

Abstract
In Alzheimer’s disease, the amyloid-beta peptide (Aβ) is implicated in neuronal toxicity via interactions with the cell membrane. Monomeric Aβ (Aβm) is intrinsically disordered, but it can adopt a range of aggregated conformations with varying toxicities from short fibrillar oligomers (FO), to globular nonfibrillar oligomers (NFO), and full-length amyloid fibrils. NFO is considered to be the most toxic, followed by fibrils, and finally Aβm. To elucidate molecular-level membrane interactions that contribute to their different toxicities, we used liquid surface X-ray scattering and Langmuir trough insertion assays to compare Aβm, FO, and NFO surface activities and interactions with anionic DMPG lipid monolayers at the air/water interface. All Aβ species were highly surface active and rapidly adopted β-sheet rich structures upon adsorption to the air/water interface. Likewise, all Aβ species had affinity for the anionic membrane. Aβm rapidly converted to β-sheet rich assemblies upon binding the membrane, and these aggregated structures of Aβm and FO disrupted hexagonally packed lipid domains and resulted in membrane thinning and instability. In contrast, NFO perturbed membrane structure by extracting lipids from the air/water interface and causing macroscale membrane deformations. Altogether, our results support two models for membrane-mediated Aβ toxicity: fibril-induced reorganization of lipid packing and NFO-induced membrane destabilization and lipid extraction. This work provides a structural understanding of Aβ neurotoxicity via membrane interactions and aids the effort in understanding early events in Alzheimer’s disease and other neurodegenerative diseases.
Graphical Abstract
■ INTRODUCTION
Alzheimer’s disease is one of the most challenging healthcare issues today, and there is no treatment or prevention currently available (Alzheimer’s Association 2019). To better understand the disease process and to find a cure, it is imperative to study the mechanisms of disease initiation and progression. The amyloid beta peptide (Aβ) is found at abnormally high concentrations in neuronal tissue prior to disease onset, and it is implicated in neurotoxicity via aberrant protein folding and aggregation.1 One mechanism of Aβ toxicity is via interactions with the cell membrane, where Aβ-membrane interactions lead to membrane disruptions and eventually cell death.2,3 In this study, we examine three structurally distinct forms of Aβ, monomeric Aβm, fibrillar oligomer FO, and nonfibrillar oligomer NFO and compare their interactions with a model cell membrane to learn the molecular-level structural details of their membrane-mediated toxicity. We find that there are two distinct modes of membrane disruption: lipid removal from the membrane by the nonfibrillar oligomers, and disruption to lipid packing by fibrillar peptide species.
Aβ is generated via cleavage from the transmembrane amyloid precursor protein (APP) to produce Aβ peptides of 36–43 amino acids in length, with Aβ1–40 and Aβ1–42 being the most abundant.4,5 Aβm follows a well-characterized aggregation pathway to form stable β-sheet rich fibrils.6,7 These fibrils eventually form amyloid plaques, a classical hallmark of Alzheimer’s disease neuropathology.8,9 Amyloid fibrils and plaques, however, are now considered to be a mostly benign species, exhibiting secondary neuronal disfunction by inducing local synaptic abnormalities10 and changing the local neuritic architecture.11 In contrast, oligomeric species of Aβ, intermediates along the aggregation pathway to fibrillation, are now thought to be the most toxic Aβ species, directly causing neuronal cell death.12,13
Aβ oligomers encompass a range of heterogeneous structures. They are stabilized by inter- and intramolecular contacts among the peptides,14 in contrast to the largely disordered Aβm.15 Among the resolved Aβ oligomer structures, FOs are small (∼15 nm in diameter) β-sheet rich structures mimicking the structure of mature fibrils.16 NMR structures of Aβ1–40 fibrils show an ordered core (amino acids 9–40) with the remaining amino acids protruding from the fibril core.17 FOs are only one-half the height of mature fibrils, but they still bind the antifibril OC antibody.16 NFOs are off-pathway, ∼20–40 nm sized aggregates that are semistable.14,16 NFOs have not been crystallized due to their amorphous and heterogeneous nature but are known to be rich in random coil and α-helices.18,19 NFOs have been well-characterized as a highly toxic species,13,20 and its toxicity has been found to be correlated with smaller size18 and increased surface hydrophobicity.21 FOs have implied toxicity as fragmentation of Aβ fibrils leads to higher toxicity and membrane disruption.22 Overall, neuronal toxicity of the Aβ species follows the trend of NFOs > fibrils > monomers with NFOs being the most toxic.23
One pathway of Aβ toxicity is through the destabilization of the cell membrane.3 Aβm favorably interacts with anionic lipid membranes, leading to accelerated fibrillation from nucleated β-sheet rich structures on the membrane surface.24,25 This membrane-induced fibrillation process was shown to lead to cell membrane permeability,25 causing cellular ion dysregulation. Aβ oligomers also have destructive consequences for lipid membranes,26 resulting in immediate toxicity and membrane disruption.13
It is clear that membranes are involved in the toxicity mechanism of Aβm, FO, and NFO. However, the structural basis for inducing that toxicity is debated. There are several hypothesized modes of Aβ-induced membrane disruption including membrane thinning, the carpet model, the detergent-like mechanism, and protein pore formation.3 Oligomer-induced membrane thinning has been shown to increase ion permeability, resulting in the disruption of ion homeostasis.27,28 The carpet model has been proposed as a mechanism of toxicity for a number of toxic membrane-binding proteins, including helical antimicrobial peptides.29 In this model, protein coats the surface of the cell membrane to cause asymmetric pressure on two membrane leaflets, which leads to leakage of small molecules across the membrane.30 Another facet of the carpet model is the ability of intrinsically disordered proteins to shape membrane curvature via mechanical stresses,31 such as membrane blebbing and severe distortion of liposomes from spherical to a pointed tear-drop shape.32 Aβ pore formation allows calcium flux across the membrane but could be inhibited by the addition of zinc.33–35 The pore hypothesis is controversial, as it was shown that Aβ can induce membrane permeability without forming protein pores.27,36 Aβm has been shown to form calcium-permeable pores that precedes membrane-induced fibrillation.37 In that case, fibrillation drove membrane damage and formation of micelle-like structures, an example of the detergent-like mechanism wherein Aβ extracts lipids from the membrane and disperses them into the surrounding solution. This has been observed for tau, resulting in formation of stable protein/lipid complexes.38 Because there are several distinct structures of Aβ, they may induce membrane toxicity through different mechanisms.
In this study, we compare the membrane interactions of Aβm, FO, and NFO to resolve structural details of membrane-mediated toxicity for each form of Aβ. Surface activity and membrane affinity were evaluated using surface pressure isotherm studies on a Langmuir trough. Molecular-level details of lipid membrane structure were elucidated using complementary synchrotron X-ray scattering techniques. We conclude that FOs and NFOs have two distinct modes of membrane interactions, with fibrillar structures disrupting native lipid packing and nonfibrillar aggregates causing toxicity via a lipid removal mechanism.
■ RESULTS
Characterization of Aβ Monomers, Fibrillar Oligomers, and Nonfibrillar Oligomers
The goal of this study is to compare the membrane affinity and toxicity of three forms of Aβ1–40: monomeric (Aβm), fibrillar oligomers (FO), and nonfibrillar oligomers (NFO). These differently aggregated states of Aβ, while all composed of the same peptide, have very different structural characteristics. Aβm is largely disordered, only adopting transiently stable secondary structures.15,39,40 FO is a small, semistable, β-sheet rich oligomer that adopts a structure that mimics that of fibrils with an ordered core of amino acids stabilized by β-sheets and a largely disordered N-terminus.14 FO has been observed to induce the formation of mature fibrils. NFOs comprise a heterogeneous population of large, transiently stable oligomers with a globular conformation rich in random coils with some α-helices.19 As Aβ oligomers are believed to the primary toxic species in human brains leading to neurodegeneration,41,42 it is imperative to understand their mechanism for neuronal toxicity. In this work, we provide angstrom-level resolution structural details for the Aβ interactions that drive specific modes of membrane-mediated toxicity.
We first characterized our preparations of Aβm, FOs, and NFOs to verify that their sizes and structural characteristics resemble those previously described.14 With TEM imaging, Aβm was nearly undetectable, FOs appeared as small, ∼20 nm sized particles, and NFOs were the largest in size and varied from ∼40 to 60 nm (Figure 1A). Dot blot assay results (Figure 1B) confirmed the fibrillar structure of FOs as they are detected by the antifibrillar OC antibody.43 Note that Aβm and NFO also show reactivity against the antibody. Aside from the fibrillar structure, OC “may also show weak reactivity against Aβ monomers”, as noted in its product application notes. Thus, the dots observed for Aβm and NFO are likely nonspecific binding of the OC antibody to monomeric Aβ in the Aβm sample and any residual monomeric Aβ in the NFO preparation. The nonfibrillar oligomer structure of NFOs are clearly detected by the A11 antibody13 (Figure 1B) as it is known to bind to nonfibrillar toxic oligomers made of a number of amyloid proteins, including Aβ, tau, and α-synuclein. The strong binding of the antibody to the NFOs imply that these oligomers are likely neurotoxic.
