

Abstract
Background
Mycosis fungoides (MF) is the most common type of cutaneous T‐cell lymphoma, a malignant, chronic disease initially affecting the skin. Several therapies are available, which may induce clinical remission for a time. This is an update of a Cochrane Review first published in 2012: we wanted to assess new trials, some of which investigated new interventions.
Objectives
To assess the effects of interventions for MF in all stages of the disease.
Search methods
We updated our searches of the following databases to May 2019: the Cochrane Skin Specialised Register, CENTRAL, MEDLINE, Embase, and LILACS. We searched 2 trials registries for additional references. For adverse event outcomes, we undertook separate searches in MEDLINE in April, July and November 2017.
Selection criteria
Randomised controlled trials (RCTs) of local or systemic interventions for MF in adults with any stage of the disease compared with either another local or systemic intervention or with placebo.
Data collection and analysis
We used standard methodological procedures expected by Cochrane. The primary outcomes were improvement in health‐related quality of life as defined by participants, and common adverse effects of the treatments. Key secondary outcomes were complete response (CR), defined as complete disappearance of all clinical evidence of disease, and objective response rate (ORR), defined as proportion of patients with a partial or complete response. We used GRADE to assess the certainty of evidence and considered comparisons of psoralen plus ultraviolet A (PUVA) light treatment as most important because this is first‐line treatment for MF in most guidelines.
Main results
This review includes 20 RCTs (1369 participants) covering a wide range of interventions. The following were assessed as either treatments or comparators: imiquimod, peldesine, hypericin, mechlorethamine, nitrogen mustard and intralesional injections of interferon‐α (IFN‐α) (topical applications); PUVA, extracorporeal photopheresis (ECP: photochemotherapy), and visible light (light applications); acitretin, bexarotene, lenalidomide, methotrexate and vorinostat (oral agents); brentuximab vedotin; denileukin diftitox; mogamulizumab; chemotherapy with cyclophosphamide, doxorubicin, etoposide, and vincristine; a combination of chemotherapy with electron beam radiation; subcutaneous injection of IFN‐α; and intramuscular injections of active transfer factor (parenteral systemics).
Thirteen trials used an active comparator, five were placebo‐controlled, and two compared an active operator to observation only. In 14 trials, participants had MF in clinical stages IA to IIB. All participants were treated in secondary and tertiary care settings, mainly in Europe, North America or Australia. Trials recruited both men and women, with more male participants overall. Trial duration varied from four weeks to 12 months, with one longer‐term study lasting more than six years. We judged 16 trials as at high risk of bias in at least one domain, most commonly performance bias (blinding of participants and investigators), attrition bias and reporting bias.
None of our key comparisons measured quality of life, and the two studies that did presented no usable data. Eighteen studies reported common adverse effects of the treatments. Adverse effects ranged from mild symptoms to lethal complications depending upon the treatment type. More aggressive treatments like systemic chemotherapy generally resulted in more severe adverse effects.
In the included studies, CR rates ranged from 0% to 83% (median 31%), and ORR ranged from 0% to 88% (median 47%). Five trials assessed PUVA treatment, alone or combined, summarised below.
There may be little to no difference between intralesional IFN‐α and PUVA compared with PUVA alone for 24 to 52 weeks in CR (risk ratio (RR) 1.07, 95% confidence interval (CI) 0.87 to 1.31; 2 trials; 122 participants; low‐certainty evidence). Common adverse events and ORR were not measured.
One small cross‐over trial found once‐monthly ECP for six months may be less effective than twice‐weekly PUVA for three months, reporting CR in two of eight participants and ORR in six of eight participants after PUVA, compared with no CR or ORR after ECP (very low‐certainty evidence). Some participants reported mild nausea after PUVA but no numerical data were given. One participant in the ECP group withdrew due to hypotension. However, we are unsure of the results due to very low‐certainty evidence.
One trial comparing bexarotene plus PUVA versus PUVA alone for up to 16 weeks reported one case of photosensitivity in the bexarotene plus PUVA group compared to none in the PUVA‐alone group (87 participants; low‐certainty evidence). There may be little to no difference between bexarotene plus PUVA and PUVA alone in CR (RR 1.41, 95% CI 0.71 to 2.80) and ORR (RR 0.94, 95% CI 0.61 to 1.44) (93 participants; low‐certainty evidence).
One trial comparing subcutaneous IFN‐α injections combined with either acitretin or PUVA for up to 48 weeks or until CR indicated there may be little to no difference in the common IFN‐α adverse effect of flu‐like symptoms (RR 1.32, 95% CI 0.92 to 1.88; 82 participants). There may be lower CR with IFN‐α and acitretin compared with IFN‐α and PUVA (RR 0.54, 95% CI 0.35 to 0.84; 82 participants) (both outcomes: low‐certainty evidence). This trial did not measure ORR.
One trial comparing PUVA maintenance treatment to no maintenance treatment, in participants who had already had CR, did report common adverse effects. However, the distribution was not evaluable. CR and OR were not assessable.
The range of treatment options meant that rare adverse effects consequently occurred in a variety of organs.
Authors' conclusions
There is a lack of high‐certainty evidence to support decision making in the treatment of MF. Because of substantial heterogeneity in design, missing data, small sample sizes, and low methodological quality, the comparative safety and efficacy of these interventions cannot be reliably established on the basis of the included RCTs. PUVA is commonly recommended as first‐line treatment for MF, and we did not find evidence to challenge this recommendation. There was an absence of evidence to support the use of intralesional IFN‐α or bexarotene in people receiving PUVA and an absence of evidence to support the use of acitretin or ECP for treating MF.
Future trials should compare the safety and efficacy of treatments to PUVA, as the current standard of care, and should measure quality of life and common adverse effects.
Plain language summary
Treatments for mycosis fungoides (a malignant cancerous condition of immune cells in the blood that affects the skin)
What was the aim of this review?
This Cochrane Review compared treatments for mycosis fungoides (also called cutaneous T‐cell lymphoma, Alibert‐Bazin syndrome or granuloma fungoides).
What was studied in the review?
Mycosis fungoides (MF) typically starts as flat and scaly pink or red areas (patches) on the torso, upper thighs or buttocks. At this stage, life expectancy is unaffected. As the disease develops, life expectancy reduces. Patches can turn into raised, itchy plaques. Plaques can become thicker, deeper, and develop into tumours. In rare cases, the disease spreads to other organs.
Many treatments exist for MF; these target specific body areas (local therapy) or the entire body (systemic therapy). Treatments include creams, ointments, oral or injected medicines, light therapy, radiotherapy (radiation that kills cancer cells) and chemotherapy (medicines that kill cancer cells).
We compared the benefits and harms of different treatments in adults, at different disease stages. We identified 20 studies published up to May 2019.
The studies included 1369, mainly male, adults. Most ran from 4 weeks to 12 months. Only five studies investigated the later stages of disease. All were set in specialised healthcare centres in Europe (12 studies), North America (11 studies), Australia (three studies), Brazil and Japan (one study each; satellite centres for studies already listed). Treatments were compared with another treatment (13 studies); an inactive treatment (placebo) (five studies); or no treatment (two studies).
Five studies did not report their funding. Eleven studies were funded by pharmaceutical companies and four by academic institutions or hospitals.
Key results
We do not know how different treatments for MF affect quality of life. Very few studies assessed this outcome and they presented no usable data.
Unwanted (adverse) effects ranged from mild symptoms to severe life‐threatening complications. More aggressive treatments (such as chemotherapy) generally caused more severe adverse effects.
PUVA (a light treatment) is the first treatment used for MF. Results from five studies provided low‐certainty evidence:
There may be little to no difference between giving PUVA alone and PUVA plus injected interferon‐α (IFN‐ α) (a messenger substance of the immune system) for 24 to 52 weeks for making the disease disappear completely. No studies investigated adverse events in these treatments or disappearance of at least 50% of the disease.
There may be little to no difference between an oral vitamin A derivative (bexarotene) plus PUVA, and PUVA alone, for complete or at least 50% disease disappearance (treatment duration: up to 16 weeks). Extreme sensitivity to ultraviolet (UV) rays occurred in some people who received bexarotene and PUVA, but not PUVA alone.
There may be little to no difference between IFN‐ α plus PUVA and IFN‐α plus acitretin (another oral vitamin A derivative) on flu‐like symptoms, when treatment is given for up to 48 weeks or until complete disease disappearance. However, there may be a lower rate of complete disease disappearance with IFN‐α plus acitretin. No studies investigated the effect on partial disappearance.
It is not clear how PUVA maintenance treatment (to prevent the disease from reappearing after it has disappeared) compares with no maintenance treatment, since the only study on this reported very limited information.
One small trial (eight people) compared extracorporeal photopheresis (ECP, a light therapy) once monthly for six months with twice‐weekly PUVA for three months. It reported complete or at least 50% disappearance of MF in some participants treated with PUVA and none who received ECP. Common side effects were reported with each treatment (PUVA may be associated with mild nausea, and ECP with hypotension). However, the very‐low certainty evidence means we are not sure of these results.
How confident are we in the results of this review?
Our confidence in the results of this review is mainly low, but very low for one set of key results. The review is based on small and poorly designed studies. Further research is likely to change its message.
Conclusion
We found no evidence to challenge or support the standard treatment (PUVA). In the absence of a cure, treatment of MF should be based on disease stage, with a focus on limiting severe adverse effects.
Summary of findings
Background
Please see Table 6 for definitions of the clinical stages of the disease, our glossary in Table 7 for an explanation of medical terms used throughout the text, and Table 8 for definitions of acronyms.
1. Clinical staging system.
|
2007 MF and Sézary syndrome |
1979 CTCL |
Disease‐specific survival rates in % (Agar 2010) | |||
| 5 year | 10 year | ||||
| IA | T1 | IA | T1 | 98 | 95 |
| N0 | N0 | ||||
| M0 | M0 | ||||
| B0‐1 | ‐ | ||||
| IB | T2 | IB | T2 | 89 | 77 |
| N0 | N0 | ||||
| M0 | M0 | ||||
| 0‐1 | ‐ | ||||
| IIA | T1‐2 | IIA | T1‐2 | 89 | 67 |
| N1‐2 | N1 | ||||
| M0 | M0 | ||||
| B0‐1 | ‐ | ||||
| IIB | T3 | IIB | T3 | 56 | 42 |
| N0‐2 | N0,1 | ||||
| M0 | M0 | ||||
| B0‐1 | ‐ | ||||
| III | T4 | III | T4 | ||
| N0‐2 | N0,1 | ||||
| M0 | M0 | ||||
| B0‐1 | ‐ | ||||
| IIIA | T4 | 54 | 45 | ||
| N0‐2 | |||||
| M0 | |||||
| B0 | |||||
| IIIB | T4 | 48 | 45 | ||
| N0‐2 | |||||
| M0 | |||||
| B1 | |||||
| IVA1 | T1‐4 | IVA | T1‐4 | 41 | 20 |
| N0‐2 | |||||
| M0 | N2‐3 ‐ |
||||
| B2 | |||||
| IVA2 | T1‐4 | M0 | 23 | 20 | |
| N3 | |||||
| M0 | ‐ | ||||
| B0‐2 | |||||
| IVB | T1‐4 | IVB | T1‐4 | 18 | not reached |
| N0‐3 | N0‐3 | ||||
| M1 | M1 | ||||
| B0‐2 | ‐ | ||||
2. Glossary of terms.
| General medical terms | Explanation |
| Apoptosis | Programmed cell death |
| Cutaneous T‐cell lymphoma | Group of skin‐directed T‐cell neoplasms with diverse clinical and histological features and prognosis |
| Cutaneous B‐cell lymphoma | Group of skin‐directed B‐cell neoplasms with diverse clinical and histological features and prognosis |
| Lesional skin atrophy | Death of the cells in the damaged area of skin |
| Lymph nodes | Small organs in the human body which are part of the immune system |
| Neoplasm | Any new and abnormal growth |
| NK‐cell lymphoma | Group of neoplasms derived from the natural killer cells (NK‐cells) with diverse clinical and histological features and prognosis |
| Plaques | A solid elevated area on the skin that is more broad than it is high |
| Pleomorphic | Variability in size and shape |
| Poikiloderma | Skin that demonstrates adjacent hyper‐ and hypopigmented areas with widened capillaries (telangiectasia) in the affected area |
| Precursor haematologic neoplasm | Clinically‐aggressive neoplasm with a high incidence of cutaneous involvement and risk of leukaemic dissemination |
| Primary cutaneous lymphoma | Cutaneous T‐ and B‐cell lymphoma that primarily affect the skin |
| T‐cell | A type of lymphocyte (white cell) |
| Adverse effects | Explanation |
| Acute myocardial infarction | Death of myocardial tissue due to blocked blood supply |
| Acute non‐lymphatic leukaemia | A quickly progressive malignant disease of too many immature non‐lymphatic leucocytes cells in the blood and bone marrow |
| ALT | The alanine transaminase is a liver enzyme (SGPT) |
| Anaemia | Low count of red blood cells |
| Anaemia hypochromic | Low count of red blood cells with low amount of haemoglobin, the red molecule that transports oxygen within the blood vessels |
| Anaphylactoid reactions | Very acute systemic allergic reaction often accompanied with flushing, angioedema,urticaria, difficulty breathing, lowered blood pressure, nausea |
| Arthralgia | Joint pain |
| AST | The aspartate transaminase is an enzyme mainly present in the liver but also in the blood, muscle cells, and bones (SGOT) |
| Asthenia | Lack of energy or physical weakness or both |
| Cardiomyopathy | Structural or functional disease of the cardiac muscle |
| Cardiopulmonary syndrome | Adverse effect where the heart and the lung are involved |
| Chill | Feeling of cold, resulting in shivering |
| CNS syndrome | Adverse effect where the central nervous system (brain, spinal cord) is involved |
| Cutaneous hypersensitivity | Altered reactivity to a specific antigen leading to cutaneous alterations |
| Cutaneous toxicity | Cutaneous adverse effect of an agent used in therapeutic dosages |
| Constipation | Hard and/or difficult bowel movements |
| Dermatitis exfoliation | Inflammation and detachment of the skin |
| Diarrhoea | Many fluid stools |
| Dyspnea | Bad breathing |
| Erythema | Redness of the skin |
| Fatigue | To be exhausted |
| Flu‐like symptoms | Symptoms which are often seen with influenza, such as fever, chills, and muscular pain |
| Gastrointestinal syndromes | Adverse effects affecting the digestive system (oesophagus, stomach, bowel) |
| Hair loss | Pathological increased loss of hair |
| Hepatotoxicity | Capacity of a substance to have damaging effects on the liver |
| Hospitalisation | Admission of a patient in a hospital |
| Hypercholesterolaemia | Elevated levels of cholesterol in the blood |
| Hyperlipidaemia | Elevated levels of lipids in the blood |
| Hypotension | Low blood pressure |
| Hypothyroidism | Low function of thyroid gland |
| Impotentia | Inability to engage in sexual intercourse |
| Insomnia | Not being able to sleep |
| LDH | Lactate dehydrogenase, an enzyme which helps to produce energy in the body when oxygen is absent |
| Leukopenia | Low count of white blood cells |
| Malaise | To feel ill |
| Mucositis | Inflammation of mucosa |
| Myalgia | Muscle pain |
| Nasopharyngitis | Inflammation of the inner nose and the throat |
| Nausea | An unpleasant sensation associated with the feeling one is going to vomit |
| Neuropathy | A problem of the nervous system or nerves, which can result in abnormal sensations, pain, or muscle weakness |
| Non‐melanoma skin cancer | Skin cancer which does not originate from melanocytes |
| Photosensitivity | Enhanced responsibility to light or ultraviolet light |
| Pruritus | Itching of the skin |
| Radiodermatitis | Dermatitis resulting from overexposure to sources of radiant energy |
| Rash | An abnormal change in the skin often affecting colour (increased redness), texture, and/or sensation |
| SGOT/SGPT | Liver enzymes, see also AST and ALT |
| T4 | Enzyme of the thyroid gland |
| Thrombopenia | Low count of thrombocytes |
| Thrombotic syndrome | Blood coagulation and clotting within blood vessels, obstructing blood flow |
| Triglycerid | Lipid |
| Vascular leak syndrome | When the blood vessels dilate and become more porous, allowing blood components to leak into the surrounding tissue |
| Vasodilatation | Widening of the blood vessels |
3. List of abbreviations and acronyms.
| Acronym | Description (letters used for acronym in capitals) |
| ADF | Arbeitsgemeinschaft Dermatologische Forschung |
| BCNU | Carmustine, a nitrogen mustard related alkylating agent |
| CENTRAL | Cochrane Central Register of Controlled Trials |
| CI | Confidence interval |
| CTCL | Cutaneous T‐Cell Lymphoma |
| DDG | German Dermatologic Society |
| EORTC | European Organization of Research and Treatment of Cancer |
| ISCL | International Society for Cutaneous Lymphoma (ISCL) |
| ITT | Intention‐to‐treat |
| LILACS | Latin American and Caribbean Health Science Information database |
| MF | Mycosis Fungoides |
| PICOS | Participants, Interventions, Controls, Outcomes and Study |
| PRISMA | Preferred Reporting Items of Systematic Reviews and Meta‐Analyses |
| RCT | Randomised‐Controlled Trial |
| RR | Risk Ratio |
| TSEB | Total Skin Electron Beam |
| TNMB | Tumour, lymph Node, Metastasis and Blood |
| UK | United Kingdom |
| USA | United States of America |
| USCLC | United States Cutaneous Lymphoma Consortium |
| WHO | World Health Organization |
Description of the condition
Mycosis fungoides (MF) is the most common type of cutaneous T‐cell lymphoma (Korgavkar 2013). It is a malignant condition with clonal T‐helper cells primarily affecting the skin. The course of the condition is chronic, so its description depends upon the stage at which it presents for clinical examination.
Typically, at first there are multiple eczematous patches on the trunk and extremities, which may be accompanied by lesional skin atrophy. After some years, these patches frequently develop into plaques and may progress to solid skin tumours (Figure 1). Skin tumours can also develop on the face and head region, but these are uncommon locations for mycosis fungoides at the early patch or plaque stage. In the more advanced stages, lymph nodes and eventually solid organs may also be involved, but progress is usually slow. Pruritus (itching) is infrequent at the patch stage but can become more frequent at the plaque and skin‐tumour stages. Clinical diagnosis needs to be confirmed by histology, but often multiple skin biopsies are necessary to establish diagnosis since histological findings are often ambiguous (Cerroni 2018).
1.

