

Abstract
Camptothecin (CPT) derivatives are clinically effective poisons of DNA topoisomerase I (Top1) able to form a ternary complex with the Top1–DNA complex. The aim of this investigation was to examine the dynamic aspects of the ternary complex formation by means of site-directed spin labeling electron paramagnetic resonance (SDSL-EPR). Two semisynthetic CPT derivatives bearing the paramagnetic moiety were synthesized, and their biological activity was tested. A 22-mer DNA oligonucleotide sequence with high affinity cleavage site for Top1 was also synthesized. EPR experiments were carried out on modified CPT in the presence of DNA, of Top1, or of both. In the last case, a slow motion component in the EPR signal appeared, indicating the formation of the ternary complex. Deconvolution of the EPR spectrum allowed to obtain the relative drug amounts in the complex. It was also possible to demonstrate that the residence time of CPT “trapped” in the ternary complex is longer than hundreds of microseconds.
Introduction
DNA topoisomerase I (Top1a) can promote changes of DNA topology by altering the linking number of the double helix1–3 through a mechanism which involves transient DNA strand breakage and religation. After binding to the double helix Top1 transiently cleaves one strand, via a nucleophilic attack of a tyrosine residue that becomes covalently linked to the 3′ end of the cut strand. This arrangement serves to maintain the energy of the original phosphodiester backbone bond and creates a protein-linked breakage in the DNA. Then, removal of the torsional tension is achieved by rotation of the free 5′ end of the cut strand around the intact one. Finally, the reaction cycle is terminated by religation of the cut strand, restoring DNA integrity. Because of the crucial role in cellular replication process, several efforts have been made to design and synthesize potential Top1 poisons. Camptothecin (CPT),4,5 a natural alkaloid extracted from the bark of the Camptotheca acuminata tree,6 is the archetype of Top1 interfacial inhibitors.7,8 CPT and its analogues interact with the Top1–DNA cleavable intermediate, thus forming a CPT–DNA–Top1 ternary cleavable complex.5,8–11 X-ray diffraction experiments10,12 clarified many details of the ternary complex structure in terms of the amino acids involved in the ternary interaction. In the presence of CPT, cleavage induced by Top1 is not affected, although the religation step is inhibited (or significantly slowed down) due to misalignment of the 5′-hydroxyl group and the scissile tyrosine–DNA linkage.4,8,10 This process leads to the accumulation of single-stranded DNA breaks. Collision of DNA replication forks with the ternary complexes produce irreversible double-strand ends, which ultimately leads to cell death.4 Recent work by Koster et al.13,14 also showed how the damage could be ascribed to the accumulation of positive supercoils in the replicating DNA. In the presence of CPT, Top1 relaxes negative supercoils faster than the positive ones, suggesting that the poison inhibits preferentially the relaxation of positive compared to negative supercoils. However, the dynamical interactions of the drug within the ternary complex are not fully known. Thus, we planned to explore the dynamical properties of the ternary Top1 complex with site-directed spin-labeling electron paramagnetic resonance (SDSL-EPR) spectroscopy that constitutes a novel and powerful approach to study protein complexes of RNA/DNA.15–21 SDSL-EPR allows the study of large protein/DNA complexes in solution without being limited by the complex dimensions. The utilization of paramagnetic labels enables the specific probing of regions of interest without altering the structure and the function of the object due to the limited steric hindrance of the probes. A large body of information can be obtained from SDSL-EPR.22 Our present work mainly focuses on revealing and analyzing the changes in the EPR spectra of labeled compounds determined by the formation of the ternary complex. CPT derivatives as well as DNA oligonucleotides both bearing the 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) paramagnetic probe were synthesized. These derivatives were then tested and evaluated to obtain the best candidate for the final EPR experiments.
Results and Discussion
Chemistry.
The first step in our synthetic strategy was the selection of a proper paramagnetic probe among all the possibilities.23 We decided to investigate the 4-amino/2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPOamine, 6) spin probe due to the high stability of its paramagnetic nitroxide function and to the reactivity of the primary amine present in the 4 position of its ring. The spin probe was synthesized according to a reported procedure,24 improving the oxime reduction step, as depicted in Scheme 1. Reaction of commercially available triacetonamine (1) with hydroxylamine hydrochloride gave the oxime (2), which was then reduced to the corresponding amine (3) in presence of LiAlH4. Acetylation of the resulting primary amine with acetic anhydride allowed the oxidation of the tetramethylpiperidine function in the presence of sodium tungstate, hydrogen peroxide, and EDTA-disodium salt.24 A final base catalyzed hydrolysis of the 4-acetamido group provided the required TEMPOamine derivative (6).
Scheme 1.
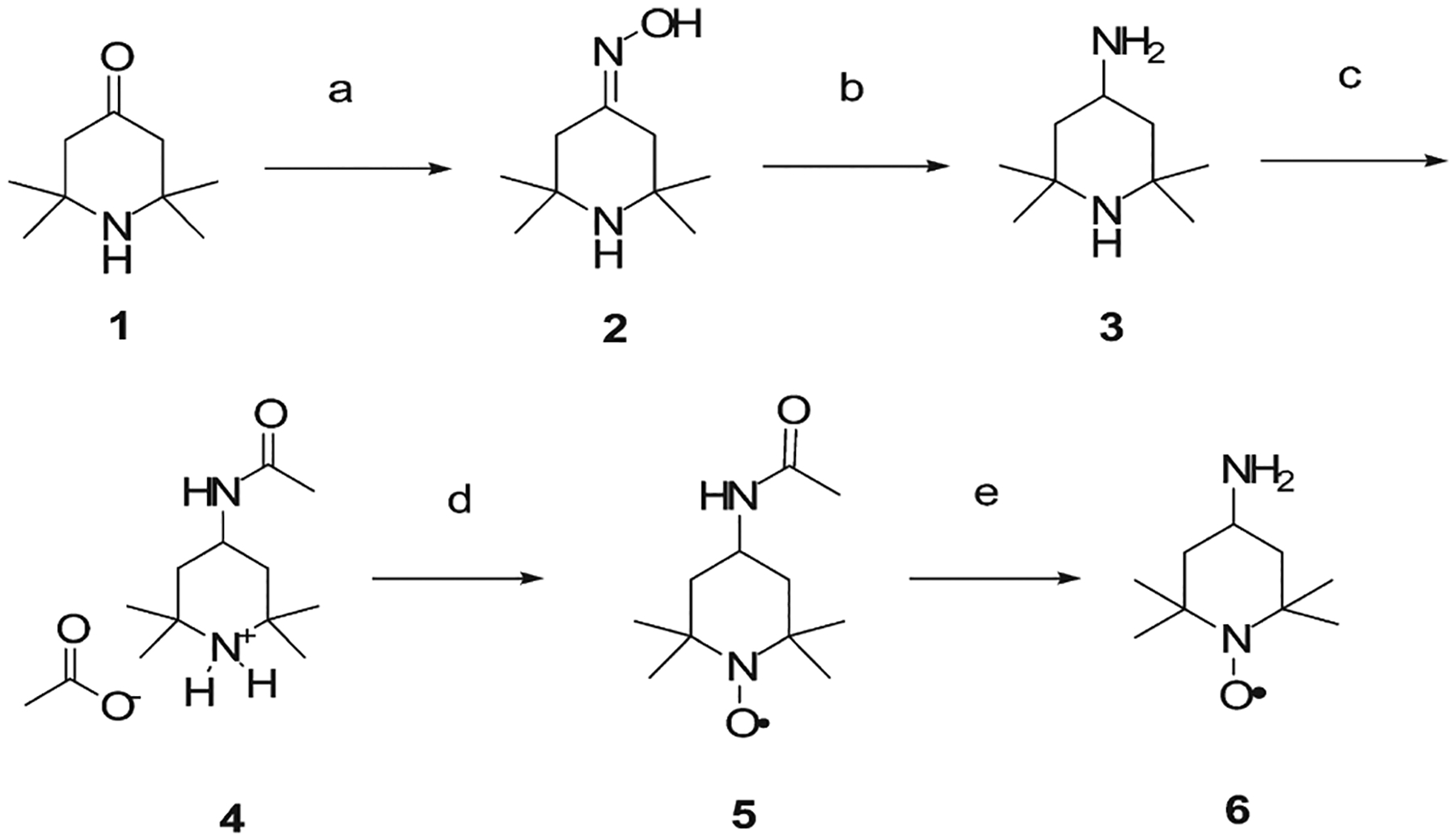
Synthesis Procedure for the Preparation of the 4-Amino-2,2,6,6-tetramethylpiperidin-1-oxyl Probea
a (a) NH2OH·HCl, sodium acetate, H2O; (b) LiAlH4, THF; (c) Ac2O, Et2O; (d) H2O2, Na2WO4, EDTA·2Na+, Na2CO3, H2O, RT, 2 days; (e) KOH 15% in H2O.
