

Abstract
Circadian rhythm affects drug-induced reward behaviour and the innate immune system. Peaks in reward-associated behaviour and immune responses typically occur during the active (dark) phase of rodents. While the role of the immune system, specifically, Toll-like receptor 4 (TLR4, an innate immune receptor) in drug-induced reward is becoming increasingly appreciated, it is unclear whether its effects vary according to light-cycle. Therefore, the aim of this study was to characterise the effects of the phase of the light-cycle and the state of the innate immune system on alcohol reward behaviour and subsequently determine whether the efficacy of targeting the immune component of drug reward depends upon the light-cycle.
This study demonstrates that mice exhibit greater alcohol-induced conditioned place preference and alcohol two-bottle choice preference during the dark cycle. This effect overlapped with elevations in reward-, thirst- and immune-related genes. Administration of (+)-Naltrexone, a biased TLR4 antagonist, reduced immune-related gene expression and alcohol preference with its effects most pronounced during the dark cycle. However, (+)-Naltrexone, like other TLR4 antagonists exhibited off-target side effects, with a significant reduction in overall saccharin intake – an effect likely attributable to a reduction in tyrosine hydroxylase (Th) mRNA expression levels. Collectively, the study highlights a link between a time-of-day dependent influence of TLR4 on natural and alcohol reward-like behaviour in mice.
Keywords: Toll-like receptor 4, TRIF, alcohol, circadian, tyrosine hydroxylase, interferon, time-of-day
3.2. Introduction
Alcohol is the most widely consumed drug globally (WHO, 2015). The initial consumption of alcohol is characterised by its rewarding, hedonic properties. These properties assist in the development of repetitive/habitual use, which can lead to misuse, loss of control of intake, and addiction (Koob & Le Moal, 2001). The rewarding properties of alcohol are attributable to its actions on the brains mesolimbic system (Imperato & Di Chiara, 1986). Specifically, dopaminergic neurons projecting from the ventral tegmental area (VTA) to the nucleus accumbens (NAcc) are thought to mediate reward and attach salient cues (Koob & Volkow, 2009; Wise, 2004). Alcohol activates these neurons via multiple pathways for example, alcohol; causes the release of GABA, or directly activates the GABAA receptors in VTA; increases opioid peptides in the VTA and NAcc; and alters glutamate signalling which innervates the NAcc’s dopaminergic projections (see Nestler, (2005) for review). Collectively, these processes control dopaminergic neurotransmission thereby influencing reward, specifically, the likability of, and the motivation to consume alcohol (Robinson et al., 2013).
The extent to which alcohol initially activates the mesolimbic system is dependent upon the time-of-day (circadian rhythm, and associated diurnal or nocturnal behaviours). Within mammals, circadian rhythm is generated and maintained, at a circuit level via, the suprachiasmatic nucleus (SCN), and on a cellular level, by a feedback loop involving a group of transcription factors (Reppert & Weaver, 2002). The SCN functions as the master regulator, linking the external (via the retinal-hypothalamic tract) and internal environments (Moore & Lenn, 1972; Stephan & Zucker, 1972). Consequently, the SCN sends neural and endocrine signals to regulate the function of organs and cells according to the time-of-day (for example Moore & Eichler, 1972). However, most cells can generate their own rhythm via an auto-regulatory feedback loop involving transcription factors. These transcription factors include two activator (CLOCK and BMAL1) and two repressor proteins (PER and CRY). In brevity, the cellular clock begins when CLOCK and BMAL1 dimerise and bind to enhancer box (E-box; DNA response elements) in the promoter region of CRY and PER, initiating their transcription. PER and CRY accumulate, dimerise and suppress the transcription of CLOCK and BMAL1. The subsequent decline in CLOCK and BMAL1 decreases the transcription of PER and CRY. This rhythmic interaction generates the cellular circadian rhythm over a 24 h period (Partch et al., 2014; Dardente & Cermakian, 2007), which is estimated to influence approximately 2 – 10 per cent of the mammalian transcriptome (Miller et al., 2007; Akhtar et al., 2002; Duffield et al., 2002).
The circadian influence on the reward pathway varies according to brain region and cell type. In the NAcc, dopaminergic and non-dopaminergic cell activity peaks during the active (dark) phase of rodents (Baltazar et al., 2013). This is attributable to transient elevations in transcription and translation of genes pertinent to the function of the mesolimbic system, such as tyrosine hydroxylase and the dopamine transporter (Chung et al., 2014; Ferris et al., 2014). Behaviourally, this manifests as a heightened sensitivity towards rewarding experiences to alcohol and other drugs of abuse during the active phase and a lower sensitivity during the inactive phase. For example, alcohol intake, alcohol preference and self-intracranial electrical stimulation of the reward pathway are greatest during the active (dark) phase relative to the inactive (light) phase (Perreau-Lenz et al., 2012; Gauvin et al., 1997; Terman & Terman, 1975). However, each drug of abuse appears to be unique, as cocaine-induced conditioned place preference is greatest during the light cycle (Kurtuncu et al., 2004).
Alcohol-induced reward-like behaviours are additionally influenced by the neuroimmune system (Crews et al., 2017; Lacagnina et al., 2016; Cui et al., 2014). Particular emphasis has been placed on Toll-like receptor 4 (TLR4), a pattern recognition receptor as a key mediator of reward induced by alcohol and other drugs of abuse (Bachtell et al., 2015). Activation of TLR4 results in the induction of two signalling pathways (MyD88 or TRIF) that culminates in the expression of classical pro-inflammatory cytokines or type-one interferons respectively (Akira & Takeda, 2004). Alcohol and other drugs of abuse activate TLR4 (either directly or indirectly) resulting in the induction of inflammatory mediators (Fernandez-Lizarbe et al., 2013; 2009). It is hypothesised that inflammatory mediators act on neighbouring neurons within the mesolimbic system (tetrapartite synapse), culminating in altered neuronal function and potentially influencing the presentation of reward-like behaviours (Lacagnina et al., 2016; Jacobsen et al., 2014).
Translationally, the effects of TLR4 on alcohol reward-like behaviour are mixed with studies demonstrating either attenuation or no effect on alcohol intake and preference (Harris et al., 2017; Aurelian et al., 2016; Bajo et al., 2016; June et al., 2015; Liu et al., 2011; Pascual et al., 2011). These discrepancies are likely due to methodological differences between the studies including brain region examined and method of drinking. Interestingly, despite TLR4 being implicated in alcohol drinking behaviour, few studies have considered which TLR4-signaling pathway (MyD88 or TRIF) is driving the alterations in reward behaviour (Blednov et al., 2017; Harris et al., 2017), and they have not considered the time of day associated rhythmicity of TLR4 expression and the subsequent impact this may have on behaviour.
Similar to the cellular and molecular components of the mesolimbic system, the expression of the TLR4-signaling pathway oscillates according to the time-of-day (Bass and Lazar, 2016). In the periphery, peak expression of TLR4-related signalling and inflammatory molecules are observed at the onset of their active (dark, nocturnal) phase and nadirs at the beginning of the inactive (light, diurnal) phase (Keller et al., 2009). Within the brain however, the opposite response is observed. Ex vivo microglial cells exhibit peak TLR4-related gene expression during the light phase compared to the dark phase (Fonken et al., 2015). However, it is unclear whether the MyD88 or TRIF pathway fluctuates according to circadian rhythm within the brain and periphery and whether these signalling pathways are involved in alcohol-induced reward-like behaviour. Therefore, this study sought to determine whether light cycle (dark vs light) differences exist in the expression of MyD88, TRIF and their downstream signalling molecules in naïve mice and mice following alcohol exposure. This study also investigated whether the efficacy of a biased TLR4 antagonist on attenuating reward-like behaviour is dependent on the light-cycle. The results presented suggest that the preference for rewarding and aversive compounds peaks and nadirs during the dark cycle with reward-, thirst-, hunger-, and immune-related genes following a similar pattern. We demonstrate that attenuating TLR4 via (+)-Naltrexone reduces alcohol drinking and conditioned place preference (key indicators of reward) with the degree of attenuation greater during the dark cycle. However, we found that (+)-Naltrexone additionally reduces saccharin preference. These effects coincide with a reduction in Tlr4 and Ifnb and Th mRNA in the nucleus accumbens. Collectively these results suggest TLR4 may play a role in dopamine synthesis and natural reward-like behaviour.
3.3. Methods
3.3.1. Animals
Male (8 – 10-week-old) Balb/c mice, obtained from the University of Adelaide Laboratory Animal Services (Adelaide, SA, Australia) were used for the following experiments. Mice were housed in light/dark (12:12 hours) and temperature controlled rooms (23±3°C) with food and water available ad libitum. The light cycle began at 7am (ZT0) and concluded at 7pm (ZT12). Following seven days of acclimatisation, mice were handled by the experimenter for five days prior to experimentation. Mice were weighed daily throughout the handling and experimental periods. All animal care and experiments complied with the principles of the Australian Code of Practice for the care and use of animals for scientific purposes and were approved by the University of Adelaide’s Animal Ethics Committee.
3.3.2. Drugs
Saccharin and quinine were purchased from Sigma Aldrich (St Louis, MO, USA). Ethanol (99.5%) (herein referred to as alcohol) was purchased from Chemsupply (Gliman, SA, Australia). Oral gavages of alcohol were dosed at 1.5g/kg and 3.2g/kg (25 per cent v/v) for conditioned place preference and molecular studies respectively. Saline oral gavages were volume-matched. The dose of alcohol used in conditioned place preference was based upon the effective dose 50 from an unpublished conditioned place preference dose response curve. 3.2g/kg was derived from the mean 2 h intake of alcohol from mice on the first day of drinking in the dark tests.
(+)-Naltrexone, a TLR4-TRIF antagonist was synthesised and kindly supplied by Dr Kenner Rice (Chemical Biology Research Branch, National Institute on Drug Abuse and National Institute of Alcohol Abuse and Alcoholism, Bethesda, MD, USA). (+)-Naltrexone was administered via intraperitoneal injections with doses ranging from 1 to 75 mg/kg (dose volume 10 ml/kg). Saline intraperitoneal injections were volume-matched.
3.3.3. Rational of behavioural tests
This study implemented a range of paradigms to assess alcohol reward. Specifically, this study was designed to assess two components of reward, likability and seeking behaviour (motivation to obtain alcohol “wanting”). The likability of alcohol was assessed using the two-bottle choice paradigm. Despite its relative coarseness in obtaining accurate drinking information, the two-bottle choice test can inform researchers about the general avidity of alcohol (Tabakoff & Hoffman, 2000). For example, the consumption of low concentration alcohol is largely driven by taste. By contrast, the consumption of alcohol at higher concentrations are attributable to its actions on the mesolimbic pathway, as increasing the concentration of alcohol imparts an increasingly bitter and aversive taste (Spanagel, 2000; Tabakoff & Hoffman, 2000). Consequently, when testing the likability of alcohol, a range of concentrations must be assessed.
The two-bottle choice test is limited in its assessment of the motivational properties of alcohol (Tabakoff & Hoffman, 2000). To infer this component of alcohol, conditioned place preference was utilised. Conditioned place preference is a paradigm in which mice learn to associate alcohol in one particular environment. If alcohol is hedonic (reinforcing), the mouse will choose to spend more time in that environment over another when given free access to both. By contrast, if the mouse finds alcohol aversive, it will spend less time in the environment. Thus, the motivation to seek alcohol is illustrated by the time spent in the paired environment in the absence of receiving alcohol (Bardo & Bevins, 2000).
To control for taste, and basal hedonic tone, the preference for quinine and saccharin were assessed. Quinine, a bitter compound is thought to reflect the higher concentrations of alcohol while saccharin, a sweet compound is thought to reflect the lower concentrations of alcohol. Additionally, saccharin is innately reinforcing, thus if mice exhibit deficits in basal hedonic behaviour, this will become evident during the test.
3.3.4. Experimental design
Testing began at ZT2 and ZT14 for mice undergoing tests during the light (inactive) and dark (active) phase respectively (figure 1). The behavioural experiments ranged from 2 – 24 h. For studies evaluating (+)-Naltrexone (and saline), mice were injected 30 min prior to undergoing behavioural testing (ZT1:30 and ZT13:30 for light and dark phases respectively) (figure 1).
Figure 1. Experimental timeline.
