

Abstract
The Conserved Oligomeric Golgi (COG) complex, a multi-subunit vesicle tethering complex of the CATCHR (Complexes Associated with Tethering Containing Helical Rods) family, controls several aspects of cellular homeostasis by orchestrating retrograde vesicle traffic within the Golgi. The COG complex interacts with all key players regulating intra-Golgi trafficking, namely SNAREs, SNARE-interacting proteins, Rabs, coiled-coil tethers, and vesicular coats. In cells, COG deficiencies result in the accumulation of non-tethered COG-complex dependent (CCD) vesicles, dramatic morphological and functional abnormalities of the Golgi and endosomes, severe defects in N- and O- glycosylation, Golgi retrograde trafficking, sorting and protein secretion. In humans, COG mutations lead to severe multi-systemic diseases known as COG-Congenital Disorders of Glycosylation (COG-CDG). In this report, we review the current knowledge of the COG complex and analyze COG-related trafficking and glycosylation defects in COG-CDG patients.
Keywords: COG complex, Golgi, Glycosylation, CDG, COG-CDG, Vesicle tethering
Abbreviations: AP, adaptor protein; ARF, ADP ribosylation factor; BFA, Brefeldin A; CATCHR, Complexes associated with tethering containing helical rods; CCD, COG complex dependent; CCT, Coil-coiled tethers; CDG, Congenital disorders of glycosylation; CHO, Chinese hamster ovary; COG, Conserved oligomeric Golgi; COPI/COPII, Coat protein complex I/complex II; EARP, Endosome-associated recycling protein; EELS, Enlarged endolysosomal structure; EM, Electron microscopy; ER, Endoplasmic reticulum; GARP, Golgi-associated retrograde protein; GEARs, COG-sensitive, integral membrane Golgi proteins; GSL, Glycosphingolipis; HEK, Human embryonic kidney; HLH, Hemophagocytic lymphohistiocytosis; HOPS, Homotypic fusion and vacuole Protein Sorting complex; HPA, Helix pomatia agglutinin; IEF, Isoelectric focusing; KD, Knock-down; KO, Knockout; LVH, Left ventricular hypertrophy; MS, mass spectrometry; MTC, Multi-subunit Tethering Complexes; NIHF, Non-Immune Hydrops Fetalis; PI4P, Phosphatidylinositol 4-phosphate; PM, Plasma membrane; PNA, Peanut agglutinin; RCA, Ricinus communis agglutinin; SNAP, Soluble NSF attachment protein; SNARE, Soluble NSF (N-ethylmaleimide sensitive factor) attachment proteins (SNAP) receptor; TGN, Trans-Golgi Network; TM, Transmembrane; TRAPP, Transport Protein Particle complex; WT, Wild type; Y2H, Yeast two-hybrid
Graphical abstract
Highlights
-
•
Intracellular membrane trafficking and glycosylation.
-
•
COG complex structure and functions.
-
•
COG-CDGs: clinical presentation and molecular analysis.
1. Membrane trafficking at the Golgi
The Golgi apparatus is the central hub of the secretory and endocytic pathways. More than one-third of cell proteins are transported via these pathways [1,2]. In anterograde trafficking, newly synthesized secretory cargo translocates to the endoplasmic reticulum (ER) and is then transported to the Golgi apparatus for further processing, modification, and sorting to the final cellular and extracellular destinations. Retrograde trafficking transports cargo in a reverse fashion from the plasma membrane (PM) down to the endosomes, Golgi and even to ER (Fig. 1 ). A balance between anterograde and retrograde transport is vital for cell physiology and cell survival. Each step of intra-organellar membrane trafficking is facilitated and controlled by specialized protein trafficking machinery, which is modular and is conserved from yeast to mammals. The major players of the trafficking machinery can broadly be categorized as cargo adaptors, vesicular coat proteins, small GTPases of Arf and Rab families, vesicular tethers, SNAREs, and SNARE-associated proteins [3]. Each compartment of the cell's secretory/endocytic system has its own set of these trafficking components, which contributes to compartment identity.
Fig. 1.

The COG complex orchestrates retrograde membrane trafficking at the Golgi. In the anterograde secretory pathway (blue arrows), biosynthetic cargo is transported from the ER to PM through the Golgi by cisternal maturation. In the retrograde endocytic pathway (purple arrows), endocytic cargo is transported from PM via the endocytic system and Golgi back to the ER. The octameric COG complex (lobe A, red and lobe B, green) mostly tethers intra-Golgi recycling vesicles at Golgi rims. Transparent COG represents possible involvement in LE-TGN, ERGIC-ER and Golgi-autophagosome pathways. EE- Early endosome, LE- Late endosome, Lys- Lysosome, MVB- Multivesicular bodies, Auto- Autophagosome, ER- Endoplasmic Reticulum, ERGIC- ER Golgi intermediate compartment.
The initial step of intracellular membrane trafficking begins with cargo concentration into emerging membrane carriers (vesicles, tubules, or megavesicles). This is achieved by the coordinated action of small GTPases, vesicular coat proteins, and cargo adaptors [4,5]. Newly formed trafficking carriers then travel to the acceptor organelle/compartment by diffusion or using the cytoskeletal “rail system” which comprises of microtubules, actin filaments and motor proteins of the dynein, kinesin and myosin families [6]. The vesicle-associated Rab GTPases cycle between an “on-and-off” state [7]. In the “on” GTP-bound state Rabs interact with molecular motors and vesicular tethers to facilitate tethering and fusion. Coiled-coil tethers (CCTs) capture vesicles to initiate tethering. Their length and flexibility facilitate long-range capture, which brings the membrane carriers close to the acceptor organelle [8,9]. Multi-subunit tethering complexes (MTCs) have multiple binding sites on their subunits. Key members of the fusion machinery interact with MTCs to finalize the tethering step and set the stage for the membrane fusion, which is brought about by the interaction of SNAREs on the donor and acceptor membranes [3,10,11].
1.1. Glycosylation and Golgi glycosylation machinery
Proteins transported by the secretory pathway play both structural (e.g. matrix proteins like collagen, elastin, α-dystroglycan) and functional roles (e.g. ligands and receptors in cell-cell signaling, immunoglobulins, neurotransmitters, enzymes, hormones). Most of these proteins undergo modifications in the ER and Golgi. One major modification, glycosylation, is the covalent linkage of proteins/lipids with oligosaccharide moieties. The process of N-linked glycosylation, wherein attachment of sugars to the amide nitrogen of asparagine residues occurs, begins in the ER, and continues in the Golgi. On the other hand, O-linked glycosylation begins in the Golgi and sugar attachment occurs at serine or threonine's hydroxyl group. O-glycans are more diverse in comparison to N-glycans and include shorter mucin-type glycans and longer glycosaminoglycan (GAG) chains on proteoglycans (for review see [137]
Glycosyltransferases and glycosidases are enzymes that transfer sugar donors onto acceptor substrates (proteins and lipids) and modify glycans, respectively. Sugar-nucleotide precursors of glycans are synthesized in the cytosol and are brought into the ER/Golgi lumen by sugar transporters. In a steady-state, each Golgi cisternae is enriched with specific sets of glycosylation enzymes, sugar transporters and other proteins, which collectively confer cisternal identity. Golgi compartmentalization and glycosylation go hand-in-hand with membrane trafficking. The proper localization of Golgi resident proteins is maintained, at least in part, by retrograde vesicular trafficking. Segregation of early acting glycosylation enzymes into cis and medial Golgi compartments and later acting enzymes into the trans-Golgi is achieved within the framework of the cisternal maturation model wherein, the enzymes and sugar transporters are actively cycled back to newly formed cisterna as cis to trans maturation continues, carrying forward biosynthetic cargo. Recycling of Golgi resident proteins within the Golgi mostly occurs in COPI coated vesicles, although COPI-independent recycling via the ER has been proposed [12]. Also, in yeast cells, some of trans-Golgi retrograde trafficking is facilitated by AP1/clathrin coated vesicles [13]. COPI coat interacts with its cargo either directly or via adapter proteins. Many Golgi enzymes have N-terminal cytoplasmic tails. A small subset of enzymes contains the ϕ-(K/R)-X-L-X-(K/R) consensus motif on their cytoplasmic tails and bind directly to COPI components [14]. A few other enzymes interact with COPI subunits through GOLPH3, an abundant peripheral membrane protein [15]. The exact recycling mechanism for the majority of the Golgi's glycosylation machinery is currently unknown and likely requires several novel adaptor proteins. Apart from cytoplasmic sorting signals, length of the transmembrane domains (TMDs) of glycosylation enzymes might contribute to their retention and sorting within the Golgi. cis-Golgi resident proteins have shorter TMDs compared to late Golgi residents. Membrane thickness increases from ER to the PM due to the increasing concentration of sphingolipids and sterols. It is energetically favorable for shorter TMDs to be retained in thinner membranes and sorted into phospholipid-rich COPI vesicles rather than cholesterol-rich clathrin-coated vesicles targeted to the PM [16,17]. Another proposed mechanism of Golgi enzyme retention is the formation of multimeric complexes. Sequentially acting glycosylation enzymes in the same cisternae can form dimers and higher-order oligomers. This increases the size of the protein preventing it from entering vesicles, leaving the Golgi. Oligomerization of sequentially acting enzymes also enhances the efficiency of glycosylation [18,19].
Also, the luminal environment within the Golgi governs glycosylation and Golgi homeostasis. Each Golgi cisternae has a unique pH and ionic environment, which contributes to correct enzyme localization [20] and also regulates enzyme activity [21,22]. The contribution of these factors to Golgi homeostasis has been reviewed by Kellokumpu [136] ]. Furthermore, reduced luminal volume in flattened Golgi stacks facilitates enzymes-substrate encounters [23].
2. Conserved oligomeric Golgi complex – The major Golgi MTC
Multi-subunit tethering complexes (MTCs) are a group of evolutionarily conserved vesicular tethers consisting of three to ten subunits. They are loosely sub-grouped into CATCHRs and non-CATCHRs [24]. CATCHR family includes MTCs functioning along the secretory pathway, namely, Dsl1/ZW10 complex, COG, GARP, EARP and the exocyst; non-CATCHR group includes CORVET and HOPS complexes which are involved in the endosomal trafficking. TRAPP, a unique MTC in structure and function is involved in ER to Golgi trafficking and autophagy [25]. The subunits of Dsl1/ZW10, COG, GARP, and the exocyst complexes share similarities in their α-helical structures [26]. COG's closest relative is the exocyst, which is also octameric and the structure of its subunits SEC3/EXOC1, SEC5/EXOC2, SEC10/EXOC5, SEC15/EXOC6 and EXO70/EXOC7 are similar to COG subunits [[27], [28], [29]]. MTCs are peripheral membrane proteins that lack membrane anchoring domains and they can only associate with membranes indirectly. So far, the GARP complex, Dsl1/ZW10 and the exocyst have been shown to be membrane associated via their interactions with Rab GTPases, SNAREs, and/or membrane lipids respectively [[30], [31], [32]].
2.1. Structure and interactions of the COG complex
The COG complex has eight subunits (Fig. 2 ), encoded by eight different genes, and exists as a hetero-octameric complex or as lobe A (COG1, COG2, COG3, COG4) and lobe B subcomplexes (COG5, COG6, COG7, COG8) [33]. Interestingly, four subunits, COG3, COG4, COG6, and COG8, are more evolutionarily conserved, as are the other four subunits. COG1-COG8 interaction bridges the two subcomplexes together [[34], [35], [36]]. Mutations that disrupt COG1/8 interactions are detrimental for COG complex functions [37,38]. The two subcomplexes are observed as lobes in deep-etch electron microscopy (EM) images of the purified COG complex from bovine brain [39]. Lobe A predominantly associates with the Golgi membrane and can be found on every cisternae [40], while lobe B preferentially interacts with vesicular membranes [38]. The observed cytoplasmic pool of COG is mainly octameric [29,36,38,39], while membrane-bound COG can be an octamer or a subcomplex [38]. COG subunits are possibly always bound to the membranes via weak protein-protein interactions with different peripheral or integral membrane proteins. Disruption of these interactions during cell lysis releases COG from the membranes and the soluble pool is likely to be an experimental artifact resulting from the cell lysis procedure. The different arrangements of the COG complex are tied to its functional state, which is described in the ‘Current models for COG tethering’ section. A slow recovery after photobleaching in Fluorescence Recovery After Photobleaching (FRAP) experiments show that the on/off cycling of COG subunits from the Golgi membrane is very slow [41], and artificially membrane-glued COG is fully functional [42].
Fig. 2.

COG complex compositions and human mutations. The left-hand side depicts vesicle tethering by the COG complex and its interactions with the Golgi trafficking machinery. This model is based on the assembly-disassembly model for the COG complex. Lobe A (red) is associated with the Golgi and lobe B (green) is associated with vesicles. Interaction between lobe A and B brings the vesicle close to the Golgi membrane and facilitates SNARE-mediated fusion. CCT: Coli-coiled tethers, SM: Sec1/Munc18-like, SNARE: soluble NSF (N-ethylmaleimide sensitive factor) attachment proteins (SNAP) receptor. The known human COG mutations in each subunit are depicted on the right Lobe A subunits COG1, COG2, COG3 and COG4 are in red and lobe B subunits COG5, COG6, COG7 and COG8 are in green.
Structural information is limited and only available for yeast COG2 [43], N-terminal domains of yeast COG5, COG7 [44] and the C-terminal domain of human COG4 [45]. Yeast COG complex has been reconstituted in vitro and investigated by single-particle EM microscopy. Investigation of lobe A revealed Y-shaped objects with three long, spindly “legs” [46]. Fluorescent microscopy-based studies on the 3D structure of the octameric COG complex predicted that COG exists as ~25 nm particles with a central “core” and multiple (4 to 8) peripheral extensions (“arms” or “legs”) [47].
