

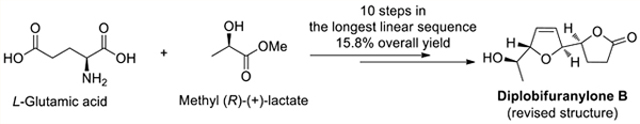
Abstract
An asymmetric total synthesis of diplobifuranylone B was achieved in 10 steps for the longest linear sequence and in 15.8% overall yield from commercially available methyl (R)-(+)-lactate and l-glutamic acid. This synthesis features a stereoselective construction of the key 2,5-dihydrofuran ring in the natural product via a recently developed asymmetric gold catalysis. The stereochemical flexibility offered by the catalysis enables an expedient revision of the reported structure of diplobifuranylone B, where the relative stereochemistry of the 2,5-dihydrofuran moiety was previously misassigned as cis instead of trans.
Graphical abstract
INTRODUCTION
Diplobifuranylone B (2) was isolated from Diplodia corticola, a fungi species well known as a phytotoxin producer, in 2006.1 Compounds of its family also include diplobifuranylone A (1)1 and diplobifuranylone C (3).2 The structures of these natural products (Figure 1) were proposed based on UV, 1H NMR, 13C NMR, COSY, HMQC, HMBC, and NOESY spectroscopic experiments. The absolute configuration of the stereogenic carbinol C6 was deduced via the Mosher’s method.3 The other stereogenic centers were determined using chiroptical methods in 2017.4 To date, the total synthesis of these secondary metabolites has not been reported.
Figure 1.

Diplobifuranylones produced by Diplodia corticola.
Recently, our group reported a stereoselective strategy for the construction of chiral 2,5-dihydrofurans from chiral propargylic alcohols via asymmetric gold catalysis. In this chemistry, a designed chiral bifunctional biphenyl-2-yl phosphine ligand (L) enables a gold-catalyzed asymmetric isomerization of an alkyne into an allene.5 With chiral propargylic alcohol as the substrate, the gold-catalyzed isomerization generates an alcohol intermediate featuring a chiral allene motif, which undergoes tandem gold-catalyzed stereospecific cyclization to form a chiral 2,5-dihydrofuran with excellent diastereoselectivity (Scheme 1). Herein, we report the application of this chemistry as a key step toward a concise total synthesis of nominal diplobifuranylone B (2), which possesses a cis 2,5-dihydrofuran ring. The stereochemical flexibility offered by our gold catalysis permits a rapid structural revision of this reported natural product, which instead features a trans-dihydrofuran moiety.
Scheme 1.

Gold-Catalyzed Asymmetric Cycloisomerization of Chiral Propargylic Alcohol
RESULTS AND DISCUSSION
Our retrosynthetic analysis of nominal diplobifuranylone B (2) is shown in Scheme 2. A key step is the installation of the cis-2,5-dihydrofuran ring by the gold-catalyzed asymmetric isomerization/cyclization of lactone 4 at the final stage. The lactone motif of 4 could be easily constructed through lactonization of dihydroxyester 5, which could, in turn, be prepared through an epoxide ring opening reaction between the epoxide 6 and the alkyne 7.
Scheme 2.

Retrosynthetic Analysis
We commenced the synthesis with the preparation of the two fragments 6 and 7 (Scheme 3). While the fragment 6 was prepared from l-glutamic acid (8) by following a reported four-step sequence,6 7 was synthesized from methyl (R)-(+)-lactate (9) in five steps. The synthetic sequence began with a TBS group protection of 9, followed by a DIBAL-H reduction. α-siloxypropanal 10 was isolated in 78% overall yield. An asymmetric nucleophilic addition to 10 by ethynyltrimethylsilane was achieved in the presence of a stoichiometric Ti(OiPr)4-BINOL complex7 to afford propargylic alcohol 11 in 70% isolated yield and with d.r. > 50:1 upon careful column separation. THP protection of 11 followed by selective removal of the TMS group gave the desired terminal alkyne 7 in a combined 94% yield.
Scheme 3.

Preparation of the Chiral Epoxide 6 and the Alkyne 7
With these two chiral fragments in hand, we attempted to implement the initially designed synthetic route. As shown in Scheme 4, the BF3-mediated ring opening8 of the chiral epoxide 6 by deprotonated 7 delivered γ-hydroxy ester 12 with the requisite carbon skeleton of the natural product. The removal of the THP group and lactonization were achieved in one step to give lactone 13 in 90% overall yield. However, when 13 was subjected to the asymmetric gold catalysis for the construction of 2,5-dihydrofuran ring, the reaction was sluggish with most of the starting material 13 remaining after 12 h; moreover, desired product 14 was not found in the reaction mixture.
Scheme 4.