Figure 1.

Transmission electron microscopy (TEM) images (A), dot blot assay results (B), and circular dichroism (CD) spectra (C) of monomeric Aβ40 (Aβm), fibrillar Aβ40 oligomers (FOs), and nonfibrillar Aβ40 oligomer (NFOs). Aβ species were probed with antibodies for Aβ sequence (6E10), fibril structure (OC), and nonfibrillar oligomers (A11) in the dot blot assay. CD spectra shown are averages of 3–5 replicate scans.
By CD spectroscopy (Figure 1C), Aβm appeared as random coils with a peak minimum at ∼195 nm, while FO was clearly β-sheet rich with the peak minimum shifted to ∼220 nm. NFO appeared as mostly random coil but was distinguishable from Aβm. Altogether, our results confirm the expected structural characteristics for these Aβ conformers. Some researchers14 refer to the globular Aβ oligomers as prefibrillar oligomers, while most others simply use the terminology “oligomers.” For clarity, we are distinguishing between the two different oligomer conformations prepared in this study as nonfibrillar NFOs and fibrillar FOs.
Air/Water Interface Induced Aβ Fibrillar Structure
To investigate membrane-mediated toxicity of the different Aβ species, we first evaluated their inherent surface activities as protein–membrane interactions are strongly influenced by protein hydrophobicity and nonpolar residues that contribute to protein surface activity. Due to their structural differences, Aβm, FO, and NFO may differ in their surface-exposed amino acids that either enhance or dampen surface activity or membrane affinity. Surface activities of the Aβ species were evaluated by assessing the adsorption of the peptides in water to a bare air/water interface. Surface pressure (π) profiles of the air/water interface were measured in a Langmuir trough after the injection of the peptides into the water subphase. π is the magnitude by which surface tension of an air/water interface (γo) is reduced by the presence of an adsorbate: π = γo − γ, where γ is the surface tension of the air/water interface with an adsorbate. Thus, π values are correlated to the extent of adsorption of peptides to the air/water interface and are an indication of the peptide’s surface activity.
Surface pressure profiles showed that all forms of Aβ tested spontaneously adsorbed to the air/water interface after injection into the water subphase, reducing its surface tension as shown by increases in surface pressure (Figure 2). All Aβ species are thus surface active. NFO and Aβm exhibited slow but steady adsorption reaching π values of around 8 mN/m after 4 h. FO rapidly adsorbed to the air/water interface and reached π values of around 13 mN/m after 30 min. We note that the fast initial increase in π may be due to residual HFIP in the FO sample from the oligomer preparation protocol; HFIP is highly surface active and may partition to the air/water interface and reduce surface tension or enhance FO’s surface activity by binding to the oligomers. Nonetheless, FO adsorbed to the air/water interface and formed a stable peptide film.
Figure 2.

Surface pressure isotherm measurements of Aβm, FO, and NFO adsorption to an air/water interface at room temperature. Peptide concentration in the water subphase was 250 nM. Oscillations in the surface pressure were caused by small temperature fluctuations in the room. Triplicate measurements were collected and one representative data set is shown.
We next determined the angstrom-level structure of Aβ adsorbed to the air/water interface using in situ complementary liquid surface X-ray reflectivity (XR) and grazing incidence X-ray diffraction (GIXD). The reflectivity of X-rays from an interface is modulated by variation in electron density at that interface; deviations from the Fresnel reflection (RF) from an ideal interface can be used to distinguish between regions of material with different electron densities. A model of the protein film was constructed by building “slabs” of electron density, each with a fitted value for thickness, electron density (normalized to water, ρ/ρwater), and interfacial roughness (σ) between adjacent slabs. This information was used to construct a 1D structure in the z-direction, averaged over the coherent area of the X-ray beam, of the peptide film adsorbed to the air/water interface as previously described.24
Background subtracted XR data were normalized to RF (R/RF) and plotted as a function of the scattering vector qz (Figure 3A). Fitted ρ/ρwater profiles are shown in Figure 3B. R/RF profiles were sufficiently modeled using a single slab that was 18.9 Å in thickness for Aβm and FO but a slightly thinner 15.9 Å for NFO (Figure 3 and Table S1 in Supporting Information). ρ/ρwater values of the peptide films were similar and around 1.3, indicating a well packed or folded protein film as the value resembles the density of folded proteins. Moreover, the thickness of the adsorbed protein layer was not concentration dependent; experiments performed with double or half concentration of Aβm did not change the thickness of the protein layer (data not shown). Taken together, our results show that the spontaneous adsorption of the different Aβ species to the air/water interface resulted in well-defined films composed of conformationally compact peptides. Adsorption thus was not a random accumulation of Aβ at the interface but was likely governed by more specific interactions and orientational effects.
Figure 3.

(A) XR measurements of Aβm, FO, or NFO adsorbed to an air/water interface. Normalized by Fresnel reflectivity, XR data is plotted as a function of the scattering vector (qz), shown as points with experimental error. The fitted model is overlaid as a solid black line. (B) The electron density profile fitted results, normalized to water (ρ/ρwater), for Aβm, FO, and NFO adsorbed to the air/water interface are plotted as a function of depth into the liquid subphase (zero is the air/water interface). Slab model fits with and without interslab roughnesses are shown with black and colored lines, respectively. Data and results are offset for clarity. Measurements were collected one time.
To further analyze the structures of the adsorbed peptide films, GIXD was used to detect ordered peptide structures. Diffraction peaks from semicrystalline structures at the air/water interface obtained from GIXD can be used to calculate the lattice spacing and average size of the diffracting structures. Aβ peptides are intrinsically disordered in solution, but upon aggregation into amyloid fibrils, Aβ misfolds and assembles into a highly ordered and β-sheet structure. This structure is evidenced by a diffraction peak at 1.32 Å−1 that arises from a lattice spacing of 4.7 Å between adjacent β-strands in the amyloid fiber.44 Strikingly, GIXD measurements of all three adsorbed peptide films yielded diffraction peaks at 1.32 Å−1 (Figure 4 and Table S2 in Supporting Information), indicating the presence of an ordered β-sheet rich structure. The average size of the coherently scattering peptide protofibrils (Lc) is about 80 Å. The result from Aβm is consistent with a previous report24 and confirms that adsorption and accumulation of Aβm at the air/water interface caused the disordered peptide to misfold and self-assemble into β-sheet-rich crystalline structures. The fibrillar structure of FOs appear to be preserved upon absorption to the air/water interface. The unexpected result is that NFO was converted from a predominantly random coil structure to a β-sheet rich protofibrillar structure when adsorbed to the air/water interface. Whether NFOs are on- or off-pathway intermediates for fibrillation has been a long-debated issue.18 Our results indicate that NFO can be converted to fibrillar structures through interactions and accumulation at a hydrophilic/hydrophobic interface.
Figure 4.

Grazing incidence X-ray diffraction (GIXD) results of Aβ species adsorbed to the air/water interface. (A) Background-subtracted integrated diffraction values (points) with experimental error are overlaid with the model fit to the data (solid line). A GIXD peak at 1.32 Å−1 is attributed to β-sheet rich protein diffraction. (B) An additional diffraction peak was observed for Aβm centered around qxy = 0.152 Å−1. X-ray scattering from FO and NFO resulted in weak diffraction in this region; however, reliable fits were difficult to obtain. A thin dashed line between points was included as a guide. This peak represents higher-order protein organization with 41.2 Å spacing between repeating units. Measurements were collected one time.
For Aβm, a second diffraction peak was observed at 0.152 ± 0.007 Å−1, which corresponds to an ordered structure with a lattice spacing of 41.2 Å and a Lc value of 180 ± 30 Å (Figure 4B and Table S2 in Supporting Information). This high qxy diffraction peak was also observed for FO and NFO. But due to their very weak intensities, the peaks could not be reliably fitted and quantified. This large spacing may arise from higher order packing of Aβ40 fibrillar oligomers.24 An amyloid intersheet packing of 10.5 Å has been reported;44 we estimate that the diffracting unit could be, on average, four β-sheets separated, approximately, by the standard intersheet packing distance.