COG protein-protein interactions within subcomplexes are mainly achieved via N-terminal alpha-helical domains that are present in all COG subunits [34]. Stability of COG subunits have varying degrees of dependency on partner subunits in the complex, but, in general, the stability of lobe A and lobe B subunits are affected by manipulations with other subunits in that lobe [48]. However, COG8 (lobe B subunit) is the exception - its stability is influenced by COG1 [37]. Through specific interaction sites on its subunits, the COG complex interacts with major players of the vesicle docking/fusion machinery (coats, Rabs, CCTs, and SNAREs as depicted in Fig. 2) on both the donor and acceptor membrane [49]. Still, the majority of these predicted interactions are not mapped or characterized in detail, yet. The best-characterized protein-protein interactions include COG4-STX5 [50], COG4-SCFD1/Sly1 [51], COG2-USO1/p115 [52] and COG7-GOLGA5/Golgin-84 [53].
2.2. COG function
The COG complex facilitates the tethering and fusion of intra-Golgi recycling vesicles at the Golgi cisternae (Fig. 1) [33]. The initial data on COG's function came from both genetic and EM studies of yeast COG mutants [29,54,55] and from biochemical and in vitro studies in mammalian cells [39,56,57]. Further studies using knock-down (KD) or knock-sideways approaches unraveled details of the COG's function and interactions. Massive accumulation of ~60 nm vesicles is a major morphological phenotype in cells acutely depleted of COG complex subunits [54,58]. These freely-diffusible CCD vesicles are enriched with medial/trans-Golgi enzymes and intra-Golgi v-SNAREs [59] and most likely represent non-tethered intra-Golgi recycling vesicles. Importantly, purified COG can bind and capture isolated CCD vesicles [58]. These studies provided evidence for COG's function as a vesicle tethering complex [38]. Another major phenotype of COG malfunction is the onset of glycosylation defects [56]. All individual COG subunit KDs display altered glycosylation of both N- and O-linked glycans. A closer investigation into glycosylation defects revealed the destabilization and mislocalization of multiple components of the Golgi glycosylation machinery [35,[59], [60], [61], [62]].
Prolonged depletion or complete knockout of individual COG subunits causes morphological changes in the Golgi (severe fragmentation and dilation of all Golgi cisternae) (Fig. 3a) that further alters glycosylation [48,59], and also affects the endolysosomal system (accumulation of Enlarged Endo-Lysosomal Structures, EELSs) [63] (Fig. 3b), delays retrograde protein trafficking [64], causes protein missorting and changes the secretion profile [65]. Formation of intra-Golgi SNAREs complex STX5/GOSR1/BET1L/YKT6 [50] and trans-Golgi SNARE complex STX16/STX6/VTI1A/VAMP4 [66] is reduced in COG deficient cells. Another COG-related trafficking abnormality is the delayed reaction to the fungal metabolite Brefeldin A (BFA) [60,67]. BFA inhibits the exchange factor GBF1 for small GTPase Arf1, causing COPI coat dysfunction and an artificial fusion of Golgi and ER membranes [68]. The kinetics of both, BFA-induced Golgi “collapse” and subsequent Golgi restoration after BFA wash-out are severely delayed in COG deficient cells. This alteration is often interpreted as COG's requirement for Golgi-ER anterograde and retrograde trafficking [69,70]. We believe that this interpretation is erroneous and that the altered response to BFA treatment is a secondary manifestation of GBF1, COPI, and/or SNARE malfunction in COG deficient cells [67]. Importantly, many types of COG depleted mammalian cells have wild-type growth and division [48], emphasizing that the COG complex is generally dispensable for intracellular anterograde trafficking.
Fig. 3.

Cellular phenotype of COG KO cells. Upper panel: (A) EM of HeLa COG3 KO cells shows a fragmented Golgi with dilated cisternae, unlike the well-organized stacks in the WT. (B) Superresolution microscopy of HEK293T WT and COG4 KO cells transfected with ST6Gal1-RFP and GFP-STX5 (WT; Golgi marker) or Lamp2-GFP (COG4 KO; Endolysosomal marker). Golgi enzyme ST6Gal1 is mislocalized to Lamp2 labeled endolysosomes and EELSs. Asterisks indicate the Golgi. Arrows point to EELSs, which are 1-10 μM in diameter and accumulate in COG KO cells.
The deletion of any of the COG subunits in human cells renders the entire complex non-functional and results in the degradation of several Golgi resident proteins. Proteins whose stability and function depend on the COG complex are termed COG sensitive proteins, or GEARS [71]. These include many Golgi glycosylation enzymes (MGAT1, MAN2A, B4GALT1, ST6GAL1) [60,61] and certain members of the trafficking machinery (GS15/BET1L, GS28/GOSR1, giantin/GOLGB1, golgin-84/GOLGA5) [71]. Importantly, not every Golgi protein is COG-sensitive; for instance, both the amount and localization of the SNARE STX5, CCT GM130/GOLGA2 and many of the Golgi Rabs are normal in COG-depleted cells [48,71] (and our unpublished observations). This indicates the existence of COG-independent Golgi recycling/retention mechanism. Analysis of cell lines partially (siRNA) or completely (COG KOs) depleted for COG subunits laid a foundation for the analysis of COG complex deficiencies in humans, described in section 3.
Another important evidence supporting the central role of the COG complex in cellular physiology comes from pathogen-host interaction research. Many intracellular pathogens hijack the host's membrane trafficking machinery to survive within them. Importantly, the COG complex is manipulated by viruses (SARS-Cov-2 [72], Rift Valley fever [73], HIV [74], and Orthopoxvirus [75]), pathogenic bacteria (Chlamydia [76] and Brucella [77]), and toxins (SubAB [48,64], Shiga [58], and Cholera [48]). It is not clear how these diverse groups of pathogens have evolved to rely on COG function.
2.3. The current model for COG tethering
The exact mechanism of COG complex function is still a mystery. Over the years, several models were put forward to explain various mutant phenotypes observed in COG-depleted cells (for review see [33]). According to the most recent “assembly/disassembly” model [38], lobe A and lobe B of the COG complex are always engaged in a dynamically regulated interaction. In this model, lobe A preferentially associates with the Golgi cisternae, while lobe B preferentially associates with recycling intra-Golgi CCD vesicles. Lobe A and lobe B are bridged via COG1-COG8 interactions when the vesicle is tethered to the Golgi membrane by CCTs. This COG1-COG8 interaction forms the octameric COG complex, which, in turn, aligns and possibly proofreads the fusion machinery on both membranes. After the formation of the trans-SNARE complex, COG is displaced from the vesicle-cisternae interspace, allowing vesicle fusion to occur.
The assembly/disassembly model would predict that the majority of Golgi associated octameric COG is inert. This then poses the question of what dissociates the two lobes to make them available for another round of vesicle tethering and fusion. While the exact mechanism of COG “recycling” is not understood, one possibility is that the specific binding to vesicular factors (coat, Rabs, SNAREs, CCTs) causes COG disassembly and association of lobe B with the vesicle membrane.
The COG complex assembly/disassembly model is in good agreement with the models proposed for the mammalian exocyst complex [78,79]. However, another recent model for the yeast exocyst function [80] suggests that exocyst subcomplexes are always physically interacting with each other and that vesicle tethering is achieved by intra-molecular changes in the complex.
3. COG-CDGs
Since the past two decades, over a hundred individuals with COG mutations have been identified (see Fig. 2 for the location of known mutations in COG subunits). As mentioned in the earlier section, the fidelity of glycosylation is largely dependent on the proper localization of the glycosylation machinery. So, it is not surprising that altered intra-Golgi trafficking due to COG malfunction leads to glycosylation defects [81]. Glycosylation disorders belong to a group of autosomal recessive disorders termed as Congenital Disorders of Glycosylation (CDGs), with largely no known cure and are categorized as type-I (CDG-I) or type-II (CDG-II) depending on the affected protein. CDG-I involves defects in the synthesis and/or transfer of a lipid-linked oligosaccharide while CDG-II involves abnormalities in the glycan modification and processing machinery, including, but not limited to, faulty glycosidases and glycosyltransferases. CDGs are multi-systemic disorders associated with poor quality of life. A list of recognizable symptoms includes global developmental defects, dysmorphic features, microcephaly, and failure to thrive, accompanied by the liver and neurological involvement to varying degrees [82,83]. A CDG diagnosis is made based on the isoelectric focusing (IEF) pattern of N-glycosylated protein transferrin. In CDG-I, there is an abundance of asialylated and/or disialylated transferrin due to defects in the transfer and assembly of glycans. Defective glycan processing, as in the case of CDG-II, results in increased mono-, di- tri- and/or asialotransferrin. But, not all CDG-II patterns can be detected by testing transferrin's IEF. Glycan mass spectrometry and/or ApoCIII IEF are often used to complement transferrin IEF testing [[84], [85], [86]]. COG mutations cause CDG-II. In COG-CDGs, the IEF pattern of serum transferrin indicates hyposialylated species, while IEF of apoC-III show increased non-glycosylated apoC-III and decreased monosialo glycoforms [81]. Galactosylation and sialylation defects are also evident by glycan mass-spectrometry in COG-CDGs. Following diagnosis, genotyping is usually done to identify CDG-causing mutations. The next section summarizes the clinical findings of COG-CDG patients and tries to reconcile these findings with current knowledge on the COG complex's cellular functions.
3.1. Mutations associated with individual COG subunits
3.1.1. COG1
3.1.1.1. Clinical presentation
CDG causing mutation in COG1 was first reported in 2006. The patient was admitted to the hospital with the feeding problem and failure to thrive. Clinical examination revealed that the patient showed signs of hypotonia, left ventricular hypertrophy (LVH), progressive microcephaly, mild hepatosplenomegaly and slight cerebral and cerebellar atrophy [87]. Zeevaert et .al [88] described two other patients with a cerebrocostomandibular-like syndrome and a novel mutation in COG1. These patients also exhibited growth retardation, moderate mental retardation, feeding difficulties and cerebellar atrophy. Costovertebral dysplasia, including rib fusion, was a clinical feature unique to these two patients. The other symptoms were cryptorchidism, recurrent infections and hemolytic uremic syndrome. Clinical examination mostly revealed dysmorphic features and a more severe phenotype compared to the earlier described COG1-CDG patient.
3.1.1.2. Molecular analysis
In the case of the first described patient, a single nucleotide insertion (2659-2660insC) in the gene encoding the COG1 subunit created a stop codon leading to a loss of 80C-terminus amino acid residues. As expected, this truncation affected the stability of COG1, all other lobe A subunits and COG8 [87]. Truncated COG1 could still localize to the Golgi but to a much lesser extent compared to WT COG1. The association of partner subunits with the Golgi was affected to varying degrees. The protein levels of tested glycosylation enzymes MAN2A and B4GALT1 were diminished. The other COG1 mutation identified by Zeevaert et al. [88] was c.1070 + 5G > A, an intronic mutation at a splice site. It resulted in the skipping of exon 6, and a frameshift that produced a premature stop codon in exon 7, leading to loss of 659 amino acids. The mutant protein was not expressed but there was about 20% WT protein expressed possibly because this mutation is leaky. The protein level of COG8 was slightly decreased, but MAN2A was unaffected. In the case of the intronic insertion 2659-2660insC, only the truncated protein was expressed. However, it is quite surprising that this leaky mutation produces a more severe clinical phenotype than the 2659-2660insC mutation [88].
3.1.2. COG2
3.1.2.1. Clinical presentation
The COG2 mutation causing CDG was reported in 2015 [89]. The patient was a 6-year old male child of healthy unrelated parents. He exhibited signs of growth retardation and microcephaly, similar to COG1-CDG patients. The other features included spastic quadriplegia, liver dysfunction without hepatosplenomegaly, hypocupremia, hypoceruloplasminemia, cerebral atrophy, and tonic seizures. The patient responded well to treatment for some symptoms.
3.1.2.2. Molecular analysis
The patient had two mutations – a de novo c.701dup (p.Y234*) change in the paternal allele and an inherited c.1900 T > G (p.W634G) mutation in the maternal allele. c.701dup (p.Y234*) is a null mutation and the mutant mRNA undergoes degradation. W634 is highly conserved in all vertebrates and the authors speculated this to be the reason COG2 was affected. COG2 was absent at the protein level. Consistent with previous findings pertaining to the stability of the other COG subunits, both COG3 and COG4 were affected.
3.1.3. COG3
At present, there are no known CDG causing mutations associated with the COG3 subunit. This may be due to the central role of COG3 in the organization of lobe A COG subcomplex.
3.1.4. COG4
3.1.4.1. Clinical presentation
So far, two patients have been described with CDG causing mutations in COG4. Both patients were 2 (P2) and 3 (P1) year old male children [90]. P1 presented with profound developmental delay, hypotonia, failure to thrive, seizures, coagulopathy, liver cirrhosis and recurrent infections. He exhibited a more severe clinical phenotype compared to P2 and died at 2 years of life. P1 and P2 shared some common clinical features which included dysmorphia, microcephaly, and moderate psychomotor delay. Moreover, P2 had axial hypotonia, mild ataxia, uncoordinated movements, bilateral cerebral atrophy of frontotemporal regions and recurrent respiratory infections. Surprisingly, a recently discovered COG4 mutation results in a rare skeletal dysplasia, named Saul-Wilson syndrome (SWS) [91] and is not categorized as CDG causing mutation per se. These patients present with speech and developmental delay, dysmorphic features, skeletal dysplasia and hearing loss. SWS newborns are often misdiagnosed with the microcephalic primordial dwarfism spectrum due to extreme short stature.
3.1.4.2. Molecular analysis
P1 was heterozygous for two mutations, de novo c.697G > T (p.E233X) and c.2318 T > G (p.L773R) in the COG4 gene [92]. The c.697G > T transcript underwent nonsense-mediated decay. COG4 protein was depleted by 70%, but, curiously, no other subunits were significantly reduced. Glycan mass-spectrometry showed protein sialylation and galactosylation defects. COG4 deficiency hampered COG's function and the Golgi failed to relocate to the ER upon BFA treatment. P2 carried a C > T point mutation at position 2185 in the COG4 gene resulting in R729W change. Though the child was born to unrelated parents - a father heterozygous for the aforementioned mutation and a normal mother, the child seemed to be homozygous for the C > T point mutation. This led the authors to speculate that the maternal allele, which was supposed to be normal, underwent deletion between intron 2 and exon 5. At the molecular level, only 20% of the COG4 protein is expressed. The deficiency of the COG4 subunit affected the stability of all the remaining lobe A subunits and COG5. Sequence and crystal structure analysis of COG4's C-terminal fragment by Richrdson et al. [45] found that the mutated arginine is highly conserved. Replacement by bulkier and rigid tryptophan disrupts intramolecular interactions. R729 forms a salt bridge network with E688 and E764 on the neighboring domain. This network stabilizes the C-terminal domain of COG4 and the mutation possibly affects the stability of the domain whose residues have interactions with arginine. The affected domain was speculated to be important in COG complex function. Patients with SWS [91] have a de novo heterozygous mutation in COG4 (c.1546G > A, or c.1546G > C) resulting in the p.G516R amino acid substitution. Glycan analysis in the mutant fibroblasts, serum/plasma and glycosylation status of serum transferrin was the same as controls. But, modification of decorin, a secreted proteoglycan, was found to be altered. Furthermore, the stability of the mutant COG4 protein was increased. The authors implicated this change in COG4 protein turnover to have a negative effect on COG function.