This investigation required a CPT skeleton based molecule (i) bearing a paramagnetic moiety linked with a rigid bond and (ii) retaining its poisonous activity for Top1. Previous work25–27 showed that bulky substituents could be inserted in position 7 of the quinoline ring of the CPT; this was successfully accomplished by rigid imino linkage, which did not affect the activity against the binary complex. Moreover, the mobility of the nitroxide label attached to this position should be greatly affected by the formation of the ternary complex. For these reasons, we exploited the amino group of the TEMPOamine probe to synthesize a 7-imino derivative of CPT. In addition, an amidic bond was further chosen to load the CPT with the spin probe, although this substitution could produce a less potent derivative.28 The syntheses of CPT derivatives are depicted in Scheme 2. Commercially available CPT is converted to the 7-dimethoxymethyl CPT (7) through a Minisci radical hydroxymethylation,29 followed by in situ oxidation with manganese dioxide. Hydrolysis of the obtained acetal with acetic acid gave the corresponding 7-formyl CPT (8), which was then converted into the desired 7-TEMPO imino CPT (9) with 6 in the presence of a Lewis acid such as ytterbium triflate [Yb(OTf)3].25 Further oxidation of 8 with hydrogen peroxide and formic acid provided the 7-carboxy CPT (10); standard FMOC-peptide coupling via HBTU/HOBt between 6 and 10 leads to the 7-TEMPO amido CPT derivative (11).
Scheme 2.
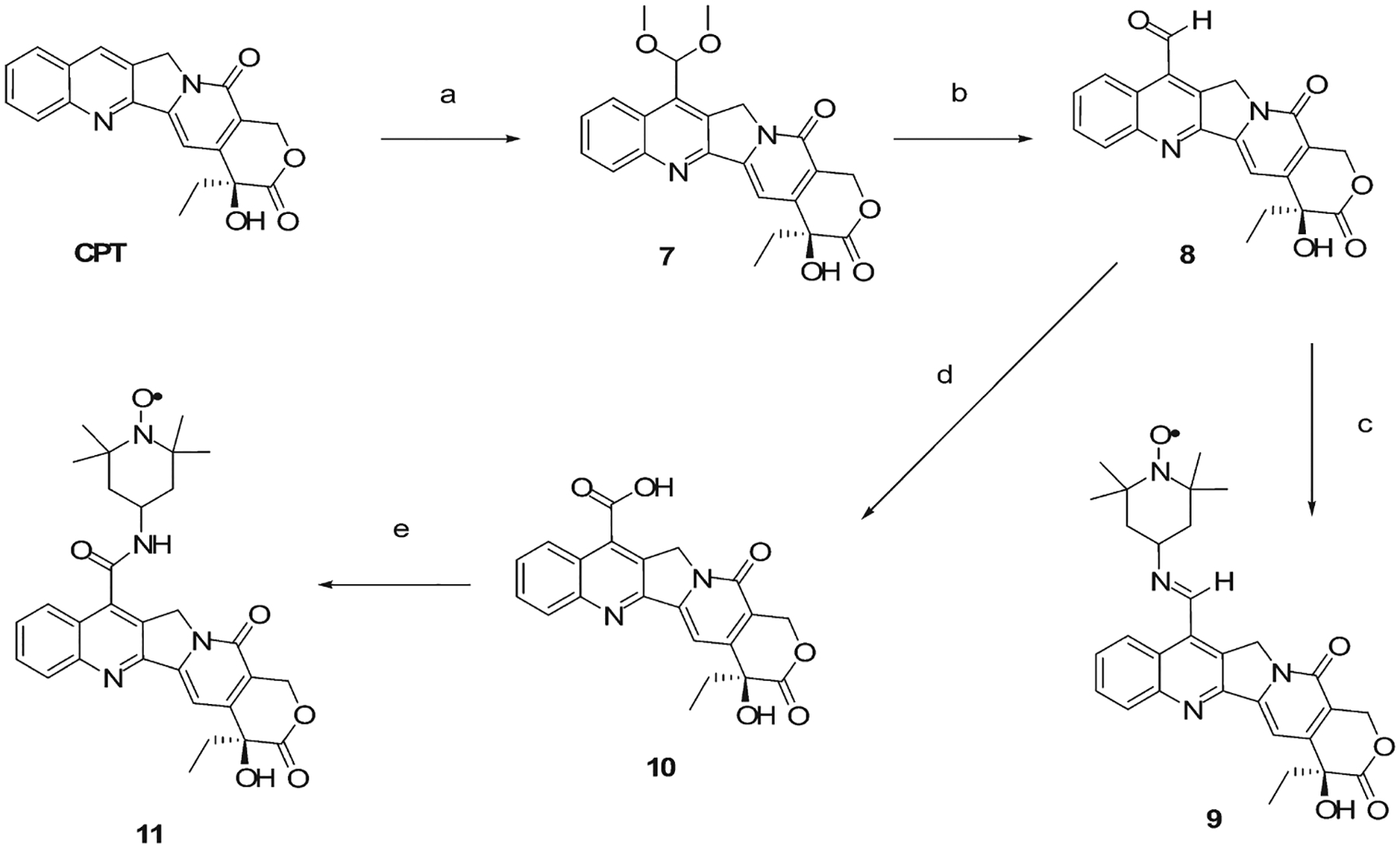
Synthesis Routes of the TEMPO-Substituted Campthotecin Derivativesa
a (a) H2SO4, MeOH, FeSO4, H2O2 30%, then Mn2O, RT, 2 h (yield 74%); (b) CH3COOH, H2O, reflux, 1 h (yield 75%); (c) TEMPOamine (6), ytterbium triflate, molecular sieves 4 Å, CH2Cl2, RT, 16 h (yield 22%); (d) HCOOH, H2O2 30%, 0 °C, 16 h (yield 75%); (e) 6, HBTU, HOBt, DIPEA, THF, RT, 16 h (yield 67%).
To generate a ternary complex, we also needed a suitable DNA substrate possessing: a minimal length and a high affinity site recognized and processed by the Top1. Previous works showed that the 22-mer DNA oligonucleotide 5′-AAAAAGACTT↓GGAAAAATTTTT and its complementary sequence bore an high affinity cleavage site (marked with a down arrow) for the Top1 enzyme.7,8,30,31
The syntheses of unmodified and modified 22-mer oligonucleotides were performed automatically via phosphite triester approach synthesis32 on 1.0 μmol scale using a controlled pore glass (CPG) resin previously functionalized with the first nucleoside. The strand syntheses direction were always from the 3′ terminus to the 5′ terminus, and all the coupling reactions gave a 98.5% average yield measured from the trityl carbocation released during the deprotection step. All the synthetic details are extensively described in the Supporting Information.
In an attempt to increase the number of useful tools for the investigation of the ternary complex, two TEMPO substituted nucleosides, a cytidine and an adenine derivative, were synthesized and inserted inside the previous oligonucleotide sequences, in the proximity of the cleavage site, to measure the internitroxide distance with continuous wave (CW) or pulsed EPR experiments. Also two singly labeled DNA were synthesized for comparative purposes.