All Aβ Conformers Bind the Model Lipid Membrane and Cause Defects
To test Aβ interactions with lipid membranes, surface pressure (π) isotherm experiments were conducted using a Langmuir trough to measure protein insertion into lipid monolayers at the air/water interface. A DMPG lipid monolayer was compressed to an initial π of 25 mN/m to mimic the bilayer cell membrane,45 and the surface area of the monolayer was kept constant after injecting the Aβ samples into the subphase. Favorable Aβ-membrane interactions that result in insertion of Aβ into the membrane were measured as an increase in π. Anionic DMPG lipids were used for this study because previous studies have shown that Aβm has preferential affinity for anionic over zwitterionic lipids.24,46 Saturated lipids were chosen to avoid oxidation in the Langmuir trough.
Our results showed that all three conformers of Aβ spontaneously interacted with and increased π of the membrane (Figure 5A). Aβm and FO yielded similar maximum π values of 37 to 38 mN/m and Aβm exhibited a 1.5 h lag time that preceded membrane surface pressure increase. NFO caused an immediate increase in π, reaching a maximum value of ∼32 mN/m. Interestingly, the lipid monolayer with NFO was not stable and slowly lost π with time, indicating removal of material from the air/water interface during the first ∼1−2 h post injection. This behavior is consistent with a model for Aβ toxicity wherein the peptide binds the membrane and extracts lipids into the subphase. The small π decrease observed for Aβm, FO, and NFO at the end of the experiment is likely due to monolayer instability as it was also observed in the absence of peptides (data not shown).
Figure 5.

Isotherms (A) and fluorescence microscopy (FM) images (B) of Aβ insertion into a DMPG monolayer containing 0.5 mol % Texas Red-DHPE at the air/water interface at 23.5 °C at constant monolayer surface area. Time in the FM images corresponds to hours post Aβ injection. The scale bar represents 50 μm for all images. Measurements were collected in triplicates, and one representative data set is shown.
Fluorescence microscopy (FM) imaging (Figure 5B) was used to directly visualize monolayer morphology. To provide contrast, 0.5 mol % of DHPE headgroup labeled with Texas Red (TR-DHPE) was included in the DMPG monolayer. In a monolayer where disordered liquid expanded (LE) and ordered lipid condensed (LC) phases are in coexistence, the bulky TR-DHPE dye is excluded from the LC phase. As a result, LC domains appear as dark patches and the surrounding LE phase appears bright. Three-dimensional perturbations to the membrane (e.g., monolayer collapse, membrane invagination, etc.) can appear as bright spots with fluorescence higher than the LE phase.47 The temperature and pressure of these experiments were chosen to produce a monolayer containing a coexistence of LE and LC phases, which allows us to detect changes to lipid morphology and packing as a result of Aβ interactions.
For a DMPG monolayer compressed to 25 mN/m at room temperature, a coexistence of bright LE and dark LC phases were observed (Figure 5B, 0 h). During lipid-only investigations, we observed that small temperature oscillations did not impact the appearance of the LE and LC phases observed over time. Three hours after injecting Aβm, the edges of the LC domains became less defined. At 6 h, where the insertion plateau was reached (Figure 5A), the smooth outline of the LC domains became rough with small bright patches dotting the edges of many LC domains. At 7 h after injection, the LC patches shrunk significantly in size. Lipid packing in the membrane thus became significantly disrupted with the insertion and incubation of Aβm. FO follows a similar trend; after having reached the insertion plateau 3 h post injection, the monolayer showed many more bright fluorescence patches on the perimeter of the LC domains.
In contrast, NFO interactions had significantly different impacts to the DMPG film. At the plateau of NFO interaction (1 h), NFO converted the small round LC domains into a network of fused oblong-shaped LC domains, indicating a reduction of line tension in the film and a likely interaction of NFO at the LC-LE interface. Three hours after NFO injection, the π had decreased by ∼3 mN/m from the maximum. Concurrently, a number of bright fluorescence patches are observed on the FM images. Interestingly, these bright fluorescent patches appeared within LC domains in contrast to the bright domains located at the LC/LE domain perimeters caused by Aβm and FO.
One likely explanation for the bright fluorescent patches observed in the FM images is three-dimensional perturbation to the membrane.47 This could include, for example, formation of tubular aggregates via membrane collapse that pushes the fluorophore out of the plane of focus. This is likely the case for NFO where the appearance of bright patches coincides with a loss of surface pressure indicating material loss from the interface. However, this cannot be the case for FO because there was no accompanying surface pressure decrease to indicate material loss at the interface. For FO and Aβm, the origin of the bright patches is unclear, but one possible explanation is peptide binding induced lipid reorganization and concentration of TR-DHPE.
In summary, all forms of Aβ favorably interacted with an anionic lipid membrane, suggesting that protein/phospholipid interactions are maintained despite the drastically differing structures. Aβm and FO exhibit similar interactions with the membrane, reaching stable insertion plateaus that resulted in bright membrane patches at the interface between LE and LC domains. In contrast, NFO removed materials from the air/water interface within 2 h of incubation with the DMPG monolayer. Membrane invaginations were formed in the center of LC lipid domains, causing loss of film surface pressure. Our observations are consistent with a previously published study which showed that nonfibrillar Aβ oligomers interacted with liposomes to form small round structures that are potentially micelles or NFO–lipid complexes.48
Aβm and FO Stably Bind to Anionic Model Phospholipid Membranes
XR data were modeled to provide ρ/ρwater profiles of the lipid-peptide film at angstrom-level resolution. The DMPG monolayer was modeled with 2 slabs: a 15.9 ± 0.2 Å thick layer of lipid tails with a ρ/ρwater value of 0.97 and a 9.1 ± 0.3 Å thick lipid headgroup layer with a ρ/ρwater value of 1.58 (Figure 6 and Table S3 in Supporting Information). After the addition of Aβm, a third slab underneath the lipid monolayer that extends into the water subphase was required to fit the XR data. This is a layer, 34.0 ± 0.3 Å in thickness with a ρ/ρwater value of 1.154 ± 0.002, of protein associated with lipid monolayer (Figure 6B). ρ/ρwater of this layer is consistent with previously published results.24 FO also formed a layer attached to the lipid headgroups with similar thickness and ρ/ρwater as Aβm, 37.4 ± 0.6 Å and 1.171 ± 0.004, respectively. Fitting of XR data with alternate models that accounted for the presence of protein in the lipid monolayer, e.g., associated with the lipid headgroups or tails, were also explored. These more complex models did not produce better fitting and thus did not support a model with Aβ significantly inserted into the lipid monolayer or adsorbed at the air/water interrace. Note that the thicknesses of the Aβm and FO layers associated with the lipid membrane are about two times that of the peptide layers adsorbed to the air/water interface. This shows that Aβm and FO adopt different structures at the membrane interface compared to the air/water interface, indicating that the lipid membrane exerts specific effects on the structure and assembly of the accumulated peptides. Also, we cannot preclude the possibility of the formation of two peptide layers, each ∼20 Å thick, on the membrane surface. However, our observation that increasing or decreasing peptide concentration did not affect the thickness of the adsorbed protein layer points to a single peptide layer formed from specific interactions with the anionic lipid membrane. Association of both FO and Aβm to the membrane caused moderate decreases in lipid tail thickness, from 15.9 ± 0.2 to 13.21 ± 0.04 and 14.67 ± 0.03 Å, respectively, possibly stemming from peptide induced disruption of lipid packing observed from FM imaging and GIXD results.
Figure 6.

X-ray reflectivity measurements of Aβ interactions with DMPG. (A) XR data was collected for a DMPG monolayer (black), and 8 h after the addition of Aβm (blue), FO (green), or NFO (red). Reflectivity, normalized to Fresnel reflectivity (R/RF), is plotted as points with experimental error, and the fitted model is overlaid as a solid line. Data are offset for clarity. (B) Electron density profiles normalized to water, ρ/ρwater, for DMPG before and after addition of Aβm, FO, or NFO. The depth is plotted with zero marking the air/lipid tails interface; negative values are in the air, and positive values extend into the water subphase. The solid lines represent slab model fits and the dashed lines represents a smoothed slab model fit where the slabs were fit with roughness parameters at interfaces. Schematic lipids and protein (Aβm, blue; FO, green) are overlaid to help with the visualization of the modeled electron density. Duplicate measurements were made during the experiment as peptide insertion leveled. The final measurement of the equilibrated system is shown.
