

Abstract
Controlling inflammation is critical for preventing many diseases including cancer, autoimmune disorders and hypersensitivity reactions. NF-E2-related factor 2 (Nrf2) is a key transcription factor that controls the cellular antioxidant and cytoprotective response. Moreover, Nrf2 has been implicated in the regulation of inflammatory processes, although the ultimate mechanism by which this is achieved is unknown. Here, we investigated mechanisms of inflammation and cell death pathways induced by a variety of Nrf2 activators including dimethyl fumarate (DMF) and the endogenous metabolite itaconate. We found that exposure of bone marrow-derived dendritic cells (BMDCs) to low concentrations of a variety of electrophilic Nrf2 activators including itaconate prior to Toll-like receptor (TLR) stimulation inhibits transcription of pro-inflammatory cytokines (such as interleukin [IL]-12 and IL-1β) by activation of Nrf2. By contrast, high doses of these electrophilic compounds after TLR activation promote inflammatory apoptosis and caspase-8-dependent IL-1β processing and release independently of Nrf2. Interestingly, tert-butylhydroquinone (tBHQ), a non-electrophilic Nrf2-activator, failed to induce IL-1β production. These results have important implications for clinical application of electrophilic compounds.
Keywords: Nrf2 activators, Itaconate, Inflammatory apoptosis, Mitochondria, Caspase-8, IL-1β
Graphical abstract
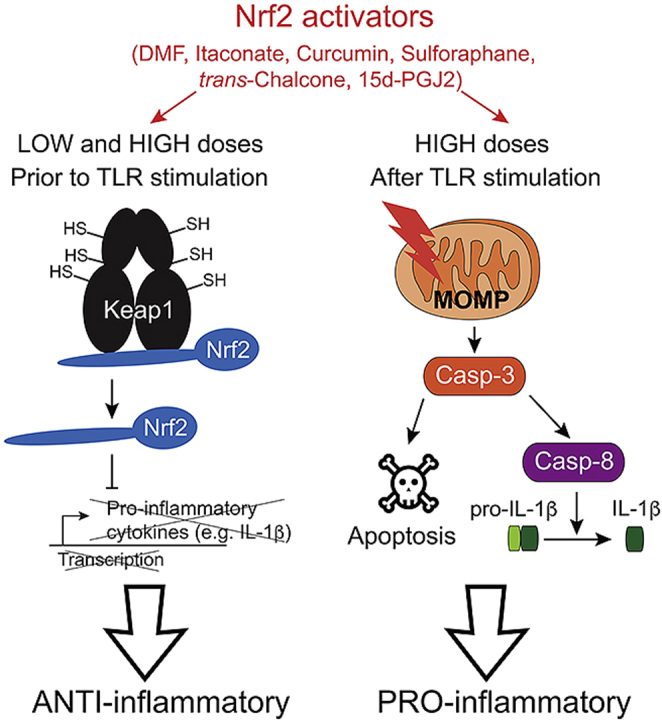
1. Introduction
Antioxidant systems scavenge reactive oxygen species (ROS) to maintain redox homeostasis in the cell. The nuclear factor erythroid 2-related factor 2 (Nrf2) is a key transcription factor that induces numerous antioxidant and cytoprotective genes in response to oxidative and xenobiotic stresses [1,2]. Under physiological conditions, Nrf2 is constitutively targeted for degradation through binding to Kelch-like ECH-associated protein 1 (Keap1), a substrate adaptor of E3 ubiquitin ligase. In the presence of oxidative stress, however, Nrf2 dissociates from Keap1, resulting in rapid Nrf2 stabilization, translocation into the nucleus, heterodimerization with small musculoaponeurotic fibrosarcoma (Maf) proteins, and transcription of antioxidant and cytoprotective genes [3,4]. In various murine models, Nrf2-deficiency exacerbates inflammatory responses and renders animals highly susceptible to septic shock [5], asthma [6], and emphysema [7]. In keeping with this, small molecules that activate Nrf2 by disrupting the interaction with Keap1 have been developed to reduce inflammation. For instance, the Nrf2 inducer Tecfidera, also known as dimethyl fumarate (DMF), is used in the clinics for treatment of multiple sclerosis (MS) [8]. Moreover, the endogenous metabolite itaconate has been proposed to activate Nrf2 and inhibit production of pro-inflammatory cytokines by macrophages [9], although the Nrf2-dependency of the mechanism has been challenged by others [10]. Therefore, Nrf2 activators open up new therapeutic strategies that can be exploited to treat inflammatory diseases.
Macrophages, monocytes, and dendritic cells (DCs) secrete a panel of inflammatory cytokines that orchestrate immune responses. The pro-inflammatory cytokines interleukin (IL)-1β and IL-18 are produced as biologically inactive precursors (pro-IL-1β and pro-IL-18, respectively) in response to sensing of pathogens or danger associated self-molecules via a variety of pattern recognition receptors (PRRs) of the innate immune system, such as Toll-like receptors (TLRs). Proteolytic cleavage of the pro-forms of IL-1β/IL-18 and subsequent secretion of the active forms require a second signal leading to the assembly of large multiprotein complexes known as inflammasomes [11,12]. The canonical NLRP3 inflammasome consists of a sensor (NLRP3 itself), an adaptor protein (ASC) and an effector (caspase-1) which gets activated and ultimately cleaves pro-IL-1β to its mature form [11]. In contrast to this canonical inflammasome pathway, lipopolysaccharide (LPS) from intracellular bacteria can be sensed by caspase-11, the mouse ortholog of human caspase-4 and caspase-5, resulting in non-canonical NLRP3 inflammasome activation [13,14]. Inflammasome activation and IL-1β release are generally associated with a programmed form of necrosis called pyroptosis [11]. The caspase-1 substrate gasdermin-D (GSDMD) has been shown to execute pyroptosis [[15], [16], [17]] and participate in IL-1β release by building pores on the plasma membrane [18,19]. Although the ultimate events leading to IL-1β production have been extensively studied in the last decade, how exactly inflammasomes are activated remains to be better defined. Interestingly, despite a protective role of Nrf2 in multiple inflammatory diseases, Nrf2-deficient mice display attenuated NLRP3 inflammasome responses due to impaired ASC speck formation [20,21]. In keeping with this pro-inflammatory role, Nrf2 has also been described to play a pathogenic role in diseases associated with chronic inflammation, such as atherosclerosis [20] and obesity [22,23]. Thus, a better understanding of this paradoxical opposite role of Nrf2 in inflammation may be beneficial for the utilization of Nrf2 activators in the treatment of inflammatory diseases.
In this work, we tested several classical Nrf2 activators including the metabolite itaconate for their potential role in regulating inflammatory responses by myeloid cells. We found that pulsing with low concentrations of various Nrf2 activators before TLR-dependent priming inhibits the transcription of pro-inflammatory cytokines, such as IL-1β, demonstrating an Nrf2-dependent anti-inflammatory bioactivity. By contrast, Nrf2 activators bearing electrophilic sites provided at high concentrations after TLR stimulation induce inflammatory apoptosis and result in caspase-8-dependent IL-1β maturation independently of Nrf2 itself. Taken together, we describe here two opposing bioactivities of electrophilic Nrf2 activators in inflammation.
2. Materials and methods
2.1. Mice
Nrf2−/− mice [1], NLRP3−/− mice [24], GSDMD−/− mice [25], GSDME−/− mice [26], Caspase-8fl/fl;Cre-ERT2 mice [27,28] (kindly provided by Olaf Gross), Caspase-11−/− mice [13], and Caspase-1/11−/− mice [29] (kindly provided by Wolf-Dietrich Hardt), Irg1−/− mice [30] (provided by Wolf-Dietrich Hardt with the permission of Rudi Balling) and C57BL/6J wild-type mice were housed in individually ventilated cages under specific pathogen free conditions at ETH Phenomics Center (EPIC; Zurich, Switzerland). GSDMD−/−GSDME−/− mice were generated by crossing GSDMD−/− and GSDME−/− animals. About 6-14 week-old age- and sex-matched mice (either male or female) were used for the experiments. All animal experiments were approved by the local animal ethics committee (Kantonales Veterinärsamt Zürich) and performed according to local guidelines (TschV, Zurich) and the Swiss animal protection law (TschG).