Skin changes in mycosis fungoides. Left: Patch; Middle: Plaque; Right: Tumour
Copyright © 2018. Department of Dermatology, University Hospital Frankfurt am Main: reproduced with permission.
Incidence and demographics
Mycosis fungoides accounts for about half of all cutaneous T‐cell lymphomas (CTCL), but it still remains a rare disease due to the low incidence of CTCL (Willemze 2005). Age‐adjusted incidence rates for CTCL have been reported for several countries. These equate to a yearly incidence per 10 million people of 13 in Norway and England/Wales, 14 in the Netherlands, 15 in Western Australia, and 41 to 64 in the USA (Bradford 2009; Criscione 2007; Morales 2000). Differences between countries have been assumed to be a result of variable diagnostic criteria in the past (Morales 2000).
Onset of symptoms, generally, occurs in late middle age with a median of 50 to 60 years (Kim 2003; Lorincz 1996; van Doorn 2000; Zackheim 1999), but cases in children and adolescents are also known (Criscione 2007; Wain 2003; Weinstock 1999). The disease occurs more often in men than in women with a ratio of 2:1 (Bradford 2009; Weinstock 1988). Ethnicity also affects incidence rates. Black populations have the highest reported incidence rates, followed by white populations. The lowest incidence rates have been reported in Asian and Hispanic populations (Bernstein 1989; Bradford 2009; Weinstock 1988). Moreover, African‐American race seems to be associated with a poorer overall survival (Nath 2014).
The median time to diagnosis is found to be about four years. This may be due to its pleomorphic presentation and often slow disease progression with non‐specific eczematous patch lesions for some years (Kim 2003).
Classification
The classification specific for primary cutaneous lymphoma made by the European Organization of Research and Treatment of Cancer (EORTC) was published in 1997 (Willemze 1997). Together with the 2001 Classification of Tumours by the World Health Organization (WHO) (Jaffe 2001), this classification was succeeded in 2004 by the commonly‐used WHO‐EORTC classification for cutaneous lymphoma, which constituted the standard classification (Willemze 2005) until 2018. This classification was updated in 2018 and was published in the 4th edition of the WHO classification for Skin Tumours Blue Book (Elder 2018).
WHO‐EORTC classification
The WHO‐EORTC classification for cutaneous lymphoma distinguishes two main entities: cutaneous T‐cell and cutaneous B‐cell lymphomas.
Cutaneous T‐cell lymphomas are further grouped into different subcategories, of which classical mycosis fungoides is one. Mycosis fungoides represents the most common type and is usually defined as classical 'Alibert‐Bazin' type with evolution of patches, plaques, and tumours. Mycosis fungoides variants and subtypes, as well as Sézary syndrome, are distinctive conditions with separate clinical, histological, and haematological findings. Therefore, they are not included in analyses done for this review (Willemze 2019).
TNMB (tumour, lymph node, metastasis, and blood) classifications for mycosis fungoides
In 2007, the International Society for Cutaneous Lymphoma (ISCL) and the cutaneous lymphoma task force of the EORTC revised the 1979 TNMB classification for CTCL to adapt to recent advances and develop a more specific classification of mycosis fungoides, as well as Sézary syndrome (Bunn 1979; Olsen 2007). In 2011, Olsen and colleagues further updated this classification (Olsen 2011). Because of overall similarity and the fact that only the most recent articles have incorporated the revised TNMB classification, both classifications are accepted in this review. Table 9 shows the original and currently‐revised TNMB classification.
4. TNMB classifications.
| Modified ISCL/EORTC classification of MF and Sézary Syndrome according toOlsen 2011 | CTCL 1979 | ||
| T: Skin1 | |||
| T0 | N.E. | Clinically and/or histopathologically suspicious lesions | |
| T1 | Limited patches, papules, and/or plaques covering < 10% of the skin surface; may further stratify into T1a (patch only) vs. T1b (plaque ± patch) | Limited plaques, papules, or eczematous patches covering < 10% of the skin surface | |
| T2 | Patches, papules, or plaques covering ≥ 10% of the skin surface; may further stratify into T2a (patch only) vs. T2b (plaque patch) | Generalised plaques, papules, or erythematous patches covering ≥ 10% of the skin surface | |
| T3 | One or more tumours (≥ 1 cm diameter) | Tumours, 1 or more | |
| T4 | Confluence of erythema covering ≥ 80% body surface area | Generalised erythroderma | |
| N: Node2 | |||
| N0 | No clinically abnormal peripheral lymph nodes; biopsy not required | No clinically abnormal peripheral lymph nodes palpable, histopathology negative for CTCL | |
| N1 | Clinically abnormal lymph nodes; histopathology Dutch grade 1 or NCI LN0-2 | Palpable Clinically abnormal peripheral lymph nodes, histopathology negative for CTCL | |
| N1a | Clone negative | ‐ | |
| N1b | Clone positive | ‐ | |
| N2 | Clinically abnormal peripheral lymph nodes, histopathology Dutch grade 2 or NCI LN3 | No clinically abnormal peripheral lymph nodes, histopathology positive for CTCL | |
| N2a | Clone negative | ‐ | |
| N2b | Clone positive | ‐ | |
| N3 | Clinically abnormal lymph nodes; histopathology Dutch grade 3‐4 or NCI LN4; clone positive or negative |
Palpable clinically abnormal peripheral lymph nodes, pathology positive for CTCL | |
| Nx | Clinically abnormal lymph nodes without histologic confirmation or inability to fully characterize the histologic subcategories | ‐ | |
| M: Visceral | |||
| M0 | No visceral organ involvement | No visceral organ involvement | |
| M1 | Visceral involvement (must have pathology confirmation and organ involved should be specified) | Visceral involvement (must have pathology confirmation and organ involved should be specified) | |
| B: Blood | |||
| B0 | Absence of significant blood involvement: ≤ 5% of peripheral blood lymphocytes are atypical (Sézary) cells | Atypical circulating cells not present (less than 5%) | |
| B0a | Clone negative | ‐ | |
| B0b | Clone positive | ‐ | |
| B1 | Low blood tumour burden: > 5% of peripheral blood lymphocytes are atypical (Sézary) cells but does not meet the criteria of B2 | Atypical circulating cells present (more than 5%), record total white blood count and total lymphocyte counts, and number of atypical cells/100 lymphocytes | |
| B1a | Clone negative | ‐ | |
| B1b | Clone positive | ‐ | |
| B2 | High blood tumour burden: ≥ 1,000/L Sézary cells with positive clone3; one of the following can be substituted for Sézary cells: CD4/CD8 ≥ 10, CD4CD7‐ cells ≥ 40% or CD4CD26‐ cells ≥ 30% | ‐ | |
1 Patch any size lesion without induration or significant elevation above the surrounding uninvolved skin: poikiloderma may be present. Plaque any size lesion that is elevated or indurated: crusting or poikiloderma may be present. Tumour any solid or nodular lesion ≥ 1 cm in diameter with evidence of deep infiltration in the skin and/or vertical growth.
2 Lymph node classification has been modified from 2007 ISCL/EORTC consensus revisions1 to include central nodes. Lymph nodes are qualified as abnormal if ≥ 1.5 cm in diameter.
3 The clone in the blood should match that of the skin. The relevance of an isolated clone in the blood or a clone in the blood that does not match the clone in the skin remains to be determined.
Staging and Prognosis
Clinical staging of mycosis fungoides was derived from the TNMB classification and can help when predicting survival (Sausville 1988; van Doorn 2000; Vonderheid 2006). Independent prognostic factors are described as age; gender; T‐cell classification; presence of extracutaneous disease; response to initial treatment; the presence of follicular mucinosis, poikilodermatous or hypopigmented mycosis fungoides; or the association with lymphomatoid papulosis (Agar 2010; Kim 2003; van Doorn 2000). The risk for disease progression is also related to the clinical stage of the disease. In stage IA, the risk is reported to be 12% within 10 years. In stage IB, the risk for progression increases to 38%, and in stage IIA, to 33% within the same period of time. In stage IIB, the risk elevates markedly to 58% within 10 years, and in stages IIIA and IIIB, it rises to 62% and 73%, respectively. Stages IVA1 and IVA2 show the highest risk for disease progression within 10 years at 83% and 80%, respectively (Agar 2010).
Description of the intervention
Treatment of mycosis fungoides is stage‐adapted aiming at the following:
complete response of lesions (i.e. remission induction);
maintaining or improving quality of life; and
prolonging disease‐free survival and overall survival (Hwang 2008).
In early stages of the disease skin‐directed treatment approaches, including topical therapies, skin‐directed phototherapies, and radiotherapy, are favoured (Dummer 2008; Trautinger 2006; Whittaker 2003). Also, an expectant policy with careful monitoring is recommended, since life expectancy is similar to age‐matched control groups (Kim 1996; Zackheim 1999).
In later stages of the disease, systemic treatment approaches are recommended. These include chemotherapy, extracorporeal photochemotherapy, biological response modifiers and combinations of these therapies (Dummer 2008; Gilson 2019; Trautinger 2006; Whittaker 2003). Extracorporeal photochemotherapy is a procedure by which leucocytes are first sensitised to ultraviolet‐A light (UVA) via 8‐methoxypsoralen followed by irradiation with UVA leading to modulation of the immune system. Biological response modifiers represent a group of substances which modify the immune response in manifold ways.
How the intervention might work
Mycosis fungoides is a rare disease affecting the skin, lymph nodes and blood. Based on severity of the disease (clinical stage), there are different treatment options (Trautinger 2017). Since most people present with early stage disease, with the disease appearing as patches and plaques on the skin, topical and skin‐directed therapies are commonly used. Disease progression with involvement of the blood, lymph nodes, or other organs is possible. At this stage of disease more aggressive therapies are often required. Potential mechanisms of action are induction of apoptosis (programmed cell death) in malignant T‐lymphocytes (Yoo 1996) and modulation of the immune system (Spaccarelli 2015).
We hereby present a brief overview of the different treatment options, which were investigated in the included trials.
Topical glucocorticoids
Topical glucocorticoids are commonly used for early stage disease. Glucocorticoids are known for their manifold effects in the human body. Treatment with topical glucocorticoids may lead to the induction of apoptosis in neoplastic lymphocytes (Schwartzman 1994).
Topical peldesine
Peldesine is a pyrimidine analogue and inhibitor of nucleoside phosphorylase. By acting as a pyrimidine analogue, it inhibits T‐cell proliferation, which presumably eases the symptoms of mycosis fungoides patients (Duvic 2001a).
Topical imiquimod
Topical treatment with imiquimod causes a local immune response modification by acting as an agonist of toll‐like receptor 7. This leads to a local cytokine shift and modification of the immune response. The stimulated immune system then eliminates the altered T‐cells causing mycosis fungoides (Sauder 2003).
Topical hypericin
Hypericin is a photosensitising agent which produces oxidative stress via superoxide radicals. In combination with visible light or UVA, it induces apoptosis in malignant T‐cells (Rook 2010).
Interferon‐α
Interferon‐α is known to have antiproliferative effects on malignant T‐cells (Wolff 1985). It is approved by European Medicines Agency (EMA) for the treatment of CTCL.
Mechlorethamine/nitrogen mustard
Mechlorethamine is an alkylating agent with cytotoxic effects preventing cell duplication via binding to deoxyribonucleic acid (DNA) (Vonderheid 1987; Wolff 1985).
Psoralen + UVA (PUVA)
There are several modes of action attributed to PUVA like generation of reactive oxygen species, inhibition of DNA synthesis and mitochondrial dysfunction (Wozniak 2008).
Total skin electron beam therapy (TSEBT)
TSEBT results in cytotoxic effects such as DNA damage, which ultimately induces cellular death within the radiation field (Kaye 1989).
Denileukin diftitox
Denileukin diftitox influences protein synthesis in cells that express the IL‐2 receptor, which in the case of T‐cell leads to downregulation of proliferation and differentiation (Olsen 2001).
Bexarotene
Bexarotene belongs to the group of retinoids which modify cellular differentiation and growth via activation of retinoid X receptors (Whittaker 2012). It was approved by the EMA for CTCL because its benefits were evaluated greater than its risks.
Lenalidomide
Lenalidomide is an immunomodulator and has multiple mechanisms of action, e.g. inhibition of proliferation of certain haematopoietic tumour cells (Bagot 2017).
Brentuximab vedotin
CD30 is frequently expressed in cutaneous T‐cell lymphoma making it targetable by the CD30‐antibody brentuximab vedotin (Prince 2017). Its approval was based on a significant benefit over treatment with bexarotene or methotrexate with an acceptable safety profile.
Mogamulizumab
Mogamulizumab is a monoclonal antibody targeting C‐C chemokine receptor 4. The mechanisms of action include antibody opsonisation of malignant T‐cells leading to the elimination of said cells by Natural killer cells. Mogamulizumab is FDA and EMA approved for patients with mycosis fungoides or Sézary syndrome who received a previous systemic treatment (Kim 2018).
Extracorporeal photopheresis
Extracorporeal photopheresis is a procedure by which leucocytes are first sensitised to ultraviolet‐A light (UVA ) via 8‐methoxypsoralen followed by irradiation with UVA leading to modulation of the immune system (Child 2004).
Stem cell transplantation
Stem cell transplantation is a complex therapy where bone marrow containing healthy lymphocytes is implanted in the patient. When stem cells from other people are used (allogeneic stem cell transplantation), there is a possibility that the donor lymphocytes eliminate the neoplastic lymphocytes causing mycosis fungoides (Duarte 2010).
Why it is important to do this review
As described above, there is a wide variety of available treatment options for mycosis fungoides. However, published reports on treatment options differ in terms of trial design, risk of bias, internal and external validity of the results and assessment of adverse effects. A systematic evaluation of these different characteristics is therefore warranted. As Humme 2014 pointed out, mycosis fungoides is an uncommon disease, which leads to difficulties recruiting patients for well‐designed randomised controlled trials (RCTs). Furthermore, it is likely that costs and logistical difficulties discourage investigators to initiate new RCTs. For these reasons, it is particularly important to assess the already available evidence. A systematic review of the evidence for benefits and harms will help decision‐making in individual clinical situations. It will also help in the process of developing evidence‐based clinical guidelines for the treatment of this disease. Since the initial review (Weberschock 2012), several potentially relevant RCTs have been published investigating new interventions, which makes an update necessary.
Objectives
To assess the effects of interventions for mycosis fungoides in all stages of the disease.
Methods
Criteria for considering studies for this review
Types of studies
We included randomised controlled trials (RCTs) of adults in which at least 90% were diagnosed with histologically‐confirmed mycosis fungoides (classical "Alibert‐Bazin" type). In contrast to the protocol (Weberschock 2011), we included studies with different investigated diseases (e.g. mycosis fungoides and other lymphomas), but separate outcome data for the mycosis fungoides cohort meeting our inclusion criteria had to be available.
For the analysis of the efficacy of interventions for mycosis fungoides, we excluded quasi‐randomised studies (e.g. alternate treatment allocation or by date of birth), as we considered this study design to be poor quality and likely to lead to unreliable study results. However, for the qualitative analysis of the safety of interventions, we included quasi‐randomised RCTs or non‐randomised studies since RCTs are known to have limited statistical power to detect rare adverse effects (Higgins 2011). For these studies we did not perform a formal qualitative assessment. A tabulated presentation of the rare adverse effects can be found in Table 10.
5. Rare adverse effects detected by separate adverse event search.
| Intervention | Rare severe adverse effects |
| PUVA | transient lymphomatoid papulosis (Aronsson 1982) basal cell carcinoma/squamous cell carcinoma (Herrmann 1995) squamous cell carcinoma (Molin 1981) cataract (Rupoli 2005) |
| extracorporeal photopheresis | sarcoma (Korpusik 2007) |
| imiquimod | no reported SAEs found |
| electron beam | total epilation (Braverman 1987, Desai 1988), nail dystrophy/oedema of hands and feet/bullae dorsum and feet/conjunctivitis/hospitalisation due to skin ulcers (Desai 1988) diffuse permanent telangiectasia/linear sclerosis/Ischaemic ulceration of finger tips (Micaily 1983) |
| cyclophosphamide, doxorubicin, etoposide, vincristine | haematological toxicity: febrile neutropenia/staphylococcal bacteraemia/disseminated herpes infection/pneumocystis carinii pneumonia/neurologic toxicity grade 3/decreased left ventricular ejection fraction (Akpek 1999) pulmonary embolism and death due to drug‐related cardiac infarction reported for doxorubicin monotherapy (Dummer 2012) nephrotic syndrome (Kairouani 2012) acute nonlymphocytic leukaemia reported for combination of CHOP with TSEBT (Kaye 1989) reticulated generalised skin pigmentation reported for combination of cyclophosphamide with prednisone (Youssef 2013) |
| active transfer factor | no reported SAEs found |
| methotrexate | severe oedematous erythroderma/denudation on the trunk and extremities/Interstitial pulmonary fibrosis (Zackheim 1996) epidermal necrosis (Mna 2016) |
| interferon‐alpha | acrocyanosis (Campo‐Voegeli 1998)
oropharyngeal lichen planus (Kütting 1997)
seizures (Legroux‐Crespel 2003)
fatal neutropenia and sepsis (Vegna 1990)
liver toxicity (Rupoli 2005, Simoni 1987, Vegna 1990) generalised urticaria in association with angio‐oedema (Hüsken 2012) |
| bexarotene | neutropenia/non‐ST‐elevation myocardial infarction/elevated liver enzymes (Abbott 2009)
lethal sepsis (Bohmeyer 2003) pancreatitis (Duvic 2001b) bleeding gastric ulcer (Sokolowska‐Wojdylo 2016) |
| peldesine (BCX‐34) | no reported SAEs found |
| denileukin diftitox (ONTAK) | capillary leak syndrome (Duvic 2002 and Duvic 2013, Talpur 2012) lethal vascular leak syndrome with rhabdomyolysis (Avarbock 2008) grade 4 infusion event (Foss 2001) visual changes (McCann 2012) |
| nitrogen mustard | urticaria and anaphylactoid reaction (Daughters 1973, Grunnet 1976, Sanchez 1977) local bullous reaction (Goday 1990) perforating follicular mucinosis (Guilhou 1980) Stevens‐Johnson‐Syndrome (Newman 1997) cutaneous squamous/basal cell carcinoma (Hoppe 1987) |
| brentuximab vedotin | peripheral neuropathy (Duvic 2015, Corbin 2017) unstable angina or myocardial infarction, pulmonary embolism (Duvic 2015) progressive multifocal leukoencephalopathy (Carson 2014) acute renal failure (Kim 2015) |
| lenalidomide | thrombocytopenia, pneumonitis, fatigue, dyspnoea, cognitive disturbance, respiratory failure, seizure (Dueck 2010a) anaemia, lymphopenia, neutropenia, cardiac ischaemia/infarction‐acute myocardial infarction, hypoxia‐respiratory failure, rash, supraventricular arrhythmia (Toumishey 2015) pneumonitis, fatigue, cognitive disturbance, dyspnoea (Dueck 2010a, Toumishey 2015) |
Types of participants
We included studies of adults (aged 18 years or more) diagnosed with histologically‐confirmed mycosis fungoides of the classical "Alibert‐Bazin" type.
We excluded studies from this review that included more than 10% of participants with variants and subtypes of mycosis fungoides, such as folliculotropic mycosis fungoides, pagetoid reticulosis, or granulomatous slack skin.
Types of interventions
We were interested in comparisons of any local or systemic therapy with either another local or systemic therapy or with placebo. Types of interventions included the following:
topical therapies;
skin‐directed phototherapies;
total skin electron beam;
radiotherapy;
chemotherapy;
extracorporeal photochemotherapy;
biological response modifiers;
combination therapies (of the interventions listed above);
other skin‐directed treatment approaches; and
other systemic treatment approaches.
We made comparisons according to the stage of the disease, whereas the TNMB (tumour, lymph node, metastasis, and blood) classification was used primarily for consideration of the applicability of interventions to be used at a certain disease stage.
Types of outcome measures
We investigated the following primary and secondary outcomes.
Primary outcomes
Improvement in health‐related quality of life as defined by participant questionnaires (all self‐completed).
Common adverse effects of the treatments, presented as proportions of participants.
Secondary outcomes
Percentage of participants demonstrating complete response (CR), defined as complete disappearance of all clinical evidence of disease.
Relapse defined as recurrence of the disease in prior CR.
Disease‐free survival.
Overall survival.
Objective response rate (ORR) defined as proportion of patients with CR or partial response (PR). A PR is considered as a regression of measurable disease of at least 50% in one of the categories T, N, M and B without any progression of disease.
Rare adverse effects.
Search methods for identification of studies
We aimed to identify all relevant randomised controlled trials (RCTs) regardless of language or publication status (published, unpublished, in press, and in progress).
Electronic searches
For this update, we revised all our search strategies in line with current Cochrane Skin practices. Details of the previous search strategies are available in Weberschock 2012.
The Cochrane Skin Information Specialist searched the following databases up to 13 May 2019:
the Cochrane Skin Group Specialised Register using the search strategy in Appendix 1;
the Cochrane Central Register of Controlled Trials (CENTRAL); 2019, Issue 5, in the Cochrane Library using the strategy in Appendix 2;
MEDLINE via Ovid (from 1946) using the strategy in Appendix 3;
Embase via Ovid (from 1974) using the strategy in Appendix 4; and
LILACS (Latin American and Caribbean Health Science Information database, from 1982) using the strategy in Appendix 5.
Trials registers
Review authors (AV and MJ) searched the following trials registers for reports of trials using the terms 'mycosis fungoides' and 'cutaneous T‐cell lymphoma' on 20 May 2019:
ClinicalTrials.gov (www.clinicaltrials.gov); and
the World Health Organization International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch/).
Adverse effects
We examined the included and excluded RCTs for common adverse effects.
To find rare but potentially serious side‐effects in non‐RCTs, we conducted a separate search in MEDLINE (using the strategy in Appendix 6) up to 13 April 2017. We qualitatively summarised findings from non‐RCTs in Table 10.
We ran two separate additional searches for specific drugs not included in the April 2017 searches: brentuximab vedotin on 18 July 2017, and lenalidomide on 8 November 2017. We used the same terms as in Appendix 6 combined with these drug terms.
Searching other resources
Searching reference lists and handsearching
We examined the citation lists of the reports of identified trials and other relevant review articles to identify further references to relevant trials.
We examined the conference proceedings of the German Dermatologic Society (DDG) for the years 2013, 2015 and 2017, and the Arbeitsgemeinschaft Dermatologische Forschung (ADF) between 2012 and 2018. These conferences are not covered by online database searches.
Correspondence with trialists/experts/organisations
We contacted the corresponding authors of potentially relevant studies for additional information.
Data collection and analysis
Selection of studies
At least two review authors (MJ and AV) independently screened titles and abstracts of studies identified from the above sources for the eligibility criteria stated previously. If this could not be done satisfactorily from the title and abstract, we obtained a full‐text version for assessment.
We assessed studies that displayed characteristics meeting the inclusion criteria by screening for eligibility using an eligibility form. This eligibility form contained the following questions.
Is the study described as randomised?
Did at least 90% of the participants in the study have biopsy‐proven classical mycosis fungoides?
Is the stage of the mycosis fungoides given?
Are the participants under investigation 18 years of age or older?
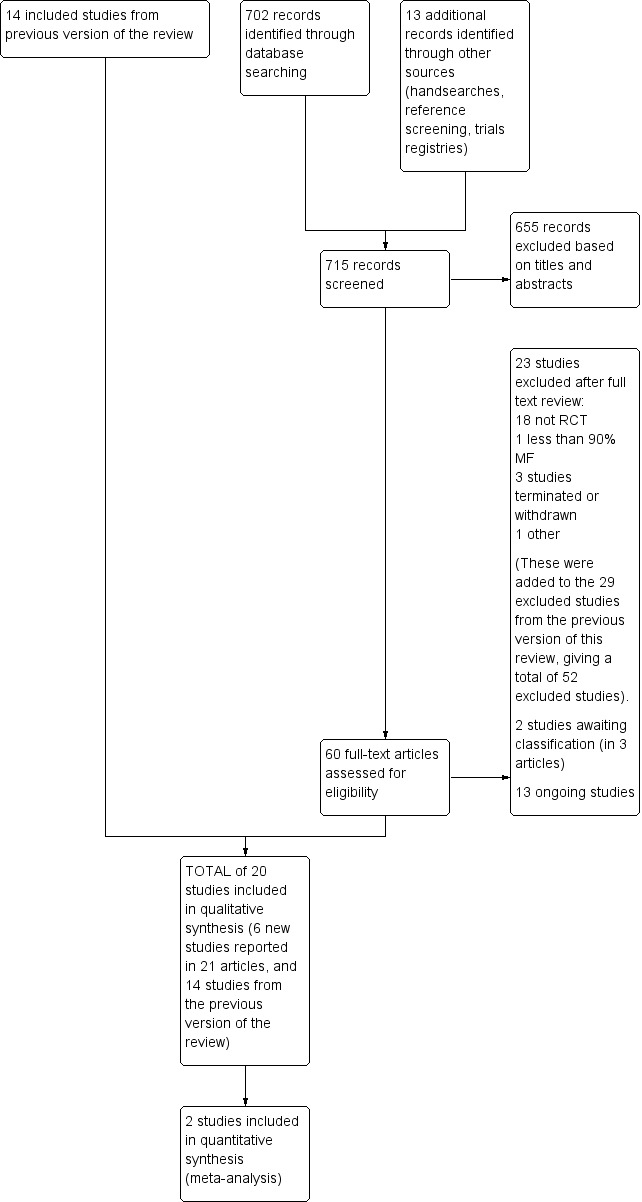
To be eligible, studies had to meet all of the criteria stated above. We included abstracts and unpublished data if sufficient information on study design, characteristics of participants, interventions and outcomes was available; otherwise, we excluded them or included them with reservations following discussion with the review authors. If there was insufficient information to judge eligibility, we tried to contact the first author of the report for clarification. This process is described in more detailed in the section 'Dealing with missing data'. We resolved any disagreements between the review authors (AV and MJ) by discussion and consensus with a third party (TW). We identified any duplicate reports. We obtained full‐text versions of all eligible studies for quality assessment and data collection, where available. At every stage of searching and screening of the literature, we documented ‐ with reasons ‐ the overall number of studies identified and the number excluded and included in a flow diagram (Figure 2) as suggested by the Preferred Reporting Items of Systematic Reviews and Meta‐Analyses (PRISMA) (Moher 2009).