Initial Attempt on the Total Synthesis of Diplobifuranylone B
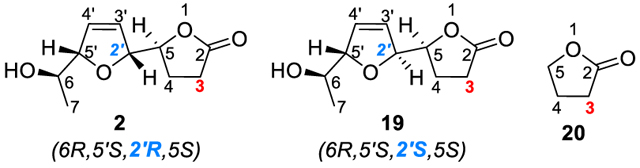
We speculated that the strong electron-withdrawing nature of the lactone motif might affect the gold-catalyzed isomerization of alkyne to allene and hence decided to modify the synthetic sequence to have the 2,5-dihydrofuran ring installed before the lactonization step. To this end, we converted γ-hydroxy esters 12 to the desired propargylic alcohol 16 in 76% combined yield through a two-step sequence, i.e., TBS protection of the free hydroxyl group to avoid potential gold-catalyzed 5-endo-dig cyclization and subsequent selective deprotection of the THP group in the presence of magnesium bromide.9 To our delight, subjecting 16 to our asymmetric gold catalysis with (S)-L as the ligand afforded smoothly the 2,5-dihydrofuran product 17 in 60% yield and with a diastereomeric ratio of 95:5. One-pot removal of both TBS groups of 17 and lactonization in the presence of PTSA/MeOH completed the synthesis of the nominal diplobifuranylone B (2) in 80% yield (Scheme 5). The absolute stereochemistry of C2′ is assigned based on our previous report on chiral 2,5-dihydrofuran synthesis.5 To our surprise, there are obvious discrepancies between the 1H and 13C NMR spectra (Table 1) as well as the specific optical rotation {[α]20D = +24.7 (c = 1.06, CHCl3); lit. [α]20D = −90.7 (c = 0.55, CHCl3)} of our synthetic compound (2) and what were reported.1
Scheme 5.

Revised Route of Total Synthesis of Nominal Diplobifuranylone B
Table 1.
13C NMR Comparison
 | ||||||
|---|---|---|---|---|---|---|
| position | 2 | 19 | 20 | lit. | Δ(2-lit.)a | Δ(19-lit.)a |
| 5 | 81.04 | 80.12 | 68.49 | 80.1 | +0.94 | +0.02 |
| 4 | 23.54 | 23.75 | 22.06 | 23.7 | −0.16 | +0.05 |
| 3 | 27.74 | 27.97 | 27.70 | 22.9 | +4.84 | +5.07 |
| 2 | 176.39 | 177.24 | 177.81 | 177.2 | −0.85 | +0.04 |
| 2′ | 87.11 | 87.97 | 87.9 | −0.79 | +0.07 | |
| 3′ | 129.50 | 128.79 | 128.7 | +0.80 | +0.09 | |
| 4′ | 127.20 | 127.33 | 127.3 | −0.10 | +0.03 | |
| 5′ | 91.00 | 91.01 | 90.9 | +0.10 | +0.11 | |
| 6 | 69.08 | 69.09 | 69.1 | −0.02 | −0.01 | |
| 7 | 18.92 | 17.94 | 17.9 | +1.02 | +0.04 | |
Calculated with the accuracy of the literature data extended to 0.01 ppm.
Our asymmetric gold catalysis permits easy access to the C2′-epimer of 2, i.e., the trans-2,5-dihydrofuran 19 (Scheme 5) by simply employing the ligand enantiomer, i.e., (R)-L, in the conversion of 16. Indeed, by following the same two-step endgame, compound 19 was synthesized with comparable diastereoselectivity and efficiency to 2. Much to our delight, the spectroscopic data including 1H NMR, 13C NMR, 1H spin decouple, COSY, HMQC, and HMBC and the HR-MS measurement of 19 match those reported for diplobifuranylone B except one surprising outlier in the 13C data. The 13C chemical shifts of 2, 19, and the parent γ-lactone10 and those reported in the isolation paper are listed in Table 1 for comparison. All of the 13C chemical shifts of 19 are within 0.11 ppm difference from the literature data, which are reported with accuracy down to the 0.1 ppm level and hence can be considered as identical except that of the lactone C3. For this outlier, the reported value is 22.9 ppm, but our measured value is 27.97 ppm, which is very close to that of the parent γ-lactone (i.e., 27.7 ppm).10 It is noted that the chemical shift of the lactone C3 of diplobifuranylone A1 is 28.2 ppm. We also performed a rather comprehensive literature search of γ-lactones possessing only one substituent at C5, as in the case of diplobifuranylone B and found that the chemical shifts of the lactone C3 range from 26.9 to 29.5 ppm.11 In none of the example, the 13C signal could shift to as high a field as 22.9 ppm. Unfortunately, we could not obtain the original 1H and 13C spectra of diplobifuranylone B. Considering that all of the other 13C chemical shifts are essentially identical between the reported data and those of 19, we feel confident to conclude that the reported C3 13C chemical shift is a typo and should be 27.9 ppm.