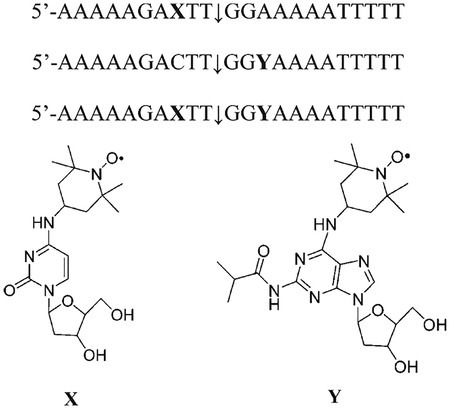
Substitution in different positions were chosen on the basis of two main considerations: (1) substitutions at the +1 or −1 position with respect to the cleavage point are known to affect the Top1 activity and poisoning of Top1 by CPT;7 (2) for doubly labeled strands, the interprobe distance r should be in the range of sensitivity of either CW EPR (0.8 nm < r < 2 nm) or pulsed EPR (2 nm < r < 6 nm). TEMPO-substituted phosphoramidites and TEMPO-substituted oligonucleotides syntheses schemes and procedures are discussed in detail in the Supporting Information.
Top1-Mediated DNA Cleavage Reactions.
Compounds 9 and 11 were evaluated by using a normal 22-mer DNA oligonucleotide as substrate for the recombinant human Top1. CPT was the reference compound. The inhibition data are expressed semiquantitatively as follows: 0, no cleavage stimulation; +, weak activity (between 20% and 50% of CPT cleavage activity); ++, modest activity (between 50% and 75% of CPT cleavage activity); +++, good activity (between 75% and 100% of CPT cleavage activity). All drugs were tested at several concentrations (100, 10, 1, 0.1 μM).
Compound 9 (Top1 +++) shows excellent Top1 inhibition activity (Figure 1), more or less comparable to that of CPT, in agreement with published data,25–27 showing that substituents at position 7 do not impair drug activity. The preference for the prominent cleavage site is maintained. The activity of compound 11 (Top1, +) is weaker in comparison with CPT and compound 9 (Figure 1).
Figure 1.
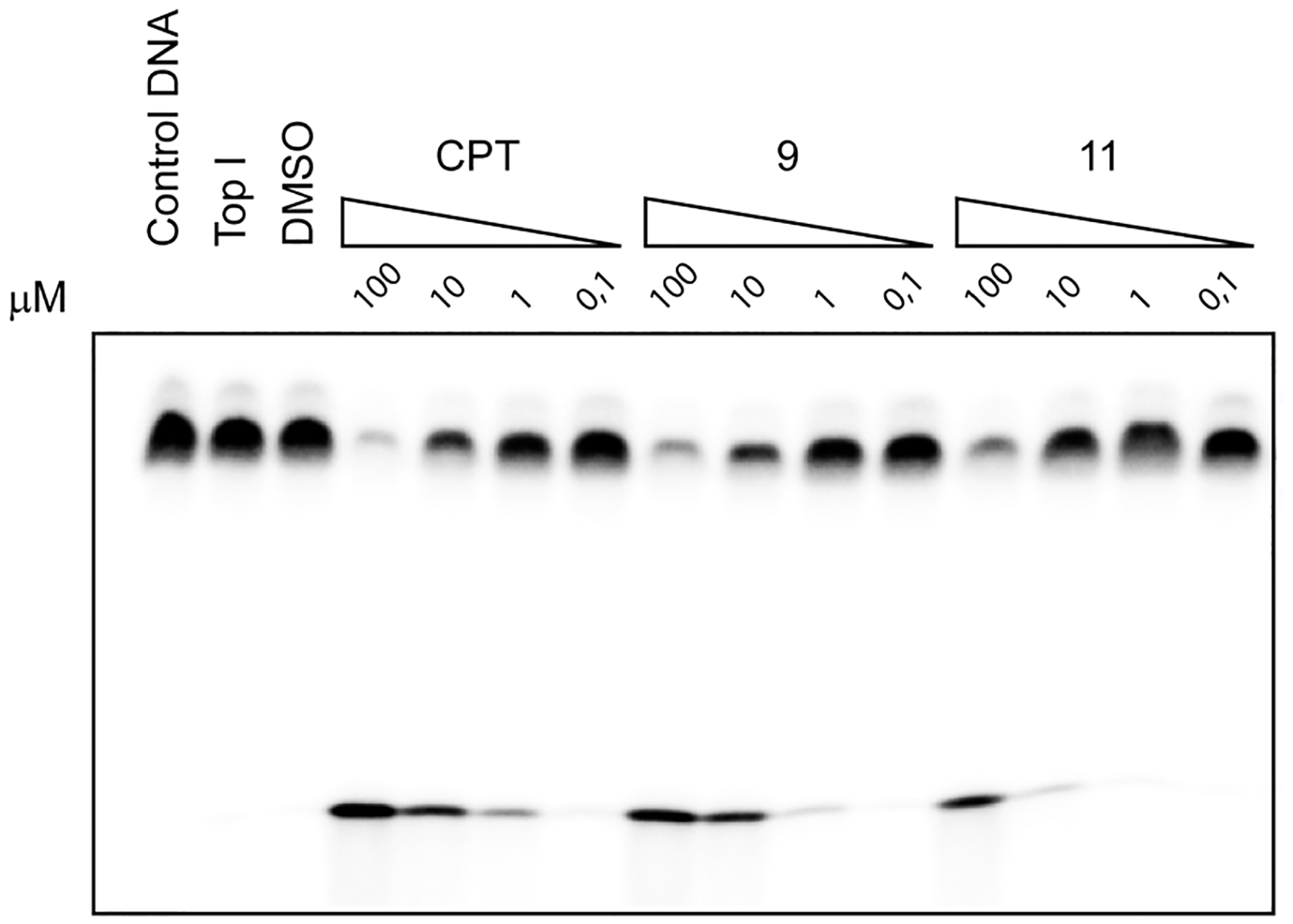
Top1-mediated DNA cleavage induced by CPT, compound 9, and compound 11. First lane, DNA substrate alone; second lane, DNA substrate in the presence of Top1; third lane, DNA substrate in the presence of Top1 and DMSO (drug solvent). Other lanes show the results obtained with three different compounds at the indicated concentrations (100, 10, 1, and 0.1 μM).
DNA oligonucleotides bearing the paramagnetic probe were tested as plausible substrates for Top1 activity. Interestingly, DNA cleavage stimulated by compound 9 was less reversible than that promoted by CPT (data not shown).
Figure 2 shows that the TEMPO-adenine strand is a good substrate for the cleavage reaction. Conversely, the insertion of a TEMPO substitution on cytidine as well as the double substituted oligonucleotide results in completely inadequate substrates, which is consistent with biochemical analyses carried out in oligonucleotides.26,33
Figure 2.
