

Summary
Ubiquitin specific protease 39 (USP39), an ortholog of Sad1p in yeast, is essential for spliceosome assembly during pre-mRNA splicing in human. Although it is known that USP39 is upregulated and plays an oncogenic role in hepatocellular carcinoma (HCC), the underlying mechanism remains unknown. The results of this study demonstrated that USP39 can be acetylated by the histone acetyltransferase MYST1, which is required for its proteasome-mediated degradation by Von Hippel-Lindau protein. In HCC cells, USP39 interacts with and is deacetylated by the lysine deacetylase sirtuin 7 (SIRT7). Notably, the deacetylation of USP39 by SIRT7 promotes its stability and thereby accelerates HCC cell proliferation and tumorigenesis in vitro and in vivo. Our data demonstrated a novel mechanism by which SIRT7 modulates the deacetylation of USP39 to promote HCC development, thus providing an effective anti-tumor therapeutic strategy for HCC.
Subject Areas: Molecular Biology, Cell Biology, Cancer
Graphical Abstract
Highlights
-
•
SIRT7 modulates the deacetylation of USP39
-
•
MYST1 promotes the acetyl binding of USP39
-
•
USP39 acetylation induces its instability
Molecular Biology; Cell Biology; Cancer
Introduction
According to 2018 global cancer statistics, liver cancer is the sixth most commonly diagnosed cancer and the fourth leading cause of cancer-related deaths worldwide (Bray et al., 2018). Hepatocellular carcinoma (HCC) is the predominant type of primary liver cancer, accounting for 75%–85% of all cases (Parkin, 2001). Although the mechanisms underlying HCC are largely understood, the lack of effective treatment options as well as multidrug resistance contributes to the high mortality rate in patients, especially those with advanced disease. Therefore, it is crucial to improve our knowledge of the carcinogenic process and the processes that mediate physiological processes including tumor cell proliferation, angiogenesis, differentiation, metastasis, and invasion, in order to identify therapeutic targets that might drive the development of targeted molecular therapies.
Mass spectrometry (MS)-based methods have shown that more than 1,000 proteins can be dynamically acetylated, which is a reversible post-translational modification (Choudhary et al., 2009; Mertins et al., 2013). Histone acetyltransferases (HATs) and histone deacetylases (HDACs) regulate gene expression by modifying core histone N-terminal tails (Lee and Workman, 2007; Shahbazian and Grunstein, 2007). Acetylation also exerts broad regulatory functions by changing the acetylation level of many non-histone proteins including p53, forkhead box transcription factors, E2F Transcription Factor 1, and c-Myc oncogene, which are involved in diverse biological processes ranging from cell growth, and apoptosis, to aging, calorie restriction, and tumorigenesis. Sirtuin 7 (SIRT7), which belongs to the SIRT family, requires NAD+ as a cofactor (Wu et al., 2018). Compared with other family members such as SIRT1 and SIRT6, SIRT7 is predominantly localized in the nucleus and is the least understood (Qi et al., 2018). SIRT7 plays critical roles in protein synthesis, chromatin remodeling, cell survival, DNA damage repair, and lipid metabolism by deacetylating a wide variety of transcription factors, metabolic enzymes, and signaling kinases, including p53, NRF1, GABP-β1, Smad4, and ATM (Barber et al., 2012; Mohrin et al., 2015; Nahalkova, 2015; Ryu et al., 2014; Tang et al., 2017b, 2019; Lu et al., 2020). SIRT7 is also involved in the process of pre-ribosomal RNA (rRNA) synthesis and maturation. SIRT7 deacetylates upstream binding factor (Grob et al., 2009) and associated factor 53 to facilitate RNA polymerase I-dependent transcription (Chen et al., 2013). Deacetylation of the nucleolar protein U3 small nuclear riboprotein factor 55K (U3-55K) by SIRT7 is required for 18S rRNA maturation (Chen et al., 2016). In addition, SIRT7 expression is regulated post-transcriptionally by microRNA (MiR-) -125a-5p and MiR-125b in HCC, which reportedly suppress SIRT7 oncogenic potential in HCC (Kim et al., 2013). SIRT7 can be downregulated by dephosphorylated TBP1 upon 5-fluorouracil treatment (Tang et al., 2017a), and its enzymatic activity was repressed via deubiquitinating by USP7 (Jiang et al., 2017). Besides, HDAC8 Cooperates with SMAD3/4 Complex to suppress SIRT7 transcription and then further activates TGF-β signaling to promote cell survival and migration (Tang et al., 2020).
USP39 is a 65-kDa SR-related protein involved in RNA splicing. It also functions as a deubiquitinating enzyme without protease activity (Lygerou et al., 1999; Makarova et al., 2001). The spliceosome comprises five small nuclear ribonucleoproteins (snRNPs), U1, U2, U4, U5, and U6, along with many non-snRNP proteins. While the yeast protein Sad1p was found to contribute to assembly U4 snRNP into U6 snRNP and pre-mRNA splicing, human USP39 protein is essential for recruitment of the tri-snRNP to the pre-spliceosome (Lygerou et al., 1999). Meanwhile, an USP39 mutant can induce splicing defects by removing the intron between exon 3 and exon 4 of retinoblastoma 1 to downregulate it in zebrafish (Rios et al., 2011). In addition, USP39 is required to maintain the spindle checkpoint and successful cytokinesis, and silencing of USP39 reduces Aurora B mRNA expression (Van Leuken et al., 2008). Increasing evidence demonstrates that USP39 plays an oncogenic role in various human cancers. Knockdown of USP39 prevents glioma cell progression by decreasing TAZ pre-mRNA splicing efficiency in vitro (Ding et al., 2019). In addition, decreasing USP39 expression is critical for the viability of KRAS-dependent cells (Fraile et al., 2017). However, the clinical relevance of USP39 in HCC and its molecular mechanisms have rarely been reported.
Here, we investigated the relationship among USP39, MYST1, and SIRT7 deacetylase in human HCC. The results showed that USP39 can be acetylated by HAT MYST1. The acetylation of USP39 promotes its degradation by the E3 ubiquitin ligase VHL-mediated proteasomal degradation. SIRT7 deacetylates USP39, which elevates its stability and stimulates its oncogenic activities in HCC. Thus, USP39 may serve as a prognostic biomarker and therapeutic target in HCC.