3.1.5. COG5
3.1.5.1. Clinical presentation
Twelve COG5-CDG patients have been identified over a span of 20 years. The first COG5-CDG patient (P1) was described in 2009 by Paesold-Burda et al. [93]. At 8 years of age, she presented with a mild neurological phenotype including hypotonia, psychomotor retardation, delayed speech development and truncal ataxia. The patient did not exhibit signs of developmental delay and dysmorphism like most of the other COG-CDG patients and at 14 years she showed significant improvement in speech. The second COG5-CDG patient (P2), described by Fung et al. in 2011 [94] also had a mild neurological phenotype including mental retardation, hypotonia, delayed speech development and microcephaly. She showed a global developmental delay, which is common in most COG-CDG patients. Moreover, she had liver cirrhosis with splenomegaly by the age of 1. At 8, she too made significant developmental progress in speech and had only mild mental retardation. In 2012, Rymen et al. described 5 more COG5-CDG patients [69]. These included three female siblings (P3.1, P3.2 and P3.3) that presented with microcephaly, delayed speech, and motor development, moderate to severe mental retardation, hypotonia, mild dysmorphia, microcephaly, short stature and walking difficulty. The two other children (P4 and P5) were unrelated and had similar clinical presentations to the three siblings with additional blindness and deafness.
Kim et al. [95] described a family where three siblings (P6.1, P6.2, P6.3) presented with Friedreich's-ataxia-like phenotypes that include cerebellar atrophy, mild to moderate intellectual disability, and scoliosis. Unlike the previous COG5-CDG cases, these siblings did not have hypotonia, microcephaly and short stature. In 2019, Yin et al. [96] described a COG5-CDG patient (P7) with smaller feet and liver involvement in addition to severe neurological involvement such as mental retardation, delayed motor and speech development, cerebral and cerebellar atrophy, microcephaly, hypotonia and recurrent seizures.
Wang et al. [97] described a patient (P8) with postural instability, walking difficulty, psychomotor delay, hypohidrosis, hyperkeratosis of the skin, macular hypoplasia, coagulation defects, liver lesions and ulnar deviation of the right-hand fingers. All the clinical features were mild but consistent with the features of other COG5-CDG patients.
3.1.5.2. Molecular analysis
P1's gDNA sequencing revealed an intronic homozygous mutation, c.1669-15 T > C 15 bps upstream of exon 15. Altered splicing caused skipping exons 15 and 16. Truncated COG5 mRNA was expressed but the mutant protein, which was predicted to have lost 58 amino acids, was not expressed. Very low levels of full-length COG5 were detected in P1's fibroblasts. Mutational analysis of P2 showed two heterozygous mutations in COG5 gene, c.556_560delAGTAAinsCT (p.S186_K187delinsL) in the maternal allele and c.1856 T > C (p.I619T) [c.95 T > G; p.M32R] in the paternal allele. P3.1, P3.2 and P3.3 had a homozygous nonsense mutation c.2518G > T (p.E840X) in COG5. P4 was heterozygous for two mutations, c.189delG (p.C64Vfs*6) and c.2338_2340dupATT; (p.I780dup) and P5 was homozygous for a missense mutation c.1780G > T (p.V594F). This mutation led to the skipping of exon 16. COG5 mutation resulted in about 80% depletion of COG7 protein levels in all these patients. From the whole-exome sequencing of P6.1, P6.2, P6.3 [95] a heterozygous mutation (c.1209delG; p.M403IfsX3) in COG5 was found. Curiously, though this was a heterozygous mutation, protein levels of full-length COG5 appeared to be severely depleted. Truncated COG5 was also expressed at low levels. P8 [96] was heterozygous for a missense mutation c.2324C > T (p.P775L) gene and a novel frameshift mutation c.330delT (p.V111Lfs*22) in the COG5 gene, each inherited from one parent. The author believes that these COG5 compound heterozygous mutations may cause glycosylation defects like other COG5-CDG. The frameshift mutation created a premature stop codon which generated a truncated 133-amino acid protein causing a loss of function in COG5. P8 had two novel compound heterozygous mutations. One was a nonsense mutation c.1290C > A (p.Y430X) and the other one was a missense mutation c.2077A > C (p.T693P). Low levels of COG5 protein were expressed and Golgi collapse into the ER was delayed upon BFA treatment [98].
Yong Ha et al. 2015 described the crystal structure of the complex between COG5 and COG7 subunits [44]. One of the features of the COG5–COG7 interaction is the involvement of the COG7 α1′, α2′ and α3′ helices to support the long COG5 helix α1. They also reported that disruption of the interaction between COG5 and COG7 subunit by site-directed mutations or in fibroblasts from patients with CDGs causes a drastic loss of COG function.
3.1.6. COG6
3.1.6.1. Clinical presentation
COG6-CDG was first (P1) described by Lübbehusen et al., in 2010 [70]. The patient suffered from a severe neurologic disease characterized by intractable focal seizures, vomiting, intracranial bleedings causing loss of consciousness, cholestasis, and vitamin K deficiency. A second COG6-CDG (P2) was reported, by Huybrechts et al. in 2012 [99]. Though both patients had the same mutation, they presented with different clinical features. P2 suffered from chronic inflammatory bowel disease, liver cirrhosis, mild psychomotor retardation, and microcephaly, associated with life-threatening and recurrent infections due to combined T and B immunodeficiency and neutrophil dysfunction. At the age of 6 years the patient suffered from immunodeficiency and enteropathy resulting in persistent infections, gastrointestinal inflammation, and malabsorption causing failure to thrive and died of brain edema. In 2013 Shaheen et al. [100] described a 12-year old male (P3) with COG6-CDG belonging to a large consanguineous family with a history of intellectual disability, hypohidrosis, abnormal teeth, acquired microcephaly, palmoplantar hyperkeratosis, and hepatitis. This was called Shaheen syndrome. Another COG6-CDG girl child (P4), with Shaheen syndrome was described by Althonaian et al. in 2018 [101]. Her clinical presentation included a global developmental disability, microcephaly, hypohydrosis, enamel hypoplasia, strabismus, splenomegaly and hypotonia. Additionally, the patient was also reported to have hemophagocytic lymphohistiocytosis (HLH) at four years of age as she exhibited the clinical signs including fever, splenomegaly, cytopenia, hypertriglyceridemia, hemophagocytosis in bone marrow, low NK cells and high ferritin. In 2015 Rymen et al. [102] assessed 7 new and 3 previously reported COG6-CDG patients, including two related to P1. They described the core features of COG6-CDG as liver involvement (9/10), microcephaly (8/10), developmental disability (8/10), recurrent infections (7/10), early lethality (6/10), and hypohidrosis predisposing for hyperthermia (6/10) and hyper-keratosis (4/10) as ectodermal signs. In 2019, Li et al. [103] reported another COG6-CDG (P5) that presented with growth retardation, developmental disability, microcephaly, liver and gastrointestinal disease, recurrent infections and. The expression of partially functional COG6 was thought to cause milder phenotypes than the previous reported patients and to avert early lethality. A retrospective study by Makhamreh et al. [104] reviewed 21 prenatal CDG cases having nonimmune hydrops fetalis (NIHF). From the 17 live births out of 21 cases, one was COG6-CDG. There are numerous factors that cause NIHF, and the authors suggested that developmental defects due to COG malfunction may be one possibility.
Though COG6-related pathologies include a whole spectrum of disorders of variable severity, the majority of patients display core features common to all COG-CDGs. 50% of the patients were deceased within the first 2 years of life and mortality was mainly due recurrent infections, hyperthermia and liver disease. The unique features of the COG6 clinical phenotype include hyperkeratosis, heart defects and liver involvement [102]. No immunodeficiency has previously been described in other COG-CDGs; however recurrent infections have been reported in COG4 and COG8 CDGs [99].
3.1.6.2. Molecular analysis
Lübbehusen et al. [70], found a single amino acid substitution (p.G549V) caused by a homozygous mutation c.1646G > T in P1. The mutated glycine residue is highly conserved in all eukaryotes. The mutation resulted in 80% depletion of COG6 protein. Protein levels of lobe B subunits, COG5 and COG7, were also affected. Overall, this COG6 mutation did not severely affect the COG complex's assembly; however, there was an abnormal accumulation of COG subcomplexes. P2 also had the same mutation. Detailed cellular and molecular analysis of P2 was not performed [99]. However, the clinical differences in both patients were attributed to variations in the level of mRNA degradation. Furthermore, glycan analysis of serum transferrin by mass-spectrometry revealed less hyposialylation. In P3 (Shaheen syndrome), through genome sequencing and autozygosity mapping a region spanning 6 genes on chromosome 13 was identified. A mutant variant in COG6 was identified among other variants in this region. An intronic mutation in COG6, c.1167-24A > G, created a new splice site resulting in a premature stop codon, p.G390FfsX6. Reverse-transcription PCR analysis of affected individuals' samples showed two bands, WT and mutant COG6, with a stark depletion in mutant COG6 mRNA. Only 30% of WT COG6 protein was expressed in the patient. However, there was no detectable glycosylation defect and IEF of serum transferrin showed normal N-glycosylation. STX6 protein was depleted by about 30%, indicating some loss of COG6 function. Thus, this is not a CDG causing COG mutation per se and the authors suggest wild-type COG6 depletion is the cause of CDG rather than the p.G549V mutation may be the cause of previously reported COG6-CDG. In other words, Shaheen et al. [100] believe that the mutation itself is not CDG causing. The mild developmental disability without liver involvement or glycosylation defects could be due to residual expression of the wild-type COG6 protein. P4, who had HLH, also carried the same mutation as P3, c.1167-24A > G [101]. Usually, biallelic mutations in genes such as STX11, UNC13D, STXBP2 and RAB27A cause primary HLH, but when no mutations were found in these genes, secondary HLH was diagnosed. Four novel COG6 mutations were identified by Rymen et al. in 2015 [102]. The first patient carried a homozygous c.511C > T (p.R171*) mutation. The second patient had a mutation c.1746 + 2 T > G resulting in altered splicing. He died at 1 year of age. The third patient had c.1238_1239insA (p.F414Lfs*4) mutation. She died at 15 months from liver failure. Two other patients, male siblings, both having c.785A > G (p.Y262C) mutation, were born to healthy non-consanguineous Moroccan parents. One of the siblings was 21 years old when reported. His brother died at 14 months from liver transplant complications. The fifth patient was a female child. She died at 5 months from necrotizing enterocolitis with disseminated intravascular coagulation and no CDG testing was performed. However, she was the sister of the patient described by Lübbehusen et al. in 2010. The sixth patient was also a child of non-consanguineous Turkish parents. She developed an infection at 15 days of birth and liver biopsy revealed fibrosis and cirrhotic changes. She carried two mutations c.511CNT (p.R171*) and c.1746 + 2G > T, the latter causing altered splicing. Her parents had another girl child who showed similar clinical features and died at 15 months; her genetic analysis was not performed. A detailed clinical analysis by Rymen et al., 2015 [102] showed that p.R171* and p.F41Lfs*4 loss-of-function mutations cause a severe clinical phenotype and early lethality while the G594V missense mutation involves variable clinical phenotypes. Impaired glycosylation is a common phenomenon of all of those patients, like other COG-CDG. P5 was a male child [103] heterozygous for mutations c.388C > T (G130*) and c.1A > G (p.?) in the COG6 gene, the former inherited from the father and the latter from the mother. The c.388C > T (G130*) is a nonsense loss-of-function mutation. Being the first base in the initiation codon, c.1A > G mutation was thought to inhibit translation or activate a downstream translation start site. The latter, which would result in the expression of a truncated but partially functionally COG6 subunit is believed to be the case here, but western blot analysis and the ability of the COG complex to assemble were not tested.
3.1.7. COG7
3.1.7.1. Clinical presentation
The first identified COG-CDG causing mutation was in COG7, and it would later come to be known as the most severe of all COG-CDGs. In 2004, in the Mediterranean area, two siblings, male (P1) and female (P2), of healthy consanguineous parents presented with the CDG phenotypes [105,106]. Both siblings had clinical features of CDG patients, which included perinatal asphyxia, dysmorphia, skeletal deformities, hypotonia and hepatosplenomegaly. Postnatally, they developed jaundice, recurrent epilepsies, infections and died from cardiac failure. In 2007, 4 children from 3 Morrocan families were reported with similar lethal mutation in COG7 gene. In all the 3 families, the parents were consanguineous [107]. Two were female patients, and the other two were male and female siblings, all died within the first year of birth. The patients exhibited clinical presentations including growth retardation, progressive, severe microcephaly, hypotonia, adducted thumbs, feeding problems by gastrointestinal pseudo-obstruction, failure to thrive, cardiac anomalies, wrinkled skin and episodes of extreme hyperthermia.