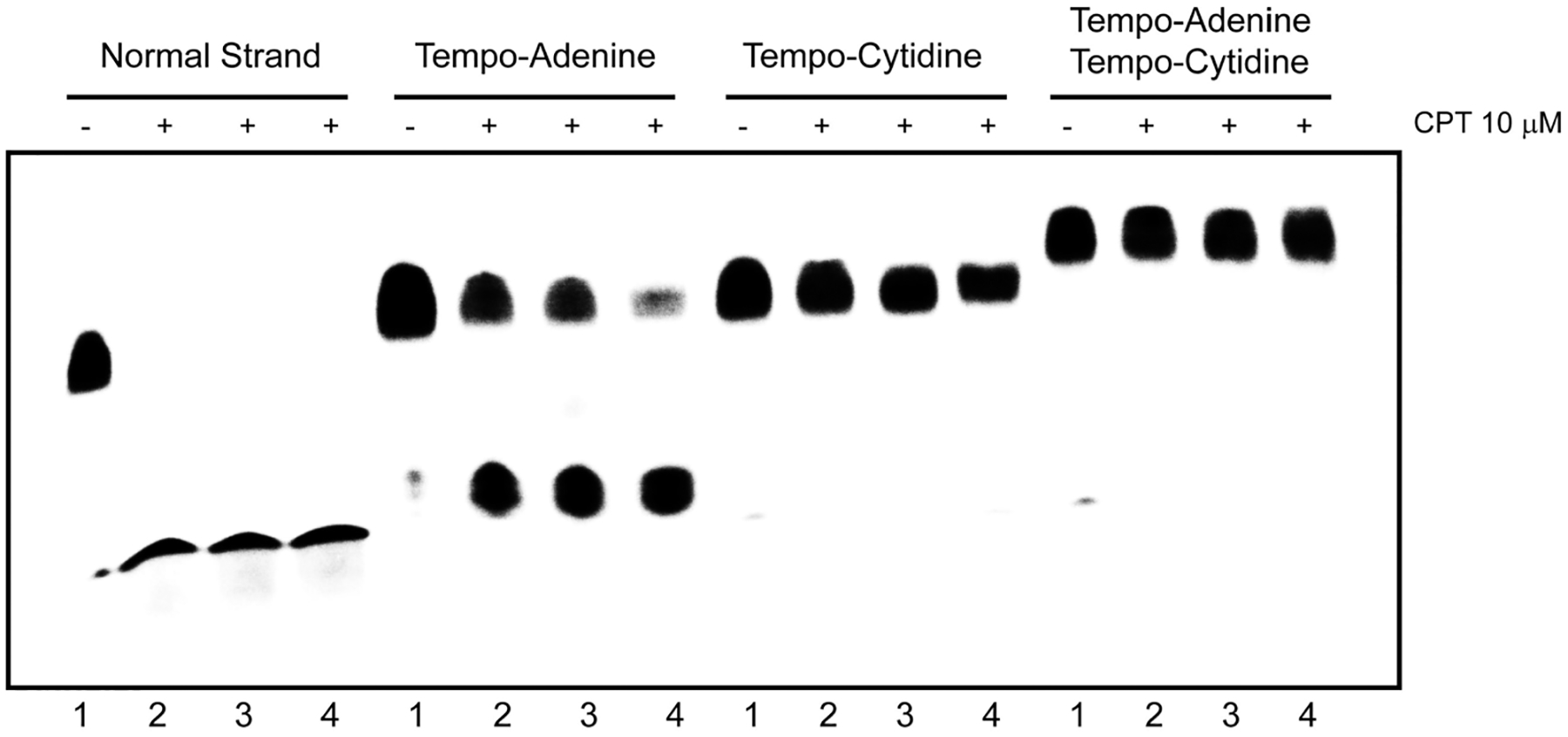
Top1-mediated DNA cleavage induced by 10 μM CPT on DNA oligonucleotides bearing the 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) paramagnetic probe. Four different concentration of enzyme were tested (lane 1, without enzyme; lane 2, dilution 1:20; lane 3, dilution 1:10; lane 4, dilution 1:2).
Our data demonstrate that the synthesized CPT derivatives could represent useful tools for the study of Top1 cm3 by EPR. On the contrary, double-substituted TEMPO-oligonucleotides and oligonucleotides containing the TEMPO moiety on the 3′ side of DNA cleavage site were unsuitable substrates for Top1.
EPR Experiments.
Preliminary EPR experiments performed on derivatives 9 and 11 confirmed the presence of the nitroxide function on both CPT derivatives (data not shown). As stated before, compound 9 was chosen for further EPR experiments as it demonstrated the highest activity (vide infra).
Figure 3 shows the CW-EPR spectra of 9 alone (A), in the presence of 5-fold excess DNA (B), and in the presence of equimolar amounts of Top1 (C); (D) is the spectrum of 9 in the presence of both DNA and Top1 in a 9:DNA:Top1 1:5:1 molar ratio.
Figure 3.
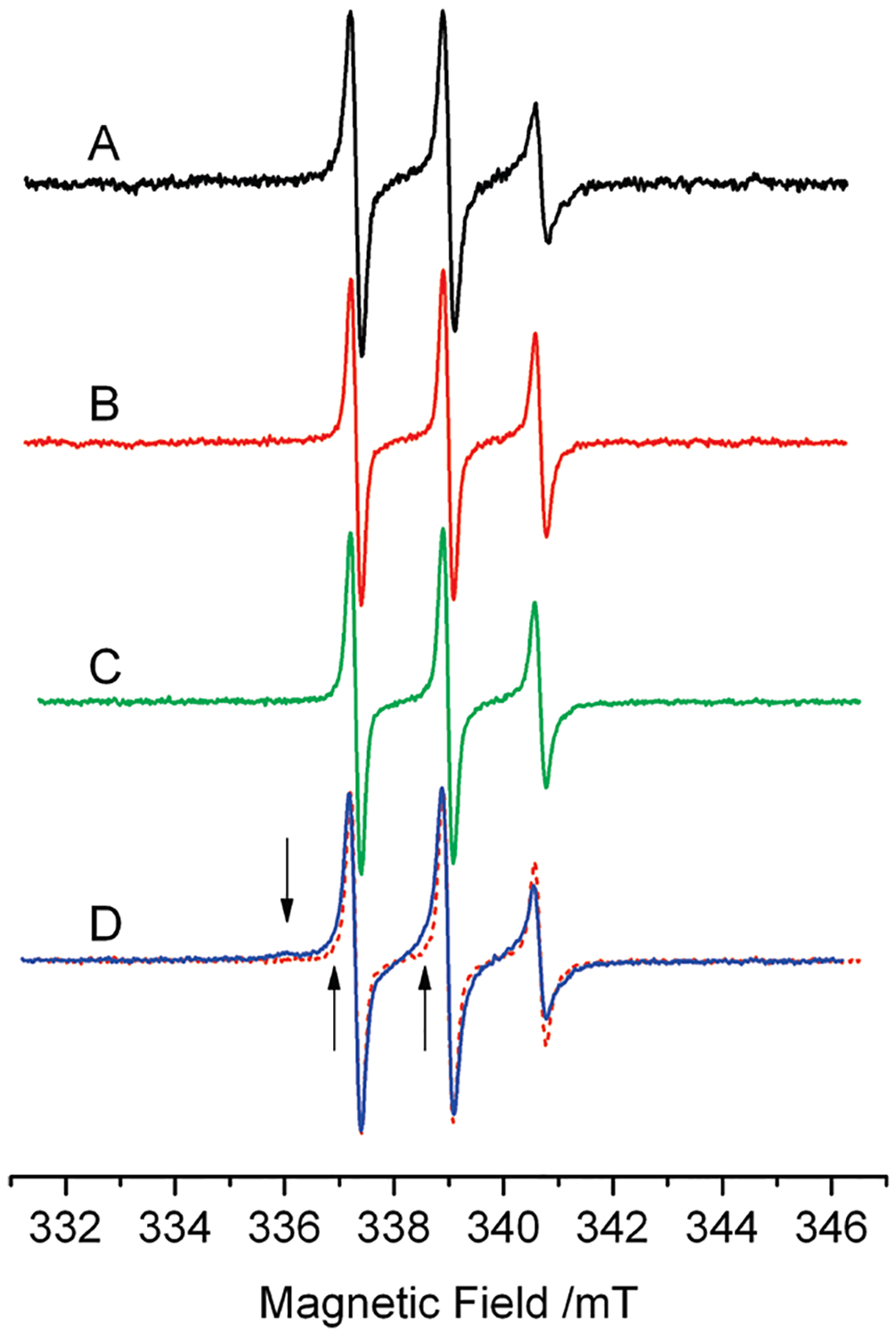
EPR spectra of 20 μM 9 at 277 K. (A) 9 in buffer. (B) 9: DNA at molar ratio 1:5. (C) 9:Top1 at molar ratio 1:1. (D) 9:DNA: Top1 at molar ratio 1:5:1; this spectrum shows an additional spectral component, absent in spectra A–C, highlighted by the arrows and by the superposition of spectrum B in dashed line.