Results
Interactome Screening Identified USP39 as a SIRT7 Interactor
HCC is the most common type of liver cancer and has one of the highest mortality rates of solid cancers. SIRT7 plays an important role in the development of many cancers. To determine the molecular mechanism underlying the upregulation of SIRT7 in HCC (Kim et al., 2013), we used a proteomic approach to identify SIRT7-interacting proteins in 293T cells. In short, whole-cell lysate from cells transfected with 3× FLAG-SIRT7 was collected and subjected to immunoprecipitation (IP) with anti-FLAG-conjugated agarose beads and eluted with 3× FLAG peptide in elution buffer (Figure 1A). Partial eluent was subjected to SDS-PAGE and Coomassie brilliant blue staining. A strong SIRT7 protein band was visualized, as shown in Figure 1B. According to mass spectrometry (MS) analysis, 15 proteins involved in a variety of biological process, including protein degradation, cell cycle, transcriptional regulation, stress factor, RNA splicing, and protein degradation, were identified as SIRT7 interacting candidates (Figures 1C and 1D). These genes in black were reported to interact with SIRT7 among the previous researches (Fan et al., 2019; Grob et al., 2009; Iyer-Bierhoff et al., 2018; Jiang et al., 2017; Qi et al., 2018), and the rest need to be verified by IP methods. USP39, an SR-related protein and deubiquitinating enzyme, was detected in the SIRT7 pull-down products, suggesting that USP39 might interact with SIRT7.
Figure 1.

USP39 Acts as an Interactor of SIRT7
(A) Flow chart and pictures of an in vivo screen to identify candidates that interact with SIRT7 in 293T cells.
(B) Coomassie brilliant blue staining of FLAG-SIRT7 precipitates.
(C) SIRT7-interacting proteins were identified by mass spectrometry analysis. The total peptide numbers of each protein are indicated.
(D) MS analysis of SIRT7 precipitates and classification of SIRT7-associated proteins. The interaction candidates in black color were reported in the literature, and candidates in green and red were new.
(E) USP39 or SIRT7 plasmids were transfected alone or with the other into 293T cells, and their interaction was determined by IP and immunoblotting with indicated antibodies.
(F) Co-immunoprecipitation of USP39 and SIRT7 was detected in Hep3B cells as in (E).
(G) Schematic picture of SIRT7 and its truncated mutants. NLS, nuclear localization signal; NoLS, nucleolus localization signal. The interaction between USP39 and SIRT7 domains is indicated by the plus signs (+).
(H) Bacterially expressed GST fusion proteins of wild-type (WT), ΔN (1–89), ΔCD (90–330), ΔC (331–400) mutants of SIRT7 were bound to glutathione Sepharose beads and incubated with cell lysates of HEK293T cells transfected with FLAG-USP39 plasmids. Bound FLAG-USP39 was subjected to western blotting analysis with anti-FLAG antibody.
(I) USP39 and its various deletion mutation constructs are shown schematically.
(J) GST-fused full length or fragments of USP39 proteins were incubated with lysates of cells transfected with SIRT7, and western blotting was performed to detect the bound protein after precipitation with GST beads.
(K) Co-localization of SIRT7 and USP39 in Hep3B cells. DAPI was used to stain the DNA. DAPI, 4, 6-diamidino-2-phenylindole. Scale bars, 10 μm.
(L) The quantitative analysis of the co-localization of USP39 and SIRT7 in Figure 1K. The quantitative values are expressed as the mean ± SD (n = 20 cells) obtained from three independent experiments.
(M) USP39 expression in nuclear and cytoplasmic fractions in control and SIRT7 KD Hep3B cells was examined. GAPDH and histone H3 were used as cytoplasmic and nuclear controls, respectively.
USP39 is frequently upregulated in human glioma cancers (Ding et al., 2019). Thus, it was plausible that USP39 was identified as a SIRT7-interacting partner, contributing to SIRT7 function by interaction. To further test this hypothesis, myc-USP39 was transfected alone or together with SIRT7 into HEK293T cells for protein interaction analysis by IP and western blotting. As shown in Figure 1E, SIRT7 and USP39 could be reciprocally pulled down. The interaction of endogenous USP39 and SIRT7 proteins was also detected in Hep3B cells by a co-immunoprecipitation (coIP) assay with anti-USP39 or anti-SIRT7 antibody (Figure 1F). These results suggest that USP39 and SIRT7 could interact with each other. Deacetylase SIRT7 comprises an N-terminal nuclear localization signal peptide, catalytic domain, and C-terminal nucleolar localization domain (Kiran et al., 2013). To map the region within SIRT7 that interacts with USP39, we generated and purified the full-length (FL) and three deletion fragments of GST-SIRT7 and then performed a GST affinity isolation assay (Figure 1G). As seen in Figure 1H, the results suggested that the catalytic domain of SIRT7 binds USP39. In addition, the USP39 protein harbors a zinc finger (ZnF) and C-terminal ubiquitin hydrolase domain (peptidase C19 domain) (Komander et al., 2009; Lin et al., 2012), and the peptidase C19 domain but not the ZnF domain of USP39 interacted with SIRT7 (Figures 1I and 1J). Finally, we found that SIRT7 co-localized with USP39 in the nucleus (Figure 1K). The quantitative analysis further confirmed extensive overlap between USP39 and SIRT7 (Figure 1L). In summary, these results indicate that SIRT7 interacts with USP39 and the interaction is probably mediated by the catalytic domain of SIRT7 and N terminus of USP39. To investigate the relationship between SIRT7 and USP39, endogenous SIRT7 was knocked down by shRNA (Figure S1). Subcellular fractionation revealed a reduction in the levels of total and nuclear USP39 from SIRT7-deficient cells (Figure 1M). These data indicated that SIRT7 knockdown (KD) was accompanied by downregulation of USP39 protein in nucleus.
USP39 Is Stabilized by SIRT7-Mediated Deacetylation
The interaction between USP39 and SIRT7 prompted us to determine whether USP39 can act as a deubiquitinase for SIRT7. To gain further insights into how SIRT7 interacts with USP39, we first investigated the possibility that, as a deubiquitinase, USP39 might remove ubiquitin from SIRT7. As shown in Figure 2A, after transfection of SIRT7 and ubiquitin with or without USP39 in 293T cells, the ubiquitination of SIRT7 in the presence of USP39 expression was not changed. Therefore, we examined whether USP39 is a novel target of SIRT7. To this end, we transfected FLAG-USP39 with or without myc-SIRT7 into 293T cells and 48 h later, the acetylation of USP39 was evaluated by coIP and western blotting. Treatment of Hep3B cells with the specific sirtuin inhibitor NAM but not the class I and II HDAC inhibitor TSA increased acetylation levels of USP39 (Figures 2B and S2A). As shown in Figures S2Band S2C, the acetylation of USP39 protein was largely inhibited in the presence of SIRT7 expression. To gain further insight into the role of SIRT7 on the acetylation of USP39, a rescue experiment was performed by transfecting wild-type (WT) or enzyme-inactivated H187Y SIRT7 mutant into SIRT7 knockdown (KD) cells. SIRT7 knockdown led to increased USP39 acetylation, which was repressed by reintroduction of SIRT7 WT but not HY mutant (Figure 2C), revealing that SIRT7-regulated USP39 acetylation is dependent on its enzymatic activity. To determine whether SIRT7 specifically deacetylates USP39, the acetylation level of USP39 in SIRT7 KD cells was compared with the level in SIRT6 KD cells by coIP assay. Notably, SIRT6 knockdown did not elevate USP39 acetylation, whereas depletion of SIRT7 led to a marked increase in the USP39 acetylation level (Figure 2D). Consistent with previous research, we found that the USP39 protein level significantly decreased upon SIRT7 deficiency in Hep3B cells. To determine the role of SIRT7 on USP39 acetylation in vitro, we transfected FLAG USP39 into 293T cells and incubated the cells with 10 mM NAM for an additional 6 h before harvesting. Then the cell lysates were precipitated using anti-FLAG-conjugated agarose beads and eluted by FLAG peptide to collect the target protein. Here, WT but not a catalytically dead mutant SIRT7 deacetylated USP39 in the presence of NAD+ (Figure 2E).