2.
Study flow diagram.
Data extraction and management
Two review authors (AV, MJ) independently extracted the following data from the studies which met the inclusion criteria of this review.
information about the treatment and outcome for each participant, which included information on the diagnosis and stage of mycosis fungoides, received treatment, additional therapy, quality of life, objective response rate/complete response, duration of remission, overall survival, and toxicity and adverse effects;
potentially significant participant‐related prognostic factors, which included information on age (birth date) and gender; and
potentially significant tumour‐related prognostic factors, which included information on histological subtype, clinical stage (patch, plaque, tumour), blood tumour burden: atypical T lymphocytes (Lutzner cells), elevated eosinophilic cells, and systemic involvement (lymph nodes, bone marrow, internal organs).
We tried to obtain any missing data from the trial authors, where possible. We developed a data collection form and piloted it in order to summarise the trials.
Assessment of risk of bias in included studies
At least two review authors (MJ, AV and TW) independently assessed quality by doing a 'Risk of bias' assessment using the new features of Review Manager 5 and as described in Table 8.5c of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). The same authors assessed the domains, which included the following:
(a) sequence generation (selection bias); (b) allocation concealment (selection bias); (c) blinding (performance and detection bias); (d) whether incomplete outcome data were addressed (attrition bias); (e) whether the study was free of selective reporting (reporting bias); and (f) whether the study was free of other bias.
We discussed any disagreements until consensus was obtained. We assessed quality using an assessment form designed for the topic of this review (sources used: Hollis 1999; Jüni 2001; Moher 1995; Verhagen 1998).
Assessment of external validity
The 'Risk of bias' domains described so far help to investigate the potentially lowered internal validity of studies. But when study data need to be incorporated into daily practice, there is also considerable risk of bias that potentially influences the external validity.
We assessed external validity of all included trials by addressing: study population and eligibility criteria; temporal, ethnic, socio‐economic and geographical aspects; and generalisability as proposed by Dekkers 2010.
The reference population for this aspect was the middle‐aged population between 50 and 60 years old with mycosis fungoides (classical "Alibert Bazin" type) treated in secondary and tertiary referral centres, since this seemed most likely to represent the overall largest group of people with the disease and possible access to the treatment options (Kim 2003; Weinstock 1988; Weinstock 1999).
More details can be found in Appendix 7.
Measures of treatment effect
The effect measures of choice were the mean difference (MD) for continuous outcomes and the risk ratio (RR) for binary outcomes. For time‐to‐event effect measures we used the hazard ratio (HR). For all measures of effect, we reported 95% confidence intervals (CIs) and corresponding P values. In order to gain more accurate analyses for smaller sample sizes, we added Fisher tests, which is a deviation from the review protocol. This change was made in order to avoid spurious (non‐)significance in studies with small sample sizes or low numbers of events. P values ≤ 0.05 were considered to be significant.
Unit of analysis issues
The standard unit of analysis in the review is the participant. This also applies to cross‐over trials. In case of within‐participant trials with lesional treatments, comparable lesions of participants were also accepted as the unit of analysis, thus, allowing us to include within‐participant trials. For cross‐over trials and within‐participant trials, we extracted the data as reported in the trials. This approach is prone to a carry‐over effect and to over‐ or underestimation of the precision of results.
Dealing with missing data
We attempted to obtain data that were not reported directly from the original researchers.
We dealt with missing data by contacting the corresponding author of the paper with missing data and asking him or her to provide these data. This was done for inclusion and exclusion criteria, possible sources of bias (as described in the section Assessment of risk of bias in included studies), and for outcome data. If the corresponding author did not reply after the third approach via letter or email within four weeks, or they did not provide the requested data, we classified the data as missing. Because of the low number of comparable trials, we could not perform reliable sensitivity analyses to assess the potential effects of missing data on the meta‐analysis.
Assessment of heterogeneity
In order to address clinical diversity between studies, we tabulated the included studies in terms of study characteristics and outcomes, and then carefully examined them for quality, similarities and differences. We addressed statistical heterogeneity between individual studies reporting on outcomes for the same intervention using the I² statistic. Pooling data was considered in case of two or more trials reporting on the same intervention with clinical similarity, and comparable study quality. In such case, we planned to use a fixed‐effect model for meta‐analysis. If clinically and methodological heterogeneity was suspected, then we would use the random‐effects model. Substantial statistical heterogeneity was defined as an I² statistic with a value greater than or equal to 50% and we would undertake subgroup analyses to investigate the clinical and methodological heterogeneity in these circumstances. If extreme levels of statistical heterogeneity existed between the studies (I² statistic > 80%) which could not be explained by subgroup analyses, we intended to report the results of the studies individually and explore heterogeneity using subgroup analyses..
Assessment of reporting biases
In order to assess for possible reporting bias, we planned to examine a funnel plot for asymmetry where feasible (e.g. more than 10 studies for an outcome). Because of the low number of comparable trials, we could not reliably investigate reporting bias.
Data synthesis
In case of clinical and methodological similarity we pooled data for meta‐analysis using the fixed‐effect model. Meta‐analysis comprised risk ratios (RR) with corresponding confidence intervals (95% CIs) and were presented as forest plots. Where HRs were available we used the generic inverse variance method (and random‐effects model) to report the pooled estimates and 95% CIs. If meta‐analysis was not possible, we used a narrative approach. Review Manager 2014 was used to conduct the analyses. For continuous outcomes we used the mean difference (MD) or the standardised mean difference (SMD) as appropriate in order to establish comparability among different scales.
Subgroup analysis and investigation of heterogeneity
We planned to explore potential causes of heterogeneity by performing subgroup analyses: stage of mycosis fungoides (patch, plaque, or tumour) and different interventions. However, we were unable to undertake any subgroup analyses due to low number of comparable trials.
Sensitivity analysis
We planned to perform sensitivity analyses to explore the influence of the following factors on effect size.
Study quality according to risk of bias (sequentially).
Largest trials in the review measured by number of randomised participants.
Excluding studies using the following filters: language of publication (English versus other), funding sources (yes/no), etc.
Investigation of the origin (individual participant data or publication) of the data/information.
Because of the low number of comparable trials, we could not perform reliable sensitivity analyses.
'Summary of findings' tables
The "Summary of findings tables" provide key outcomes, the magnitude of effects and the certainty of the evidence presented in Cochrane systematic reviews. For creating our 'Summary of findings tables' we used the GRADE approach (GRADE pro GDT 2015). In order to evaluate the certainty of the evidence, we assessed the data in the categories risk of bias, inconsistency, indirectness, imprecision and publication bias. Based on these criteria the quality of evidence was rated as high, moderate, low or very low (Guyatt 2008).
Due to the variety of the interventions, a rational summarisation of all interventions proved to be difficult. Therefore, we focused on a treatment which is essential for the therapy of mycosis fungoides. Patients with mycosis fungoides are mostly treated by dermatologists in a tertiary care setting in which PUVA is readily available. The treatment is easy to perform and has manageable adverse effects. Reflecting the importance of PUVA therapy, current international guidelines recommend PUVA as a first‐line therapy in early stage of disease (Olsen 2016). Furthermore, PUVA is combined with systemic therapies in more advanced stages of disease (Trautinger 2017). For these reasons we focused on RCTs comparing PUVA with other interventions.
The primary end points of this review (quality of life, common adverse events) were not adequately measured, thus the following secondary end points were also reported in the tables: complete response (CR), and objective response rate (ORR).
Results
Description of studies
Please see the 'Characteristics of included studies', Characteristics of excluded studies', 'Characteristics of studies awaiting classification', and Characteristics of ongoing studies' tables for full descriptions.
Results of the search
In this update, the Electronic searches retrieved 702 records. We searched a number of other sources: handsearching (no records identified), examination of the reference lists of relevant studies and reviews (one record identified), and searches of trials databases (12 records identified). We therefore had a total of 715 records.
We excluded 655 records based on titles and abstracts. We obtained the full text of the remaining 60 records. We excluded 23 studies (see Characteristics of excluded studies). We added three records to two studies awaiting classification (see Characteristics of studies awaiting classification). We identified 13 ongoing studies (see Characteristics of ongoing studies).
We included six new studies reported in 21 references (see Characteristics of included studies). We combined these studies with the 14 previously included in this review, and for this update we included a total of 20 trials (1369 participants). We have excluded a total of 52 studies (23 new, 29 from the previous review). Figure 2 shows a flow diagram summarising our study selection process.
Dealing with missing data
After inclusion of all publications, we tried to contact the authors of included publications by sending an individualised missing data contact form to them via email.
Included studies
Designs
In the initial review we included 14 randomised controlled trial (RCTs) (Child 2004; Chong 2004; Duvic 2001; Duvic 2001a; Guitart 2002; Kaye 1989; Olsen 2001; Rook 2010; Stadler 1998; Stadler 2006; Thestrup‐Pedersen 1982; Vonderheid 1987; Wolff 1985; Wozniak 2008). Two were within‐participant designs assessing efficacy of topically applied agents (Rook 2010) or intralesional injections (Vonderheid 1987), one had a cross‐over design (Child 2004), and 11 had a parallel‐group design. In this update we identified six more parallel RCTs (Bagot 2017; Kim 2018; Lessin 2013; Prince 2017; Vieyra‐Garcia 2019; Whittaker 2012). All studies randomly assigned participants or comparable lesions of participants to one of the treatment groups.
Sample Size
The number of participants evaluated in the studies varied from four to 260 participants. Seven of the included RCTs consisted of a very small sample size of less than 20 participants (Child 2004; Chong 2004; Rook 2010; Thestrup‐Pedersen 1982; Vieyra‐Garcia 2019; Vonderheid 1987; Wolff 1985). Three studies had a sample size of 20 to 49 participants (Bagot 2017; Guitart 2002; Wozniak 2008), seven studies enrolled 50 to 99 participants (Duvic 2001; Duvic 2001a; Olsen 2001; Prince 2017; Stadler 1998; Stadler 2006; Whittaker 2012), and three studies (Kaye 1989; Kim 2018; Lessin 2013) had more than 100 participants.
Population
Fourteen of the included trials exclusively assessed participants with cutaneous T‐cell lymphomas (CTCL) in clinical stages IA to IIB (Child 2004; Chong 2004; Duvic 2001; Duvic 2001a; Guitart 2002; Lessin 2013; Rook 2010; Stadler 1998; Stadler 2006; Vieyra‐Garcia 2019; Vonderheid 1987; Whittaker 2012; Wolff 1985; Wozniak 2008). Three studies also included participants with CTCL in clinical stage III (Kaye 1989; Olsen 2001; Thestrup‐Pedersen 1982), and five studies (Bagot 2017; Kaye 1989; Kim 2018; Olsen 2001; Prince 2017) also assessed participants in stage IV.
Child 2004, Chong 2004,Guitart 2002,Kaye 1989,Lessin 2013, Olsen 2001, Thestrup‐Pedersen 1982,Vieyra‐Garcia 2019, Vonderheid 1987,Whittaker 2012, Wolff 1985, and Wozniak 2008 enrolled participants with mycosis fungoides (MF) only.
All studies except one (Chong 2004), enrolled men and women, although most of the studies enrolled more men than women. When information on participants' origin was available, most enrolled participants were white. Participants' ages ranged from 18 to >75 years. In Lessin 2013, one of 260 participants was a minor (11 years old). In an effort to not withhold the interested reader of this review from the results of this trial, we sought further information to exclude this patient from our analysis. Unfortunately, we did not receive a reply within four weeks. After careful consideration, we still included this trial. No other published RCT was excluded because of this criterion.
Setting
All included studies took place in secondary and tertiary care skin tumour settings. Twelve studies enrolled participants in Europe (Bagot 2017; Child 2004; Chong 2004; Duvic 2001; Kim 2018; Prince 2017; Stadler 1998; Stadler 2006; Thestrup‐Pedersen 1982; Vieyra‐Garcia 2019; Whittaker 2012; Wozniak 2008), 11 studies in North America (Duvic 2001; Duvic 2001a; Guitart 2002; Kaye 1989; Kim 2018; Lessin 2013; Olsen 2001; Prince 2017; Rook 2010; Vonderheid 1987; Wolff 1985), and three studies in Australia (Duvic 2001; Kim 2018; Prince 2017). Prince 2017 also included patients from South America (Brazil). Kim 2018 included patients from Japan. Fifteen RCTs were multicentre trials (Bagot 2017; Duvic 2001; Duvic 2001a; Guitart 2002; Kaye 1989; Kim 2018; Lessin 2013; Olsen 2001; Prince 2017; Rook 2010; Stadler 1998; Stadler 2006; Vieyra‐Garcia 2019; Whittaker 2012; Wozniak 2008). Fifty‐five per cent of the included trials were completely or partially funded by pharmaceutical companies. Non‐commercial sponsors funded 20% of the studies. Further details are provided in the Characteristics of included studies section.
Interventions
The studies were conducted with a wide range of interventions including:
invasive and non‐invasive topical treatments;
light therapies, including extracorporeal photopheresis;
oral treatments;
parenteral applied systemic agents; and
radiation therapies.
Comparators comprised of active ingredients, placebo or observation after having achieved complete response (CR).
Nine RCTs combined two or more therapies for at least one treatment group (Guitart 2002; Kaye 1989; Rook 2010; Stadler 1998; Stadler 2006; Thestrup‐Pedersen 1982; Whittaker 2012; Wolff 1985; Wozniak 2008).
The topical treatments, used as interventions or comparators, consisted of:
topical application of hypericin (Rook 2010);
imiquimod 5% (Chong 2004);
intralesional injections of IFN‐α without (Vonderheid 1987) or with the combined use of topical steroids (Wolff 1985);
mechlorethamine 0.02% gel versus mechlorethamine 0.02% ointment (comparator) (Lessin 2013);
nitrogen mustard (comparator: Kaye 1989; intervention: Thestrup‐Pedersen 1982); or
peldesine (Duvic 2001a).
The light therapies investigated, as interventions or comparators were:
psoralen plus ultraviolet A light (PUVA) used as intervention and comparator (Child 2004; Kaye 1989; Stadler 1998; Stadler 2006; Thestrup‐Pedersen 1982; Vieyra‐Garcia 2019; Wozniak 2008), or PUVA combined with bexarotene (Guitart 2002; Whittaker 2012),
extracorporeal photopheresis (intervention and comparator) (Child 2004); and
visible light (intervention and comparator) (Rook 2010).
The oral treatments assessed included:
acitretin (comparator) (Stadler 1998);
bexarotene (intervention and comparator) (Duvic 2001; Guitart 2002; Whittaker 2012);
lenalidomide (Bagot 2017);
vorinostat (comparator) (Kim 2018); and
methotrexate (comparator) (Kaye 1989; Prince 2017).
Treatment with parenteral systemic agents consisted of:
the infusion of brentuximab vedotin (Prince 2017), denileukin diftitox (Olsen 2001), and mogamulizumab (Kim 2018);
the parenteral chemotherapy with cyclophosphamide, doxorubicin, etoposide, and vincristine (Kaye 1989);
the subcutaneous injection of IFN‐α (intervention and comparator) (Stadler 2006; Stadler 1998; Wozniak 2008); and
the intramuscular injections of active transfer factor (Thestrup‐Pedersen 1982).
Finally, we assessed one RCT using electron beam therapy (Kaye 1989).
Outcomes
Only three studies used standardised written questionnaires to assess quality of life (QoL) during treatment: Duvic 2001 used the Spitzer QoL questionnaire validated for survivors in palliative care and hospice settings (Spitzer 1981) and a non‐validated CTCL‐patient questionnaire. Kim 2018, Olsen 2001 and Prince 2017 used the Functional Assessment in Cancer Therapy‐general (FACT‐G) questionnaire developed by Cella 1993.
Eighteen studies reported common adverse effects or their absence. This outcome was assessed by physicians (Bagot 2017; Child 2004; Chong 2004; Duvic 2001; Duvic 2001a; Guitart 2002; Kaye 1989; Kim 2018; Lessin 2013; Olsen 2001; Prince 2017; Rook 2010; Stadler 1998; Thestrup‐Pedersen 1982; Vieyra‐Garcia 2019; Vonderheid 1987; Whittaker 2012; Wolff 1985).
All studies but Bagot 2017 and Rook 2010 assessed clearance (either directly or indirectly) in at least one of the following aspects of disease: lesion surfaces or lesion size of all lesions or target lesions only, blood tumour burden, or tumour size. In Vieyra‐Garcia 2019 CR was a condition for randomisation in one of the study arms (PUVA maintenance versus no maintenance) and could therefore not be attributed to one of the interventions.
Relapse was investigated in seven studies (Duvic 2001; Guitart 2002; Kaye 1989; Kim 2018; Vieyra‐Garcia 2019; Whittaker 2012; Wozniak 2008). In two of those, relapse was not assessable (Kim 2018; Whittaker 2012). Due to the special study design of Vieyra‐Garcia 2019, relapse equates to disease‐free survival (see below).
Kaye 1989, Vieyra‐Garcia 2019 and Wozniak 2008 examined disease‐free survival.
Survival rates were the subject of eight studies (Child 2004; Duvic 2001a; Guitart 2002; Kaye 1989; Olsen 2001; Rook 2010; Thestrup‐Pedersen 1982; Whittaker 2012). However, this outcome was not evaluable in Whittaker 2012.
Objective response rate (ORR) was measured in 13 studies (Chong 2004; Duvic 2001; Duvic 2001a; Guitart 2002; Kaye 1989; Kim 2018; Lessin 2013; Olsen 2001; Prince 2017; Rook 2010; Stadler 1998; Thestrup‐Pedersen 1982; Whittaker 2012). In Stadler 1998, this outcome was not assessable due to not fulfilling the criteria of Olsen 2011.
Rare adverse effects were described in five studies (Child 2004; Chong 2004; Duvic 2001; Guitart 2002; Olsen 2001).
Definition of complete or partial response and quantitative assessment of clearance were very heterogeneous among the included studies.
Outcomes were generally assessed by physicians. Improvement of quality of life was the only patient‐reported outcome.
Time point of outcome assessment varied among the included studies due to the number and variety of interventions, their different administration routes as well as their different mechanisms of action.
Study duration
The duration of the studies varied widely (four weeks to 12 months, except one study lasting more than six years). This was, in part, related to the clinical stage of disease and outcomes to be observed, for example, time to first response or survival. Some studies continued treatment until an optimal response was achieved. The longest study was Kaye 1989, with a median period from enrolment to analysis of 75.3 months (range 25.9 to 118.2 months).
Excluded studies
In the initial review we excluded 150 studies after reading the full text. These were mostly (122) excluded because they were not RCTs, which was identified in the full text (121) or after author contact (Olsen 1986). We excluded 15 studies as they enrolled < 90% participants with Alibert‐Bazin type MF with no subgroup analysis available (Cooper 1994; Currie 1980; Dang 2007; Doan 1958; Dueck 2010; Fisher 1993; Kaung 1969; Kuzel 2010; Neering 1972; Anonymous 1982; Prince 2010; Simon 2010; Thomsen 1977; Wiernik 1998; Zubrod 1960). We excluded four RCTs since they did not report any relevant outcome as set in the trial protocol (Argyropoulos 1979; Breneman 1991; Lansigan 2010; Schrag 1997). We excluded eight studies after attempts to contact corresponding authors (because of insufficient data for abstraction) were unsuccessful (Fawzi 2010; JapicCTI‐050041; Kujawska 2003; NCT00054171; Negro‐Vilar 2007; Pan 2007; Plettenberg 2001; Wain 2005). We excluded the RCT by Peugeot 1995 because of scientific fraud (Grant 2009).
In this update we excluded 23 studies after reading the full text (Figure 2). We excluded 18 studies because they were not RCTs (Anonymous 2000; Aviles 2015; Bazex 1975; Duvic 2010; Foss 2011; Heald 2003; Lambert 1986; Loescher 1984; Marsden 1968; Moog 2008; NCT00091208; O'Neill 2013; Serri 1990; Shi 2015; Thomsen 1979; Thomsen 1989; Touraine 1978; Wilson 1995). We excluded one study (Groth 1979) as it enrolled < 90% participants with Alibert‐Bazin type MF. From the ongoing trials in the initial review, one trial (NCT01187446) was terminated with the following reason given: "business decision". We contacted the principal investigator for results of the trial but did not receive a reply. Another clinical trial (NCT01625455) was terminated due to difficulty recruiting. One trial was excluded due to being withdrawn prior to enrolment (NCT01386398). We excluded NCT01007448 due to insufficient data as we contacted the authors but were unable to obtain any results. Further details are provided in the 'Characteristics of excluded studies' tables.
Ongoing trials
Five of the ongoing trials identified in the initial review have been published in the meantime and were included in this update (NCT01098656 ➝ Bagot 2017; NCT01728805 ➝ Kim 2018; NCT00168064 ➝ Lessin 2013; NCT01686594 ➝ Vieyra‐Garcia 2019; NCT00056056 ➝ Whittaker 2012).
Furthermore, we were able to identify 13 new ongoing trials.
NCT01738594 (carfilzomib IV versus carfilzomib IV and romidepsin IV)
NCT02213861 (1.0% SHAPE gelled solution once daily versus 0.5% SHAPE gelled solution twice daily versus 1.0% SHAPE gelled solution twice daily)
NCT02301494 (fluocinonide (Vanos) cream 0.1% versus 3.75% imiquimod (Zyclara) cream)
NCT02323659 (methotrexate versus interferon alfa‐2b)
NCT02448381 (topical SGC301, a topical photosensitising agent, versus placebo)
NCT02811783 (naloxone hydrochloride lotion 0.5% versus placebo)
NCT02943642 (A‐dmDT390‐bisFv(UCHT1) versus vorinostat)
NCT02953301 (resminostat versus placebo)
NCT03011814 (durvalumab IV versus durvalumab IV plus lenalidomide)
NCT03292406 (placebo followed by CD11301 (0.03%) topical gel versus CD11301 (0.03%) topical gel versus CD11301 (0.06%) topical gel)
NCT03454945 (vibramycin versus UVA + psoralen)
NCT03713320 (cobomarsen versus vorinostat)
UMIN000029537 (bexarotene alone versus bexarotene plus phototherapy)
Furher details are presented in the 'Characteristics of ongoing studies' section.
Studies awaiting classification
In the initial review, we identified two studies (Foss 2011; Lessin 2011), which were detailed in the Characteristics of studies awaiting classification table. Ultimately, we excluded Foss 2011 because it did not have a RCT design. Lessin 2011 is an excerpt from Lessin 2013, which was included in this update.
We identified three studies (Bashey 2014; Kim 2014), which we detailed in the Characteristics of studies awaiting classification table. One study that we initially included in the ongoing trials section has been completed (Kim 2014). The author stated that the results had not been published yet and that a further trial would be underway. Unpublished data were not provided. These studies will be assessed in the next update of this review.
Risk of bias in included studies
Regarding the risk of bias in the included studies, we looked at the following seven possible sources of bias: generation of the randomisation sequence, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective reporting, and other source of bias. Further details are provided in the 'Characteristics of included studies' tables and the 'Risk of bias' tables for each study. See also Figure 3 and Figure 4 for a graphical summary of the 'Risk of bias' components.
3.