The spectra in Figure 3A–C are typical of a spin-labeled molecule freely tumbling in viscous solution; these spectra were simulated considering an anisotropic axial motion, and the fitting gave a rotational correlation time τ = 1.2 ns. Therefore, 9 shows no effective interactions with either DNA or Top1 alone (Figure 3B–C), at the molar ratios used in this study, as was previously reported for the unmodified camptotecin.34
The line shape of 9 changes significantly in the presence of both DNA and Top1 (Figure 3D); apparently the line width increases and additional features appear in the spectrum as indicated by the arrows in Figure 3D. Here is also reported, for comparison, the spectrum of 9:DNA in the dashed line to highlight these additional features. These features arise from the presence of a second spectral component characterized by a correlation time much longer than that of 9 free in solution. The line shape of the slow component was obtained from a weighed subtraction of the line shape of 9 free in solution (Figure 3A) from the spectrum in Figure 3D; the simulation of the resulting line shape yielded the broad spectrum in Figure 4B. This component is typical of a molecule in slow, hindered anisotropic motion;35 we obtained from the simulation a rotational correlation time τ = 17 ns, about 10 times the correlation time found for the molecule free in solution. This result identifies the second component with the fraction of 9 participating in the ternary complex; the mobility of 9 is strongly reduced by the interactions with other residues from the protein or DNA.36 The relative amounts of the two components in the spectrum of the ternary mixture were obtained by fitting the experimental spectrum with a weighed sum of the simulations of the two components (Figure 4A). The relative amount of 9 in the ternary complex was 40 ± 10% of the total at the steady state, while the remaining 60 ± 10% was free in solution.
Figure 4.
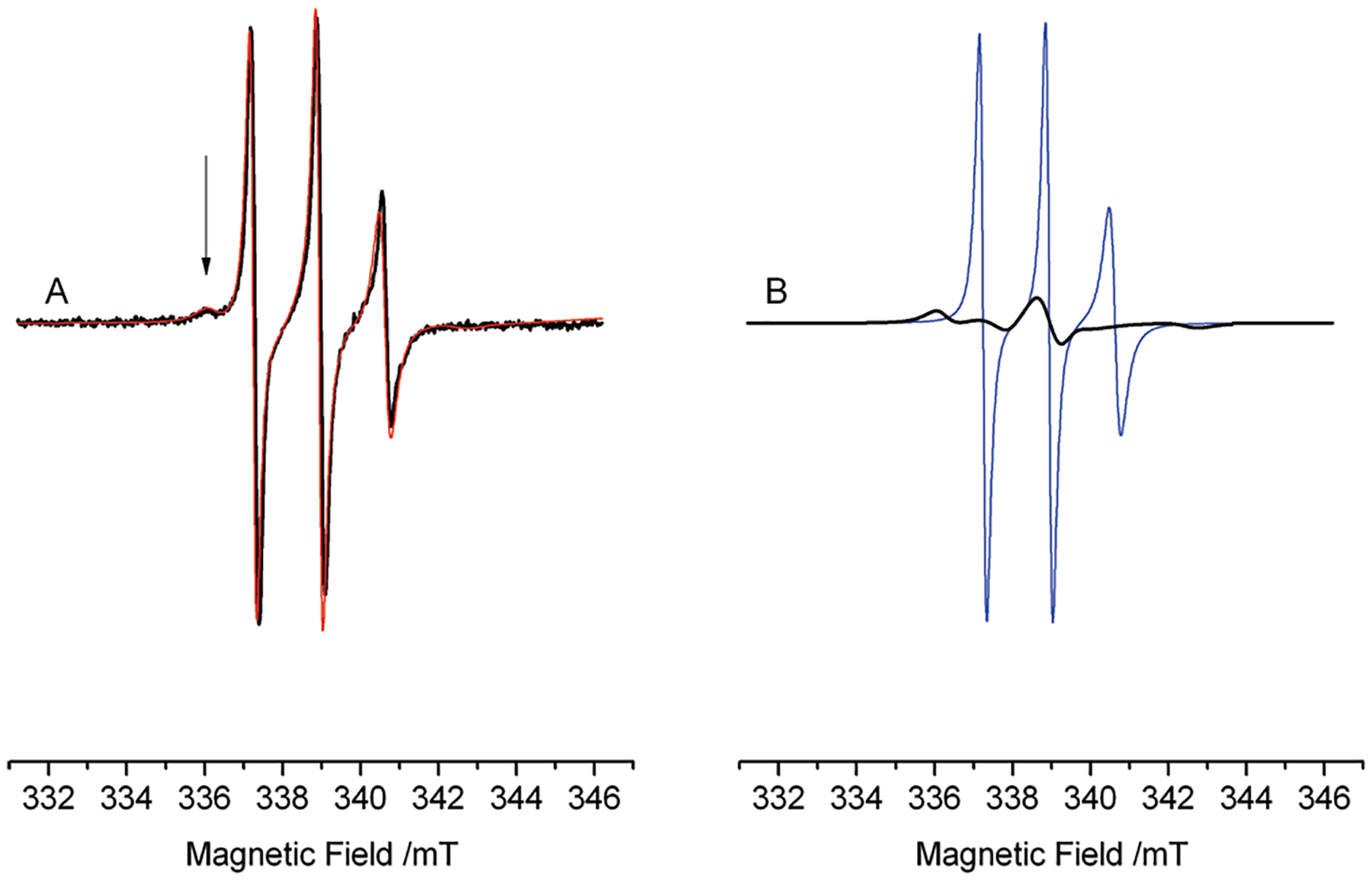
(A) EPR spectrum of the ternary complex (Figure 3D), together with its simulation (thin red line) obtained as a sum of a component arising from 9 in the ternary complex (40%) and a component arising from 9 free in solution (60%). (B) Individual components of the simulation in (A): 9 free in solution (thin blue line) and in the ternary complex (thick black line).
The presence of two individual components implies that 9 does not quickly exchange (on the EPR time scale) between the ternary complex and the solution, therefore the mean residence time of the drug in the complex must be longer than hundreds of microseconds. This lower time limit depends on the CW-EPR sensitivity to molecular motions that modulate the magnetic interactions. The residence time of CPT in the ternary complex that we estimate from the EPR spectra is compatible with the kinetics of anthracycline–DNA interaction,37,38 which shows times on the order of milliseconds to seconds. Moreover, our data is not in contrast with the time reported in ref 14. (Δt = 121 ± 11 s) for the Topotecan–DNA–Top1 complex; we wish to note that the EPR data implies that, even though the complete DNA uncoiling might be due to a series of fast successive steps, these cannot be faster than hundreds of microseconds.
Conclusions
In summary, we used for the first time the site-directed spin labeling electron paramagnetic resonance (SDSL-EPR) to reveal further information about the ternary complex between CPT, DNA, and Top1. This goal was attained through the preparation of two semisynthetic CPT derivatives functionalized in the 7-position with the paramagnetic TEMPO-amine probe. In particular, the 7-iminoCPT derivative (9) showed good poisonous ability against Top1 activity. Furthermore, a 22-mer DNA sequence with a high affinity cleavage for Top1 was produced via solid phase oligonucleotide synthesis. Two single and a doubly modified DNA oligonucleotides were also synthesized to gain potential information but, unfortunately, two of them were not processed by the enzyme.
EPR experiments were initially performed in the presence of 9 with DNA alone and Top1 alone showing the inability of the alkaloid to interact with the different substrates. When the EPR experiments were carried out in the presence of all three components, a general broadening of the spectrum was observed and a second component in the EPR signal appeared. This new signal could be due to the reduced motion of the paramagnetic probe inside the ternary complex. The deconvolution of the EPR spectrum allowed us to obtain the relative drug amounts in the complex or not free in solution, which was 40 ± 10%. The presence of two components implies also a slow exchange between TEMPO–CPT stabilized into the complex and free in solution. Finally, these data confirm how the EPR technique could be used as a potential new tool for the investigation of the interaction between complex macromolecules and small substrates because it is able to simplify complex systems enlightening only the desired hotspot region.
Experimental Section
Chemistry General Information.