Figure 2.

SIRT7 Deacetylates and Stabilizes USP39
(A) HA-ubiquitin, myc-SIRT7, and FLAG-USP39 plasmids were transfected alone or co-transfected in 293T cells and the ubiquitination of SIRT7 was tested by western blotting. The expression of SIRT7 and USP39 was confirmed with the indicated antibodies.
(B) Hep3B cells were treated with NAM (10 mM), TSA (1 μM), or NAM (10 mM) and TSA (1 μM) for 6 h before harvesting. The effects of TSA and NAM on USP39 acetylation were detected as indicated.
(C) SIRT7 knockdown cells were transfected with the indicated plasmids and the acetylation of USP39 was tested as in (B).
(D) Hep3B cells were transfected with SIRT6 small hairpin RNA (shRNA) or SIRT7 shRNA and treated with NAM for 6 h before harvesting. USP39 acetylation was examined as in (B).
(E) FLAG-USP39 and FLAG-SIRT7 were purified from 293T cells, followed by in vitro deacetylation assays in the presence or absence of NAD+.
(F) SIRT7 KD hep3B cells were transfected with increasing amounts of FLAG-SIRT7 plasmid, followed by deacetylation assays.
(G) SIRT7 KD cells or control cells were treated with the proteasome inhibitor MG132 for 4 h before collection and the protein expression was tested by western blotting.
(H) The ubiquitination of endogenous USP39 in Hep3B cells was analyzed by IP and western blotting. GAPDH was used as a loading control.
(I) The prediction of E3 ubiquitin ligase candidates for USP39 (http://ubibrowser.ncpsb.org/home/index).
(J) Identification of USP39 E3 ligase. Cells were transfected with myc-USP39 and HA ubiquitin with or without FLAG E3, and IP and immunoblotting experiments were performed with the indicated antibodies.
(K) The endogenous interaction of USP39 and VHL was tested in Hep3B cells.
(L) The protein level of USP39 was detected in control or SIRT7-depleted Hep3B cells transfected with control or VHL knockdown plasmids.
Moreover, with increasing amounts of SIRT7 in SIRT7 KD cells, the expression levels of USP39 increased in a dose-dependent manner (Figure 2F). The results suggest that deacetylation of USP39 by SIRT7 might mediate the degradation of USP39. To further confirm SIRT7 KD-mediated USP39 degradation, the expression level of USP39 in SIRT7 KD cells was restored with treatment of MG132, a proteasome inhibitor (Figure 2G). Next, we examined the endogenous ubiquitination of USP39 in SIRT7-deficient cells. As shown in Figures 2H, S2D and S2E, SIRT7 KD in Hep3B cells mainly led to elevated Lys48-ubiquitination of USP39 in the presence of MG132 compared with the nonspecific sgRNA control. Importantly, reintroducing the WT form of SIRT7 rescued USP39 from ubiquitination. We employed the web tool ubibrowser (http://ubibrowser.ncpsb.org/home/index) to screen USP39 predicted E3 ubiquitin ligases. To identify the relationship between USP39 and its putative partners, we attempted to verify the association by coIP followed by western blotting analysis. We tested the ubiquitination of USP39 with 10 high-scoring E3 ligase candidates including SMURF2, VHL, STUB1, NEDD4, SYVN1, MDM2, March9, FZR1, TRIM25, and RING1 (Figure 2I). Except VHL, the ubiquitination level of USP39 with the remaining candidates was not elevated in transiently transfected 293T cells (Figures 2J and S2F), suggesting that VHL may be the true E3 ligase for USP39. To further confirm that VHL promotes proteasome-mediated degradation of USP39, we detected the endogenous interaction of VHL and USP39 in Hep3B cells by performing coIP with anti-VHL antibody compared with anti-IgG antibody (Figure 2K). Consistently, USP39 protein stability was increased with VHL knockdown in SIRT7 KD cells (Figures 2L and S2G). Together, these data indicate that USP39 deacetylation mediated by SIRT7 was required for USP39 stability.
MYST1 Acts as a Specific Acetyltransferase for USP39
To identify the lysine acetyl transferase (KAT) of USP39, we transfected KATs and USP39 into 293T cells and then detected their interaction and USP39 acetylation. The results showed that many KATs interacted with USP39, including MYST1, Tip60, GCN5, and PCAF (Figure S3A), but only co-expression of MYST1 significantly increased the acetylation level of USP39 (Figure 3A). Furthermore, the interaction between MYST1 and USP39 was tested by transfecting MYST1/USP39 alone or together into 293T cells followed by coIP. As shown in Figure 3B, MYST1 was pulled down by USP39. Besides, similar result was obtained in a reciprocal coIP assay using anti-myc antibody. In addition, endogenous MYST1 and USP39 could be reciprocally pulled down (Figures 3C and S3B–S3D). These results suggest that USP39 interacts with MYST1. Moreover, the ZnF domain of USP39 was very important for its interaction with MYST1, as deletion of this domain completely abrogated the interaction (Figures 1I and 3D). According to the mapped regions of MYST1, we generated truncated mutants of MYST1 and found that 1–177 domain of MYST1 was required for the interaction with USP39 (Figures 3E and 3F). Furthermore, MYST1 and USP39 were largely co-localized in the nucleus of Hep3B cells (Figure 3G). The quantitative analysis further showed an extensive overlap between USP39 and MYST1 (Figure 3H).
Figure 3.

MYST1 Specifically Acetylates USP39
(A) USP39 plasmid was transfected alone or with MYST1, MYST2, Tip60, GCN5, or PCAF into 293T cells and the acetylation of USP39 was tested by performing IP and immunoblotting with the indicated antibodies.
(B) MYST1 or USP39 plasmids were transfected alone or with the other one into 293T cells and the interaction was determined by IP and immunoblotting as indicated.
(C) The endogenous interaction between USP39 and SIRT7 in Hep3B cells was detected as in (B).
(D) MYST1 interaction with USP39 and its mutants were detected as indicated.
(E) Schematic representation of MYST1 and its truncated mutants.
(F) Interaction of USP39 with MYST1 and its mutants were tested as indicated.
(G) Co-localization of MYST1 and USP39 in Hep3B cells. Scale bars, 10 μm.
(H) The quantitation analysis of the co-localization of USP39 and MYST1 in Figure 3G. The quantitative values are expressed as the mean ± SD (n = 20 cells) obtained from three independent experiments.
