

Abstract
As one of the fundamental requirements for cell growth and proliferation, nitrogen acquisition and utilization must be tightly regulated. Nitrogen can be generated from amino acids and utilized for biosynthetic processes through transamination and deamination reactions. Importantly, limitations of nitrogen availability in cells can disrupt the synthesis of proteins, nucleic acids and other important nitrogen-containing compounds. Rewiring cellular metabolism to support anabolic processes is a feature common to both cancer and proliferating immune cells. In this review, we will discuss how nitrogen is utilized in biosynthetic pathways and highlight different metabolic and oncogenic programs that alter the flow of nitrogen in order to sustain biomass production and growth - an important emerging feature of cancer and immune cell proliferation.
Keywords: Nitrogen Metabolism, Cancer cells, T cells, Non-essential Amino Acids, Transaminases, Ammonia, Tumor Microenvironment
Overview of Nitrogen Utilization
Nitrogen is essential for de novo synthesis of a variety of biomolecules including nucleotides, amino acids, polyamines, hexosamines, glutathione, porphyrins, ammonia, creatine, nitric oxide and many other critical biological compounds. Amino acids and nucleotides are the predominant sources of nitrogen in cells and undergo rapid turnover to support the growth of cancer and immune cells [1–3]. Many cancer cells have increased demand for nitrogen sources, particularly amino acids, and recent studies have shown that depletion of specific amino acids in the tumor milieu can serve as a metabolic checkpoint for antitumor immunity [4, 5]. Lack of amino acid tryptophan has long been known to promote immune suppression with recent studies highlighting roles of other nitrogen sources in directing immune cell fate and function [6]. Accordingly, the circuits that control the acquisition of nitrogen from the extracellular environment are often rewired at multiple levels in cancers and immune cells [5, 7, 8].
Nitrogen is primarily acquired in its reduced state through amino acids [1, 8]. Amino acids account for the majority of the biomass of proliferating cells and are a major reservoir for cellular nitrogen [1, 8]. These amino acids (AA) can be nutritionally characterized as being essential (EAA) or non-essential (NEAA). While EAAs cannot be synthesized de novo due to lack of their carbon backbone, NEAAs can be adequately synthesized from existing carbon and nitrogen sources [9]. Nevertheless, all of the 20 proteinogenic amino acids are needed as substrates to charge tRNAs for mRNA translation, and deficiency of any one amino acid or a negative nitrogen balance can limit protein synthesis and growth of cells [9].
The use of amino acid nitrogen for biosynthetic pathways involves two processes: (1) a transamination reaction followed by (2) a deamination reaction (Box 1 and Fig. 1). Any excess amino acids must also be catabolized to ensure a tight intracellular nitrogen balance (Fig. 1) as amino acids are not disposed of in their native form [10]. Excess nitrogen in the form of ammonia is converted to urea via the urea cycle in the liver, which is excreted from the body [11]. Most ammonia leaves cells through the amide nitrogen of glutamine, as high level of ammonia is toxic [12]. Excess glutamine in the liver can be converted to ammonium ion (NH4+) and excreted in the form of non-toxic urea. However, in the case of proliferating cancer cells, most of the excess nitrogen is preserved and is diverted to various metabolic pathways [13–15]. Enzymes that are involved in recycling of nitrogen from ammonia (GLUD, GS and CPS) are regulated by oncogenes (e.g. c-MYC), tumor suppressors (e.g. LKB1), and growth signaling pathways (e.g. mTORC1, MAP kinase) [16–20]. Glutamate and glutamine, two crucial NEAAs, play a major role in preserving nitrogen by storing assimilated ammonia for anabolic processes and preventing its excretion in the form of unusable urea [13, 21, 22].
Box 1. Nitrogen Utilization: Transamination versus Deamination Reaction.
Transamination refers to the transfer of the alpha-amino group from the AA to an alpha-keto acid carrier molecule (usually an alpha-Ketoglutarate or alpha-KG). This transfer of an amino group to alpha-KG is mediated by enzymes called transaminases or aminotransferases (e.g. ALT, AST, PSAT, BCAT, OAT) [154, 155], and results in formation of glutamate (Fig. 1). Reactions catalyzed by transaminases are freely reversible and the directionality depends on the supply and demand of the local amino acid pool [154, 155].
The oxidative deamination reaction is the second catabolic reaction necessary for intracellular nitrogen processing whereby the resulting glutamate (if not used in biosynthetic processes) can undergo removal of its amino group by the enzyme glutamate dehydrogenase (GDH) [156] to produce ammonia and regenerate alpha-KG (Fig. 1). Some amino acids, such as glutamine, can directly undergo deamination to produce free ammonia by glutaminase (GLS) (Fig. 1). Serine and threonine are two other AA that can directly undergo deamination, bypassing the transamination to alpha-KG by the action of serine/threonine dehydratase [157].
The amino group produced by the deamination can have two different metabolic fates (Fig. 1). It can either be recycled in biosynthetic processes through its assimilation into glutamate and glutamine or the excess ammonia can be converted into urea for nitrogen disposal. When the amino group is shunted towards biosynthetic anabolic processes, mammalian cells rely on three major ammonia assimilating enzymes: 1) GDH, involved in synthesis of glutamate from alpha-KG; 2) Glutamine synthetase (GS), involved in the synthesis of glutamine from glutamate; and 3) mitochondrial carbamoyl phosphate synthetase (CPS I), involved in production of urea [21]. The cytosolic counterpart of CPS I, CPS II, is a part of single trifunctional protein called CAD and can also use ammonia as a substrate [158], however, it preferentially uses cytosolic glutamine for synthesis of carbamoyl phosphate.
Figure 1. Utilization of nitrogen from amino acids.
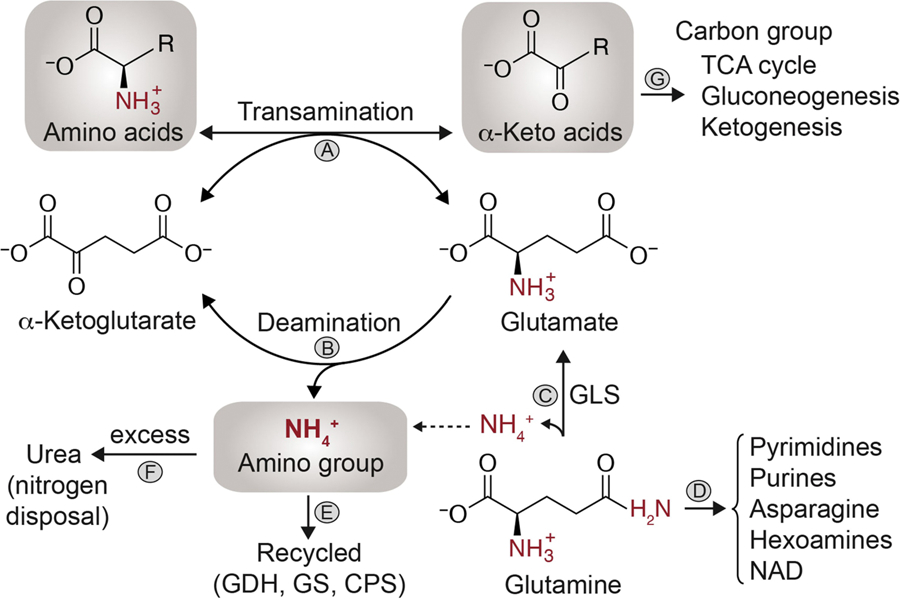
Removal of nitrogen from amino acids and addition of nitrogen to ketoacids are freely reversible transamination reactions catalyzed by aminotransferases or transaminase (A). During the transamination reactions, the alpha-amino group is transferred to alpha-Ketoglutarate (α-KG) to form glutamate. During synthesis of amino acids, transaminases can utilize glutamate as a nitrogen source and preserve the flow of nitrogen towards biosynthetic processes. When not used in biosynthesis, glutamate nitrogen is converted to free ammonium (NH4+) by an oxidative deamination reaction catalyzed by the bidirectional enzyme glutamate dehydrogenase (GDH) (B). Both transamination and oxidative deamination can therefore convert glutamate back to α-KG. Glutamate acts as a prominent intermediate in nitrogen metabolism and is usually formed by direct deamination of glutamine by glutaminase (GLS) (C). Glutamine terminal γ-nitrogen is used in the synthesis of pyrimidines, purines, asparagine, hexosamines and nicotinamide-adenine dinucleotide (NAD) (D). In mammalian cells, NH4+ can be recycled (E) by reductive activity of GDH, glutamine synthetase (GS), and by carbamoyl phosphate synthetase (CPS). Excess ammonia can be converted to urea by the urea cycle enzymes (F). The remaining carbon skeleton left as alpha-ketoacids from amino acid deamination can have three different fates (G). They can be used to fuel the TCA cycle, synthesize glucose/glycogen in liver cells (glucogenic AA) or be converted to acetyl-CoA/acetoacetyl-CoA (ketogenic AA).
Proliferating cells must balance the increased consumption of carbon sources with coordinated acquisition of nitrogen in order to fulfill the metabolic requirements for biomass production. In recent years, therapeutic interventions for cancers have focused on interfering with carbon utilization for tumor growth. For example, various studies have used glutaminase inhibitors [23], which block the deamination of glutamine to prevent glutamine carbon from being used as an anaplerotic substrate for the TCA cycle. However, the contribution of the terminal amide group of glutamine as a direct nitrogen source towards many anabolic reactions is a process independent of glutaminolysis (Fig. 1). Therefore, analogous to the carbon utilizing pathways, defining the metabolic and regulatory pathways that control the flow of nitrogen towards anabolic processes and growth should be a major focus of research.
In this review, we will discuss the flow of nitrogen towards synthesis of nucleotides, NEAAs and other important nitrogenous compounds, while highlighting the importance of glutamine and glutamate as both the central collection point and a major donor of nitrogen in the context of cancer and immune cells. Amino acids such as glutamine, arginine, aspartate, alanine, glycine and serine are important nitrogen sources shared by cancer cells, immune cells, endothelial cells, and stromal cells in the tumor microenvironment. We will highlight some of these important amino acids that play a role in the tumor microenvironment and specific differences that exist in nitrogen handling between some of the cell types within the tumor. Finally, we will discuss how the metabolic composition of the tumor microenvironment can influence various cellular adaptive and recycling processes. Knowledge on the basic principles of nitrogen handling in cancer and immune cells will help in understanding the requirements and metabolic constraints in these cell types and may ultimately aid in identifying areas of intervention for cancer therapy.