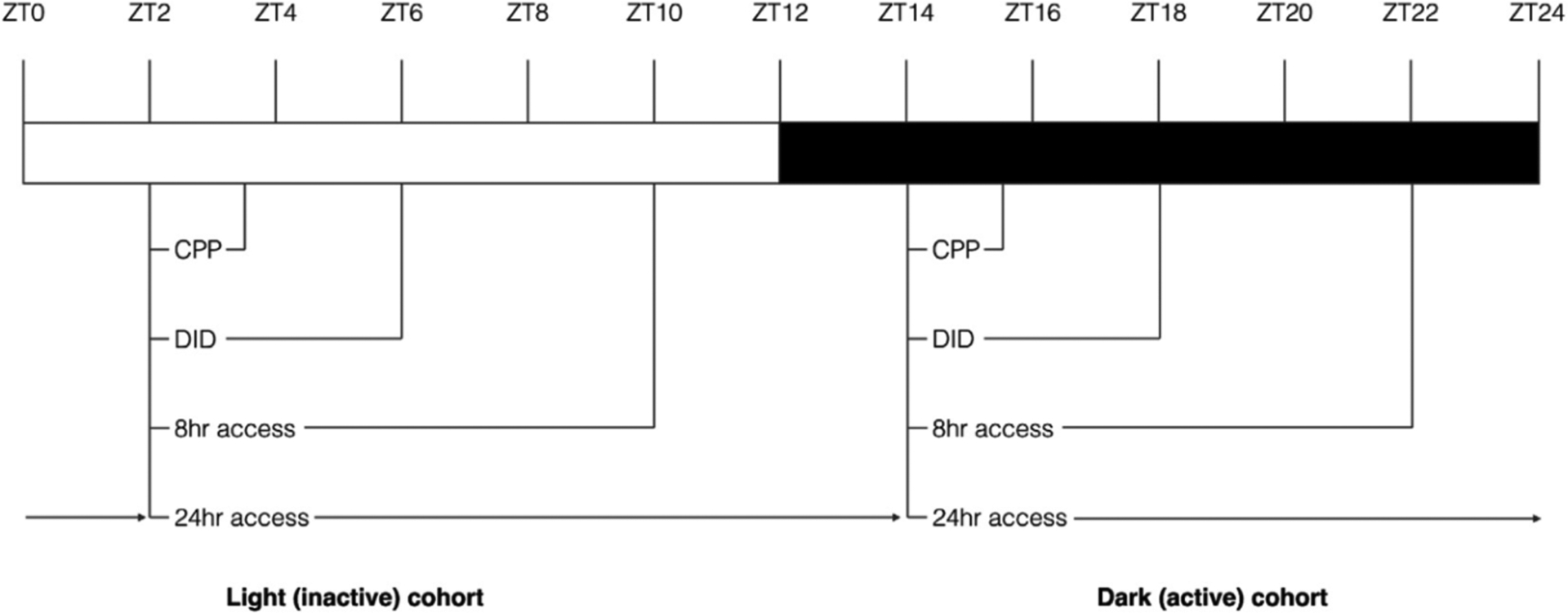
All behavioural testing began 2 h into the light or dark phase. Conditioned place preference occurred between ZT2 to ZT3 and ZT14 to ZT15 for mice in the light or dark phase respectively. Drinking in the dark (days 1 – 3) occurred between ZT2 to ZT4 and ZT14 to ZT16 for the light and dark cohorts respectively. On the final day of testing, the test concluded at ZT6 and ZT18 for the light and dark cohorts respectively.
3.3.5. Alcohol drinking tests
Three alcohol-drinking paradigms were used for the following experiments: 24 h and 8 h two-bottle choice and drinking in the dark.
3.3.5.1. Alcohol two-bottle choice
Alcohol drinking and preference was assessed using an 8 or 24 h two-bottle choice paradigm. Following 14 days of acclimatisation and handling, mice were placed into individual cages. After a further week of acclimatisation, mice were presented with two bottles containing water 2 h after the beginning of the light or dark cycle (ZT2 or ZT14 respectively) for 8 or 24 h. Two bottles of water were initially presented to mice in order to control for novelty-induced drinking. For the 8 h test, the bottles were removed at ZT10 or ZT22, weighed and replaced with a single bottle of water randomised to either the left or right side of the cage for the remaining 16 h. For the 24 h two-bottle choice test, after the test period had elapsed the bottles were removed, weighed and replaced with two new bottles.
Following five days of drinking water from two bottles, mice were offered one bottle containing water and the other 3, 6, 9, 12, 15, 18, 21 or 42 per cent of alcohol (v/v). The concentration and bottle position was randomised daily to prevent the immediate acquisition of alcohol drinking and side preferences respectively. After 8 h bottles were removed weighed and replaced by a water bottle randomly allocated to either side of the cage lid. For mice in the longer test, the bottles were replaced with two new bottles (one containing alcohol the other water) after 24 h.
The amount of alcohol consumed was determined by the difference in bottle weights before and after drinking sessions. This enabled the calculation of the amount of alcohol consumed per kilogram bodyweight (grams/kilogram) and the preference ratio (alcohol intake ÷ (total water + alcohol intake)) for each mouse and averaged for each group per concentration of alcohol. An empty cage with two bottles was used to determine the rate of evaporation. The rate of evaporation was subtracted from the final weight of test bottles.
3.3.5.2. Saccharin and quinine two-bottle choice
Mice were also tested using the same 8 h paradigm above for saccharin (1, 15, 30, 45 and 60 mM) and quinine (0.001, 0.01, 0.1, 1 and 5 mM) preference. However, instead of the bottle of alcohol, mice received a bottle of saccharin or quinine and a bottle of water.
For studies evaluating the dose of (+)-Naltrexone, the protocol for the 8 h two-bottle choice protocol was followed. However, the concentration of the alcohol, saccharin and quinine were fixed at 12 per cent, 15 mM, 0.1 mM respectively.
3.3.5.3. Drinking in the dark (alcohol and saccharin)
Drinking in the dark, a limited access alcohol intake test was additionally used. Following two weeks of acclimatisation and handling, mice were individually housed. After one week of further acclimatisation, the single bottle of water was replaced with a bottle of 20 per cent (v/v) alcohol 2 h into the beginning of the light or dark cycle (ZT2 and ZT14). After 2 h, the bottle of alcohol was removed weighed and replaced with a bottle of water. This was repeated for the following two days. On the fourth day, mice were offered the bottle for 4 h.
Mice were additionally tested for saccharin (15 mM) consumption using a 2 h limited access paradigm as outlined above. However, this test was not repeated for 4 consecutive days.
3.3.6. Conditioned place preference
Conditioned place preference was used to infer alcohol-seeking behaviour (Bardo & Bevins, 2000).
3.3.6.1. Apparatus
The conditioning apparatus consisted of two conditioning chambers (10.9 (length) × 9.3 (width) × 35 (height) cm) separated by a neutral chamber (16.6 × 4.8 × 35 cm). The neutral chamber contained black walls with grey flooring. The conditioning chambers differed in tactile and visual cues. The flooring of the conditioning chambers were either black plexiglass perforated holes (5 mm apart) or black plexiglass grids (5 mm apart). The walls of each chamber were white or black. The combination of wall colour to floor texture was randomised for each cohort to prevent any inherent biases mice have for a specific texture x colour combination.
During conditioning, a sliding partition restricted access to only one chamber. Movement and time spent in each chamber was recorded using Logitech Quickcam Pro 5000s and AnyMaze (Stoelting co., Wooddale, IL, USA).
3.3.6.2. Procedure
Day 1: Pretest. Mice were placed into the neutral chamber and allowed to explore all three chambers for 30 min.
Day 2 to 9: Conditioning. Mice received an oral gavage of alcohol (1.5 g/kg) and placed within their conditioning chamber for 30 min on days 1, 3, 5 and 7. On days 2, 4, 6 and 8, mice received an oral gavage of saline and placed within the unconditioned chamber for 30 min. Mice received a total of four conditioning sessions with each drug (alcohol or saline).
Day 10: Test. Mice received an oral gavage of saline, were placed into the neutral chamber and allowed to explore all three chambers for 30 min.
To infer whether the conditioning was successful, the time spent in the conditioned chamber during the post-test was subtracted from the time spent in the conditioned chamber during the pre-test.
3.3.7. Blood alcohol concentration assay
Serum alcohol concentration was measured using a commercial kit (ADH-NAD Reagent Multiple Test Vial; Sigma-Aldrich) and performed as per the manufacturer instructions. In brief, it estimates alcohol induced reduction of nicotinamide adenine dinucleotide (NAD+) to NADH in the presence of alcohol dehydrogenase. The reaction is observed by recording the absorbance of 340 nM by the solution. For two-bottle choice tests blood was acquired immediately after behavioral testing by creating a small incision into the tail of the mouse. To determine blood alcohol concentration following conditioned place preference, a separate cohort of mice underwent the conditioned place preference procedure. However, 30 minutes after the last alcohol conditioning session tail blood was collected. Blood was subsequently collected and spun down (1500g) at 4°C for 10 mins, thereby separating serum from the residual pellet.
3.3.8. Molecular analysis
3.3.8.1. Gavage model
The dose and duration of alcohol administered for the molecular studies was designed to model the drinking in the dark tests. 3.2g/kg was derived from the mean 2 h intake of alcohol from all cohorts of mice on the first day of drinking in the dark tests.
In brief, mice were injected for four consecutive days with either (+)-Naltrexone or saline followed by a gavage of alcohol or saline 30 min later. The injections of (+)-Naltrexone or saline commenced at either ZT1 or ZT13. The gavages of alcohol occurred at ZT1:30 or ZT13:30. On the final day of testing mice were culled at ZT2 and ZT14.
3.3.9. RNA Isolation and qPCR
Brain regions were isolated by placing the brain into an acrylic matrix (Able Scientific, Canning Vale, WA, AUS) and subsequently cutting them into 1 to 2 mm thick sections. The nucleus accumbens and the hypothalamus was subsequently microdissected using micropunches (Kai Medical, Seki City, Japan) and submerged in RNAlater® ICE (ThermoFisher Scientific, Waltham, MA, USA) prior to performing RNA isolation. RNA was isolated using Maxwell® 16 LEV simply RNA Tissue Kit (Promega, Madison, WI, USA) as per manufacturer instructions. RNA was quantified using spectrophotometric analysis, with the quality of RNA verified by the OD260/280 ratio. 900 ng of RNA was reversed transcribed into cDNA using iScript™ cDNA reverse transcription kit (BioRad, Hercules, CA, USA) as per manufacturer instructions.
Gene expression was assessed using iTaq™ Universal SYBR® Green Supermix (as per manufacturer instructions). Real time PCR was performed using the CFX96 Touch™ Real-Time PCR Detection System (BioRad). All primers were synthesised by Integrated DNA Technologies Pte. Ltd. (Baulkham Hills, NSW, Australia) with their sequences are outlined in the supplementary material (Table 1).
The relative difference in expression level of each of the genes of interest were normalised to the CT of GAPDH for both the test and control sample. The ΔCT of the test sample was normalised to the ΔCT a control sample (equal amount of cDNA from all samples), and then expressed as a ratio (2−ΔΔCT).
3.3.10. Statistical analysis
Experiment 1: The effect of light-cycle on the intake and preference for alcohol, saccharin and quinine was analysed using a two-way ANOVA with Tukey post hoc (figure 2). The effect of light-cycle on conditioned place preference and relative conditioned place preference was analysed using a two-way ANOVA with Tukey post hoc and a paired two-tail t-test respectively (figure 3).
Figure 2. Circadian timing affects the intake and preference of alcohol (a - b) and saccharin (c - d) but not quinine (e - f).
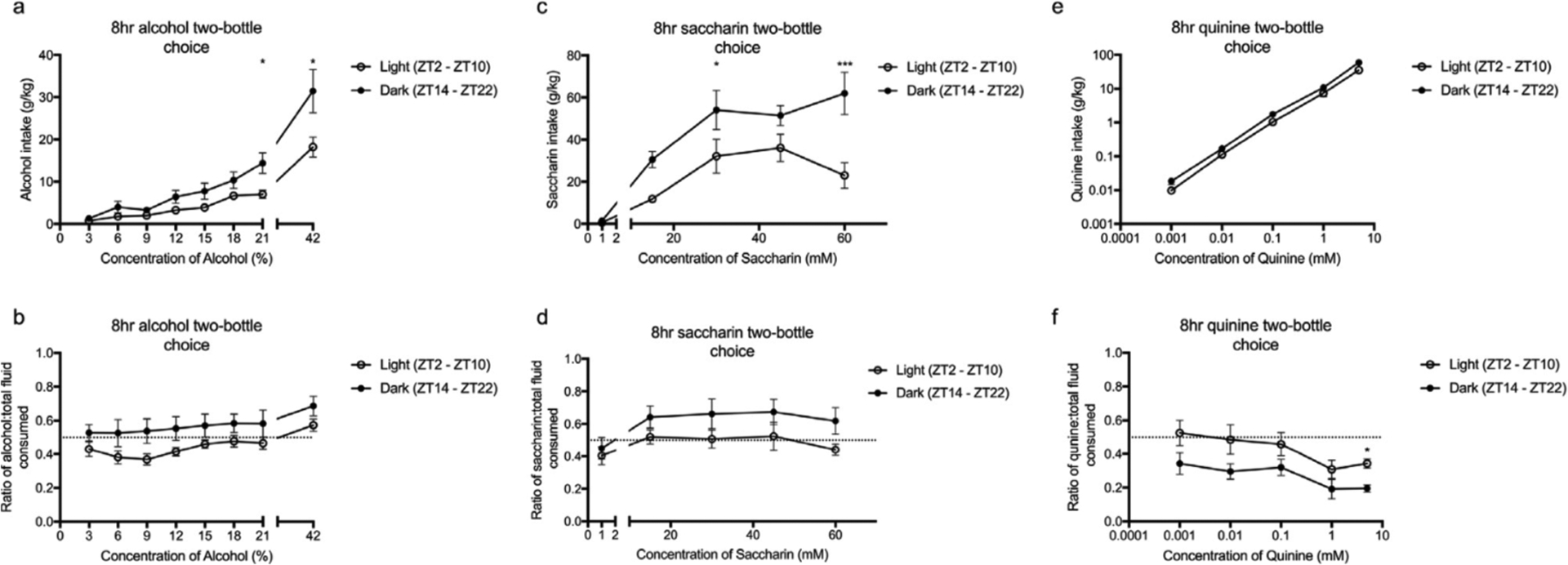
There was a main effect of light-cycle on the intake and preference for alcohol (3 – 42%) and saccharin (1 – 60mM) and the intake ratio of quinine (0.001 – 5mM). However, the intake of quinine was independent of light cycle. Post hoc analysis determined significant differences between light and dark at 21 – 42 per cent alcohol (a), 30 – 60mM of saccharin (c) and 5mM quinine (f). All data was analysed using a two-way ANOVA with Tukey post hoc. Summary values represented as mean±SEM; n=10, *p < 0.05; **p < 0.01, *** p < 0.001.