(I) Acetylation of USP39 in Hep3B cells transfected with MYST1 or its inactive mutant was tested.
(J) Hep3B cells were transfected with FLAG-USP39, myc- MYST1 with SIRT7 or SIRT6 and the acetylation of USP39 was examined as in (A).
(K) The ubiquitination of USP39 was tested in cells transfected with MYST1 or its inactive mutant.
Next, we generated an enzyme inactive mutant (G327E) of MYST1 (Valerio et al., 2017; Akhtar and Becker, 2000) (Figure S3E) and evaluated the effect of the mutation on USP39 acetylation. As seen in Figure 3I, the MYST1 mutant failed to elevate USP39 acetylation levels, suggesting that acetyltransferase activity is necessary for MYST1 to preserve the acetyl form of USP39. In addition, MYST1 was co-transfected with SIRT7 or SIRT6 into cells, and IP was performed using the cell lysate. The acetylation of USP39 stimulated by MYST1 was inhibited only by SIRT7, indicating that SIRT7 specifically removed acetyl group from USP39 (Figure 3J). The acetylation of USP39 elevated by wild-type (WT) MYST1 but not the mutant MYST1 promoted the ubiquitination of USP39 (Figure 3K). Taken together, these data suggest that MYST1 interacts with and acetylates USP39.
Acetylation of USP39 at Lysine 466 and 508 Residues Is Required for Acetylation-Mediated Degradation
To identify the exact acetylation sites of USP39, USP39 and MYST1 were co-transfected into Hep3B cells and maintained for 2 days followed by a treatment with NAM for an additional 6 h. The cell lysate was purified with M2 beads, and all the eluted proteins were used for western blotting and Coomassie brilliant blue staining (Figure 4A). According to MS analysis, lysine residues 466 and 508 (K466 and K508) were acetylated (Figure 4B). It was previously reported that USP39 lysine residue 428 (K428) could be acetylated (Choudhary et al., 2009; Mertins et al., 2013) (Figure 4C). By transfecting K428R-, K466R- and K508R-mutated FLAG-USP39 constructs into Hep3B cells, the acetylation level of various USP39 mutants were detected by IP with anti-FLAG antibody and western blotting with various anti-acetyl antibody and the K508R-mutated USP39 shows a sharp reduction of acetylation (Figure 4D). Since K466R-mutated USP39 also shows a comparable reduction of acetylation when detected by K466ac antibody, we generated 2KR (K466R and K508R) mutant of USP39. As shown in Figure 4E, the acetylation level of USP39 WT but not USP39 mutant especially 2KR was downregulated in the presence of SIRT7. Moreover, the 2KR mutant of USP39 with co-expressed SIRT7 and VHL in cells showed less ubiquitination level than WT (Figure 4F).
Figure 4.

Identification of USP39 Acetylation Sites
(A) Coomassie brilliant blue staining of FLAG-USP39 precipitates.
(B) MS analysis of USP39 acetylated lysine residues.
(C) Sequence alignment of conserved K428-, K466-, and K508-containing regions in USP39 orthologs of various species.
(D) Hep3B cells were transfected with the indicated mutant constructs and incubated with 10 mM NAM for an additional 6 h. The levels of acetylation and total USP39 protein were confirmed by IP and/or western blotting.
(E) Immunoblots showing the acetylation levels of USP39 WT and its mutants in the absence or presence of SIRT7 as indicated.
(F) Immunoblots showing the ubiquitination status of USP39 WT or 2KR mutant in the presence of SIRT7 or VHL or together.
SIRT7 or USP39 Depletion Induces the Expression of Cell Growth and Cancer-Associated Genes
To clarify the function of SIRT7 and USP39 on cancer cell transcriptome, depletion of endogenous SIRT7 or USP39 in Hep3B cells by single guide RNA (sgRNA) was generated (Figure S4A) and examined by western blot (Figure S4B) and single clone was picked (Figure S4C) and then these cells were subjected to transcriptome analysis. Venn matrix diagrams illustrated distinct subsets of differentially expressed genes (DEGs) that were regulated by depletion of USP39 or SIRT7 (Figure 5A). For functional pathway analysis, these genes were involved in 27 pathways, mainly mediating signal transduction, human diseases, and metabolism (Figure 5B). To investigate the expression of cancer-related genes, we counted transcriptional relevant inducers, stemness, and cell cycle, cell proliferation associated regulators in the transcriptome (Figure 5C). As seen in Figure 5D, 120 USP39-activated genes in blue and 46 USP39-repressed genes in red, and 196 SIRT7-activated genes in blue and 123 SIRT7-repressed genes in red are shown in the volcano plot. We used RT-qPCR (Figure 5E) and western blot analysis (Figure 5F) to determine proliferation-related genes expression in cancer cell transcriptome (Figure 5C). Likewise, SIRT7 or USP39 knockout in Hep3B cells led to upregulation of cell cycle-related genes/proteins (Figures 5E and 5F) and tumor suppressor PTEN. How the expressions of these genes/proteins are regulated by SIRT7 or USP39 needs further investigation.
Figure 5.

Transcriptome Identification of Cancer-Associated Genes upon SIRT7 or USP39 Depletion
(A) Transcriptome analysis of DEGs in all distinctive cell lines. DEG number in different color columns is presented after being compared with WT and the indicated cell lines. DEGs with a fold change ≥2 or ≤0.5 and diverge probability ≥0.8 (compared with the control group) were selected.
(B) KEGG classification of DEGs in the indicated cell lines.
(C) Heat maps showing color-coded expression levels of cell proliferation-related genes in transcriptome analysis.
(D) USP39 activated genes in blue and repressed genes in red (left panel) and SIRT7-activated genes in blue and repressed genes in red are shown in the volcano plot.
(E) Expression levels of multiple genes in the transcriptome analysis were confirmed by RT-qPCR: control versus SIRT7 and USP39 knockout cells.
(F) Protein levels of multiple genes in the above assay, including cancer-related genes, were determined by western blotting.