To understand the misassignment of the natural product’s 2,5-dihydrofuran stereochemistry, we carefully examined the original reports.1,4 It was based on the NOESY, ROESY, and double decoupling experiments. Our DFT calculations reveal that the distances between H-2′ and H-5′ in the optimized structures of the cis (2) and the trans (19) isomers at the B3LYP//cc-pVDZ level are 3.50 and 3.99 Å, respectively. With both of these measurements being less than 4 Å, the observed nOe might not be a reliable indicator of a cis configuration. The double decoupling experiment revealed that the long-distance coupling constant between H-2′ and H-5′ is 5.5 Hz, which was suggested to corroborate the cis stereochemistry. However, this large long-range coupling constant is characteristic of trans-2,5-dihydrofurans.12 Typically, the J values of trans-2,5-dihydrofurans are >5 Hz, whereas those of cis-2,5-dihydrofurans are <4 Hz. Some of the examples we found in the literature are listed in Figure 2.13–17 Our decoupling experiments reveal that the coupling constants between H-2′ and H-5′ are 3.8 and 5.8 Hz for 2 and 19, respectively, which is consistent with our stereochemistry assignments. A related case is furanomycin (30) (Figure 3). The relative stereochemistry of its featured 2,5-dihydrofuran ring was initially assigned incorrectly as cis in 31 based on the coupling constant of 5.7 Hz between H-2 and H-5.18 However, it was later determined to be trans upon its total synthesis13 and X-ray diffraction studies.19
Figure 2.

Long-range coupling constant between H-2 and H-5 (J2,5) in the 2,5-dihydrofuran systems.
Figure 3.

Structure of (+)-furanomycin and its initially misassigned cis-structure 31.
CONCLUSIONS
In conclusion, an asymmetric total synthesis of diplobifuranylone B was accomplished in 10 steps for the longest linear sequence and in 14 total steps from the commercially available methyl (R)-(+)-lactate and l-glutamic acid. The overall yield was 15.8%. The key 2,5-dihydrofuran moiety of the natural product is constructed via a recently published asymmetric gold catalysis. This work allows the revision of the structure of diplobifuranylone B, in which the relative stereochemistry of its 2,5-dihydrofuran moiety is established as trans instead of the originally assigned cis. The total synthesis is convergent and should be applicable to the synthesis of other diplobifuranylones.
EXPERIMENTAL SECTION
General Information.
Ethyl acetate (ACS grade), hexanes (ACS grade), and diethyl ether (ACS grade) were purchased from Fisher Scientific and used without further purification. Anhydrous dichloro-methane (HPLC grade) and 1,2-dichloroethane (HPLC grade) were purified by distillation over calcium hydride. Tetrahydrofuran, toluene, and o-xylene were distilled over sodium/benzophenone. Commercially available reagents were used without further purification. Reactions were monitored by thin layer chromatography (TLC) using silicycle-precoated silica gel plates. Flash column chromatography was performed over Silicycle silica gel (230–400 mesh). 1H NMR and 13C NMR spectra were recorded on Varian 400, 500, and 600 MHz spectrometers using residue solvent peaks as internal standards (CDCl3, 1H: 7.26 ppm; 13C: 77.00 ppm).
(R)-2-((tert-Butyldimethylsilyl)oxy)propanal (10). Aldehyde (10) was synthesized from methyl (R)-(+)-lactate (9) according to the literature procedure.20 The yield was 78%, colorless oil; 1H NMR (600 MHz, CDCl3) δ 9.61 (d, J = 1.3 Hz, 1H), 4.09 (qd, J = 6.8, 1.3 Hz, 1H), 1.28 (d, J = 6.8 Hz, 3H), 0.92 (s, 9H), 0.10 (s, 3H), 0.09 (s, 3H); 1H NMR is in accordance with the literature.20