2.2. Flow cytometry
For analysis of cell death, the fixable viability dyes eFluor® 780 (eBioscience), propidium iodide (PI; Thermo Scientific) and Annexin-V-APC (BD Bioscience) were used for staining according to manufacturer's instructions. For stainings of cleaved caspase-3 or caspase-8, cells were fixed with 4% formalin for 10min, permeabilized with permeabilization buffer, stained for 20min at room temperature with anti-cleaved caspase-3 (5A1E; Cell Signalling Technology) or anti-cleaved caspase-8 (D5B2; Cell Signalling Technology) antibodies, and detected with an anti-rabbit secondary antibody labeled with the FITC fluorophore (Southern Biotech). For detection of caspase-9 activation, the CaspGLOW Fluorescein Active Caspase-9 (FITC-LEHD-FMK probe) Staining Kit was utilized according to the manufacturer's instructions. To address mitochondrial mass and membrane potential, cells were stained with MitoTracker™ Green FM and MitoTracker™ Deep Red FM (20nM; Thermo Scientific) for 30min at 37°C. Flow cytometry analysis was performed on a FACSCanto II (BD Bioscience), and data were then analyzed with FlowJo software (Tree Star).
2.3. Nrf2 activators, lipids and other reagents
The compounds dimethyl fumarate (DMF), trans-chalcone, curcumin, sulforaphane, tert-butylhydroquinone (tBHQ) and dimethyl itaconate (DMI) were purchased from Sigma-Aldrich. 4-OI was chemically synthetized as described elsewhere [9]. The prostaglandins 15d-PGJ2, PGE2 and PGD2 were purchased from Cayman Chemicals, while epoxycyclopentenone (EC), cyclo-epoxycyclopentenone (cEC), EC-reduced were synthetized as previously described [[31], [32], [33]]. The antioxidant N-acetylcysteine (NAC) and the pan-caspase inhibitor Q-VD-OPh (QVD) were purchased from Sigma-Aldrich, whereas the necroptosis inhibitor Nec-1s was from Merck Millipore.
2.4. Generation of bone marrow-derived dendritic cells (BMDCs) in vitro
Bone marrow-derived dendritic cells (BMDCs) were generated as previously described [34] and differentiated in RPMI-1640 medium (Gibco) supplemented with GM-CSF (supernatant from X63-GMCSF cell line), 2 mM l-glutamine (GE Healthcare), 10 mM HEPES (Lonza), 100 U/mL Penicillin, 100 μg/mL Streptomycin (Gibco), 10% FCS (Gibco). Fresh medium was supplemented on day 3 and day 6 of culture, and non-adherent cells were harvested and used in experiments on day 7 or 8. To delete caspase-8 in cells from the bone marrow of Caspase-8fl/fl;Cre-ERT2 mice, 4-OH-TAM (Sigma) at a final concentration of 25 μM was added at day 0. On day 3 and on day 6 on culture, fresh medium was given containing the same concentration of 4-OH-TAM.
2.5. Cytokine secretion assays in vitro
To assess the anti-inflammatory action of the Nrf2 activators, BMDCs were pulsed with various compounds for the indicated time, washed and stimulated with R837 (5 μg/ml; Tocris Bioscience) or LPS (400 ng/ml; InvivoGen) for the indicated times. To measure IL-1β secretion, cells were additionally stimulated with ATP (2 mM; Sigma) or nigericin (5 μM; Sigma) for 1h. For non-canonical inflammasome activation, BMDCs were electroporated with 30 μg/ml LPS using the Amaxa Mouse Macrophage Nucleofector Kit following the manufacturer's instructions. To address the pro-inflammatory effect of the Nrf2 activators, BMDCs were first primed with either LPS (400 ng/ml; InvivoGen) for 3 h before the addition of the Nrf2 activators for the indicated time. To measure secreted cytokines, supernatants were collected, clarified by centrifugation and stored at −20°C. Cell pellets were instead used for analysis by flow cytometry.
2.6. RNA analysis by real-time quantitative PCR
Total RNA was extracted using TRIzol (Life Technologies), followed by reverse transcription using GoScript Reverse Transcriptase (Promega) according to the manufacturer's instructions. Real-time quantitative PCR (RT-PCR) was performed using Brilliant SYBR Green (Stratagene) on an i-Cycler (Bio-Rad Laboratories) according to manufacturer's protocol. Expression was normalized to the housekeeping gene Tbp. The sequences of all used primers are listed in Supplementary Table 1.
2.7. Cytokine detection by ELISA
Culture supernatants were harvested at the indicated times, and the concentration of the indicated cytokines was assessed by sandwich ELISA. The following antibody pairs were used: IL-1β (B122 & 13-7112-85; eBioscience), IL-12/23p40 (C15.6 & C17.8; eBioscience), IL-6 (MP5-20F3 & MP5-32C11; Thermo Scientific), and TNF-α (G281-2626 [BD pharmingen] & 13-7341-85 [Thermo Scientific]). To assess IL-1β secretion upon stimulation with various Nrf2 activators (IL-1β levels in the pg/ml range), the IL-1β Mouse Uncoated ELISA Kit (88-7013-76, Thermo Scientific) was used.
2.8. Western blotting
Cells were lysed on ice with RIPA buffer (20 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1 mM Na3VO4, 1% Triton X-100, supplemented with protease inhibitor [Sigma-Aldrich]). Samples were then spun for 10min at 4°C to remove all cell debris. Protein concentrations were determined using the PierceTM BCA Protein Assay Kit (Thermo Scientific). For the quantification of IL-1β in the supernatants, serum-free supernatants were precipitated with 10% TCA (vol/vol) for 30min on ice. Precipitated proteins were pelleted, washed with ice-cold acetone, air-dried, and resuspended in SDS-PAGE sample buffer. Whole cell extracts (30 μg of proteins) and precipitated supernatants were fractionated by SDS-PAGE and transferred to a Polyvinylidene difluoride (PVDF) membrane using a transfer apparatus according to manufacturer's instructions (Bio-Rad). After blocking with 4% nonfat milk in TBST (50mM Tris, pH 8.0, 150 mM NaCl, 0.1% Tween 20) for 45min, the membranes were washed once with TBST and incubated with primary antibodies (1:1000 in TBST with 4% BSA) at 4°C for 15 h. Membranes were washed three times for 10min and incubated with horseradish peroxidase-conjugated anti-rabbit or anti-goat antibodies for 1h. After washing with TBST, blots were developed with the ECL system (Thermo Scientific) according to manufacturer's instructions. Antibodies for Caspase-8 (#4927; Cell Signalling Technology; 1:1′000), IL-1β (AF-401-NA, R&D; 1:1′000), GSDMD (EPR19828; Abcam; 1:1′000), GSDME (EPR19859; Abcam; 1:1′000), and β-Actin (AC-15; Sigma-Aldrich) were used in this study.
2.9. Statistical analysis
Two-group comparisons were assessed with a Student's t-test (two-tailed, unpaired). Multi-group comparisons were assessed by one-way ANOVA followed by either Tukey's or Dunnett's corrections. The method of statistical evaluation and the significance levels are indicated in each figure legend.
3. Results
3.1. Nrf2 activators and the itaconate surrogate 4-OI inhibit transcription of pro-inflammatory cytokines such as IL-1β and IL-12
The role of Nrf2 and its activators in inflammation and the mechanism by which they regulate inflammatory responses remain controversially discussed and incompletely understood. To study this question, we tested various Nrf2 activators for their capacity to regulate the production of pro-inflammatory cytokines by myeloid cells. For this purpose, we made use of the classical Nrf2 activators DMF, trans-chalcone, sulforaphane, tert-butylhydroquinone (tBHQ) and curcumin (Fig. 1A–E). As a control, we also included cyclo-epoxycyclopentenone (cEC) (Fig. 1F), which had previously been identified as a potent anti-inflammatory component arising from the oxidation of the phospholipid 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine (PAPC) [35]. Experimentally, we pulsed bone marrow-derived dendritic cells (BMDCs) for 1 h with these Nrf2 activators before stimulation with either the TLR7 ligand R837 or the TLR4 ligand LPS followed by ATP to investigate IL-1β production (Fig. 1G). We found that all the tested Nrf2 activators inhibit IL-1β production in a concentration-dependent manner without affecting cell survival, although the oxidized lipid cEC displayed the highest anti-inflammatory bioactivity (Fig. 1H–J and Supplementary Fig. 1A–F).
Fig. 1.

Classical Nrf2 activators and the itaconate surrogate 4-OI inhibit the production of pro-inflammatory cytokines. (A-F) Chemical structures of the utilized Nrf2 activators: dimethyl fumarate (DMF) (A), trans-chalcone (B), sulforaphane (C), tBHQ (D), curcumin (E), and cEC (F). (G) Experimental setup for IL-1β detection. (H) Dose-response curves of IL-1β secretion by ELISA in BMDCs pulsed for 1 h with the indicated Nrf2 activators followed by stimulation with R837 and ATP (n = 3). (I) IC50 values of the compounds shown in (H) (n = 3). (J) BMDCs were pulsed for 1h with the indicated compounds and subsequently stimulated for 5 h with LPS and for 1h with ATP. IL-1β secretion was then assessed by ELISA (n = 3). (K) Chemical structure of 4-octyl itaconate (4-OI). (L) Dose-response curves of IL-1β secretion by ELISA in BMDCs pulsed for 1 h or 3 h or 8 h with 4-OI followed by stimulation with R837 and ATP. cEC was used as a control (n = 3). (M) Chemical structure of dimethyl itaconate (DMI). (N) IL-1β secretion measured by ELISA in BMDCs treated with DMI and 4-OI for 3 h followed by stimulation with R837 and ATP according to the experimental setup in (G) (n = 3). Bar graphs show mean + standard deviation. Data are representative of two independent experiments. For each panel, a representative experiment with replicates of in vitro culture conditions is shown. One-way ANOVA followed by Dunnett's correction (comparison to the control 0 μM) was used in (J,N): *p ≤ 0.0332; **p ≤ 0.0021; ***p ≤ 0.0002; ****p ≤ 0.0001.