'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
4.

'Risk of bias' summary: review authors' judgements about each 'Risk of bias' item for each included study.
Allocation
Sequence generation and allocation concealment
We contacted the authors asking for further details about the random sequence generation and allocation concealment by the same contact procedure as when dealing with missing data, since procedures of randomisation and concealment were missing or incomplete in most included studies. In most cases we did not obtain any further information, which we classified according to the categories in the 'Risk of bias' table. The authors of one study stated that the randomisation list was generated by a statistician, who was provided by the sponsor and was not involved in the remainder of the trial (Prince 2017).
The method of generation of the randomisation sequence was described in only eight of the studies. Child 2004 used envelopes randomly allocating participants to treatment groups created by a statistician. However, regarding concealment of allocation, it was not stated whether those envelopes were sealed and opaque. Kaye 1989 used stratified block randomisation without any information on concealment, while Olsen 2001 only mentioned stratification of participants by stage of CTCL for this multicentre trial. The statements that data management (Duvic 2001a) or randomisation through a central institution stratified by pre‐treatment (Stadler 1998) were conducted by third parties led us to conclude that randomisation was concealed for these two studies. Two studies used an interactive voice response system for randomly assigning the patients into the treatment arms (Kim 2018 and Prince 2017) and one study used a computerised randomisation service not otherwise specified (Vieyra‐Garcia 2019), which we evaluated as a concealed allocation.
In total, we assessed nine trials to be of low risk of selection bias (Bagot 2017; Child 2004; Kaye 1989; Kim 2018; Olsen 2001; Prince 2017; Stadler 1998; Vieyra‐Garcia 2019; Whittaker 2012). The remaining 11 trials were considered to be of unclear risk (Chong 2004; Duvic 2001; Duvic 2001a; Guitart 2002; Lessin 2013; Rook 2010; Stadler 2006; Thestrup‐Pedersen 1982; Vonderheid 1987; Wolff 1985; Wozniak 2008). No trial was rated to be of high risk of selection bias.
In terms of allocation concealment, six studies were considered to be of low risk (Bagot 2017; Duvic 2001a; Kim 2018; Prince 2017; Stadler 1998; Vieyra‐Garcia 2019), 13 of unclear (Child 2004; Chong 2004; Duvic 2001; Guitart 2002; Kaye 1989; Lessin 2013; Olsen 2001; Rook 2010; Stadler 2006; Vonderheid 1987; Whittaker 2012; Wolff 1985; Wozniak 2008, and one study to be of high risk (Thestrup‐Pedersen 1982). Downgrading the validity of Thestrup‐Pedersen 1982 is due to the trial author confirming that the randomisation list was open, which we rated as high risk.
Further details are provided in the 'Risk of bias' tables in the Characteristics of included studies section.
Blinding
Although six studies (Chong 2004; Duvic 2001a; Rook 2010; Thestrup‐Pedersen 1982; Vonderheid 1987; Wolff 1985) were described as 'double‐blind' or implied double‐blinding, only one of them (Duvic 2001a) provided details about how double‐blinding of e.g. participants, investigators, statisticians and other study personnel was maintained. If studies used different modalities (e.g. PUVA versus capsules) and blinding participants or clinicians was hardly possible, we judged lack of blinding to be an unclear risk (Child 2004; Kaye 1989; Lessin 2013; Prince 2017; Stadler 1998; Vieyra‐Garcia 2019). If studies used different treatment intervals (e.g. once‐weekly injection versus twice‐weekly injections) and blinding was not provided by placebo treatment (e.g. placebo injections), we judged lack of blinding, which was described or assumed by the description of the study, to represent a high risk of performance bias (Duvic 2001; Guitart 2002; Olsen 2001; Stadler 2006; Wozniak 2008). Kim 2018 was an open‐label study. The authors of one study comparing PUVA + bexarotene versus PUVA alone (Whittaker 2012) described that study personnel were not blinded to the treatment arms, which we ranked as a high risk of performance bias, since a placebo pill in the control arm could have been easily administered.
Outcome assessors were described as blinded in eight studies (Duvic 2001; Duvic 2001a; Kim 2018; Lessin 2013; Olsen 2001; Prince 2017; Thestrup‐Pedersen 1982; Wolff 1985). In all other studies (Bagot 2017; Child 2004; Chong 2004; Guitart 2002; Kaye 1989; Rook 2010; Stadler 1998; Stadler 2006; Vonderheid 1987; Whittaker 2012; Wozniak 2008), it remained unclear whether assessors were blinded or not. Final assessment of Prince 2017 and Bagot 2017 was performed after contacting the respective author teams.
Further details are provided in the 'Risk of bias' tables in the Characteristics of included studies section.
Incomplete outcome data
The overall number of participants lost to follow‐up was 185/1369 (13.5%) of the total number of study participants included in the review. Dropout rates varied from 0% to 72.4%. Eighteen of the included studies analysed data on an intention‐to‐treat (ITT) basis. We regarded studies to have a low risk of bias if either analyses were ITT analyses with complete outcome data for the participants analysed in the randomised group (Chong 2004; Kim 2018; Prince 2017; Rook 2010; Vieyra‐Garcia 2019; Vonderheid 1987; Wolff 1985; Wozniak 2008) or dropout rates were < 10% and data from dropouts were explicitly included in the ITT analysis (Guitart 2002; Lessin 2013; Whittaker 2012). We judged studies to have an unclear risk of bias if dropouts were 10% to 20% and data were analysed on an ITT basis (Kaye 1989), or if the number of participants randomised was unclear (Thestrup‐Pedersen 1982). Studies with dropouts > 10% that were analysed by per‐protocol analysis (Stadler 1998) or with dropouts > 20% that were analysed by ITT (Bagot 2017; Child 2004; Duvic 2001; Duvic 2001a; Olsen 2001; Stadler 2006) were regarded as having a high risk of bias. In particular, the high dropout rate of 72.4% in the trial of Duvic 2001 made it difficult to draw reasonable conclusions.
Further details are provided in the 'Risk of bias' tables in the Characteristics of included studies section.
Selective reporting
We contacted the corresponding authors of included studies to provide data about outcomes not reported in their publications, although this method has methodological limitations (Chan 2004). With regard to selective reporting, if an included study was previously registered in a screened database of ongoing trials, we compared all stages of the published with the reported outcomes.
Three authors responded and stated that all of the outcomes within their studies were reported (Bagot 2017; Guitart 2002; Thestrup‐Pedersen 1982). These studies were judged to have a low risk of reporting bias. Since some studies reported results for some end points for responders and non‐responders instead of comparisons between treatment groups (Olsen 2001; Wozniak 2008), only reported on a single end point (Stadler 2006), or used mean differences instead of absolute results (Wolff 1985), we regarded these studies to have a high risk of bias for selective reporting. In Kim 2018 and Prince 2017, only a proportion of the outcomes were reported, therefore we rated these trials to be of high risk of reporting bias as well. Furthermore, we identified two studies which did not report the prespecified secondary outcomes listed in the trials registry (Lessin 2013; Vieyra‐Garcia 2019). Hence, we judged these studies to be at high risk of reporting bias.
Further details are provided in the 'Risk of bias' tables in the Characteristics of included studies section.
Other potential sources of bias
Details of other aspects that are likely to have impact on validity or may be sources of other bias are mentioned in the 'Risk of bias' tables in the Characteristics of included studies section. Only four studies were considered free of other sources of bias (Chong 2004; Duvic 2001aLessin 2013; Prince 2017). Ten studies were considered high risk of other biases (Bagot 2017; Child 2004; Duvic 2001; Guitart 2002; Kim 2018; Thestrup‐Pedersen 1982; Vieyra‐Garcia 2019; Whittaker 2012; Wolff 1985; Wozniak 2008); reasons included that the study was prematurely closed, discontinued randomisation, and use of a concomitant treatment. In the remaining six studies it was unclear whether other biases were presented. Unclear risk was assigned to six studies (Kaye 1989; Olsen 2001; Rook 2010; Stadler 1998; Stadler 2006; Vonderheid 1987).
Assessment of external validity
Appendix 7 shows the checklist we used to assess external validity. After careful consideration, no major limitations for the external validity could be observed. Since MF is a rare disease which is normally treated in specialised medical centres (tertiary care setting), the results of the studies were considered applicable to daily practice.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5
Summary of findings 1. IFN‐α + PUVA compared to PUVA alone for mycosis fungoides.
| IFN‐α + PUVA compared to PUVA alone for mycosis fungoides | ||||||
|
Patient or population: people with mycosis fungoides
Setting: tertiary care setting
Intervention: IFN‐α + PUVA
Comparison: PUVA alone Number of trials included: 2 (Stadler 2006; Wozniak 2008) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with PUVA alone | Risk with IFN‐α + PUVA | |||||
| Improvement of quality of life | ‐ | ‐ | ‐ | ‐ | ‐ | Not measured |
| Common adverse effects | ‐ | ‐ | ‐ | ‐ | ‐ | Not measured |
| Complete response (CR)
assessed with: outcome assessment not described Time point of measurement Stadler 2006: up to week 52 Wozniak 2008: up to week 24 |
Study population | RR 1.07 (0.87 to 1.31) | 122 (2 RCTs) | ⊕⊕⊝⊝ Low a | ‐ | |
| 731 per 1000 | 783 per 1000 (636 to 958) | |||||
| Objective response rate (ORR) | ‐ | ‐ | ‐ | ‐ | ‐ | Not measured |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). The assumed risk is based on the number of events/number of participants in the control groups in Analysis 5.1. CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
a Downgraded by two levels to low‐certainty evidence. One level because of low internal validity (risk of bias ‐ performance bias in both studies, attrition bias in Stadler 2006) and one level because of low sample size (imprecision)
Summary of findings 2. Extracorporeal photopheresis compared to PUVA for mycosis fungoides.
| Extracorporeal photopheresis compared to PUVA for mycosis fungoides | ||||||
|
Patient or population: people with mycosis fungoides
Setting: tertiary care setting
Intervention: Extracorporeal photopheresis (ECP)
Comparison: PUVA Number of trials included: 1 (Child 2004) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with PUVA | Risk with Extracorporeal photopheresis | |||||
| Improvement of quality of life | ‐ | ‐ | ‐ | ‐ | ‐ | Not measured |
| Common adverse effects (Time point of measurement: three months of PUVA or six months of ECPc) |
Some participants reported mild nausea after PUVA. However, incidences and time points were not stated. One participant starting in the ECP group had hypotension leading to withdrawal from the study. | ‐ | 16 (1 RCT) b | ⊕⊝⊝⊝ Very low a | ‐ | |
| Complete response (Time point of measurement: three months of PUVA or six months of ECPc) |
Study population | RR 0.20 (0.01 to 3.61) | 16 (1 RCT) b | ⊕⊝⊝⊝ Very low a | ‐ | |
| 250 per 1000 | 50 per 1000 (3 to 903) | |||||
| Objective response rate (Time point of measurement: three months of PUVA or six months of ECPc) |
Study population | RR 0.08 (0.01 to 1.17) | 16 (1 RCT) b | ⊕⊝⊝⊝ Very low a | Additionally computed 95% CI by the use of the method described by Miettinen 1985 (adverse effects requiring discontinuation): 95% CI by Miettinen 0.00 to 0.40, Fisher test P = 0.002 | |
| 750 per 1000 | 53 per 1000 (0 to 750) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
a Downgraded by three levels to very low‐certainty evidence. One level because of low internal validity (risk of bias ‐ high risk of attrition bias) and two levels because of very low sample size (imprecision)
b Cross‐over design, no carry‐over effect suspected due to long washout phase of three months
c The PUVA‐first group was given PUVA twice a week for 3 months followed by ECP once monthly for 6 months (doses not reported). The ECP‐first group was given ECP once monthly for 6 months followed by PUVA twice a week for 3 months (doses not reported).
Summary of findings 3. Bexarotene + PUVA compared to PUVA alone for mycosis fungoides.
| Bexarotene + PUVA compared to PUVA alone for mycosis fungoides | ||||||
|
Patient or population: people with mycosis fungoides
Setting: tertiary care setting
Intervention: Bexarotene + PUVA
Comparison: PUVA alone Number of trials included: 1 (Whittaker 2012) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with PUVA alone | Risk with Bexarotene + PUVA | |||||
| Improvement of quality of life | ‐ | ‐ | ‐ | ‐ | ‐ | Not measured |
| Common adverse effects ‐ Photosensitivity (Time point of measurement: up to 16 weeks) | Zero events in with PUVA alone, so unable to calculate absolute effects. | RR 2.68 (0.11 to 64.04) | 87 (1 RCT) | ⊕⊕⊝⊝ Low a | ‐ | |
| Complete response (Time point of measurement: up to 16 weeks) | Study population | RR 1.41 (0.71 to 2.80) | 93 (1 RCT) | ⊕⊕⊝⊝ Low a | ‐ | |
| 222 per 1000 | 313 per 1000 (158 to 622) | |||||
| Objective response rate (Time point of measurement: up to 16 weeks) | Study population | RR 0.94 (0.61 to 1.44) | 93 (1 RCT) | ⊕⊕⊝⊝ Low a | ‐ | |
| 489 per 1000 | 460 per 1000 (298 to 704) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
a Downgraded by two levels to low‐certainty evidence. One level because of low internal validity (risk of bias ‐ high risk of performance bias) and one level because of low sample size (imprecision)
Summary of findings 4. IFN‐α + acitretin compared to IFN‐α + PUVA for mycosis fungoides.
| IFN‐α + acitretin compared toIFN‐α + PUVA for mycosis fungoides | ||||||
|
Patient or population: people with mycosis fungoides
Setting: tertiary care setting
Intervention: IFN‐α + acitretin
Comparison: IFN‐α + PUVA Number of trials included: 1 (Stadler 1998) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with IFN‐α + PUVA | Risk with IFN‐α + acitretin | |||||
| Improvement of quality of life | ‐ | ‐ | ‐ | ‐ | ‐ | Not measured |
| Common adverse effects ‐ Flu‐like symptoms (Time point of measurement: up to 48 weeks or until complete response) |
Study population | RR 1.32 (0.92 to 1.88) | 82 (1 RCT) | ⊕⊕⊝⊝ Low a | ‐ | |
| 525 per 1000 | 693 per 1000 (483 to 987) | |||||
| Complete response (Time point of measurement: up to 48 weeks or until complete response) |
Study population | RR 0.54 (0.35 to 0.84) | 82 (1 RCT) | ⊕⊕⊝⊝ Low a | ‐ | |
| 700 per 1000 | 378 per 1000 (245 to 588) | |||||
| Objective response rate | ‐ | ‐ | ‐ | ‐ | ‐ | Not measured |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate;the true effect is likely to be substantially different from the estimate of effect. | ||||||
a Downgraded by two levels to low‐certainty evidence. One level because of low internal validity (risk of bias ‐ high risk of attrition bias) and one level because of low sample size (imprecision)
Summary of findings 5. PUVA maintenance compared to no maintenance for mycosis fungoides.
| PUVA maintenance compared to no maintenance for mycosis fungoides | ||||||
|
Patient or population: mycosis fungoides
Setting: tertiary care setting
Intervention: PUVA maintenance
Comparison: no maintenance Number of trials included: 1 (Vieyra‐Garcia 2019) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with no maintenance | Risk with PUVA maintenance | |||||
| Improvement of quality of life | Study population | not estimable | (1 RCT) | ‐ | Not measured | |
| 0 per 1.000 | 0 per 1.000 (0 to 0) | |||||
| Common adverse effects | Study population | not estimable | (1 RCT) | ‐ | Measured but distribution in treatment arms was not reported | |
| 0 per 1.000 | 0 per 1.000 (0 to 0) | |||||
| Complete response | Study population | not estimable | (1 RCT) | ‐ | Complete response was a condition for randomisation and not an outcome | |
| 0 per 1.000 | 0 per 1.000 (0 to 0) | |||||
| Objective response rate | Study population | not estimable | (1 RCT) | ‐ | Not measured | |
| 0 per 1.000 | 0 per 1.000 (0 to 0) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect. Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different. Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect. Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect. | ||||||
In this section, we have presented the results for the effects of interventions for included studies that examined at least one primary or secondary outcome of interest in this review. Only two studies (Stadler 2006; Wozniak 2008) were similar enough to allow meta‐analysis. We have presented all included studies using the PICO scheme to describe included Participants, Interventions, Controls, and Outcomes. The outcomes are displayed separately for each reported primary and secondary outcome of interest below the PICO description. For each intervention, we mention the relevant studies, and the study itself is presented in full only once, in the section where the treatment under investigation fits best. This is primarily the treatment for which it was randomised. If a study was randomised to two different treatments, we chose the section for reporting the trial data according to the newer treatment. The following treatment modalities are sorted by their level of invasiveness, starting from least to most, and differ from their sequence of appearance in the Data and analyses section.
This section is laid out as follows.
I. Topical therapies
I.1. Topical peldesine versus placebo
I.2. Topical Imiquimod versus placebo
I.3. Topical hypericin versus placebo
I.4. Interferon‐α (intralesionally‐injected) versus placebo
I.5. Mechlorethamine gel versus mechlorethamine ointment
II. Skin‐directed phototherapies
II.1. PUVA maintenance versus no maintenance
III. Total skin electron beam
IV. Radiotherapy
V. Chemotherapies and biological response modifiers
V.1. IFN‐α + PUVA versus PUVA alone
V.2. Denileukin diftitox high versus low dose
V.3. Bexarotene
V.4. Lenalidomide maintenance versus observation
V.5. Brentuximab vedotin versus physician's choice (oral methotrexate or bexarotene)
V.6. Mogamulizumab versus vorinostat
VI. Extracorporeal photochemotherapy
VI.1. Extracorporeal photopheresis
VII. Combination therapies
VII.1. Electron‐beam radiation and parenteral chemotherapy compared to topical treatment
VII.2. IFN‐α (subcutaneously‐injected) combined with either PUVA or acitretin capsules
VII.3. Active or inactivated transfer factor (injected intramuscularly) under concomitant therapy with topically‐applied nitrogen mustard
Results from separate rare or serious adverse effects search in non‐randomised studies
I. Topical therapies
Three included studies (Kaye 1989; Lessin 2013; Thestrup‐Pedersen 1982) examined the effect of topical nitrogen mustard, either as monotherapy or in combination with other interventions. Kaye 1989 compared nitrogen mustard and a series of four sequential escape therapies (an escape therapy (see also escape medication) is a therapy that may be taken by a participant in the event of a treatment failure of the investigational therapy during the trial) to a combination of electron‐beam radiation and parenteral chemotherapy. Lessin 2013 contrasted two different base formulations for mechlorethamine, gel versus compounded ointment. The results are described in this section. Thestrup‐Pedersen 1982 assessed the effect of active or inactivated transfer factor (injected intramuscularly) under concomitant therapy with nitrogen mustard. PUVA was administered instead of nitrogen mustard in participants with severe hypersensitivity to nitrogen mustard or relapse after treatment. The trials, in which topical nitrogen mustard is not used as monotherapy, are described in section 'VIII. Combination therapies'.
Other included studies examined the effect of topical peldesine 1% (Duvic 2001a), topical imiquimod 5% (Chong 2004), hypericin 0.05% to 0.25%, in combination with visible light (Rook 2010), or the effect of intralesionally‐injected IFN‐α (Vonderheid 1987; Wolff 1985).
Rook 2010 and Vonderheid 1987 have a within‐participant design. We extracted the data as reported and did not adjust for unit of analysis issues.
I.1. Topical peldesine (one trial)
PICO: In participants with MF Stage I (patches with or without plaques), Duvic 2001a investigated the effect of topical peldesine 1% compared to placebo cream given for 24 weeks. End points were evaluated at the end of this period.
Primary outcomes
Common adverse effects of the treatments
In Duvic 2001a, no differences between peldesine and placebo were seen for pruritus, albeit there is imprecision of the result due to the small sample size. There were nine cases of pruritus in the topical peldesine group and six cases in the placebo group risk ratio ((RR) 1.81, 95% confidence intervals (Cs)I 0.73 to 4.49, 64 participants, Analysis 1.1, Fisher test P = 0.24).
1.1. Analysis.