(3S,4R)-4-((tert-Butyldimethylsilyl)oxy)-1-(trimethylsilyl)pent-1-yn-3-ol (11). Propargylic alcohol (11) was synthesized from aldehyde (10) according to a modified literature procedure.21 Under N2 protection, TMS acetylene (5.54 mL, 40 mmol) and 14 mL toluene were added into a Schlenk flask. Et2Zn (1.5 M) in toluene (26.7 mL, 40 mmol) was added to the solution carefully. The mixture was heated to reflux for 1 h, during which time a large amount of white precipitate formed in the reaction flask. The mixture was cooled to room temperature, and (S)-BINOL (1.14 g, 4 mmol), Et2O (80 mL), and Ti(OiPr)4 (2.96 mL, 10 mmol) were added. After stirring for 1 h, aldehyde 9 (1.88 g, 10 mmol) was added, and the mixture was stirred overnight. Tartaric acid (1.0 M) was slowly added into the reaction mixture to quench the reaction and further stirring for 30 min. The mixture was partitioned in a separatory funnel and the aqueous portion was extracted three times with Et2O. The combined organic extracts were washed with brine and dried over MgSO4. Filtration and concentration, followed by flash column chromatography on the silica gel (hexane/Et2O = 100:1 to hexane/Et2O = 10:1 gradient), afforded propargylic alcohol 11 (2.03 g, 70% yield, d.r. > 50:1) as a white solid. 1H NMR (500 MHz, CDCl3) δ 4.23 (dd, J = 5.1, 3.8 Hz, 1H), 3.91 (qd, J = 6.2, 3.9 Hz, 1H), 2.34 (d, J = 5.4 Hz, 1H), 1.22 (d, J = 6.2 Hz, 3H), 0.90 (s, 9H), 0.17 (s, 9H), 0.10 (s, 3H), 0.09 (s, 3H). 13C{1H} NMR (126 MHz, CDCl3) δ 103.7, 90.8, 71.0, 67.5, 25.8, 18.2, 18.0, −0.2, −4.4, −4.8. These data are in accordance with the literature.21
tert-Butyldimethyl(((2R,3S)-3-((tetrahydro-2H-pyran-2-yl)oxy)-5-(trimethylsilyl)pent-4-yn-2-yl)oxy)silane (11–1). Propargylic alcohol (11) (631.5 mg, 2.2 mmol) was dissolved in 10 mL of DCM. 3,4-Dihydro-2H-pyran (0.3 mL, 3.3 mmol) and pyridinium p-toluenesulfonate (28 mg, 0.11 mmol) were added into this solution and stirred overnight. When the reaction was near to completion, sodium bicarbonate solid was added into the reaction mixture and stirred for 30 min. The solvent was removed under reduced pressure and the residue was purified by flash chromatography on the silica gel (hexane/Et2O = 50:1), afforded compound 11–1 (805.6 mg, 99% yield, d.r. = 1:1) as a colorless liquid. 1H NMR (500 MHz, CDCl3) δ 5.03 (t, J = 3.1 Hz, 1H), 4.85 (t, J = 3.2 Hz, 1H), 4.15 (d, J = 5.9 Hz, 1H), 4.13 (d, J = 5.1 Hz, 1H), 4.07−4.01 (m, 1H), 3.94−3.90 (m, 2H), 3.92−3.87 (m, 1H), 3.83−3.76 (m, 2H), 1.97−1.46 (m, 12H), 1.24 (d, J = 6.1 Hz, 3H), 1.21 (d, J = 6.2 Hz, 3H), 0.89 (s, 9H), 0.88 (s, 9H), 0.15 (s, 9H), 0.15 (s, 9H), 0.10−0.06 (m, 12H); 13C{1H} NMR (126 MHz, CDCl3) δ 104.4, 103.2, 99.4, 94.6, 90.7, 89.8, 72.9, 71.3, 70.4, 61.9, 61.6, 30.22, 30.18, 25.84, 25.80, 25.52, 25.51, 20.4, 19.1, 18.9, 18.8, 18.1, 18.0, −0.08, −0.11, −4.51, −4.53, −4.7.
tert-Butyldimethyl(((2R,3S)-3-((tetrahydro-2H-pyran-2-yl)oxy)-pent-4-yn-2-yl)oxy)silane (7). Compound 11–1 (0.80g, 2.2 mmol) was dissolved in 10 mL of MeOH, followed by the addition of K2CO3 (450 mg, 3.3 mmol) into this reaction mixture. The solution was stirred at room temperature for 1 h. Et2O (30 mL) was added into this reaction mixture, and then the solid was removed via filtration through the silica gel pad (Et2O as eluent). The solvent was removed under reduced pressure to afford terminal alkyne (7) (612.1 mg, 94% yield) as a colorless liquid. 1H NMR (600 MHz, CDCl3) δ 5.02 (t, J = 3.3 Hz, 1H), 4.84 (t, J = 3.4 Hz, 1H), 4.21 (t, J = 4.5 Hz, 1H), 4.21 (t, J = 4.6 Hz, 1H), 4.02 (ddd, J = 11.4, 9.5, 3.1 Hz, 1H), 3.98−3.94 (m, 1H), 3.94−3.90 (m, 1H), 3.82 (ddd, J = 11.0, 9.7, 2.9 Hz, 1H), 3.57−3.49 (m, 2H), 2.41 (d, J = 2.2 Hz, 1H), 2.33 (d, J = 2.1 Hz, 1H), 1.91−1.48 (m, 12H), 1.26 (d, J = 6.2 Hz, 3H), 1.23 (d, J = 6.2 Hz, 3H), 0.89 (s, 9H), 0.88 (s, 9H), 0.09 (s, 3H), 0.09 (s, 3H), 0.08 (s, 3H), 0.07 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 99.7, 94.8, 82.2, 81.1, 74.1, 73.4, 72.0, 71.3, 70.3, 69.9, 62.2, 61.7, 30.24, 30.20, 25.84, 25.78, 25.5, 25.4, 20.2, 20.0, 18.9, 18.7, 18.2, 18.0, −4.55, −4.59, −4.59, −4.8.