Nitrogen Flow Towards Pyrimidines and Purines
Nucleotides are almost exclusively synthesized within cells. Due to their relatively small cellular pool [8] (except ATP), rapidly proliferating cells must continue to acquire reduced nitrogen to meet the demand for nucleotides for DNA replication, transcription, and ribosomal RNA content. Nitrogen from the three major NEAAs (glutamine, aspartate and glycine) is transferred and incorporated into the structure of purines and pyrimidines (Fig. 2). Importantly, de novo synthesis of nucleotides is a target for an array of widely used chemotherapeutic agents (e.g. 6-mercaptopurine, pemetrexed, 5-fluorouracil, methotrexate, gemcitabine) and immunosuppressive drugs (e.g. leflunomide, brequinar, azathioprine, mycophenolic acid), highlighting its importance in both cancer and immune cell proliferation [3, 23, 24].
Figure 2. Nitrogen requirement for pyrimidine and purine synthesis.
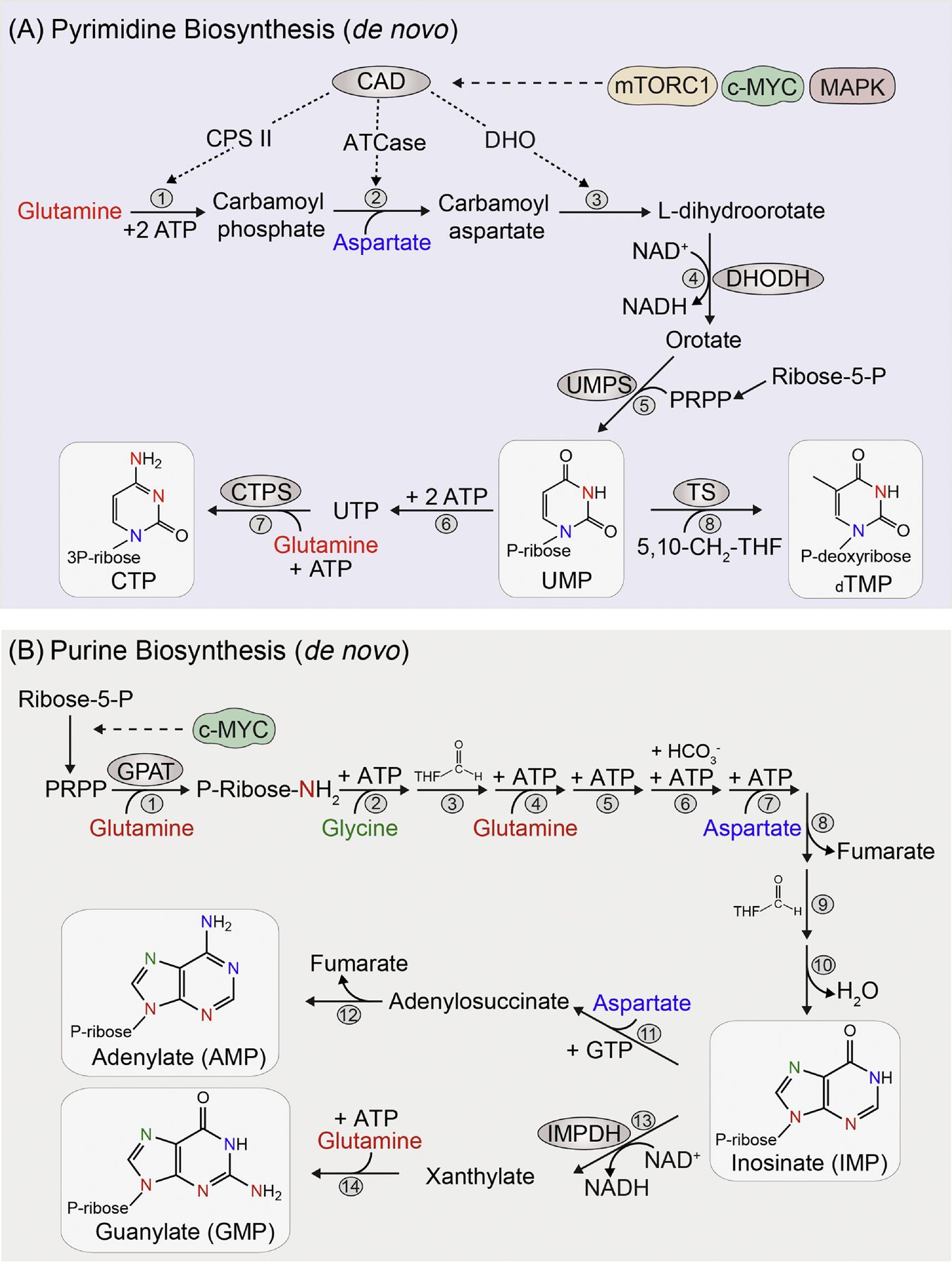
De novo synthesis of pyrimidines (A) starts with the construction of a six-membrane nitrogenous base, which is later attached to an activated ribose sugar molecule called 5-phosphoribosyl-1-pyrophosphate (PRPP). Glutamine and aspartate serve as a source of nitrogen for pyrimidines. The first step of pyrimidine synthesis begins with the cytosolic enzyme carbamoyl-phosphate synthetase II (CPS II) which uses glutamine nitrogen for the synthesis of carbamoyl phosphate (1). Carbamoyl-phosphate is combined with aspartate by the enzyme aspartate transcarbamoylase (ATCase), donating the second nitrogen atom and forming carbamoylaspartate (2). This is then circularized to dihydroorotate by the enzyme dihydroorotase (DHO) (3). The first three enzymes (CPSII, ATCase and DHO) are part of a single large multienzyme protein called CAD and which is regulated by mTORC1, c-MYC and growth factor signaling (MAPK). Dihydroorotate is then oxidized to orotate by the mitochondrial enzyme dihydroorotate dehydrogenase (DHODH), which transfers electrons from dihydroorotate to mitochondrial ubiquinone (4). Finally, orate is coupled with the activated ribose PRPP by the bifunctional enzyme (phosphoribosyltransferase and decarboxylase) called uridine monophosphate synthetase (UMPS) to eventually form uridine monophosphate (UMP) (5). Other nucleotide species are subsequently derived from UMP. Cytidine triphosphate (CTP) acquires its third nitrogen atom via the amination of UTP by glutamine and this step is catalyzed by the enzyme cytidylate synthetase (CTPS) (6–7). For generation of thymidylate (TMP), deoxy-UMP is modified by the enzyme thymidylate synthase (TS), requiring a methyl group from 5,10-CH2-trtrahydrofolate (5,10-CH2-THF) (8).
Purines are two ring compounds containing 5 nitrogen groups, derived from glutamine, aspartate and glycine. Unlike pyrimidines, de novo synthesis of purines (B) starts with the activated ribose sugar as a foundation and then purine bases are constructed on the ribose ring in a stepwise fashion. The generation of activated ribose sugar (PRPP) from ribose-5-phosphate is regulated by c-MYC. The first committed step towards purine synthesis is catalyzed by the enzyme glutamine phosphoribosyl amidotransferase (GPAT). The pyrophosphate group of PRPP is displaced by glutamine-derived ammonia, adding the first nitrogen to form highly unstable 5-phosphoribosylamine (1). After this, two carbon and a second nitrogen atoms are added from the amino acid glycine onto the 5-phosphoribosylamine (2). The amino group of glycine is then formylated by the N10-formyl-THF (3) and the third nitrogen atom from glutamine is added to form the first ring structure (4). Following this, carbon atom from bicarbonate (6) and a fourth nitrogen from the aspartate (7) is added. The TCA intermediate fumarate is generated as the remaining carbon skeleton of aspartate after donating its amino group (8). Finally, one carbon atom from N10-formyl-THF (9) is added to form the second ring of purine base (9–10). Inosinate (IMP) is the first intermediate with a complete ring which can give rise to both adenylate (AMP) and guanylate (GMP). For AMP synthesis (11–12), the fifth nitrogen group is added from aspartate and requires GTP as an energy source. GMP, on the other hand, first requires oxidation of IMP to xanthylate (XMP) by NAD+, which is mediated by the enzyme IMP dehydrogenase (IMPDH) (13). This is followed by addition of the last nitrogen group from glutamine and this step requires ATP as an energy source (14). The requirement of the reciprocal nucleotides GTP and ATP for generation of AMP and GMP is an important regulatory mechanism in ensuring a balance between adenine and guanine synthesis. The colors of the nitrogen atoms in the structure of pyrimidines and purines correspond to their amino acid donor color.
The nitrogen groups for pyrimidines are derived from glutamine and aspartate (Fig. 2A). The first step of pyrimidine synthesis begins with the synthesis of carbamoyl phosphate by the cytosolic enzyme CPS II. In mammals, the first three enzymatic reactions of pyrimidine synthesis (CPSII, ATCase and DHO) are part of a single large multi-enzyme protein abbreviated as CAD, which is subject to regulation by growth signaling pathways [16, 17]. For instance, the ribosomal S6 kinase 1, which is an immediate downstream target of mTORC1 (mechanistic target of rapamycin complex 1), directly phosphorylates and activates CAD [25], connecting the sensing of amino acids through mTORC1 to nucleotide biosynthesis. Also, one of the earliest known targets of c-MYC is trifunctional CAD enzyme [26], highlighting the importance of oncogenic signaling in orchestrating pyrimidine synthesis.