Depletion of USP39 or Its Deacetylase SIRT7 Inhibits Cell Growth and Tumorigenesis
Next, we investigated whether the protein level upregulated by USP39 deacetylation would contribute to cell growth and tumorigenesis. To test this possibility, we evaluated the role of depletion of USP39/SIRT7 (Figures S5A and S5B) or rescue with USP39 WT/2KR in SIRT7 KO (Figures S5C and S5D) on the viability and foci formation of Hep3B and SMMC7721 cells. As expected, depletion of USP39 expression blocked the growth rate of Hep3B and SMMC7721 cells, but replenishment with USP39 WT, USP39 2KR mutant, or USP39 2KR mutant + SIRT7 in the USP39-KD cells rescued malignant phenotype and USP39 WT + SIRT7 aggravated malignancy, indicating a promoter function of USP39 and its deacetylation on cell proliferation in Hep3B and SMMC7721 cells (Figures 6A and 6B). Next, we established MYST1 single knockdown (Figure S5E), double depletion of MYST1 and USP39 (Figure S5F), and replenished USP39 WT/2KR in SIRT7 KO cells (Figure S5G) and compared their effect on growth rate. As observed in Figure 6C, the cell line with MYST1-KD had a higher growth rate than control, whereas depletion of SIRT7 resulted in a retarded cell growth. Importantly, knockout of USP39 in shMYST1 cells rescued shMYST1-induced malignant cell proliferation, and overexpression of USP39 2KR in SIRT7-depleted cells got a worse malignancy than USP39 WT, indicating a vital role of deacetylation on USP39 stabilization and pro-oncogenic properties. In in vitro foci formation assay, USP39-deficiency attenuated cloning ability of Hep3B cells, whereas overexpression of USP39 WT and its mutant led to an increase in colony numbers in USP39-KD cells, and SIRT7 reinforced the function of WT but not mutant of USP39 (Figures 6D and 6E). Similar results were obtained in SMMC7721 cells (Figures 6F and 6G). Using a mouse xenograft model, we found a much smaller tumor size in SIRT7-KO cells and larger tumor size in MYST1 knockdown cells than control (Figures 6H–6J). Further depletion of USP39 in MYST1 KD cells rescued the malignant phenotype caused by MYST1 knockdown. Consistent with the results in Figure 6C, replenishing USP39 2KR in SIRT7 KO cells caused a worse malignancy than USP39 WT (Figures 6H–6J). Taken together, these results reveal a novel mechanism of USP39 or SIRT7 depletion leads to restriction of cell cycle-related genes expression and thereby limited HCC cell growth.
Figure 6.

Depletion of USP39 or Its Deacetylase SIRT7 Inhibits Cells Proliferation and Tumorigenesis
(A and B) The growth rate of Hep3B (A) and SMMC7721 (B) cells stably expressing shUSP39, shUSP39 + USP39 WT, shUSP39 + USP39 2KR, shUSP39 + USP39 WT + SIRT7, shUSP39 + USP39 2KR + SIRT7 or their corresponding control vector was examined at the indicated times. Results presented represent the means of triplicate experiments ±SEM. ∗p < 0.05; ∗∗p < 0.01.
(C) The growth rate of Hep3B cells stably expressing indicated single plasmids or combination was examined as in (A).
(D–G) Clonogenic assay was performed to measure the capacity of foci formation in Hep3B (C and D) or SMMC7721 cells (F and G) stably expressing indicated plasmids or combination. The error bar represents the SEM of triplicate experiments. ∗∗p < 0.01, two-tailed Student's t test.
(H–J) Representative photos showing xenograft tumors from euthanized mice (H) at 4 weeks post subcutaneous injection (n = 3). Tumor sizes were measured and depicted as tumor volume (I) or tumor weight (J). Results presented represent the means of triplicate experiments ±SEM. ∗p < 0.05; ∗∗p < 0.01.
SIRT7 and USP39 Expression Are Positively Correlated in HCC
To investigate the expression pattern of SIRT7 and USP39 in HCC cells, we used the web tool Kaplan-Meier database and found that the aberrant expression of SIRT7 and USP39 transcripts were reversely correlated with overall survival (OS) of patients, whereas MYST1 showed a positive correlation (Figure 7A). To further investigate the relationship among SIRT7, USP39, and MYST1 in human malignant tissues, we analyzed their expression levels in HCC tissues and non-tumor liver tissues using the GEO (GSE25097, GSE4108, GSE22405, and GSE50579) database. The results shown in Figures 7B and S6A–S6C implied that USP39 and SIRT7 were upregulated in histopathological process, whereas MYST1 showed a downregulation. Similarly, after performing immunohistochemical staining in HCC tissues array, SIRT7 and USP39 protein levels were detected significantly upregulated in HCC tissue compared with normal parts, whereas USP39 K508Ac and MYST1 were decreased in cancer tissues when compared with normal tissues (Figures 7C and 7D). In addition, more than 50% of tumor tissues had higher levels of SIRT7 and USP39 relative to the paired adjacent normal tissues from the same individuals. On the contrary, the expression level of USP39 K508Ac and MYST1 in tumors was lower than in paired normal tissues (Figure 7E). Collectively, our results indicated that SIRT7 expression is positively correlated with USP39 expression and inversely related to USP39 K508Ac and MYST1 expression and that USP39 and SIRT7 expression levels may serve as prognostic markers in HCC (Figure 7F).
Figure 7.

Both USP39 and SIRT7 Are Upregulated in HCC
(A) The relationship between SIRT7, USP39, or MYST1 expression with OS of patients with HCC based on the Kaplan-Meier database.
(B) Microarray analysis of SIRT7, USP39, and MYST1 expression in HCC tissues and matched adjacent normal tissues. N, normal tissue; T, tumor tissue; GEO database (Accession No. GSE25097), unpaired Student's t test, ∗∗p < 0.01, ∗p < 0.05.
(C) Representative immunohistochemical (IHC) staining of SIRT7, USP39, USP39 K508Ac and MYST1 in liver cancer tissues and paired liver tissues. Scale bar, 100 μm.
(D) Average IHC scores of SIRT7, USP39, USP39 K508Ac, and MYST1 in liver tumors and normal tissues (n = 24).
(E) Expression distribution (up, down, or equal) in liver tumor tissue relative to the paired adjacent normal tissue.
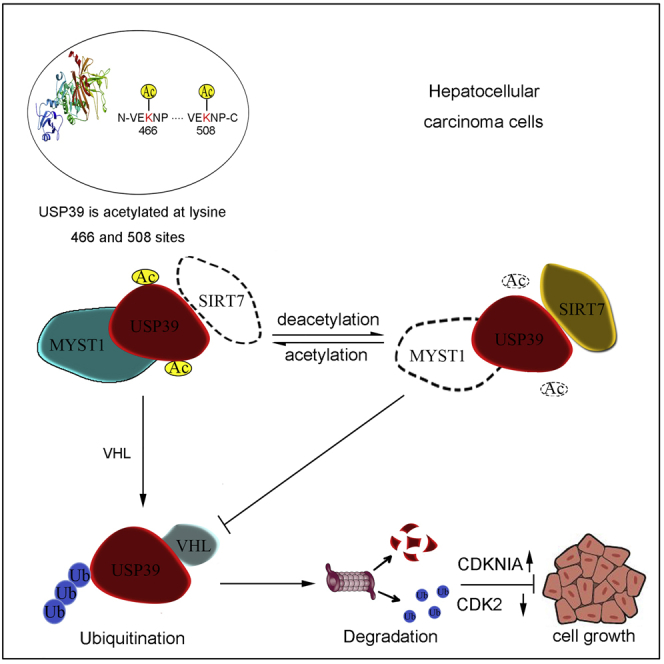
(F) Schematic model of the function of SIRT7, MYST1, and USP39 interplay in the cell growth and hepatocellular carcinoma progression. Decreased acetylation level of USP39 by upregulation of SIRT7 and/or downregulation of MYST1 expression in patients with liver cancer leads to inhibition of VHL-mediated USP39 degradation and subsequent transcriptional activation of cell proliferation associated genes, thus resulting in enhanced cell growth and liver tumorigenesis.