The electrophilic properties of the endogenous metabolite itaconate have been shown to be critical for the suppression of inflammation [9,10], but it remains unclear whether this is mediated by the activation of Nrf2 [9] or by the inhibition of NF-κB inhibitor zeta (IκBζ) [10]. As cEC also possesses electrophilic properties incorporated in the endo- and exocyclic enones, we next aimed at comparing the potency of the anti-inflammatory bioactivity of itaconate and the known Nrf2 activator cEC. To this end, we synthetized the cell-permeable derivative 4-octyl itaconate (4-OI) (Fig. 1K), which has been recently described by the group of Luke O'Neill to be a suitable itaconate surrogate [9]. We observed that cEC displays a more potent anti-inflammatory bioactivity in inhibiting IL-1β responses compared to 4-OI (Fig. 1L). Notably, 4-OI treatment for 1 h had only very moderate anti-inflammatory effects without affecting cell survival (Supplementary Fig. 1G), and longer incubations were required to achieve increased bioactivities (Fig. 1L). We additionally tested the doubly esterified itaconate derivative dimethyl itaconate (DMI; Fig. 1M), which is not metabolized into itaconate in macrophages [36]. Similar to 4-OI, DMI also inhibited IL-1β production, but to a slightly lower extent than 4-OI (Fig. 1N and Supplementary Fig. 1H).
3.2. Nrf2 activators and the itaconate surrogate 4-OI activate Nrf2 to inhibit inflammation
We next aimed to understand how Nrf2 activators inhibit inflammation. We first confirmed that treatment of BMDCs with these compounds results in increased expression of the Nrf2 target gene Nqo1, which did not occur in Nrf2-deficient BMDCs (Fig. 2A). Furthermore, we found that all tested compounds inhibit TLR-induced transcription of the pro-IL-1β gene (Fig. 2B) and production of the pro-IL-1β protein (Fig. 2C). This anti-inflammatory activity was abolished in Nrf2-deficient BMDCs (Fig. 2B), and this was not limited to IL-1β, as IL-12p40 production was inhibited by the tested Nrf2 activators in a similar concentration-dependent manner in WT but not Nrf2-deficient cells (Fig. 2D–I). While the mechanism of the itaconate activity has been controversially discussed [9,10], our data are consistent with the requirement of Nrf2 signaling for the bioactivity of itaconate, as the anti-inflammatory function of 4-OI was abolished in the absence of Nrf2 (Fig. 2B and J). Altogether, these results indicate that classical Nrf2 activators and the metabolite itaconate interfere with the transcription of pro-inflammatory cytokines in an Nrf2-dependent manner.
Fig. 2.

Nrf2 activators and the itaconate surrogate 4-OI inhibit transcription of pro-inflammatory cytokines in a Nrf2-dependent manner. (A,B) Nqo1 (A) and IL-1β (B) expression levels determined by real-time PCR in WT and Nrf2-deficient BMDCs pulsed with the indicated compounds (cEC, 1.1 μM; 4-octyl itaconate [4-OI], 320 μM; curcumin, 20 μM; dimethyl fumarate [DMF], 40 μM; sulforaphane, 20 μM; tBHQ, 20 μM; and trans-chalcone, 80 μM) for 1 h and stimulated with R837 for 4 h. The control “no R837” represents no R837 stimulation (nd, not detected). The expression was normalized to the housekeeping gene Tbp (n = 2–3). (C) Immunoblotting for pro-IL-1β in cell lysates of BMDCs, which were pulsed with the indicated compounds (cEC, 2 μM; tBHQ, 100 μM; curcumin [Curc.], 50 μM; DMF, 100 μM; and sulforaphane [Sulf.], 50 μM) for 1 h and stimulated with R837 for 6 h. β-Actin was utilized as a loading control. (D-J) WT and Nrf2-deficient BMDCs were pulsed with cEC (D), DMF (E), trans-chalcone (F), sulforaphane (G), curcumin (H), tBHQ (I) and 4-OI (J) for 1 h and stimulated with R837 for 17 h. IL-12p40 in the supernatants was then detected by ELISA (n = 3). Bar graphs show mean + standard deviation. Data are representative of two independent experiments. For each panel, a representative experiment with replicates of in vitro culture conditions is shown. One-way ANOVA followed by Dunnett's correction (comparison to the control 0 μM) was used in (A): *p ≤ 0.0332; **p ≤ 0.0021; ***p ≤ 0.0002; ****p ≤ 0.0001. Student's t-test (two-tailed, unpaired) was used for the comparison of two groups (B,D-J): *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001; ****, P ≤ 0.0001.
3.3. High concentrations of Nrf2 activators including the itaconate surrogate 4-OI induce non-toxic cell death and concomitant IL-1β cleavage
We previously reported that Nrf2-activating epoxyisoprostanoids such as cEC possess a dual role in inflammation by inhibiting IL-1β transcription at low doses and inducing IL-1β processing and release at high doses [37]. Therefore, we next wondered whether the two opposing activities are restricted to epoxyisoprostanoids or are generally shared by Nrf2 activators. Above we showed that exposure of BMDCs to a variety of well-established Nrf2 activators prior TLR-triggering inhibited pro-inflammatory cytokine (i.e. IL-12 and IL-1β) production. We next stimulated BMDCs with LPS prior to treatment with increasing concentrations of the Nrf2 activators described above (Fig. 1A–F) and measured IL-1β release (Fig. 3A). Indeed, similarly to cEC (Fig. 3B), we found that DMF, curcumin, sulforaphane and trans-chalcone (Fig. 3C–F) induced IL-1β release at high doses. Moreover, IL-1β production was associated with a partial loss in cell viability (Fig. 3G–K), consistent with the well-established association of IL-1β release with pyroptotic cell death [[15], [16], [17]]. IL-1β processing was also validated by western blot (Fig. 3L). Importantly, production of TNF-α, IL-12p40 and IL-6 was not increased upon cEC treatment (Supplementary Fig. 2A–C), demonstrating that high doses of Nrf2 activators do not generally increase pro-inflammatory responses but the activity is specific for IL-1β. Finally, we wondered whether 4-OI, which has been extensively described as a potent anti-inflammatory compound (9, 10), also possesses the capacity to promote IL-1β responses. Indeed, 4-OI at high doses resulted in IL-1β release and loss in cell viability (Fig. 3M and N). Although slightly less potent than 4-OI, DMI also displayed this pro-inflammatory function at high concentrations (Fig. 3O).
Fig. 3.

High concentrations of electrophilic Nrf2 activators and the itaconate surrogate 4-OI upon LPS priming induce cell death and IL-1β release. (A) Experimental setup used to study the induction of IL-1β release by Nrf2 activators. (B–K) IL-1β secretion measured by ELISA (B–F) and cell survival addressed by the incorporation of PI by flow cytometry (G-K) in BMDCs stimulated with cEC (B,G), DMF (C,H), curcumin (D,I), sulforaphane (E,J), and trans-chalcone (F,K) according to (A) (n = 3). (L) Immunoblotting for IL-1β in cell lysates and supernatants of BMDCs, which were stimulated according to the experimental setup in (A). β-Actin was utilized as a loading control. DMF, 120 μM; Curcumin (Curc.), 240 μM; 4-octyl itaconate (4-OI), 480 μM. (M,N) IL-1β secretion measured by ELISA (M) and cell survival addressed by the incorporation of PI by flow cytometry (N) in BMDCs stimulated with 4-OI according to (A) (n = 3). (O) IL-1β secretion measured by ELISA in BMDCs stimulated with either 4-OI or dimethyl itaconate (DMI) according to (A) (n = 3). (P,Q) Nqo1 (P) and IL-1β (Q) expression levels determined by real-time PCR in WT BMDCs, which were treated with high doses of cEC (8 μM), 4-OI (480 μM), DMF (120 μM) and sulforaphane (Sulf.; 60 μM) either before or after LPS stimulation as indicated. The expression was normalized to the housekeeping gene Tbp (n = 3). Bar graphs show mean + standard deviation. Data are representative of two independent experiments. For each panel, a representative experiment with replicates of in vitro culture conditions is shown. One-way ANOVA followed by Dunnett's correction (comparison to the control 0 μM) was used in (B–K,M-Q): *p ≤ 0.0332; **p ≤ 0.0021; ***p ≤ 0.0002; ****p ≤ 0.0001.