Figure 3. Circadian timing alters the relative preference for an alcohol-induced conditioned place preference.
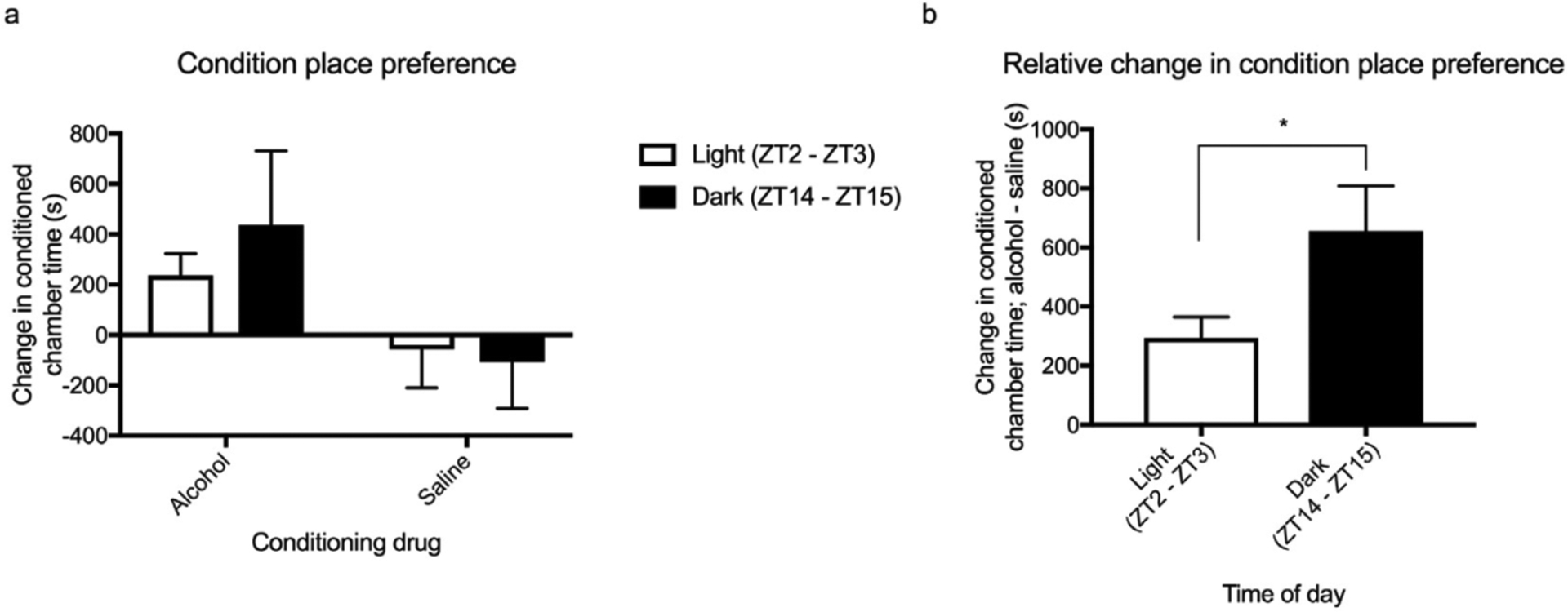
Light-cycle did not alter alcohol-induced conditioned place preference (a). However, when the relative change in conditioned chamber time was assessed, there was significantly greater preference towards alcohol during the dark compared to the light cycle (b). All data was analysed using a two-way ANOVA with Tukey post hoc (a) and a paired two-tail t-test (b). Summary values represented as mean±SEM; n=8, *p < 0.05.
Experiment 2: The effect of light-cycle on hunger-, reward-, thirst- and TLR4-related gene expression was analysed using a paired two-tail t-test (figures 4 and 5).
Figure 4. Circadian timing effects the expression of genes relating to reward (a), thirst and hunger (b).
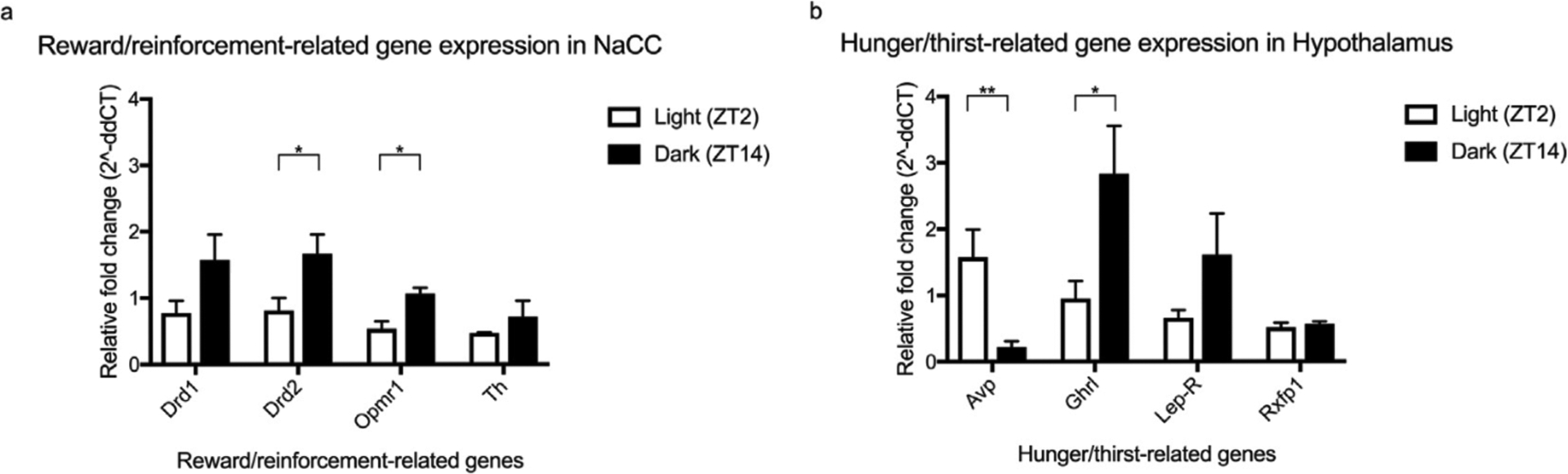
The expression of Drd2, Oprml and Ghrl were significantly elevated during the dark cycle compared to the light cycle. By contrast, the expression of Avp, was significantly elevated during the light compared to the dark cycle. The expression of Drd1, Th, Lepr and Rxfpl was unaffected by light-cycle. All data was analysed using a paired two-tail t-test. Summary values represented as mean±SEM; n=3, *p < 0.05; **p < 0.01.
Figure 5. Circadian timing effects the expression of TLR4-related genes in the nucleus accumbens (a) and hypothalamus (b).
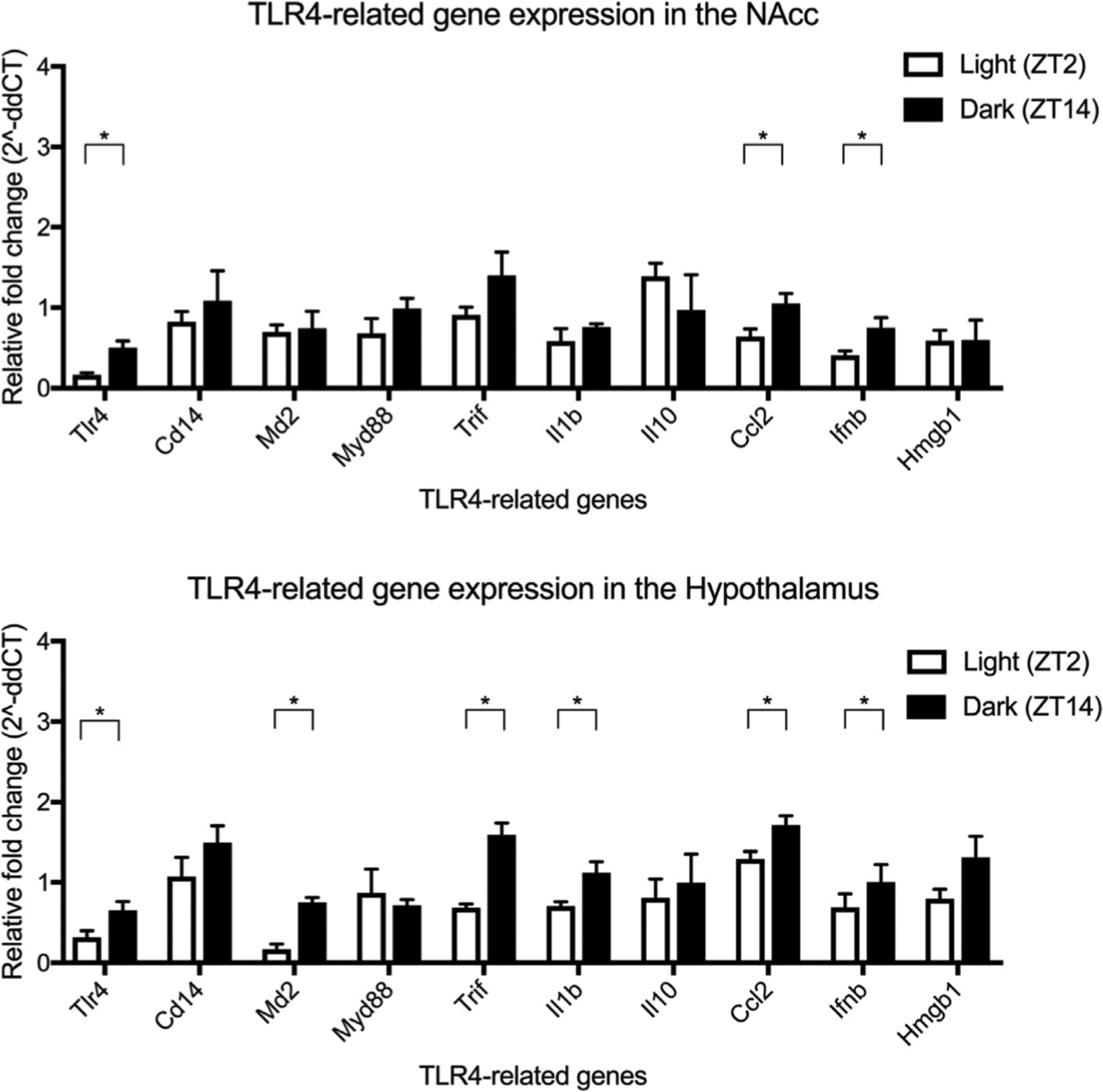
The expression of Tlr4, Ccl2 and Ifnb was significantly greater during the dark cycle compared to the light cycle in the nucleus accumbens. By contrast, the hypothalamus exhibited a more pronounced effect of circadian timing with greater expression of Tlr4, Md2, Trif, 111 b, Ccl2 and Ifnb during the dark cycle compared to the light cycle. All data was analysed using a paired two-tail t-test. Summary values represented as mean±SEM; n=3–4, *p < 0.05.
Experiment 3: The effects of light-cycle and the dose of (+)-Naltrexone on alcohol, saccharin and quinine intake and preference was analysed using two-way ANOVA with Tukey post hoc (figure 6). The effects of light-cycle and (+)-Naltrexone on the intake and preference for varying concentrations of alcohol, saccharin and quinine was analysed using a three-way ANOVA with Tukey post hoc (figure 7). The effects of light-cycle and (+)-Naltrexone on conditioned place preference and relative conditioned place preference was assessed using three-way AN OVA with Tukey post hoc and two-way ANOVA with Tukey post hoc respectively (figure 8 and s5).
Figure 6. Circadian timing influences the efficacy of (+)-Naltrexone on the intake and preference for alcohol (12%) (a – b), saccharin (30 mM) (c – d) and quinine (0.1 mM) (e – f).