Discussion
Our study showed that SIRT7 and MYST1 mediate USP39 acetylation. This conclusion was supported by various extrinsic signals. IP and immunofluorescence assays confirmed that USP39 is a substrate for SIRT7. The acetylation of USP39 mediated by MYST1 is a prerequisite for its degradation by E3 ligase through the proteasome. USP39 deficiency decelerated HCC cell proliferation and tumor formation, and reintroducing USP39 WT or acetylation-defective mutant in USP39-KD cells restored the cell proliferative phenotype. Collectively, our study demonstrated that SIRT7 promotes HCC cell proliferation and development by downregulating USP39 acetylation. In addition, both USP39 and SIRT7 protein levels are elevated and might involve in human HCCs.
Liver cancer is one of the cancers with highest mortality according to 2018 statistics, and HCC accounts for the largest proportion of cases (Bray et al., 2018); thus, it is urgent to uncover the potential mechanism of HCC pathogenesis. SIRT7 is an NAD+-dependent deacetylase of the SIRT family and located in nucleolus. SIRT7 plays an essential role in controlling of gene expression, RNA polymerase I transcription activation, promoting pre-rRNA processing, and participating in DNA damage repair by deacetylating histone 3 lysine residue 18, p53, U3-55k, and ATM (Barber et al., 2012; Chen et al., 2013, 2016; Tang et al., 2019). In our study, SIRT7 interacted with and deacetylated USP39, which shows great correlation with USP39 deubiquitination. Unexpectedly, we did not find the deubiquitination of SIRT7 or mRNA level change induced by USP39, suggesting a possible role of SIRT7 regulation on USP39 function. Although the acetylation of USP39 promotes its degradation by VHL E3 ubiquitin ligase through the proteasome pathway, the mechanism by which USP39 acetylation promotes the ubiquitination is still not clear, and the relationship between SIRT7 and VHL needs further study.
The oncogenic properties of SIRT7 in HCC (Kim et al., 2013; Zhao et al., 2019) and thyroid cancer (Li et al., 2019) and USP39 in prostate cancer (Wen et al., 2014) were well studied, which are closely related to poor prognosis. Thus, we speculated that SIRT7 promotes tumorigenesis through regulating the acetylation of USP39. From transcriptomic database, on the one hand, we found SIRT7 or USP39 deficiency influences many genes in the PI3K/AKT pathway, which was consistent with previous study that SIRT7 induced deacetylation of p70S6K1 to active AKT by an indirect mechanism. Besides, we reintroduced the USP39 without or with SIRT7 into USP39 knockdown cells to detect the growth viability. The above observations demonstrated that SIRT7 might likely deacetylate USP39 to modulate cell growth through PI3K/AKT pathway, which needs further exploration. What's more, considering that USP39 and SIRT7 correlated with cyclins and cyclin-dependent kinases (CDKs) in transcriptomic database and evidences show different cyclins not only play critical roles in cell cycle progression to accelerate tumor development but also influence EMT through Slug, Smad4 (Wang et al., 2015), the relationship of SIRT7-USP39 axis with metastasis in HCC needs further exploration.
MYST1 (KAT8) is a member of the MYST acetyltransferase family, which is homologous to Drosophila males absent on the first (MOF) protein. Deletion of MYST1 in cells leads to genomic instability, spontaneous chromosomal aberration, cell cycle defects, reduced transcription of certain genes, defective DNA damage repair, and early embryonic lethality by transfer acetyl group on histone protein and other proteins (Carrozza et al., 2003; Gupta et al., 2008; Rea et al., 2007; Sharma et al., 2010). In our study, we found that the deficiency of MYST1 in HCC cell lines facilitated cell growth rate and tumorigenesis than control. In addition, our data identified USP39 as a novel target of MYST1 and was acetylated by MYST1 at K466 and K508 residues, indicating a direct role of MYST1 in mediating USP39 acetylation and function. USP39-KD cells replenished with USP39 2KR displayed reduced ability to be ubiquitinated compared with USP39 WT. The decrease of USP39 ubiquitination contributed to cancer cell development; however, it remains to be determined if the pathogenesis of patients with HCC is due to mutations of USP39 at K466 and K508. If true, it is possible that a special inhibitor could be developed as a treatment strategy for HCC.
HDAC inhibitors were widely used in cell culture for deacetylases catalytic inactivation, including TSA for HDAC classes I and II, vorinostat for HDAC classes I, II, and IV, and NAM for HDAC class III. HDACs are important drug targets in cancer and neurodegenerative diseases, such as Parkinson's and Alzheimer's diseases, and two HDAC inhibitors, suberoylanilide hydroxamic acid (SAHA) and valproic acid, are in clinical use. Furthermore, HDAC inhibitors are potent reprogramming agents for the generation of induced pluripotent stem cells. Thus, we speculate that an inhibitor designed to specifically arrest the activation of SIRT7 may stimulate the degradation of USP39, ultimately inducing inhibition of cancer cell development.
Limitations of the Study
Our study presents data about the interacting partners and deacetylase and acetyltransferase of USP39, mainly identified by mass spectrometry and coIP. Data were generated with 293T and HCC cell lines (Hep3B and MHCC97H), whereas follow-up experiments were conducted with limited HCC tissues. In this article, we investigated USP39 and the effect of its acetylation level on HCC cell growth, which could be extended by the analysis of USP39 acetylation on cell metastasis and invasion, and USP39 site-specific acetylation in tumor tissues.
Resource Availability
Lead Contact
Further information should be directed to and will be fulfilled by the Lead Contact, Zhenghong Lin (zhenghonglin@cqu.edu.cn).
Materials Availability
Materials are available upon request from Dr. Zhenghong Lin.
Data and Code Availability
RNA-seq information including all raw data has been deposited at Gene Expression Omnibus under GSE153783: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE153783. All raw data of western blotting were submitted to Mendeley Data: https://data.mendeley.com/datasets/c7gn2djxn8/2.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (Grant No. 31571454 to Z.Lin), the Fundamental Research Funds for the Central Universities (Project no. 106112017CDJQJ298833 to Z.Lin.), the 61st Batch of China Postdoctoral Science Fund (Grant No.2017M610588 to H. Li), the Special Fund for Postdoctoral Research Projects of Chongqing City (Grant No. Xm2017080 to H.Li), and startup funds from the “100 talent scholar program” at Chongqing University (to Z.Lin). We thank LetPub (www.letpub.com) for linguistic assistance during the preparation of this manuscript. We also thank the Analytical and Testing Center of Chongqing University for confocal fluorescence microscopy assistance.
