DeVane and Nemeroff66 |
| Risperidone | 10‐82 | In vitro | Isolated rat liver mitochondria | 20‐60 | Yes | Modica‐Napolitano et al65; Heykants et al67 | |
|
Salicylates a Acetylsalicylic acid
Salsalate |
90‐1802 |
In vitro |
Isolated subsarcolemmal mitochondria |
1.0‐4.8 |
300
300 |
No |
Schulz and Schmoldt13; Nulton‐Persson et al68; Nagelschmitz et al69 |
|
Statins a Atorvastatin Lovastatin Pravastatin Rosuvastatin Simvastatin |
16‐56 13‐41 36‐42 25‐48 19‐84 |
In vitro |
Murine myoblasts (C2C12) |
27‐66 10‐20 45‐55 37 10‐34 |
Yes Yes Yes Yes Yes |
Bellosta et al70; Schirris et al71 | |
| Topiramateb | 18‐90 | In vivo | Isolated mitochondria from treated Sprague‐Dawley rats | 3.7‐7.7 | No | Kudin et al72; Doose et al73; Matar and Tayem74 | |
| Valproic acid | 3.6‐29 | In vitro | Isolated rat liver mitochondria | 98‐113 | 150 | No | Schulz and Schmoldt13; Nunez et al75; Jafarian et al76 |
Note: The concentration range used in in vitro studies (eg, cellular studies) or peak plasma concentrations (C max) of animal studies were compared with the corresponding human peak plasma concentrations of drugs evaluated. When no peak plasma concentrations were available, they were calculated using standard pharmacokinetic equations (ie, C max = F × D/Vd) or obtained from studies using the same dosing regimen and for animal studies the same species and strain. For pharmacokinetic calculations an average rat weight of 190 mg was assumed.
Abbreviation: ND, no data available.
For drug classes, up to five representative members were selected based on UpToDate. Waltham, MA: UpToDate Inc. https://www.uptodate.com (Accessed on May 6, 2019).
Pre‐clinical C max values were calculated using available pharmacokinetic data.
Plasma concentrations after subcutaneous articaine injection. Low plasma levels are expected as this drug is intended for local anaesthesia where much higher concentrations can be reached.
Based on a single case‐report.
Total free enalapril concentrations, C max including the main metabolite: 1.49 μg⋅mL‐1.
C max after consumption of one alcoholic 20% (v/v) drink.
Indicated concentrations show the clinical plasma concentration range, not C max.
No C max, but concentration at loss of consciousness.
Limitations were noticed as well in drawing recommendations from published clinical series and case reports, due to several factors. Many studies describe patients with a biochemical defect in OXPHOS without mentioning a specific mitochondrial genetic defect, that is, without confirmation of primary mitochondrial disease. Furthermore, patients often have co‐morbidities and/or are using polypharmacy making it impossible to determine whether the effects reported are directly related to the drug in question or are consequences of one of the other diseases or other drugs.
We plan to revise the existing list regularly and to extend the list of drugs in the future using the same Delphi process. The list of drugs studied will be freely available on the web pages of centres of mitochondrial disease expertise and the IMP. This provides the opportunity to easily revise and update knowledge about the included drugs. Furthermore, given the frequent disparities between pre‐clinical studies and clinically relevant treatment doses, future pre‐clinical toxicity studies that more closely model patient doses and conditions (ie, mitochondrial deficiencies) are needed for improved drug‐induced mitochondrial dysfunction assessments in mitochondrial patients.
5. CONCLUSION
This study details the consensus guidelines of a mitochondrial expert panel on the safe use of medications in patients with mitochondrial disease. We acknowledge that the quality of available evidence or published literature used to accredit these recommendations is currently limited. The key recommendations are: (a) valproate should be avoided in patients with POLG‐related mitochondrial disease and alternative anticonvulsants considered as first‐line therapeutic strategies; (b) prolonged use of specific drugs may have negative consequences and should be avoided if good alternative treatment options are available; and (c) the usual standards of good practice prevail when prescribing any drug, irrespective of the drug's mitochondrial toxicity potential or profile. We do emphasise that drug prescriptions should always be tailored to the individual patient after careful consideration of their specific needs.