3.1.7.2. Molecular analysis
In the case of the first two patients, COG7 cDNA sequencing revealed deletion of 19 bases of the first exon. gDNA sequencing indicated a homozygous intronic point mutation IVS1 + 4 A → C (c.169 + 4 A > C) that impaired splicing. 19 bp deletion in the COG7 mRNA occurred due to the use of alternative splice site close to the first intron/exon boundary and resulted in dramatic reduction at mRNA and protein levels (95% in P1 and 85% in P2). Hence, a small amount of normal COG7 mRNA was detectable in the patients' fibroblasts. However, N- and O- glycosylation defects were confirmed by the IEF pattern of serum transferrin and apolipoprotein C-III. Staining of patient samples with Concanavalin A, which binds to early branched mannose N-glycan, indicated normal ER glycan assembly. Moreover, only P1 fibroblasts showed differences in glycan processing. Differences in RCA-1 binding between P1 and control fibroblasts indicated defects in Golgi glycan processing, particularly sialylation because in N-glycosylation, galactose is the penultimate glycan and is masked by terminal sialic acid. Further analysis showed sialylation was impaired due to decreased activity of the nucleotide-sugar transporter as well as the glycosyltransferase in P1 but not in P2. Based on the clinical presentation, and defects in multiple glycosylation pathways, the COG complex's integrity and functionality were assessed. Lobe A was Golgi localized but all lobe B subunits had diffused cytosolic staining and COG7 staining was undetectable [105]. In addition, BFA-induced Golgi collapse into the ER was delayed. Further analysis of the patients' fibroblasts revealed that COG sensitive proteins, GOSR1 and BET1L, were mislocalized and depleted. ERGIC-53 was mislocalized to the Golgi, indicating that ER-to-Golgi retrograde recycling was impaired [108]. Glycosylation defects, both N- and O- linked, were confirmed by IEF profiles of serum transferrin, apoCIII [107] as well as by increased PNA lectin binding to the COG7 deficient fibroblasts. A different intronic mutation, c.170-7A > G in COG7 was identified in two COG7-CDG siblings of consanguineous Moroccan parents. This mutation also activated a cryptic splice site upstream of the canonical one. As a result, 6 base pairs were inserted between exon 1 and 2 in the mutant mRNA. The mutant protein subunit contained additional amino acids, alanine and threonine. The mutation had milder physiological effects compared to the first c.169 + 4 A > C mutation. N- and O- linked glycosylation effects were confirmed by IEF of serum transferrin and apoC-III. The protein levels of COG6, COG7 and COG8 were depleted by 20–30% and COG5 was depleted by about 60%. Also, the Golgi largely failed to redistribute to the ER upon BFA treatment [109].
3.1.8. COG8
3.1.8.1. Clinical presentation
In 2007, an 8-year old girl child, born to consanguineous Spanish parents was presented with acute encephalopathy, loss of psychomotor abilities, mild dysmorphia, hypotonia, alternating esotropia, pseudo-ptosis, unregulated coagulation, mental retardation, gradually worsening cerebellar ataxia and PFAPA syndrome (Periodic Fever, Aphthous Stomatitis, Pharyngitis, Adenitis) during infancy. She showed oculomotor apraxia with dysinergia oculocephalica, pseudo-ptosis and alternating esotropia at 7 years of age [37]. In the same year, another COG8-CDG patient was reported [110]. He presented with hypotonia, decreased reflexes, chronic axonal neuropathy, ventriculomegaly ex vacuo with atrophy, high levels of lactate in the gray matter at 7 months, seizures, and esotropia/amblyopia in the first two years of birth. Severe retardation was evident at 8 years of age. He also had keratosis pilaris, mild spasticity and contractures, lacked bowel/bladder control and appeared malnourished due to lack of muscle form all areas. Yang et al. 2017 [111] described another patient with COG8 mutation presenting the clinical features including hypotonia, failure to thrive, elevated serum liver enzymes, microcephaly, skeletal deformities and mild psychomotor retardation. This patient exhibited milder psychomotor retardation without a seizure history or severe ocular symptoms, unlike other COG8-CDG patients. Arora et al. 2019 [112] reported a patient who was severely affected compared to the 3 earlier COG8-CDG patients. Antenatal conditions included increased nuchal translucency, arthrogryposis multiplex congenita, Dandy-Walker malformation and fetal growth restrictions. At birth he presented with facial dysmorphia, bilateral camptodactyly and required ventilator support. He died 2 days later. He had an elder sibling who also died shortly after birth. Autopsy revealed in neurological defects, including dilated ventricles and missing corpus callosum, vermian agenesis and enlarged cisterna magna.
3.1.8.2. Molecular analysis
In the first reported patient, cDNA sequencing of all COG subunits revealed a nonsense mutation (c.1611C > G) in COG8 gene. The premature stop codon resulted in 76 amino acids missing at the C-terminus (Y537X) and the expression of the truncated protein was 25% of wild type COG8 protein. Glycosylation defects in this patient were detected by IEF profiles of serum transferrin and ApoC-III and increased PNA lectin staining in patient fibroblasts which led to a CDG diagnosis. AA glycan mass-spectrometry confirmed the diagnosis. Loss of COG function had different effects on the abundance of two Golgi glycosylation enzymes - MAN2A and B4GalT1. The former was unaffected while the latter was reduced. However, both lobe A and lobe B subunits were depleted except for COG4 and COG5. Co-immunoprecipitation and velocity gradient fractionation showed that the mutant COG8 subunit was unable to interact with COG1, which precluded octameric COG assembly. Furthermore, COG8, COG1, COG2, COG6 and COG7 did not localize to the Golgi, but localization of COG3, COG4 and COG5 were unaffected [37]. The second patient carried two heterozygous mutations, IVS3 + 1G > A, in intron 3 and a two-nucleotide deletion, TT1687–1688, in exon 5. IVS3 + 1G > A alters the conserved splice sequence from GT to AT. This activates an alternate splice site in exon 3 truncating the mRNA by 500 bases and the protein by 306C-terminal amino acids. In the second case, the protein is truncated by 47 amino acids due a premature stop codon produced by the frameshift. Hyposialylation, due to reduced ST3Gal1 activity, was observed by IEF of serum transferrin, lectin binding and mass spectrometry. In the patient's fibroblasts COG8 protein was severely depleted and not detectable by western blot. COG1, COG5, COG6 and COG7 were also depleted by 60–70% [113]. Only COG2, COG3 and COG4 had perinuclear localization. Loss of COG8 affected the assembly of the octameric COG complex. In addition, the abundance of GOSR1 and BETL1 was reduced, and the formation of the STX5 Golgi SNARE complex was impaired. COG8 depletion also affected the abundance of VAMP4 and the assembly of the STX16 SNARE complex, negatively affecting endosome to TGN retrograde trafficking. In 2017, Yang et al. found two novel frameshift mutations, c.171dupG (p.L58Afs*29) and c.1656dupC (p.A553Rfs*15) in COG8 [111]. The patient described by Arora et al. 2019, was homozygous for the c.1583-1G > A mutation present at the boundary of intron 4 and exon 5. It is interesting that four out of six reported mutations in COG8 were present in exon 5 suggesting that it may be a hotspot for mutations [112].
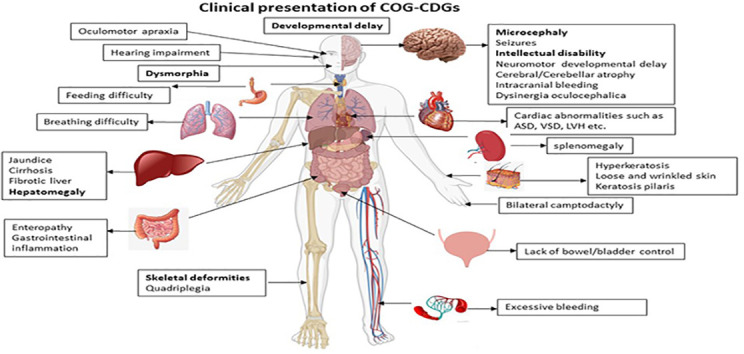
4. Summary
The cellular phenotype and clinical presentations of COG-CDGs are heterogeneous, but the patients mainly exhibit neurological, morphological, skeletal, and hepatic abnormalities [94,102,114] (Fig. 4 ). One of the main reasons for heterogeneity could be because the mutations have varying effects on both stability and specific function of the mutant COG subunits (see Table 1 ) and the COG complex as a whole. Defects in Golgi glycosylation, particularly reduced sialylation and galactosylation, is the most common feature between COG-CDG patients and COG KO cell lines. Importantly, complete depletion of individual COG subunits in human cell lines resulted in more homogenous and striking cellular phenotypes as described earlier. To get a complete understanding of the clinical phenotypes and to design a working treatment protocol, COG-CDG patients should be tested for all COG-related abnormalities, specifically in cells obtained from biopsies of most affected organ systems.
Fig. 4.

The COG-CDG phenotype in humans. Effect of COG-CDGs on various organs/organ-systems. The most common and prominent COG-CDG clinical phenotypes are in bold. This figure was made using BioRender.
Table 1.
Molecular and clinical presentations of COG complex mutations.
| Gene | Mutation | Effect on mutant subunit | Effect on COG complex | Clinical presentation | Refs |
|---|---|---|---|---|---|
| COG1 | Homozygous 2659-2660insC (p.P888fsX900) |
Truncated by 80aa at C-terminus due to premature stop codon Around 15% of mutant COG1 expressed and weakly localized to the Golgi |
Depletion of all lobe A subunits and COG8 | Feeding problems since birth, failure to thrive, hypotonia, psychomotor and growth retardation, short stature, microcephaly, liver and spleen enlargement, small hands and feet, straightened bitemporal space, antimongoloid eyelids, cardiac abnormalities included ventricular hypertrophy | [87] |
| Homozygous c.1070 + 5G > A |
Mutant COG1 truncated by 321aa due to loss of exon 6 and new stop codon in exon 7. Truncated COG1 not expressed. Low protein levels of WT COG1 present |
Decreased COG8 protein levels | More severe than the previous COG1-CDG. Cerebrocostomandibular syndrome including mental retardation, cerebellar atrophy, renal and cardiac abnormalities, arthrogryposis, hearing loss and skeletal abnormalities. |
[114] | |
| COG2 | Heterozygous mutations:
|
c.701dup mRNA is not expressed. c.1900 T > G mRNA expressed is low levels. COG2 protein is non-detected. |
Decreased COG3, COG4 levels | Growth retardation, microcephaly, spastic quadriplegia, liver dysfunction, hypocupremia, hypoceruloplasminemia, cerebral atrophy and tonic seizures | [89] |
| COG4 |
c.2185C > T (p.R729W) 16q22 deletion |
80% depletion of COG4 protein | Decreased COG1, COG2, COG3 COG5 levels | Mild psychomotor retardation, dysmorphia, epilepsia | [90] |
Heterozygous
|
Degradation of c.697G > T mRNA 70% depletion of COG4 |
No significant effect on other COG subunits | [92] | ||
Heterozygous de novo
|
– | No effect on other subunits | Skeletal dysplasia caused by Saul-Wilson Syndrome, developmental delay and dysmorphic features | [91] | |
| COG5 | Homozygous c.1669-15 T > C |
Intronic substitution caused skipping of exons 15 & 16. Truncated COG5 (by 58aa) not expressed. Low levels of WT COG5 expressed |
– | Mild neurological phenotype including hypotonia, psychomotor retardation, delayed speech development and truncal ataxia | [93] |
Heterozygous
|
Deletion predicted to be in frame Affected M is in a highly conserved region 75% depletion of COG5 |
About 90% depletion of COG5 | Same as above with cirrhotic liver and hepatosplenomegaly | [94] | |
| Homozygous c.2518G > T (p.E840X) |
75% depletion of COG5 | Microcephaly, delayed speech, and motor development, moderate to severe mental retardation, hypotonia, mild dysmorphia, microcephaly, short stature and walking difficulty | [69] | ||
Heterozygous
|
Same as above with blindness and deafness | ||||
| Homozygous c.1780G > T (p.V594F) |
Skipping of exon 16 75% depletion of COG5 |
Same as above with blindness and deafness. | |||
| Heterozygous c.1209delG (p.M403IfsX3) |
Depletion of WT COG5. Low levels of mutant COG5 also present. |
– | Friedreich's-ataxia-like phenotype including cerebellar atrophy, mild to moderate intellectual disability, and scoliosis. | [95] | |
Heterozygous
|
P775 and V111 are highly conserved in mammals. Truncated p.V111Lfs*22 (by 690aa) non- functional |
– | Most severe COG5-CDG phenotype including severe mental retardation, delayed speech and motor development, microcephaly, cerebral and cerebellar atrophy, hypotonia, recurrent seizures, liver involvement and small feet. | [96] | |
Heterozygous
|
T693 is highly conserved 80% depletion of WT COG5. Truncated COG5 present at very low levels |
– | Neonatal jaundice, recurrent upper respiratory tract infections, hypohidrosis, hyperkeratosis, ulnar deviation and delayed psychomotor development. | [97] | |
Heterozygous
|
– | – | Neurodevelopmental disorders, non-syndromic intellectual disability | [134] | |
| COG6 |
Homozygous c. 1646G > T (p.G549V) |
80% depletion of COG6 | Decreased COG5, COG7 levels. Assembly of octameric COG unaffected |
Fatal (2 siblings died within 2 months of birth.) Major neurologic involvement, intractable focal seizures, vomiting, intracranial bleedings causing loss of consciousness, cholestasis, vitamin K deficiency, brain oedema, necrotizing enterocolitis with disseminated intravascular coagulation. |
[70,135] |
| – | – | Fatal. Died at the age of 6 Chronic inflammatory bowel disease, gastrointestinal malabsorption, liver cirrhosis, mild psychomotor retardation, and microcephaly, recurrent infections due to immunodeficiency, failure to thrive. |
[99] | ||
| c.1167-24A > G (p.G390FfsX6) | Intronic mutation created a new splice site. Mutant COG6 not expressed 30% of WT COG6 protein present |
– | Shaheen syndrome (mild intellectual disability, hypohidrosis, abnormal teeth, and acquired microcephaly, palmoplantar hyperkeratosis) | [100] | |
| – | – | Shaheen syndrome, strabismus, splenomegaly, hypotonia, HLH | [101] | ||
| Homozygous c.511C > T (p.R171*) |
– | Fatal. Died a few days after birth. Respiratory failure extreme hypotonia |
[102] | ||
| Homozygous c.1746 + 2 T > G |
Splice site mutation | – | Fatal. (All patients died within 1 year) Developmental disability, chronic diarrhea with failure to thrive, splenomegaly, hypotonia and recurrent infections, bilateral sensorineural hearing loss, enlarged extra-axial CSF spaces and asymmetric lateral ventricles. |
||
| Homozygous c.1238_1239insA (p.F414Lfs*4) |
– | – | Fatal. Died at the age of 1 year. Psychomotor disability, failure to thrive, chronic diarrhea and hepatosplenomegaly, recurrent infections from immune dysfunction, liver failure |
||
Heterozygous
|
– | – | Microcephaly, splenomegaly, and delay in motor development, gastroenteritis, high fever, and convulsions, recurrent infections from immune dysfunction | ||
Heterozygous
|
– | – | 2 siblings, one died at 15 months Diarrhea, fever at birth due to viral infection, hepatosplenomegaly, cirrhotic liver, psychomotor disability, hypotonia, scoliosis, microcephaly, dysmorphia, and growth retardation recurrent infections and hyperthermia |
||
| COG7 | Homozygous IVS1 + 4 A → C aka c.169 + 4A > C |
Intronic mutation in splice site 19 bp deletion due to altered splicing and premature stop codon Degradation of COG7 mRNA Less than 20% protein expressed |
Decreased COG5 (95%), COG6 (70%), and COG8 (71%) Lobe A associated with the Golgi, not lobe B |
Fatal. Cardiac failure and other complications within the first year of birth. Perinatal asphyxia, dysmorphia, skeletal deformities, hypotonia, hepatosplenomegaly, neonatal jaundice, recurrent epilepsies, infections |
[[105], [106], [107]] |
| Homozygous c.170-7A > G (p.56–57insAT) |
Activation of upstream cryptic splice site inserted 6 bps between exon 1 and 2 About 70% protein expressed |
COG6 and COG8 levels decreased by 20% COG5 decreased by 60% |
Milder clinical phenotype including growth retardation, failure to thrive, feeding problems, hypotonia, recurrent hyperthermia and cerebral atrophy | [109] | |
| COG8 | Homozygous c.1611C > G (p.Y537X) |
Loss of 76-C terminus amino acids due to a premature stop codon. 25% of truncated COG8 is expressed |
Decreased COG1, COG2, COG3, COG6 and COG7 Golgi localization of COG subunits unaffected. Octameric COG assembly affected |
Acute encephalopathy, loss of psychomotor abilities, mild dysmorphia, hypotonia, alternating esotropia, pseudo-ptosis, unregulated coagulation, mental retardation, cerebellar ataxia, PFAPA syndrome during infancy, oculomotor apraxia with dysinergia oculocephalica and pseudo-ptosis | [37] |
Heterozygous
|
G > A in intron 3 alters splicing leads to protein truncation by 306aa at the C-terminal TT deletion in exon 5 creates a premature stop codon and truncates the protein by 76aa COG8 protein is undetected |
Decreased COG1, COG5, COG6 and COG7 by 60–70% COG2, COG3 and COG4 associated with the Golgi |
Severe growth retardation, Hypotonia, decreased reflexes, chronic axonal neuropathy, ventriculomegaly ex vacuo with atrophy, seizures, esotropia/amblyopia, keratosis pilaris, mild spasticity and contractures, and no bowel/bladder control. | [110] | |
Heterozygous
|
– | – | Psychomotor retardation, hypotonia, failure to thrive, microcephaly, skeletal deformities, and mild psychomotor retardation. | [111] | |
| Homozygous c.1583-1G > A |
G > A at intron-exon boundary of intron 4 | – | Fatal. Died 2 days after birth with severe neurological defects. Antenatal conditions included increased nuchal translucency, arthrogryposis multiplex congenita, Dandy-Walker malformation and fetal growth restrictions. |
[112] |
In all reported COG-CDGs, there is a significant impact on the nervous system. This underscores the importance of the COG complex in the development and function of this system. Key players in synaptic transmission are glycosylated and glycans are involved in neurotransmitter release, receptor binding and uptake [115]. Severe changes in copper homeostasis cause neurodegeneration and COG KO cells show altered stability and localization of copper transporters ATP7A and SLC31A1 [116]. Furthermore, altered glycosphingolipids synthesis in the COG-CDGs could be another possible explanation for neuronal involvement [117]. A complete block in N-glycosylation in mice leads to embryonic lethality. Yet, COG mutations do not lead to embryonic lethality suggesting that there is some degree of tolerance to COG malfunction during embryonic development.