Comparison 1: Topical peldesine versus placebo, Outcome 1: Common adverse effects
In Duvic 2001a, there were differences between peldesine and placebo for rash. There were six cases of rash in the peldesine group and one case in the placebo group (RR 7.24, 95% CI 0.92 to 56.76 (95% CI according to Miettinen: 1.22 to 45.1), 64 participants, Analysis 1.1). Although the number of events was low resulting in wide 95% CIs, the risk ratio showed a higher risk for peldesine (Fisher test P = 0.041).
Secondary outcomes
Complete response (CR)
Defined as complete disappearance of all clinical evidence of disease.
In Duvic 2001a, there were no differences between the peldesine and placebo groups for complete response. Each group reported 1 CR (RR 1.21, 95% CI 0.08 to 18.46, 64 participants, Analysis 1.2, Fisher test P = 1.00).
1.2. Analysis.

Comparison 1: Topical peldesine versus placebo, Outcome 2: Complete response
Overall survival
In Duvic 2001a, no deaths were reported (the follow‐up period was not reported).
Objective response rate (ORR) (defined as proportion of patients with CR or partial response (PR). A PR is considered as a regression of measurable disease of at least 50% in one of the categories T, N, M and B without any progression of disease.)
In Duvic 2001a, the ORR was not different between peldesine and placebo. Each group reported 11 ORRs (RR 1.21, 95% CI 0.61 to 2.37, 64 participants, Analysis 1.3, Fisher test P = 0.61).
1.3. Analysis.

Comparison 1: Topical peldesine versus placebo, Outcome 3: Objective response rate
I.2. Topical Imiquimod (one trial)
PICO: In participants with MF plaque stage 1B MF (T2N0M0), Chong 2004 investigated the effect of topical imiquimod 5% compared to placebo cream administered for 16 weeks. End points were evaluated at the end of this period.
Primary outcomes
Common adverse effects of the treatments
The only adverse effect reported by Chong 2004 was mild lesional irritation in the imiquimod group (numbers were not reported). No participant discontinued the study because of adverse effects.
Secondary outcomes
Complete response (CR)
In Chong 2004, none of the four participants treated with either imiquimod or placebo showed CR (assessment was at 16 weeks after the end of the intervention).
Objective response rate (ORR)
In Chong 2004, none of the four participants treated with either imiquimod or placebo showed ORR (assessment was at 16 weeks after the end of the intervention).
Rare adverse effects
Chong 2004 explicitly stated that no rare adverse events occurred.
I.3. Topical hypericin (one trial)
PICO: In participants with stable patch or plaque phase CTCL, Rook 2010 investigated the effect of hypericin 0.05% to 0.25% compared to placebo cream, both in combination with visible light administered for six weeks. End points were evaluated at the end of this period.
Primary outcomes
Common adverse effects of the treatments
Rook 2010 described mild to moderate burning, itching, erythema, and pruritus at the application site, which are typical phototoxic reactions expected from the study drug (numbers not reported).
Secondary outcomes
Overall survival (OS)
In Rook 2010, no deaths were reported (information about follow‐up was not reported).
Objective response rate (ORR)
In Rook 2010, the ORR is better in the topical hypericin 0.05% to 0.25% group compared to the placebo cream group. There were seven cases of ORR in the topical hypericin group and one in the placebo group (RR 7.00, 95% CI 1.01 to 48.54, 24 lesions, Analysis 2.1; Fisher test P = 0.028).
2.1. Analysis.

Comparison 2: Topical hypericin versus placebo, Outcome 1: Objective response rate
I.4. Interferon‐α (intralesionally‐injected) (two trials)
PICO: In participants with MF stages IA to IIA, Vonderheid 1987 investigated the effect of injections of 1 million units of IFN‐α 2b three times weekly compared to injections of isotonic sterile water administered for four weeks. End points for this study were evaluated at different time points.
PICO: In participants with histologically‐proven MF stages IA to IB, Wolff 1985 investigated the effect of intralesional injections of two million units of IFN‐α in the superficial dermis three times weekly compared to intralesional injections of buffered glycine serum human albumin in the superficial dermis given for four weeks. End points were evaluated at the end of this period.
Primary outcomes
Common adverse effects of the treatments
Vonderheid 1987 described systemic adverse effects probably due to the injection of IFN‐α: five of six participants had mild fatigue; four of six had low grade fever; three of six had chills or headaches or generalised myalgias. Since this was a within‐participant trial, differences with regard to systemic effects were not detectable between treatment groups. Locally, there were differences between IFN‐α and sterile water for lesional mild erythema. There were five cases of erythema in the IFN‐α group and zero in the placebo group (RR 11.00, 95% CI 0.74 to 163.49 (95% CI according to Miettinen: 1.83 to not estimable), 12 lesions, Analysis 3.1, assessed after three weeks of intervention). The risk ratio showed a higher risk for IFN‐α (Fisher test P = 0.016).
3.1. Analysis.

Comparison 3: IFN‐α versus placebo, Outcome 1: Common adverse effects
In Wolff 1985, there were differences between IFN‐α and placebo injections for mild and transient fevers. There were five cases of fever in the IFN‐α group and zero in the placebo group (RR 11.00, 95% CI 0.70 to 173.66 (95% CI according to Miettinen: 1.59 to not estimable), 18 participants, Analysis 3.1). The risk ratio showed a higher risk for IFN‐α (Fisher test P = 0.03).
For the other reported adverse effects in Wolff 1985, there were no differences between IFN‐α and placebo injections (18 participants each) in terms of myalgia. There were three cases of myalgia in the IFN‐α group and zero cases in the placebo group (RR 7.00, 95% CI 0.41 to 118.69, Analysis 3.1, Fisher test P = 0.21). Due to low number of events, the result was very imprecise with a wide 95% CI. Wolff 1985 reported on two cases of chills or weakness in the IFN‐α group and zero in the control group (RR 5.00, 95% CI 0.27 to 91.52, Analysis 3.1, Fisher test P =0.47); for nausea, arthralgia, and malaise there was one case in IFN‐α group and zero in the placebo group (RR 3.00, 95% CI 0.14 to 65.16, Analysis 3.1, Fisher test P = 1.00).
Secondary outcomes
Complete response (CR)
In Vonderheid 1987, there were differences between intralesional IFN‐α and intralesional placebo for CR assessed 4 weeks after the end of the intervention. There were 10 cases of CR in the IFN‐α group and one in the placebo group (RR 10.00, 95% CI 1.51 to 66.43, 24 lesions, Analysis 3.2). Although the 95% confidence interval was very wide, the risk ratio favoured IFN‐α (Fisher test P = 0.001).
3.2. Analysis.

Comparison 3: IFN‐α versus placebo, Outcome 2: Complete response
In Wolff 1985, the CR did not differ between intralesional IFN‐α and intralesional placebo. There were three cases of CR in the IFN‐α group and zero in the placebo group (RR 2.80, 95% CI 0.18 to 42.80, 12 participants, Analysis 3.2, Fisher test P = 0.51). Due to the low number of events, the result was very imprecise with a wide 95%CI including 1.
Since Vonderheid 1987 was a within‐participant trial and Wolff 1985 had a parallel‐group design with intralesional injections, we did not perform a meta‐analysis of these results.
I.5. Topical Mechlorethamine (one trial)
PICO: In participants with MF stage IA to IIA, Lessin 2013 investigated the effect of topical mechlorethamine 0.02% gel compared to 0.02% mechlorethamine ointment administered for up to 12 months.
Primary outcomes
Common adverse effects of the treatments
Lessin 2013 described skin irritation, pruritus, erythema, contact dermatitis, skin hyperpigmentation and folliculitis as common adverse effects. There were more skin irritations in the mechlorethamine gel arm. The authors reported on 32 cases of skin irritation in the mechlorethamine gel arm and 18 cases in the ointment arm (RR 1.78, 95% CI 1.05 to 3.00, 260 participants, Analysis 4.1, Fisher test P = 0.04). No difference was found in the other categories:
4.1. Analysis.

Comparison 4: Mechlorethamine gel vs mechlorethamine ointment, Outcome 1: Common adverse event
pruritus: gel 25 cases, ointment 20 cases (RR 1.25, 95% CI 0.73 to 2.14, Analysis 4.1, Fisher test P = 0.51)
erythema: gel 22 cases, ointment 18 cases (RR 1.22, 95% CI 0.69 to 2.17, Analysis 4.1, Fisher test P = 0.61)
contact dermatitis: each group 19 cases (RR 1.00, 95% CI 0.56 to 1.80, Analysis 4.1, Fisher test P = 1.00)
skin hyperpigmentation: gel seven cases, ointment nine cases (RR 0.78, 95% CI 0.30 to 2.03, Analysis 4.1, Fisher test P = 0.80)
folliculitis: gel seven cases, ointment five cases (RR 1.40, 95% CI 0.46 to 4.30, Analysis 4.1, Fisher test P = 0.77)
Secondary outcomes
Complete response (CR)
In Lessin 2013, CR did not differ between mechlorethamine gel versus mechlorethamine ointment (18 and 15 cases, respectively, RR 1.20, 95% CI 0.63 to 2.28, 260 participants, Analysis 4.2, Fisher test P = 0.71).
4.2. Analysis.

Comparison 4: Mechlorethamine gel vs mechlorethamine ointment, Outcome 2: Complete response
Objective response rate (ORR)
In Lessin 2013, the ORR did not differ between both study arms (58 and 47 cases, respectively, RR 1.23, 95% CI 0.92 to 1.66, 260 participants, Analysis 4.3, Fisher test P = 0.21).
4.3. Analysis.