CONFLICT OF INTEREST
M. A. reports grants from Virginia Tech Interdisciplinary Graduate Education Program, outside the submitted work.
D. B. reports grants from National Institutes of Health, grants and personal fees from Stealth BioTherapeutics, grants from vTv Therapeutics, grants from Catabasis Pharmaceuticals, grants from United States Dept of Agriculture, outside the submitted work.
A. K. reports that she is the current President of the Mitochondrial Medicine Society.
R. M. reports personal fees from Eisai, outside the submitted work.
S. R. reports personal fees from BioMedical, personal fees from NeuroVive, personal fees from Partners 4 Access, outside the submitted work, and that she is an Editor of the Journal of Inherited Metabolic Disease.
T. S. reports grants from Princes Beatrix Muscle Foundation (Prinses Beatrix Spierfonds), grants from European Molecular Biology Organization, during the conduct of the study.
L. B., G. G., N. K., C. L., M. M., R. M., Y. S. N., M. O., R. P., F. R., K. V., and M. C. De V. declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
All authors contributed to the acquisition of data and preparation of critical revision of manuscript.
M. C. De V. and M. M. contributed to the study conception and design, and analysis and interpretation of data.
All authors except M. E. A. contributed to workshop participation.
M. C. De V., M. M., D. A. B., M. E. A., and T. J. J. S. contributed to drafting of manuscript.
Supporting information
Data S1: Supplementary data
ACKNOWLEDGMENTS
M.M. is grateful to Drs Vincenzo Montano, Costanza Simoncini, and Elena Ferrari for their help in literature search. M.O. is grateful to Dr Rafael Artuch, Juan Darío Ortigoza‐Escobar, and Leticia Pías for their help in literature search. G.S.G., Y.S.N., and R.M. would like to acknowledge the contribution of their Newcastle colleagues, Professor Sir Doug M Turnbull, Dr Andrew M. Schaefer, and Dr Rhys Thomas for sharing their clinical experience on treating patients with mitochondrial disease, which has underpinned aspects of these consensus recommendations.
The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors. The workshop was sponsored by the patient foundations: AEPMI, DGM, Eurordis, International Mito Patients (IMP), the Lily Foundation, Mitocanada, Mitocon, Muscular Dystrophy UK, and the Mito Foundation.
Michelangelo Mancuso was partially supported by Telethon Grant GUP09004 and Telethon‐MITOCON grant GSP16001. Shamima Rahman is supported by research grant funding from Great Ormond Street Hospital Children's Charity, the NIHR Great Ormond Street Hospital Biomedical Research Centre, and the Lily Foundation. Nandaki Keshavan is supported by Action Medical Research. RDSP is supported by a Medical Research Council Clinician Scientist Fellowship (MR/S002065/1). D. A. B. is supported by National Institutes of Health NHLBI R01 (HL123647) and a USDA National Institute of Food and Agriculture Hatch Project (1017927). M. E. A. is supported by a Translational Obesity Fellowship from the Virginia Tech Interdisciplinary Graduate Education Program. M. O. and the team of Hospital Sant Joan de Déu of Barcelona is supported by Instituto de Salud Carlos III (ISCIII‐FIS PI17/00109), the FEDER Funding Program from the European Union, and CIBERER‐ISCIII. R. M., G. S. G., and Y. S. N. are supported by The Wellcome Trust (203105/Z/16/Z and 204709/Z/16/Z), Newcastle University Centre for Ageing and Vitality (supported by the Biotechnology and Biological Sciences Research Council and Medical Research Council L016354) UK NIHR Biomedical Research Centre for Ageing and Age‐related disease award to the Newcastle upon Tyne Hospitals NHS Foundation Trust, National Institute for Health Research (NIHR), and UK NHS Specialist Commissioners which funds the ‘Rare Mitochondrial Disorders of Adults and Children’ Service in Newcastle upon Tyne; in addition to infrastructure support from the UK Medical Research Council (MRC) Centre Mitochondrial Disease Patient Cohort: A Natural History Study and Patient Registry (REC ref 13/NE/0326), NIHR Biomedical Research Centre, Newcastle and North Tyneside Comprehensive Local Research Network. Y. S. N. holds an NIHR Clinical Lectureship in Neurology (CL‐2016‐01‐003). T. J. J. S. is supported by a grant from the Princes Beatrix Muscle Foundation (Prinses Beatrix Spierfonds (WO.R16‐19) and a Long‐Term EMBO Fellowship grant of the European Molecular Biology Organisation (ALTF 268‐2016).