Reynders et al. 2009 [90] compared three COG4-CDG patients with COG1, COG7 and COG8-CDG patients. They concluded that COG1 and COG4 patients present milder problems in comparison to COG7 and COG8 patients. Among all of the COG-CDG, COG7 mutation is most severe. In the case of COG6-CDG, 50% of patients died within the first 2 years of life which indicates it is nearly as severe as COG7-CDG deficiency. There is some additional clinical overlap between COG6 and COG7 patients but, surprisingly, less similarity is observed for CDGs associated with the other lobe B subunit, COG5 [102]. Zeevaert et al. [114] observed that COG8 deficiency mostly had neurological involvement, whereas growth retardation was a predominant feature in COG1 and COG7-CDG patients and dysmorphic features of face and hands are common in all CDG patients. Given the small cohort of COG-CDGs that have been reported, it is difficult to make further correlations between the affected subunit and clinical manifestations. Comparisons between the clinical phenotype of COG-CDGs should take into consideration the effect of the mutation on the affected COG subunit. In the recent retrospective analysis of COG-CDGs, Haijes et al. [118] made such a comparison and concluded that lobe A mutations are more detrimental than lobe B mutation, but the discussion is not settled mainly because of the small number of known mutations and lack of robust data on these patients.
Treating congenital multi-systemic disorders is challenging and one single approach could be insufficient. A good example is the treatment of MPI-CDG. Clinical presentation of MPI-CDG includes but is not limited to liver fibrosis and gastrointestinal complications, thrombosis, and hypoglycemia. With oral mannose intake and liver transplantation, the patient improved dramatically [119]. Similarly, galactose supplementation has been used to alleviate some of the pathologies of PGM1-CDG [120]. Patients with mutations in TMEM165 have CDG-II and their glycan profiles reveal hypogalactosylation. Two patients with TMEM165-CDG responded positively to galactose supplementation [121]. COG mutations cause a wide range of defects that directly (glycosylation enzymes, sugar transporters) and indirectly (Golgi homeostasis, cisternal compartmentalization, and stability of SNAREs and GEARs) impact glycosylation and other trafficking processes. Interestingly, data from our lab indicates that TMEM165 is dependent on the COG complex for its stability and its depletion in COG KO cells may contribute to misglycosylation (unpublished observations). If this prediction is correct, galactose therapy may partially alleviate hypoglycosylation in COG-CDGs. It is important to note that COG mutations not only affect the Golgi, but also the endolysosomal system, causing altered secretion and potential malfunction of the lysosomal degradation machinery [65]; therefore, treatment of COG-CDG patients should combine strategies used for the treatment of both CDG and lysosomal disorders. Antisense therapy and gene therapy have not yet been well fully explored for the treatment of CDGs [122]. In TMEM165-CDG patient fibroblast, antisense therapy successfully rescued exon skipping due to an intronic mutation and drove expression of the WT protein [123]. This approach is applicable where an intronic mutation leads to the activation of a cryptic splice site upstream of the canonical splice site. Antisense morpholino oligonucleotides block the cryptic splice site and promote canonical splicing. With the advancement in research on gene therapy, it could soon become a reality for the treatment of COG-CDGs. Theoretically, gene therapy should have promising outcomes in the treatment of COG-CDGs since the introduction of the WT copy of the affected COG subunit fully rescues COG-CDG defects in cell lines [63]. At present, the significant challenges/risks include safe vehicular delivery and targeted integration of the delivered gene. Gene therapy, through intravenous and intramuscular liposomal delivery, rescued GNE-CDG (UDP-GlcNAc2 epimerase-CDG) in mice [124]. The establishment of a reliable animal COG disease model will benefit the development of therapeutics. There are some encouraging attempts to explore fly [125] and fish [91] models to study COG disorders. To date, all major COG studies have come from yeast and mammalian cell lines, including COG-CDG patient fibroblasts. Patient fibroblasts are not well representative of all the organ systems and cell types that are affected by COG mutations; therefore iPS (induced pluripotent stem) cells made from patients' fibroblasts will be a good starting point to explore potential therapeutics.
5. Future questions
From more than 30 years of research, the COG complex has emerged as the major player in Golgi trafficking. COG has been studied in single-celled yeast, higher eukaryotes, mammalian cell lines and COG-CDG patients. Its tethering function, interactions with the Golgi trafficking machinery, the impact of its deficiency on glycosylation and Golgi trafficking are well established.
Nevertheless, the molecular mechanism of COG-mediated vesicle tethering is only speculative and the exact molecular reactions and sequence of events leading to docking and fusion of retrograde membrane carriers at the Golgi are not fully understood. COG has multiple interactions with other components of a vesicle fusion machinery, but the temporal sequence of these protein-protein interactions is not clear. The prediction is that at least some COG interactions with the Golgi trafficking machinery are sequential rather than simultaneous since individual subunits interact with more than one trafficking partner. In vitro reconstitution of the COG complex function will undoubtedly help in answering this and other mechanistic questions. So far, successful in vitro reconstitution was achieved only for one MTC, yeast HOPS complex [126].
Which molecules are responsible for the COG complex's recruitment to the Golgi membrane? CATCHRs employ diverse strategies for membrane association: Dsl1 is membrane-associated by ER anchored SNAREs [127]. The GARP complex is recruited by Arl5 [128] and the exocyst interacts with both Rabs and PM lipid PIP2 [32]. Are COG interacting Rabs or SNAREs responsible for its recruitment to the Golgi rims, or is it Golgi membrane lipids? It is likely that COG uses several membrane attachment strategies since the deletion of individual Rabs fails to dislocate COG to the cytosol (our unpublished observation).
Members of the CATCHR complexes have three (Dsl1), four (GARP and EARP), or eight (COG and exocyst) subunits. They all share structural homology and have common α-helical compact folds despite low sequence homology. This indicates that they evolved from a common ancestor [129]. Why did the exocyst and COG acquire double the number of subunits? Do they perform any additional functions than the Dsl1 and GARP? Dsl1 and COG tether COPI coated vesicles at the ER and Golgi, respectively. Is it possible that the COG also tethers recycling membrane intermediates in a COPI independent pathway?
Does the COG complex solely function in intra-Golgi retrograde trafficking? Several publications suggest that the COG complex has other, possibly subordinate, roles outside of the Golgi arena. At the TGN, COG1 shows specific interaction with RINT-1, a component of the Dsl1/ZW10 complex [130]. Meanwhile, COG6 interacts with GARP partner SNARE STX6 [131] and we have detected multiple COG-GARP and COG-exocyst protein-protein interactions in cell lysates (unpublished observations). Does it mean that CATCHR complexes can share their subunits to perform new functions? Is there a cross-talk between the CATCHRs and other vesicle tethering factors?
In yeast, the cytoplasm to vacuole targeting pathway is dependent on the activity of COG lobe A subunits. Autophagosome formation and the sorting of Atg proteins to the site of nucleation is affected in COG mutant cells [132]. This suggests that the COG complex might be involved in tethering events leading to autophagosome formation. Furthermore, one of the Atg proteins, Atg6 is dispersed in COG2 yeast mutants while several other Atg proteins have been shown to interact with COG subunits in yeast two-hybrid assays. At the TGN, COG8 controls Atg9 trafficking in collaboration with Arl1 and Arl3 GTPases [133]. If indeed COG is involved in autophagy, it would be worth studying the status of autophagy in COG-CDG patients and if found faulty, weigh in its impact on the COG-CDG phenotype.
6. Conclusion
The first COG subunits were discovered by the Monty Krieger lab 35 years ago [56]; it took almost 20 years to realize that these proteins act together as an evolutionarily conserved vesicle tethering complex [29,39] and that mutations in COG subunits are responsible for a whole family of human CDG-II diseases [105,114]. We hope that the potent combination of molecular (gene editing and stem cell manipulations), biochemical (purification of active proteins and complexes and in vitro reconstitution), microscopical (superresolution and cryo-EM microscopy) techniques and development of advanced animal models will allow not only for the complete understanding of COG complex cellular function(s) but also for the efficient diagnostics and treatment of COG-related human disorders.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
We would like to thank Irina Pokrovskaya for the EM pictures and all members of Lupashin's lab for helpful discussion. This work was supported by the NIH grant GM083144. The authors declare no competing financial interests.