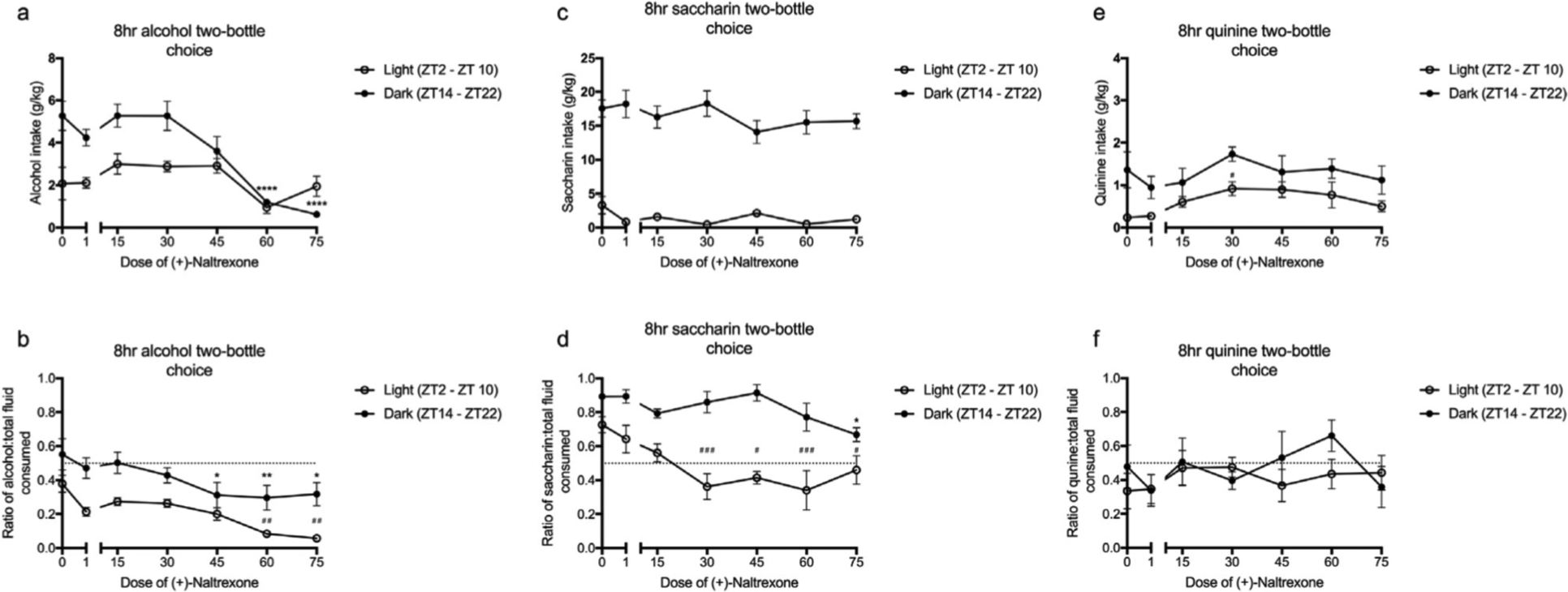
(+)-Naltrexone decreased the intake and preference for alcohol with a greater effect observed during the dark cycle compared to the light cycle as inferred by post hoc differences. (+)-Naltrexone did not affect the intake of saccharin, however, the drug significantly decreased the preference for saccharin between 30 – 75mgkg and 75mg/kg in the light and dark cycles respectively. The response to quinine was the opposite, with (+)-Naltrexone altering intake but not intake ratio. All data was analysed using a two-way ANOVA with Tukey post hoc. Summary values represented as meaniSEM; n=8–10, *p < 0.05, **p < 0.01, ***p <0.001, ****p <0.0001 compared to saline (dark); #p < 0.05 , ##p < 0.01, ###p <0.001, ####p <0.0001 compared to saline (light).
Figure 7. Circadian timing influences the efficacy of (+)-Naltrexone (60 mg/kg) on decreasing the intake and preference for alcohol (a – b) and saccharin (c – d) but not quinine (e – f).
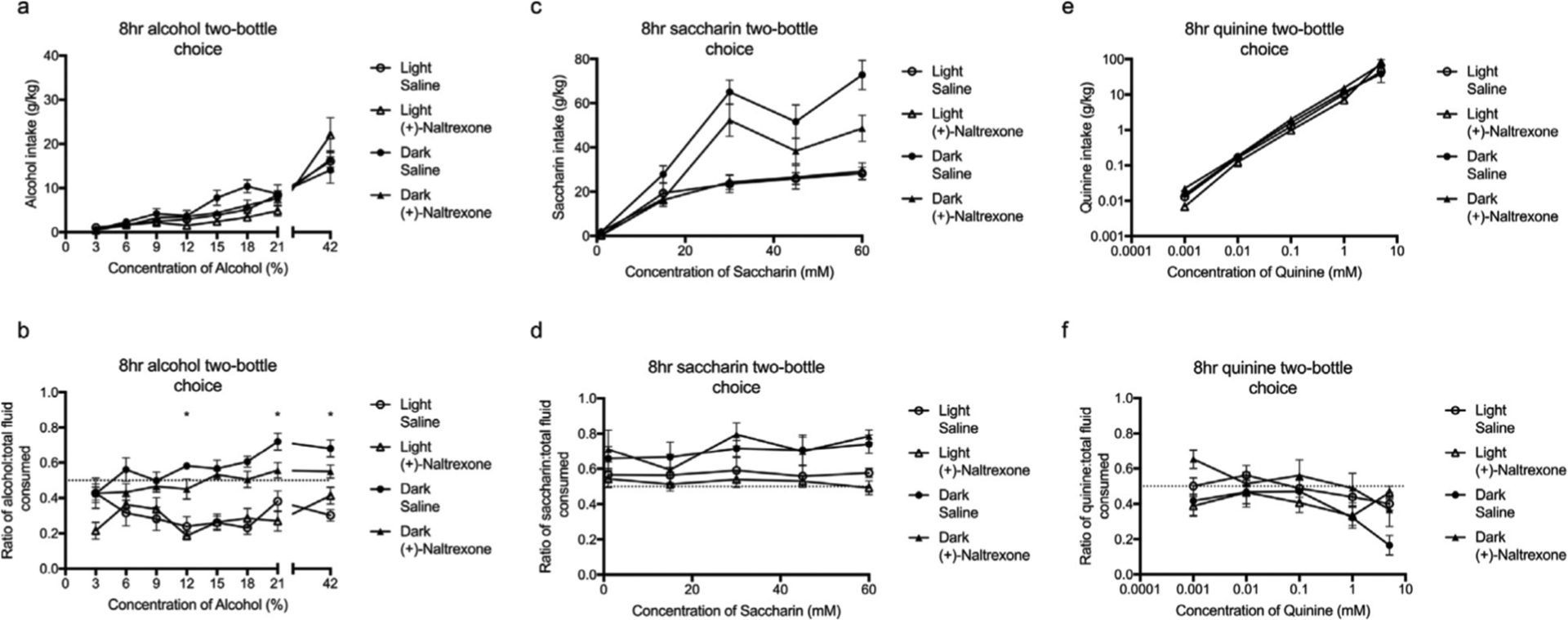
There was a main effect of pretreatment on the intake (a) and preference (b) for alcohol (3 – 42%) with (+)-Naltrexone exhibiting a greater effect during the dark cycle (post hoc analysis). Similarly, there was a main effect of pretreatment on intake (c) but not preference (d) for saccharin (1 – 60mM). Pretreatment had no effect on quinine intake (e) or intake ratio (f) (0.001 – 10mM). All data was analysed using a three-way ANOVA with Tukey post hoc Summary values represented as mean±SEM; n=10.
Figure 8. Circadian timing influences efficacy of (+)-Naltrexone on relative change in conditioned chamber time.
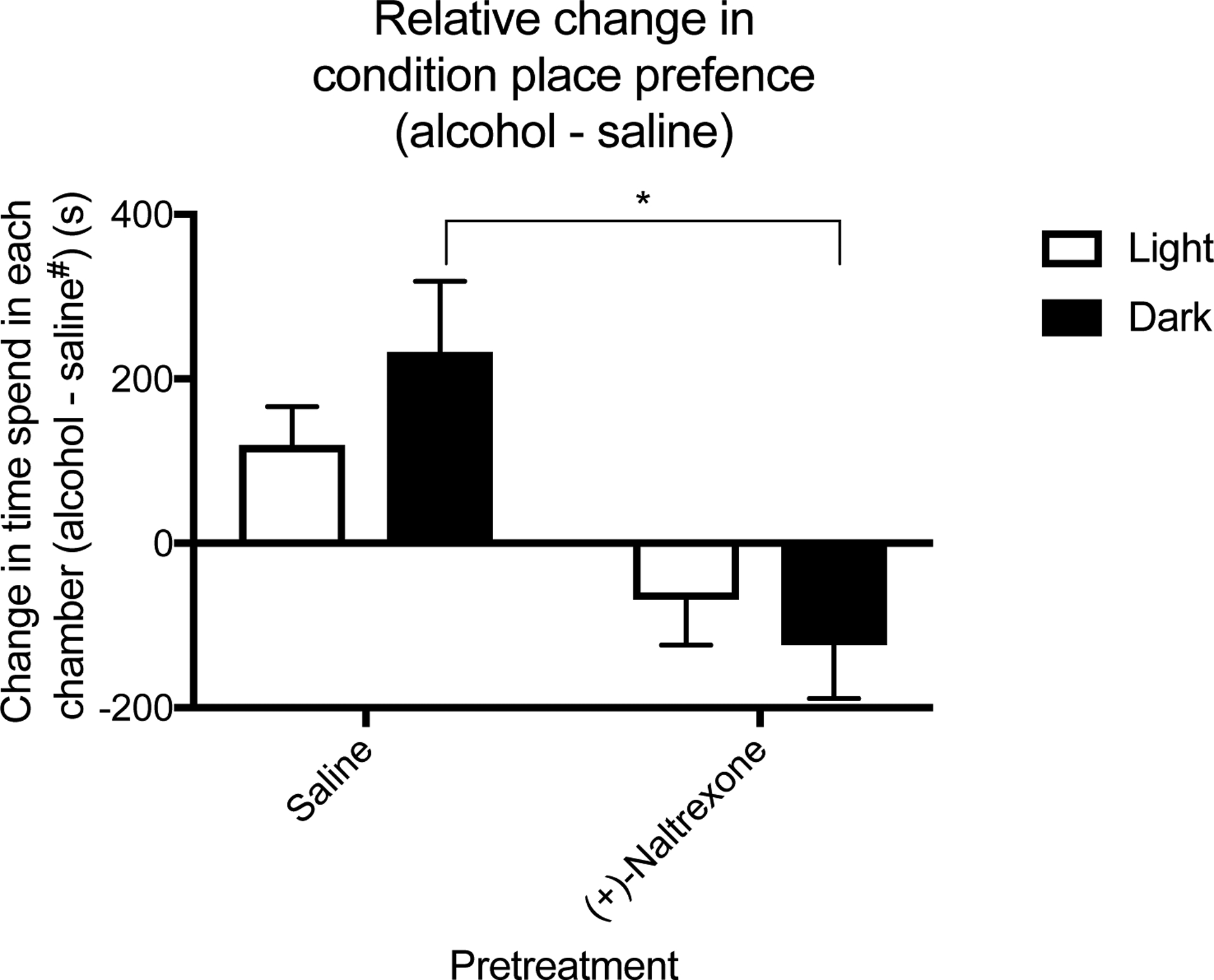
There was a significant effect of pretreatment on alcohol-induced conditioned place preference time with mice in the dark cycle exhibiting a significant reduction between saline and (+)-Naltrexone in conditioned place preference time. All data was analysed using a two-way ANOVA with Tukey post hoc. Summary values represented as mean±SEM; n=8, *p < 0.05. saline# = saline conditioned mice.
Experiment 4: The effect of light-cycle and (+)-Naltrexone on hunger-, reward-, thirst- and TLR4-related gene expression was analysed using a two-way ANOVA with Bonferonni post hoc (figures 9 and 10).
Figure 9. Circadian timing influences efficacy of (+)-Naltrexone on decreasing the mRNA expression of the TLR4-signaling pathway.