References used for the drug reviews are mentioned in the summaries of the drug reviews, published as Supplementary data.
De Vries MC, Brown DA, Allen ME, et al. Safety of drug use in patients with a primary mitochondrial disease: An international Delphi‐based consensus. J Inherit Metab Dis. 2020;43:800–818. 10.1002/jimd.12196
Communicating Editor: Areeg El‐Gharbawy
Funding information European Molecular Biology Organization, Grant/Award Number: ALTF 268‐2016; Fondazione Telethon, Grant/Award Numbers: GUP09004, GSP16001; Instituto de salud carlos, Grant/Award Number: ISCIII‐FIS PI17/00109; Medical Research Council (GB), Grant/Award Number: MR/S002065/1; National Institute Food and Agriculture, Grant/Award Number: 1017927; UK Medical Research Council, Grant/Award Number: 13/NE/0326; Wellcome Trust, Grant/Award Numbers: 203105/Z/16/Z, 204709/Z/16/Z; European Molecular Biology Organisation; Prinses Beatrix Spierfonds, Grant/Award Number: WO.R17‐19; National Institute for Health Research, Grant/Award Number: CL‐2016‐01‐003; Newcastle upon Tyne Hospitals NHS Foundation Trust; Biotechnology and Biological Sciences Research Council; Newcastle University; Wellcome; European Union; Instituto de Salud Carlos III; Virginia Tech; National Institute of Food and Agriculture; National Institutes of Health, Grant/Award Number: HL123647; Medical Research Council, Grant/Award Number: L016354; Action Medical Research; Biomedical Research Centre; Muscular Dystrophy UK
REFERENCES
- 1. Rahman J, Rahman S. Mitochondrial medicine in the omics era. Lancet. 2018;391:2560‐2574. [DOI] [PubMed] [Google Scholar]
- 2. Gorman GS, Chinnery PF, DiMauro S, et al. Mitochondrial diseases. Nat Rev Dis Primers. 2016;2:16080. [DOI] [PubMed] [Google Scholar]
- 3. Parikh S, Goldstein A, Karaa A, et al. Patient care standards for primary mitochondrial disease: a consensus statement from the mitochondrial medicine society. Genet Med. 2017;19:1380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Hargreaves IP, Al Shahrani M, Wainwright L, et al. Drug‐induced mitochondrial toxicity. Drug Saf. 2016;39:661‐674. [DOI] [PubMed] [Google Scholar]
- 5. Chan K, Truong D, Shangari N, O'Brien PJ. Drug‐induced mitochondrial toxicity. Expert Opin Drug Metab Toxicol. 2005;1:655‐669. [DOI] [PubMed] [Google Scholar]
- 6. McMillan SS, King M, Tully MP. How to use the nominal group and Delphi techniques. Int J Clin Pharmacol. 2016;38:655‐662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bitner‐Glindzicz M, Rahman S. Ototoxicity caused by aminoglycosides. BMJ. 2007;335(7624):784‐785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Guan MX. Mitochondrial 12S rRNA mutations associated with aminoglycoside ototoxicity. Mitochondrion. 2011;11:237‐245. [DOI] [PubMed] [Google Scholar]
- 9. Wolf NI, Rahman S, Schmitt B, et al. Status epilepticus in children with Alpers' disease caused by POLG1 mutations: EEG and MRI features. Epilepsia. 2009;50:1596‐1607. [DOI] [PubMed] [Google Scholar]
- 10. Ng YS, Alston CL, Diodato D, et al. The clinical, biochemical and genetic features associated with RMND1‐related mitochondrial disease. J Med Genet. 2016;53:768‐775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rudnicki M, Mayr JA, Zschocke J, et al. MELAS syndrome and kidney disease without Fanconi syndrome or proteinuria: a case report. Am J Kidney Dis. 2016;68:949‐953. [DOI] [PubMed] [Google Scholar]
- 12. Seidowsky A, Hoffmann M, Glowacki F, et al. Renal involvement in MELAS syndrome—a series of 5 cases and review of the literature. Clin Nephrol. 2013;80:456‐463. [DOI] [PubMed] [Google Scholar]