References
- 1.Pfeffer S.R. A prize for membrane magic. Cell. 2013;155:1203–1206. doi: 10.1016/j.cell.2013.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dacks J.B., Field M.C. Evolutionary origins and specialisation of membrane transport. Curr. Opin. Cell Biol. 2018;53:70–76. doi: 10.1016/j.ceb.2018.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonifacino J.S., Glick B.S. The mechanisms of vesicle budding and fusion. Cell. 2004;116:153–166. doi: 10.1016/s0092-8674(03)01079-1. [DOI] [PubMed] [Google Scholar]
- 4.Rout M.P., Field M.C. The evolution of Organellar coat complexes and Organization of the Eukaryotic Cell. Annu. Rev. Biochem. 2017;86:637–657. doi: 10.1146/annurev-biochem-061516-044643. [DOI] [PubMed] [Google Scholar]
- 5.Bethune J., Wieland F.T. Assembly of COPI and COPII vesicular coat proteins on membranes. Annu. Rev. Biophys. 2018;47:63–83. doi: 10.1146/annurev-biophys-070317-033259. [DOI] [PubMed] [Google Scholar]
- 6.Cross J.A., Dodding M.P. Motor-cargo adaptors at the organelle-cytoskeleton interface. Curr. Opin. Cell Biol. 2019;59:16–23. doi: 10.1016/j.ceb.2019.02.010. [DOI] [PubMed] [Google Scholar]
- 7.Pylypenko O., Hammich H., Yu I.M., Houdusse A. Rab GTPases and their interacting protein partners: structural insights into Rab functional diversity. Small Gtpases. 2018;9:22–48. doi: 10.1080/21541248.2017.1336191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Viotti C. ER to Golgi-dependent protein secretion: the conventional pathway. Methods Mol. Biol. 2016;1459:3–29. doi: 10.1007/978-1-4939-3804-9_1. [DOI] [PubMed] [Google Scholar]
- 9.Gillingham A.K., Munro S. Transport carrier tethering - how vesicles are captured by organelles. Curr. Opin. Cell Biol. 2019;59:140–146. doi: 10.1016/j.ceb.2019.04.010. [DOI] [PubMed] [Google Scholar]
- 10.Wang H., Zhang C., Xiao H. Mechanism of membrane fusion: protein-protein interaction and beyond. Int. J. Physiol. Pathophysiol. Pharmacol. 2019;11:250–257. [PMC free article] [PubMed] [Google Scholar]
- 11.Rothman J.E., Krishnakumar S.S., Grushin K., Pincet F. Hypothesis - buttressed rings assemble, clamp, and release SNAREpins for synaptic transmission. FEBS Lett. 2017;591:3459–3480. doi: 10.1002/1873-3468.12874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Storrie B., Pepperkok R., Nilsson T. Breaking the COPI monopoly on Golgi recycling. Trends Cell Biol. 2000;10:385–391. doi: 10.1016/s0962-8924(00)01818-3. [DOI] [PubMed] [Google Scholar]
- 13.Casler J.C., Papanikou E., Barrero J.J., Glick B.S. Maturation-driven transport and AP-1-dependent recycling of a secretory cargo in the Golgi. J. Cell Biol. 2019;218:1582–1601. doi: 10.1083/jcb.201807195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu L., Doray B., Kornfeld S. Recycling of Golgi glycosyltransferases requires direct binding to coatomer. Proc. Natl. Acad. Sci. U. S. A. 2018;115:8984–8989. doi: 10.1073/pnas.1810291115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eckert E.S., Reckmann I., Hellwig A., Rohling S., El-Battari A., Wieland F.T., Popoff V. Golgi phosphoprotein 3 triggers signal-mediated incorporation of glycosyltransferases into coatomer-coated (COPI) vesicles. J. Biol. Chem. 2014;289:31319–31329. doi: 10.1074/jbc.M114.608182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pelham H.R., Munro S. Sorting of membrane proteins in the secretory pathway. Cell. 1993;75:603–605. doi: 10.1016/0092-8674(93)90479-A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Holthuis J.C., Pomorski T., Raggers R.J., Sprong H., Van Meer G. The organizing potential of sphingolipids in intracellular membrane transport. Physiol. Rev. 2001;81:1689–1723. doi: 10.1152/physrev.2001.81.4.1689. [DOI] [PubMed] [Google Scholar]
- 18.Welch L.G., Munro S. A tale of short tails, through thick and thin: investigating the sorting mechanisms of Golgi enzymes. FEBS Lett. 2019;593:2452–2465. doi: 10.1002/1873-3468.13553. [DOI] [PubMed] [Google Scholar]
- 19.Banfield D.K. Mechanisms of protein retention in the Golgi. Cold Spring Harb. Perspect. Biol. 2011;3:a005264. doi: 10.1101/cshperspect.a005264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Axelsson M.A., Karlsson N.G., Steel D.M., Ouwendijk J., Nilsson T., Hansson G.C. Neutralization of pH in the Golgi apparatus causes redistribution of glycosyltransferases and changes in the O-glycosylation of mucins. Glycobiology. 2001;11:633–644. doi: 10.1093/glycob/11.8.633. [DOI] [PubMed] [Google Scholar]
- 21.Hassinen A., Pujol F.M., Kokkonen N., Pieters C., Kihlstrom M., Korhonen K., Kellokumpu S. Functional organization of Golgi N- and O-glycosylation pathways involves pH-dependent complex formation that is impaired in cancer cells. J. Biol. Chem. 2011;286:38329–38340. doi: 10.1074/jbc.M111.277681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hassinen A., Khoder-Agha F., Khosrowabadi E., Mennerich D., Harrus D., Noel M., Dimova E.Y., Glumoff T., Harduin-Lepers A., Kietzmann T., Kellokumpu S. A Golgi-associated redox switch regulates catalytic activation and cooperative functioning of ST6Gal-I with B4GalT-I. Redox Biol. 2019;24:101182. doi: 10.1016/j.redox.2019.101182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X., Wang Y. Glycosylation quality control by the Golgi structure. J. Mol. Biol. 2016;428:3183–3193. doi: 10.1016/j.jmb.2016.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ungermann C., Kummel D. Structure of membrane tethers and their role in fusion. Traffic. 2019;20:479–490. doi: 10.1111/tra.12655. [DOI] [PubMed] [Google Scholar]
- 25.Kim J.J., Lipatova Z., Segev N. TRAPP complexes in secretion and autophagy. Front. Cell Dev. Biol. 2016;4:20. doi: 10.3389/fcell.2016.00020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu I.-M., Hughson F.M. Tethering factors as organizers of intracellular vesicular traffic. Annu. Rev. Cell Dev. Biol. 2010;26:137–156. doi: 10.1146/annurev.cellbio.042308.113327. [DOI] [PubMed] [Google Scholar]
- 27.Dong G., Hutagalung A.H., Fu C., Novick P., Reinisch K.M. The structures of exocyst subunit Exo70p and the Exo84p C-terminal domains reveal a common motif. Nat. Struct. Mol. Biol. 2005;12:1094–1100. doi: 10.1038/nsmb1017. [DOI] [PubMed] [Google Scholar]
- 28.Sivaram M.V., Furgason M.L., Brewer D.N., Munson M. The structure of the exocyst subunit Sec6p defines a conserved architecture with diverse roles. Nat. Struct. Mol. Biol. 2006;13:555–556. doi: 10.1038/nsmb1096. [DOI] [PubMed] [Google Scholar]
- 29.Whyte J.R., Munro S. The Sec34/35 Golgi transport complex is related to the exocyst, defining a family of complexes involved in multiple steps of membrane traffic. Dev. Cell. 2001;1:527–537. doi: 10.1016/s1534-5807(01)00063-6. [DOI] [PubMed] [Google Scholar]
- 30.Zhang X., Orlando K., He B., Xi F., Zhang J., Zajac A., Guo W. Membrane association and functional regulation of Sec3 by phospholipids and Cdc42. J. Cell Biol. 2008;180:145–158. doi: 10.1083/jcb.200704128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.He B., Xi F., Zhang X., Zhang J., Guo W. Exo70 interacts with phospholipids and mediates the targeting of the exocyst to the plasma membrane. EMBO J. 2007;26:4053–4065. doi: 10.1038/sj.emboj.7601834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pleskot R., Cwiklik L., Jungwirth P., Zarsky V., Potocky M. Membrane targeting of the yeast exocyst complex. Biochim. Biophys. Acta. 2015;1848:1481–1489. doi: 10.1016/j.bbamem.2015.03.026. [DOI] [PubMed] [Google Scholar]
- 33.Blackburn J.B., D’Souza Z., Lupashin V.V. Maintaining order: COG complex controls Golgi trafficking, processing, and sorting. FEBS Lett. 2019;593:2466–2487. doi: 10.1002/1873-3468.13570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ungar D., Oka T., Vasile E., Krieger M., Hughson F.M. Subunit architecture of the conserved oligomeric Golgi complex. J. Biol. Chem. 2005;280:32729–32735. doi: 10.1074/jbc.M504590200. [DOI] [PubMed] [Google Scholar]
- 35.Oka T., Vasile E., Penman M., Novina C.D., Dykxhoorn D.M., Ungar D., Hughson F.M., Krieger M. Genetic analysis of the subunit organization and function of the conserved oligomeric golgi (COG) complex: studies of COG5- and COG7-deficient mammalian cells. J. Biol. Chem. 2005;280:32736–32745. doi: 10.1074/jbc.M505558200. [DOI] [PubMed] [Google Scholar]
- 36.Fotso P., Koryakina Y., Pavliv O., Tsiomenko A.B., Lupashin V.V. Cog1p plays a central role in the organization of the yeast conserved oligomeric Golgi complex. J. Biol. Chem. 2005;280:27613–27623. doi: 10.1074/jbc.M504597200. [DOI] [PubMed] [Google Scholar]
- 37.Foulquier F., Ungar D., Reynders E., Zeevaert R., Mills P., Garcia-Silva M.T., Briones P., Winchester B., Morelle W., Krieger M., Annaert W., Matthijs G. A new inborn error of glycosylation due to a Cog8 deficiency reveals a critical role for the Cog1-Cog8 interaction in COG complex formation. Hum. Mol. Genet. 2007;16:717–730. doi: 10.1093/hmg/ddl476. [DOI] [PubMed] [Google Scholar]
- 38.Willett R., Blackburn J.B., Climer L., Pokrovskaya I., Kudlyk T., Wang W., Lupashin V. COG lobe B sub-complex engages v-SNARE GS15 and functions via regulated interaction with lobe a sub-complex. Sci. Rep. 2016;6:29139. doi: 10.1038/srep29139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ungar D., Oka T., Brittle E.E., Vasile E., Lupashin V.V., Chatterton J.E., Heuser J.E., Krieger M., Waters M.G. Characterization of a mammalian Golgi-localized protein complex, COG, that is required for normal Golgi morphology and function. J. Cell Biol. 2002;157:405–415. doi: 10.1083/jcb.200202016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vasile E., Oka T., Ericsson M., Nakamura N., Krieger M. IntraGolgi distribution of the conserved oligomeric Golgi (COG) complex. Exp. Cell Res. 2006;312:3132–3141. doi: 10.1016/j.yexcr.2006.06.005. [DOI] [PubMed] [Google Scholar]
- 41.Willett R., Pokrovskaya I., Kudlyk T., Lupashin V. Multipronged interaction of the COG complex with intracellular membranes. Cell Logist. 2014;4 doi: 10.4161/cl.27888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Climer L.K., Pokrovskaya I.D., Blackburn J.B., Lupashin V.V. Membrane detachment is not essential for COG complex function. Mol. Biol. Cell. 2018;29:964–974. doi: 10.1091/mbc.E17-11-0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cavanaugh L.F., Chen X., Richardson B.C., Ungar D., Pelczer I., Rizo J., Hughson F.M. Structural analysis of conserved oligomeric Golgi complex subunit 2. J. Biol. Chem. 2007;282:23418–23426. doi: 10.1074/jbc.M703716200. [DOI] [PubMed] [Google Scholar]
- 44.Ha J.Y., Pokrovskaya I.D., Climer L.K., Shimamura G.R., Kudlyk T., Jeffrey P.D., Lupashin V.V., Hughson F.M. Cog5-Cog7 crystal structure reveals interactions essential for the function of a multisubunit tethering complex. Proc. Natl. Acad. Sci. U. S. A. 2014;111:15762–15767. doi: 10.1073/pnas.1414829111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Richardson B.C., Smith R.D., Ungar D., Nakamura A., Jeffrey P.D., Lupashin V.V., Hughson F.M. Structural basis for a human glycosylation disorder caused by mutation of the COG4 gene. Proc. Natl. Acad. Sci. U. S. A. 2009;106:13329–13334. doi: 10.1073/pnas.0901966106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ha J.Y., Chou H.T., Ungar D., Yip C.K., Walz T., Hughson F.M. Molecular architecture of the complete COG tethering complex. Nat. Struct. Mol. Biol. 2016;23:758–760. doi: 10.1038/nsmb.3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Picco A., Irastorza-Azcarate I., Specht T., Boke D., Pazos I., Rivier-Cordey A.S., Devos D.P., Kaksonen M., Gallego O. The in vivo architecture of the exocyst provides structural basis for exocytosis. Cell. 2017;168:400–412. doi: 10.1016/j.cell.2017.01.004. (e418) [DOI] [PubMed] [Google Scholar]
- 48.Bailey Blackburn J., Pokrovskaya I., Fisher P., Ungar D., Lupashin V.V. COG complex complexities: detailed characterization of a complete set of HEK293T cells lacking individual COG subunits. Front. Cell Dev. Biol. 2016;4:23. doi: 10.3389/fcell.2016.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Willett R., Ungar D., Lupashin V. The Golgi puppet master: COG complex at center stage of membrane trafficking interactions. Histochem. Cell Biol. 2013;140:271–283. doi: 10.1007/s00418-013-1117-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shestakova A., Suvorova E., Pavliv O., Khaidakova G., Lupashin V. Interaction of the conserved oligomeric Golgi complex with t-SNARE Syntaxin5a/Sed5 enhances intra-Golgi SNARE complex stability. J. Cell Biol. 2007;179:1179–1192. doi: 10.1083/jcb.200705145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Laufman O., Kedan A., Hong W., Lev S. Direct interaction between the COG complex and the SM protein, Sly1, is required for Golgi SNARE pairing. EMBO J. 2009;28:2006–2017. doi: 10.1038/emboj.2009.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sohda M., Misumi Y., Yoshimura S., Nakamura N., Fusano T., Ogata S., Sakisaka S., Ikehara Y. The interaction of two tethering factors, p115 and COG complex, is required for Golgi integrity. Traffic. 2007;8:270–284. doi: 10.1111/j.1600-0854.2006.00530.x. [DOI] [PubMed] [Google Scholar]