As we showed that low doses of Nrf2 activators inhibit transcription of IL-1β, while high concentrations promote its processing into the mature form, we next wondered whether high doses of these compounds would still activate Nrf2 or would suppress it to allow apoptosis and IL-1β release. To address this, we treated BMDCs with 4-OI or DMF at high doses either before or after LPS stimulation. We observed that high concentrations of these compounds, which we had previously shown to induce inflammatory apoptosis, resulted in the activation of the Nrf2 pathway measured as the induction of Nqo1 expression independently of whether cells were treated before or after LPS stimulation (Fig. 3P). Similarly to what we have described at low doses, 4-OI and DMF also inhibited the transcription of IL-1β at high concentrations (Fig. 3Q). Therefore, these results indicate that in our experimental assay, in which we first prime cells with LPS and subsequently stimulate them with various Nrf2 activators at high doses (Fig. 3A), the inhibitory effect on IL-1β transcription is masked by the 3-h treatment with LPS. Indeed, the latter induces IL-1β transcription before the addition of the Nrf2 activators that would otherwise inhibit it.
3.4. Electrophilic properties are required for the induction of IL-1β responses by Nrf2 activators at high doses
In contrast to DMF, curcumin, sulforaphane, trans-chalcone, 4-OI and DMI, which bear electrophilic sites such as α,β-unsaturated carbonyl moieties, we found that the Nrf2 activator tBHQ at high doses did not promote IL-1β secretion (Fig. 4A and B). Additionally, we observed that tBHQ does not display an intermediate degree of cell death, as at 240 μM cells only marginally died, while at 480 μM their survival was massively reduced (Fig. 4C). tBHQ lacks electrophilic sites shared by the other Nrf2 activators, and it has been proposed to induce Nrf2 activation by the induction of mitochondrial oxidative stress [38] or by its auto-oxidation to tert-butylbenzoquinone (TBQ), which bears electrophilic sites and can then covalently modify Keap1 [39]. We observed that treatment with the antioxidant N-acetylcysteine (NAC) partially inhibited the induction of the Nrf2 target gene Nqo1 (Fig. 4D), possibly suggesting that tBHQ activates Nrf2 via ROS induction. Alternatively, a quinone metabolite of tBHQ may directly react with NAC, and the resulting conjugate loses its electrophilicity towards other intracellular proteins such as Keap1.
Fig. 4.

Electrophilic sites of Nrf2 activators are required to trigger IL-1β release at high doses. (A) Chemical structure of tBHQ. (B,C) IL-1β secretion measured by ELISA (B) and cell survival addressed by the incorporation of PI by flow cytometry (C) in BMDCs primed with LPS for 3 h and subsequently stimulated with tBHQ for additional 17 h (n = 3). (D) Nqo1 expression levels determined by real-time PCR in WT BMDCs, which were pre-treated with N-acetyl cysteine (NAC) 2 h prior stimulation with tBHQ at the indicated concentrations for 2 additional hours. The expression was normalized to the housekeeping gene Tbp (n = 3). (E) Chemical structures of EC (left) and EC-reduced (right). (F) IL-1β secretion measured by ELISA in BMDCs primed with LPS for 3 h and subsequently stimulated with the indicated compounds for additional 17 h (n = 3). (G) Chemical structures of the prostaglandins 15d-PGJ2 (top-left), PGE2 (bottom-left), and PGD2 (right). (H) IL-1β secretion measured by ELISA in BMDCs primed with LPS for 3 h and subsequently stimulated with the indicated compounds for additional 17 h (n = 3). Bar graphs show mean + standard deviation. Data are representative of two independent experiments. For each panel, a representative experiment with replicates of in vitro culture conditions is shown. One-way ANOVA followed by Dunnett's correction (comparison to the control 0 μM) was used in (B,C,F,H): *p ≤ 0.0332; **p ≤ 0.0021; ***p ≤ 0.0002; ****p ≤ 0.0001. Student's t-test (two-tailed, unpaired) was used for the comparison of two groups (D): *, P ≤ 0.05.
To better expand on why tBHQ does not induce IL-1β responses, we made use of the previously described oxidized lipid epoxycyclopentenone (EC) [35] and its reduced variant in which both the endo- and exocyclic enones have been removed (Fig. 4E). Interestingly, treatment with high doses of EC but not EC-reduced resulted in IL-1β accumulation in the culture supernatant (Fig. 4F). Similarly, the prostaglandin 15d-PGJ2 that contains endo- and exocyclic enones induced IL-1β secretion [37], while the prostaglandins PGE2 and PGD2 lacking electrophilic sites did not show this bioactivity (Fig. 4G and H). Overall, our data indicate that Nrf2 activators bearing electrophilic sites have the capacity to trigger IL-1β production upon LPS priming. Moreover, this effect is exclusively observed at higher concentrations than the ones required to inhibit IL-1β transcription.
3.5. The itaconate surrogate 4-OI at high dose induces apoptotic cell death and IL-1β release independently of the classical inflammasome pathway
Itaconate has recently emerged as an important anti-inflammatory metabolite [9,10]. Since the results shown above (Fig. 2B and J) and findings by Luke O'Neill and coworkers [9] suggest that the electrophilicity of itaconate mediates Nrf2 activation, we decided to focus mainly on this metabolite to study the shared molecular mechanism by which electrophilic Nrf2 activators trigger IL-1β secretion. Interestingly, we found that WT and Nrf2-deficient BMDCs undergo 4-OI-induced cell death to a similar extent (Fig. 5A), suggesting that the pro-inflammatory pathway is Nrf2-independent. The heat shock factor 1 (Hsf1), a transcription factor that coordinates stress-induced responses, has been shown to be activated by electrophiles [40,41]. Therefore, we investigated whether the Nrf2 activators used in our assay could also act through Hsf1. While Hsf1 expression was unchanged upon treatment with DMF and 4-OI (Supplementary Fig. 3A), consistent with other studies [40], expression of Hsf1 target genes (i.e. Hspb8 and Hspa1a) was upregulated after 20 h of stimulation, but not at earlier time points, indicating Hsf1 activity (Supplementary Fig. 3B and C). Since we generally observed cell death at earlier time points, these results suggest that the late Hsf1 activation (at 20 h) is likely a consequence of cellular stress due to massive death rather than playing any role in the induction of inflammatory apoptosis.
Fig. 5.

High concentrations of the itaconate surrogate 4-OI induce apoptosis and IL-1β release independently of the inflammasome. (A) The survival of WT and Nrf2-deficient BMDCs, which were primed with LPS for 3 h and stimulated with 4-octyl itaconate (4-OI) for additional 17 h, was assessed by PI incorporation by flow cytometry. Cells were treated with 2-fold increasing concentrations of 4-OI (0, 15, 30, 60, 120, 240, 480 μM) (n = 3). (B) Canonical inflammasome activation in Caspase-1/11-, Caspase-11-, NLRP3- and GSDMD-deficient BMDCs was investigated by LPS priming (3 h) followed by stimulation with ATP for 1 h (n = 3). (C) Non-canonical inflammasome activation in Caspase-1/11-, Caspase-11-, NLRP3- and GSDMD-deficient BMDCs was assessed by LPS electroporation (30 μg/ml; n = 3). (D,E) IL-1β secretion in the supernatant measured by ELISA (D) and cell survival assessed by PI incorporation by flow cytometry (E) in BMDCs primed with LPS for 3 h and stimulated with 4-OI for additional 17 h. BMDCs from the indicated genotypes were stimulated with 2-fold increasing concentrations of 4-OI (0, 2, 4, 8, 15, 30, 60, 120, 240, 480 μM; n = 3). (F,G) IL-1β release in the supernatant measured by ELISA (F) and cell survival assessed by PI incorporation by flow cytometry (G) in BMDCs primed with LPS for 3 h in the presence of either the apoptosis inhibitor QVD (100 μM) or the necroptosis inhibitor Nec-1s (100 μM) and stimulated with 4-OI for additional 17 h (n = 3). (H) BMDCs were primed with LPS for 3 h and treated with 4-OI for additional 8 h. Shown is the degree of apoptosis assessed by the viability dye eFluor780 and Annexin-V. Bar graphs show mean + standard deviation. Numbers in the FACS plots indicate the percentage of the depicted gates. Data are representative of three (A-E) and two (F–H) independent experiments. For each panel, a representative experiment with replicates of in vitro culture conditions is shown (A-G). One-way ANOVA adjusted by Tukey's multiple comparison test was used in (F,G): *p ≤ 0.0332; **p ≤ 0.0021; ***p ≤ 0.0002; ****p ≤ 0.0001.