Comparison 4: Mechlorethamine gel vs mechlorethamine ointment, Outcome 3: Objective response rate
II. Skin‐directed phototherapies
Eight studies (Child 2004; Guitart 2002; Kaye 1989; Stadler 1998; Stadler 2006; Thestrup‐Pedersen 1982; Whittaker 2012: Wozniak 2008) investigated the effect of psoralen plus ultraviolet A light (PUVA) in combination with other therapies. Child 2004 compared extracorporeal photopheresis (ECP) to PUVA in a cross‐over design randomising participants either to start with three months of PUVA or six months of extracorporeal photopheresis. This trial is described in section 'VI. Extracorporeal photochemotherapy'. While Guitart 2002 assessed the effect of two‐dose regimens of bexarotene in combination with PUVA, Whittaker 2012 compared bexarotene in combination with PUVA to PUVA alone. The results of these trials are described in section 'V. chemotherapy'. Kaye 1989 used PUVA as one of four sequential escape therapies, while comparing electron‐beam radiation and parenteral chemotherapy to topical treatment. This trial is described in section 'VIII. Combination therapies'. Stadler 1998 investigated the effect of IFN‐α (subcutaneously‐injected) combined with either acitretin capsules or PUVA. This trial is described in section 'VIII. Combination therapies'. Thestrup‐Pedersen 1982 assessed the effect of active or inactivated transfer factor (injected intramuscularly) under concomitant therapy with either nitrogen mustard or PUVA in combination. This trial is described in section 'VIII. Combination therapies'.
Stadler 2006 and Wozniak 2008 compared IFN‐α (subcutaneously‐injected) to placebo under concomitant therapy with PUVA. These studies are described in section 'V. Chemotherapy' of this section.
One study (Rook 2010) assessing the effect of visible light in combination with topical hypericin is described in section 'I. Topical therapies'.
Another study (Vieyra‐Garcia 2019) compared PUVA maintenance therapy to no maintenance after having achieved a complete response to PUVA.
II.1 PUVA maintenance (one trial)
PICO: In participants with MF CTCL stages IA to IIB, Vieyra‐Garcia 2019 investigated the effect of PUVA maintenance therapy compared to no maintenance after achieving a complete response with PUVA therapy. Patients were treated or observed for up to nine months.
Please see Table 5.
Primary outcomes
Common adverse effects of the treatments
Vieyra‐Garcia 2019 measured adverse effects but the distribution in the treatment arms was not reported.
Secondary outcomes
Relapse
Since all randomised patients started with a complete response, relapse automatically equates to disease‐free survival. We present the data for disease‐free survival (see below).
Disease‐free survival
In Vieyra‐Garcia 2019, the hazard ratio (HR) for recurrence when comparing patients with PUVA maintenance with those without maintenance indicated that the chance of extending the duration of disease‐free survival is approximately three times higher in the PUVA maintenance group (HR = 3.25, CI: 1.14 ‐ 9.29, 8 participants, Analysis 16.1). Maximum follow‐up time was 60 months. The certainty of evidence is moderate as we downgraded once due to low sample size (imprecision).
16.1. Analysis.

Comparison 16: PUVA maintenance vs. no maintenance, Outcome 1: Disease‐free survival
III. Total skin electron beam
One study (Kaye 1989) compared a combination of electron‐beam radiation and parenteral chemotherapy to topical treatment and a series of four sequential escape therapies. This trial is described in section 'VIII. Combination therapies'.
IV. Radiotherapy
We did not identify any RCTs assessing the effect of radiotherapy other than total skin electron beam for participants with mycosis fungoides.
V. Chemotherapies and biological response modifiers
Ten studies examined the effect of chemotherapy. Kaye 1989 assessed parenteral chemotherapy with cyclophosphamide, doxorubicin, etoposide, and vincristine in combination with total skin electron‐beam radiation to topical treatment. This trial is described in section 'VIII. Combination therapies'.
Stadler 2006 and Wozniak 2008 compared IFN‐α (subcutaneously‐injected) to placebo under concomitant therapy with PUVA.
Treatment with parenteral systemic agents, such as brentuximab vedotin (Prince 2017), denileukin diftitox (Olsen 2001) or oral systemic agents like bexarotene (Duvic 2001; Guitart 2002; Whittaker 2012) and lenalidomide (Bagot 2017), are also described in this section. Topical mechlorethamine treatment (Lessin 2013) is described in the topical treatment section.
V.1. IFN‐α + PUVA (two trials)
PICO: In participants with MF (CTCL stages lA to IIA), Stadler 2006 investigated the effect of subcutaneous injections of IFN‐α in combination with PUVA compared to treatment with PUVA alone given for up to 52 weeks or until complete response. End points were evaluated at the end of this timeframe.
PICO: In participants with histologically‐proven MF (CTCL stages lA to IIA), Wozniak 2008 investigated the effect of subcutaneous injections of IFN‐α in combination with PUVA compared to treatment with PUVA alone administered for 24 weeks. End points were evaluated at the end of this period.
The studies were similar enough to perform a meta‐analysis ‐ see Table 1.
Secondary outcomes
Complete response (CR)
Combining Stadler 2006 and Wozniak 2008 in a meta‐analysis indicated no difference between the groups for CR (43 and 49 cases, respectively, RR 1.07, 95% CI 0.87 to 1.31, 122 participants, Analysis 5.1, Figure 5, Z‐test P = 0.51). Statistical heterogeneity I² statistic was 0%; no clinical heterogeneity could be observed in the two trials. The certainty of evidence is low. We downgraded by two levels to low‐certainty evidence: one level due to low internal validity (risk of bias ‐ performance bias in both studies, attrition bias in Stadler 2006) and one level due to low sample size (imprecision).
5.1. Analysis.

Comparison 5: IFN‐α + PUVA versus PUVA alone, Outcome 1: Complete response
5.

Forest plot of comparison: 6 IFN‐α + PUVA versus PUVA alone, outcome: 6.1 Complete response.
Relapse
In Wozniak 2008, 20 of 22 participants who obtained CR experienced a relapse (the median duration of complete response was 66 weeks). However, no distribution between treatment groups were reported.
Stadler 2006 did not assess this outcome.
Disease‐free survival
In Wozniak 2008, median duration of complete response was reported to be 93 weeks among all participants. However, no distribution between treatment groups were reported.
Stadler 2006 did not assess this outcome.
V.2. Denileukin diftitox (one trial)
PICO: In participants with MF with ≥ 20% of lymphocytes within the skin biopsy stain positively for CD25 by immunohistochemistry stages IB‐IVA, Olsen 2001 investigated the effect of denileukin diftitox intravenous infusions of 9 µg/kg/day (low‐dose group) compared to denileukin diftitox intravenous infusions of 18 µg/kg/day (high‐dose group) given for up to six months. End points were evaluated at the end of this period.
Primary outcomes
Improvement in quality of life as defined by participant questionnaires
The study of Olsen 2001 investigated quality of life using the Functional Assessment in Cancer Therapy‐general (FACT‐G) questionnaire (version 3), as completed by participants at baseline, before the start of each treatment cycle, and when the study was discontinued. Participants were excluded from the end point FACT‐G analyses if they did not complete a post‐baseline FACT‐G questionnaire or answer a sufficient number of questions for calculation of their post‐baseline FACT‐G composite and subscale scores.
Sixty participants completed a FACT‐G questionnaire (scale 0 to 112 points, higher scores indicating better quality of life) at the end point of the study. Changes in the FACT‐G composite score were only given for 42 participants from baseline to the end of treatment. The mean difference for the 23 participants receiving the low dose was +7 points and for the 19 participants in the high‐dose group, +4 points.
However, the results were sub‐divided into responders and non‐responders. The mean difference from baseline for eight responders receiving the low dose was +14 points compared to a difference of +2 points in 13 responders in the high‐dose group. Among non‐responders, for the 15 participants receiving the low dose the mean difference from baseline was +3 points compared to +7 points in the six non‐responders in the high‐dose group. The study did not provide data on variability.
Common adverse effects of the treatments
The study of Olsen 2001 reported on common adverse effects, such as acute infusion events, constitutional symptoms, gastrointestinal syndromes, vascular leak syndrome, thrombotic events, cardiopulmonary and central nervous system (CNS) syndromes, as well as laboratory abnormalities. Of all adverse events, 87% occurred for the first time during the first cycle.
The most common adverse effects were constitutional symptoms like chills, fever, asthenia, arthralgia, myalgia; headaches followed by infections; gastrointestinal and CNS syndromes; as well as rash and vascular leak syndrome.
A spectrum of adverse effects and higher‐grade adverse effects (grade 3 or 4) have been reported, but we found no difference between the low‐ and high‐dose groups (71 participants):
constitutional symptoms and grade 3 to 4 constitutional symptoms: RR 0.85, 95% CI 0.70 to 1.04, Analysis 6.1, Fisher test P = 0.19; and RR 1.27, 95% CI 0.73 to 2.21, Analysis 6.1, Fisher test P = 0.47, respectively;
infections and grade 3 to 4 infections: RR 0.80, 95% CI 0.53 to 1.20, Analysis 6.1, Fisher test P = 0.34; and RR 0.78, 95% CI 0.43 to 1.42, Analysis 6.1, Fisher test P = 0.47, respectively;
gastrointestinal syndromes and grade 3 or 4 syndromes: RR 1.05, 95% CI 0.81 to 1.36, Analysis 6.1, Fisher test P = 0.79; and RR 0.63, 95% CI 0.38 to 1.06, Analysis 6.1, Fisher test P = 0.10, respectively;
CNS syndromes and grade 3 or 4 CNS syndromes: RR 1.14, 95% CI 0.73 to 1.79, Analysis 6.1, Fisher test P = 0.64; and RR 1.46, 95% CI 0.58 to 3.67, Analysis 6.1, Fisher test P = 0.56, respectively;
rash and grade 3 or 4 rashes: RR 0.76, 95% CI 0.40 to 1.45, Analysis 6.1, Fisher test P = 0.46; and RR 0.85, 95% CI 0.35 to 2.10, Analysis 6.1, Fisher test P = 0.78, respectively;
vascular leak syndrome and grade 3 to 4 vascular leak syndrome: RR 0.97, 95% CI 0.44 to 2.16, Analysis 6.1, Fisher test P = 1.00; and RR 1.46, 95% CI 0.58 to 3.67, Analysis 6.1, Fisher test P = 0.56, respectively;
thrombotic events and grade 3 to 4 thrombotic events: RR 0.97, 95% CI 0.26 to 3.59, Analysis 6.1, Fisher test P = 1.00; and RR 2.92, 95% CI 0.32 to 26.72, Analysis 6.1, Fisher test P = 0.61, respectively;
cardiopulmonary events, such as increased cough or right heart failure, and grade 3 or 4 cardiopulmonary events: RR 0.52, 95% CI 0.24 to 1.16, Analysis 6.1, Fisher test P = 0.12; and RR 0.65, 95% CI 0.12 to 3.65, Analysis 6.1, Fisher test P = 0.67, respectively;
acute infusion‐related events, such as back pain, chest pain, hypotension, pruritus, vasodilatation or dyspnoea and grade 3 or 4 acute infusion‐related events: RR 0.92, 95% CI 0.80 to 1.04, Analysis 6.1, Fisher test P = 0.36; and RR 1.13, 95% CI 0.61 to 2.10, Analysis 6.1, Fisher test P = 0.81, respectively;
grade 3 to 4 laboratory abnormalities: RR 1.25, 95% CI 0.74 to 2.10, Analysis 6.1, Fisher test P = 0.48.
6.1. Analysis.

Comparison 6: Denileukin diftitox high versus low dose, Outcome 1: Common adverse effects
Secondary outcomes
Complete response (CR)
In Olsen 2001, no differences were observed between the low‐ and high‐dose group for CR: RR 1.30, 95% CI 0.31 to 5.38, 71 participants, Analysis 6.2, Fisher test P = 1.00. All responders in the low‐dose group had stage IB disease, whereas responders in the high‐dose group had stage IB (1), IIB (2), or IVA (1) disease.
6.2. Analysis.

Comparison 6: Denileukin diftitox high versus low dose, Outcome 2: Complete response
Overall survival (OS)
There were two deaths reported within 90 days of the last study drug administration in the 18 µg/kg/day group: One participant died of sepsis at day 58 after the last study drug. One participant died of myocardial infarction at day 26.
Objective response rate (ORR)
In Olsen 2001, ORR was did not differ between the low‐ and high‐dose group: RR 1.58, 95% CI 0.75 to 3.34, 71 participants, Analysis 6.3, Fisher test P = 0.30. Responders in the low‐dose group had stage IB (1), IIA (2), IIB (1), or IVA (1) disease, whereas responders in the high‐dose group had stage IB (2), IIA (1), IIB (3), III (2), or IVA (1) disease.
6.3. Analysis.

Comparison 6: Denileukin diftitox high versus low dose, Outcome 3: Objective response rate
Rare adverse effects
Olsen 2001 reported one case of thyrotoxicosis. However, the authors did not state to which group this participant was allocated.
V.3. Bexarotene (three trials)
PICO: In participants with MF CTCL stages I to IIA, Duvic 2001 investigated the effect of bexarotene capsules dosed 300 mg/m²/day to 650 mg/m²/day versus bexarotene capsules dosed 6.5 mg/m²/day given for 16 weeks. End points were evaluated at the end of this period. The study of Duvic 2001 reduced the dose of bexarotene twice during the study and partly discontinued randomisation for an uncertain period of time in the middle of the study; the dropout rate was 72.4%. Therefore, the results of this study are only presented qualitatively.
PICO: In participants with MF CTCL stages IB and IIA, Guitart 2002 investigated the effect of 300 mg/day bexarotene in combination with PUVA and 54 mg/day fenofibrate to 150 mg/day bexarotene in combination with PUVA and 54 mg/day fenofibrate given for 24 weeks. End points were evaluated at the end of this period.
PICO: In participants with MF CTCL stages IB to IIA, Whittaker 2012 investigated the effect of bexarotene in combination with PUVA to PUVA alone for up to 16 weeks. Treatment continued until a patient achieved complete response, there was progressive disease or unacceptable toxicity. Therefore, end points were evaluated individually. Please see Table 3.
Primary outcomes
Improvement in quality of life as defined by participant questionnaires
Duvic 2001 investigated quality of life using a non‐validated CTCL quality of life questionnaire and the Spitzer quality of life questionnaire (six items) at baseline and each month. However, results were divided into responders and non‐responders rather than treatment groups.
Guitart 2002 did not assess this outcome.
Whittaker 2012 did not assess this outcome.
Common adverse effects of the treatments
Duvic 2001 reported on adverse effects in the body as a whole, the digestive and endocrine systems, the skin and appendages as well as laboratory abnormalities of the haematological and metabolic systems. Adverse effects were compared between the low‐dose group before cross‐over (L1) and after the cross‐over period (L1‐>H1) as well as with the high‐dose groups taking 300 mg/m²/day of bexarotene (H1) and the replaced high‐dose group taking 650 mg/m²/day (H2). In the low‐dose group, 11 of 15 participants crossed over to the high‐dose therapy group resulting in increased incidences of almost every adverse event.
Common adverse effects including systemic reactions: affection of digestive, endocrine, haematological/lymphatic, and metabolic systems; and skin reactions are shown in the Table 11.
6. Common adverse events from Duvic 2001.
| Body as a whole |
| Altered hormone level, asthenia, chills, headache, infection, pain, abdominal pain |
| Digestive system |
| Diarrhoea, nausea |
| Endocrine system |
| Hypothyroidism |
| Haematological and lymphatic system |
| Anaemia, hypochromic anaemia, leukopenia |
| Metabolic and nutritional system |
| Hypercholesterolaemia, hyperlipidaemia, lactate dehydrogenase (LDH) increase, aspartate aminotransferase (AST) increase, alanine aminotransferase (ALT) increase |
| Skin and appendages |
| Dermatitis exfoliation, pruritus, rash, skin disorder |
Guitart 2002 reported on the following common adverse effects: photosensitivity, nausea, constipation, fatigue, pruritus, arthralgias, nasopharyngitis, headaches, and insomnia for which there was no difference between the low‐ and the high‐dose group (39 participants each):
photosensitivity: RR 1.40, 95% CI 0.36 to 5.46, Analysis 7.1, Fisher test P = 0.69;
nausea: RR 0.82, 95% CI 0.38 to 1.75, Analysis 7.1, Fisher test P = 0.75;
constipation: RR 3.16, 95% CI 0.36 to 27.78, Analysis 7.1, Fisher test P = 0.34;
fatigue: RR 1.20, 95% CI 0.54 to 2.67, Analysis 7.1, Fisher test P = 0.75;
pruritus: RR 0.70, 95% CI 0.31 to 1.59, Analysis 7.1, Fisher test P = 0.51;
arthralgias: RR 1.40, 95% CI 0.36 to 5.46, Analysis 7.1, Fisher test P = 0.69;
nasopharyngitis: RR 0.53, 95% CI 0.11 to 2.55, Analysis 7.1, Fisher test P = 0.66;
headaches: RR 0.84, 95% CI 0.27 to 2.67, Analysis 7.1, Fisher test P = 1.00;
insomnia: RR 1.05, 95% CI 0.16 to 6.74, Analysis 7.1, Fisher test P = 1.00.
7.1. Analysis.

Comparison 7: Bexarotene high versus low dose, Outcome 1: Common adverse effects
There were differences in laboratory abnormalities for free T4, cholesterol, triglycerides, and liver enzymes SGOT/SGPT between low‐ and high‐dose groups. The results of Guitart 2002 indicate that cholesterol and triglyceride levels were higher in the high‐dose group (39 participants each):
free T4 abnormalities: RR 1.26, 95% CI 0.46 to 3.46, Analysis 7.1, Fisher test P = 0.73;
cholesterol abnormalities: 13 cases in high‐dose arm, three cases in low‐dose arm, RR 4.56, 95% CI 1.54 to 13.53, Analysis 7.1. The risk ratio showed a higher risk for the high‐dose group (Fisher test P = 0.002);
triglyceride abnormalities: 15 cases in high‐dose arm, five cases in low‐dose arm, RR 3.16, 95% CI 1.43 to 6.98, Analysis 7.1. The risk ratio showed a higher risk for the high‐dose group (Fisher test P = 0.002);
abnormalities of SGOT/SGPT: RR 5.25, 95% CI 0.27 to 102.74, Analysis 7.1, Fisher test P = 0.23.
Whittaker 2012 reported on the following adverse effects: liver toxicities, renal toxicities, haematological toxicities, increased fasting cholesterol, photosensitivity, pruritus, rash and hypertriglyceridaemia. Bexarotene and PUVA lead to a increase of liver toxicities and fasting cholesterol (87 participants):
liver toxicities: six cases in combination arm, zero cases in PUVA alone arm, RR 11.62, 95% CI 0.7 to 200.07 (95% CI according to Miettinen: 1.46 to not estimable), Analysis 8.1, Fisher test P = 0.03;
Increased fasting cholesterol: six cases combination arm, zero cases in PUVA‐alone arm, RR 11.62, 95% CI 0.67 to 200.07 (95% CI according to Miettinen: 1.46 to not estimable), Analysis 8.1, Fisher test P = 0.03.
8.1. Analysis.