- 53.Sohda M., Misumi Y., Yamamoto A., Nakamura N., Ogata S., Sakisaka S., Hirose S., Ikehara Y., Oda K. Interaction of Golgin-84 with the COG complex mediates the intra-Golgi retrograde transport. Traffic. 2010;11:1552–1566. doi: 10.1111/j.1600-0854.2010.01123.x. [DOI] [PubMed] [Google Scholar]
- 54.Wuestehube L.J., Duden R., Eun A., Hamamoto S., Korn P., Ram R., Schekman R. New mutants of Saccharomyces cerevisiae affected in the transport of proteins from the endoplasmic reticulum to the Golgi complex. Genetics. 1996;142:393–406. doi: 10.1093/genetics/142.2.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suvorova E.S., Duden R., Lupashin V.V. The Sec34/Sec35p complex, a Ypt1p effector required for retrograde intra-Golgi trafficking, interacts with Golgi SNAREs and COPI vesicle coat proteins. J. Cell Biol. 2002;157:631–643. doi: 10.1083/jcb.200111081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kingsley D.M., Kozarsky K.F., Segal M., Krieger M. Three types of low density lipoprotein receptor-deficient mutant have pleiotropic defects in the synthesis of N-linked, O-linked, and lipid-linked carbohydrate chains. J. Cell Biol. 1986;102:1576–1585. doi: 10.1083/jcb.102.5.1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Walter D.M., Paul K.S., Waters M.G. Purification and characterization of a novel 13 S hetero-oligomeric protein complex that stimulates in vitro Golgi transport. J. Biol. Chem. 1998;273:29565–29576. doi: 10.1074/jbc.273.45.29565. [DOI] [PubMed] [Google Scholar]
- 58.Zolov S.N., Lupashin V.V. Cog3p depletion blocks vesicle-mediated Golgi retrograde trafficking in HeLa cells. J. Cell Biol. 2005;168:747–759. doi: 10.1083/jcb.200412003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shestakova A., Zolov S., Lupashin V. COG complex-mediated recycling of Golgi glycosyltransferases is essential for normal protein glycosylation. Traffic. 2006;7:191–204. doi: 10.1111/j.1600-0854.2005.00376.x. [DOI] [PubMed] [Google Scholar]
- 60.Pokrovskaya I.D., Willett R., Smith R.D., Morelle W., Kudlyk T., Lupashin V.V. Conserved oligomeric Golgi complex specifically regulates the maintenance of Golgi glycosylation machinery. Glycobiology. 2011;21:1554–1569. doi: 10.1093/glycob/cwr028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Willett R.A., Pokrovskaya I.D., Lupashin V.V. Fluorescent microscopy as a tool to elucidate dysfunction and mislocalization of Golgi glycosyltransferases in COG complex depleted mammalian cells. Methods Mol. Biol. 2013;1022:61–72. doi: 10.1007/978-1-62703-465-4_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Peanne R., Legrand D., Duvet S., Mir A.M., Matthijs G., Rohrer J., Foulquier F. Differential effects of lobe a and lobe B of the conserved oligomeric Golgi complex on the stability of {beta}1,4-galactosyltransferase 1 and {alpha}2,6-sialyltransferase 1. Glycobiology. 2011;21:864–876. doi: 10.1093/glycob/cwq176. [DOI] [PubMed] [Google Scholar]
- 63.D’Souza Z., Blackburn J.B., Kudlyk T., Pokrovskaya I.D., Lupashin V.V. Defects in COG-mediated Golgi trafficking Alter Endo-lysosomal system in human cells. Front. Cell Dev. Biol. 2019;7:118. doi: 10.3389/fcell.2019.00118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith R.D., Willett R., Kudlyk T., Pokrovskaya I., Paton A.W., Paton J.C., Lupashin V.V. The COG complex, Rab6 and COPI define a novel Golgi retrograde trafficking pathway that is exploited by SubAB toxin. Traffic. 2009;10:1502–1517. doi: 10.1111/j.1600-0854.2009.00965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Blackburn J.B., Kudlyk T., Pokrovskaya I., Lupashin V.V. More than just sugars: conserved oligomeric Golgi complex deficiency causes glycosylation-independent cellular defects. Traffic. 2018;19:463–480. doi: 10.1111/tra.12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Laufman O., Hong W., Lev S. The COG complex interacts directly with Syntaxin 6 and positively regulates endosome-to-TGN retrograde transport. J. Cell Biol. 2011;194:459–472. doi: 10.1083/jcb.201102045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Flanagan-Steet H., Johnson S., Smith R.D., Bangiyeva J., Lupashin V., Steet R. Mislocalization of large ARF-GEFs as a potential mechanism for BFA resistance in COG-deficient cells. Exp. Cell Res. 2011;317:2342–2352. doi: 10.1016/j.yexcr.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Niu T.K., Pfeifer A.C., Lippincott-Schwartz J., Jackson C.L. Dynamics of GBF1, a Brefeldin A-sensitive Arf1 exchange factor at the Golgi. Mol. Biol. Cell. 2005;16:1213–1222. doi: 10.1091/mbc.E04-07-0599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rymen D., Keldermans L., Race V., Regal L., Deconinck N., Dionisi-Vici C., Fung C.W., Sturiale L., Rosnoblet C., Foulquier F., Matthijs G., Jaeken J. COG5-CDG: expanding the clinical spectrum. Orphanet Jo. Rare Dis. 2012;7:94. doi: 10.1186/1750-1172-7-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lubbehusen J., Thiel C., Rind N., Ungar D., Prinsen B.H., de Koning T.J., van Hasselt P.M., Korner C. Fatal outcome due to deficiency of subunit 6 of the conserved oligomeric Golgi complex leading to a new type of congenital disorders of glycosylation. Hum. Mol. Genet. 2010;19:3623–3633. doi: 10.1093/hmg/ddq278. [DOI] [PubMed] [Google Scholar]
- 71.Oka T., Ungar D., Hughson F.M., Krieger M. The COG and COPI complexes interact to control the abundance of GEARs, a subset of Golgi integral membrane proteins. Mol. Biol. Cell. 2004;15:2423–2435. doi: 10.1091/mbc.E03-09-0699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stukalov A., Girault V., Grass V., Bergant V., Karayel O., Urban C., Haas D., Huang Y., Oubraham L., Wang A., Hamad S., Piras A., Tanzer M., Hansen F., Enghleitner T., Reinecke M., Lavacca T., Ehmann R., Wölfel R., Jores J., Kuster B., Protzer U., Rad R., Ziebuhr J., Thiel V., Scaturro P., Mann M., Pichlmair A. bioRxiv. 2020. Multi-level proteomics reveals host-perturbation strategies of SARS-CoV-2 and SARS-CoV. [DOI] [PubMed] [Google Scholar]
- 73.Riblett A.M., Blomen V.A., Jae L.T., Altamura L.A., Doms R.W., Brummelkamp T.R., Wojcechowskyj J.A. A haploid genetic screen identifies Heparan sulfate proteoglycans supporting Rift Valley fever virus infection. J. Virol. 2015;90:1414–1423. doi: 10.1128/JVI.02055-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu S., Dominska-Ngowe M., Dykxhoorn D.M. Target silencing of components of the conserved oligomeric Golgi complex impairs HIV-1 replication. Virus Res. 2014;192:92–102. doi: 10.1016/j.virusres.2014.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Realegeno S., Priyamvada L., Kumar A., Blackburn J.B., Hartloge C., Puschnik A.S., Sambhara S., Olson V.A., Carette J.E., Lupashin V., Satheshkumar P.S. Conserved Oligomeric Golgi (COG) complex proteins facilitate orthopoxvirus entry, fusion and spread. Viruses. 2020;12 doi: 10.3390/v12070707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pokrovskaya I.D., Szwedo J.W., Goodwin A., Lupashina T.V., Nagarajan U.M., Lupashin V.V. Chlamydia trachomatis hijacks intra-Golgi COG complex-dependent vesicle trafficking pathway. Cell. Microbiol. 2012;14:656–668. doi: 10.1111/j.1462-5822.2012.01747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Miller C.N., Smith E.P., Cundiff J.A., Knodler L.A., Bailey Blackburn J., Lupashin V., Celli J. A Brucella type IV effector targets the COG tethering complex to remodel host secretory traffic and promote intracellular replication. Cell Host Microbe. 2017;22:317–329. doi: 10.1016/j.chom.2017.07.017. (e317) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Moskalenko S., Tong C., Rosse C., Mirey G., Formstecher E., Daviet L., Camonis J., White M.A. Ral GTPases regulate exocyst assembly through dual subunit interactions. J. Biol. Chem. 2003;278:51743–51748. doi: 10.1074/jbc.M308702200. [DOI] [PubMed] [Google Scholar]
- 79.Ahmed S.M., Nishida-Fukuda H., Li Y., McDonald W.H., Gradinaru C.C., Macara I.G. Exocyst dynamics during vesicle tethering and fusion. Nat. Commun. 2018;9:5140. doi: 10.1038/s41467-018-07467-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rossi G., Lepore D., Kenner L., Czuchra A.B., Plooster M., Frost A., Munson M., Brennwald P. Exocyst structural changes associated with activation of tethering downstream of Rho/Cdc42 GTPases. J. Cell Biol. 2020;219 doi: 10.1083/jcb.201904161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Foulquier F. COG defects, birth and rise! Biochim. Biophys. Acta. 2009;1792:896–902. doi: 10.1016/j.bbadis.2008.10.020. [DOI] [PubMed] [Google Scholar]
- 82.Peanne R., de Lonlay P., Foulquier F., Kornak U., Lefeber D.J., Morava E., Perez B., Seta N., Thiel C., Van Schaftingen E., Matthijs G., Jaeken J. Congenital disorders of glycosylation (CDG): quo vadis? Eur. J. Med. Genet. 2018;61:643–663. doi: 10.1016/j.ejmg.2017.10.012. [DOI] [PubMed] [Google Scholar]
- 83.Jaeken J. Congenital disorders of glycosylation. Handb. Clin. Neurol. 2013;113:1737–1743. doi: 10.1016/B978-0-444-59565-2.00044-7. [DOI] [PubMed] [Google Scholar]
- 84.Lefeber D.J., Morava E., Jaeken J. How to find and diagnose a CDG due to defective N-glycosylation. J. Inherit. Metab. Dis. 2011;34:849–852. doi: 10.1007/s10545-011-9370-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Guillard M., Morava E., van Delft F.L., Hague R., Korner C., Adamowicz M., Wevers R.A., Lefeber D.J. Plasma N-glycan profiling by mass spectrometry for congenital disorders of glycosylation type II. Clin. Chem. 2011;57:593–602. doi: 10.1373/clinchem.2010.153635. [DOI] [PubMed] [Google Scholar]
- 86.Wopereis S., Morava E., Grunewald S., Adamowicz M., Huijben K.M., Lefeber D.J., Wevers R.A. Patients with unsolved congenital disorders of glycosylation type II can be subdivided in six distinct biochemical groups. Glycobiology. 2005;15:1312–1319. doi: 10.1093/glycob/cwj017. [DOI] [PubMed] [Google Scholar]
- 87.Foulquier F., Vasile E., Schollen E., Callewaert N., Raemaekers T., Quelhas D., Jaeken J., Mills P., Winchester B., Krieger M., Annaert W., Matthijs G. Conserved oligomeric Golgi complex subunit 1 deficiency reveals a previously uncharacterized congenital disorder of glycosylation type II. Proc. Natl. Acad. Sci. U. S. A. 2006;103:3764–3769. doi: 10.1073/pnas.0507685103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zeevaert R., Foulquier F., Dimitrov B., Reynders E., Van Damme-Lombaerts R., Simeonov E., Annaert W., Matthijs G., Jaeken J. Cerebrocostomandibular-like syndrome and a mutation in the conserved oligomeric Golgi complex, subunit 1. Hum. Mol. Genet. 2009;18:517–524. doi: 10.1093/hmg/ddn379. [DOI] [PubMed] [Google Scholar]
- 89.Kodera H., Ando N., Yuasa I., Wada Y., Tsurusaki Y., Nakashima M., Miyake N., Saitoh S., Matsumoto N., Saitsu H. Mutations in COG2 encoding a subunit of the conserved oligomeric golgi complex cause a congenital disorder of glycosylation. Clin. Genet. 2015;87:455–460. doi: 10.1111/cge.12417. [DOI] [PubMed] [Google Scholar]
- 90.Reynders E., Foulquier F., Leao Teles E., Quelhas D., Morelle W., Rabouille C., Annaert W., Matthijs G. Golgi function and dysfunction in the first COG4-deficient CDG type II patient. Hum. Mol. Genet. 2009;18:3244–3256. doi: 10.1093/hmg/ddp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ferreira C.R., Xia Z.J., Clement A., Parry D.A., Davids M., Taylan F., Sharma P., Turgeon C.T., Blanco-Sanchez B., Ng B.G., Logan C.V., Wolfe L.A., Solomon B.D., Cho M.T., Douglas G., Carvalho D.R., Bratke H., Haug M.G., Phillips J.B., Wegner J., Tiemeyer M., Aoki K., Diseases N. Undiagnosed, Genome P. Scottish, Nordgren A., Hammarsjo A., Duker A.L., Rohena L., Hove H.B., Ek J., Adams D., Tifft C.J., Onyekweli T., Weixel T., Macnamara E., Radtke K., Powis Z., Earl D., Gabriel M., Russi A.H.S., Brick L., Kozenko M., Tham E., Raymond K.M., Phillips J.A., 3rd, Tiller G.E., Wilson W.G., Hamid R., Malicdan M.C.V., Nishimura G., Grigelioniene G., Jackson A., Westerfield M., Bober M.B., Gahl W.A., Freeze H.H. A recurrent de novo heterozygous COG4 substitution leads to saul-wilson syndrome, disrupted vesicular trafficking, and altered proteoglycan glycosylation. Am. J. Hum. Genet. 2018;103:553–567. doi: 10.1016/j.ajhg.2018.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ng B.G., Sharma V., Sun L., Loh E., Hong W., Tay S.K., Freeze H.H. Identification of the first COG-CDG patient of Indian origin. Mol. Genet. Metab. 2011;102:364–367. doi: 10.1016/j.ymgme.2010.11.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Paesold-Burda P., Maag C., Troxler H., Foulquier F., Kleinert P., Schnabel S., Baumgartner M., Hennet T. Deficiency in COG5 causes a moderate form of congenital disorders of glycosylation. Hum. Mol. Genet. 2009;18:4350–4356. doi: 10.1093/hmg/ddp389. [DOI] [PubMed] [Google Scholar]