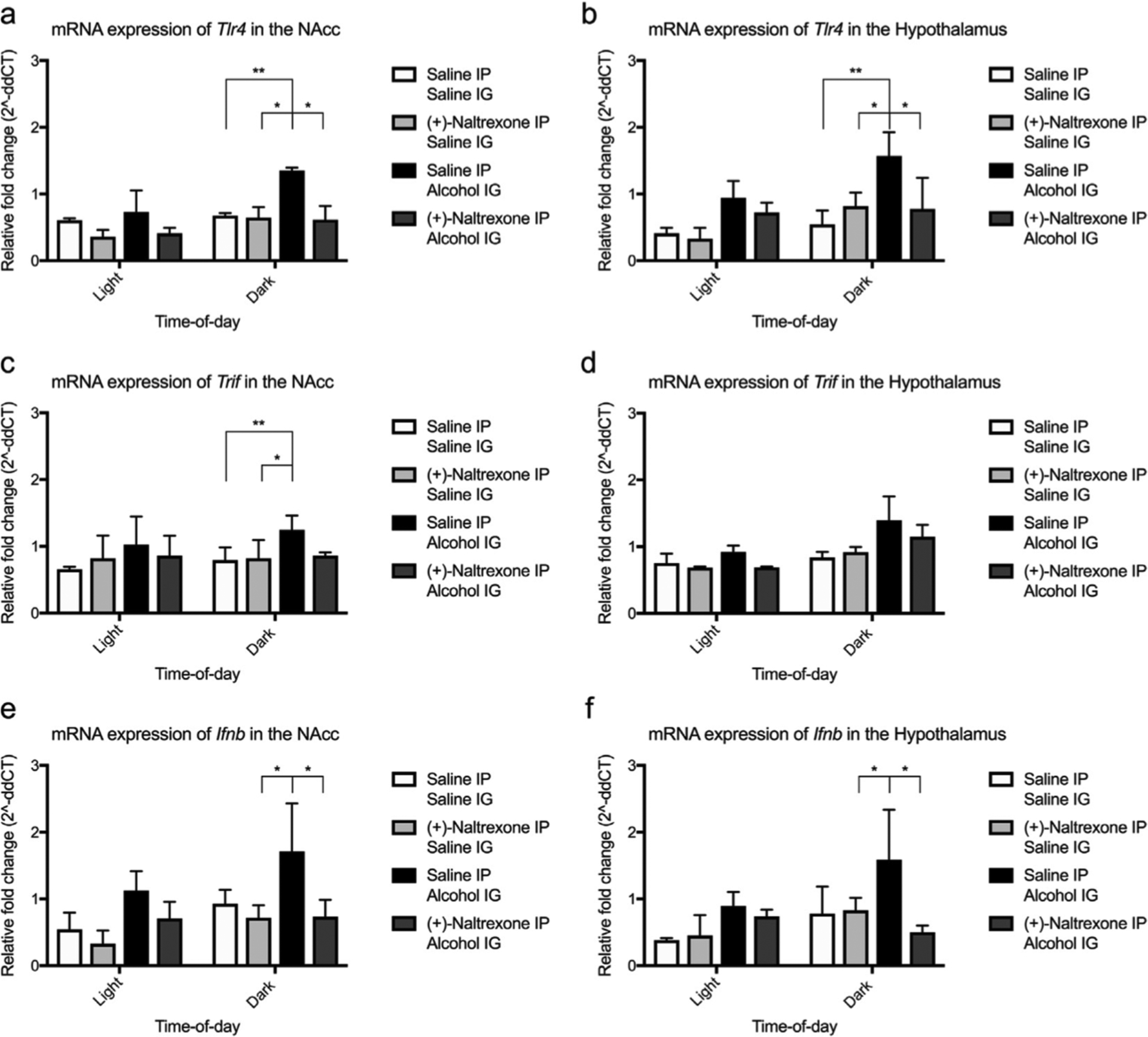
There was a significant effect of pretreatment on the expression of Tlr4 and Ifnb in the nucleus accumbens and Ifnb in the hypothalamus. Post hoc analyses determined differences between the groups were observed in the dark cycle only. All data was analysed using a two-way ANOVA with Bonferonni post hoc. Summary values represented as mean±SEM; n=3, *p < 0.05, **p < 0.01.
Figure 10. Circadian timing influences the efficacy of (+)-Naltrexone on decreasing the mRNA expression of tyrosine hydroxylase.
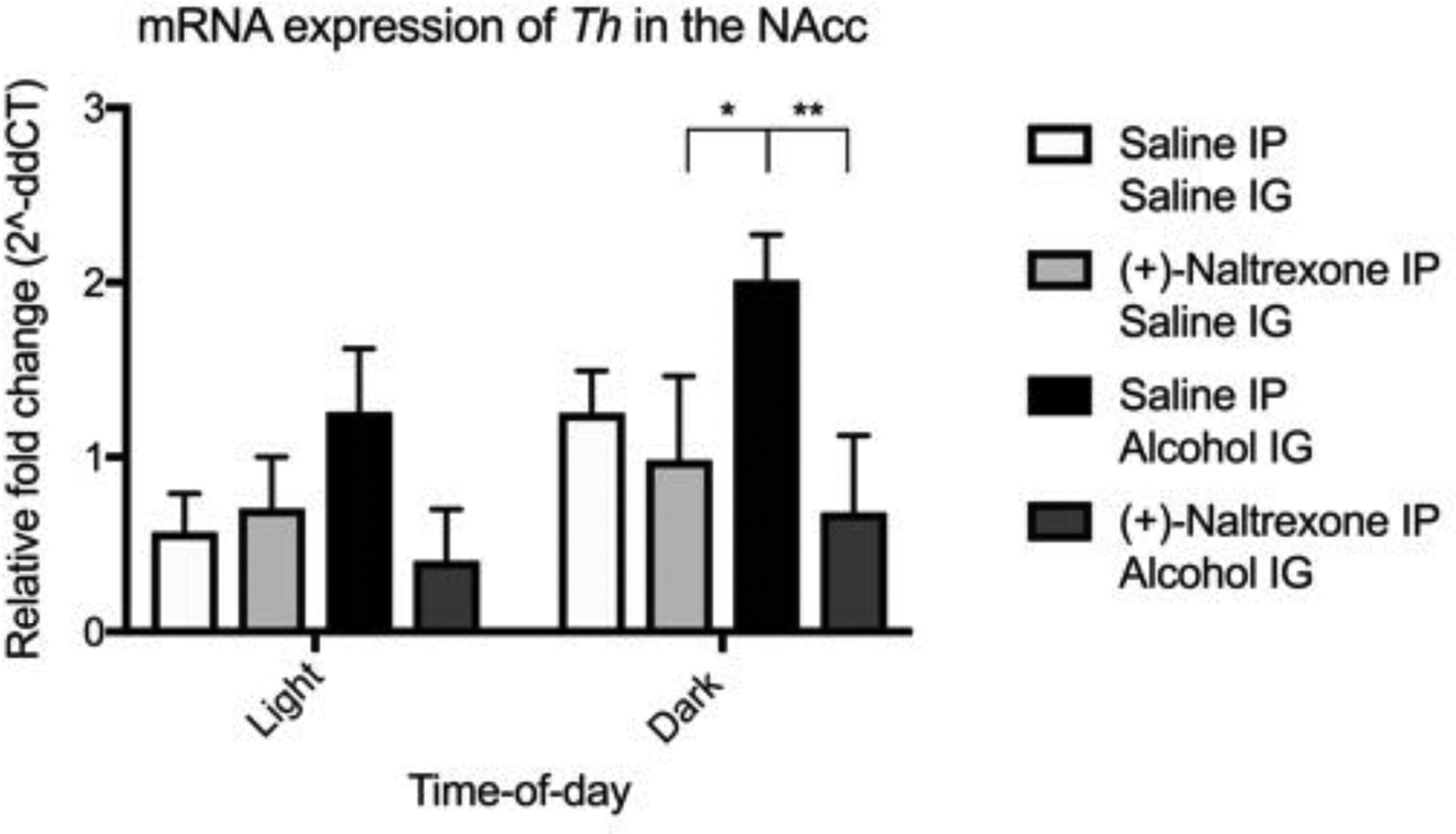
(+)-Naltrexone significantly reduced the expression of Th with its greatest effect observed during the dark cycle. All data was analysed using a two-way ANOVA with Bonferonni post hoc. Summary values represented as mean±SEM; n=3, *p < 0.05, **p < 0.01.
All summary values presented as mean ± standard error of mean (SEM). p-values ≤ 0.05 were considered statistically significant.
3.4. Results
3.4.1. Experiment 1: Are there light-cycle differences in reward-like behaviour?
To determine whether alcohol reward-like behaviour was dependent on light-cycle, mice underwent the two-bottle choice paradigm for 8 h during the light (ZT2 – ZT10) or dark cycle (ZT14 – ZT22) (figure 2a – b). A two-way ANOVA determined a significant effect of light-cycle on alcohol intake and preference (effect of light-cycle, F(1, 9) = 5.21, p =0.048 and F(1, 9) = 9.16, p =0.014, respectively). Post hoc analysis determined that mice exposed to alcohol during the dark cycle exhibited significantly greater intake and preference for alcohol compared to those in the light cycle at 21 and 42 per cent.
To determine whether the light-cycle dependent effect on alcohol intake and preference was due to an increased reward or decreased aversion, the preference for two compounds: saccharin, a sweet-tasting and rewarding compound; and quinine, a bitter-tasting and aversive compound, was assessed (figures 2c – f). Similar to alcohol, saccharin intake and preference was dependent on light-cycle (effect of light-cycle, F(1, 9) = 15.53, p = 0.0034 and F(1, 9) = 8.32, p = 0.015, respectively) with mice in the dark cycle exhibiting potentiated intake and preference compared to the light cycle at 30 and 60mM (post hoc analysis). However, the behavioural response to quinine was inconsistent. There was a significant effect of light-cycle for intake and the preference ratio (intake ratio) (effect of light-cycle, F(1, 9) = 4.72, p = 0.052 and F(1, 9) = 20.31, p = 0.0009, respectively). Overall, mice in the dark cycle exhibited greater intake but a reduced intake ratio for quinine. For all preceding tests the concentration of the solution (alcohol, saccharin or quinine) was a significant variable. The statistical values can be found in the supplementary material.
The light-cycle dependent effects observed in the alcohol, saccharin and quinine drinking tests may be attributable to thirst rather than reward. Indeed, a light-cycle dependent effect was found for water intake, with mice in the dark cycle exhibiting greater intake (supplementary material, figure s1, p = 0.0057). Thus, to control for this confounding variable, conditioned place preference, a reward/memory paradigm which is independent of thirst was used (figure 3a – b). The change in conditioned-chamber time was significantly affected by conditioning drug (effect of drug, F(1, 9) = 50.45, p = 0.004). However, there was no effect of light-cycle or an interactive effect (effect of light-cycle, F(1, 9) = 2.41, p =0.17; and interaction, F(1, 9) = 0.95, p = 0.37, respectively). However, if the relative change in conditioned place preference is considered (alcohol chamber time – saline chamber time), a significant light-cycle effect emerges. Mice in dark cycle exhibited a greater change in conditioned preference than those in the light cycle (effect of light-cycle, t= 2.17 df = 16, p = 0.047). Therefore, for the remaining experiments, relative conditioned chamber time was evaluated.
3.4.2. Experiment 2: Are there light-cycle differences in the molecular basis of reward-like behaviour?
The light-cycle dependent variations in preference and intake are potentially explained by the circadian oscillations in reward-, hunger- and thirst-related genes. Thus, the expression of these genes in the nucleus accumbens and hypothalamus was compared between light (ZT2) and dark (ZT14) cycles (figure 4a – b). A two-tail t-test determined significant light-dark cycle differences in the expression of: Drd2, Oprm1, Avp and Ghrl (effect of light-cycle, t = 1.77 df = 4, p = 0.11 and t = 3.96 df = 4, p = 0.0033; t = 3.18 df = 4, p = 0.0097 and t = 3.67 df = 4, p = 0.021, respectively). This effect was not ubiquitous among reward- and hunger-related genes, as light-cycle had no effect on the expression of Drd1, Th, Lepr and Rxfp1 (effect of light-cycle, t = 1.77 df = 9, p = 0.11; t = 1.00 df = 4, p = 0.071; t = 2.45 df = 4, p = 0.69 and t = 0.65 df = 4, p = 0.55, respectively).
Given the emerging role of the neuroimmune system in the manifestation of reward-like behaviour, the expression of the TLR4-signaling pathway was additionally assessed (figures 5a – b). Light cycle significantly influenced the expression of Tlr4 mRNA in the nucleus accumbens but not hypothalamus (effect of light-cycle, t = 3.9 df = 4, p = 0.019; and t = 2.97 df = 4, p = 0.069, respectively). By contrast, the expression of Ccl2 and Ifnb mRNA was significantly influenced by light-cycle in the nucleus accumbens (effect of light-cycle, t = 2.27 df = 4, p = 0.05; and t = 2.49 df = 4, p = 0.047, respectively) and hypothalamus (effect of light-cycle, t = 2.57 df = 4, p = 0.049 and t = 2.85 df = 4, p = 0.049, respectively). The effect of light-cycle was more pronounced in the hypothalamus with Md2, Trif and Il1b exhibiting light-cycle dependent effects as well (effect of light-cycle, t = 6.76 df = 4, p = 0.0025; t = 6.18 df = 4, p = 0.0035 and t = 4.0 df = 4, p = 0.043 respectively). No differences were observed in either the nucleus accumbens or hypothalamus for Cd14, Myd88, Il10, or Hmgb1 mRNA (see supplementary material for results of the statistical analysis).
3.4.3. Experiment 3: Does the efficacy of (+)-Naltrexone on attenuating the reward-like behaviour depend on the light-cycle?