- 13. Schulz M, Schmoldt A. Therapeutic and toxic blood concentrations of more than 800 drugs and other xenobiotics. Pharmazie. 2003;58:447‐474. [PubMed] [Google Scholar]
- 14. Quan Y, Xia L, Shao J, et al. Adjudin protects rodent cochlear hair cells against gentamicin ototoxicity via the SIRT3‐ROS pathway. Sci Rep. 2015;5:8181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hodiamont CJ, Janssen JM, de Jong MD, et al. Therapeutic drug monitoring of gentamicin peak concentrations in critically ill patients. TDM. 2017;39:522‐530. [DOI] [PubMed] [Google Scholar]
- 16. Oertel R, Rahn R, Kirch W. Clinical pharmacokinetics of articaine. Clin Pharmacokinet. 1997;33:417‐425. [DOI] [PubMed] [Google Scholar]
- 17. Günaydin B, Demiryürek AT. Effects of prilocaine and articaine on human leucocytes and reactive oxygen species in vitro. Acta Anaesthesiol Scand. 2001;45:741‐745. [DOI] [PubMed] [Google Scholar]
- 18. Dalmora SL, Sangoi MDS, Nogueira DR, et al. Determination of phenobarbital in human plasma by a specific liquid chromatography method: application to a bioequivalence study. Quím Nova. 2010;33:124‐129. [Google Scholar]
- 19. Santos NAG, Medina WSG, Martins NM, Mingatto FE, Curti C, Santos AC. Aromatic antiepileptic drugs and mitochondrial toxicity: effects on mitochondria isolated from rat liver. Toxicol in Vitro. 2008;22:1143‐1152. [DOI] [PubMed] [Google Scholar]
- 20. Mitchell DY, Heise MA, Pallone KA, et al. The effect of dosing regimen on the pharmacokinetics of risedronate. Br J Clin Pharmacol. 1999;48:536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yun MH, Woo JS, Kwon KI. Bioequivalence and pharmacokinetics of 70 mg alendronate sodium tablets by measuring alendronate in plasma. Arch Pharm Res. 2006;29:328‐332. [DOI] [PubMed] [Google Scholar]
- 22. Nagano Y, Matsui H, Shimokawa O, et al. Bisphosphonate‐induced gastrointestinal mucosal injury is mediated by mitochondrial superoxide production and lipid peroxidation. J Clin Biochem Nutr. 2012;51:196‐203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li J, Duan R, Zhang Y, et al. Beta‐adrenergic activation induces cardiac collapse by aggravating cardiomyocyte contractile dysfunction in bupivacaine intoxication. PloS One. 2018;13(10):e0203602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bethea JW. Clinical anesthesia. Anesthesiology. 2010;112:767‐768. [Google Scholar]
- 25. Mahmood I, Chamberlin N. A limited sampling method for the estimation of AUC and C max of carbamazepine and carbamazepine epoxide following a single and multiple dose of a sustained‐release product. Br J Clin Pharmacol. 1998;45:241‐246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pochini L, Galluccio M, Scumaci D, et al. Interaction of beta‐lactam antibiotics with the mitochondrial carnitine/acylcarnitine transporter. Chem Biol Interact. 2008;173:187‐194. [DOI] [PubMed] [Google Scholar]
- 27. Kelly JG, Doyle G, Donohue J, et al. Pharmacokinetics of enalapril in normal subjects and patients with renal impairment. Br J Clin Pharmacol. 1986;21:63‐69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Holenarsipur VK, Gaud N, Sinha J, et al. Absorption and cleavage of enalapril, a carboxyl ester prodrug, in the rat intestine: in vitro, in situ intestinal perfusion and portal vein cannulation models. Biopharm Drug Dispos. 2015;36:385‐397. [DOI] [PubMed] [Google Scholar]