IL-1β processing and release have been reported to require cytoplasmic multi-protein complexes called inflammasomes [42]. To test whether itaconate-induced IL-1β release requires inflammasome activation, we primed WT, NLRP3−/−, Caspase-1/11−/− and Caspase-11−/− BMDC with LPS followed by 4-OI stimulation. Although canonical and non-canonical inflammasome activations result in the expected phenotype (Fig. 5B and C), we surprisingly found that 4-OI-triggered IL-1β release and cell death were independent of the classical inflammasome components (Fig. 5D and E). Moreover, the pyroptosis effector protein gasdermin D (GSDMD) (15–17) was also dispensable for itaconate-induced IL-1β production and cell death (Fig. 5D and E). Overall, these results indicate that itaconate induces IL-1β release independently of the classical inflammasome pathway.
We next wondered whether cell death inhibitors might prevent IL-1β secretion by itaconate. Interestingly, we found that incubation of BMDCs with the pan-caspase inhibitor Q-VD-OPh (QVD) completely abolished 4-OI-induced IL-1β release by BMDCs, while inhibition of necroptosis using the RIPK1 inhibitor Necrostatin-1s (Nec-1s) showed no effect (Fig. 5F). Furthermore, the presence of QVD but not Nec-1s increased cell survival (Fig. 5G). As QVD is a well-established inhibitor of apoptosis, our data are in line with itaconate triggering apoptotic cell death at high concentrations. In keeping with this, we observed accumulation of phosphatidylserine residues on the outer leaflet of the cytoplasmic membrane (Annexin-V staining), which is a typical feature of cells undergoing apoptosis, upon treatment of BMDCs with 4-OI at high doses (Fig. 5H). Altogether, these results indicate that the Nrf2 activator and itaconate surrogate 4-OI induces IL-1β production during apoptotic cell death independently of the classical inflammasome pathway and of Nrf2 itself.
3.6. Itaconate-triggered apoptosis is characterized by loss of mitochondrial membrane potential leading to caspase-3-dependent caspase-8 activation
Our data so far indicate that high concentrations of itaconate result in apoptosis. To further expand on this, we measured the activation of the executioner apoptotic caspase-3 [43]. In keeping with the appearance of phosphatidylserine residues on the outer leaflet of the cytoplasmic membrane (Fig. 5H), LPS-primed BMDCs treated with high concentrations of 4-OI displayed caspase-3 activation (Fig. 6A).
Fig. 6.

High concentrations of the itaconate surrogate 4-OI trigger mitochondrial apoptosis leading to caspase-3 and caspase-8 activation. (A-C) Cleavage of caspase-3 (A) and of caspase-8 (B), and loss in mitochondrial membrane potential (C) were assessed by flow cytometry in BMDCs primed with LPS for 3 h and treated with 4-octyl itaconate (4-OI) for additional 10 h. (D) Cleavage of caspase-3 (top) and caspase-8 (bottom) in BMDCs primed with LPS for 3 h in the presence of QVD (100 μM) and subsequently stimulated with 4-OI for additional 8 h. (E) Loss in mitochondrial membrane potential assessed by flow cytometry in BMDCs primed with LPS for 3 h in the presence or absence of QVD (100 μM) and stimulated with 4-OI for additional 8 h. (F) Caspase-9 activation with the FITC-LEHD-FMK probe in BMDCs primed with LPS for 3 h and subsequently stimulated with 4-OI for additional 6 h. (G) Western blot showing the deletion of GSDME in GSDME−/− and GSDME−/−GSDMD−/− BMDCs, and of GSDMD in GSDME−/−GSDMD−/− BMDCs. β-Actin was utilized as a loading control. (H) Cell survival addressed by the incorporation of PI by flow cytometry (left) and IL-1β secretion measured by ELISA (right) in WT, GSDME−/− and GSDME−/−GSDMD−/− BMDCs primed with LPS for 3 h and treated with 4-OI for additional 17 h (n = 3). Bar graphs show mean + standard deviation. Numbers in the FACS plots indicate the percentage of the depicted gates. Data are representative of three (A-E) and two (F–H) independent experiments. For each panel, a representative experiment with replicates of in vitro culture conditions is shown. One-way ANOVA adjusted by Tukey's multiple comparison test was used in (H): ns, not significant.
Mechanistically, caspase-3 cleavage and apoptosis execution can be elicited by caspase-8 or the mitochondrial pathway [43]. Interestingly, we found that stimulation of LPS-primed BMDCs with 4-OI resulted in the cleavage of caspase-8 (Fig. 6B). Concomitantly, we also measured a reduction in mitochondrial membrane potential, which is a typical feature of mitochondrial outer membrane permeabilization (MOMP) during the engagement of the mitochondrial pathway (Fig. 6C). Treatment with QVD completely abolished the activation of caspase-3 and -8, in line with a potential role of these caspases in 4-OI-induced cell death and IL-1β release (Fig. 6D). The fact that both the caspase-8 and the mitochondrial pathways are activated by 4-OI is consistent with the observation that both pathways can feed into each other as an amplification mechanism [43,44]. Treatment with QVD, which prevented caspase-3 and -8 activation (Fig. 6D), only marginally influenced MOMP, indicating that the induction of the mitochondrial pathway occurs before and independently of caspase-8 activation (Fig. 6E). Concomitantly, we detected the activation of the initiator caspase-9, thus suggesting that the latter is the upstream activator of caspase-3 in response to high doses of 4-OI (Fig. 6F). Finally, we also verified that other electrophilic Nrf2 activators, namely DMF, trans-chalcone, curcumin and sulforaphane, display a similar potential to activate caspase-3/-8 and to cause MOMP (Supplementary Fig. 4A–C), thus indicating a shared mechanism that induces IL-1β production among several well-known Nrf2-activating compounds. Taken together, these data demonstrate that Nrf2 activators and in particular the metabolite itaconate at high concentrations can engage the mitochondrial pathway of apoptosis by inducing MOMP, thus resulting in caspase-3-dependent caspase-8 activation.
By contrast, we observed that tBHQ at 480 μM, the only concentration at which we observed cell death, caused a massive loss in mitochondrial membrane potential in the absence of caspase-3 and caspase-8 activation (Supplementary Fig. 4A–C). This is reminiscent of what we have previously reported for extremely high concentrations of cEC [37], where rapid primary necrosis is induced even before there would be the time to cleave caspases in the cell. This indicates that tBHQ does not display a concentration window at which an intermediate degree of cell death is induced, characterized by inflammatory apoptosis and IL-1β release, but it directly becomes toxic at higher concentrations leading to primary necrosis.
While GSDMD is well known to be cleaved by caspase-1, gasdermin E (GSDME) has been shown to possess a caspase-3 cleavage motif [45]. Therefore, GSDME processing occurs in response to apoptotic stimuli, leading to pyroptosis or secondary necrosis [46,47]. As high doses of 4-OI resulted in caspase-3 activation, we next wondered whether the genetic ablation of GSDME would restore cell survival and inhibit IL-1β release in response to 4-OI. However, we found that deletion of GSDME in both WT and GSDMD-deficient animals did not affect cell survival and IL-1β production upon 4-OI treatment (Fig. 6G and H). Thus, these results indicate that high concentrations of Nrf2 activators induce secondary necrosis independently of GSDME and GSDMD. This observation is consistent with a recent study, which showed that, although it is properly cleaved, GSDME does not contribute to cell death in bone marrow-derived macrophages (BMDMs) lacking GSDMD [48].
3.7. 4-OI-mediated anti- and pro-inflammatory functions are independent of Irg1
4-OI has been reported to be hydrolyzed to itaconate by cellular esterases in LPS-activated macrophages, thus making it a suitable cell-permeable surrogate of itaconate [9]. However, a recent study proposed that 4-OI does not generate free itaconate and that any increase is due to endogenous itaconate being generated (for instance through immune‐responsive gene 1 [Irg1] function) [49]. To test this latter hypothesis in our system, we generated BMDCs from WT and Irg1-deficient animals, pulsed them with 4-OI followed by stimulation with LPS and ATP (Fig. 7A). We found that the anti-inflammatory property of 4-OI was not abolished in the absence of Irg1 (Fig. 7B). Therefore, we next decided to address whether this was also the case for the pro-inflammatory activity of 4-OI at high doses (Fig. 7C). We observed that WT and Irg1-deficient cells displayed a similar degree of MOMP (Fig. 7D), of activation of caspase-8 (Fig. 7E) and caspase-3 (Fig. 7F), and underwent cell death to a similar extent (Fig. 7G). Moreover, 4-OI-induced IL-1β release was also unchanged in the absence of Irg1 (Fig. 7H). Taken together, these data indicate that 4-OI does not act by boosting endogenous itaconate production via Irg1 but suggest that it exerts its functions by itself or by a direct conversion into itaconate via cellular esterases.