Interestingly, the addition of NFO showed minimal impact on the R/RF of the DMPG monolayer and no associated peptide layer was observed as a good fit was achieved with a 2-slab lipid membrane model (Figure 6). This observation seems surprising, as NFOs clearly interacted with the monolayer increasing the π of the film. However, considering that fast and then slow loss of material from the film, likely NFO–lipid complexes, was observed as shown by decreases of π at longer incubation times (Figure 5A), the film measured by XR could be predominantly lipids, with few NFOs bound. Combining surface pressure isotherm and XR results, we thus conclude that the interaction of NFOs with the monolayer likely resulted in the extraction of lipids from the monolayer, and at the time of measurement, little protein was found associated with the lipid monolayer. However, the rate of extraction is slow, and a lipid monolayer was clearly still present.
Protofibrillar Aβ Disrupts Phospholipid Packing
As described earlier, a DMPG monolayer at 25 mN/m and 23.5 °C contains both disordered LE and ordered LC phases where the lipid acyl tails arrange into crystalline domains. A major advantage of studying crystalline lipid domains in monolayers is the ability to measure X-ray diffraction from the LC phase. GIXD was therefore used to monitor changes in membrane lipid packing with the association of the different Aβ species. Diffraction peaks arise from ordered lipid tail domains in the lipid monolayer. In addition to obtaining lattice spacings of the ordered lipid tails from the qxy positions of the peaks, the intensity of the diffraction peak obtained from GIXD can be used to quantify the amount of diffracting material, for example, lipid tails in the LC domains.
The pure DMPG monolayer at 25 mN/m produced a single diffraction peak, indicating that the lipid tails are arranged in a 2D hexagonal unit cell structure with the tails oriented perpendicular to the liquid surface (Figure 7). The Bragg peak was centered at a qxy value of 1.491 ± 0.006 Å−1, corresponding to a lattice spacing of 4.87 ± 0.02 Å (Table S3 in Supporting Information). From the FWHM of the diffraction peak, the average length of the packed lipid domains (Lc) was 382 ± 10 Å (Table S3 in Supporting Information).
Figure 7.

Grazing incidence X-ray diffraction (GIXD) of Aβ interacting with a DMPG monolayer. GIXD measurements were collected on a pure DMPG monolayer compressed to 25 mN/m, and after addition of Aβm, FO, or NFO. Background-subtracted integrated diffraction values (points) with experimental error are overlaid with the model fit to the data (solid line). GIXD peaks around 1.48 Å−1 are attributed to condensed lipid tail diffraction, and peaks around 1.32 Å−1 (inset) are attributed to β-sheet rich protein diffraction. GIXD scans were obtained 8 h after protein injection. Duplicate measurements were made during the experiment as peptide insertion leveled. The final measurement of the equilibrated system is shown.
After addition of the Aβ conformers, lattice spacing of ordered lipid tails remained unchanged, but the size and amount of diffracting lipid domains significantly decreased as indicated by decreases in Lc and integrated Bragg peak intensities, respectively (Table S3 in Supporting Information). The addition of Aβm caused a decrease in the integrated intensity of the diffraction peak by ∼50%, indicating a 50% decrease in the amount of ordered lipids. This loss occurs despite the higher π of 35.8 mN/m due to protein association, which in the absence of Aβm, would have led to more ordered lipids compared to the monolayer at 25 mN/m. FO interaction with the membrane resulted in a 68% decrease in lipid packing and reduced the size of lipid domains by nearly half compared to the pure DMPG monolayer. NFO induced the greatest reduction in lipid diffraction, about 80%. This result confirms FM and surface pressure measurements (Figure 5) that NFO interacted with and caused significant disruptions to the lipid monolayer. Although no NFO was detected at the membrane surface at this 8 h post injection time point based on XR results, the association and extraction of lipids from the monolayer significantly disrupted membrane lipid packing. Interestingly, NFO interactions with the membrane did not reduce Lc of the ordered lipid domains compared to the monolayer at 25 mN/m (Table S3 in Supporting Information). For Aβm and FO, a second diffraction peak is observed at 1.33 Å−1, which corresponds to a d-spacing of 4.7 Å. As discussed earlier, this spacing exactly matches the distance between β-strands in amyloid fibrils.44 Thus, the disordered Aβm assembled into a β-sheet rich protofibrillar structure at the anionic lipid membrane surface. FO retains its original structure, which is readily accommodated by the membrane. The Lc values of the β-sheet diffraction peaks (79 ± 11 and 67 ± 17 Å) corresponds to, approximately, 14–17 β-strands in registry and is similar to the Lc values obtained for protofibrils formed at the air/water interface.
From our experience, it was explicitly necessary to remove the HFIP used during the preparation of the FO sample for membrane studies. HFIP is surface active, and we observed drastically different results in the surface activity, XR, and GIXD data when HFIP from preparation (<17%) was present in the sample. In X-ray scattering studies with DMPG and FO containing HFIP, we observed no protein binding to the DMPG film, no reduction in lipid diffraction, and no formation of β-sheet rich protein structure (data not shown). Furthermore, previous studies have shown that HFIP alone is capable of inducing membrane thinning,49 thus obscuring results from studies that evaluate protein impacts on membrane structure. Overall, we stress the importance of removing HFIP from protein samples when testing surface activity or interactions with lipid membranes.
The strength of the lipid monolayer membrane model is its simplicity and control over lipid composition density. However, a lipid monolayer does not capture several important features of bilayer membranes, including membrane curvature. Saturated DMPG lipids were used for the Langmuir trough studies to avoid lipid oxidation. However, a more biologically relevant system would include unsaturated lipids with a varied composition. As such, we tested Aβ interactions with 100 nm size large unilamellar vesicles (LUVs) composed of 70 mol % POPC and 30 mol % POPG. This proportion of anionic to zwitterionic lipids mimics the composition found on the intracellular leaflet of human erythrocyte membranes.50 Protein secondary structures were monitored by circular dichroism spectroscopy to determine the impacts of LUVs on Aβ structure. Consistent with GIXD results, a large secondary structural transition was observed when Aβm binds to the vesicles (Figure 8). Aβm in solution is largely random coil, but after binding to LUVs, the peptide adopted a mix of random coil and β-sheet structures (Figure 8A). FO and NFO retained their original β-sheet and random coil structures, respectively, after incubation with the LUVs. Notably, the spectrum of NFO became significantly noisier after the addition of LUVs. This is perhaps due to the extraction of lipids from the vesicles that resulted in significant light scattering from protein/lipid complexes in the solution. The CD measurements corroborate that Aβm, FO, and NFO undergo similar secondary structural changes, or the lack thereof, when interacting with vesicles and monolayers. Thus, we expect that Aβ structural changes are a common feature among interactions with bilayers composed of unsaturated lipids and, in our studies, with monolayers assembled from saturated lipids.
Figure 8.

Circular dichroism spectra of Aβm (A), FO (B), and NFO (C) incubation with 7:3 POPC/POPG LUVs. CD spectra shown are averages of 3–5 replicate scans.
Altogether, our results show that Aβm, FO, and NFO all interact with anionic-containing lipid membranes. For Aβm, membrane interactions result in a rapid conversion of the unstructured Aβm to a β-sheet rich fibrillar structure. The binding and incorporation of the Aβ species disrupts lipid packing, as evidenced by reductions in both the amount of lipids in ordered domains and in the average size of these domains. NFO did not remain stably bound to the membrane after initial insertion, yet it caused the largest reduction in lipid diffraction. Furthermore, there was minimal impact to the size of the lipid domains, indicating that NFO does not interfere with membrane structure via rearrangement of the lipids. Coupled with the loss of surface pressure and the absence of proteins detected at the membrane surface by XR, it is apparent that NFO binding results in the removal of lipids from the membrane, potentially by forming stable protein/lipid complexes and detaching from the membrane surface. This finding points to an NFO toxicity mechanism where NFO induces toxicity via a detergent-like lipid removal mechanism that destabilizes the cell membrane.
Combining findings from XR and GIXD results, we constructed a model for Aβm and FO protofibril formation on the DMPG membrane or at the air/water interface (Figure 9). The protofibril formed at the DMPG membrane (Figure 9B) had a thickness of ∼36 Å, as measured by XR. In each protofibrillar assembly that spans 70 Å along the fibril growth axis, a 4.7 Å space separates each β-strand. Concomitant with protofibril binding or formation, disruption to the packing of lipid tails occurred.
Figure 9.

Model of all three Aβ40 conformers adsorbed to an air/water interface (A) and protofibrils formed from Aβm and FO adsorbed to DMPG (B). In part A, the 80 Å sized protofibrils adsorbed to the air/water interface are 19 Å in width with 4.7 Å between diffracting β-strands. The second 180 Å sized diffracting structure with a lattice spacing of 41 Å likely corresponds to packing between neighboring β-sheets. In part B, the 70 Å sized protofibrils assembled onto an anionic DMPG monolayer are 36 Å in width with 4.7 Å of spacing between diffracting β-strands. Protein binding also disrupted phospholipid tail packing.