Methyl (S)-3-(Oxiran-2-yl)propanoate (6). Epoxide (6) was synthesized from l-glutamic acid (8) according to the literature procedure.8 The overall yield was 20%, colorless oil; 1H NMR (600 MHz, CDCl3) δ 3.63 (s, 3H), 2.95−2.90 (m, 1H), 2.72−2.69 (m, 1H), 2.46−2.43 (m, 1H), 2.43−2.38 (m, 2H), 1.96−1.87 (m, 1H), 1.76−1.67 (m, 1H). 1H NMR is in accordance with the literature.8
Methyl (4S,8S,9R)-9-((tert-Butyldimethylsilyl)oxy)-4-hydroxy-8-((tetrahydro-2H-pyran-2-yl)oxy)dec-6-ynoate (12). Compound 12 was synthesized according to a modified literature procedure.8 Under nitrogen at −78 °C, terminal alkyne (7) (2.09 g, 7.0 mmol) was dissolved in dry THF(5 mL), then n-BuLi (2.5 M in hexane, 2.8 mL, 7.0 mmol) was added. After 5 min, BF3·Et2O (0.86 mL, 7.0 mmol) was added and 30 min later, epoxide (6) (650.7 mg, 5.0 mmol) was added. The reaction media was stirred at −78 °C for 3 h, then quenched with sat. NaHCO3(aq). The aqueous layer was extracted three times with Et2O. The combined organic layers were washed with brine, dried over Na2SO4. The solvent was remove under reduced pressure, and the residue was purified by flash column chromatography on the silica gel (hexane/EtOAc = 20:1 to hexane/ EtOAc = 10:1 to hexane/EtOAc = 1:1), to afford compound 12 (1.68 g, 78% yield) as a colorless liquid. 1H NMR (400 MHz, CDCl3) δ 4.97 (t, J = 3.2 Hz, 1H), 4.80 (t, J = 3.5 Hz, 1H), 4.19 (dt, J = 3.8, 1.8 Hz, 1H), 4.13 (dt, J = 4.1, 2.0 Hz, 1H), 4.02−3.92 (m, 2H), 3.88 (td, J = 6.2, 4.3 Hz, 2H), 3.79−3.70 (m, 2H), 3.66 (s, 6H), 3.53−3.46 (m, 2H), 2.53−2.26 (m, 8H), 1.96−1.44 (m, 16H), 1.20 (d, J = 6.5 Hz, 3H), 1.19 (d, J = 6.5 Hz, 3H), 0.87 (s, 9H), 0.87 (s, 9H), 0.07−0.04 (m, 12H); 13C{1H} NMR (101 MHz, CDCl3) δ 174.14, 174.12, 99.7, 94.8, 82.5, 82.0, 81.6, 80.1, 73.1, 71.2, 70.7, 70.3, 69.17, 69.15, 62.3, 61.8, 51.59, 51.57, 31.1, 31.0, 30.5, 30.38, 30.36, 30.2, 27.97, 27.93, 25.76, 25.72, 25.42, 25.33, 20.32, 19.18, 19.12, 19.01, 18.1, 18.0, −4.56, −4.67, −4.75, −4.75; HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C22H40O6SiNa 451.2492; found 451.2494.
Methyl (4S,8S,9R)-4,9-Bis((tert-butyldimethylsilyl)oxy)-8-((tetra-hydro-2H-pyran-2-yl)oxy)dec-6-ynoate (15). TBSCl (370.8 mg, 2.46 mmol) was added into a solution of 12 (878.9 mg, 2.05 mmol) and imidazole (279.1 mg, 4.10 mmol) in DMF (10 mL) at room temperature and stirred for 24 h. The reaction was quenched by the addition of sat. NaHCO3(aq) into the mixture. The aqueous phase was extracted four times with Et2O. The combined organic layer was washed with brine, dried over Na2SO4. The solvent was removed under reduced pressure, and the residue was purified by flash column chromatography on the silica gel (hexane/EtOAc = 20:1), to afford compound 15 (1.01 g, 91% yield) as a colorless liquid. 1H NMR (600 MHz, CDCl3) δ 4.99 (t, J = 3.3 Hz, 1H), 4.85 (t, J = 3.4 Hz, 1H), 4.23 (dt, J = 4.1, 2.1 Hz, 1H), 4.19 (dt, J = 4.9, 1.9 Hz, 1H), 4.00 (ddd, J = 11.4, 9.5, 3.0 Hz, 1H), 3.89 (dqd, J = 12.4, 6.2, 4.4 Hz, 2H), 3.85−3.79 (m, 3H), 3.65 (s, 6H), 3.49 (dqd, J = 12.0, 4.1, 1.5 Hz, 2H), 2.44−2.29 (m, 8H), 2.03−1.95 (m, 2H), 1.89−1.77 (m, 4H), 1.75−1.45 (m, 10H), 1.22 (d, J = 6.2 Hz, 3H), 1.20 (d, J = 6.2 Hz, 3H), 0.89−0.85 (m, 36H), 0.09−0.03 (m, 24H); 13C{1H} NMR (126 MHz, CDCl3) δ 174.0, 173.9, 99.4, 94.7, 82.6, 82.1, 79.2, 72.1, 71.7, 70.6, 70.2, 69.9, 69.8, 62.1, 61.6, 51.4, 31.4, 31.3, 30.26, 30.23, 29.62, 29.62, 27.63, 27.57, 25.80, 25.75, 25.72, 25.52, 25.46, 20.1, 19.02, 18.99, 18.5, 18.1, 18.00, 17.96, −4.56, −4.57, −4.65, −4.80, −4.88, −4.89; HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C28H54O6Si2Na 565.3357; found 565.3362.