Author Contributions
L. Dong., L. Yu., H. Li., and L. Shi. performed the experiments. Z. Luo. and Z. Liu. reviewed the manuscript and provided constructive suggestions. G. Yin., X. Yan., and H. Zhao. provided the clinical samples and directed the related experiment. L. Dong., X. Yan., and Z. Lin. analyzed data and wrote the manuscript.
Declaration of Interests
The authors have no conflicts of interest to declare.
Published: August 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101351.
Contributor Information
Xiaohua Yan, Email: yanxiaohua@ncu.edu.cn.
Zhenghong Lin, Email: zhenghonglin@cqu.edu.cn.
Supplemental Information
References
- Akhtar A., Becker P.B. Activation of transcription through histone H4 acetylation by MOF, an acetyltransferase essential for dosage compensation in Drosophila. Mol. Cell. 2000;5:367–375. doi: 10.1016/s1097-2765(00)80431-1. [DOI] [PubMed] [Google Scholar]
- Barber M.F., Michishita-Kioi E., Xi Y., Tasselli L., Kioi M., Moqtaderi Z., Tennen R.I., Paredes S., Young N.L., Chen K. SIRT7 links H3K18 deacetylation to maintenance of oncogenic transformation. Nature. 2012;487:114–118. doi: 10.1038/nature11043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F., Ferlay J., Soerjomataram I., Siegel R.L., Torre L.A., Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018;68:394–424. doi: 10.3322/caac.21492. [DOI] [PubMed] [Google Scholar]
- Carrozza M.J., Utley R.T., Workman J.L., Cote J. The diverse functions of histone acetyltransferase complexes. Trends Genet. 2003;19:321–329. doi: 10.1016/S0168-9525(03)00115-X. [DOI] [PubMed] [Google Scholar]
- Chen S., Blank M.F., Iyer A., Huang B., Wang L., Grummt I., Voit R. SIRT7-dependent deacetylation of the U3-55k protein controls pre-rRNA processing. Nat. Commun. 2016;7:10734. doi: 10.1038/ncomms10734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Seiler J., Santiago-Reichelt M., Felbel K., Grummt I., Voit R. Repression of RNA polymerase I upon stress is caused by inhibition of RNA-dependent deacetylation of PAF53 by SIRT7. Mol. Cell. 2013;52:303–313. doi: 10.1016/j.molcel.2013.10.010. [DOI] [PubMed] [Google Scholar]
- Choudhary C., Kumar C., Gnad F., Nielsen M.L., Rehman M., Walther T.C., Olsen J.V., Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Ding K., JI J., Zhang X., Huang B., Chen A., Zhang D., Li X., Wang X., Wang J. RNA splicing factor USP39 promotes glioma progression by inducing TAZ mRNA maturation. Oncogene. 2019;38:6414–6428. doi: 10.1038/s41388-019-0888-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan X., Wang Y., Tang X.Q. Extracting predictors for lung adenocarcinoma based on Granger causality test and stepwise character selection. BMC Bioinformatics. 2019;20:197. doi: 10.1186/s12859-019-2739-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraile J.M., Manchado E., Lujambio A., Quesada V., Campos-Iglesias D., Webb T.R., Lowe S.W., Lopez-Otin C., Freije J.M. USP39 deubiquitinase is essential for KRAS oncogene-driven cancer. J. Biol. Chem. 2017;292:4164–4175. doi: 10.1074/jbc.M116.762757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grob A., Roussel P., Wright J.E., Mcstay B., Hernandez-Verdun D., Sirri V. Involvement of SIRT7 in resumption of rDNA transcription at the exit from mitosis. J. Cell Sci. 2009;122:489–498. doi: 10.1242/jcs.042382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A., Guerin-Peyrou T.G., Sharma G.G., Park C., Agarwal M., Ganju R.K., Pandita S., Choi K., Sukumar S., Pandita R.K. The mammalian ortholog of Drosophila MOF that acetylates histone H4 lysine 16 is essential for embryogenesis and oncogenesis. Mol. Cell Biol. 2008;28:397–409. doi: 10.1128/MCB.01045-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer-Bierhoff A., Krogh N., Tessarz P., Ruppert T., Nielsen H., Grummt I. SIRT7-Dependent deacetylation of fibrillarin controls histone H2A methylation and rRNA synthesis during the cell cycle. Cell Rep. 2018;25:2946–2954 e5. doi: 10.1016/j.celrep.2018.11.051. [DOI] [PubMed] [Google Scholar]
- Jiang L., Xiong J., Zhan J., Yuan F., Tang M., Zhang C., Cao Z., Chen Y., Lu X., Li Y. Ubiquitin-specific peptidase 7 (USP7)-mediated deubiquitination of the histone deacetylase SIRT7 regulates gluconeogenesis. J. Biol. Chem. 2017;292:13296–13311. doi: 10.1074/jbc.M117.780130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.K., Noh J.H., Jung K.H., Eun J.W., Bae H.J., Kim M.G., Chang Y.G., Shen Q., Park W.S., Lee J.Y. Sirtuin7 oncogenic potential in human hepatocellular carcinoma and its regulation by the tumor suppressors MiR-125a-5p and MiR-125b. Hepatology. 2013;57:1055–1067. doi: 10.1002/hep.26101. [DOI] [PubMed] [Google Scholar]
- Kiran S., Chatterjee N., Singh S., Kaul S.C., Wadhwa R., Ramakrishna G. Intracellular distribution of human SIRT7 and mapping of the nuclear/nucleolar localization signal. FEBS J. 2013;280:3451–3466. doi: 10.1111/febs.12346. [DOI] [PubMed] [Google Scholar]
- Komander D., Clague M.J., Urbe S. Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 2009;10:550–563. doi: 10.1038/nrm2731. [DOI] [PubMed] [Google Scholar]
- Lee K.K., Workman J.L. Histone acetyltransferase complexes: one size doesn't fit all. Nat. Rev. Mol. Cell Biol. 2007;8:284–295. doi: 10.1038/nrm2145. [DOI] [PubMed] [Google Scholar]
- Li H., Tian Z., Qu Y., Yang Q., Guan H., Shi B., Ji M., Hou P. SIRT7 promotes thyroid tumorigenesis through phosphorylation and activation of Akt and p70S6K1 via DBC1/SIRT1 axis. Oncogene. 2019;38:345–359. doi: 10.1038/s41388-018-0434-6. [DOI] [PubMed] [Google Scholar]
- Lin Z., Yang H., Kong Q., Li J., Lee S.M., Gao B., Dong H., Wei J., Song J., Zhang D.D., Fang D. USP22 antagonizes p53 transcriptional activation by deubiquitinating Sirt1 to suppress cell apoptosis and is required for mouse embryonic development. Mol. Cell. 2012;46:484–494. doi: 10.1016/j.molcel.2012.03.024. [DOI] [PubMed] [Google Scholar]