The protofibrils nucleated from Aβm and FO at the air/water interface (Figure 9A) were roughly half the thickness compared to the Aβm- and FO-derived protofibrils formed at the DMPG membrane surface (19 Å compared to 36 Å), although similar in Lc values. While Bragg rod analysis was not performed on this data due to weak scattering intensities, Chi et al.24 previously reported that the length of the diffracting unit is 13.5 Å out of the 18.6 Å total protein layer. With 3.35 Å between neighboring cα carbons extended in a β-sheet conformation, this suggests that four amino acids participate in stable β-sheet hydrogen bonding along the width of the fibril. An additional diffraction peak was observed in the Aβm adsorbed film that indicated another characteristic lattice packing spacing of 41.2 Å. This spacing is likely from ordered packing between neighboring β-sheets, which formed average scattering domains of 180 Å in length.
■ DISCUSSION
In this study, we compared the surface activity and membrane interactions of three structurally distinct forms of Aβ40: Aβm, FOs, and NFOs. These three Aβ conformers have different structures (disordered, β-sheet rich fibrillar, and random coil/α-helix rich globular) that could potentially lead to different interactions with lipid membranes, which may account for their different toxicities. Aβ oligomers extracted from diseased brain tissue are able to induce toxicity and disease in previously healthy cells and animals51 and are thus considered to be the most toxic species of the Aβ peptide. For membrane-mediated Aβ toxicity, NFOs are the most deleterious to membrane structure, followed by fibrillar species, and then monomers.23 Previous characterization methods have included measurements of cellular ion dysregulation, vesicular dye leakage assays, and imaging. However, the molecular-level structural details of membrane disruption have not been characterized for the different Aβ species.13,25,26,37 Aβ/membrane interactions have been documented to play a key role in the peptide’s neurotoxicity,3 and it is imperative to study the mechanism and resolve molecular-level structures of these interactions.
One factor in protein/membrane interactions is the protein’s inherent surface activity. The air/water interface was used as a model hydrophilic/hydrophobic interface to evaluate the intrinsic surface activity of Aβm, FO, and NFO. All forms of the peptide adsorbed to the air/water interface (Figure 2), and all adopted a β-sheet rich protofibrillar structure that extended 19 Å below the air/water interface (Figures 3 and 4). The air/water interface induced disordered Aβm to misfold and aggregate into β-sheet rich assemblies. Importantly, adsorption to the interface also caused NFO, the most toxic Aβ species,23 to transition from a globular structure to a fibrillar structure that is associated with less toxicity. One hypothesis in the prion field is that protein aggregation can be a natural mechanism for controlling free protein concentration in the cell. In Huntington’s disease, the huntingtin protein was less toxic when present in the form of large aggregate inclusions than when the protein was soluble.52 We hypothesize that the same control mechanism may be at play here: NFO can be converted to a less toxic protofibrillar form via a hydrophilic/hydrophobic interface.
Aβm, FO, and NFO all favorably interacted with anionic lipid membranes (Figure 5). Consistent with previous findings,24 Aβm misfolded and assembled into a β-sheet rich protofibrillar structure that was indistinguishable from FO’s structure upon binding the membrane. Aβm and FO had similar deleterious impacts to the membrane structure, including disruption of lipid packing and macroscale membrane deformation. Note that the protofibril structures for Aβm and FO were different depending on whether they were scaffolded by the anionic phospholipid membrane or induced by the hydrophilic/hydrophobic air/water interface. The protofibril thickness was 36 Å for the anionic membrane-scaffolded protofibrils (for Aβm and FO) and 19 Å for the air/water interface-induced protofibrils (Aβm, FO, and NFO). Our results indicate that Aβ possess a strong propensity for an extended β-sheet structure and its formation can be catalyzed by accumulation at interfaces. There are, however, subtle differences in the way the two interfaces induced Aβ structural changes. Specific interactions with the anionic lipids, or modulation of peptide–peptide interactions by the lipids, may favor the formation of a particular type of β-sheet protofibril and one that is different than those formed at the air/water interface. Our findings are consistent with the polymorphic nature of amyloid structures, where the same protein is capable of forming fibrillar aggregates of different conformations depending on conditions under which these aggregates are formed.
Aβm and FO both induced similar disruptions to the membrane structure. In both cases, hexagonally packed lipid domains in the membrane became disrupted (Figures 7 and 10). As membrane organization plays a key role in cell homeostasis, disruptions to lipid packing, for example, in lipid rafts, are likely deleterious to many membrane-mediated functions including cell–cell communication and trafficking. Disordering of the phospholipid tails also contributed to thinning of the monolayer (Table S3 in Supporting Information), suggesting a possible mode of toxicity for Aβm and FO.
Figure 10.

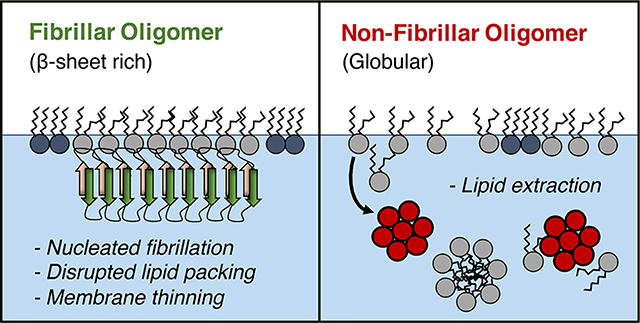
Model for two mechanisms of membrane-mediated toxicity. (A) The anionic lipid DMPG forms a stable monolayer with many phospholipids packed into condensed domains. (B) Aβ40 monomer binds the membrane and transitions to a β-sheet rich protofibrillar structure. Membrane-bound FOs retain their protofibrillar structure. The observed membrane thinning and disruption to the phospholipid packing likely contribute toward their toxicities. (C) Nonfibrillar oligomer extracts lipids from the membrane, likely resulting in micelles and formation of protein/phospholipid complexes that detach from the membrane.
More strikingly, our results support a membrane-mediated model for NFO toxicity via a detergent-like lipid removal mechanism (Figure 10). NFO was associated with the lipid monolayer during the first ∼2 h of incubation; this was followed by the formation of NFO–lipid complexes and detachment from the membrane as evidenced by decreasing surface pressure (Figure 5). There was a lack of NFO present at longer incubation times. NFOs also resulted in the greatest reduction in amount of ordered lipids among the three Aβ conformers tested. FM images (Figure 5) indicated lipid extraction into the subphase that originated in the condensed lipid domains. Overall, we found that the fibrillar and nonfibrillar Aβ conformers are both deleterious to the membrane, although mechanisms differ.
Our results closely corroborate a recent atomic force microscopy study published by Bode et al.;53 they report that oligomeric Aβ interactions with a lipid membrane bilayer resulted in “widespread lipid extraction and subsequent deposition” followed by “widespread curvature and discontinuities within lipid vesicle membranes”. They did not observe the same results for Aβm or fibrils, although those proteins did attach to the membrane. This lipid removal activity was also recently observed for full-length tau protein interactions with an anionic membrane.38 In their study, they show that tau acts via a “molecular tweezer mechanism” to extract lipids from an anionic membrane, resulting in the formation of stable tau/phospholipid complexes. These complexes were neurotoxic and displayed inter-cell trafficking behavior, suggesting a mechanism for how the disease may spread throughout the brain.
In conclusion, we show here that an anionic DMPG membrane rapidly nucleated protofibril formation from Aβm and these structures induced membrane thinning, disrupted phospholipid tail packing, and caused overall membrane instability. The globular NFO form of Aβ instead induces membrane instability by extracting lipids from the membrane and causing macroscale membrane deformations. Taken together, our results support two models for membrane-mediated Aβ toxicity: fibril-induced lipid reorganization and NFO-induced lipid extraction. These membrane perturbations are corroborated by previous membrane-permeability and cell toxicity assays finding that NFOs are the most deleterious to membrane structure, followed by fibrillar species. This work provides a molecular-level structural understanding of Aβ neurotoxicity via membrane interactions and aids the effort to understand early events in Alzheimer’s disease and other neurodegenerative diseases.