Fig. 7.

The anti- and pro-inflammatory functions of the itaconate surrogate 4-OI are independent of Irg1. (A) Experimental setup to study the anti-inflammatory properties of 4-octyl itaconate (4-OI). (B) IL-1β secretion measured by ELISA in WT and Irg1-deficient BMDCs stimulated with 4-OI according to the experimental setup in (A) (n = 3). (C) Experimental setup to study the pro-inflammatory properties of 4-OI. (D-F) Loss in mitochondrial membrane potential (D) and cleavage of caspase-8 (E) and caspase-3 (F) were assessed by flow cytometry in WT and Irg1-deficient BMDCs primed with LPS for 3 h followed by stimulation with 4-OI for additional 8 h. (G,H) Cell survival addressed by the incorporation of PI by flow cytometry (G) and IL-1β release by ELISA (H) in WT and Irg1-deficient BMDCs stimulated with 4-OI according to the experimental setup in (C) (n = 3). Bar graphs show mean + standard deviation. One-way ANOVA followed by Dunnett's correction (comparison to the control 0 μM) was used in (B): ***p ≤ 0.0002; ****p ≤ 0.0001. Student's t-test (two-tailed, unpaired) was used for the comparison of two groups (G): *, P ≤ 0.05. One-way ANOVA adjusted by Tukey's multiple comparison test was used in (H): **p ≤ 0.0021; ***p ≤ 0.0002.
3.8. Caspase-8 cleaves IL-1β in response to high doses of the itaconate surrogate 4-OI
To test whether caspase-8 is responsible for IL-1β release downstream of 4-OI, we isolated BMDCs from Caspase-8fl/fl;Cre-ERT2 and WT mice and deleted caspase-8 with 4-hydroxytamoxifen (4-OH-TAM) in vitro (Fig. 8A). Although loss of caspase-8 did not affect canonical NLRP3 activation (Fig. 8B), 4-OI-induced IL-1β production was massively reduced in the absence of caspase-8 (Fig. 8C). Furthermore, as observed with cyclopentenone isoprostanoids [37], 4-OI-driven cell death (Fig. 8D), cleavage of caspase-3 (Fig. 8E) and loss of mitochondrial membrane potential (Fig. 8F) were only partially reduced in Caspase-8-deficient BMDCs, thus implying a similar positive feedback loop where caspase-8 indirectly amplifies the induction of MOMP. The BCL-2 homology 3 (BH3)-interacting domain death agonist (BID) is very likely involved in this amplification loop [[37], [44]]. Taken altogether, these results demonstrate that Nrf2 activators, such as the endogenous metabolite itaconate, have the capacity to initiate intrinsic apoptosis, resulting in caspase-8 activation which ultimately cleaves IL-1β and promotes its release by secondary necrosis.
Fig. 8.

High concentrations of the itaconate surrogate 4-OI promote caspase-8-mediated IL-1β release. (A-F) BMDCs were differentiated in vitro from the bone marrow of Caspase-8fl/fl;Cre-ERT2 (Caspase-8 KO) and control Caspase-8fl/fl (WT) mice in the presence of 4-OH-TAM to delete the Caspase-8 gene. (A) Western blot showing the deletion efficiency of caspase-8 in BMDCs. (B) IL-1β secretion after classical NLRP3 stimulation (5 h priming with either LPS or R837 followed by 1-h stimulation with ATP) assessed by ELISA (n = 3). (C,D) IL-1β secretion measured by ELISA (C) and cell survival addressed by the PI incorporation by flow cytometry (D) in WT or Caspase-8-deficient BMDCs primed with LPS for 3 h and stimulated with 4-octyl itaconate (4-OI) for additional 17 h (n = 3). (E,F) Percentage of caspase-3 cleavage (E) and loss in mitochondrial membrane potential (F) assessed by flow cytometry in WT or Caspase-8-deficient BMDCs primed with LPS for 3 h and stimulated with 4-OI for additional 17 h (n = 3). Bar graphs show mean + standard deviation. Numbers in the FACS plots indicate the percentage of the depicted gates. Data are representative of two independent experiments. For each panel, a representative experiment with replicates of in vitro culture conditions is shown. Student's t-test (two-tailed, unpaired) was used for the comparison of two groups (B): ns, not significant. One-way ANOVA adjusted by Tukey's multiple comparison test was used in (C-E): *p ≤ 0.0332; **p ≤ 0.0021; ***p ≤ 0.0002; ****p ≤ 0.0001.
4. Discussion
Nrf2 activators are generally utilized as anti-inflammatory compounds to treat various inflammatory diseases. However, here we describe that they can both inhibit and promote inflammation depending on their dose and on the time of stimulation. Our data show that low doses of electrophilic Nrf2-stimulating compounds lead to an anti-inflammatory phenotype. This is consistent with the observation that electrophilic compounds alkylate Keap1 resulting in Nrf2 stabilization and activation of an antioxidant and anti-inflammatory transcriptional program [50]. However, the precise mechanism by which Nrf2 activators lead to inhibition of inflammation is not completely resolved. It has been proposed that Nrf2 interferes with the transcriptional upregulation of pro-inflammatory cytokines in macrophages upon LPS priming by binding to the proximity of pro-inflammatory genes and inhibiting the recruitment of RNA polymerase II [51]. Additionally, Nrf2 induction by oxidized lipid mediators inhibits the activation of the NF-κB and mitogen-activated protein kinase (MAPK) pathways and the ultimate production of pro-inflammatory cytokines [37]. Importantly, by comparing the activity of the various Nrf2 activators, our study identifies cyclopentenone-containing lipid species as the molecules possessing the most potent anti-inflammatory bioactivity and suggests that their utilization for the treatment of pro-inflammatory diseases may result in beneficial outcomes. It is important to mention, however, that our comparative analysis on the strength of the tested Nrf2 activators does not consider half-lives and bioavailability of the compounds in vivo.
In contrast to low doses, stimulation of BMDCs with high concentrations of Nrf2 activators results in an inflammatory program characterized by the induction of apoptosis. Our data indicate that these compounds, independently of their Keap1-alkylating and Nrf2-activating capabilities, induce MOMP resulting in cleavage of the executioner apoptotic caspase-3, which then triggers apoptotic cell death as well as caspase-8 activation that mediates cleavage of pro-IL-1β to bioactive IL-1β. The fact that MOMP induction and caspase-3 activation are only partially decreased in Caspase-8-deficient BMDCs is in line with the possibility that caspase-8 further amplifies the pathway by cleaving the BCL-2 homology 3 (BH3)-interacting domain death agonist (BID), consequently driving MOMP and caspase-3 processing further [37,52]. Recently, several studies reported involvement of caspase-8 in IL-1β cleavage during apoptotic cell death [26,[53], [54], [55]]. In contrast to these studies, but similarly to what we proposed earlier for cyclopentenone-containing lipid species [37], Nrf2 activators promote IL-1β release independently of the pyroptosis executioner GSDMD, the inflammasome component NLRP3 and the inflammatory caspases-1 and -11. Another interesting point of this study is that it suggests the requirement of electrophilic sites for the induction of IL-1β release. Indeed, the Nrf2 activator tBHQ, which does not bear electrophilic positions and has been suggested to activate Nrf2 indirectly by induction of ROS [38], was unable to promote IL-1β production. In line with this, we demonstrated that removal of endo- and exocyclic enones from cyclopentenone isoprostanoids also abolished their pro-inflammatory bioactivities. Additionally, we tested whether Hsf1, which is known to coordinate stress-induced pathways in response to electrophiles [40,41], was critical for the induction of inflammatory apoptosis. However, we exclusively detected Hsf1 activation after 20 h of stimulation. Since we generally detect MOMP induction by high doses of Nrf2 activators already 2 h after stimulation, these results indicate that the late Hsf1 activation (at 20 h) is likely a consequence of cellular stress due to massive death rather than playing any role in the induction of inflammatory apoptosis. Thus, further research is required to better understand the exact mechanism by which electrophilic sites of Nrf2 activators target them to mitochondria and induce MOMP.