- Lu Y.F., Xu X.P., Lu X.P., Zhu Q., Liu G., Bao Y.T., Wen H., Li Y.L., Gu W., Zhu W.G. SIRT7 activates p53 by enhancing PCAF-mediated MDM2 degradation to arrest the cell cycle. Oncogene. 2020;39:4650–4665. doi: 10.1038/s41388-020-1305-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lygerou Z., Christophides G., Seraphin B. A novel genetic screen for snRNP assembly factors in yeast identifies a conserved protein, Sad1p, also required for pre-mRNA splicing. Mol. Cell Biol. 1999;19:2008–2020. doi: 10.1128/mcb.19.3.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova O.V., Makarov E.M., Luhrmann R. The 65 and 110 kDa SR-related proteins of the U4/U6.U5 tri-snRNP are essential for the assembly of mature spliceosomes. EMBO J. 2001;20:2553–2563. doi: 10.1093/emboj/20.10.2553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertins P., Qiao J.W., Patel J., Udeshi N.D., Clauser K.R., Mani D.R., Burgess M.W., Gillette M.A., Jaffe J.D., Carr S.A. Integrated proteomic analysis of post-translational modifications by serial enrichment. Nat. Methods. 2013;10:634–637. doi: 10.1038/nmeth.2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohrin M., Shin J., Liu Y., Brown K., Luo H., Xi Y., Haynes C.M., Chen D. Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science. 2015;347:1374–1377. doi: 10.1126/science.aaa2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nahalkova J. Novel protein-protein interactions of TPPII, p53, and SIRT7. Mol. Cell Biochem. 2015;409:13–22. doi: 10.1007/s11010-015-2507-y. [DOI] [PubMed] [Google Scholar]
- Parkin D.M. Global cancer statistics in the year 2000. Lancet Oncol. 2001;2:533–543. doi: 10.1016/S1470-2045(01)00486-7. [DOI] [PubMed] [Google Scholar]
- Qi H., Shi X., Yu M., Liu B., Liu M., Song S., Chen S., Zou J., Zhu W.G., Luo J. Sirtuin 7-mediated deacetylation of WD repeat domain 77 (WDR77) suppresses cancer cell growth by reducing WDR77/PRMT5 transmethylase complex activity. J. Biol. Chem. 2018;293:17769–17779. doi: 10.1074/jbc.RA118.003629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rea S., Xouri G., Akhtar A. Males absent on the first (MOF): from flies to humans. Oncogene. 2007;26:5385–5394. doi: 10.1038/sj.onc.1210607. [DOI] [PubMed] [Google Scholar]
- Rios Y., Melmed S., Lin S., Liu N.A. Zebrafish usp39 mutation leads to rb1 mRNA splicing defect and pituitary lineage expansion. PLoS Genet. 2011;7:e1001271. doi: 10.1371/journal.pgen.1001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D., Jo Y.S., Lo Sasso G., Stein S., Zhang H., Perino A., Lee J.U., Zeviani M., Romand R., Hottiger M.O. A SIRT7-dependent acetylation switch of GABPbeta1 controls mitochondrial function. Cell Metab. 2014;20:856–869. doi: 10.1016/j.cmet.2014.08.001. [DOI] [PubMed] [Google Scholar]
- Shahbazian M.D., Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu. Rev. Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- Sharma G.G., So S., Gupta A., Kumar R., Cayrou C., Avvakumov N., Bhadra U., Pandita R.K., Porteus M.H., Chen D.J. MOF and histone H4 acetylation at lysine 16 are critical for DNA damage response and double-strand break repair. Mol. Cell Biol. 2010;30:3582–3595. doi: 10.1128/MCB.01476-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M., Li Z., Zhang C., Lu X., Tu B., Cao Z., Li Y., Chen Y., Jiang L., Wang H. SIRT7-mediated ATM deacetylation is essential for its deactivation and DNA damage repair. Sci. Adv. 2019;5:eaav1118. doi: 10.1126/sciadv.aav1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M., Lu X., Zhang C., Du C., Cao L., Hou T., Li Z., Tu B., Cao Z., Li Y. Downregulation of SIRT7 by 5-fluorouracil induces radiosensitivity in human colorectal cancer. Theranostics. 2017;7:1346–1359. doi: 10.7150/thno.18804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X., Shi L., Xie N., Liu Z., Qian M., Meng F., Xu Q., Zhou M., Cao X., Zhu W.G., Liu B. SIRT7 antagonizes TGF-beta signaling and inhibits breast cancer metastasis. Nat. Commun. 2017;8:318. doi: 10.1038/s41467-017-00396-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang X., Li G., Su F., Cai Y., Shi L., Meng Y., Liu Z., Sun J., Wang M., Qian M. HDAC8 cooperates with SMAD3/4 complex to suppress SIRT7 and promote cell survival and migration. Nucleic Acids Res. 2020;48:2912–2923. doi: 10.1093/nar/gkaa039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valerio D.G., Xu H., Chen C.W., Hoshii T., Eisold M.E., Delaney C., Cusan M., Deshpande A.J., Huang C.H., Lujambio A. Histone acetyltransferase activity of MOF is required for MLL-AF9 leukemogenesis. Cancer Res. 2017;77:1753–1762. doi: 10.1158/0008-5472.CAN-16-2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Leuken R.J., Luna-Vargas M.P., Sixma T.K., Wolthuis R.M., Medema R.H. Usp39 is essential for mitotic spindle checkpoint integrity and controls mRNA-levels of aurora B. Cell Cycle. 2008;7:2710–2719. doi: 10.4161/cc.7.17.6553. [DOI] [PubMed] [Google Scholar]
- Wang W.L., Huang H.C., Kao S.H., Hsu Y.C., Wang Y.T., Li K.C., Chen Y.J., Yu S.L., Wang S.P., Hsiao T.H. Slug is temporally regulated by cyclin E in cell cycle and controls genome stability. Oncogene. 2015;34:1116–1125. doi: 10.1038/onc.2014.58. [DOI] [PubMed] [Google Scholar]
- Wen D., Xu Z., Xia L., Liu X., Tu Y., Lei H., Wang W., Wang T., Song L., Ma C. Important role of SUMOylation of Spliceosome factors in prostate cancer cells. J. Proteome Res. 2014;13:3571–3582. doi: 10.1021/pr4012848. [DOI] [PubMed] [Google Scholar]
- Wu D., Li Y., Zhu K.S., Wang H., Zhu W.G. Advances in cellular characterization of the sirtuin isoform, SIRT7. Front. Endocrinol. (Lausanne) 2018;9:652. doi: 10.3389/fendo.2018.00652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Wozniak A., Adams A., Cox J., Vittal A., Voss J., Bridges B., Weinman S.A., Li Z. SIRT7 regulates hepatocellular carcinoma response to therapy by altering the p53-dependent cell death pathway. J. Exp. Clin. Cancer Res. 2019;38:252. doi: 10.1186/s13046-019-1246-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq information including all raw data has been deposited at Gene Expression Omnibus under GSE153783: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE153783. All raw data of western blotting were submitted to Mendeley Data: https://data.mendeley.com/datasets/c7gn2djxn8/2.