■ EXPERIMENTAL PROCEDURES
Materials
Lyophilized Aβ1–40 powder was purchased with 95.49% purity (purified by reverse-phase HPLC) from Peptide 2.0 (Chantilly, VA,) and product molecular weight was confirmed by mass spectrometry. 1,2-Dimyristoyl-sn-glycero-3-phosphorylglycerol (DMPG) was purchased from Avanti Polar Lipids (Alabaster, AL) as a dry powder; this was then dissolved in 7:3 v/v chloroform to methanol for spreading on the Langmuir trough. Lipids used for vesicle experiments, 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphorylglycerol (POPG), were obtained from Avanti Polar Lipids as presolubilized chloroform solutions. Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanoloamine triethylammonium salt (TR-DHPE) was purchased from Invitrogen-Thermo Fisher Scientific (Carlsbad, CA). All water used was prepared by Millipore Synergy UV filtration and purification. Copper grids (400 mesh), covered by a Formvar film and a 5–10 nm carbon layer, were purchased from Ted Pella (Redding, CA), and 2% aqueous uranyl acetate was purchased from Electron Microscopy Sciences (Hatfield, PA). Polyvinylidene difluoride membrane was purchased from ThermoFisher (Waltham, MA). A11 and OC antibodies from rabbit serum were purchased from Invitrogen (Carlsbad, CA) and Millipore (Burlington, MA), respectively, and 6E10 antibody from mouse serum was purchased from Convance (Princeton, NJ). Secondary antibodies, alkaline phosphatase-conjugated goat antirabbit, and goat antimouse, were purchased from Thermo Fisher Scientific (Waltham, MA) and Jackson ImmunoResearch (West Grove, PA). Horseradish peroxidase was purchased from Thermo Fisher Scientific.
Preparation of Aβm, FO, and NFO
For Aβm, the lyophilized peptide was solubilized (with sonication) in 100% DMSO at 2 mg/mL. To remove insoluble aggregates, the protein solution was centrifuged for 10 min at 14000 rpm, and the supernatant was collected. DMSO-solubilized Aβm were diluted to desired concentrations with Milli-Q water immediately prior to use. FOs and NFOs were prepared by adapting protocols published by Breydo et al.14 NFOs are referred to as prefibrillar oligomers (PFOs) in the referenced protocol. FOs were prepared by dissolving 0.3 mg of lyophilized Aβ peptide in 180 μL of hexafluoroisopropanol (HFIP). After 25 min of incubation at room temperature, the peptide was diluted to 1060 μL with Milli-Q water. FOs were grown by stirring the solution in a perforated microcentrifuge tube for 8 h at room temperature, and finally, HFIP was removed from the solution by blowing an airstream over the sample until it reached 50% of the original volume. Water was added to bring the sample to original volume, and the supernatant was collected after centrifugation to remove incidental mature fibrils. For NFOs, 0.3 mg of lyophilized Aβ powder was dissolved in 30 μL of 100 mM sodium hydroxide and incubated for 30 min at room temperature. pH 7.5, 10 mM phosphate buffer with 0.2% w/v sodium azide was added to dilute the sample to 780 μL; the sample was incubated at room temperature for 4 days and finally incubated an additional 5 days at 4 °C. In all cases, prepared samples were stored at −80 °C until use, and peptide concentration was verified by UV/vis absorbance with an Aβ extinction coefficient of 1490 M−1cm−1 at 280 nm.
Transmission Electron Microscopy (TEM)
A 4 μL portion of the 5 μM protein sample was deposited onto a glow discharged carbon-Formvar grid. Protein was allowed to bind for 5 min, and the remaining solution was then wicked away. A 4 μL portion of uranyl acetate was applied as a stain and removed after 3 min. After the initial staining, uranyl acetate was applied 3 more times with 30 s of staining for each application. Grids were imaged with a HITACHI HT7700 transmission electron microscope (Hitachi High Technologies Corp., Tokyo, Japan).
Dot Blot
Polyvinylidene difluoride membrane was prepared by washing in methanol, water, and Tris phosphate buffered saline (TPBS) before blotting 1 μL of 50 μM protein and air-drying the membrane for 15 min. Unbound protein was removed from the membrane by washing in methanol, water, and TPBS with 0.2% v/v TWEEN 20 (TBS-T). The membrane was blocked for 1 h in TBS-T and 5% nonfat dry milk, and it was washed 6 times with TBS-T. The primary antibody was diluted in TBS-T with 5% milk and incubated overnight with the membrane in 4 °C with agitation (6E10 diluted 1:500, OC 1:40000, and A11 1:2000). The membrane was washed 6 times with TBS-T before applying the secondary antibody diluted in TBS-T and milk for 1 h at room temperature. Peroxidase-conjugated goat antirabbit antibody was diluted 1:10000 for A11 blot and 1:40000 for OC blot, and peroxidase-conjugated goat antimouse antibody was diluted 1:1000 for the 6E10 blot. Finally, the membrane was washed 5 times before developing with horseradish peroxidase for 2–10 min.
Preparation of Large Unilamellar Vesicles (LUVs)
A 4 mM lipid mixture of 7:3 POPC to POPG in chloroform was dried overnight under a vacuum to form a dry film of lipids. The film was rehydrated in pH 6.0, 10 mM phosphate buffer to 2 mM lipid concentration. For vesicle formation, lipids were vortexed, incubated at room temperature for 30 min, and frozen and thawed five times. Finally, rehydrated vesicles were extruded 19 times to achieve a homogeneous 100 nm diameter sample.
Circular Dichroism (CD) Spectroscopy
Samples for CD measurements contained 50 μM protein in pH 6.0, 10 mM phosphate buffer. For samples containing a mixture of protein and LUVs, vesicles were added to 680 μM lipids (13.5:1 ratio of lipids to protein). LUVs were prepared with a mole ratio of 7:3 POPC to POPG. After mixing protein and vesicles, the sample was incubated for 30 min at room temperature before collecting data on an AVIV 410 CD spectrometer (Aviv Biomedical Inc., Lakewood, NJ) using a 0.1 cm path length quartz cell (Starna Cells, Atascaders, CA). Spectra shown are an average of 3–5 replicate scans with an averaging time of 15 s.
Lipid Monolayer Insertion and Surface Activity Assays
The insertion of Aβ into a DMPG lipid monolayer was measured using a KSV Micromini Langmuir trough (KSV Instruments Ltd., Finland) with Delrin barriers. DMPG spreading solution was 0.2 mg/mL DMPG with 0.5 mol % TR-DHPE dissolved in 9:1 v/v chloroform to methanol. This was prepared from stock solutions of 3.0 mg/mL DMPG (sonicated for 30 s in 7:3 v/v chloroform to methanol) and 0.5 mg/mL TR-DHPE solution (in 9:1 chloroform to methanol). Protein insertion experiments were performed in a 50 mL Teflon trough at room temperature (23.5 ± 0.5°C) on a pure water subphase. After spreading the lipids on the air/water interface, 10 min were allowed for solvent evaporation before lipids were compressed with symmetric barriers at 3.0 mm/sec to a surface pressure of 25 mN/m. After reaching 25 mN/m, the barrier positions were fixed so that peptide insertion resulted in surface pressure increase. Aβ was injected into the subphase to reach a final subphase concentration of 500 nM, and fluorescence microscopy images were collected with an Olympus IX51 model inverted fluorescent microscope (Tokyo, Japan) with a Teledyne QImaging Exi Aqua Bio-Imaging Camera (Surrey, BC, Canada). Surface pressure measurements to determine peptide surface activity were performed with a 25 mL Teflon trough containing a pure water subphase at room temperature. Aβ was injected to reach a final concentration of 250 nM in the subphase. Lipid monolayer insertion and surface activity assays were repeated three times; a representative profile is shown for each experimental condition.
X-ray Scattering Experiments
Synchrotron X-rays were used for liquid surface scattering experiments. Experiments were performed at the Advanced Photon Source (APS) at Argonne National Laboratories (Sector 15 NSF’s ChemMatCARS). A 20 mL Langmuir trough (6.5 × 6.5 cm2) was filled with degassed water and experiments were performed at room temperature (23.5 ± 0.5°C). Surface pressure was monitored with a Wilhelmy plate balance. DMPG lipids (0.2 mg/mL; dissolved in 9:1 v/v chloroform to methanol) were deposited to a surface pressure of 25 mN/m. Aβ was injected into the subphase to a final concentration of 2.5 μM in the trough; the high peptide concentration was chosen to accelerate protein insertion into DMPG. Data was collected after peptide insertion reached plateaus, which were 8, 4, and 11 h after peptide injection for Aβm, FO, and NFO, respectively. Protein insertion was slower in the Langmuir trough used for X-ray scattering studies. Duplicate XR and GIXD measurements were made on each sample while peptide insertion was equilibrated. Measurements of Aβ adsorbed to the air/water interface were performed with 1.25 μM protein injected into the 20 mL Langmuir trough that was filled with degassed water. The system was allowed to equilibrate at room temperature for 8–14 h prior to data collection. One set of XR and GIXD data was collected.