Purines are two ring compounds containing 5 nitrogen groups, derived from glutamine, aspartate and glycine (Fig. 2B). Since, de novo synthesis of purines starts with the activated ribose sugar as a foundation (Fig. 2B), the cellular levels of PRPP is an important factor for initiating purine synthesis. The expression of phosphoribosyl pyrophosphate synthetase, which converts ribose-5-phosphate to activated phosphoribosyl pyrophosphate (PRPP), is facilitated by c-MYC in cancers [27]. The first committed step to purine synthesis, which is catalyzed by the enzyme glutamine phosphoribosyl amidotransferase (GPAT), is inhibited by 6-mercaptopurine and 6-thioguanine [23]. Both the serine synthesis pathway (required for glycine synthesis) and folate metabolism (required for N10-formyl-THF) are needed for purine synthesis and are important regulatory points modulated by various other growth signaling pathways including mTORC1 [28, 29]. It is worth nothing that the nitrogen contained in nucleotides cannot be directly reused in biosynthetic pathways and is disposed of as free ammonia during the process of nucleotide catabolism and formation of uric acid. However, studies have shown that nucleotides can also be salvaged towards the nucleotide pool during the process of nucleotide catabolism [30], which can preserve the nitrogen from its conversion to uric acid [29, 31, 32]. The salvage pathway is therefore likely a metabolic adaptation under nitrogen limitation and further studies are needed to investigate conditions where the role of the salvage pathway in preserving nitrogen is critical in cancer and immune proliferation.
Nitrogen Flow Towards Non-Essential Amino Acids
NEAAs provide building blocks for protein and nucleotide synthesis, constitute components for redox homeostasis, one-carbon metabolism, and the urea cycle, and serve as important substrates for many nitrogenous compounds [5, 7, 9]. Therefore, when the cellular demand exceeds the local NEAA availability, proliferative cells often demonstrate metabolic adaptation towards de novo synthesis of NEAAs. This switch is noticeable near poorly vascularized regions of tumors, where the regional pools of glutamine, arginine, asparagine, aspartate, serine and glycine are often depleted [5, 33, 34]. For example, the circulating concentration of glutamine in serum is around 550 μM, while concentration in the core of some tumors has been shown to be around 100 μM [35]. However, it is important to consider that the synthesis of NEAAs might not be applicable to all cell types, as this requires reallocation of the necessary energy, carbon backbones (intermediates of glycolysis and TCA cycle) and specific transaminases to transfer the α-amino group of glutamate and glutamine as a nitrogen source (Fig. 3). In this section, we will only highlight some of the major NEAAs that play an essential role in nitrogen handling in cancers and immune cells.
Figure 3. Allocation of nitrogen for NEAAs synthesis.
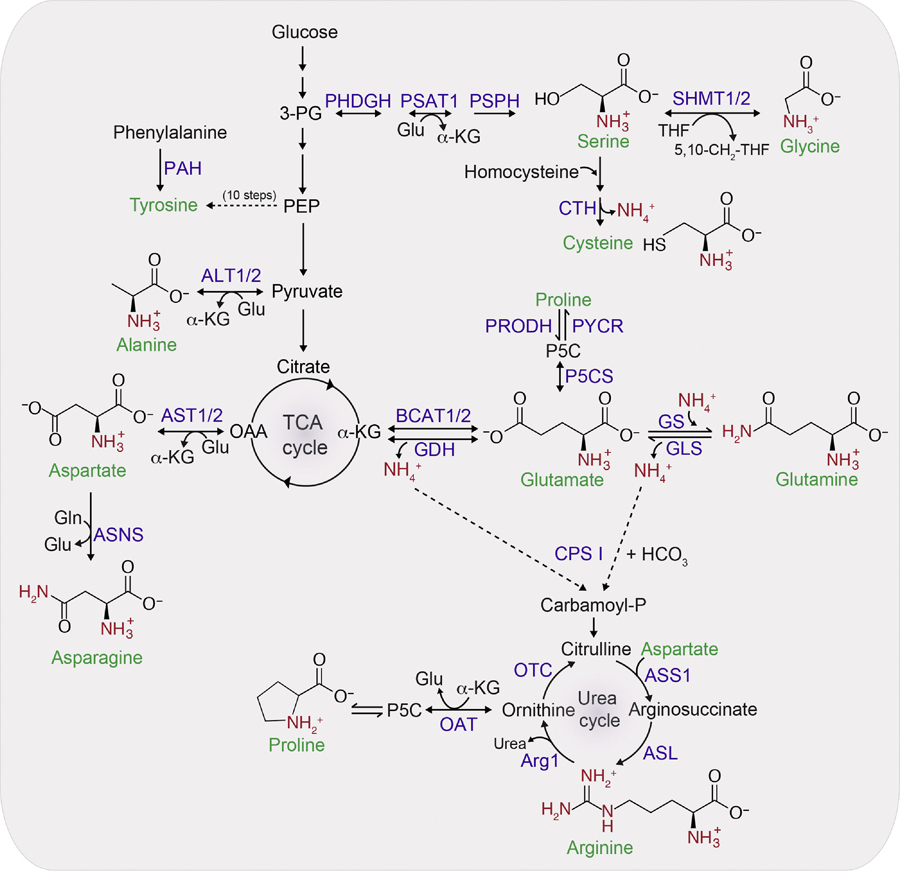
Glutamine provides the nitrogen for asparagine synthesis and glutamate provides the nitrogen for all other NEAAs synthesis. The carbon backbone for the synthesis of serine, glycine and cysteine comes from 3-phosphoglyceric acid (3-PG). Glycine can be directly generated from serine via the reversible reaction catalyzed by SHMT1/2 enzyme. One carbon (1C) units from the serine is transferred to the carrier molecule tetrahydrofolate (THF) resulting in 5,10-CH2-THF. Since this reaction is reversible, the directionality depends upon the supply and demand of 1C units. For the synthesis of cysteine from serine, the sulfur atom comes from methionine in the form of homocysteine. Cystathionase (CTH) catalyzes the removal of nitrogen as a free ammonium ion from homocysteine. Tyrosine is usually synthesized in one step from phenylalanine, but in conditions where phenylalanine is limiting, phosphoenolpyruvate (PEP) serves as a carbon backbone for tyrosine and is synthesized in 10 steps. Alanine, which is the second-most abundant amino acid in plasma after glutamine, can be synthesized from pyruvate by ALT1/2. Majority of the intracellular aspartate is generated from oxaloacetate by the enzyme aspartate aminotransefases (AST1/2 or GOT1/2). Asparagine is synthesized from aspartate and is the only amino acid that uses glutamine as a nitrogen donor. alpha-KG is used as a carbon backbone for synthesis of glutamate, glutamine, arginine and proline. Proline can be synthesized from either ornithine by OAT or glutamate by the spontaneous cyclization of the intermediate glutamate γ-semialdehyde. Beside its synthesis by GDH, glutamate can also be generated from alpha-KG by action of BCAT1/2 using nitrogen from leucine, isoleucine and valine. Arginine is synthesized from glutamate via ornithine in the urea cycle. Arginine synthesis also requires aspartate nitrogen and the enzyme ASS1 and ASL. Two nitrogen groups of urea (for disposal) comes from the carbamoyl phosphate generated in the mitochondrial matrix from free ammonia via CPS I and aspartate which is also formed in the mitochondrial matrix from OAA. Reactions that are reversible are indicated by two-sided arrow heads. Free ammonium ions are indicated by NH4+ while the amino group donated from either glutamate or glutamine are colored in red. Abbreviations: Arg1, arginine 1; ASNS, asparagine synthetase; GLS, glutaminase; GLUD1, glutamate dehydrogenase 1; GS, glutamine synthetase; OTC, ornithine transcarbamylase; P5C, 1-pyrroline-5-carboxylic acid; P5CS, pyrroline-5-carboxylate synthase; PAH, phenylalanine hydroxylase; PHDGH, D-3-phosphoglycerate dehydrogenase; PRODH, proline dehydrogenase; PSAT1, phosphoserine aminotransferase 1; PSPH, phosphoserine phosphatase; PYCR, pyrroline-5-carboxylate reductase.
Glutamine and Glutamate Play a Central Role in Nitrogen Metabolism
Glutamine is the most abundant amino acid in human plasma [5, 34] and the second principal fuel for highly proliferative cells besides glucose. Glutamine has two nitrogen atoms at the α and γ positions (Fig. 1). While the α-position nitrogen is used to synthesize NEAAs through glutamate, the terminal γ–position nitrogen is used as a nitrogen source for the synthesis of pyrimidines, purines, asparagine, hexosamines and nicotinamide-adenine dinucleotide (NAD) [36]. High levels of intracellular glutamine are indicative of a positive nitrogen balance for cell growth, and, as a result, transporters of glutamine such as SLC1A5 and SLC38A2/SLC38A5 are upregulated in proliferating cancer and immune cells [37–41]. In addition, high levels of intracellular glutamine can be exchanged for the uptake of other EAAs (leucine, isoleucine, valine and methionine) through the bidirectional antiporter SLC7A5 or LAT1 [42–45]. The increase in glutamine transporters is mediated by oncogenic signaling in various cancer types [39, 40, 46]. However, in immune cells, this is orchestrated by antigen-induced signaling through the T cell receptor (TCR) [43].
In cases where exogenous glutamine availability declines, cells can synthesize glutamine from intracellular glutamate and free ammonia by GS (Fig. 3). GS expression is not only induced during glutamine starvation [47] but also by oncogenic c-MYC in cancer [18]. Synthesis of glutamine through GS is particularly important to fuel nucleotide biosynthesis in cancers such as glioblastoma, as they have limited access to circulating glutamine [48]. Interestingly, targeting GS in stromal cells of ovarian tumors can deplete the local pool of glutamine used by adjacent cancer cells for their growth [49], further highlighting the importance of de novo glutamine synthesis. Moreover, in immune cells, targeting GS has been shown to polarize macrophages towards an M1-like phenotype, which is associated with accumulation of CD8+ T cells and diminished tumor metastasis [50, 51].
Glutamine-derived glutamate is the major nitrogen source for many transaminases (Fig. 3). Most of the intracellular glutamate is synthesized from deamination of glutamine by the enzyme GLS rather than being imported from extracellular space [52]. However, in certain cancers like glioma and AML, glutamate can also be generated from alpha-KG by action of branched-chain aminotransferases (BCAT1/2) using nitrogen from leucine, isoleucine and valine (Fig. 3) [53, 54].