All water and/or air sensitive reactions were carried out in oven-dried glassware under a nitrogen atmosphere with dry solvents, unless otherwise noted. All commercial reagents were purchased from Sigma-Aldrich and were used without further purification. All the phosphoramidites were purchased from Glen Research (22825 Davis Drive, Sterling, Virginia, 20164). The bottles containing the desired phosphoramidites were open and dried under vacuum overnight before preparing the coupling solution. The solvents were purchased from Fluka, Carlo Erba, and Glen Research; anhydrous solvents were therefore obtained as follows: tetrahydrofuran (THF) was distilled from sodium with benzophenone, pyridine was distilled over KOH, and dichloromethane (CH2Cl2) and acetonitrile (CH3CN) were distilled from calcium hydride. Reactions were usually monitored by thin-layer chromatography (TLC) carried out on 0.25 mm Merck glass plates (60 F254) using UV light at 254 nm as visualizing agent and an ethanolic solution of phosphomolybdic acid or ninhydrin as developing agents when required. Merck silica gel (230–400 mesh) was used for silica gel chromatography. 1H NMR and 31P NMR spectra were recorded on a Bruker Avance AMX 300 spectrometer. Chemical shifts (δ) were expressed in parts per million relative to tetramethylsilane (ppm), and the following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, dd = doublet doublets, t = triplet, dt = doublet triplets, tt = triplet triplets, q = quartet, m = multiplet, bm = broad multiplet bs = broad singlet. IR spectra were recorded on a Perkin-Elmer 1760 infrared Fourier transform spectrometer. Mass spectrometric data of small molecules were obtained using a Mariner API-TOF (Perseptive Biosystems Inc., Framingham MA 01701, USA), while mass spectra of oligonucleotides were obtained with an Applied Biosystem Voyager System 2081 MALDI-TOF mass spectrometer. Purities (>97%) of all tested compounds were established by HPLC using a Varian pro star equipped with a double pump, an UV–vis detector (BioRad), a Phenomenex C18 (4.6 mm × 250 mm, 5 μm) column, and H2O and CH3CN as eluents. A gradient from 5% to 95% of CH3CN over 30 min followed by 5 min more at 95% CH3CN percentage was used at a flow rate of 1.00 mL/min. The signal was detected with an UV–vis detector fixed at 254 nm.
Synthesis of 7-Dimethoxymethyl Camptothecin (7).39
A suspension of 347 mg (1 mmol) of camptothecin (B) in 21 mL of methanol was mixed with 2.1 mL of conc H2SO4 and 278 mg (1 mmol) of ferrous sulfate heptahydrate. The resulting yellow solution was heated to 50°C before the slow addition of 1 mL (9.19 mmol) of hydrogen peroxide (30% in water). The reaction was monitored by TLC (eluent CH2Cl2:MeOH 95:5) until the disappearance of the starting material; 130 mg (1.49 mmol) of manganese dioxide were then added to the mixture. The black suspension was stirred for 2 h at RT, filtered, and concentrated to ⅓ of the starting volume. Water (25 mL) was added to the residue, and the acid solution was neutralized with a 35% NaOH aqueous solution. The yellow precipitated formed was collected, rinsed with water, and dried to obtain 315 mg (0.74 mmol) of 7, which was used in the next step without further purification. An analytical sample was obtained by silica gel chromatography in order to provide complete characterization (eluent CH2Cl2: MeOH 97:3). 1H NMR (CDCl3, 300 MHz) δ (ppm): 8.31 (m, 2H), 7.80 (t, 1H, J = 8.3 Hz), 7.66 (m, 2H), 6.24 (s, 1H), 5.47 (s, 2H), 5.78 (d, 1H, J = 15.8 Hz) 5.28 (d, 1H, J = 15.8 Hz), 3.43 (s, 3H), 3.40 (s, 3H), 1.92 (m, 2H), 1.03 (t, 3H, J = 7.2). HRMS (ESI): calcd for C23H23N2O6 (M + H+) 423.1551, found 423.1558.
Synthesis of 7-Formyl Camptothecin (8).
Crude 7(300 mg, 0.71 mmol) was dissolved into 10 mL of an acetic acid/water mixture (3:2 ratio), and the solution was heated to reflux. After 30 min, the formation of a suspension was observed; after 1 h, the mixture was allowed to cool to RT, and the precipitate was collected, washed with water, and purified by silica gel chromatography (eluent CH2Cl2:MeOH 97:3). 8 (215 mg, 0.57 mmol) was obtained as a yellow solid with a 75% yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 11.20 (s, 1H), 8.77 (d, 1H, J = 8.1 Hz), 8.38 (d, 1H, J = 8.3 Hz), 7.90 (m, 2H) 7.33 (s, 1H), 5.62 (s, 2H), 5.70 (d, 1H, J = 16 Hz) 5.20 (d, 1H, J = 16 Hz), 1.90 (m, 2H), 1.05 (t, 3H, J = 7.3 Hz). IR (KBr): 1747 (v C=O, lactone), 1653 (v C=O, amide), 1590 (v C=O, aldehyde). HRMS (ESI): calcd for C21H17N2O5 (M + H+) 377.1132, found 377.1121.
Synthesis of 7-(1-Oxyl-2,2,6,6-tetramethylpiperidin-4-aminyl)-iminomethyl Camptothecin (9).
In a oven-dried flask fitted with molecular sieves (4 Å) were suspended 50 mg (0.13 mmol) of 8 into 5 mL of dry CH2Cl2; 8 mg (0.01 mmol) of ytterbium triflate and 26 mg (0.15 mmol) of 6 were further added on. The mixture was allowed to react for 48 h at RT under N2 atmosphere, after which the sieves where removed by filtration. The filter was washed with CH2Cl2, and all the organic solution were collected, washed with water, dried (Na2SO4), and finally purified by silica gel chromatography (eluent CH2Cl2:MeOH: Et2O 96:2:2). 10 (15 mg, 0.03 mmol) was obtained as a yellow solid. Yield 22%. 1H NMR (CDCl3, 300 MHz) δ (ppm): 9.69 (bs, 1H), 8.73 (bs, 1H), 8.26 (bs, 1H), 7.93 (m, 1H), 7.78 (m, 1H), 7.50 (s, 1H), 5.60 (s, 2H), 5.70–5.20 (m, 2H), 1.90 (m, 2H), 1.04 (bs, 3H), from −15 to −35 (bm, 16H). IR (KBr): 2973 (vas TEMPOCH3), 2936 (vs TEMPOCH3), 1746 (v C=O, lactone), 1658 (v C=O, amide), 1606 (v C=N, imine), 1459–1363 (δ TEMPOCH3).
Synthesis of 7-Carboxy Camptothecin (10).
8 (100 mg, 0.27 mmol) was dissolved into 1.3 mL of formic acid. The red mixture was cooled to 0–4 °C (ice bath), added with 41 μL of hydrogen peroxide and left at the same temperature overnight. The yellow suspension was filtered off, rinsed with water, and dried (Na2SO4) to give 80 mg (0.20 mmol) of 10 as a bright-yellow solid. Yield 75%. 1H NMR (CDCl3, 300 MHz) δ (ppm): 13.8 (bs, 1H), 8.73 (d, J = 7.8 Hz 1H), 8.26 (d, J = 7.9 Hz, 1H), 7.93–7.78 (m, 2H), 7.50 (s, 1H), 5.71 (d, 1H, J = 16.2 Hz), 5.60 (s, 2H), 5.23 (d, 1H, J = 16.2 Hz), 1.90 (m, 2H), 1.04 (bs, 3H). HRMS (ESI): calcd for C21H16N2O7 (M − H+) 391.0935, found 391.0936.
Synthesis of 7-(1-Oxyl-2,2,6,6-tetramethylpiperidin-4-aminyl)-amidomethyl Camptothecin (11).
A round-bottomed flask was charged with a suspension of 15 mg (0.04 mmol) of 10 in 5 mL of freshly distilled THF. HBTU (17 mg, 0.045 mmol) was then added to the stirring mixture, followed by 6 mg (0.045 mmol) of HOBt and 15.7 μL (0.09 mmol) of DIEA. After 10 min, 19.5 mg (0.11 mmol) of 6 were charged into the reaction mixture. The solution was stirred at RT overnight and monitored by TLC (eluent CH2Cl2:MeOH 97:3). After 18 h, the reaction mixture was dried (Na2SO4) and the yellow solid was dissolved again in CH2Cl2, washed two times with a 10% NaHCO3 aqueous solution, followed by a 0.3 M solution of citric acid in H2O (2 times), and finally with brine. The organic phase was dried (Na2SO4) and solvents evaporated under reduced pressure to give 14 mg (0.026 mmol) of pure 11 as a yellow solid with a 67% of yield. 1H NMR (CDCl3, 300 MHz) δ (ppm): 8.75 (bs, 1H), 8.26 (bs, 1H), 7.96 (m, 1H), 7.80 (m, 1H), 7.52 (s, 1H), 5.63 (s, 2H), 5.73–5.22 (m, 2H), 1.95 (m, 2H), 1.05 (bs, 3H), from −15 to −35 (bm, 16H). IR (KBr): 2963 (vas TEMPOCH3), 2864 (vs TEMPOCH3), 1744 (v C=O, lactone), 1655 (v C=O, amide 3a), 1603 (v C=O, amide), 1458–1363 (δ TEMPOCH3). HRMS (ESI): calcd for C30H34N4O6 (M + H+) 546.2473, found 546.2482.