For all experiments, the trough was sealed in a canister and purged with helium gas. Data collection proceeded after oxygen content was <2% to prevent background scattering and oxidative beam damage to the monolayer. Data was collected using 1.24 Å wavelength X-rays, and the dimensions of the incoming X-ray beam footprint on the liquid surface were ∼1 × 3 to ∼1 × 10 mm2 for XR and ∼1 × 29 mm2 for GIXD. Each data collection scan typically required 1 h; as an additional precaution against beam damage, the trough was systematically translated by 1 mm (horizontally) during data collection. X-ray scattering was detected with a Dectris PILATUS 100 K detector, and X-ray reflectivity and diffraction images were integrated using Python software built by beamline support scientists. (https://github.com/weibu/Liquid_Surface_ChemMatCARS). Additional theory and analysis are described in the Supporting Information.
Supplementary Material
■ ACKNOWLEDGMENTS
X-ray scattering experiments were performed at the Advanced Photon Source at Argonne National Laboratory using NSF’s ChemMatCARS Sector 15 beamline. NSF’s ChemMatCARS Sector 15 is supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE- 1834750. Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02–06CH11357. This work was supported by NSF grants 1150855 and 1605225 to E.Y.C. and a postdoctoral fellowship from ASERT IRACDA K12 GM088021 to C.M.V.Z. NSF provided support for J.M. to contribute to this project through their Independent Research and Development program. L.W. was a summer intern in the Undergraduate Pipeline Network (UPN) program supported by ASERT IRACDA K12 GM088021, UNMCCC P30 CA118100, UNM CTSC UL1 TR001449; and NM INBRE P20 GM103451. I.B. was supported by NSF award #1560058 as a summer intern for Research Experience for Undergraduates (REU) in Nano Science & Micro Systems. Any opinion, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
■ ABBREVIATIONS
- Aβ
amyloid beta
- FO
fibrillar oligomer
- NFO
nonfibrillar oligomer
- APP
amyloid precursor protein
- AFM
atomic force microscopy
- HFIP
hexafluoroisopropanol
- DMSO
dimethyl sulfoxide
- TBS
tris-buffered saline
- CD
circular dichroism
- DMPG
1,2-dimyristoyl-sn-glycero-3-phosphorylglycerol
- XR
X-ray reflectivity
- GIXD
grazing incidence X-ray diffraction
- FM
fluorescence microscopy
- LC
liquid condensed
- LE
liquid expanded
- LUV
large unilamellar vesicles
- POPC
1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine
- POPG
1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- TEM
transmission electron microscopy
- TR-DHPE
Texas Red 1,2-dihexadecanoyl-sn-glycero-3-phosphoethanoloamine triethylammonium salt
Footnotes
The authors declare no competing financial interest.
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.langmuir.9b02484.
X-ray scattering data analysis; XR fit parameters; calculated values from GIXD fitting parameters; XR fit parameters for DMPG monolayer; two-dimensional intensity, I(qxy, qz), GIXD images of diffraction peaks (PDF)
■ REFERENCES
- (1).Hamley IW The Amyloid Beta Peptide: A Chemist’s Perspective. Role in Alzheimer’s and Fibrillization. Chem. Rev. 2012, 112 (10), 5147–5192. [DOI] [PubMed] [Google Scholar]
- (2).Kotler SA; Walsh P; Brender JR; Ramamoorthy A Differences between Amyloid-β Aggregation in Solution and on the Membrane: Insights into Elucidation of the Mechanistic Details of Alzheimer’s Disease. Chem. Soc. Rev. 2014, 43 (19), 6692–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Williams TL; Serpell LC Membrane and Surface Interactions of Alzheimer’s Aβ Peptide - Insights into the Mechanism of Cytotoxicity. FEBS J. 2011, 278 (20), 3905–3917. [DOI] [PubMed] [Google Scholar]
- (4).O’Brien RJ; Wong PC Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci. 2011, 34, 185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Price DL; Sisodia SS; Gandy SE Amyloid Beta Amyloidosis in Alzheimer’s Disease. Curr. Opin. Neurol. 1995, 8, 268–274. [DOI] [PubMed] [Google Scholar]
- (6).Benzinger TLS; Gregory DM; Burkoth TS; Miller-Auer H; Lynn DG; Botto RE; Meredith SC Two-Dimensional Structure of β-Amyloid(10–35) Fibrils. Biochemistry 2000, 39 (12), 3491–3499. [DOI] [PubMed] [Google Scholar]
- (7).Wälti MA; Ravotti F; Arai H; Glabe CG; Wall JS; Böckmann A; Güntert P; Meier BH; Riek R Atomic-Resolution Structure of a Disease-Relevant Aβ(1–42) Amyloid Fibril. Proc. Natl. Acad. Sci. U. S. A. 2016, 113 (34), E4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Selkoe DJ Physiological Production of the β-Amyloid Protein and the Mechanism of Alzheimer’s Disease. Trends Neurosci. 1993, 16 (10), 403–409. [DOI] [PubMed] [Google Scholar]
- (9).Soto C Unfolding the Role of Protein Misfolding in Neurodegenerative Diseases. Nat. Rev. Neurosci. 2003, 4 (1), 49–60. [DOI] [PubMed] [Google Scholar]
- (10).Tsai J; Grutzendler J; Duff K; Gan W Fibrillar Amyloid Deposition Leads to Local Synaptic Abnormalities and Breakage of Neuronal Branches. Nat. Neurosci. 2004, 7 (11), 1181–1183. [DOI] [PubMed] [Google Scholar]
- (11).Meyer-Luehmann M; Spires-Jones TL; Prada C; Garcia-Alloza M; de Calignon A; Rozkalne A; Koenigsknecht-Talboo J; Holtzman DM; Bacskai BJ; Hyman BT Rapid Appearance and Local Toxicity of Amyloid-β Plaques in a Mouse Model of Alzheimer’s Disease. Nature 2008, 451, 720–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Glabe CC Amyloid Accumulation and Pathogensis of Alzheimer’s Disease: Significance of Monomeric, Oligomeric and Fibrillar Aβ In: Harris JR, Fahrenholz F (Eds) Alzheimer’s Disease. Subcellular Biochemistry, Vol 38 Springer: Boston, MA, 2012. [DOI] [PubMed] [Google Scholar]
- (13).Kayed R; Head E; Thompson JL; McIntire TM; Milton SC; Cotman CW; Glabe CG Common Structure of Soluble Amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science (Washington, DC, U. S.) 2003, 300 (5618), 486–489. [DOI] [PubMed] [Google Scholar]
- (14).Breydo L; Kurouski D; Rasool S; Milton S; Wu JW; Uversky VN; Lednev IK; Glabe CG Structural Differences between Amyloid Beta Oligomers. Biochem. Biophys. Res. Commun. 2016, 477 (4), 700–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Hou L; Shao H; Zhang Y; Li H; Menon NK; Neuhaus EB; Brewer JM; Byeon IL; Ray DG; Vitek MP; et al. Solution NMR Studies of the Aβ (1 – 40) and Aβ (1 – 42) Peptides Establish That the Met35 Oxidation State Affects the Mechanism of Amyloid Formation. J. Am. Chem. Soc. 2004, 126 (7), 1992–2005. [DOI] [PubMed] [Google Scholar]
- (16).Wu JW; Breydo L; Isas JM; Lee J; Kuznetsov YG; Langen R; Glabe C Fibrillar Oligomers Nucleate the Oligomerization of Monomeric Amyloid β but Do Not Seed Fibril Formation. J. Biol. Chem. 2010, 285 (9), 6071–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Paravastu AK; Leapman RD; Yau W-M; Tycko R Molecular Structural Basis for Polymorphism in Alzheimer’s β-Amyloid Fibrils. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (47), 18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Bemporad F; Chiti F Protein Misfolded Oligomers: Experimental Approaches, Mechanism of Formation, and Structure-Toxicity Relationships. Chem. Biol. 2012, 19 (3), 315–327. [DOI] [PubMed] [Google Scholar]
- (19).Fändrich M Oligomeric Intermediates in Amyloid Formation: Structure Determination and Mechanisms of Toxicity. J. Mol. Biol. 2012, 421, 427–440. [DOI] [PubMed] [Google Scholar]
- (20).Ehrnhoefer DE; Bieschke J; Boeddrich A; Herbst M; Masino L; Lurz R; Engemann S; Pastore A; Wanker EE EGCG Redirects Amyloidogenic Polypeptides into Unstructured, off-Pathway Oligomers. Nat. Struct. Mol. Biol. 2008, 15 (6), 558–566. [DOI] [PubMed] [Google Scholar]