When glutamate nitrogen is transferred to alpha-keto acids (oxaloacetate, pyruvate, and 3-phosphohydroxy pyruvate) to synthesize the corresponding NEAA (aspartate, alanine and serine), the resulting carbon backbone left after the transamination reaction is alpha-KG (Fig. 3). It is worth noting that alpha-KG, an intermediate of the TCA cycle, can also be generated by oxidative deamination of glutamate by the action of the mitochondrial enzyme GDH (Fig. 3 and Fig. 1). However, in this deamination reaction, the amino group from glutamate is liberated as a free ammonia instead of being used as an amino group for NEAA synthesis, which diminishes the flow of nitrogen towards NEAAs. Nevertheless, GDH activity is compartmentalized to the mitochondria and is needed to provide a carbon source for TCA cycle anaplerosis [55], while the production of alpha-KG by cytosolic transaminases can be utilized for subsequent transamination reactions (Fig. 1) or used as a substrate for alpha-KG-dependent dioxygenases. Studies have examined whether there is a preferential use of glutamate by transaminases or by GDH. Data support that cells in a quiescent state preferentially use GDH-mediated deamination of glutamate, while cells transitioning to a proliferative state use transaminases to preserve glutamate-derived nitrogen for synthesis of NEAAs [56]. Interestingly, recent findings have shown that free ammonia can be recycled from the tumor microenvironment both by the reductive activity of GDH to form glutamate [13] and by the subsequent higher activity GS to synthesize glutamine [57]. Recycling of ammonia by using alpha-KG as a carbon backbone was shown to be important for cancer cell growth, and inhibition of either GDH or GS can reduce tumor growth [13, 57].
Beside its conversion to alpha-KG by transaminases or GDH, glutamate is also used as a substrate to produce the antioxidant glutathione and as an exchange factor for glutamate antiporters. Glutamate is an important constituent in the synthesis of glutathione, a tripeptide made up of cysteine, glutamate and glycine that is required to buffer the ROS regenerated during T cell activation and effector function [58]. As an exchange factor, glutamate efflux through the antiporter xCT (SLC7A11) can be used to import extracellular cystine in cancers [59]. Interestingly, high levels of extracellular cystine can drive cancer cells towards increased glutamine dependency [60]. This is because high extracellular cystine concentrations promote cystine import in exchange for glutamate, making cells more dependent on the deamination of glutamine via GLS to replenish the intracellular glutamate pool. This could explain some of the discrepancies in sensitivity of glutaminase inhibitors seen during in vitro and in vivo cancer studies, as most in vitro cell culture media contain a high level of cysteine [61]. Nevertheless, use of CB-839 as a GLS inhibitor is being investigated as a cancer therapeutic in the clinic for its potential efficacy [23, 62, 63]. Cancers driven by oncogenic drivers such as c-MYC, KRAS/Keap1 or Nrf2 mutations tend to be more sensitive and likely to respond to glutaminase inhibition [41, 64, 65].
In summary, glutamine and glutamate are central to nitrogen metabolism and low levels of these two amino acids can limit the flow of nitrogen within cells and delay biomass production and growth. Alpha-KG can be used as a carbon backbone for ammonia assimilation [13, 57, 66, 67] to replenish glutamate and glutamine levels and therefore act as a critical link between carbon and nitrogen metabolism [57]. Indeed, accumulation of alpha-KG is a characteristic of nitrogen limitation in bacteria, and interestingly, high levels of alpha-KG can limit the uptake of carbon sources (e.g., glucose) in order to match the insufficient nitrogen source available for biosynthetic processes [68]. In cancer cells, the factors that regulate the net direction of ammonia metabolism must be dependent on the level of nitrogen donors as well as the availability of sufficient carbon sources. Under conditions where cellular nitrogen is abundant (e.g., high glutamine), cells might utilize amino acid carbon in the TCA cycle, resulting in net ammonia production by GLS, GDH and other deaminases. However, under conditions where nitrogen donors are limited (e.g., low glutamate and low glutamine), the carbon backbone from TCA must be utilized with reactions favoring net ammonia consumption by GDH, GS and CPS to replenish sufficient nitrogen for biosynthetic processes.
Aspartate and Asparagine Metabolism
The circulating aspartate concentration in human plasma is around 20 μM, which is among the lowest of all proteinogenic amino acids [5, 34]. In addition, import of aspartate from the extracellular space is limited due to poor membrane permeability [69] and low expression of the aspartate transporter SLC1A3 in most tissues [70]. As a result, the majority of intracellular aspartate is generated from oxaloacetate by aspartate aminotransferase enzymes (AST1/2 or GOT1/2) using glutamate as a nitrogen donor (Fig. 3). It is worth noting that, unlike glutamine-derived glutamate, aspartate cannot be synthesized from asparagine due to a lack of cytosolic asparaginase activity needed to convert asparagine to aspartate [69, 71]. Interestingly, high levels of exogenous aspartate supplementation can rescue various cancer cells’ growth inhibited by electron transport chain (ETC) deficiency, suggesting that one of the major roles of mitochondrial respiration in cancer cells is to facilitate aspartate biosynthesis [72, 73]. Besides protein synthesis, aspartate is needed for nucleotide synthesis (discussed above), the malate-aspartate shuttle, and maintenance of redox balance [72, 73]. Limiting aspartate synthesis can inhibit tumor growth in vivo [69]. However, aspartate usage appears to be cell type specific. For example, in endothelial cells, high aspartate or a cell permeable methyl aspartate supplementation does not rescue the proliferative defect mediated by ETC inhibition [74]. Similarly, in regulatory T cells (Treg), aspartate levels are not altered under ETC inhibition [75], suggesting that aspartate synthesis mediated by respiring mitochondria might be a specific metabolic adaptation in some cancer cells.
Asparagine, the circulating physiological concentration of which is around 50 μM [5, 34], is the only NEAA that requires glutamine-derived nitrogen for its biosynthesis (Fig. 3). Asparagine is synthesized from aspartate by the enzyme asparagine synthetase (ASNS) in a manner similar to that of glutamine synthesis from glutamate by GS, as both require ATP and have a final amide group added to their side chains. However, despite being structurally similar to glutamine, asparagine cannot be used in transamination reactions or for TCA cycle anaplerosis as it cannot be catabolized in mammals due to lack of cytosolic asparaginase [71, 76, 77]. Moreover, when glutamine levels are low, asparagine becomes an essential amino acid for cells due to its insufficient production from glutaminolysis [71]. Nevertheless, asparagine is important for protein translation and can also be used as an exchange factor for counter-importing EAAs into the cells [78]. Blood cancers such as acute lymphoblastic leukemia (ALL) lack the ability to synthesize asparagine endogenously, due to lack of ASNS, and are entirely dependent on extracellular asparagine [79, 80]. Depleting asparagine by a recombinant form of bacterial L-asparaginase in the plasma has been an important part of the therapeutic regimen for ALL [79, 80]. Limiting exogenous asparagine or inhibiting its synthesis has also been shown reduce metastasis in breast cancer [81] and is being clinically investigated (Table 1).
Table 1:
Drugs Targeting Nitrogen Metabolism Currently in Clinical Studies
| Drug | Target | Disease Type/indications | Clinical status |
|---|---|---|---|
| Telaglenastat (CB-839) | Glutaminase I inhibitor | Advanced/Metastatic Solid Tumors | Phase I/II (In combination with Palbociclib, Talazoparib or Cabozantinib) |
| Eryaspase | L-Asparaginase encapsulated in red blood cells (depletes plasma asparagine) | Pancreatic cancer and TNBC | Phase II/III (In combination with chemotherapy) |
| PEG-BCT-100 (ADI-PEG20) | Arginine deiminase formulated with polyethylene glycol (depletes circulating arginine) | Multiple Advanced Cancers (Hematological and Solid Tumors) | Phase I/II/III (In combination with immune checkpoint therapy or chemotherapy) |
| Pegzilarginase (AEB1102 or Co-Argl-PEG) | Pegylated Human Recombinant Arginase 1 (depletes circulating arginine) | Advanced Cancers (Hematological and Solid Tumors), Arginase I Deficiency | Phase I/II (As a single agent and in combination with immune checkpoint therapy or chemotherapy) |
| INCB001158 (CB-1158) | Arginase inhibitor (maintains circulating arginine) to enhance anti-tumor immunity | Solid Tumors, Relapsed or Refractory Multiple Myeloma | Phase I/II (As a single agent and in combination with immune checkpoint therapy or chemotherapy) |
| AMXT-1501 and DFMO | Polyamine transport and synthesis (ODC1) inhibitor | Neuroblastoma, Advanced Solid Tumors | Phase I (AMXT-1501 alone and in combination with DFMO) |
| Epacadostat (INCB024360) | Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitor (tryptophan metabolism) | Multiple Advanced Solid Tumors | Phase I/II (In combination with immune checkpoint therapy and/or chemotherapy) |
| Indoximod (Prodrug-NLG802) | Indoleamine 2,3-dioxygenase inhibitor (tryptophan metabolism) | Multiple Malignancies (Melanoma, Glioblastoma, Lung, Pancreatic, Breast Cancers and Others) | Phase I/II (In combination with immune checkpoint therapy or chemotherapy) |
| BMS-986205 | IDO1 inhibitor (tryptophan metabolism) | Advanced Cancers | Phase I/II (In combination with immunotherapy and/or chemotherapy) |
| Hominex-2 | Methionine free amino acid modified medical food | Triple Negative Breast Cancer (TNBC) | Phase II (In combination with Akt/ERK inhibitor) |
Source: Select agents are shown from ClinicalTrials.gov
Arginine Integrates Many Metabolic Pathways
Arginine, which contains four nitrogen atoms (highest among all amino acids), is an important intermediate in nitrogen metabolism. Most cancer cells usually rely on exogenous arginine, as de novo synthesis of arginine requires expression of subsets of urea cycle enzymes (ASS1 and ASL), traditionally considered to function only in liver and kidney (Fig. 3). Loss of ASS1 and ASL expression can also be beneficial to various tumor types as it can help cancer cells preserve a limited aspartate pool and increase nitrogen flow towards pyrimidine synthesis [11, 14, 82]. Interestingly, CPS I, which catalyzes the first step of the urea cycle, was shown to be expressed in a subset of non-small-cell lung cancer cells and can assimilate ammonia towards carbamoyl-phosphate production [19]. This carbamoyl-phosphate can be exported to the cytosol to enhance pyrimidine synthesis and tumor growth [19] rather than being utilized in the urea cycle due to lack of OTC enzyme in most cancers [14].