Among the various Nrf2 activators that have been tested in this work, we have particularly focused our attention on the endogenous metabolite itaconate, which is strikingly increased in LPS-activated macrophages and was recently proposed to promote an anti-inflammatory phenotype in part via Nrf2 activation [9,10]. Indeed, both 4-OI, a cell-permeable surrogate of itaconate, and other Nrf2 activators alkylate Keap1, leading to Nrf2 release followed by its nuclear translocation. In contrast to cyclopentenone isoprostanoids and other tested compounds, the anti-inflammatory potency of itaconate was lower, and longer incubation times were required to obtain an anti-inflammatory effect similar to the one observed for cEC. Importantly, we demonstrated that addition of itaconate after LPS stimulation leads to secretion of IL-1β, which warrants caution in the overall perception of itaconate as an anti-inflammatory mediator [9,10,56]. Although this pro-inflammatory activity of itaconate requires relatively high concentrations (240–480 μM), it is probably of physiological relevance considering reports showing intracellular itaconate concentrations in the mM range upon LPS activation [57,58]. Therefore, although IL-1β is a critical player in several inflammatory diseases [59] and itaconate could be used as a potential therapeutic compound, more research is required to better understand the pro-inflammatory impact of this metabolite.
How exactly 4-OI exerts its anti- and pro-inflammatory functions remains controversial. Indeed, it has been originally shown that 4-OI is hydrolyzed to itaconate by esterases in LPS-activated macrophages [9], but a recent publication proposed that any increase in itaconate upon 4-OI treatment is due to endogenous itaconate being generated, for instance through Irg1 activity [49]. Our results with Irg1-deficient BMDCs excluded the possibility that 4-OI acts by increasing Irg1-dependent production of endogenous itaconate in our system, thus in keeping with the possibility that 4-OI exerts its anti- and pro-inflammatory bioactivities by itself or by a direct conversion into itaconate via cellular esterases. In line with this latter hypothesis, flux analysis has recently demonstrated that 4-OI is directly converted into itaconate (Luke O'Neill, personal communication [60]). In contrast to 4-OI, the other esterified itaconate derivative DMI cannot be metabolized into itaconate [36], and thus our observed bioactivity is likely mediated by a direct modification of intracellular cysteines. Our results indicate that 4-OI displays a slightly increased bioactivity in comparison to DMI, but whether this is caused by the different electrophilicity between 4-OI and DMI remains unknown at present. Indeed, which of the two compounds is the strongest electrophile is still controversial in the field [9,49]. Taken together, our results are consistent with 4-OI as a cell-permeable molecule that delivers free itaconate, which then regulates inflammation as we have described here. If and especially to which extent 4-OI acts by itself remains unclear at the moment and requires further investigations in future studies.
DMF is an efficient therapy in patients suffering from MS, but the mechanism of this activity remains elusive. MS patients are treated with 240 mg (max 480 mg) DMF daily corresponding to about 3.4 mg/kg (assuming 240 mg DMF and 70 kg body weight) or 300–600 μM (assuming 5 L blood volume) by disregarding pharmacokinetics and bioavailability. Considering that the induction of inflammatory apoptosis occurs at 100 μM in vitro, our results may be clinically relevant. Similarly to the induction of cell death at high doses that we have observed here, it has been proposed that DMF acts in a Nrf2-independent manner in vivo [61]. One could thereby speculate that killing of a particular inflammatory myeloid population may play a role in the efficacy of DMF in MS. Further research, however, is needed to expand on this, especially because DMF is reported to be extremely unstable and to be rapidly metabolized into monomethylfumarate (MMF). While the neutral DMF readily diffuses across the plasma membrane, the negatively-charged MMF is largely prevented to enter cells and consequently it is thought to act via interaction with the cell surface receptor hydroxycarboxylic acid receptor 2 (HCA2) rather than by binding to intracellular cysteines [62]. Therefore, it may be that a significantly higher dose of DMF has to be exploited in vivo compared to in vitro in order to observe the here described pro-inflammatory action of DMF.
In summary, this work considerably extends a recent report [37] which demonstrated that cyclopentenone-containing lipid species share dose-dependent pro- and anti-inflammatory activities. Indeed, we show here that a large variety of electrophilic Nrf2 activators including the metabolite itaconate can broadly inhibit inflammatory responses at low doses prior to TLR stimulation and trigger Nrf2-independent caspase-8-mediated IL-1β production and apoptotic cell death at high doses after TLR activation. Specifically, we found that IL-1β is released by cells dying of secondary necrosis in a GSDMD- and GSDME-independent manner. These findings have important implications for treatment of inflammatory diseases with Nrf2 activators and for understanding the mechanisms of clinical outcome.
Declaration of competing interest
The authors declare that they have no conflict of interest.
Acknowledgements
We thank Olaf Gross for providing Caspase-8fl/fl;Cre-ERT2 mice; Wolf-Dietrich Hardt for sharing Caspase-11−/−, Caspase-1/11−/− and Irg1−/− (with the permission of Rudi Balling) mice; Masahiro Yamamoto for providing Nrf2−/− mice; the late Jürg Tschopp for sharing NLRP3−/− mice. We further thank Benjamin Demarco Vergara for the preparation of GSDME- and GSDME/D-deficient bone marrows. We are grateful for research grants from ETH Zurich (ETH-23-16-2) and SNF (310030_163443/1). H.W. acknowledges the Stipendienfonds Schweizerische Chemische Industrie (SSCI).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2020.101647.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Itoh K. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem. Biophys. Res. Commun. 1997;236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 2.Venugopal R., Jaiswal A.K. Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P)H:quinone oxidoreductase1 gene. Proc. Natl. Acad. Sci. U. S. A. 1996;93:14960–14965. doi: 10.1073/pnas.93.25.14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Itoh K. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taguchi K., Motohashi H., Yamamoto M. Molecular mechanisms of the Keap1-Nrf2 pathway in stress response and cancer evolution. Gene Cell. 2011;16:123–140. doi: 10.1111/j.1365-2443.2010.01473.x. [DOI] [PubMed] [Google Scholar]
- 5.Thimmulappa R.K. Nrf2 is a critical regulator of the innate immune response and survival during experimental sepsis. J. Clin. Invest. 2006;116:984–995. doi: 10.1172/JCI25790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rangasamy T. Disruption of Nrf2 enhances susceptibility to severe airway inflammation and asthma in mice. J. Exp. Med. 2005;202:47–59. doi: 10.1084/jem.20050538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ishii Y. Transcription factor Nrf2 plays a pivotal role in protection against elastase-induced pulmonary inflammation and emphysema. J. Immunol. 2005;175:6968–6975. doi: 10.4049/jimmunol.175.10.6968. [DOI] [PubMed] [Google Scholar]
- 8.Burness C.B., Deeks E.D. Dimethyl fumarate: a review of its use in patients with relapsing-remitting multiple sclerosis. CNS Drugs. 2014;28:373–387. doi: 10.1007/s40263-014-0155-5. [DOI] [PubMed] [Google Scholar]
- 9.Mills E.L. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature. 2018;556:113–117. doi: 10.1038/nature25986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bambouskova M. Electrophilic properties of itaconate and derivatives regulate the IkappaBzeta-ATF3 inflammatory axis. Nature. 2018;556:501–504. doi: 10.1038/s41586-018-0052-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Broz P., Dixit V.M. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016;16:407–420. doi: 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- 12.Martinon F., Burns K., Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002;10:417–426. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- 13.Kayagaki N. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479:117–121. doi: 10.1038/nature10558. [DOI] [PubMed] [Google Scholar]
- 14.Shi J. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–192. doi: 10.1038/nature13683. [DOI] [PubMed] [Google Scholar]
- 15.Kayagaki N. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–671. doi: 10.1038/nature15541. [DOI] [PubMed] [Google Scholar]
- 16.Liu X. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–158. doi: 10.1038/nature18629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi J. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–665. doi: 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- 18.Evavold C.L. Immunity; 2017. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Heilig R. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur. J. Immunol. 2018;48:584–592. doi: 10.1002/eji.201747404. [DOI] [PubMed] [Google Scholar]
- 20.Freigang S. Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 2011;41:2040–2051. doi: 10.1002/eji.201041316. [DOI] [PubMed] [Google Scholar]