- 29. Piotrkowski B, Koch OR, De Cavanagh EM, Fraga CG. Cardiac mitochondrial function and tissue remodelling are improved by a non‐antihypertensive dose of enalapril in spontaneously hypertensive rats. Free Radic Res. 2009;43:390‐399. [DOI] [PubMed] [Google Scholar]
- 30. Bonet‐Ponce L, Saez‐Atienzar S, da Casa C, et al. On the mechanism underlying ethanol‐induced mitochondrial dynamic disruption and autophagy response. Biochim Biophys Acta. 2015;1852:1400‐1409. [DOI] [PubMed] [Google Scholar]
- 31. Klockhoff H, Näslund I, Jones AW. Faster absorption of ethanol and higher peak concentration in women after gastric bypass surgery. Br J Clin Pharmacol. 2002;54:587‐591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Djafarzadeh S, Madhusudanarao V, Jeger V, et al. The effects of fentanyl on hepatic mitochondrial function. Anesth Analg. 2016;123:311‐325. [DOI] [PubMed] [Google Scholar]
- 33. Miller DB, Spence JD. Clinical pharmacokinetics of fibric acid derivatives (fibrates). Clin Pharmacokinet. 1998;34:155‐162. [DOI] [PubMed] [Google Scholar]
- 34. Abshagen U, Bablok W, Koch K, et al. Disposition pharmacokinetics of bezafibrate in man. Eur J Clin Pharmacol. 1979;16:31‐38. [DOI] [PubMed] [Google Scholar]
- 35. Bastin J, Aubey F, Rotig A, Munnich A, Djouadi F. Activation of peroxisome proliferator‐activated receptor pathway stimulates the mitochondrial respiratory chain and can correct deficiencies in patients' cells lacking its components. J Clin Endocrinol Metab. 2008;93:1433‐1441. [DOI] [PubMed] [Google Scholar]
- 36. Chen C, Han CHS, Sweeney M, Cowles VE. Pharmacokinetics, efficacy, and tolerability of a once‐daily gastroretentive dosage form of gabapentin for the treatment of postherpetic neuralgia. J Pharm Sci. 2013;102:1155‐1164. [DOI] [PubMed] [Google Scholar]
- 37. Eckland DA, Danhof M. Clinical pharmacokinetics of pioglitazone. Exp Clin Endocrinol Diabetes. 2000;108(Sup 2):234‐242. [Google Scholar]
- 38. Loi CM, Young M, Randinitis E, Vassos A, Koup JR. Clinical pharmacokinetics of troglitazone. Clin Pharmacokinet. 1999;37:91‐104. [DOI] [PubMed] [Google Scholar]
- 39. Balfour JAB, Plosker GL. Rosiglitazone. Drugs. 1999;57:921‐930. [DOI] [PubMed] [Google Scholar]
- 40. Hu D, Wu CQ, Li ZJ, et al. Characterizing the mechanism of thiazolidinedione‐induced hepatotoxicity: an in vitro model in mitochondria. Toxicol Appl Pharmacol. 2015;284:134‐141. [DOI] [PubMed] [Google Scholar]
- 41. Barrientos A, Marín C, Miró O, et al. Biochemical and molecular effects of chronic haloperidol administration on brain and muscle mitochondria of rats. J Neurosci Res. 1998;53:475‐481. [DOI] [PubMed] [Google Scholar]
- 42. Lei K, He GF, Zhang CL, et al. Investigation of the synergistic effects of haloperidol combined with Calculus Bovis Sativus in treating MK‐801‐induced schizophrenia in rats. Exp Anim. 2018;67:163‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Desai M, Tanus‐Santos JE, Li L, et al. Pharmacokinetics and QT interval pharmacodynamics of oral haloperidol in poor and extensive metabolizers of CYP2D6. Pharmacogenomics J. 2003;3:105‐113. [DOI] [PubMed] [Google Scholar]
- 44. Hanley PJ, Ray J, Brandt U, Daut J. Halothane, isoflurane and sevoflurane inhibit NADH: ubiquinone oxidoreductase (complex I) of cardiac mitochondria. J Physiol. 2002;544:687‐693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Atallah MM, Geddes IC. The gas chromatographic estimation of halothane in blood using electron capture detector unit. Br J Anaesth. 1972;44:1035‐1039. [DOI] [PubMed] [Google Scholar]