Since light-cycle differences were observed in reward-like behaviour and TLR4-related gene expression, questions arose as to whether TLR4 was causatively involved in these effects, and if it was, were these events mediated by the TRIF- or MyD88-dependent pathway and would the efficacy of a TLR4-based intervention be dependent upon light-cycle. Given the expression of Ifnb mRNA was elevated in the nucleus accumbens and hypothalamus, it was hypothesised that the TRIF-pathway may be mediating these effects. Thus, (+)-Naltrexone, a biased TLR4-TRIF antagonist, was used in the following experiments. Interestingly, there was not an effect of (+)-Naltrexone on serum alcohol concentration following 2, 8 or 24 h two-bottle choice paradigms (effect of pretreatment, F(1, 5) = 0.070, p = 0.80, F(1, 5) = 1.59, p = 0.24 and F(1, 5) = 3.76, p = 0.088 respectively) (figure 4s a – c). However, post hoc analysis determined a significant difference in serum alcohol concentration between saline and (+)-Naltrexone treated mice during the dark cycle in the 8 but not 2 or 24 h tests. Further, there was there was a significant effect of (+)-Naltrexone on serum alcohol concentration following conditioned place preference (effect of pretreatment, F(1, 5) = 29.93, p < 0.0001). Again, post hoc analysis determined there was a significant difference between (+)-Naltrexone and saline mice during the dark cycle. Therefore, caution must be used when interpreting these studies as (+)-Naltrexone may modify alcohol metabolism following a bolus gavage.
For the following experiments characterising the light-cycle effects on the behavioural pharmacology of (+)-Naltrexone, a significant effect of light-cycle was observed for alcohol, saccharin and quinine intake and preference (figure 6a – f). Like earlier experiments, mice in the dark cycle exhibited significantly greater intake and preference for alcohol and saccharin (supplementary data).
Overall, there was a significant effect of (+)-Naltrexone’s dose on the intake and preference for 12 per cent alcohol (effect of dose, F(6, 48) = 15.72, p < 0.0001 and F(6, 48) = 7.57, p < 0.0001, respectively) (figure 6a and b) with a significant interactive effect observed for alcohol intake but not preference (interaction, F(6, 48) = 4.98, p = 0.0005 and F(6, 48) = 0.63, p = 0.70, respectively). Post hoc analysis determined that mice in the dark cycle exhibited a significant reduction in intake and preference from 45 – 75 mg/kg doses of (+)-Naltrexone relative to saline. In comparison, mice in the light cycle exhibited a reduction in preference but not intake at doses of 60 – 75 mg/kg.
The preference but not intake of saccharin was significantly modified by (+)-Naltrexone’s dose (effect of dose, F(6, 48) = 3.82, p = 0.0034 and F(6, 48) = 0.56, p = 0.76 respectively) (figure 6c – d). Further, both intake and preference demonstrated interactive effects between (+)-Naltrexone and light-cycle (interaction, F(6, 48) = 2.01, p = 0.024 and F(6, 48) = 3.65 p < 0.0046, respectively). Post hoc analysis further demonstrated a significant reduction in saccharin preference in the light-cycle between 30 and 75 mg/kg dose of (+)-Naltrexone - an effect absent in the dark cycle. By contrast, quinine intake but not the intake ratio was significantly affected by (+)-Naltrexone (effect of dose, F(6, 48) = 3.05, p = 0.013 and F(6, 48) = 0.79, p = 0.58, respectively). There were no interactive effects for intake or the intake ratio (interaction, F(6, 48) = 1.7, p = 0.15 and F(6, 48) = 1.2, p = 0.28, respectively). Collectively, the results suggest (+)-Naltrexone attenuates the intake and preference of alcohol. However, this action of (+)-Naltrexone may be due to non-specific effects as saccharin and quinine intake were significantly perturbed as well.
To further the findings of light-cycle-dependent effects of (+)-Naltrexone on alcohol intake and preference, 60 mg/kg dose of (+)-Naltrexone was administered to mice and their preference for differing concentrations of alcohol was examined. These tests are important, given different mechanisms are thought to govern the responses to low and high doses of alcohol (Kiefer, 1995).
Light-cycle and concentration were found to be significant variables influencing preference and intake for alcohol, saccharin and quinine (p < 0.05) (see supplementary data). There was a significant effect of pretreatment on the intake and preference for alcohol (effect of pretreatment, F(1, 344) = 4.95, p = 0.027 and F(1, 344) = 13.58, p = 0.00027) (figure 7a – b). No interactive effect were found for intake or preference. However, post hoc analysis determined (+)-Naltrexone significantly reduced alcohol preference at 12, 21 and 42 per cent alcohol compared to saline. There were no post hoc differences between (+)-Naltrexone and saline during the light phase for intake or preference of alcohol.
There was a main effect of pretreatment on saccharin intake but not preference (effect of pretreatment, F(1, 220) = 8.95, p = 0.0031 and F(1, 220) = 0.25, p = 0.62, respectively) (figure 7c – d). A significant interactive effect was observed for saccharin intake (light-cycle x treatment, F(4, 220) = 9.11, p = 0.0026). However, this effect was not observed for saccharin preference (F(1, 220) = 1.37, p = 0.24). Further, there were no post hoc differences between the groups for intake or preference. By contrast, quinine intake and the intake ratio were unaffected by pretreatment (effect of pretreatment, F(1, 220) = 4.315, p = 0.09 and F(1, 220) = 2.01, p = 0.16, respectively) (figure 7e – f). There were no interactive effects or post hoc differences for quinine intake and the intake ratio.
To provide further evidence indicating TLR4-TRIF involvement in reward/reinforcing behaviour, conditioned place preference was assessed (figure 8). Pretreatment significantly modified relative alcohol-induced conditioned place preference (effect of pretreatment, F(1, 7) = 20.52, p = 0.0027). Post hoc analysis determined (+)-Naltrexone significantly decreased relative alcohol-induced conditioned place preference time compared saline during the dark only. There was no effect of light-cycle, nor an interaction between light-cycle and pretreatment (effect of light-cycle, F(1, 7) = 0.0011, p = 0.92; and interaction, F(1, 7) = 1.62, p = 0.24).
(+)-Naltrexone was additionally screened against a 24 h two-bottle choice and drinking in the dark paradigm. Both paradigms found a significant effect of (+)-Naltrexone with post hoc analysis demonstrating a significant effect of (+)-Naltrexone on alcohol intake during the dark but not light cycle. See supplementary materials for figures and precise statistical information.
3.4.4. Experiment 4: Does the efficacy of (+)-Naltrexone on attenuating the TLR4 pathway depend on the light-cycle?
qPCR was used to identify potential mechanisms underpinning the behavioural changes induced by alcohol and (+)-Naltrexone. There were significant effects of light-cycle, pretreatment ((+)-Naltrexone vs. saline) and drug (alcohol vs. saline) for Tlr4 and Ifnb mRNA expression in the nucleus accumbens (Tlr4, effect of light cycle, F(1, 16) = 11.79, p = 0.0034; pretreatment, F(1, 16) = 7, p = 0.022; and drug, F(1, 16) = 6.49, p = 0.021) (Ifnb light-cycle, F(1, 16) = 9.09 p = 0.0083; pretreatment, F(1, 16) = 8.26, p = 0.010; and drug, F(1, 16) = 13.69, p = 0.0019). Within the hypothalamus only Ifnb exhibited a significant effect of light-cycle (F(1, 16) = 8.92, p = 0.0087), pretreatment (F(1, 16) = 13.63, p = 0.020) and drug (F(1, 16) = 9.54, p = 0.007). Tlr4 expression was significantly influenced by light-cycle (F(1, 16) = 9.45, p = 0.0073), drug (F(1, 16) = 20.14, p = 0.004) but not pretreatment (F(1, 16) = 3.71, p = 0.072). Post hoc analysis furthered these findings as (+)-Naltrexone attenuated alcohol-induced increases in Tlr4 and Ifnb mRNA expression in the dark but not light cycle.
Interestingly, (+)-Naltrexone did not affect the expression of Trif in the nucleus accumbens or hypothalamus (NAcc, effect of light-cycle, F(1, 16) = 0.71, p = 0.41; pretreatment, F(1, 16) = 4.46, p = 0.053; and drug, F(1, 16) = 0.71, p = 0.41) (hypo light-cycle, F(1, 16) = 23.23, p = 0.0002; pretreatment, F(1, 16) = 3.21, p = 0.092; and drug, F(1, 16) = 1.36, p = 0.26). All remaining TLR4-related genes did not exhibit a significant effect of all three variables (statistical information are available in the supplementary material, see figures s5 – 6).
Interestingly, while there was an effect of pretreatment on the expression of Lepr (F(1, 16) = 6.44, p = 0.022) and Rxfp1 mRNA (F(1, 16) = 6.01, p = 0.026) there was no effect of drug nor light-cycle (see supplementary). However, there was a significant effect of light-cycle (F(1, 16) = 13.15 p = 0.0023), pretreatment (F(1, 16) = 17.76, p = 0.0007) but not drug (F(1, 16) = 2.38, p = 0.14) on the expression of Th mRNA (figure 10). Post hoc analysis determined a (+)-Naltrexone significantly reduced alcohol-potentiated Th mRNA expression compared to saline in the dark but not light cycle.
3.5. Discussion
The current study demonstrates that the intake and preference for alcohol, saccharin and quinine fluctuate according to the time-of-day. The preference for alcohol and saccharin peaked during the dark phase, while quinine preference was greatest during the light phase. This effect coincided with elevations in reward-, thirst- and immune-related genes. This study further highlighted that the efficacy of (+)-Naltrexone, a biased TLR4 antagonist, on attenuating alcohol-induced immune signalling and alcohol preference is dependent on the light-cycle, with the greatest effect again observed in the dark cycle. However, (+)-Naltrexone additionally reduced saccharin intake and preference. These effects are potentially attributable to (+)-Naltrexone’s down regulation of Th mRNA. Given T5342126, a TLR4-MD2 disruptor, additionally attenuates alcohol and saccharin intake (Bajo et al., 2016), the studies collectively indicate a pivotal link between TLR4 and natural reward-like behaviours.
The effects of the circadian influence on reward and drug seeking behaviour have recently received renewed interest (see Parekh et al., 2015; Perreau-Lenz & Spanagel, 2015; Webb et al., 2015 for review). Earlier studies indicated rodents have a higher preference and intake of alcohol during the dark cycle (Gauvin et al., 1997). Interestingly, the time of heightened sensitivity towards drugs of abuse appears to be unique to each class, as cocaine exhibits its greatest rewarding effects during the day (Kurtuncu et al., 2004). Results presented in this study reinforce the importance of the light-cycle with respect to alcohol reward-like behaviour. Despite higher water intake during the dark cycle, mice in the dark cycle exhibited greater preference and intake of alcohol compared to those in the light cycle. These findings were furthered as mice exhibited relatively higher conditioned place preference towards alcohol during the dark cycle compared to the light. The increased intake and preference for alcohol however, are potentially attributable to either an increase in the rewarding- or a decrease in the aversive properties of alcohol. To control for this possibility, the intake of saccharin; a sweet non-calorific, non-alcoholic rewarding solution, and quinine; an aversive, bitter solution, was measured. Mice displayed light-cycle-dependent differences in saccharin intake and preference, with the greatest preference observed during the dark cycle. By contrast, the lowest intake ratio for quinine was during the dark cycle. As mice in the dark cycle exhibited enhanced and reduced preference towards saccharin and quinine, respectively. It is difficult to determine whether the increased preference of alcohol was due to increased reward or reduced aversion. Further, one cannot rule out the possibility of alcohol as an energy source acting as a motivator for increase in preference and intake.
Previous studies have identified circadian differences in nucleus accumbens and ventral tegmental area (key reward-related regions) in terms of gene and protein expression and the activity of dopaminergic- and non-dopaminergic neurons (Hampp et al., 2008; Sleipness et al., 2008; Sleipness et al., 2007). Our results are in accordance with these findings; the expression of dopamine and opioid receptors and tyrosine hydroxylase mRNA in the nucleus accumbens was increased during the dark cycle. The elevated levels of reward-related genes (if translated into protein) may enhance an individual’s sensitivity towards alcohol (Mendez & Morales-Mulia, 2008; Gianoulakis, 2001). In addition, we observed light-cycle dependent expression in genes related to thirst and hunger in the hypothalamus. The fluctuations in vasopressin and leptin mRNA may additionally drive the intake of alcohol (Pickering et al., 2007; Wurst et al., 2007). The day-night differences in reward-related gene expression are attributable to multiple circuit-level and molecular mechanisms. For example, the SCN innervates the reward pathway via glutamatergic afferents from the medial prefrontal cortex regulating reward behaviour (Baltazar et al., 2014; Baltazar et al., 2013); and dopamine transporter, dopamine D1 and D2 receptors, monoamine oxidase and tyrosine hydroxylase – key proteins regulating dopamine synthesis and reward, contain BMAL1 and CLOCK binding sites in their promoter regions (Webb et al., 2009; Hampp et al., 2008; Sleipness et al., 2008; Sleipness et al., 2007; McClung et al., 2005; Akhisaroglu et al., 2005). This suggests the circadian clock controls aspects of dopaminergic activity including neurotransmitter synthesis, release, degradation and postsynaptic actions. No study has determined whether the μ opioid receptor or any of its endogenous agonists (associated with the “liking” component of reward) are under the control of clock proteins in the nucleus accumbens or VTA.