Comparison 8: Bexarotene + PUVA vs PUVA alone, Outcome 1: Common adverse events
For all the other adverse effects, no differences could be observed:
renal toxicities: RR 0.30, 95% CI 0.01 to 7.12, Analysis 8.1, Fisher test P = 0.47;
haematological toxicities: five cases combination arm, zero cases PUVA‐alone arm, RR 9.83, 95% CI 0.56 to 172.50, Analysis 8.1, Fisher test P = 0.06;
photosensitivity: RR 2.68, 95% CI 0.11 to 64.04, Analysis 8.1, Fisher test P = 1.00. The certainty of evidence is low as we downgraded twice: once due to low internal validity (risk of bias ‐ high risk of performance bias) and once due to low sample size (imprecision);
pruritus: RR 0.30, 95% CI 0.01 to 7.12, Analysis 8.1, Fisher test P = 0.47;
rash: RR 2.68, 95% CI 0.11 to 64.04, Analysis 8.1, Fisher test P = 1.00;
hypertriglyceridaemia: RR 4.47, 95% CI 0.22 to 90.44, Analysis 8.1, Fisher test P = 0.50.
Secondary outcomes
Complete response (CR)
In Duvic 2001, zero of 15 participants (0%) in the low‐dose group showed CR compared to six of 43 participants (14%) in the high‐dose therapy arm ‐ but the trial had a high dropout rate of 72.4%, which made it difficult to draw reasonable conclusions from these data.
Guitart 2002 found no differences in CR between the low‐ and high‐dose groups: RR 0.86, 95% CI 0.46 to 1.60, 39 participants, Analysis 7.2, Fisher test P = 0.75.
7.2. Analysis.

Comparison 7: Bexarotene high versus low dose, Outcome 2: Complete response
In Whittaker 2012, no difference could be observed between bexarotene in combination with PUVA versus PUVA alone for CR: RR 1.41, 95% CI 0.71 to 2.80, 93 participants, Analysis 8.2, Fisher test P = 0.36. The certainty of evidence is low as we downgraded twice: once due to low internal validity (risk of bias ‐ high risk of performance bias) and once due to low sample size (imprecision).
8.2. Analysis.

Comparison 8: Bexarotene + PUVA vs PUVA alone, Outcome 2: Complete response
Relapse
In Duvic 2001, zero of three participants achieving partial remission in the low‐dose group relapsed compared to seven of 25 participants achieving complete or partial remission in the high‐dose therapy arms. No time to event data were provided.
In Guitart 2002, no differences between the low‐ and high‐dose groups were seen six months after treatment: four cases in high‐dose arm, eight cases in low‐dose arm (RR 0.53, 95% CI 0.19 to 1.46, 39 participants, Analysis 7.3, Fisher test P = 0.30).
7.3. Analysis.

Comparison 7: Bexarotene high versus low dose, Outcome 3: Relapse
Whittaker 2012 did not allow assessment of this outcome, since relapse and progression were summarised in one category.
Disease‐free survival
In Guitart 2002, the median disease‐free survival was 155 days for the low‐dose group and 103 days for the high‐dose group (assessed six months after the end of the intervention).
No distribution measure was given for relapse.
Duvic 2001 did not assess this outcome.
Whittaker 2012 did not allow assessment of this outcome.
Overall survival (OS)
In Duvic 2001, no participant died during the study or within four weeks time after therapy discontinuation, but three participants died within three months of discontinuation of therapy, which was attributed to infection or progressive disease by the study authors. No allocation to one of the treatment groups was reported.
In Guitart 2002, no deaths reported.
Whittaker 2012 did not provide overall survival rates.
Objective response rate (ORR)
In Duvic 2001, three of 15 participants (20%) showed ORR as defined above compared to 25 of 43 participants (58%) in the high‐dose therapy arms.
In Guitart 2002, there were no differences between the low‐ and high‐dose group for ORR: RR 0.93, 95% CI 0.69 to 1.25, 39 participants, Analysis 7.4, Fisher test P = 0.69.
7.4. Analysis.

Comparison 7: Bexarotene high versus low dose, Outcome 4: Objective response rate
In Whittaker 2012, there were no differences between bexarotene combined with PUVA vs PUVA alone for ORR: RR 0.94, 95% CI 0.61 to 1.44, 93 participants, Analysis 8.3, Fisher test P = 0.84. The certainty of evidence is low as we downgraded twice: once due to low internal validity (risk of bias ‐ high risk of performance bias) and once due to low sample size (imprecision).
8.3. Analysis.

Comparison 8: Bexarotene + PUVA vs PUVA alone, Outcome 3: Objective response rate
Rare adverse effects
Duvic 2001 reported three cases of acute pancreatitis due to massive hyperlipidaemia in the high‐dose therapy arms. Furthermore, lens opacity without loss of visual acuity was detected in 2 of 58 participants (3%) in the study. However, the distribution within the therapy arms was not reported.
V.4. Lenalidomide (one trial)
PICO: In participants with MF CTCL stages I to IV, Bagot 2017 investigated the effect of lenalidomide maintenance therapy compared to observation after debulking therapy. Patients were treated or observed until disease progression. Therefore end points were evaluated individually.
Primary outcomes
Common adverse effects of the treatments
In Bagot 2017, the following adverse events were documented:
neutropenia: one case in lenalidomide arm, zero cases in observation arm, RR 4.33, 95% CI 0.20 to 94.83, 20 participants, Analysis 9.1, Fisher test P = 0.40;
hyperbilirubinaemia: one case in lenalidomide arm, zero cases in observation arm, RR 4.33, 95% CI 0.20 to 94.83, 20 participants, Analysis 9.1, Fisher test P = 0.40;
hypercalcaemia: one case in lenalidomide arm, zero cases in observation arm, RR 4.33, 95% CI 0.20 to 94.83, 20 participants, Analysis 9.1, Fisher test P = 0.40;
hypokalaemia: one case in lenalidomide arm, zero cases in observation arm, RR 4.33, 95% CI 0.20 to 94.83, 20 participants, Analysis 9.1, Fisher test P = 0.40;
hypophosphataemia: 2 cases in lenalidomide arm, zero cases in observation arm RR 7.22, 95% CI 0.39 to 133.24, 20 participants, Analysis 9.1, Fisher test P = 0.15;
erythema multiforme: one case in lenalidomide arm,zero cases in observation arm, RR 4.33, 95% CI 0.20 to 94.83, 20 participants, Analysis 9.1, Fisher test P = 0.40;
periorbital oedema: one case in lenalidomide arm, zero cases in observation arm, RR 4.33, 95% CI 0.20 to 94.83, 20 participants, Analysis 9.1, Fisher test P = 0.40;
pruritus: RR 1.50, 95% CI 0.11 to 20.68, 20 participants, Analysis 9.1, Fisher test P = 1.00;
other adverse effects: 2 cases in lenalidomide arm, zero cases in observation arm, RR 7.22, 95% CI 0.39 to 133.24, 20 participants, Analysis 9.1, Fisher test P = 0.15.
9.1. Analysis.

Comparison 9: Lenalidomide maintenance versus observation after debulking therapy, Outcome 1: Common adverse effects
Secondary outcomes
Assessment of overall survival was intended by the investigators but ultimately not measured.
V.5. Brentuximab vedotin (one trial)
PICO: In participants with CD30+ MF CTCL stages IA ‐ IVB or CD30+ primary cutaneous anaplastic large‐cell lymphoma, Prince 2017 investigated the effect of brentuximab vedotin compared to physician's choice (methotrexate or bexarotene) for up to 48 weeks. The end points were evaluated individually.
Primary outcomes
Common adverse effects of the treatments
In Prince 2017, common adverse effects were assessed but could not be retraced to the patients with CD30‐positive mycosis fungoides, since the adverse effects were summarised for all patients according to their allocated treatment.
Secondary outcomes
Complete response (CR)
In Prince 2017, differences were seen between brentuximab vedotin and physician's choice for CR. There were five cases in the brentuximab vedotin arm and zero cases in the physician's choice arm (RR 11.22, 95% CI 0.64 to 197.60 (95% CI according to Miettinen: 1.36 to not estimable), 97 participants, Analysis 10.1). The risk ratio favoured brentuximab vedotin (Fisher test P = 0.03).
10.1. Analysis.

Comparison 10: Brentuximab vedotin vs. physician's choice (MTX or bexarotene), Outcome 1: Complete response
Objective response rate (ORR)
In Prince 2017, ORR was different between brentuximab vedotin and physician's choice. The authors reported on 31 cases in the brentuximab vedotin arm and eight cases in the control group (RR 3.96, 95% CI 2.03 to 7.71, 97 participants, Analysis 10.2). The risk ratio favoured brentuximab vedotin (Fisher test P < 0.001).
10.2. Analysis.

Comparison 10: Brentuximab vedotin vs. physician's choice (MTX or bexarotene), Outcome 2: Objective response rate
V.6. Mogamulizumab (one trial)
PICO: In participants with MF stages IB ‐ IVB, Kim 2018 investigated the effect of Mogamulizumab compared to vorinostat for up to 12 months. The end points were evaluated individually.
Primary outcomes
Common adverse effects of the treatments
Kim 2018 measured adverse effects but the distribution in the treatment arms was not reported.
Objective response rate (ORR)
In Kim 2018, there were differences between mogamulizumab and vorinostat for ORR. There were 22 cases of ORR in the mogamuizumab group and 7 in the placebo group (RR 2.96, 95% CI 1.32 to 6.63 (95% CI according to Miettinen: 1.37 to 6.56), 204 participants, Analysis 15.1). The risk ratio favoured mogamulizumab (Fisher test P = 0.005).
15.1. Analysis.

Comparison 15: Mogamulizumab vs. Vorinostat, Outcome 1: Objective response rate
VI. Extracorporeal photochemotherapy
One study (Child 2004) investigated the effect of extracorporeal photopheresis compared to other therapies in a cross‐over design. We extracted the data as reported and did not adjust for unit of analysis issues.
VI.1. Extracorporeal photopheresis (one trial)
PICO: In participants with plaque stage (Bunn Lamberg stage 1B) MF and a peripheral blood T‐cell clone, Child 2004 investigated the effect of extracorporeal photopheresis compared to PUVA in a cross‐over design randomising participants either to start with three months of PUVA or six months of ECP. End points were evaluated at the end of these periods.
Please see Table 2.
Primary outcomes
Common adverse effects of the treatments
The study of Child 2004 reported that some participants reported mild nausea after PUVA. However, incidences and time points were not stated. One participant starting in the ECP group had hypotension leading to withdrawal from the study. The certainty of evidence is very low as we downgraded thrice: once due to low internal validity (risk of bias ‐ high risk of attrition bias) and twice due to very low sample size (imprecision).
Secondary outcomes
Complete response (CR)
In Child 2004, there were no differences between ECP and PUVA for complete response. There were zero cases in the ECP arm and two cases in the PUVA arm (RR 0.20, 95% CI 0.01 to 3.61, 16 participants, Analysis 11.1, Fisher test P = 0.47). The certainty of evidence is very low as we downgraded thrice: once due to low internal validity (risk of bias ‐ high risk of attrition bias) and twice due to very low sample size (imprecision).
11.1. Analysis.

Comparison 11: Extracorporeal photopheresis versus PUVA, Outcome 1: Complete response
Overall survival (OR)
Of the eight evaluable participants completing the cross‐over study of Child 2004, one participant was lost to follow‐up. All other participants were alive at the end of follow‐up, which lasted two to 21 months.
Objective response rate (ORR)
In Child 2004, differences were seen between ECP and PUVA for ORR. There were zero cases of ORR in the ECP arm and six cases in the PUVA arm (RR 0.08, 95% CI 0.01 to 1.17 (95% CI according to Miettinen: 0.00 to 0.40), eight participants, Analysis 11.2). The risk ratio favoured PUVA (Fisher test P = 0.01). The certainty of evidence is very low as we downgraded thrice: once due to low internal validity (risk of bias ‐ high risk of attrition bias) and twice due to very low sample size (imprecision).
11.2. Analysis.

Comparison 11: Extracorporeal photopheresis versus PUVA, Outcome 2: Objective response rate
VII. Combination therapies
Eight studies (Child 2004; Guitart 2002; Kaye 1989; Stadler 1998; Stadler 2006; Thestrup‐Pedersen 1982; Whittaker 2012; Wozniak 2008) investigated the effect of combination therapies. Child 2004 compared PUVA with extracorporeal photopheresis in a cross‐over design randomising participants either to start with three months of PUVA or six months of ECP. This trial is described in section 'VI. Extracorporeal photochemotherapy'. Guitart 2002 assessed the effect of two low‐dose regimens of bexarotene in combination with PUVA. The results of this trial are described in section 'V. Chemotherapy'. Stadler 2006 and Wozniak 2008 compared IFN‐α (subcutaneously‐injected) to placebo under concomitant therapy with PUVA. These trials are described in section 'V. Chemotherapy'.
Kaye 1989 investigated electron‐beam radiation and parenteral chemotherapy compared to topical treatment supported by four sequential escape therapies. Stadler 1998 investigated the effect of IFN‐α (subcutaneously‐injected) combined with either acitretin capsules or PUVA. Thestrup‐Pedersen 1982 assessed the effect of active or inactivated transfer factor (injected intramuscularly) under concomitant therapy with nitrogen mustard. PUVA was administered instead of nitrogen mustard in participants with severe hypersensitivity to nitrogen mustard or relapse after treatment. Whittaker 2012 assessed the effect of bexarotene combined with PUVA versus PUVA alone. The results of this trial are presented in the skin‐directed phototherapies section. At least one outcome of interest is reported in the three studies (Kaye 1989; Stadler 1998; Thestrup‐Pedersen 1982) included in this section.
VII.1. Electron‐beam radiation and parenteral chemotherapy (one trial)
PICO: In participants with MF of all stages, Kaye 1989 investigated the effect of a "combined therapy" consisting of electron‐beam radiation and parenteral chemotherapy with cyclophosphamide, doxorubicin, etoposide, and vincristine, given for eight to 12 weeks, versus a "conservative treatment" consisting of topical treatment with mechlorethamine supported by a stepwise escalation of the therapy according to the stage of the disease. End points were evaluated at the end of this timeframe.
Primary outcomes
Common adverse effects of the treatments
Kaye 1989 reported on common fatal and non‐fatal adverse events. Because of the variety of different therapies in the conservative group, some procedure‐specific adverse effects only occurred in one treatment arm. In others words, e.g. not every patient in the control arm received electron beam radiation therapy. These adverse effects are presented qualitatively.
Adverse effects assessable in both treatment groups were hospitalisation, fatal acute myocardial infarction, cutaneous toxicity from electron beam radiotherapy, acute non‐lymphatic leukaemia, non‐melanoma skin cancer, and occurrence of other cancers not specified more closely by Kaye 1989. These were compared for the participants of the combined‐therapy groups versus those participants of the conservative group receiving the same treatment:
hospitalisation (due to myelosuppression or radiodermatitis): RR 33.65, 95% CI 2.07 to 546.09, 101 participants, Analysis 12.1. In absolute numbers, there were 16 cases of hospitalisation in the combination arm and zero cases in the conservative arm (Fisher test: P < 0.001);
fatal myocardial infarction: RR 1.02, 95% CI 0.07 to 15.86, 101 participants, Analysis 12.1, Fisher test P = 1.00;
cutaneous toxicity from electron beam therapy: 13 cases in combination arm, one case conservative arm, RR 3.38, 95% CI 0.49 to 23.53, 63 participants, Analysis 12.1, Fisher test P = 0.26;
acute non‐lymphocytic leukaemia: RR 2.04, 95% CI 0.19 to 21.79, 101 participants, Analysis 12.1, Fisher test P = 0.62;
non‐melanoma skin cancer: RR 0.20, 95% CI 0.01 to 4.14, 101 participants, Analysis 12.1, Fisher test P = 0.50;
unspecified other cancers: RR 0.11, 95% CI 0.01 to 2.05, 101 participants, Analysis 12.1, Fisher test P =0.12.
12.1. Analysis.

Comparison 12: Combined therapy versus conservative therapy, Outcome 1: Common adverse effects
Adverse effects that were only reported for the combined‐therapy group included leucopenia (white cell count < 1.000 cells per microlitre of blood) [7/50 (14%) participants], thrombopenia (platelets < 50.000 per microlitre of blood) [1/50 (2%) participants], neuropathy [8/50 (16%) participants], and cardiomyopathy [5/50 (10%) participants].
Adverse effects that were only reported for the conservative‐treatment group included cutaneous hypersensitivity to mechlorethamine [15/51 (29%) participants], cutaneous toxicity to methoxsalen and UVA [3/26 (12%) participants], reversible hepatotoxicity due to oral methotrexate [1/16 (6%) participants], and mucositis due to oral methotrexate [3/16 (19%) participants].
Secondary outcomes
Complete response (CR)
In Kaye 1989, differences were seen between the combined‐therapy group and the conservative‐treatment group for CR: 20 cases in the combination arm and nine cases in the conservative arm, RR 2.18, 95% CI 1.10 to 4.33, 103 participants, Analysis 12.2. The risk ratio favoured the combined‐therapy group (Fisher test P = 0.03). Responders in the combined‐therapy group had stage IA (2), IB (5), IIA (2), IIB (2) IVA (7), or IVB (2) disease, whereas responders in the conservative‐treatment group had stage IA (2), IB (3), IIA (1), or IVA (3) disease.
12.2. Analysis.

Comparison 12: Combined therapy versus conservative therapy, Outcome 2: Complete response
Relapse
In Kaye 1989, no differences were seen in relapse after 48 months between the combined‐therapy group and the conservative‐treatment group: RR 0.98, CI 0.88 to 1.09, 103 participants, Analysis 12.3, Fisher test P = 1.00.
12.3. Analysis.

Comparison 12: Combined therapy versus conservative therapy, Outcome 3: Relapse
Disease‐free survival
The median disease‐free survival in Kaye 1989 was 12.9 months in the combined‐therapy group compared to 21.3 months in the conservative‐treatment group. No distribution measure was reported for this outcome.
Overall survival (OS)
In Kaye 1989, the median follow‐up was 68 months (range = 19 to 111 months). Within the follow‐up period no differences between the combined‐therapy group and the conservative‐treatment group were seen for overall survival: RR 0.95, 95% CI 0.68 to 1.32, 103 participants, Analysis 12.4, Fisher test P = 0.15. For stages I and II, overall survival was longer than five years for more than 80% of the participants in both groups; for stage III no data were provided with only few participants belonging to this group, and for stage IV the median length of overall survival was 49 months in the combined group and 68 months in the conservative group.
12.4. Analysis.

Comparison 12: Combined therapy versus conservative therapy, Outcome 4: Overall survival
Objective response rate (ORR)
In Kaye 1989, there were differences between the combined‐therapy group and the conservative‐treatment group for ORR: RR 1.40, 95% CI 1.12 to 1.74, 103 participants, Analysis 12.5. This risk ratio favoured the combined‐therapy (Fisher test P = 0.003). Responders in the combined‐therapy group had stage IA (2), IB (8), IIA (5), IIB (5), IVA (20), or IVB (7) disease, whereas responders in the conservative‐treatment group had stage IA (2), IB (6), IIA (3), IIB (4), III (1), IVA (12), or IVB (5) disease.
12.5. Analysis.

Comparison 12: Combined therapy versus conservative therapy, Outcome 5: Objective response rate
VII.2. IFN‐α (subcutaneously‐injected) combined with either PUVA or acitretin capsules (one trial)
PICO: In participants with MF Bunn Lamberg stages I and II, Stadler 1998 investigated the effect of IFN‐α subcutaneously‐injected acitretin to IFN‐α subcutaneously injected + PUVA administered for up to 48 weeks or until complete response. End points were evaluated at the end of this period.
Please see Table 4.
Primary outcomes
Common adverse effects of the treatments
Stadler 1998 reported common adverse effects divided into grades of severity (I = mild, II = moderate, III = severe).
Adverse effects of grade I or II were not different between the IFN‐α + acitretin group and the IFN‐α + PUVA group: RR 0.95, 95% CI 0.64 to 1.42, 82 participants, Analysis 13.1, Fisher test P = 0.83.
13.1. Analysis.