- 46. Venâncio C, Félix L, Almeida V, et al. Acute ketamine impairs mitochondrial function and promotes superoxide dismutase activity in the rat brain. Anesth Analg. 2015;120:320‐328. [DOI] [PubMed] [Google Scholar]
- 47. Wellington D, Mikaelian I, Singer L. Comparison of ketamine–xylazine and ketamine–dexmedetomidine anesthesia and intraperitoneal tolerance in rats. J Am Assoc Lab Anim. 2013;52:481‐487. [PMC free article] [PubMed] [Google Scholar]
- 48. Moaddel R, Sanghvi M, Ramamoorthy A, et al. Subchronic administration of (R, S)‐ketamine induces ketamine ring hydroxylation in Wistar rats. J Pharmaceut Biomed. 2016;127:3‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yanagihara Y, Ohtani M, Kariya S, et al. Plasma concentration profiles of ketamine and norketamine after administration of various ketamine preparations to healthy Japanese volunteers. Biopharm Drug Dispos. 2003;24:37‐43. [DOI] [PubMed] [Google Scholar]
- 50. Onizuka S, Yonaha T, Tamura R, Kasiwada M, Shirasaka T, Tsuneyoshi I. Lidocaine depolarizes the mitochondrial membrane potential by intracellular alkalization in rat dorsal root ganglion neurons. J Anesth. 2011;25:229‐239. [DOI] [PubMed] [Google Scholar]
- 51. Bobylev I, Maru H, Joshi AR, Lehmann HC. Toxicity to sensory neurons and Schwann cells in experimental linezolid‐induced peripheral neuropathy. J Antimicrob Chemother. 2016;71:685‐691. [DOI] [PubMed] [Google Scholar]
- 52. Shu Y, Brown C, Castro RA, et al. Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther. 2008;83:273‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Andrzejewski S, Gravel SP, Pollak M, St‐Pierre J. Metformin directly acts on mitochondria to alter cellular bioenergetics. Cancer Metab. 2014;2:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Link B, Haschke M, Grignaschi N, et al. Pharmacokinetics of intravenous and oral midazolam in plasma and saliva in humans: usefulness of saliva as matrix for CYP3A phenotyping. Br J Clin Pharmacol. 2008;66:473‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Colleoni M, Costa B, Gori E, Santagostino A. Biochemical characterization of the effects of the benzodiazepine, midazolam, on mitochondrial electron transfer. Pharmacol Toxicol. 1996;78:69‐76. [DOI] [PubMed] [Google Scholar]
- 56. Sandoval‐Acuña C, Lopez‐Alarcón C, Aliaga ME, Speisky H. Inhibition of mitochondrial complex I by various non‐steroidal anti‐inflammatory drugs and its protection by quercetin via a coenzyme Q‐like action. Chem Biol Interact. 2012;199:18‐28. [DOI] [PubMed] [Google Scholar]
- 57. Caille G, Du Souich P, Gervais P, Besner JG. Single dose pharmacokinetics of ketoprofen, indomethacin, and naproxen taken alone or with sucralfate. Biopharm Drug Dispos. 1987;8:173‐183. [DOI] [PubMed] [Google Scholar]
- 58. Araújo IM, Ambrósio AF, Leal EC, et al. Neurotoxicity induced by antiepileptic drugs in cultured hippocampal neurons: a comparative study between carbamazepine, oxcarbazepine, and two new putative antiepileptic drugs, BIA 2‐024 and BIA 2‐093. Epilepsia. 2004;45:1498‐1505. [DOI] [PubMed] [Google Scholar]
- 59. Tartara A, Galimberti CA, Manni R, et al. The pharmacokinetics of oxcarbazepine and its active metabolite 10‐hydroxy‐carbazepine in healthy subjects and in epileptic patients treated with phenobarbitone or valproic acid. Br J Clin Pharmacol. 1993;36:366‐368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sevilla‐Tirado FJ, Gonzalez‐Vallejo EB, Leary AC, Breedt HJ, Hyde VJ, Fernandez‐Hernando N. Bioavailability of two new formulations of paracetamol, compared with three marketed formulations, in healthy volunteers. Method Find Exp Clin. 2003;25:531‐536. [DOI] [PubMed] [Google Scholar]