However, there are consequences of altering nitrogen flow away from de novo arginine synthesis. First, this makes cells highly dependent on extracellular arginine [23, 83–85] and renders cells sensitive to depletion of plasma arginine, a phenomenon seen with asparagine depletion. This metabolic vulnerability is being tested in clinical trials (Table 1). The second consequence of dysregulated urea cycle enzymes and enhanced pyrimidine synthesis is an increase in mutational burden due to imbalance of the pyrimidine to purine ratio [14]. This makes cancer cells immunogenic and heightens their sensitivity to current immune checkpoint inhibitors [11, 14].
Arginine is also extensively utilized during T cell activation: providing additional arginine to CD4+ and CD8+ T cells can enhance their survival and anti-tumor immunity [86]. Arginase 1 (Arg1) produced by myeloid-derived suppressor cells, neutrophils and macrophages (M2 subtype) can degrade arginine needed for T cell metabolic fitness and expression of T-cell receptor (CD3zeta chain) needed for antigen recognition [87, 88]. Similarly, reduction in arginine, either by Arg1 or by inhibiting de novo synthesis by depleting argininosuccinate synthase 1 (ASS1), can diminish differentiation and quantity of peripheral T cells [86, 89–91]. In addition, arginine is a precursor for the production of nitric oxide by nitric oxide synthase (iNOS), which is associated with the inflammatory anti-tumor M1 macrophage phenotype and function [92, 93]. This suggests that availability of circulating arginine is beneficial to T cell responsiveness and anti-tumor activity. The immunomodulating potential of arginine is being evaluated in clinical studies (Table 1).
Besides its role in the urea cycle and nitric oxide, arginine also serves as a precursor for biosynthesis of important nitrogenous compounds such as ornithine, polyamines and creatine [11, 94]. Ornithine can be diverted towards synthesis of proline by the enzyme ornithine aminotransferase (Figure 3) as well as polycationic aliphatic amines or polyamines (putrescine, spermidine, and spermine). Ornithine is decarboxylated by the rate-limiting enzyme ornithine decarboxylase 1 (ODC1) to form putrescine. The amino-propyl moiety from decarboxylated S-adenosyl-L-methionine (SAM) is subsequently added to putrescine to synthesize spermidine and spermine. Polyamines were shown to play an important role in both cancer and immune cells physiology [95, 96]. Due to its important cellular function, oncogenes like MYC directly regulate polyamines synthesis and uptake by transcriptionally upregulating ODC1 and SLC3A2 transporter [95, 97]. In addition, arginine depletion in cells that lack urea cycle enzyme ASS1 has been shown to result in synthetic lethality with inhibition of the polyamine synthesis pathway [98]. Interestingly, inhibition of polyamine synthesis or uptake has been shown to reduce tumor growth while enhancing T cell anti-tumor responses [99], with a recent report highlighting an important role for polyamines in macrophage activation [100].
Finally, the creatine and phospho-creatine energy shuttle has been shown to be important for acute myeloid leukemia (AML) and breast cancer cell growth [101, 102], further highlighting the diversity of the roles of arginine metabolism in cancer. Since arginine sits at the crossroads of many metabolic pathways, it is not surprising that both cancer and T cells economically utilize available arginine and downregulate pathways that prevent arginine nitrogen disposal through urea [11, 14].
Adaptation to Nitrogen Availability
The allocation and utilization of nitrogen to biosynthetic pathways needs to be tightly coupled with appropriate levels of amino acids in cells. mTORC1 is a highly conserved regulator of eukaryotic nitrogen- and carbon-sensing pathways that couples the availability of nitrogen sources to cellular growth [103]. Availability of key amino acids (arginine, glutamine, leucine, lysine and methionine) can mediate the translocation of mTORC1 to the lysosome for its activation to signal anabolic processes in cells [104–108]. On the other hand, deficiency of amino acids can activate the General Control Nonderepressible 2 (GCN2)-Activating Transcription Factor 4 (ATF4) integrated stress response pathway [109–111]. The GCN-ATF4 pathway limits amino acid consumption and upregulates pathways that can help replenish the amino acid pool and maintain optimal nitrogen balance for biosynthetic processes [109–111]. These adaptive pathways include selective autophagy, upregulation of amino acid transporters and enzymes involved in de novo synthesis of NEAAs, inhibition of mTORC1 and upregulation of cytosolic amioacyl-tRNA synthetase genes in order to facilitate efficient charging of tRNAs under low amino acid levels [42, 111–113]. Deficiency of amino acids also induces cells to scavenge extracellular protein (through micropinocytosis), a process during which inhibition of mTORC1 can promote cellular growth by allowing sufficient time for restoration of cellular amino acids [114, 115]. The scavenging of extracellular proteins to acquire amino acids is an adaptation seen particularly in cancer cells [115, 116] and therapeutic agents that target scavenging in cancers are currently under investigation [115].
The mTORC1 growth pathway and the GCN-ATF4 stress response pathway are adaptive mechanisms common to both tumors and T cells [117, 118], and hence studies targeting nitrogen metabolism must consider the role of these adaptive processes when designing drugs for cancer. For instance, drugs depleting circulating arginine (Table 1) can lead to inhibition of mTORC1 while activating micropinocytosis and autophagy, processes usually upregulated in subsets of cancer types [116, 119], to replenish cellular arginine pool and provide a mechanism of resistance [84]. Therefore, combining agents that target autophagy, micropinocytosis or mTOCR1 with drugs that deplete circulating arginine, glutamine or methionine (Table 1) might provide a better therapeutic strategy.
Nitrogen Metabolism in the Tumor Microenvironment
Nitrogen metabolism in the tumor milieu is influenced by various factors, including the metabolic state at the organismal level, the nutrient supply from the circulation and the complex interaction between various cell types within the tumor. The tumor microenvironment consists of cancer cells as well as stromal cells, immune cells, and endothelial cells (Fig. 4), all with distinct metabolic phenotypes [120–122]. Numerous other cell-autonomous and non-cell-autonomous factors can affect nitrogen acquisition from the extracellular environment. For example, there will be differences in the rate of consumption and secretion of nitrogen sources between various cell types depending on transporter expression, metabolic needs, growth signals, proliferative capacity as well as varying levels of specific transaminase/deaminase expression. Cancer cells often exhibit increased uptake of local NEAAs and extracellular protein as nitrogen sources to meet their high anabolic needs [33, 35, 116]. Moreover, cancer cells also express various transaminases that can enable synthesis of NEAA during times of limited nitrogen availability in the tumor milieu [56]. This is not necessarily the case with T cells or slowly proliferating dendritic cells and macrophages [123]. For instance, recent studies have shown that alanine is important for early T cell activation and memory T cell re-stimulation, but due to limited expression of ALT1/2 (or GPT1/2) transaminases needed for alanine biosynthesis (Fig. 3), T cells mostly depend on extracellular alanine for protein synthesis [124]. This suggests that under nutrient starvation, consumption of exogenous alanine by cancer cells can be detrimental to T cell function.
Figure 4. Nitrogen metabolism within the tumor microenvironment.
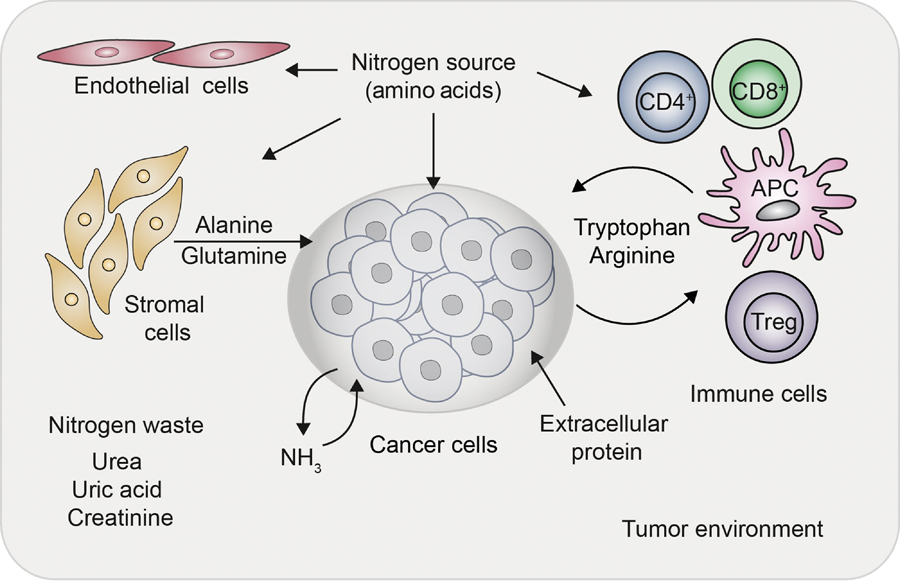
Nitrogen sources in the form of amino acids are shared by cancer cells, immune cells, endothelial cells, and stromal cells in the tumor microenvironment. Besides free amino acids, the acquisition of nitrogen in the form of extracellular protein (through micropinocytosis) is upregulated in cancer. Ammonia as a nitrogen waste can also be assimilated by the cancer cells from the microenvironment; however, whether such a phenomenon exists in other cell types remains largely unknown. Tumor cells upregulate intracellular catabolism of tryptophan and arginine, depleting their levels in the extracellular space. Depletion of tryptophan and arginine from the local space can inhibit effector CD8+ T cells proliferation and function. The catabolic derivative of tryptophan (kynurenine) has been shown to enhance T regulatory cells (Treg) phenotype. Stromal cells that reside in the vicinity of the cancer cells can release alanine through autophagy as well as glutamine through enzyme glutamine synthetase, which can be utilized by the neighboring cancer cells.
The metabolic composition in the tumor milieu can have a distinct metabolic effect on tumor cells and infiltrating T cells. Glutamine nitrogen is needed for efficient clonal expansion of activated T cells and their differentiation into effector cells [43, 125–127]. However, limiting glutamine consumption, either by SLC1A5 deficiency or limiting local glutamine availability, can favor expression Treg lineage-defining transcription factor Foxp3 [37, 128]. A decrease in glutaminolysis can release the alpha-KG-dependent demethylation of the Foxp3 locus, thereby promoting suppressive Treg cell generation and blockage of T helper 1 cell differentiation [129]. Accordingly, in another study, accumulation of 2-HG, presumably as a result of increased glutamic-oxaloacetic transaminase (GOT1)-dependent transamination, can lead to repression of the Foxp3 locus through promotor methylation and diminish the induction of Tregs [130]. Moreover, glutamine is a precursor for synthesis of hexosamines, which are needed for protein glycosylation, and was shown to be indispensable for activated T cell function [131, 132]. Increased glutamine consumption by cancer cells could therefore potentially regulate antitumor immunity and facilitate development of a suppressive Treg population by depleting the local glutamine pool needed to fuel the effector T cell response. Interestingly, a recent report showed that while cancer cells are sensitive to glutamine antagonism, effector T cells can rewire their metabolism towards a more oxidative and a long-lived activated phenotype [133].