Methyl (4S,8S,9R)-4,9-Bis((tert-butyldimethylsilyl)oxy)-8-hydrox-ydec-6-ynoate (16). MgBr2·Et2O (1.44 g, 5.57 mmol) was added into a solution of 15 (859.8 mg, 1.58 mmol) in Et2O and the reaction mixture was stirred at room temperature until the completion of the reaction. The solvent was removed and the residue was purified by flash column chromatography (hexane/EtOAc = 20:1 to hexane/EtOAc = 10:1) to give 16 (609.3 mg, 84% yield) as a colorless oil. [α]D20 = −22.7 (c = 1.19, CHCl3); 1H NMR (500 MHz, CDCl3) δ 4.27−4.22 (m, 1H), 3.89 (qd, J = 6.2, 3.6 Hz, 1H), 3.84 (tt, J = 7.6, 4.3 Hz, 1H), 3.66 (s, 3H), 2.44−2.29 (m, 5H), 2.00 (dddd, J = 13.8, 9.1, 6.7, 3.9 Hz, 1H), 1.82 (dddd, J = 13.7, 8.9, 7.4, 6.1 Hz, 1H), 1.21 (d, J = 6.2 Hz, 3H), 0.89 (s, 9H), 0.87 (s, 9H), 0.08 (s, 3H), 0.08 (s, 3H), 0.06 (s, 3H), 0.05 (s, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 174.0, 83.0, 80.0, 71.2, 69.8, 67.1, 51.5, 31.4, 29.6, 27.5, 25.8, 18.03, 17.97, −4.47, −4.55, −4.84, −4.86; HRMS (ESI-TOF) m/z: [M +Na]+ calculated for C23H46O5Si2Na 481.2781; found 481.2796.
Methyl (S)-4-((tert-Butyldimethylsilyl)oxy)-4-((2R,5S)-5-((R)-1-((tert-butyldimethylsilyl)oxy)ethyl)-2,5-dihydrofuran-2-yl)-butanoate (17). To a 2-dram vial were added sequentially 16 (68.0 mg, 0.15 mmol), 10 mol % (S)-LAuCl (13.2 mg), 20 mol % NaBAr4F (26.4 mg), and 0.75 mL of dry dichloroethane (DCE). The reaction was stirred at 60 °C for 50 h. Upon completion, the reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel flash column chromatography (hexane/Et2O = 20:1) to yield 17 (40.8 mg, 60% yield, d.r. = 95:5) as a colorless oil. [α]n20 = −13.7 (c = 1.11, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.99−5.96 (m, 1H), 5.95−5.92 (m, 1H), 4.58−4.52 (m, 1H), 4.47−4.37 (m, 1H), 3.66 (s, 3H), 3.48 (qd, J = 4.3, 2.0 Hz, 2H), 2.47 (t, J = 8.0 Hz, 2H), 2.02−1.84 (m, 2H), 1.20 (d, J = 6.0Hz, 3H), 0.89 (s, 9H), 0.89 (s, 9H), 0.06 (s, 3H), 0.05 (s, 3H), 0.04 (s, 3H), 0.03 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 174.3, 129.6, 129.5, 91.2, 88.2, 74.6, 72.4, 51.5, 29.3, 28.5, 25.8, 21.1, 18.04, 18.03, −4.23, −4.24, −4.5, −4.7; HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C23H46O5Si2Na 481.2781; found 481.2770.
Methyl (S)-4-((tert-Butyldimethylsilyl)oxy)-4-((2S,5S)-5-((R)-1-((tert-butyldimethylsilyl)oxy)ethyl)-2,5-dihydrofuran-2-yl)-butanoate (18). To a 2-dram vial were added sequentially 16 (68.0 mg, 0.15 mmol), 10 mol % (R)-LAuCl (13.2 mg), 20 mol % NaBAr4F (26.4 mg), and 0.75 mL of dry dichloroethane (DCE). The reaction was stirred at 60 °C for 50 h. Upon completion, the reaction mixture was concentrated under reduced pressure. The residue was purified by silica gel flash column chromatography (hexane/Et2O = 20:1) to yield 18 (40.8 mg, 62% yield, d.r. = 95:5) as a colorless oil. [α]n20 = −124.0 (c = 1.08, CHCl3); 1H NMR (600 MHz, CDCl3) δ 5.96 (dt, J = 6.3, 1.8 Hz, 1H), 5.85 (dt, J = 6.3, 1.8 Hz, 1H), 4.76 (tt, J = 5.2, 1.8 Hz, 1H), 4.55 (tt, J = 6.0, 1.8 Hz, 1H), 3.73−3.65 (m, 2H), 3.65 (s, 3H), 2.46 (ddd, J = 15.8, 10.2, 5.4 Hz, 1H), 2.30 (ddd, J = 16.1, 10.0, 6.1 Hz, 1H), 1.79 (tdt, J = 9.9, 6.1, 3.4 Hz, 1H), 1.64−1.54 (m, 1H), 1.15 (d, J = 6.2 Hz, 3H), 0.88 (s, 9H), 0.87 (s, 9H), 0.07 (s, 3H), 0.05 (s, 3H), 0.04 (s, 3H), 0.03 (s, 3H); 13C{1H} NMR (126 MHz, CDCl3) δ 174.2, 129.5, 127.6, 90.9, 89.4, 73.5, 71.3, 51.5, 30.5, 27.3, 25.9, 25.8, 20.3, 18.11, 18.07, −4.32, −4.43,−4.84, −4.88; HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C23H46O5Si2Na 481.2781; found 481.2786.