Given the emerging role of the immune system in reward-like behaviour, the expression of the TLR4 pathway was additionally examined. Interestingly, a light-cycle dependent effect was observed for some, but not all TLR4-related genes in the nucleus accumbens and hypothalamus. Both these regions exhibited increases in Tlr4, Ifnb and Ccl2 mRNA during the dark cycle. However, the hypothalamus reported additional light-dark differences in Md2, Trif and Il1b expression. These findings are in contrast to Fonken et al., (2015) who observed that isolated microglia exhibit peaks in inflammatory gene expression during the light cycle. There are however, numerous differences in terms of study design between the present study and Fonken et al., (2015), which may explain these differences (genes and cells examined, in vivo vs ex vivo tissue and methods of analysis). The present findings are similar to studies examining circadian influence on peripheral immune cells. For example, peripheral macrophages exhibit an increase in TLR4-related mRNA during the dark (active) cycle (Keller et al., 2009). Nevertheless, these data point to regional specific circadian control of brain innate immune reactivity. Like the reward-related gene expression, the oscillations in TLR4-related gene expression are driven by sympathetic and parasympathetic effects from the SCN and the molecular clock mechanisms. For example, BMAL1-CLOCK binds to E-boxes in the promoters of chemokine genes (Nguyen et al., 2013); CLOCK can directly interact with p65 subunit of NFκB enhancing its activity (Spengler & Kuropatwinski, 2012); and glucocorticoid receptors bind to NFκB and AP-1 repressing their activities (Dickmeis et al., 2013; Coutinho & Chapman 2011). These regulatory processes act in a concerted manner, temporally gating specific parts of the immune response to distinct times of the day.
The role of TLR4 in regulating cocaine- and opioid-induced reward is well established (Northcutt et al., 2015; Hutchinson et al., 2012). However, for TLR4’s impact on alcohol pharmacodynamics, there are conflicting evidence with studies demonstrating either no effect or a reduction in alcohol drinking and reward-like behaviour (Blednov et al., 2017; Harris et al., 2017; Aurelian et al., 2016; Bajo et al., 2016; June et al., 2015; Liu et al., 2011; Pascual et al., 2011). The differential results are likely attributable to differences in brain regions examined, models of alcohol exposure and species examined. However, only two of the preceding studies have considered whether the differences (or lack thereof) are attributable to activation of different TLR4-signaling pathways (TRIF or MyD88) (Blednov et al., 2017; Harris et al., 2017). Given the light-cycle differences in the expression of Ifnb mRNA, and previous work establishing a causal relationship between interferon signalling and excessive alcohol use (Duncan et al., 2016; Manzardo et al., 2016; Johnson et al., 2015), (+)-Naltrexone, a biased TLR4-TRIF antagonist was used to explore the role of TLR4-TRIF signalling on light-cycle dependent differences in alcohol drinking and reward behaviour.
(+)-Naltrexone significantly attenuated alcohol intake and preference across a range of doses, alcohol concentrations and testing times. However, the response was mixed. While there were significant effects regarding the dose of (+)-Naltrexone on intake and preference, post hoc analysis determined the differences were most pronounced during the dark cycle. Similarly, the reduction in relative alcohol-induced conditioned place preference by (+)-Naltrexone was only statistically significant during the dark cycle. These two paradigms would infer that (+)-Naltrexone attenuates alcohol-induced reward-like behaviour with the greatest effect during the dark cycle. However, the results are confounded. (+)-Naltrexone significantly modified saccharin (but not quinine) intake and preference. Therefore, (+)-Naltrexone may act as an antagonist towards all rewarding compounds, rather than one specific to alcohol. While this finding contrasts Northcutt et al., (2015), they are congruent with the actions of other pharmacological TLR4 antagonists such as T5342126 (Bajo et al., 2016). Hence, there appears to be a critical circadian-TLR4 signalling involvement in the rewarding properties of multiple diverse agents.
The findings presented in this manuscript add to the growing body of evidence aimed at elucidating the precise function of each of the TLR4-signaling pathways in alcohol-reward behaviour. Interestingly, our findings largely contrast those by Harris et al., (2017) who demonstrated a lack of effect of (+)-Naloxone, a chemically-related compound and TLR4-TRIF antagonist, on alcohol drinking behaviour in naïve mice. However, Harris et al., (2017) observed a significant effect of (+)-Naloxone in paradigms designed to mimic excessive drinking. This would suggest TLR4-TRIF is involved in the chronic but not acute effects of alcohol. In addition to the TRIF pathway, TLR4 signals via MyD88 raising the possibility that the acute effects of alcohol are mediated by this pathway as well. Recent evidence has shown naïve MyD88−/− mice exhibit potentiated alcohol intake compared to wildtype mice (Blednov et al., 2017). On the surface, this may suggest MyD88 is a negative regulator of alcohol-reward behaviour. However, MyD88−/− mice also display reduced saccharin intake (Blednov et al., 2017) and opioid-induced reward (Hutchinson et al., 2012), suggesting that MyD88−/− mice may find alcohol less rewarding than wildtype mice and therefore must consume greater quantities to achieve the same pharmacological effect. Collectively, these studies and ours highlights the growing appreciation that the individual TLR4-signalling pathways play a unique role in alcohol reward.
To identify potential mechanisms underlying (+)-Naltrexone’s ability to attenuate alcohol-reward like behaviour, genes relating to reward and the immune system within the nucleus accumbens and hypothalamus were again examined. Alcohol increased the expression of genes relating to the TLR4 pathway. Specifically, a rise in Tlr4, Cd14, Md2, Trif, Myd88, Ccl2, Hmgb1 and Ifnb mRNA expression was observed. This indicates an acute moderate dose of alcohol upregulates markers of the MyD88 and the TRIF pathway. Only Tlr4 and Ifnb, however, reported additional light-cycle and pretreatment effects. There was a significant decrease in the expression of Tlr4 and Ifnb mRNA following (+)-Naltrexone. However, like the behavioural tests, only significant post hoc differences were found in the dark cycle. Again, highlighting that the largest effect of (+)-Naltrexone occurred during the dark cycle. The dark cycle effect may be due to a floor effect. That is, because TLR4 expression is relatively lower during the light cycle, an antagonist may be unable to reduce the signalling and expression further. By contrast, when the expression is comparatively higher (during the dark cycle), the antagonist now appears to exert an effect. This extends to conclusions about TLR4s involvement in reward-like behaviour. During the light cycle, TLR4 expression was low and therefore, TLR4 may exert a smaller effect on reward behaviour compared to during the dark when its expression was the highest.
No genes associated with hunger or thirst were significantly altered by alcohol in the hypothalamus. By contrast, alcohol significantly potentiated the expression of Oprm1 and Th mRNA in the nucleus accumbens. These genes are pivotally involved in the manifestation of reward-like behaviour (Alves et al., 2015; Charbogne et al., 2014; Webb et al., 2009). However, only the expression of Th mRNA was significantly altered by light-cycle and pretreatment – suggesting a link between TLR4 and tyrosine hydroxylase (TH) (figure 10). This study is not the first to highlight a potential link between TLR4 and TH as Aurelian et al., (2016) determined TLR4 activation induces the expression of TH in VTA dopaminergic neurons via a PKA/pCREB signal. Work from the present study builds upon this connection suggesting that either IFNβ or CCL2 may underlie this link given both inflammatory mediators demonstrated a significant effect of pretreatment. Interestingly, all three inflammatory mediators (IFNβ, CCL2 and TLR4) can signal through PKA and CREB (Akira &Takeda, 2004), leading to altered transcription of Th mRNA. Importantly the downregulation of Th mRNA following (+)-Naltrexone potentially explains the broad effects (decreased saccharin preference) observed with TLR4 antagonists. Tyrosine hydroxylase is the rate limiting enzyme of catecholamine synthesis, catalysing the conversion of tyrosine to L-DOPA, a precursor molecule for dopamine (see Daubner et al., (2011) for review). Consequently, reducing its transcription using (+)-Naltrexone may reduce basal dopamine level. Thus, mice experience reduced rewarding sensations upon consuming saccharin and alcohol. Collectively, the results highlight the importance of TLR4 in regulating basal dopamine synthesis and implicates the TLR4 system in the rewarding properties of multiple diverse agents.
In summary, the results highlighted above suggest the preference for rewarding and aversive stimuli peak and nadir during the dark cycle respectively. This effect coincides with elevations in genes relating to dopaminergic and opioidergic transmission and the TLR4-signalling pathway. Attenuating the TRIF component of the TLR4-signalling pathway significantly reduced alcohol preference, with a greater effect during the dark cycle. Saccharin preference was additionally reduced by TLR4 blockade– an effect potentially attributable to a reduction in Th mRNA. Given that antagonism of TLR4 reduced alcohol- and saccharin preference and tyrosine hydroxylase mRNA, TLR4 may play a role in the dopamine synthesis and natural reward-like behaviour. Further research is required to establish how these preclinical studies translate to the human condition, and whether future pharmacological targeting of neuroimmune systems generally (Ray et al., 2017) or TLR4 specifically, may need to be timed specifically to a light cycle. Moreover, these data point to a significant impact on the brain of time-of-day on long term impact of alcohol exposure.
Supplementary Material
3.6. Acknowledgements
This research was support by grants Australian Research Council Research Fellowship (DP110100297). A portion of this work was supported by the NIH Intramural Research Programs of the National Institute on Drug Abuse (NIDA) and the National Institute of Alcohol Abuse and Alcoholism.
Footnotes
The authors declare no competing financial interests.