- 94.Fung C.W., Matthijs G., Sturiale L., Garozzo D., Wong K.Y., Wong R., Wong V., Jaeken J. COG5-CDG with a mild Neurohepatic presentation. JIMD Rep. 2012;3:67–70. doi: 10.1007/8904_2011_61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim Y.O., Yun M., Jeong J.H., Choi S.M., Kim S.K., Yoon W., Park C., Hong Y., Woo Y.J. A mild form of COG5 defect showing early-childhood-onset Friedreich’s-Ataxia-like phenotypes with isolated cerebellar atrophy. J. Korean Med. Sci. 2017;32:1885–1890. doi: 10.3346/jkms.2017.32.11.1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Yin S., Gong L., Qiu H., Zhao Y., Zhang Y., Liu C., Jiang H., Mao Y., Kong L.Y., Liang B., Lv Y. Novel compound heterozygous COG5 mutations in a Chinese male patient with severe clinical symptoms and type IIi congenital disorder of glycosylation: a case report. Exp. Therap. Med. 2019;18:2695–2700. doi: 10.3892/etm.2019.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wang X., Han L., Wang X.Y., Wang J.H., Li X.M., Jin C.H., Wang L. Identification of two novel mutations in COG5 causing congenital disorder of glycosylation. Front. Genet. 2020;11:168. doi: 10.3389/fgene.2020.00168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Palmigiano A., Bua R.O., Barone R., Rymen D., Regal L., Deconinck N., Dionisi-Vici C., Fung C.W., Garozzo D., Jaeken J., Sturiale L. MALDI-MS profiling of serum O-glycosylation and N-glycosylation in COG5-CDG. JMS. 2017;52:372–377. doi: 10.1002/jms.3936. [DOI] [PubMed] [Google Scholar]
- 99.Huybrechts S., De Laet C., Bontems P., Rooze S., Souayah H., Sznajer Y., Sturiale L., Garozzo D., Matthijs G., Ferster A., Jaeken J., Goyens P. Deficiency of subunit 6 of the conserved oligomeric Golgi complex (COG6-CDG): second patient, different Phenotype. JIMD Rep. 2012;4:103–108. doi: 10.1007/8904_2011_79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Shaheen R., Ansari S., Alshammari M.J., Alkhalidi H., Alrukban H., Eyaid W., Alkuraya F.S. A novel syndrome of hypohidrosis and intellectual disability is linked to COG6 deficiency. J. Med. Genet. 2013;50:431–436. doi: 10.1136/jmedgenet-2013-101527. [DOI] [PubMed] [Google Scholar]
- 101.Althonaian N., Alsultan A., Morava E., Alfadhel M. Secondary Hemophagocytic syndrome associated with COG6 gene defect: report and review. JIMD Rep. 2018;42:105–111. doi: 10.1007/8904_2018_88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rymen D., Winter J., Van Hasselt P.M., Jaeken J., Kasapkara C., Gokcay G., Haijes H., Goyens P., Tokatli A., Thiel C., Bartsch O., Hecht J., Krawitz P., Prinsen H.C., Mildenberger E., Matthijs G., Kornak U. Key features and clinical variability of COG6-CDG. Mol. Genet. Metab. 2015;116:163–170. doi: 10.1016/j.ymgme.2015.07.003. [DOI] [PubMed] [Google Scholar]
- 103.Li G., Xu Y., Hu X., Li N., Yao R., Yu T., Wang X., Guo W., Wang J. Compound heterozygous variants of the COG6 gene in a Chinese patient with deficiency of subunit 6 of the conserved oligomeric Golgi complex (COG6-CDG) Eur. J. Med. Genet. 2019;62:44–46. doi: 10.1016/j.ejmg.2018.04.017. [DOI] [PubMed] [Google Scholar]
- 104.Makhamreh M.M., Cottingham N., Ferreira C.R., Berger S., Al-Kouatly H.B. Nonimmune hydrops fetalis and congenital disorders of glycosylation: a systematic literature review. J. Inherit. Metab. Dis. 2019;43(2):223–233. doi: 10.1002/jimd.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wu X., Steet R.A., Bohorov O., Bakker J., Newell J., Krieger M., Spaapen L., Kornfeld S., Freeze H.H. Mutation of the COG complex subunit gene COG7 causes a lethal congenital disorder. Nat. Med. 2004;10:518–523. doi: 10.1038/nm1041. [DOI] [PubMed] [Google Scholar]
- 106.Spaapen L.J.M., Bakker J.A., Meer S.B.v.d., Sijstermans H.J., Steet R.A., Wevers R.A., Jaeken J. Clinical and biochemical presentation of siblings with COG-7 deficiency, a lethal multiple O- and N-glycosylation disorder. J. Inherit. Metab. Dis. 2005;28:707–714. doi: 10.1007/s10545-005-0015-z. [DOI] [PubMed] [Google Scholar]
- 107.Morava E., Zeevaert R., Korsch E., Huijben K., Wopereis S., Matthijs G., Keymolen K., Lefeber D.J., De Meirleir L., Wevers R.A. A common mutation in the COG7 gene with a consistent phenotype including microcephaly, adducted thumbs, growth retardation, VSD and episodes of hyperthermia. Eur. J. Hum. Genet. 2007;15:638–645. doi: 10.1038/sj.ejhg.5201813. [DOI] [PubMed] [Google Scholar]
- 108.Steet R., Kornfeld S. COG-7-deficient human fibroblasts exhibit altered recycling of Golgi proteins. Mol. Biol. Cell. 2006;17:2312–2321. doi: 10.1091/mbc.E05-08-0822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zeevaert R., Foulquier F., Cheillan D., Cloix I., Guffon N., Sturiale L., Garozzo D., Matthijs G., Jaeken J. A new mutation in COG7 extends the spectrum of COG subunit deficiencies. Eur. J. Med. Genet. 2009;52:303–305. doi: 10.1016/j.ejmg.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 110.Kranz C., Ng B.G., Sun L., Sharma V., Eklund E.A., Miura Y., Ungar D., Lupashin V., Winkel R.D., Cipollo J.F., Costello C.E., Loh E., Hong W., Freeze H.H. COG8 deficiency causes new congenital disorder of glycosylation type IIh. Hum. Mol. Genet. 2007;16:731–741. doi: 10.1093/hmg/ddm028. [DOI] [PubMed] [Google Scholar]
- 111.Yang A., Cho S.Y., Jang J.H., Kim J., Kim S.Z., Lee B.H., Yoo H.W., Jin D.K. Further delineation of COG8-CDG: A case with novel compound heterozygous mutations diagnosed by targeted exome sequencing. Clin. Chimica Acta. Int. J. Clin. Chem. 2017;471:191–195. doi: 10.1016/j.cca.2017.06.010. [DOI] [PubMed] [Google Scholar]
- 112.Arora V., Puri R.D., Bhai P., Sharma N., Bijarnia-Mahay S., Dimri N., Baijal A., Saxena R., Verma I. The first case of antenatal presentation in COG8-congenital disorder of glycosylation with a novel splice site mutation and an extended phenotype. Am. J. Med. Genet. A. 2019;179:480–485. doi: 10.1002/ajmg.a.61030. [DOI] [PubMed] [Google Scholar]
- 113.Laufman O., Freeze H.H., Hong W., Lev S. Deficiency of the Cog8 subunit in normal and CDG-derived cells impairs the assembly of the COG and Golgi SNARE complexes. Traffic. 2013;14:1065–1077. doi: 10.1111/tra.12093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Zeevaert R., Foulquier F., Jaeken J., Matthijs G. Deficiencies in subunits of the conserved oligomeric Golgi (COG) complex define a novel group of congenital disorders of glycosylation. Mol. Genet. Metab. 2008;93:15–21. doi: 10.1016/j.ymgme.2007.08.118. [DOI] [PubMed] [Google Scholar]
- 115.Scott H., Panin V.M. N-glycosylation in regulation of the nervous system. Adv. Neurobiol. 2014;9:367–394. doi: 10.1007/978-1-4939-1154-7_17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Comstra H.S., McArthy J., Rudin-Rush S., Hartwig C., Gokhale A., Zlatic S.A., Blackburn J.B., Werner E., Petris M., D'Souza P., Panuwet P., Barr D.B., Lupashin V., Vrailas-Mortimer A., Faundez V. The interactome of the copper transporter ATP7A belongs to a network of neurodevelopmental and neurodegeneration factors. eLife. 2017;6 doi: 10.7554/eLife.24722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Climer L.K., Hendrix R.D., Lupashin V.V. Conserved oligomeric Golgi and neuronal vesicular trafficking. Handb. Exp. Pharmacol. 2018;245:227–247. doi: 10.1007/164_2017_65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Haijes H.A., Jaeken J., Foulquier F., van Hasselt P.M. Hypothesis: lobe a (COG1-4)-CDG causes a more severe phenotype than lobe B (COG5-8)-CDG. J. Med. Genet. 2018;55:137–142. doi: 10.1136/jmedgenet-2017-104586. [DOI] [PubMed] [Google Scholar]
- 119.Janssen M.C., de Kleine R.H., van den Berg A.P., Heijdra Y., van Scherpenzeel M., Lefeber D.J., Morava E. Successful liver transplantation and long-term follow-up in a patient with MPI-CDG. Pediatrics. 2014;134:e279–e283. doi: 10.1542/peds.2013-2732. [DOI] [PubMed] [Google Scholar]
- 120.Wong S.Y., Gadomski T., van Scherpenzeel M., Honzik T., Hansikova H., Holmefjord K.S.B., Mork M., Bowling F., Sykut-Cegielska J., Koch D., Hertecant J., Preston G., Jaeken J., Peeters N., Perez S., Nguyen D.D., Crivelly K., Emmerzaal T., Gibson K.M., Raymond K., Bakar N. Abu, Foulquier F., Poschet G., Ackermann A.M., He M., Lefeber D.J., Thiel C., Kozicz T., Morava E. Oral D-galactose supplementation in PGM1-CDG, Genetics in medicine. Off. J. Am. College Med. Gene. 2017;19:1226–1235. doi: 10.1038/gim.2017.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Morelle W., Potelle S., Witters P., Wong S., Climer L., Lupashin V., Matthijs G., Gadomski T., Jaeken J., Cassiman D., Morava E., Foulquier F. Galactose supplementation in patients with TMEM165-CDG rescues the glycosylation defects. J. Clin. Endocrinol. Metab. 2017;102:1375–1386. doi: 10.1210/jc.2016-3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Brasil S., Pascoal C., Francisco R., Marques-da-Silva D., Andreotti G., Videira P.A., Morava E., Jaeken J., Dos Reis Ferreira V. CDG therapies: from bench to bedside. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19051304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yuste-Checa P., Medrano C., Gamez A., Desviat L.R., Matthijs G., Ugarte M., Perez-Cerda C., Perez B. Antisense-mediated therapeutic pseudoexon skipping in TMEM165-CDG. Clin. Genet. 2015;87:42–48. doi: 10.1111/cge.12402. [DOI] [PubMed] [Google Scholar]
- 124.Phadke A.P., Jay C., Chen S.J., Haddock C., Wang Z., Yu Y., Nemunaitis D., Nemunaitis G., Templeton N.S., Senzer N., Maples P.B., Tong A.W., Nemunaitis J. Safety and in vivo expression of a GNE-transgene: a novel treatment approach for hereditary inclusion body myopathy-2. Gene Regulation Syst. Biol. 2009;3:89–101. doi: 10.4137/grsb.s2210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Frappaolo A., Sechi S., Kumagai T., Robinson S., Fraschini R., Karimpour-Ghahnavieh A., Belloni G., Piergentili R., Tiemeyer K.H., Tiemeyer M., Giansanti M.G. COG7 deficiency in Drosophila generates multifaceted developmental, behavioral and protein glycosylation phenotypes. J. Cell Sci. 2017;130:3637–3649. doi: 10.1242/jcs.209049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Song H., Orr A.S., Lee M., Harner M.E., Wickner W.T. HOPS recognizes each SNARE, assembling ternary trans-complexes for rapid fusion upon engagement with the 4th SNARE. eLife. 2020;9 doi: 10.7554/eLife.53559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Travis S.M., Kevin D.A., Yu I.M., McMahon C., Hamid S., Ramirez-Arellano G., Jeffrey P.D., Hughson F.M. Structural basis for the binding of SNAREs to the multisubunit tethering complex Dsl1. J. Biol. Chem. 2020;295:10125–10135. doi: 10.1074/jbc.RA120.013654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ishida M., Bonifacino J.S. ARFRP1 functions upstream of ARL1 and ARL5 to coordinate recruitment of distinct tethering factors to the trans-Golgi network. J. Cell Biol. 2019;218:3681–3696. doi: 10.1083/jcb.201905097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hughson F.M., Reinisch K.M. Structure and mechanism in membrane trafficking. Curr. Opin. Cell Biol. 2010;22:454–460. doi: 10.1016/j.ceb.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Arasaki K., Takagi D., Furuno A., Sohda M., Misumi Y., Wakana Y., Inoue H., Tagaya M. A new role for RINT-1 in SNARE complex assembly at the trans-Golgi network in coordination with the COG complex. Mol. Biol. Cell. 2013;24:2907–2917. doi: 10.1091/mbc.E13-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kudlyk T., Willett R., Pokrovskaya I.D., Lupashin V. COG6 interacts with a subset of the Golgi SNAREs and is important for the Golgi complex integrity. Traffic. 2013;14:194–204. doi: 10.1111/tra.12020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Yen W.L., Shintani T., Nair U., Cao Y., Richardson B.C., Li Z., Hughson F.M., Baba M., Klionsky D.J. The conserved oligomeric Golgi complex is involved in double-membrane vesicle formation during autophagy. J. Cell Biol. 2010;188:101–114. doi: 10.1083/jcb.200904075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Wang I.H., Chen Y.J., Hsu J.W., Lee F.J. The Arl3 and Arl1 GTPases co-operate with Cog8 to regulate selective autophagy via Atg9 trafficking. Traffic. 2017;18:580–589. doi: 10.1111/tra.12498. [DOI] [PubMed] [Google Scholar]
- 134.Cherot E., Keren B., Dubourg C., Carre W., Fradin M., Lavillaureix A., Afenjar A., Burglen L., Whalen S., Charles P., Marey I., Heide S., Jacquette A., Heron D., Doummar D., Rodriguez D., de Villemeur T. Billette, Moutard M.L., Guet A., Xavier J., Perisse D., Cohen D., Demurger F., Quelin C., Depienne C., Odent S., Nava C., David V., Pasquier L., Mignot C. Using medical exome sequencing to identify the causes of neurodevelopmental disorders: Experience of 2 clinical units and 216 patients. Clin. Genet. 2018;93:567–576. doi: 10.1111/cge.13102. [DOI] [PubMed] [Google Scholar]
- 135.Ciftci H., Celepkolo B., Dilmec F., Koksal M., Yeni E., Yagmur I., Gumus K. Genetic polymorphisms of hspa1b and hspa1l in infertile men. JPMA. 2015;65:701–704. [PubMed] [Google Scholar]
- 136.Kellokumpu Sakari. Golgi pH, Ion and Redox Homeostasis: How Much Do They Really Matter? Front. Cell Dev. Biol. 2019 doi: 10.3389/fcell.2019.00093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Moremen Kelley W, Tiemeyer Michael, Nairn Alison V. Vertebrate protein glycosylation: diversity, synthesis and function. Nat Rev Mol Cell Biol. 2012;13:448–462. doi: 10.1038/nrm3383. [DOI] [PMC free article] [PubMed] [Google Scholar]