- (21).Ladiwala ARA; Litt J; Kane RS; Aucoin DS; Smith SO; Ranjan S; Davis J; Van Nostrand WE; Tessier PM Conformational Differences between Two Amyloid β Oligomers of Similar Size and Dissimilar Toxicity. J. Biol. Chem. 2012, 287 (29), 24765–24773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Xue W; Hellewell AL; Gosal WS; Homans SW; Hewitt EW; Radford SE J. Biol. Chem. 2009, 284 (49), 34272–34282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Dahlgren KN; Manelli AM; Blaine Stine W; Baker LK; Krafft GA; Ladu MJ Oligomeric and Fibrillar Species of Amyloid-β Peptides Differentially Affect Neuronal Viability. J. Biol. Chem. 2002, 277 (35), 32046–32053. [DOI] [PubMed] [Google Scholar]
- (24).Chi EY; Ege C; Winans A; Majewski J; Wu G; Kjaer K; Lee KYC Lipid Membrane Templates the Ordering and Induces the Fibrillogenesis of Alzheimer’s Disease Amyloid-β Peptide. Proteins: Struct., Funct., Genet. 2008, 72 (1), 1–24. [DOI] [PubMed] [Google Scholar]
- (25).Williams TL; Day IJ; Serpell LC The Effect of Alzheimer’s Aβ Aggregation State on the Permeation of Biomimetic Lipid Vesicles. Langmuir 2010, 26 (22), 17260–17268. [DOI] [PubMed] [Google Scholar]
- (26).Canale C; Seghezza S; Vilasi S; Carrotta R; Bulone D; Diaspro A; San Biagio PL; Dante S Different Effects of Alzheimer’s Peptide Aβ(1–40) Oligomers and Fibrils on Supported Lipid Membranes. Biophys. Chem. 2013, 182, 23–29. [DOI] [PubMed] [Google Scholar]
- (27).Sokolov Y; Kozak JA; Kayed R; Chanturiya A; Glabe C; Hall JE J. Gen. Physiol. 2006, 128 (6), 637–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Dong X; Sun Y; Wei G; Nussinov R; Ma B Binding of Protofibrillar Aβ Trimers to Lipid Bilayer Surface Enhances Aβ Structural Stability and Causes Membrane Thinning. Phys. Chem. Chem. Phys. 2017, 19, 27556–27569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Sato H; Feix JB Biochim. Biophys. Acta, Biomembr. 2006, 1758, 1245–1256. [DOI] [PubMed] [Google Scholar]
- (30).Hebda JA; Miranker AD The Interplay of Catalysis and Toxicity by Amyloid Intermediates on Lipid Bilayers: Insights from Type II Diabetes. Annu. Rev. Biophys. 2009, 38, 125–152. [DOI] [PubMed] [Google Scholar]
- (31).Busch DJ; Houser JR; Hayden CC; Sherman MB; Lafer EM; Stachowiak JC Intrinsically Disordered Proteins Drive Membrane Curvature David. Nat. Commun. 2015, 6 (7875), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Milanesi L; Sheynis T; Xue W-F; Orlova EV; Hellewell AL; Jelinek R; Hewitt EW; Radford SE; Saibil HR Direct Three-Dimensional Visualization of Membrane Disruption by Amyloid Fibrils. Proc. Natl. Acad. Sci. U. S. A. 2012, 109 (50), 20455–20460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Quist A; Doudevski I; Lin H; Azimova R; Ng D; Frangione B; Kagan B; Ghiso J; Lal R Amyloid Ion Channels: A Common Structural Link for Protein-Misfolding Disease. Proc. Natl. Acad. Sci. U. S. A. 2005, 102 (30), 10427–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Lashuel HA; Hartley D; Petre BM; Walz T; Lansbury PT Amyloid Pores from Pathogenic Mutations. Nature 2002, 418, 291. [DOI] [PubMed] [Google Scholar]
- (35).Prangkio P; Yusko EC; Sept D; Yang J; Mayer M Multivariate Analyses of Amyloid-Beta Oligomer Populations Indicate a Connection between Pore Formation and Cytotoxicity. PLoS One 2012, 7 (10), e47261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Eliezer DJ Gen. Physiol. 2006, 128 (6), 631–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Sciacca MFM; Kotler SA; Brender JR; Chen J; Lee DK; Ramamoorthy A Two-Step Mechanism of Membrane Disruption by Aβ through Membrane Fragmentation and Pore Formation. Biophys. J. 2012, 103 (4), 702–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Ait-Bouziad N; Lv G; Mahul-Mellier AL; Xiao S; Zorludemir G; Eliezer D; Walz T; Lashuel HA Discovery and Characterization of Stable and Toxic Tau/Phospholipid Oligomeric Complexes. Nat. Commun. 2017, 8 (1678), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Vivekanandan S; Brender JR; Lee SY; Ramamoorthy A A Partially Folded Structure of Amyloid-Beta(1–40) in an Aqueous Environment. Biochem. Biophys. Res. Commun. 2011, 411 (2), 312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Sgourakis NG; Yan Y; McCallum SA; Wang C; Garcia AE The Alzheimer’s Peptides Aβ40 and 42 Adopt Distinct Conformations in Water: A Combined MD/NMR Study. J. Mol. Biol. 2007, 368 (5), 1448–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Yang T; Li S; Xu XH; Walsh DM; Selkoe DJ Large Soluble Oligomers of Amyloid β-Protein from Alzheimer Brain Are Far Less Neuroactive Than the Smaller Oligomers to Which They Dissociate. J. Neurosci. 2017, 37 (1), 152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Esparza TJ; Wildburger NC; Jiang H; Gangolli M; Cairns NJ; Bateman RJ; Brody DL Soluble Amyloid-Beta Aggregates from Human Alzheimer’s Disease Brains. Sci. Rep. 2016, 6 (1), 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Kayed R; Head E; Sarsoza F; Saing T; Cotman CW; Necula M; Margol L; Wu J; Breydo L; Thompson JL; Rasool S; Gurlo T; Butler P; Glabe CG; et al. Fibril Specific, Conformation Dependent Antibodies Recognize a Generic Epitope Common to Amyloid Fibrils and Fibrillar Oligomers That Is Absent in Prefibrillar Oligomers. Mol. Neurodegener. 2007, 2 (1), 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Serpell LC Alzheimer’s Amyloid Fibrils: Structure and Assembly. Biochim. Biophys. Acta, Mol. Basis Dis. 2000, 1502, 16–30. [DOI] [PubMed] [Google Scholar]
- (45).Seelig A Local Anesthetics and Pressure- a Comparison of Dibucaine Binding to Lipid Monolayers and Bilayers. Biochim. Biophys. Acta, Biomembr. 1987, 899 (2), 196–204. [DOI] [PubMed] [Google Scholar]
- (46).Ege C; Lee KYC Insertion of Alzheimer’s A Beta 40 Peptide into Lipid Monolayers. Biophys. J. 2004, 87 (3), 1732–1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Lee KYC Collapse Mechanisms of Langmuir Monolayers. Annu. Rev. Phys. Chem. 2008, 59, 771–791. [DOI] [PubMed] [Google Scholar]
- (48).Cheng Q; Hu Z; Doherty KE; Tobin-miyaji YJ; Qiang W The On-Fibrillation-Pathway Membrane Content Leakage and off-Fibrillation-Pathway Lipid Mixing Induced by 40-Residue β-Amyloid Peptides in Biologically Relevant Model Liposomes. Biochim. Biophys. Acta, Biomembr. 2018, 1860 (9), 1670–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Capone R; Quiroz FG; Prangkio P; Saluja I; Sauer AM; Bautista MR; Turner RS; Yang J; Mayer M Amyloid-β-Induced Ion Flux in Artificial Lipid Bilayers and Neuronal Cells: Resolving a Controversy. Neurotoxic. Res. 2009, 16, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Dodge JT; Phillips GB Composition of Phospholipids and Phospholipid Fatty Acids and Aldehydes in Human Red Cells. J. Lipid Res. 1976, 8, 667–675. [PubMed] [Google Scholar]
- (51).Cline EN; Bicca MA; Viola KL; Klein WL The Amyloid-β Oligomer Hypothesis: Beginning of the Third Decade. J. Alzheimer’s Dis. 2018, 64, S567–S610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Arrasate M; Mitra S; Schweitzer ES; Segal MR; Finkbeiner S Inclusion Body Formation Reduces Levels of Mutant Huntingtin and the Risk of Neuronal Death. Nature 2004, 431, 805–810. [DOI] [PubMed] [Google Scholar]
- (53).Bode DC; Freeley M; Nield J; Palma M; Viles JH Amyloid-β Oligomers Have a Profound Detergent-like Effect on Lipid Membrane Bilayers, Imaged by Atomic Force and Electron Microscopy. J. Biol. Chem. 2019, 294 (19), 7566–7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.