(S)-5-((2R,5S)-5-((R)-1-Hydroxyethyl)-2,5-dihydrofuran-2-yl)-dihydrofuran-2(3H)-one (2). p-Toluenesulfonic acid monohydrate (1.0 mg, 0.0055 mmol) was added into a solution of 17 (12.5 mg, mol) in MeOH (0.3 mL). The mixture was stirred at room temperature until the starting material 17 was consumed. The solvent was removed under reduced pressure and the residue was dissolved in DCM and stirred for 1 h. Then, the solvent was removed again, and the residue was purified by flash column chromatography (hexane/ EtOAc = 1:2) to yield 2 (4.5 mg, 83% yield) as a colorless oil. [α]D20 = +24.7 (c = 1.06, CHCl3); 1H NMR (600 MHz, CDCl3) δ 6.07 (dt, J = 6.6, 1.7 Hz, 1H), 5.96 (dt, J = 6.3, 1.8 Hz, 1H), 4.85−4.81 (m, 1H), 4.74−4.71 (m, 1H), 4.53 (dt, J = 7.3, 5.8 Hz, 1H), 3.89 (dq, J = 6.5, 3.8 Hz, 1H), 2.61−2.49 (m, 2H), 2.39−2.31 (m, 1H), 2.19−2.11 (m, 1H), 1.22 (d, J = 6.5 Hz, 3H); 13C{1H} NMR (101 MHz, CDCl3) δ 176.4, 129.5, 127.2, 91.0, 87.1, 81.0, 69.1, 27.7, 23.5, 18.9; HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C10H14O4Na 221.0790; found 221.0789; J(H2′−H5′) = 3.8 Hz.
(S)-5-((2S,5S)-5-((R)-1-Hydroxyethyl)-2,5-dihydrofuran-2-yl)-dihydrofuran-2(3H)-one (19). p-Toluenesulfonic acid monohydrate (2.0 mg, 0.01 mmol) was added into a solution of 18 (22.4 mg, 0.05 mol) in MeOH (0.5 mL). The mixture was stirred at room temperature until the starting material 18 has been consumed. The solvent was removed under reduced pressure, and the residue was dissolved in DCM and stirred for 1 h. Then, the solvent was removed again, and the residue was purified by flash column chromatography (hexane/EtOAc = 1:2) to yield diplobifuranylone B (19) (7.0 mg, 73% yield) as a colorless oil. [α]n20 = −132.4 (c = 0.55, CHCl ); literature1: [α]D 20 = −90.7 (c = 0.55, CHCl3); 1H NMR (600 MHz, CDCl3) δ 6.01 (dt, J = 6.3, 2.0 Hz, 1H), 5.90 (dt, J = 6.3, 2.0 Hz, 1H), 4.97 (dtd, J = 6.1, 2.5, 1.7 Hz, 1H), 4.79 (dddd, J = 5.9, 3.7, 2.3, 1.5 Hz, 1H), 4.54 (ddd, J = 8.0, 5.3, 2.8 Hz, 1H), 3.90 (dq, J = 6.5, 3.4 Hz, 1H), 2.66 (ddd, J = 17.7, 10.1, 7.0, 1H), 2.47 (ddd, J = 17.7, 10.3, 6.4, 1H), 2.34−2.27 (m, 1H), 1.64 (br, s, 1H), 1.17 (d, J = 6.5 Hz); 13C NMR{1H} (126 MHz, CDCl3) δ 177.2, 128.8, 127.3, 91.0, 88.0, 80.1, 69.1, 28.0, 23.8, 17.9; HRMS (ESI-TOF) m/z: [M + Na]+ calculated for C10H14O4Na 221.0790; found 221.0785; J(H2′−H5′) = 5.8 Hz, J(H5−H2′) = 2.8 Hz; literature1: J(H5−H2′) = 5.5 Hz; J(H2−H2′) = 2.8 Hz.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank NSF (CHE 1800525) and NIGMS (R01GM123342) for generous financial support, and NIH shared instrument grant S10OD012077 for the acquisition of a 400 MHz NMR spectrometer. The authors thank Dr Hongjun Zhou for NMR assistance. We also acknowledge support from the Center for Scientific Computing at the California NanoSystems Institute and Materials Research Laboratory, an NSF MRSEC (DMR1720256).
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.joc.9b01613.