Additionally, modulating nitrogen sources in the tumor can lead to different metabolic responses between various cells types. For example, limiting extracellular nitrogen can initiate an autophagic mode of survival in surrounding stromal cells by degrading their limited intracellular protein [134]. Interestingly, this can benefit cancer cells, as stromal cells secrete alanine during autophagic degradation to the local space, which can be sequestered by the cancer cells to be used as additional fuel (Fig. 4). Moreover, restricting dietary serine and glycine has been shown to be effective in reducing tumor growth in mouse models of intestinal cancer and lymphoma [135, 136]. Studies have shown that the enzymes in the de novo serine synthesis pathway, PHGDH, PSAT1 and PSPH (Fig. 3), are amplified and upregulated in many cancer types [137–140]. However, de novo synthesis of serine does not seem to be sufficient for maintaining optimal redox state and nucleotide production in cancer cells [141]. Extracellular serine also maintains glycine levels and one-carbon metabolism needed for nucleotide biosynthesis and mitochondrial biogenesis for T cell expansion and effector function [142, 143]. Since both cancer and T cells share this metabolic feature of benefiting from local serine levels, it will be important to consider cell type specificity in designing metabolic therapies that target serine metabolism.
Finally, depletion of tryptophan from the tumor microenvironment can suppress anti-tumor immune function of infiltrating lymphocytes. Cancer cells, tumor-associated macrophages and certain dendritic cells can deplete the local pool of tryptophan by upregulating the metabolic enzyme indoleamine 2,3-dioxygenase-1 (IDO1) or tryptophan-2,3-dioxygenase (TDO2) [144–146]. An increase in tryptophan catalyzing enzymes can enhance the conversion of tryptophan to its derivative kynurenine. Depletion of tryptophan from the local space can inhibit effector T cell function while the catabolic derivative kynurenine can exert a suppressive effect on anti-tumor immunity by favoring the generation of Foxp3+ Treg cells [147, 148]. Since IDO1 was shown to be immunosuppressive in the tumor microenvironment [149], inhibitors of IDO1 are currently being tested in clinical trials (Table 1).
Concluding Remarks
Many studies have advanced our understanding of how nitrogen is acquired and utilized towards various anabolic processes in proliferating cells. There are many successful examples of treatment which have targeted nitrogen utilization for cancer therapy, such as targeting nucleotide metabolism with gemcitabine, 5-fluorouracil and cytarabine for treatment of various malignancies as well as the use of L-asparaginase for ALL [150, 151]. In addition, several ongoing clinical trials are evaluating the efficacy of targeting nitrogen metabolism for cancer therapy (Table 1). While the carbon backbone of amino acids can be utilized as fuel (Fig.1), important considerations must also be made regarding the distinct roles of amino acids’ carbon and nitrogen contributions before attempting to therapeutically target amino acid metabolism. The oversight of nitrogen contribution and usage to cell growth and function might explain some of the inconsistencies seen in cancer models with current GLS inhibitors in vivo [61]. In addition, adaptive pathways that can restore or recycle nitrogen sources must also be considered.
New interest in understanding metabolic processes in a cell non-autonomous fashion and recent advances in mass spectrometry and nuclear magnetic resonance techniques have also led us to link specific alterations in the nitrogen metabolome to the biology of distinct tumor types [152, 153]. Ex vivo sampling and measurement of metabolites have shown striking differences in amino acid compositions between tissue, blood, interstitial fluid, and laboratory cell culture media, and even within different regions of the same tumor [5, 33, 34]. Therefore, the metabolic pressure in the tumor milieu combined with the complex nature of cancer cell number, mutational load, heterogeneity and metabolic preferences is likely to create a regional competition for the available nitrogen between cancer cells, immune cells and other cells in the stroma. Identifying cell type specific pathway redundancy of nitrogen metabolism can provide us with strategies in developing targeted therapies with less toxicity. We also need to better understand the metabolic needs of immune cells before modulating nitrogen metabolism in tumors for therapy (see Outstanding Questions). Future work on nitrogen handling is likely to expand towards a more physiological in vivo system, where the preferences, acquisition and utilization rate of reduced nitrogen can be measured simultaneously in various cell types, while gaining both spatial and temporal resolution that occurs in the normal and tumor tissue.
Outstanding Questions.
Glutamine can be either directly used as a nitrogen donor for anabolic reactions or catabolized to glutamate by glutaminase liberating free ammonia. What determines the flux towards these two processes? Under which cellular states are glutamine and glutamate preferentially required?
What are the consequences of using glutamate as nitrogen donor for NEAAs synthesis? Can other nitrogen-containing molecules be used as nitrogen donor (beside glutamine and glutamate)? Is glutamate compartmentalized or are the levels between cytosol and mitochondria in equilibrium?
Alpha-KG is used as carbon backbone to assimilate nitrogen by GDH and GS. Can high levels of alpha-KG indicate lack of nitrogen in mammalian cells?
Do cells have specific sensors for glutamine, glutamate or alpha-KG?
While some amino acids (leucine, arginine, lysine, glutamine and methionine) can activate the mTORC1 (mechanistic target of rapamycin complex 1) growth pathway, other amino acids cannot. Which amino acids are indicative of nitrogen levels in mammalian cells?
Aspartate is a precursor for both asparagine and arginine biosynthesis. The balance between utilization and synthesis of aspartate and its regulation within different cell types in a tumor is still incompletely understood.
Are there differences in utilization of particular amino acids between various cells types within the tumor microenvironment? Are the differences in utilization prominent under specific metabolic perturbations?
Do immune cells (especially CD8+ T cells) possess the necessary transaminases for synthesizing all or most NEAAs?
Besides cancer cells, can activated T cells and other cell types in the tumor microenvironment recycle ammonia?
How does recycling of ammonia or urea by the microbiome contribute to nitrogen availability for tumors or immune cells?
Highlights.
Transaminases and deaminases regulate nitrogen availability for various biosynthetic pathways and are often altered to support cancer and immune cell growth.
Glutamine and glutamate, which are the major reservoirs of nitrogen in cells, are used as nitrogen donors for synthesizing many nitrogenous compounds in both cancer and immune cells.
Ammonia, liberated during deamination reactions, can be recycled by GS, GDH and CPS in cancer cells; however, whether such metabolic adaptation exists in other cell types remains largely unknown.
The circuits that control the acquisition and maintenance of cellular nitrogen are often rewired at multiple levels in cancer and immune cells.
Each cell type within the tumor microenvironment have a unique requirement for nitrogen sources. Expression of specific transaminases for NEAAs synthesis are differentially regulated in cancer and T cells during amino acid deficiencies.