Biological Assays.
Human recombinant Top1 was purified from Baculovirus as previously described.40 DNA cleavage reactions were performed using a 22-bp DNA oligonucleotide with a prominent topoisomerase I cleavage site. Single-stranded oligonucleotide was labeled according to the manufacturers’ instructions by using terminal deoxynucleotidyl transferase (USB Corporation, Cleveland, OH) that added a single labeled cordycepin molecule (β-32P, 5000 Ci/mmol, PerkinElmer Life and Analytical Sciences, MA) to the 3′ terminus. Unincorporated nucleotides were removed by QIAquick nucleotide removal kit (Qiagen, Hilden, Germany). The duplex DNA oligonucleotide was annealed by addition of an equal concentration of the complementary strand, heated to 95 °C, and slow cooled to room temperature. For the topoisomerase I cleavage reaction, DNA oligonucleotides were reacted for 20 min at 25 °C with a 12 ng/mL solution of human topoisomerase I and the desired amount of drugs in 10 mM Tris-HCl, pH 7.5, 50 mM KCl, 5 mM MgCl2, 0.1 mM EDTA, and 15 μg/mL bovine serum albumin. Reactions were stopped by adding 0.5% SDS and formamide containing 0.25% bromophenol blue and xylene cyanol, heated at 95 °C for 5 min, and chilled on ice. Reaction products were separated in 20% polyacrylamide denaturing sequencing gels. Dried gels were visualized using a B40 Storm phosphor imager (Amersham Biosciences, GE Healthcare, UK). Experiments with DNA oligonucleotides bearing the 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO) paramagnetic probe were performed as above-described replacing the normal 22-bp DNA oligonucleotide with those of interest.
EPR Experiments.
All samples were prepared in the following buffer: 10 mM phosphate buffer, 50 mM KCl, 5 mM MgCl, 100 μM EDTA, 150 mg/mL BSA, and ~45% by volume of glycerol. Final buffer solution was maintained at pH = 7.4. For each sample, 20 μL of solution were loaded into a 1 mm inner diameter quartz tube and placed in the spectrometer. EPR spectra were recorded using a Bruker ER200D at X-band (~9.5 GHz), equipped with a rectangular cavity ER4102ST and the relative cryostat, and a variable-temperature controller Bruker ER4111VT. The frequency was measured by a microwave frequency counter HP5342A. All spectra were obtained using the following parameters: microwave power 21 mW; modulation amplitude 0.1 mT; modulation frequency 100 kHz; time constant 41 ms; conversion time 82 ms; scan width 15 mT; 1024 points; temperature 277 K. In the Supporting Information, we report the spectrum of the ternary complex recorded at 298 K.
EPR Spectral Simulations.
The simulations of all EPR spectra were performed using the MOMD formalism for the simulation of slow-motion EPR spectra41 in the implementation for Matlab by Prof. P. Fajer, Florida State University, Tallahassee, FL, USA; all spectra were simulated using either isotropic or axial motion in the absence of any orientation potential. The hyper-fine and g tensor principal values used in the simulations were obtained by fitting the spectrum of 9 in frozen solution at 220 K: Axx = 0.73 mT, Ayy = 0.71 mT, Azz = 3.64 mT, gxx = 2.0085, gyy = 2.0058, gzz = 2.0021. The rotational correlation time was calculated from the principal values of the diffusion tensor (R) obtained from the simulations according to the formula τ = 1/(6(R‖ R⊥)1/2),15 where R‖ and R⊥ are respectively the parallel and perpendicular components of the diffusion tensor (R‖ = R⊥ for an isotropic rotation). When performing sums or differences of spectra and simulations, all lineshapes were first normalized to the same number of spins.
Supplementary Material
Acknowledgment.
This work was supported by the “Associazione Italiana Ricerca sul Cancro”, Milan (IG 4494 to G.C.), and the University of Bologna, Bologna, Italy (to G.C.). We wish to acknowledge support from the NIH Intramural Program, Center for Cancer Research, National Cancer Institute (to Y.P.), and from the grants CTQ2007-68014-C02-01 (Ministerio de Ciencia e Innovación, Spain) and 2009SGR-208 (Generalitat de Catalunya).
Footnotes
Supporting Information Available: Extended experimental section, including the synthetic details of the TEMPO-nucleosides and oligonucleotides. EPR spectrum of the ternary complex at 298 K. This material is available free of charge via the Internet at http://pubs.acs.org.
Abbreviations: EPR, electron paramagnetic resonance; SDSL-EPR, site-directed spin labeling electron paramagnetic resonance; TEMPO, 2,2,6,6-tetramethylpiperidine-1-oxyl; Top1, DNA topoisomerase I; CPT, camptothecin; CW, continuous wave; Yb(OTf)3, ytterbium triflate
References
- (1).Champoux JJ DNA topoisomerases: structure, function, and mechanism. Annu. Rev. Biochem 2001, 70, 369–413. [DOI] [PubMed] [Google Scholar]
- (2).Wang JC DNA Topoisomerases. Annu. Rev. Biochem 1996, 635–692. [DOI] [PubMed] [Google Scholar]
- (3).Wang JC Cellular roles of DNA topoisomerases: a molecular perspective. Nature Rev. Mol. Cell Biol 2002, 3, 430–440. [DOI] [PubMed] [Google Scholar]
- (4).Pommier Y Topoisomerase I inhibitors: camptothecins and beyond. Nature Rev. Cancer 2006, 6, 789–802. [DOI] [PubMed] [Google Scholar]
- (5).Pommier Y DNA Topoisomerase I Inhibitors: Chemistry, Biology, and Interfacial Inhibition. Chem. Rev 2009, 109, 2894–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wall MEW; Wani MC; Cook CE; Palmer KH; McPhail AT; Sim GA Plant antitumor agents. I. The isolation and structure of Camptothecin, a novel alkaloid leukemia and tumor inhibitor, from Camptotheca acuminata. J. Am. Chem. Soc 1966, 88, 3888–3890. [Google Scholar]
- (7).Jaxel C; Capranico G; Kerrigan D; Kohn KW; Pommier Y Effect of local DNA sequence on topoisomerase I cleavage in the presence or absence of camptothecin. J. Biol. Chem 1991, 266 (30), 20418–20423. [PubMed] [Google Scholar]
- (8).Ioanoviciu A; Antony S; Pommier Y; Staker BL; Stewart L; Cushman M Synthesis and mechanism of action studies of a series of norindenoisoquinoline topoisomerase I poisons reveal an inhibitor with a flipped orientation in the ternary DNA–enzyme-inhibitor complex as determined by X-ray crystallographic analysis. J. Med. Chem 2005, 48 (15), 4803–4814. [DOI] [PubMed] [Google Scholar]
- (9).Hsiang YH; Hertzberg R; Hecht S; Liu LF Camptothecin induces protein-linked DNA breaks via mammalian DNA topoisomerase I. J. Biol. Chem 1985, 260 (27), 14873–14878. [PubMed] [Google Scholar]
- (10).Staker BL; Hjerrild K; Feese MD; Behnke CA; Burgin AB Jr.; Stewart L The mechanism of topoisomerase I poisoning by a camptothecin analog. Proc. Natl. Acad. Sci. U.S.A 2002, 99, 15387–15392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hsiang YH; Liu LF Identification of mammalian DNA topoisomerase I as an intracellular target of the anticancer drug camptothecin. Cancer Res. 1988, 48 (7), 1722–1726. [PubMed] [Google Scholar]