- 61. Burcham PC, Harman AW. Acetaminophen toxicity results in site‐specific mitochondrial damage in isolated mouse hepatocytes. J Biol Chem. 1991;266:5049‐5054. [PubMed] [Google Scholar]
- 62. Suthisisang C, Payakachat N, Chulavatnatol S, Towanabut S. Bioavailability of phenytoin sodium capsules available in Thailand. Part II: in vivo study. J Med Assoc Thai. 1998;81:64‐70. [PubMed] [Google Scholar]
- 63. Branca D, Roberti MS, Lorenzin P, Vincenti E, Scutari G. Influence of the anesthetic 2, 6‐diisopropylphenol on the oxidative phosphorylation of isolated rat liver mitochondria. Biochem Pharmacol. 1991;42:87‐90. [DOI] [PubMed] [Google Scholar]
- 64. Khan MS, Zetterlund EL, Gréen H, et al. Pharmacogenetics, plasma concentrations, clinical signs and EEG during propofol treatment. Basic Clin Pharmacol Toxicol. 2014;115:565‐570. [DOI] [PubMed] [Google Scholar]
- 65. Modica‐Napolitano JS, Lagace CJ, Brennan WA, Aprille JR. Differential effects of typical and atypical neuroleptics on mitochondrial functionin vitro. Arch Pharm Res. 2003;26:951‐959. [DOI] [PubMed] [Google Scholar]
- 66. DeVane CL, Nemeroff CB. Clinical pharmacokinetics of quetiapine. Clin Pharmacokinet. 2001;40:509‐522. [DOI] [PubMed] [Google Scholar]
- 67. Heykants J, Huang ML, Mannens G, et al. The pharmacokinetics of risperidone in humans: a summary. J Clin Psychiatry. 1994;55:13‐17. [PubMed] [Google Scholar]
- 68. Nulton‐Persson AC, Szweda LI, Sadek HA. Inhibition of cardiac mitochondrial respiration by salicylic acid and acetylsalicylate. J Cardiovasc Pharmacol. 2004;44:591‐595. [DOI] [PubMed] [Google Scholar]
- 69. Nagelschmitz J, Blunck M, Kraetzschmar J, Ludwig M, Wensing G, Hohlfeld T. Pharmacokinetics and pharmacodynamics of acetylsalicylic acid after intravenous and oral administration to healthy volunteers. Clin Pharmacol. 2014;6:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Bellosta S, Paoletti R, Corsini A. Safety of statins: focus on clinical pharmacokinetics and drug interactions. Circulation. 2004;109(23_suppl_1):III‐50. [DOI] [PubMed] [Google Scholar]
- 71. Schirris TJ, Renkema GH, Ritschel T, et al. Statin‐induced myopathy is associated with mitochondrial complex III inhibition. Cell Metab. 2015;22:399‐407. [DOI] [PubMed] [Google Scholar]
- 72. Kudin AP, Debska‐Vielhaber G, Vielhaber S, Elger CE, Kunz WS. The mechanism of neuroprotection by topiramate in an animal model of epilepsy. Epilepsia. 2004;45:1478‐1487. [DOI] [PubMed] [Google Scholar]
- 73. Doose DR, Walker SA, Gisclon LG, Nayak RK. Single‐dose pharmacokinetics and effect of food on the bioavailability of topiramate, a novel antiepileptic drug. J Clin Pharmacol. 1996;36:884‐891. [DOI] [PubMed] [Google Scholar]
- 74. Matar KM, Tayem YI. Effect of experimentally induced hepatic and renal failure on the pharmacokinetics of topiramate in rats. BioMed Res Int. 2014;2014:570910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Nunez DA, Schiaffino S, Roldán EJA. Comparison of two formulations of sodium valproate plasma concentrations after a single 500 mg oral dose in healthy subjects, and stochastic sub‐analysis of the individual “clinical perceptible” levels. J Bioequiv Availab. 2013;5:197‐200. [Google Scholar]
- 76. Jafarian I, Eskandari MR, Mashayekhi V, Ahadpour M, Hosseini MJ. Toxicity of valproic acid in isolated rat liver mitochondria. Toxicol Mech Methods. 2013;23:617‐623. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1: Supplementary data