Acknowledgments
We apologize for the omission of any primary literature and work we could not cite due to space limitation. We would like to thank Jaime Laurel Schneider, Jefte Drijvers, Jessica Spinelli, Giulia Notarangelo, and Liam Kelley for their careful reading and feedback on the manuscript. K.K. is a Gilead Sciences fellow of the Life Sciences Research Foundation. M.C.H. is supported by the Ludwig Center at Harvard, the Glenn Foundation for Medical Research, and National Institutes of Health grant RO1CA213062.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hosios AM et al. (2016) Amino Acids Rather than Glucose Account for the Majority of Cell Mass in Proliferating Mammalian Cells. Dev. Cell 36 (5), 540–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jang C et al. (2018) Metabolomics and Isotope Tracing. Cell 173 (4), 822–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lane AN and Fan TW (2015) Regulation of mammalian nucleotide metabolism and biosynthesis. Nucleic Acids Res 43 (4), 2466–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kishton RJ et al. (2017) Metabolic Regulation of T Cell Longevity and Function in Tumor Immunotherapy. Cell Metab 26 (1), 94–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lukey MJ et al. (2017) Targeting amino acid metabolism for cancer therapy. Drug Discov Today 22 (5), 796–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.O’Sullivan D et al. (2019) Metabolic interventions in the immune response to cancer. Nat Rev Immunol 19 (5), 324–335. [DOI] [PubMed] [Google Scholar]
- 7.Pavlova NN and Thompson CB (2016) The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23 (1), 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palm W and Thompson CB (2017) Nutrient acquisition strategies of mammalian cells. Nature 546 (7657), 234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu G (2009) Amino acids: metabolism, functions, and nutrition. Amino Acids 37 (1), 1–17. [DOI] [PubMed] [Google Scholar]
- 10.Hoffer LJ (2016) Human Protein and Amino Acid Requirements. JPEN J Parenter Enteral Nutr 40 (4), 460–74. [DOI] [PubMed] [Google Scholar]
- 11.Keshet R et al. (2018) Rewiring urea cycle metabolism in cancer to support anabolism. Nat Rev Cancer 18 (10), 634–645. [DOI] [PubMed] [Google Scholar]
- 12.Li L et al. (2019) p53 regulation of ammonia metabolism through urea cycle controls polyamine biosynthesis. Nature 567 (7747), 253–256. [DOI] [PubMed] [Google Scholar]
- 13.Spinelli JB et al. (2017) Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 358 (6365), 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lee JS et al. (2018) Urea Cycle Dysregulation Generates Clinically Relevant Genomic and Biochemical Signatures. Cell 174 (6), 1559–1570 e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lie S et al. (2019) The ability to utilise ammonia as nitrogen source is cell type specific and intricately linked to GDH, AMPK and mTORC1. Sci Rep 9 (1), 1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Graves LM et al. (2000) Regulation of carbamoyl phosphate synthetase by MAP kinase. Nature 403 (6767), 328–32. [DOI] [PubMed] [Google Scholar]
- 17.Robitaille AM et al. (2013) Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 339 (6125), 1320–3. [DOI] [PubMed] [Google Scholar]
- 18.Bott AJ et al. (2015) Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab 22 (6), 1068–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim J et al. (2017) CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546 (7656), 168–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jin L et al. (2018) The PLAG1-GDH1 Axis Promotes Anoikis Resistance and Tumor Metastasis through CamKK2-AMPK Signaling in LKB1-Deficient Lung Cancer. Mol Cell 69 (1), 87–99 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adeva MM et al. (2012) Ammonium metabolism in humans. Metabolism 61 (11), 1495–511. [DOI] [PubMed] [Google Scholar]
- 22.Takeuchi Y et al. (2018) Glutamate production from ammonia via glutamate dehydrogenase 2 activity supports cancer cell proliferation under glutamine depletion. Biochem Biophys Res Commun 495 (1), 761–767. [DOI] [PubMed] [Google Scholar]
- 23.Luengo A et al. (2017) Targeting Metabolism for Cancer Therapy. Cell Chem Biol 24 (9), 1161–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Valvezan AJ et al. (2017) mTORC1 Couples Nucleotide Synthesis to Nucleotide Demand Resulting in a Targetable Metabolic Vulnerability. Cancer Cell 32 (5), 624–638 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ben-Sahra I et al. (2013) Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 339 (6125), 1323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stine ZE et al. (2015) MYC, Metabolism, and Cancer. Cancer Discov 5 (10), 1024–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cunningham JT et al. (2014) Protein and nucleotide biosynthesis are coupled by a single rate-limiting enzyme, PRPS2, to drive cancer. Cell 157 (5), 1088–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ben-Sahra I et al. (2016) mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 351 (6274), 728–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Villa E et al. (2019) Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers (Basel) 11 (5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maiuolo J et al. (2016) Regulation of uric acid metabolism and excretion. Int J Cardiol 213, 8–14. [DOI] [PubMed] [Google Scholar]
- 31.Sykes DB et al. (2016) Inhibition of Dihydroorotate Dehydrogenase Overcomes Differentiation Blockade in Acute Myeloid Leukemia. Cell 167 (1), 171–186 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Austin WR et al. (2012) Nucleoside salvage pathway kinases regulate hematopoiesis by linking nucleotide metabolism with replication stress. J Exp Med 209 (12), 2215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kamphorst JJ et al. (2015) Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75 (3), 544–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cantor JR et al. (2017) Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 169 (2), 258–272 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pan M et al. (2016) Regional glutamine deficiency in tumours promotes dedifferentiation through inhibition of histone demethylation. Nat Cell Biol 18 (10), 1090–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bott AJ et al. (2019) The Pleiotropic Effects of Glutamine Metabolism in Cancer. Cancers (Basel) 11 (6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nakaya M et al. (2014) Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40 (5), 692–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ren W et al. (2017) Amino-acid transporters in T-cell activation and differentiation. Cell Death Dis 8 (3), e2655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kandasamy P et al. (2018) Amino acid transporters revisited: New views in health and disease. Trends Biochem. Sci 43 (10), 752–789. [DOI] [PubMed] [Google Scholar]
- 40.van Geldermalsen M et al. (2016) ASCT2/SLC1A5 controls glutamine uptake and tumour growth in triple-negative basal-like breast cancer. Oncogene 35 (24), 3201–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Romero R et al. (2017) Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 23 (11), 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nicklin P et al. (2009) Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136 (3), 521–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sinclair LV et al. (2013) Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat Immunol 14 (5), 500–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Elorza A et al. (2012) HIF2alpha acts as an mTORC1 activator through the amino acid carrier SLC7A5. Mol Cell 48 (5), 681–91. [DOI] [PubMed] [Google Scholar]
- 45.Hayashi K et al. (2013) LAT1 is a critical transporter of essential amino acids for immune reactions in activated human T cells. J Immunol 191 (8), 4080–5. [DOI] [PubMed] [Google Scholar]
- 46.White MA et al. (2017) Glutamine Transporters Are Targets of Multiple Oncogenic Signaling Pathways in Prostate Cancer. Mol Cancer Res 15 (8), 1017–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nguyen TV et al. (2016) Glutamine Triggers Acetylation-Dependent Degradation of Glutamine Synthetase via the Thalidomide Receptor Cereblon. Mol Cell 61 (6), 809–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tardito S et al. (2015) Glutamine synthetase activity fuels nucleotide biosynthesis and supports growth of glutamine-restricted glioblastoma. Nat Cell Biol 17 (12), 1556–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yang L et al. (2016) Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab 24 (5), 685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jha AK et al. (2015) Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42 (3), 419–30. [DOI] [PubMed] [Google Scholar]
- 51.Palmieri EM et al. (2017) Pharmacologic or Genetic Targeting of Glutamine Synthetase Skews Macrophages toward an M1-like Phenotype and Inhibits Tumor Metastasis. Cell Rep 20 (7), 1654–1666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Eagle H et al. (1956) The growth response of mammalian cells in tissue culture to L-glutamine and L-glutamic acid. J Biol Chem 218 (2), 607–16. [PubMed] [Google Scholar]
- 53.Raffel S et al. (2017) BCAT1 restricts alphaKG levels in AML stem cells leading to IDHmut-like DNA hypermethylation. Nature 551 (7680), 384–388. [DOI] [PubMed] [Google Scholar]
- 54.McBrayer SK et al. (2018) Transaminase Inhibition by 2-Hydroxyglutarate Impairs Glutamate Biosynthesis and Redox Homeostasis in Glioma. Cell 175 (1), 101–116 e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yang C et al. (2014) Glutamine oxidation maintains the TCA cycle and cell survival during impaired mitochondrial pyruvate transport. Mol Cell 56 (3), 414–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Coloff JL et al. (2016) Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell Metab 23 (5), 867–80. [DOI] [PubMed] [Google Scholar]
- 57.Bott AJ et al. (2019) Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism. Cell Rep 29 (5), 1287–1298 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mak TW et al. (2017) Glutathione Primes T Cell Metabolism for Inflammation. Immunity 46 (4), 675–689. [DOI] [PubMed] [Google Scholar]
- 59.Timmerman LA et al. (2013) Glutamine sensitivity analysis identifies the xCT antiporter as a common triple-negative breast tumor therapeutic target. Cancer Cell 24 (4), 450–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Muir A et al. (2017) Environmental cystine drives glutamine anaplerosis and sensitizes cancer cells to glutaminase inhibition. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Davidson SM et al. (2016) Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab 23 (3), 517–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Biancur DE et al. (2017) Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat Commun 8, 15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Momcilovic M et al. (2017) Targeted Inhibition of EGFR and Glutaminase Induces Metabolic Crisis in EGFR Mutant Lung Cancer. Cell Rep 18 (3), 601–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wise DR and Thompson CB (2010) Glutamine addiction: a new therapeutic target in cancer. Trends Biochem Sci 35 (8), 427–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shin CS et al. (2017) The glutamate/cystine xCT antiporter antagonizes glutamine metabolism and reduces nutrient flexibility. Nat Commun 8, 15074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yuan J et al. (2009) Metabolomics-driven quantitative analysis of ammonia assimilation in E. coli. Mol Syst Biol 5, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hart Y et al. (2011) Robust control of nitrogen assimilation by a bifunctional enzyme in E. coli. Mol Cell 41 (1), 117–27. [DOI] [PubMed] [Google Scholar]
- 68.Doucette CD et al. (2011) alpha-Ketoglutarate coordinates carbon and nitrogen utilization via enzyme I inhibition. Nat Chem Biol 7 (12), 894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sullivan LB et al. (2018) Aspartate is an endogenous metabolic limitation for tumour growth. Nat Cell Biol 20 (7), 782–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Garcia-Bermudez J et al. (2018) Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol 20 (7), 775–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pavlova NN et al. (2018) As Extracellular Glutamine Levels Decline, Asparagine Becomes an Essential Amino Acid. Cell Metab 27 (2), 428–438 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Birsoy K et al. (2015) An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162 (3), 540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sullivan LB et al. (2015) Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162 (3), 552–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Diebold LP et al. (2019) Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat Metab 1 (1), 158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Weinberg SE et al. (2019) Mitochondrial complex III is essential for suppressive function of regulatory T cells. Nature 565 (7740), 495–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Su AI et al. (2002) Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci U S A 99 (7), 4465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Uhlen M et al. (2015) Proteomics. Tissue-based map of the human proteome. Science 347 (6220), 1260419. [DOI] [PubMed] [Google Scholar]