- (12).Staker BL; Feese MD; Cushman M; Pommier Y; Zembower D; Stewart L; Burgin AB Structures of Three Classes of Anti-cancer Agents Bound to the Human Topoisomerase I–DNA Covalent Complex. J. Med. Chem 2005, 48, 2336–2345. [DOI] [PubMed] [Google Scholar]
- (13).Koster DA; Croquette V; Dekker C; Shuman S; Dekker NH Friction and torque govern the relaxation of DNA supercoils by eukaryotic topoisomerase IB. Nature 2005, 434, 671–674. [DOI] [PubMed] [Google Scholar]
- (14).Koster DA; Palle K; Bot ESM; Bjornsti MA; Dekker NH Antitumor drugs impede DNA uncoiling by topoisomerase I. Nature 2007, 448, 213–217. [DOI] [PubMed] [Google Scholar]
- (15).Goldman SA; Bruno GV; Freed JH Estimating Slow-Motional Rotational Correlation Times for Nitroxides by ESR. J. Phys. Chem 1972, 76 (15), 1858–1860. [Google Scholar]
- (16).Huang H; Cafiso DS Conformation and Membrane Position of the Region Linking the Two C2 Domains in Synaptotagmin 1 by Site-Directed Spin Labeling. Biochemistry 2008, 47 (47), 12380–12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Ranaldi S; Belle V; Woudstra M; Rodriguez J; Guigliarelli B; Sturgis J; Carriere F; Fournel A Lid Opening and Unfolding in Human Pancreatic Lipase at Low pH Revealed by Site-Directed Spin Labeling EPR and FTIR Spectroscopy. Biochemistry 2009, 48 (3), 630–638. [DOI] [PubMed] [Google Scholar]
- (18).Sarewicz M; Borek A; Daldal F; Froncisz W; Osyczka A Demonstration of short-lived complexes of cytochrome c with cytochrome bc(1) by EPR spectroscopy—Implications for the mechanism of interprotein electron transfer. J. Biol. Chem 2008, 283 (36), 24826–24836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Sarewicz M; Szytuza S; Dutka M; Osyczka A; Froncisz W Estimation of binding parameters for the protein–protein interaction using a site-directed spin labeling and EPR spectroscopy. Eur. Biophys. J 2008, 37 (4), 483–493. [DOI] [PubMed] [Google Scholar]
- (20).Xi XM; Sun Y; Karim CB; Grigoryants VM; Scholes CP HIV-1 nucleocapsid protein NCp7 and its RNA stem loop 3 partner: Rotational dynamics of spin-labeled RNA stem loop 3. Biochemistry 2008, 47 (38), 10099–10110. [DOI] [PubMed] [Google Scholar]
- (21).Tong JS; Borbat PP; Freed JH; Shin Y-K A scissors mechanism for stimulation of SNARE-mediated lipid mixing by cholesterol. Proc. Natl. Acad. Sci. U.S.A 2009, 106 (3), 5141–5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Berliner LJ, Spin Labeling. Theory and Applications; Academic Press Inc: New York, 1976. [Google Scholar]
- (23).Likhtenshtein G; Yamauchi J; Nakatsuji S; Smirnov AI; Tamura R, Nitroxides: Applications in Chemistry, Biomedicine, and Materials Science; Wiley: New York, 2008. [Google Scholar]
- (24).Rozantsev EG, Free Nitroxyl Radicals; Springer: New York, 1970. [Google Scholar]
- (25).Dallavalle S; Merlini L; Morini G; Musso L; Penco S; Beretta GL; Tinelli S; Zunino F Synthesis and cytotoxic activity of substituted 7-aryliminomethyl derivatives of camptothecin. Eur. J. Med. Chem 2004, 39, 507–513. [DOI] [PubMed] [Google Scholar]
- (26).Jaxel C; Kohn KW; Wani MC; Wall ME; Pommier Y Structure–activity study of the actions of camptothecin derivatives on mammalian topoisomerase I: evidencefor aspecific receptor siteand a relation to antitumor activity. Cancer Res. 1989, 49 (6), 1465–1469. [PubMed] [Google Scholar]
- (27).Tanizawa A; Fujimori A; Fujimori Y; Pommier Y Comparison of topoisomerase I inhibition, DNA damage, and cytotoxicity of camptothecin derivatives presently in clinical trials. J. Natl. Cancer Inst 1994, 86 (11), 836–842. [DOI] [PubMed] [Google Scholar]
- (28).Sawada S; Nokata K; Furuta T; Yokokura T; Miyasaka T Chemical modification of an antitumor alkaloid camptothecin: synthesis and antitumor activity of 7-C-substituted camptothecins. Chem. Pharm. Bull 1991, 39 (10), 2574–2580. [DOI] [PubMed] [Google Scholar]
- (29).Citterio A; Gentile A; Minisci F; Serravalle M; Ventura S α-Hydroxyalkylation of Heteroaromatic Bases by Alcohols and Hydroxylamine-O-sulphonic Acid. Tetrahedron 1984, 41 (3), 617–620. [Google Scholar]
- (30).Andersen AH; Gocke E; Bonven BJ; Nielsen OF; Westergaard O Topoisomerase I has a strong binding preference for a conserved hexadecameric sequence in the promotor region of the rRNA gene from Tetrahymena pyriformis. Nucleic Acids Res. 1985, 13 (5), 1543–1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lesher D-TT; Pommier Y; Stewart L; Redinbo MR 8-Oxoguanine rearranges the active site of human topoisomerase I. Proc. Natl. Acad. Sci. U.S.A 2002, 99 (19), 12102–12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Agrawal S, Protocols for Oligonucleotides and Analogs: Synthesis and Properties; Humana Press Inc.: Totowa, NJ, 1993; p 33. [Google Scholar]
- (33).Pommier Y; Laco GS; Kohlhagen G; Sayer JM; Kroth H; Jerina DM Position-specific trapping of topoisomerase I–DNA cleavage complexes by intercalated benzo[a]-pyrene diol epoxide adducts at the 6-amino group of adenine. Proc. Natl. Acad. Sci. U. S.A 2000, 97 (20), 10739–10744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Leteurtre F; Fesen M; Kohlhagen G; Kohn KW; Pommier Y Specific interaction of camptothecin, a topoisomerase I inhibitor, with guanine residues of DNA detected by photoactivation at 365 nm. Biochemistry 1993, 32, 8955–8962. [DOI] [PubMed] [Google Scholar]
- (35).Berliner LJ, Biological Magnetic Resonance; Springer: New York, 1998; Vol. 14. [Google Scholar]
- (36).McHaourab HS; Lietzow MA; Hideg K; Hubbell WL Motion of spin-labeled side chains in T4 lysozyme, correlation with protein structure and dynamics. Biochemistry 1996, 35 (24), 7692–7704. [DOI] [PubMed] [Google Scholar]
- (37).Phillips DR Daunomycin–DNA Dissociation Kinetics. Mol. Pharmacol 1987, 225–230. [PubMed] [Google Scholar]
- (38).Rizzo V Kinetic Studies of Anthracycline–DNA Interaction. Biochemistry 1989, 28, 274–282. [DOI] [PubMed] [Google Scholar]
- (39).Giannini G; Marzi M; Cabri W; Marastoni E; Battistuzzi G; Vesci L; Pisano C; Beretta GL; Cesare MD; Zunino F E-Ring-modified 7-oxyiminomethyl camptothecins: synthesis and preliminary in vitro and in vivo biological evaluation. Bioorg. Med. Chem. Lett 2008, 18 (9), 2910–2915. [DOI] [PubMed] [Google Scholar]
- (40).Pourquier P; Ueng LM; Fertala J; Wang D; Park HJ; Essigmann JM; Bjornsti MA; Pommier Y Induction of reversible complexes between eukaryotic DNA topoisomerase I and DNA-containing oxidative base damages: 7,8-dihydro-8-oxoguanine and 5-hydroxycytosine. J. Biol. Chem 1999, 274 (13), 8516–8523. [DOI] [PubMed] [Google Scholar]
- (41).Budil DE; Lee S; Saxena S; Freed JH Nonlinear-Least-Squares Analysis of Slow-Motion EPR Spectra in One and Two Dimensions Using a Modified Levenberg–Marquardt Algorithm. J. Magn. Reson., Ser. A 1996, 120, 155–189. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.