- 21.Zhao C., Gillette D.D., Li X., Zhang Z., Wen H. Nuclear factor E2-related factor-2 (Nrf2) is required for NLRP3 and AIM2 inflammasome activation. J. Biol. Chem. 2014;289:17020–17029. doi: 10.1074/jbc.M114.563114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chartoumpekis D.V. Nrf2 represses FGF21 during long-term high-fat diet-induced obesity in mice. Diabetes. 2011;60:2465–2473. doi: 10.2337/db11-0112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pi J. Deficiency in the nuclear factor E2-related factor-2 transcription factor results in impaired adipogenesis and protects against diet-induced obesity. J. Biol. Chem. 2010;285:9292–9300. doi: 10.1074/jbc.M109.093955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Martinon F., Petrilli V., Mayor A., Tardivel A., Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature. 2006;440:237–241. doi: 10.1038/nature04516. [DOI] [PubMed] [Google Scholar]
- 25.Santos J.C. LPS targets host guanylate-binding proteins to the bacterial outer membrane for non-canonical inflammasome activation. EMBO J. 2018;37 doi: 10.15252/embj.201798089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chen K.W. Extrinsic and intrinsic apoptosis activate pannexin-1 to drive NLRP3 inflammasome assembly. EMBO J. 2019;38 doi: 10.15252/embj.2019101638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beisner D.R., Ch'en I.L., Kolla R.V., Hoffmann A., Hedrick S.M. Cutting edge: innate immunity conferred by B cells is regulated by caspase-8. J. Immunol. 2005;175:3469–3473. doi: 10.4049/jimmunol.175.6.3469. [DOI] [PubMed] [Google Scholar]
- 28.Hameyer D. Toxicity of ligand-dependent Cre recombinases and generation of a conditional Cre deleter mouse allowing mosaic recombination in peripheral tissues. Physiol. Genom. 2007;31:32–41. doi: 10.1152/physiolgenomics.00019.2007. [DOI] [PubMed] [Google Scholar]
- 29.Kuida K. Altered cytokine export and apoptosis in mice deficient in interleukin-1 beta converting enzyme. Science. 1995;267:2000–2003. doi: 10.1126/science.7535475. [DOI] [PubMed] [Google Scholar]
- 30.Cordes T. Immunoresponsive gene 1 and itaconate inhibit succinate dehydrogenase to modulate intracellular succinate levels. J. Biol. Chem. 2016;291:14274–14284. doi: 10.1074/jbc.M115.685792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Egger J., Bretscher P., Freigang S., Kopf M., Carreira E.M. Synthesis of epoxyisoprostanes: effects in reducing secretion of pro-inflammatory cytokines IL-6 and IL-12. Angew Chem. Int. Ed. Engl. 2013;52:5382–5385. doi: 10.1002/anie.201300739. [DOI] [PubMed] [Google Scholar]
- 32.Egger J., Bretscher P., Freigang S., Kopf M., Carreira E.M. Discovery of a highly potent anti-inflammatory epoxyisoprostane-derived lactone. J. Am. Chem. Soc. 2014;136:17382–17385. doi: 10.1021/ja509892u. [DOI] [PubMed] [Google Scholar]
- 33.Wolleb H. Synthesis and structure-activity relationship studies of anti-inflammatory epoxyisoprostane analogues. Org. Lett. 2018;20:3014–3016. doi: 10.1021/acs.orglett.8b01042. [DOI] [PubMed] [Google Scholar]
- 34.Muri J., Thut H., Feng Q., Kopf M. Thioredoxin-1 distinctly promotes NF-kappaB target DNA binding and NLRP3 inflammasome activation independently of Txnip. Elife. 2020;9 doi: 10.7554/eLife.53627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bretscher P. Phospholipid oxidation generates potent anti-inflammatory lipid mediators that mimic structurally related pro-resolving eicosanoids by activating Nrf2. EMBO Mol. Med. 2015;7:593–607. doi: 10.15252/emmm.201404702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.ElAzzouny M. Dimethyl itaconate is not metabolized into itaconate intracellularly. J. Biol. Chem. 2017;292:4766–4769. doi: 10.1074/jbc.C117.775270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Muri J. Cyclopentenone prostaglandins and structurally related oxidized lipid species instigate and share distinct pro- and anti-inflammatory pathways. Cell Rep. 2020;30:4399–4417. doi: 10.1016/j.celrep.2020.03.019. e4397. [DOI] [PubMed] [Google Scholar]
- 38.Imhoff B.R., Hansen J.M. Tert-butylhydroquinone induces mitochondrial oxidative stress causing Nrf2 activation. Cell Biol. Toxicol. 2010;26:541–551. doi: 10.1007/s10565-010-9162-6. [DOI] [PubMed] [Google Scholar]
- 39.Abiko Y., Miura T., Phuc B.H., Shinkai Y., Kumagai Y. Participation of covalent modification of Keap1 in the activation of Nrf2 by tert-butylbenzoquinone, an electrophilic metabolite of butylated hydroxyanisole. Toxicol. Appl. Pharmacol. 2011;255:32–39. doi: 10.1016/j.taap.2011.05.013. [DOI] [PubMed] [Google Scholar]
- 40.Kansanen E. Nrf2-dependent and -independent responses to nitro-fatty acids in human endothelial cells: identification of heat shock response as the major pathway activated by nitro-oleic acid. J. Biol. Chem. 2009;284:33233–33241. doi: 10.1074/jbc.M109.064873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y. HSF1-dependent upregulation of Hsp70 by sulfhydryl-reactive inducers of the KEAP1/NRF2/ARE pathway. Chem. Biol. 2011;18:1355–1361. doi: 10.1016/j.chembiol.2011.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Petrilli V., Dostert C., Muruve D.A., Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr. Opin. Immunol. 2007;19:615–622. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 43.Fuchs Y., Steller H. Live to die another way: modes of programmed cell death and the signals emanating from dying cells. Nat. Rev. Mol. Cell Biol. 2015;16:329–344. doi: 10.1038/nrm3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tait S.W., Green D.R. Mitochondria and cell death: outer membrane permeabilization and beyond. Nat. Rev. Mol. Cell Biol. 2010;11:621–632. doi: 10.1038/nrm2952. [DOI] [PubMed] [Google Scholar]
- 45.Broz P., Pelegrin P., Shao F. The gasdermins, a protein family executing cell death and inflammation. Nat. Rev. Immunol. 2020;20:143–157. doi: 10.1038/s41577-019-0228-2. [DOI] [PubMed] [Google Scholar]
- 46.Rogers C. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat. Commun. 2017;8:14128. doi: 10.1038/ncomms14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. doi: 10.1038/nature22393. [DOI] [PubMed] [Google Scholar]
- 48.Heilig R. Caspase-1 cleaves Bid to release mitochondrial SMAC and drive secondary necrosis in the absence of GSDMD. Life Sci Alliance. 2020;3 doi: 10.26508/lsa.202000735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Swain A. Comparative evaluation of itaconate and its derivatives reveals divergent inflammasome and type I interferon regulation in macrophages. Nat. Metabol. 2020 doi: 10.1038/s42255-020-0210-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Robledinos-Anton N., Fernandez-Gines R., Manda G., Cuadrado A. Activators and inhibitors of NRF2: a review of their potential for clinical development. Oxid Med Cell Longev. 2019;2019:9372182. doi: 10.1155/2019/9372182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kobayashi E.H. Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription. Nat. Commun. 2016;7:11624. doi: 10.1038/ncomms11624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gurung P., Kanneganti T.D. Novel roles for caspase-8 in IL-1beta and inflammasome regulation. Am. J. Pathol. 2015;185:17–25. doi: 10.1016/j.ajpath.2014.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chauhan D. BAX/BAK-Induced apoptosis results in caspase-8-dependent IL-1beta maturation in macrophages. Cell Rep. 2018;25:2354–2368 e2355. doi: 10.1016/j.celrep.2018.10.087. [DOI] [PubMed] [Google Scholar]
- 54.Vince J.E. The mitochondrial apoptotic effectors BAX/BAK activate caspase-3 and -7 to trigger NLRP3 inflammasome and caspase-8 driven IL-1beta activation. Cell Rep. 2018;25:2339–2353 e2334. doi: 10.1016/j.celrep.2018.10.103. [DOI] [PubMed] [Google Scholar]
- 55.Zewinger S. Apolipoprotein C3 induces inflammation and organ damage by alternative inflammasome activation. Nat. Immunol. 2020;21:30–41. doi: 10.1038/s41590-019-0548-1. [DOI] [PubMed] [Google Scholar]
- 56.Lampropoulou V. Itaconate links inhibition of succinate dehydrogenase with macrophage metabolic remodeling and regulation of inflammation. Cell Metabol. 2016;24:158–166. doi: 10.1016/j.cmet.2016.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Meiser J. Itaconic acid indicates cellular but not systemic immune system activation. Oncotarget. 2018;9:32098–32107. doi: 10.18632/oncotarget.25956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Strelko C.L. Itaconic acid is a mammalian metabolite induced during macrophage activation. J. Am. Chem. Soc. 2011;133:16386–16389. doi: 10.1021/ja2070889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dinarello C.A. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hooftman A. 2020. The Immunomodulatory Metabolite Itaconate Modifies NLRP3 and Inhibit the Inflammasome Activation (In Revision) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schulze-Topphoff U. Dimethyl fumarate treatment induces adaptive and innate immune modulation independent of Nrf2. Proc. Natl. Acad. Sci. U. S. A. 2016;113:4777–4782. doi: 10.1073/pnas.1603907113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mrowietz U., Morrison P.J., Suhrkamp I., Kumanova M., Clement B. The pharmacokinetics of fumaric acid esters reveal their in vivo effects. Trends Pharmacol. Sci. 2018;39:1–12. doi: 10.1016/j.tips.2017.11.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.