DFT calculation and spectra data (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Evidente A; Andolfi A; Fiore M; Spanu E; Maddau L; Franceschini A; Marras F; Motta A. Diplobifuranylones A and B, 5′-Monosubstituted Tetrahydro-2H-bifuranyl-5-ones Produced by Diplodia corticola, a Fungus Pathogen of Cork Oak. J. Nat. Prod 2006, 69, 671–674. [DOI] [PubMed] [Google Scholar]
- (2).Masi M; Maddau L; Linaldeddu BT; Cimmino A; D’Amico W; Scanu B; Evidente M; Tuzi A; Evidente A. Bioactive Secondary Metabolites Produced by the Oak Pathogen Diplodia corticola. J. Agric. Food Chem 2016, 64, 217–225. [DOI] [PubMed] [Google Scholar]
- (3).Hoye TR; Jeffrey CS; Shao F. Mosher Ester Analysis for the Determination of Absolute Configuration of Stereogenic (Chiral) Carbinol Carbons. Nat. Protoc 2007, 2, 2451–2458. [DOI] [PubMed] [Google Scholar]
- (4).Mazzeo G; Cimmino A; Masi M; Longhi G; Maddau L; Memo M; Evidente A; Abbate S. Importance and Difficulties in the Use of Chiroptical Methods to Assign the Absolute Configuration of Natural Products: The Case of Phytotoxic Pyrones and Furanones Produced by Diplodia corticola. J. Nat. Prod 2017, 80, 2406–2415. [DOI] [PubMed] [Google Scholar]
- (5).Cheng X; Wang Z; Quintanilla CD; Zhang L. Chiral Bifunctional Phosphine Ligand Enabling Gold-Catalyzed Asymmetric Isomerization of Alkyne to Allene and Asymmetric Synthesis of 2,5-Dihydrofuran. J. Am. Chem. Soc 2019, 141, 3787–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Haynes SW; Sydor PK; Corre C; Song L; Challis GL Stereochemical Elucidation of Streptorubin B. J. Am. Chem. Soc 2011, 133, 1793–1798. [DOI] [PubMed] [Google Scholar]
- (7).Marshall JA; Bourbeau MP Synthesis of Enantioenriched Propargylic Alcohols Related to Polyketide Natural Products. A Comparison of Methodologies. Org. Lett 2003, 5, 3197–3199. [DOI] [PubMed] [Google Scholar]
- (8).Peru A; Flourat A; Gunawan C; Raverty W; Jevric M; Greatrex B; Allais F. Chemo-Enzymatic Synthesis of Chiral Epoxides Ethyl and Methyl (S)-3-(Oxiran-2-yl)propanoates from Renewable Levoglucosenone: An Access to Enantiopure (S)-Dairy Lactone. Molecules 2016, 21, 988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Kim S; Park JH Selective Removal of Tetrahydropyranyl Ethers in the Presence of tert-Butyldimethylsilyl Ethers with Magnesium Bromide in Ether. Tetrahedron Lett. 1987, 28, 439–440. [Google Scholar]
- (10).Das A; Chaudhuri R; Liu R-S Gold-Catalyzed Oxidative Cleavage of Aryl-Substituted Alkynyl Ethers Using Molecular Oxygen. Simultaneous Degradation of C−H and Single and Triple Carbon–Carbon Bonds under Ambient Conditions. Chem. Comm 2009, 4046–4048. [DOI] [PubMed] [Google Scholar]
- (11).For details, please see the Supporting Information.
- (12).Barfield M; Spear R; Sternhell S. Interproton Spin-Spin Coupling across a Dual Path in 2, 5-Dihydrofurans and Phthalans. J. Am. Chem. Soc 1975, 97, 5160–5167. [Google Scholar]
- (13).Semple JE; Wang PC; Lysenko Z; Joullie MM Total Synthesis of (+)-Furanomycin and Stereoisomers. J. Am. Chem. Soc 1980, 102, 7505–7510. [Google Scholar]
- (14).Cheng JCY; Hacksell U; Daves GD Facile Synthesis of 2′-Deoxy-3′-keto- and 2′-Deoxypseudouridine Derivatives and Analogues. Palladium(II)-mediated Coupling Reactions of Furanoid Glycals. J. Org. Chem 1986, 51, 3093–3098. [Google Scholar]
- (15).Outten RA; Daves GD Benzo[d]naphtho[1,2-b]pyran-6-one C-glycosides. Aryltri-n-butylstannanes in Palladium-mediated Coupling with 2,3-Dihydropyran and Furanoid Glycals. J. Org. Chem 1989, 54, 29–35. [Google Scholar]
- (16).Napolitano JG; Norte M; Padróń JM; Fernández JJ; Hernández Daranas A. Belizeanolide a Cytotoxic Macrolide from the Dinoflagellate Prorocentrum Belizeanum. Angew. Chem., Int. Ed 2009, 48, 796–799. [DOI] [PubMed] [Google Scholar]
- (17).Jain TC; Jenkins ID; Russell AF; Verheyden JPH; Moffatt JG Reactions of 2-Acyloxyisobutyryl Halides with Nucleosides. 1V.l A Facile Synthesis of 2,3-Unsaturated Nucleosides Using Chromous Acetate. J. Org. Chem 1974, 39, 30–38. [Google Scholar]
- (18).Katagiri K; Tori K; Kimura Y; Yoshida T; Nagasaki T; Minato H. A New Antibiotic. Furanomycin, an Isoleucine Antagonist. J. Med. Chem 1967, 10, 1149–1154. [DOI] [PubMed] [Google Scholar]
- (19).Shiro M; Nakai H; Tori K; Nishikawa J; Yoshimura Y; Katagiri K. X-Ray Crystal Structure Determination of Furanomycin. J. Chem. Soc., Chem. Commun 1980, No. 375a. [Google Scholar]
- (20).Gibson SM; Lanigan RM; Benhamou L; Aliev AE; Sheppard TD A Lactate-Derived Chiral Aldehyde for Determining the Enantiopurity of Enantioenriched Primary Amines. Org. Biomol. Chem 2015, 13, 9050–9054. [DOI] [PubMed] [Google Scholar]
- (21).Marshall JA; Bourbeau MP Synthesis of Enantioenriched Propargylic Alcohols Related to Polyketide Natural Products. A Comparison of Methodologies. Org. Lett 2003, 5, 3197–3199. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.