3.7 References
- Akhtar RA, Reddy AB, Maywood ES, Clayton JD, 2002. Circadian cycling of the mouse liver transcriptome, as revealed by cDNA microarray, is driven by the suprachiasmatic nucleus. Current Biology. [DOI] [PubMed] [Google Scholar]
- Akira S, Takeda K, 2004. Toll-like receptor signalling. Nat Rev Immunol 4, 499–511. doi: 10.1038/nri1391 [DOI] [PubMed] [Google Scholar]
- Aurelian L, Warnock KT, Balan I, Puche A, June H, 2016. TLR4 signaling in VTA dopaminergic neurons regulates impulsivity through tyrosine hydroxylase modulation 6, e815–9. doi: 10.1038/tp.2016.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachtell R, Hutchinson MR, Wang X, Rice KC, Maier SF, Watkins LR, 2015. Targeting the Toll of Drug Abuse: The Translational Potential of Toll-Like Receptor 4. CNS Neurol Disord Drug Targets 14, 692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajo M, Montgomery SE, Cates LN, Nadav T, Delucchi AM, Cheng K, Yin H, Crawford EF, Roberts AJ, Roberto M, 2016. Evaluation of TLR4 Inhibitor, T5342126, in Modulation of Ethanol-Drinking Behavior in Alcohol-Dependent Mice. Alcohol and Alcoholism 51, 541–548. doi: 10.1093/alcalc/agw026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltazar RM, Coolen LM, Webb IC, 2013. Diurnal rhythms in neural activation in the mesolimbic reward system: critical role of the medial prefrontal cortex. European Journal of Neuroscience 38, 2319–2327. doi: 10.1111/ejn.12224 [DOI] [PubMed] [Google Scholar]
- Bardo MT, Bevins RA, 2000. Conditioned place preference: what does it add to our preclinical understanding of drug reward? Psychopharmacology 153, 31–43. doi: 10.1007/s002130000569 [DOI] [PubMed] [Google Scholar]
- Bass J, Lazar MA, 2016. Circadian time signatures of fitness and disease. Science 354, 994–999. doi: 10.1126/science.aah4965 [DOI] [PubMed] [Google Scholar]
- Blednov YA, Black M, Chernis J, Da Costa A, Mayfield J, & Harris RA 2017. Ethanol Consumption in Mice Lacking CD14, TLR2, TLR4, or MyD88. Alcoholism: Clinical and Experimental Research, 1–15. 10.1111/acer.13316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Lee EJ, Yun S, Choe HK, Park S-B, Son HJ, Kim K-S, Dluzen DE, Lee I, Hwang O, Son GH, Kim K, 2014. Impact of Circadian Nuclear Receptor REV-ERBα on Midbrain Dopamine Production and Mood Regulation. Cell 157, 858–868. doi: 10.1016/j.cell.2014.03.039 [DOI] [PubMed] [Google Scholar]
- Crews FT, Lawrimore CJ, Walter TJ, Coleman LG Jr, 2017. The role of neuroimmune signaling in alcoholism. Neuropharmacology 1–18. doi: 10.1016/j.neuropharm.2017.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui C, Shurtleff D, Harris RA, 2014. Neuroimmune Mechanisms of Alcohol and Drug Addiction, in: Neuroimmune Signaling in Drug Actions and Addictions, International Review of Neurobiology. Elsevier, pp. 1–12. doi: 10.1016/B978-0-12-801284-0.00001-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dardente H, Cermakian N, 2007. Molecular circadian rhythms in central and peripheral clocks in mammals. Chronobiol. Int 24, 195–213. doi: 10.1080/07420520701283693 [DOI] [PubMed] [Google Scholar]
- Daubner SC, Le T, Wang S, 2011. Tyrosine hydroxylase and regulation of dopamine synthesis. Archives of Biochemistry and Biophysics 508, 1–12. doi: 10.1016/j.abb.2010.12.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffield GE, Best JD, Meurers BH, Bittner A, Loros JJ, Dunlap JC, 2002. Circadian programs of transcriptional activation, signaling, and protein turnover revealed by microarray analysis of mammalian cells. Curr. Biol 12, 551–557. [DOI] [PubMed] [Google Scholar]
- Duncan JW, Johnson S, Zhang X, Zheng B, Luo J, Ou X-M, Stockmeier CA, Wang JM, 2016. Up-Regulation of PKR Signaling Pathway by Ethanol Displays an Age of Onset-Dependent Relationship. Alcohol Clin Exp Res 40, 2320–2328. doi: 10.1111/acer.13209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Lizarbe S, Montesinos J, Guerri C, 2013. Ethanol induces TLR4/TLR2 association, triggering an inflammatory response in microglial cells. Journal of Neurochemistry 126, 261–273. doi: 10.1111/jnc.12276 [DOI] [PubMed] [Google Scholar]
- Fernandez-Lizarbe S, Pascual M, Guerri C, 2009. Critical Role of TLR4 Response in the Activation of Microglia Induced by Ethanol. The Journal of Immunology 183, 4733–4744. doi: 10.4049/jimmunol.0803590 [DOI] [PubMed] [Google Scholar]
- Ferris MJ, España RA, Locke JL, Konstantopoulos JK, Rose JH, Chen R, Jones SR, 2014. Dopamine transporters govern diurnal variation in extracellular dopamine tone. Proc Natl Acad Sci USA 111, E2751–E2759. doi: 10.1073/pnas.1407935111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonken LK, Frank MG, Kitt MM, Barrientos RM, Watkins LR, Maier SF, 2015. Microglia inflammatory responses are controlled by an intrinsic circadian clock. Brain Behavior and Immunity 45, 171–179. doi: 10.1016/j.bbi.2014.11.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauvin DV, Baird TJ, Vanecek SA, Briscoe RJ, Vallett M, Holloway FA, 1997. Effects of time-of-day and photoperiod phase shifts on voluntary ethanol consumption in rats. Alcohol Clin Exp Res 21, 817–825. [PubMed] [Google Scholar]
- Gianoulakis C 2001. Influence of the endogenous opioid system on high alcohol consumption and genetic predisposition to alcoholism. Journal of Psychiatry & Neuroscience : JPN, 26(4), 304–318. [PMC free article] [PubMed] [Google Scholar]
- Harris RA, Bajo M, Bell RL, Blednov YA, Varodayan FP, Truitt JM, de Guglielmo G, Lasek AW, Logrip ML, Vendruscolo LF, Roberts AJ, Roberts E, George O, Mayfield J, Billiar TR, Hackam DJ, Mayfield RD, Koob GF, Roberto M, Homanics GE, 2017. Genetic and Pharmacologic Manipulation of TLR4 Has Minimal Impact on Ethanol Consumption in Rodents. J. Neurosci 37, 1139–1155. doi: 10.1523/JNEUROSCI.2002-16.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hampp G, Ripperger JA, Houben T, Schmutz I, Blex C, Perreau-Lenz S, et al. 2008. Regulation of Monoamine Oxidase A by Circadian-Clock Components Implies Clock Influence on Mood. Current Biology, 18(9), 678–683. 10.1016/j.cub.2008.04.012 [DOI] [PubMed] [Google Scholar]
- Hutchinson MR, Northcutt AL, Hiranita T, Wang X, Lewis SS, Thomas J, van Steeg K, Kopajtic TA, Loram LC, Sfregola C, Galer E, Miles NE, Bland ST, Amat J, Rozeske RR, Maslanik T, Chapman TR, Strand KA, Fleshner M, Bachtell RK, Somogyi AA, Yin H, Katz JL, Rice KC, Maier SF, Watkins LR, 2012. Opioid Activation of Toll-Like Receptor 4 Contributes to Drug Reinforcement. J. Neurosci 32, 11187–11200. doi: 10.1523/JNEUROSCI.0684-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperato A, Di Chiara G, 1986. Preferential stimulation of dopamine release in the nucleus accumbens of freely moving rats by ethanol. Journal of Pharmacology and Experimental …. [PubMed] [Google Scholar]
- Jacobsen JHW, Watkins LR, Hutchinson MR, 2014. Discovery of a Novel Site of Opioid Action at the Innate Immune Pattern-Recognition Receptor TLR4 and its Role in Addiction, 1st ed, Neuroimmune signaling in drug actions and addictions. Elsevier Inc. doi: 10.1016/B978-0-12-801284-0.00006-3 [DOI] [PubMed] [Google Scholar]
- Johnson S, Duncan J, Hussain SA, Chen G, Luo J, Mclaurin C, May W, Rajkowska G, Ou X-M, Stockmeier CA, Wang JM, 2015. The IFN γ-PKR Pathway in the Prefrontal Cortex Reactions to Chronic Excessive Alcohol Use. Alcohol Clin Exp Res 39, 476–484. doi: 10.1111/acer.12650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- June HL, Liu J, Warnock KT, Bell KA, Balan I, Bollino D, Puche A, Aurelian L, 2015. CRF-amplified neuronal TLR4/MCP-1 signaling regulates alcohol self-administration. Neuropsychopharmacology 40, 1549–1559. doi: 10.1038/npp.2015.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller M, Mazuch J, Abraham U, Eom GD, Herzog ED, Volk H-D, Kramer A, Maier B, 2009. A circadian clock in macrophages controls inflammatory immune responses. Proc Natl Acad Sci USA 106, 21407–21412. doi: 10.1073/pnas.0906361106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer SW, Taste reactivity in high alcohol drinking and low alcohol drinking rats. 1995. Taste reactivity in high alcohol drinking and low alcohol drinking rats, 19(2), 279–284. [DOI] [PubMed] [Google Scholar]
- Koob GF, Le Moal M, 2001. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology 24, 97–129. doi: 10.1016/S0893-133X(00)00195-0 [DOI] [PubMed] [Google Scholar]
- Koob GF, Volkow ND, 2009. Neurocircuitry of Addiction. Neuropsychopharmacology 35, 217–238. doi: 10.1038/npp.2009.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtuncu M, Arslan AD, Akhisaroglu M, Manev H, Uz T, 2004. Involvement of the pineal gland in diurnal cocaine reward in mice. European Journal of Pharmacology 489, 203–205. doi: 10.1016/j.ejphar.2004.03.010 [DOI] [PubMed] [Google Scholar]
- Lacagnina MJ, Rivera PD, Bilbo SD, 2016. Glial and Neuroimmune Mechanisms as Critical Modulators of Drug Use and Abuse 42, 156–177. doi: 10.1038/npp.2016.121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yang AR, Kelly T, Puche A, Esoga C, Elnabawi A, Merchenthaler I, Sieghart W, June HL, Aurelian L, 2011. Binge alcohol drinking is associated with GABAA alpha2-regulated Toll-like receptor 4 (TLR4) expression in the central amygdala. Proc. Natl. Acad. Sci. U.S.A 108, 4465–4470. doi: 10.1073/pnas.1019020108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzardo A, Poje A, Penick E, Butler M, 2016. Multiplex Immunoassay of Plasma Cytokine Levels in Men with Alcoholism and the Relationship to Psychiatric Assessments. IJMS 17, 472–14. doi: 10.3390/ijms17040472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez M, & Morales-Mulia M 2008. Role of mu and delta opioid receptors in alcohol drinking behaviour. Current Drug Abuse Reviews, 1(2), 239–252. [DOI] [PubMed] [Google Scholar]
- Miller BH, McDearmon EL, Panda S, Hayes KR, Zhang J, Andrews JL, Antoch MP, Walker JR, Esser KA, Hogenesch JB, Takahashi JS, 2007. Circadian and CLOCK-controlled regulation of the mouse transcriptome and cell proliferation. Proc Natl Acad Sci USA 104, 3342–3347. doi: 10.1073/pnas.0611724104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore RY, Eichler VB, 1972. Loss of a circadian adrenal corticosterone rhythm following suprachiasmatic lesions in the rat. Brain research. [DOI] [PubMed] [Google Scholar]
- Moore RY, Lenn NJ, 1972. A retinohypothalamic projection in the rat. J. Comp. Neurol 146, 1–14. doi: 10.1002/cne.901460102 [DOI] [PubMed] [Google Scholar]
- Northcutt AL, Hutchinson MR, Wang X, Baratta MV, Hiranita T, Cochran TA, Pomrenze MB, Galer EL, Kopajtic TA, Li CM, Amat J, Larson G, Cooper DC, Huang Y, O’Neill CE, Yin H, Zahniser NR, Katz JL, Rice KC, Maier SF, Bachtell RK, Watkins LR, 2015. DAT isn’t all that: cocaine reward and reinforcement require Toll-like receptor 4 signaling. Mol Psychiatry 20, 1525–1537. doi: 10.1038/mp.2014.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh PK, Ozburn AR, McClung CA, 2015. Circadian clock genes: Effects on dopamine, reward and addiction. Alcohol 49, 341–349. doi: 10.1016/j.alcohol.2014.09.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partch CL, Green CB, Takahashi JS, 2014. Molecular architecture of the mammalian circadian clock. Trends in Cell Biology 24, 90–99. doi: 10.1016/j.tcb.2013.07.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual M, Baliño P, Alfonso-Loeches S, Aragón CMG, Guerri C, 2011. Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage. Brain Behavior and Immunity 25 Suppl 1, S80–91. doi: 10.1016/j.bbi.2011.02.012 [DOI] [PubMed] [Google Scholar]
- Perreau-Lenz S, Spanagel R, 2015. Clock genes × stress × reward interactions in alcohol and substance use disorders. Alcohol 49, 351–357. doi: 10.1016/j.alcohol.2015.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perreau-Lenz SEP, Vengeliene V, Noori HR, Merlo-Pich EV, Corsi MA, Corti C, Spanagel R, 2012. Inhibition of the Casein-Kinase-1-Epsilon/Delta Prevents Relapse-Like Alcohol Drinking. Neuropsychopharmacology 37, 2121–2131. doi: 10.1038/npp.2012.62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickering C, Avesson L, Liljequist S, Lindblom J, Schiöth HB, 2007. The role of hypothalamic peptide gene expression in alcohol self-administration behavior. Peptides 28, 2361–2371. doi: 10.1016/j.peptides.2007.09.011 [DOI] [PubMed] [Google Scholar]
- Ray LA, Bujarski S, Shoptaw S, Roche DJ, Heinzerling K, & Miotto K 2017. Development of the Neuroimmune Modulator Ibudilast for the Treatment of Alcoholism: A Randomized, Placebo-Controlled, Human Laboratory Trial, 1–13. 10.1038/npp.2017.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reppert SM, Weaver DR, 2002. Coordination of circadian timing in mammals. Nature 418, 935–941. doi: 10.1038/nature00965 [DOI] [PubMed] [Google Scholar]
- Sleipness EP, Jansen HT, Schenk JO, & Sorg BA 2008. Time-of-day differences in dopamine clearance in the rat medial prefrontal cortex and nucleus accumbens. Synapse, 62(12), 877–885. 10.1002/syn.20552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleipness EP, Sorg BA, & Jansen HT 2007. Diurnal differences in dopamine transporter and tyrosine hydroxylase levels in rat brain: dependence on the suprachiasmatic nucleus. Brain Research, 1129(1), 34–42. 10.1016/j.brainres.2006.10.063 [DOI] [PubMed] [Google Scholar]
- Spanagel R, 2000. Recent animal models of alcoholism. Alcohol Research & Health. [PMC free article] [PubMed] [Google Scholar]
- Stephan FK, Zucker I, 1972. Circadian rhythms in drinking behavior and locomotor activity of rats are eliminated by hypothalamic lesions. Proc Natl Acad Sci USA 69, 1583–1586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabakoff B, Hoffman PL, 2000. Animal models in alcohol research. Alcohol Res Health 24, 77–84. [PMC free article] [PubMed] [Google Scholar]
- Terman M, Terman JS, 1975. Control of the rat’s circadian self-stimulation rhythm by light-dark cycles. Physiology & Behavior 14, 781–789. [DOI] [PubMed] [Google Scholar]
- Webb IC, Lehman MN, Coolen LM, 2015. Diurnal and circadian regulation of reward-related neurophysiology and behavior. Physiology & Behavior 143, 58–69. doi: 10.1016/j.physbeh.2015.02.034 [DOI] [PubMed] [Google Scholar]
- Wise RA, 2004. Dopamine, learning and motivation. Nat Rev Neurosci 5, 483–494. doi: 10.1038/nrn1406 [DOI] [PubMed] [Google Scholar]
- Wurst FM, Rasmussen DD, Hillemacher T, Kraus T, Ramskogler K, Lesch O, Bayerlein K, Schanze A, Wilhelm J, Junghanns K, Schulte T, Dammann G, Pridzun L, Wiesbeck G, Kornhuber J, Bleich S, 2007. Alcoholism, Craving, and Hormones: The Role of Leptin, Ghrelin, Prolactin, and the Pro-Opiomelanocortin System in Modulating Ethanol Intake. Alcohol Clin Exp Res 31, 1963–1967. doi: 10.1111/j.1530-0277.2007.00531.x [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.