- 78.Krall AS et al. (2016) Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nat Commun 7, 11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Koprivnikar J et al. (2017) Safety, efficacy, and clinical utility of asparaginase in the treatment of adult patients with acute lymphoblastic leukemia. Onco Targets Ther 10, 1413–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Marini BL et al. (2017) Catalyzing improvements in ALL therapy with asparaginase. Blood Rev 31 (5), 328–338. [DOI] [PubMed] [Google Scholar]
- 81.Knott SRV et al. (2018) Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 554 (7692), 378–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rabinovich S et al. (2015) Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 527 (7578), 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Fultang L et al. (2016) Molecular basis and current strategies of therapeutic arginine depletion for cancer. Int J Cancer 139 (3), 501–9. [DOI] [PubMed] [Google Scholar]
- 84.Poillet-Perez L et al. (2018) Autophagy maintains tumour growth through circulating arginine. Nature 563 (7732), 569–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Riess C et al. (2018) Arginine-Depleting Enzymes - An Increasingly Recognized Treatment Strategy for Therapy-Refractory Malignancies. Cell Physiol Biochem 51 (2), 854–870. [DOI] [PubMed] [Google Scholar]
- 86.Geiger R et al. (2016) L-Arginine Modulates T Cell Metabolism and Enhances Survival and Anti-tumor Activity. Cell 167 (3), 829–842 e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Taheri F et al. (2001) L-Arginine regulates the expression of the T-cell receptor zeta chain (CD3zeta) in Jurkat cells. Clin Cancer Res 7 (3 Suppl), 958s–965s. [PubMed] [Google Scholar]
- 88.Rodriguez PC and Ochoa AC (2008) Arginine regulation by myeloid derived suppressor cells and tolerance in cancer: mechanisms and therapeutic perspectives. Immunol Rev 222, 180–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Rodriguez PC et al. (2007) L-arginine availability regulates T-lymphocyte cell-cycle progression. Blood 109 (4), 1568–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tarasenko TN et al. (2015) Impaired T cell function in argininosuccinate synthetase deficiency. J Leukoc Biol 97 (2), 273–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rodriguez PC et al. (2004) Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res 64 (16), 5839–49. [DOI] [PubMed] [Google Scholar]
- 92.Rath M et al. (2014) Metabolism via Arginase or Nitric Oxide Synthase: Two Competing Arginine Pathways in Macrophages. Front Immunol 5, 532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Garcia-Ortiz A and Serrador JM (2018) Nitric Oxide Signaling in T Cell-Mediated Immunity. Trends Mol Med 24 (4), 412–427. [DOI] [PubMed] [Google Scholar]
- 94.Morris SM Jr. (2016) Arginine Metabolism Revisited. J Nutr 146 (12), 2579S–2586S. [DOI] [PubMed] [Google Scholar]
- 95.Casero RA Jr. et al. (2018) Polyamine metabolism and cancer: treatments, challenges and opportunities. Nat Rev Cancer 18 (11), 681–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Hesterberg RS et al. (2018) Role of Polyamines in Immune Cell Functions. Med Sci (Basel) 6 (1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Gamble LD et al. (2019) Inhibition of polyamine synthesis and uptake reduces tumor progression and prolongs survival in mouse models of neuroblastoma. Sci Transl Med 11 (477). [DOI] [PubMed] [Google Scholar]
- 98.Locke M et al. (2016) Inhibition of the Polyamine Synthesis Pathway Is Synthetically Lethal with Loss of Argininosuccinate Synthase 1. Cell Rep 16 (6), 1604–1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hayes CS et al. (2014) Polyamine-blocking therapy reverses immunosuppression in the tumor microenvironment. Cancer Immunol Res 2 (3), 274–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Puleston DJ et al. (2019) Polyamines and eIF5A Hypusination Modulate Mitochondrial Respiration and Macrophage Activation. Cell Metab 30 (2), 352–363 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fenouille N et al. (2017) The creatine kinase pathway is a metabolic vulnerability in EVI1-positive acute myeloid leukemia. Nat Med 23 (3), 301–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kurmi K et al. (2018) Tyrosine Phosphorylation of Mitochondrial Creatine Kinase 1 Enhances a Druggable Tumor Energy Shuttle Pathway. Cell Metab 28 (6), 833–847 e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liu GY and Sabatini DM (2020) mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chantranupong L et al. (2016) The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 165 (1), 153–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Gu X et al. (2017) SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 358 (6364), 813–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wolfson RL et al. (2016) Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 351 (6268), 43–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wyant GA et al. (2017) mTORC1 Activator SLC38A9 Is Required to Efflux Essential Amino Acids from Lysosomes and Use Protein as a Nutrient. Cell 171 (3), 642–654 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jewell JL et al. (2015) Metabolism. Differential regulation of mTORC1 by leucine and glutamine. Science 347 (6218), 194–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Dong J et al. (2000) Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol Cell 6 (2), 269–79. [DOI] [PubMed] [Google Scholar]
- 110.Harding HP et al. (2003) An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell 11 (3), 619–33. [DOI] [PubMed] [Google Scholar]
- 111.Pakos-Zebrucka K et al. (2016) The integrated stress response. EMBO Rep 17 (10), 1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ye J et al. (2015) GCN2 sustains mTORC1 suppression upon amino acid deprivation by inducing Sestrin2. Genes Dev 29 (22), 2331–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Nakamura A et al. (2018) Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proc Natl Acad Sci U S A 115 (33), E7776–E7785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Nofal M et al. (2017) mTOR Inhibition Restores Amino Acid Balance in Cells Dependent on Catabolism of Extracellular Protein. Mol Cell 67 (6), 936–946 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Finicle BT et al. (2018) Nutrient scavenging in cancer. Nat Rev Cancer 18 (10), 619–633. [DOI] [PubMed] [Google Scholar]
- 116.Commisso C et al. (2013) Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497 (7451), 633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Yang X et al. (2018) ATF4 Regulates CD4+ T Cell Immune Responses through Metabolic Reprogramming. Cell Rep. 23 (6), 1754–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ye J et al. (2010) The GCN2-ATF4 pathway is critical for tumour cell survival and proliferation in response to nutrient deprivation. EMBO J 29 (12), 2082–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Palm W et al. (2015) The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 162 (2), 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Turley SJ et al. (2015) Immunological hallmarks of stromal cells in the tumour microenvironment. Nat Rev Immunol 15 (11), 669–82. [DOI] [PubMed] [Google Scholar]
- 121.Buck MD et al. (2017) Metabolic Instruction of Immunity. Cell 169 (4), 570–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Olenchock BA et al. (2017) Biochemical Underpinnings of Immune Cell Metabolic Phenotypes. Immunity 46 (5), 703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.O’Neill LA et al. (2016) A guide to immunometabolism for immunologists. Nat Rev Immunol 16 (9), 553–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ron-Harel N et al. (2019) T Cell Activation Depends on Extracellular Alanine. Cell Rep 28 (12), 3011–3021 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Johnson MO et al. (2018) Distinct Regulation of Th17 and Th1 Cell Differentiation by Glutaminase-Dependent Metabolism. Cell 175 (7), 1780–1795 e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang R et al. (2011) The transcription factor Myc controls metabolic reprogramming upon T lymphocyte activation. Immunity 35 (6), 871–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Carr EL et al. (2010) Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J Immunol 185 (2), 1037–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Metzler B et al. (2016) Restricting Glutamine or Glutamine-Dependent Purine and Pyrimidine Syntheses Promotes Human T Cells with High FOXP3 Expression and Regulatory Properties. J Immunol 196 (9), 3618–30. [DOI] [PubMed] [Google Scholar]
- 129.Klysz D et al. (2015) Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci Signal 8 (396), ra97. [DOI] [PubMed] [Google Scholar]
- 130.Xu T et al. (2017) Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature 548 (7666), 228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Swamy M et al. (2016) Glucose and glutamine fuel protein O-GlcNAcylation to control T cell self-renewal and malignancy. Nat Immunol 17 (6), 712–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Araujo L et al. (2017) Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Leone RD et al. (2019) Glutamine blockade induces divergent metabolic programs to overcome tumor immune evasion. Science 366 (6468), 1013–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Sousa CM et al. (2016) Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536 (7617), 479–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Maddocks OD et al. (2013) Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493 (7433), 542–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Maddocks ODK et al. (2017) Modulating the therapeutic response of tumours to dietary serine and glycine starvation. Nature 544 (7650), 372–376. [DOI] [PubMed] [Google Scholar]
- 137.Locasale JW et al. (2011) Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 43 (9), 869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Possemato R et al. (2011) Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476 (7360), 346–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Ding J et al. (2013) The histone H3 methyltransferase G9A epigenetically activates the serine-glycine synthesis pathway to sustain cancer cell survival and proliferation. Cell Metab 18 (6), 896–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.DeNicola GM et al. (2015) NRF2 regulates serine biosynthesis in non-small cell lung cancer. Nat Genet 47 (12), 1475–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Diehl FF et al. (2019) Cellular redox state constrains serine synthesis and nucleotide production to impact cell proliferation. Nat Metab 1 (9), 861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ron-Harel N et al. (2016) Mitochondrial Biogenesis and Proteome Remodeling Promote One-Carbon Metabolism for T Cell Activation. Cell Metab 24 (1), 104–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ma EH et al. (2017) Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab 25 (2), 482. [DOI] [PubMed] [Google Scholar]
- 144.Munn DH et al. (2002) Potential regulatory function of human dendritic cells expressing indoleamine 2,3-dioxygenase. Science 297 (5588), 1867–70. [DOI] [PubMed] [Google Scholar]
- 145.Uyttenhove C et al. (2003) Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med 9 (10), 1269–74. [DOI] [PubMed] [Google Scholar]
- 146.Opitz CA et al. (2020) The therapeutic potential of targeting tryptophan catabolism in cancer. Br J Cancer 122 (1), 30–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sharma MD et al. (2007) Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes directly activate mature Tregs via indoleamine 2,3-dioxygenase. J Clin Invest 117 (9), 2570–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Fallarino F et al. (2006) The combined effects of tryptophan starvation and tryptophan catabolites downregulate T cell receptor zeta-chain and induce a regulatory phenotype in naive T cells. J Immunol 176 (11), 6752–61. [DOI] [PubMed] [Google Scholar]
- 149.Adams JL et al. (2015) Big opportunities for small molecules in immuno-oncology. Nat Rev Drug Discov 14 (9), 603–22. [DOI] [PubMed] [Google Scholar]
- 150.Ananieva E (2015) Targeting amino acid metabolism in cancer growth and anti-tumor immune response. World J Biol Chem 6 (4), 281–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vander Heiden MG and DeBerardinis RJ (2017) Understanding the Intersections between Metabolism and Cancer Biology. Cell 168 (4), 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kang YP et al. (2018) Recent advances in cancer metabolism: a technological perspective. Exp Mol Med 50 (4), 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Bhinderwala F et al. (2018) Expanding the Coverage of the Metabolome with Nitrogen-Based NMR. Anal Chem 90 (7), 4521–4528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Hirotsu K et al. (2005) Dual substrate recognition of aminotransferases. Chem Rec 5 (3), 160–72. [DOI] [PubMed] [Google Scholar]
- 155.Cooper AJ and Meister A (1989) An appreciation of Professor Alexander E. Braunstein. The discovery and scope of enzymatic transamination. Biochimie 71 (4), 387–404. [DOI] [PubMed] [Google Scholar]
- 156.Smith HQ et al. (2019) Glutamate Dehydrogenase, a Complex Enzyme at a Crucial Metabolic Branch Point. Neurochem Res 44 (1), 117–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ogawa H et al. (1989) Human liver serine dehydratase. cDNA cloning and sequence homology with hydroxyamino acid dehydratases from other sources. J Biol Chem 264 (27), 15818–23. [PubMed] [Google Scholar]
- 158.Shaw SM and Carrey EA (1992) Regulation of the mammalian carbamoyl-phosphate synthetase II by effectors and phosphorylation. Altered affinity for ATP and magnesium ions measured using the ammonia-dependent part reaction. Eur J Biochem 207 (3), 957–65. [DOI] [PubMed] [Google Scholar]