Comparison 13: IFN‐α + acitretin versus IFN‐α + PUVA, Outcome 1: Common adverse effects
There was a difference in adverse effects of grade III between IFN‐α + acitretin, and between IFN‐α + PUVA: RR 3.10, 95% CI 1.10 to 8.70, 82 participants, Analysis 13.1. The risk ratio for adverse events grade III showed a higher risk for the IFN‐α + acitretin group (Fisher test P = 0.03).
Adverse effects requiring discontinuation of the study differed between IFN‐α + acitretin and IFN‐α + PUVA: nine cases in the IFN‐α + acitretin arm and two cases in the IFN‐α + PUVA arm, RR 4.29, 95% CI 0.99 to 18.63 (95% CI according to Miettinen: 1.13 to 17.1), 82 participants, Analysis 13.1. The risk ratio indicated a higher risk for the IFN‐α + acitretin group (Fisher test P = 0.049).
Side‐effects being compared for the treatment with IFN‐α + acitretin and IFN‐α + PUVA were flu‐like symptoms; dryness/redness of the skin, hair loss, or both; neurological disorders; psychiatric disorders; gastrointestinal disorders; elevated liver or biliary tract enzymes; elevated triglycerides; anaemia; leukopenia; impotentia and redness; and infiltration at application site, but differences were only found for neurological disorders (82 participants):
flu‐like symptoms: 29 cases in the IFN‐α + acitretin and 21 cases in the IFN‐α + PUVA, RR 1.32, 95% CI 0.92 to 1.88, Analysis 13.1, Fisher test P = 0.17. The certainty of evidence is low as we downgraded twice: once due to low internal validity (risk of bias ‐ high risk of attrition bias) and once due to low sample size (imprecision);
dryness/redness of the skin, hair loss, or both: RR 1.48, 95% CI 0.72 to 3.03, Analysis 13.1, Fisher test P = 0.33;
neurological disorders: 11 cases in the IFN‐α + acitretin arm and three in the IFN‐α + PUVA arm, RR 3.49, 95% CI 1.05 to 11.60, Analysis 13.1. The risk ratio showed a higher risk for the acitretin group (Fisher test P = 0.04);
psychiatric disorders: RR 1.43, 95% CI 0.25 to 8.11, Analysis 13.1, Fisher test P = 1.00;
gastrointestinal disorders: RR 0.76, 95% CI 0.33 to 1.73, Analysis 13.1, Fisher test P = 0.60;
elevated liver or biliary tract enzymes: RR 2.38, 95% CI 0.49 to 11.58, Analysis 13.1, Fisher test P = 0.43;
elevated triglycerides: five cases in the IFN‐α + acitretin arm and zero cases in the IFN‐α + PUVA arm, RR 10.49, 95% CI 0.60 to 183.74, Analysis 13.1, Fisher test P = 0.55;
anaemia: RR 0.95, 95% CI 0.14 to 6.44, Analysis 13.1, Fisher test P = 1.00;
leukopenia: RR 0.95, 95% CI 0.42 to 2.15, Analysis 13.1, Fisher test P = 1.00;
impotentia: RR 0.48, 95% CI 0.04 to 5.05, Analysis 13.1, Fisher test P = 0.61;
redness and infiltration at application site: RR 0.14, 95% CI 0.01 to 2.56, Analysis 13.1, Fisher test P = 0.11.
Secondary outcomes
Complete response (CR)
In Stadler 1998, differences were observed between the IFN‐α + acitretin and the IFN‐α + PUVA groups for CR: 16 cases in the IFN‐α + acitretin arm and 28 in the IFN‐α + PUVA arm (RR 0.54, 95% CI 0.35 to 0.84, 82 participants, Analysis 13.2). The risk ratio favoured the IFN‐α + PUVA group (Fisher test P = 0.005). The certainty of evidence is low as we downgraded twice: once due to low internal validity (risk of bias ‐ high risk of attrition bias) and once due to low sample size (imprecision).
13.2. Analysis.

Comparison 13: IFN‐α + acitretin versus IFN‐α + PUVA, Outcome 2: Complete response
Objective response rate (ORR)
Stadler 1998 did not provide ORR.
VII.3. Active or inactivated transfer factor (injected intramuscularly) under concomitant therapy with topically‐applied nitrogen mustard (one trial)
PICO: In participants with MF van Scott stage II‐IV (van Scott 1973), Thestrup‐Pedersen 1982 investigated the effect of topically‐applied nitrogen mustard with active transfer factor to topically‐applied nitrogen mustard with inactivated transfer factor given for one year. PUVA was administered instead of nitrogen mustard in participants with severe hypersensitivity to nitrogen mustard or relapse after treatment. Van Scott stage II‐IV is best transferred to clinical stage I‐III according to Table 6.
Primary outcomes
Common adverse effects of the treatments
Thestrup‐Pedersen 1982 reported that some participants reported slight or moderate pain at the site of injection, which according to the study author, was probably due to the hyperosmolarity of the solution. Furthermore, fatigue and unrest for two to four hours in one participant and fever and malaise in three participants was observed. However, allocation to treatment groups and time points were not stated.
Secondary outcomes
Complete response (CR)
In Thestrup‐Pedersen 1982, differences were seen between the active transfer factor group and the inactivated transfer factor group for CR: zero cases of CR in the active transfer factor group and five cases in the inactive transfer factor group, RR 0.09, 95% CI 0.01 to 1.41 (95% CI according to Miettinen: 0.00 to 0.61), 16 participants, Analysis 14.1 (assessed one year after the end of the intervention). The risk ratio favoured the inactivated transfer factor group (Fisher test P = 0.03). The authors of this study speculate that the participants in the inactivated transfer factor group may have had a better prognosis initially.
14.1. Analysis.

Comparison 14: Topical nitrogen mustard with active transfer factor versus topical nitrogen mustard with inactivated transfer factor, Outcome 1: Complete response
Overall survival (OS)
In Thestrup‐Pedersen 1982, the overall survival at the end of the two‐year follow‐up did not differ between the active transfer factor group and the inactivated transfer factor group: RR 1.00, 95% CI 0.69 to 1.45, 16 participants, Analysis 14.2, Fisher test P = 1.00 (assessed one year after the end of the intervention).
14.2. Analysis.

Comparison 14: Topical nitrogen mustard with active transfer factor versus topical nitrogen mustard with inactivated transfer factor, Outcome 2: Overall survival
Objective response rate (ORR)
In Thestrup‐Pedersen 1982, no differences were seen between the active transfer factor group and the inactivated transfer factor for ORR: four cases in the active transfer factor group and seven cases in the inactive transfer factor arm, RR 0.57, 95% CI 0.27 to 1.20, 16 participants, Analysis 14.3, Fisher test P = 0.28 (assessed one year after the end of the intervention).
14.3. Analysis.

Comparison 14: Topical nitrogen mustard with active transfer factor versus topical nitrogen mustard with inactivated transfer factor, Outcome 3: Objective response rate
Results from separate adverse effects search in non‐randomised studies
In addition to rare adverse effects found in the included studies, we were able to extract information on severe rare adverse effects from a separate adverse effect search. Most of the rare adverse effects, which are listed in Table 10, were reported in case reports and other non‐randomised trials. Due to the large spectrum of different treatment options, rare adverse effects consequently occurred in a variety of organs.
Discussion
Summary of main results
This review identified 20 studies that met the inclusion criteria. Although these studies assessed a wide range of interventions, we were only able to meta‐analyse two studies. Most interventions were evaluated by only one study, which limits the evidence base.
Topical treatments included an immune response modifier (imiquimod), a photosensitising agent (hypericin), an inhibitor of purine nucleoside phosphorylase (BCX‐34 dermal cream), and the cytotoxin nitrogen mustard. Oral treatments included an immunomodulatory drug (lenalidomide) and the chemotherapy drug bexarotene, which was assessed by three studies. Electron beam radiation combined with chemotherapy was assessed by one study. Parenteral systemic agents included the monoclonal antibody mogamulizumab, the cytotoxin denileukin diftitox intravenous infusion, the targeted therapy brentuximab vedotin, and transfer factor. Light therapies included extracorporeal photopheresis (ECP) and psoralen plus ultraviolet A) (PUVA); PUVA was the intervention that was most often a part of study interventions, but no single trial investigated the effect of PUVA versus placebo, and only five studies assessed PUVA given alone or in combination. However, interferon‐α (IFN‐α, given alone or combined with additional therapy, was assessed by 30% of the studies. Only one trial investigated the effect of topical steroids, despite its widespread recommended use (Wolff 1985).
Our primary end point improvement of quality of life was only measured in four studies. Eighteen out of 20 studies reported common adverse effects. Complete response (CR) and common adverse effects were the most frequently reported outcomes. Objective response rate (ORR) was measured in 13 studies (although we could only assess the outcome in 12 studies). Other outcomes of interest were poorly addressed.
We created 'Summary of findings tables' for our main comparisons.
IFN‐α + PUVA compared to PUVA alone (Table 1)
ECP compared to PUVA (Table 2)
Bexarotene + PUVA compared to PUVA alone (Table 3)
IFN‐α + acitretin compared to IFN‐α + PUVA (Table 4)
PUVA maintenance compared to no maintenance (Table 5)
In terms of our outcomes of interest, our primary outcome quality of life was not assessed by any of our key comparisons, and of the two studies that did report it, results were divided into responders and non‐responders rather than treatment groups. Ninety per cent of all studies reported on common or rare serious adverse effects or their absence. The studies reported a wide range of adverse effects, from mild symptoms to potentially lethal complications. Severity of the common adverse effects was mostly dependent on the invasiveness of the intervention, with local therapies generally resulting in less severe adverse effects.
Eighty per cent of the trials included in our key comparisons reported on our secondary outcome CR, whereas only 33% addressed ORR. Only one of five studies comprising our key comparisons investigated disease‐free survival. Other secondary outcomes were not assessed in the trials comprising our key comparisons.
The studies assessing PUVA alone versus PUVA plus interferon‐α did not measure common adverse events or ORR. For CR, there may be little to no difference between groups (low‐certainty evidence).
We found very low‐certainty evidence for the comparison of ECP versus PUVA. Although the study in this comparison did report ORR and CR in some participants treated with PUVA and none treated with ECP, and common adverse events with each treatment (some participants reported mild nausea after PUVA, and one participant in the ECP group withdrew due to hypotension), due to the certainty of the evidence, we cannot be sure of the results.
There may be little to no difference in CR and ORR in participants given bexarotene in combination with PUVA compared to PUVA alone (low‐certainty‐evidence). In the bexarotene plus PUVA group, one participant reported photosensitivity compared to no participants in the PUVA‐alone group (low‐certainty evidence).
The comparison of IFN‐α combined with either acitretin capsules or PUVA indicated there may be little to no difference in flu‐like symptoms (low‐certainty evidence). However, IFN‐α + PUVA may lead to a higher CR rate than IFN‐α + acitretin (low‐certainty evidence). ORR was not measured.
Another trial compared common adverse effects of PUVA maintenance versus no maintenance in participants with CR. However, the distribution between study arms was not provided. CR and OR were, by study design, not assessable.
We found low‐certainty evidence, so could not draw conclusions from the studies included in this review.
Overall completeness and applicability of evidence
We were only able to include studies for a limited number of commonly‐used types of treatment for mycosis fungoides (MF) because of the low number of randomised controlled trials (RCTS) found.
Included trials investigated participants with all stages of disease. The more recent trials were conducted as multicentre and multinational studies, facilitating the application of the study results.
We did not find any RCTs exclusively assessing the effect of topical steroids compared to placebo. Most of the trials used an active comparator instead of placebo or watchful waiting. This may limit the certainty of therapeutic value as overall survival or quality of life might have been equal or even worse for some treatments compared to placebo or watchful waiting.
Treatments like carmustine (BCNU) for the early stages of MF or stem cell transplantation for stage IV participants have not been evaluated in RCTs so far.
Most of the included studies did not assess our primary outcome patient‐reported quality of life, and when reported, the data was not usable; this hindered our conclusions in terms of this outcome. However, another primary outcome, common adverse effects, was widely assessed.
The applicability of the complete response (CR) and objective response rate (ORR) outcome was limited by the heterogeneous measures of assessment. Relapse, disease‐free survival, survival rates and rare adverse effects were poorly reported in the included studies. Drawing applicable conclusions was limited.
A separate rare adverse effects search highlighted important, but in the case of RCTs rarely reported, adverse effects.
Study duration was variable and reflected the types of outcomes assessed by different trials (e.g. first response outcomes will require shorter follow‐up than survival outcomes). Very few studies provided a long enough follow‐up. Due to small sample sizes, reliable survival analysis was not possible. Long‐term benefit (i.e. clearance of all symptoms of disease lasting at least two years) was not widely assessed.
After decades of discussions about different classifications of cutaneous lymphoma, the joint publication of the WHO‐EORTC classification in 2005 established defined disease entities. This helps both clinicians and researchers in the management and the exploration of MF. For the first time, commonly‐accepted criteria exist that define cutaneous lymphoma not only by means of histological findings but also by giving information on management, treatment and prognosis. Accordingly, diagnosis can only be made when clinical and histological findings are available. The classification was updated in 2018 (Elder 2018; Willemze 2019). Several risk factors for the development of MF have been established, amongst them ethnicity, which might imply a genetic predisposition, since incidences differ between Hispanic, Asian, Black and White populations.
The review does not have any major limitations regarding external validity; since MF is a rare disease which is normally treated in specialised medical centres (tertiary care setting), we consider the results of the studies applicable to daily practice.
Quality of the evidence
The quality of the included studies was very variable. All results expressed regarding the interventions, except the comparison of PUVA with or without IFN‐α, were based on single, often underpowered studies limiting the robustness of the findings.
In all the included studies, the risk of bias concerning random sequence generation was low or unclear. Only one study (Thestrup‐Pedersen 1982) had a high risk of selection bias for allocation concealment due to an open‐list randomisation procedure, and risk of bias was unclear for many of the other included studies.
We considered seven studies to be at high risk of bias concerning incomplete outcome data (Bagot 2017; Child 2004; Duvic 2001; Duvic 2001a; Olsen 2001; Stadler 1998; Stadler 2006). A common reason for assigning a high risk were high dropout rates. Other studies with small sample sizes did not report dropouts at all, so we had to assume dropout rates were zero, which may be untrue.
We rated the evidence of the studies displayed in the 'Summary of findings' tables as low certainty due to low internal validity and imprecision. This was based on a high risk of bias and a low event rate. We downgraded the results in Table 2 further to very low certainty due to very low sample size.
Potential biases in the review process
We consider our search strategy to be comprehensive and sensitive enough to identify relevant trials regarding our efficacy outcomes as we did not apply any language restrictions. Despite not applying any language restrictions, synonyms for mycosis fungoides in other languages can differ, which can lead to the omission of potential relevant trials published in other languages. Allthough we followed Cochrane Handbook for Systematic Reviews of Interventions, potential biases in the review may have occurred. The fact that two studies have not yet been incorporated may be a source of potential bias. In order to confirm inclusion criteria for studies retrieved by our search strategy, when necessary, we asked the corresponding authors how many of their included participants actually had MF of the Alibert‐Bazin type. Some authors answered that all participants included in their publications had this type of MF, although they defined cutaneous T‐cell lymphomas (CTCL) as inclusion criteria. Furthermore, missing efficacy data may be a possible source for bias as we were not able to obtain all relevant data of the included studies (e.g. improvement in quality of life in Prince 2017) by contacting the authors. In order to detect all dropouts, even those occurring during the study, we contacted the authors for further information. In case of no reply within four weeks of contact, we assumed there were no dropouts during the study, which probably does not reflect the truth. Data from cross‐over and within‐participant trials were presented without adjustments. A limitation of this approach is the possibility of a carry‐over effect and the proneness to over‐ or underestimation of the precision of results.
Agreements and disagreements with other studies or reviews
The evidence‐based Joint British Association of Dermatologists and UK Cutaneous Lymphoma Group guidelines for the management of primary cutaneous T‐cell lymphomas of 2018 (Gilson 2019) concluded that skin‐directed treatments represent a fundamental treatment option for all stages of MF. Skin‐directed therapies including phototherapy, local radiotherapy as well as topical treatments with steroids, carmustine, mechlorethamine or bexarotene are recommended in the early stages of disease (IA‐IIA). The use of PUVA is recommended by Gilson 2019 for early‐stage disease to obtain complete remission, but not for maintenance therapy, although it is unclear whether disease‐specific survival is affected. The authors state that Total Skin Electron Beam (TSEB) causes short‐term complete response (up to 50 months) and should be reserved for those people for whom topical treatments and phototherapy have failed (stage IB) or as a first‐line treatment in patients with extensive cutaneous disease (stage T2b). The use of IFN‐α is not recommended for early stages as there is no evidence that IFN‐α positively influences long‐term outcome. The combination of IFN‐α and PUVA is considered superior to the combination of IFN‐α and acitretin based on Stadler 1998 for therapy‐resistant early stages or those people with thick plaques. Chemotherapies are recommended for advanced stages of the disease.
Concordant with Gilson 2019, we were not able to identify any curative intervention for MF even for participants with early‐stage disease. According to Gilson 2019, PUVA in combination with additional IFN‐α may be beneficial. However, our meta‐analysis of the data of Stadler 2006 and Wozniak 2008 did not support a high grade of evidence.
The EORTC Cutaneous Lymphoma Task Force consensus workshop summarised recommendations for the treatment of mycosis fungoides/Sézary syndrome based on the best practice of each national group that was involved (Trautinger 2017). Recommendations for treatment of each stage of MF were given.
Based on limited evidence, recommendations for first‐line treatment of early stage disease (IA, IB, and IIA) include expectant policy and skin‐directed therapies like PUVA, UVB, topical corticosteroids, localised radiotherapy, or mechlorethamine.
Second‐line treatments for these stages include systemic therapies like retinoids, IFN‐α, TSEB and low dose methotrexate (MTX).
Recommendations for first‐line treatment of stage IIB patients include systemic therapies such as retinoids and IFN‐α, TSEB, monochemotherapy (gemcitabine, pegylated liposomal doxorubicin), low‐dose MTX or localised radiotherapy.
Second‐line treatments for stage IIB include polychemotherapy (CHOP is the most widely used regimen: it is a specific regimen of chemotherapy, including the drugs cyclophosphamide, doxorubicin hydrochloride (hydroxydaunorubicin), vincristine sulphate and prednisone) and allogeneic stem cell transplantation.
Treatment recommendations for stage III disease include systemic therapies (retinoids, IFN‐α), ECP, low‐dose MTX and TSEB.
Second‐line treatments for stage III include monochemotherapy (gemcitabine, pegylated liposomal doxorubicin) and allogeneic stem cell transplantation.
Trautinger 2017 state that stage IV patients should be treated with chemotherapy (gemcitabine, pegylated liposomal doxorubicin, CHOP and CHOP‐like polychemotherapy), radiotherapy (TSEB and localised), alemtuzumab or allogeneic stem cell transplantation.
Since these guidelines are determined with all levels of evidence, we cannot comment on most of the recommendations made as we identified only a comparatively small number of RCTs. As noted in the previously‐mentioned guidelines, we agree with the authors that focus should be on quality of life‐targeted skin‐directed therapy for the early stages of disease to avoid the severe adverse effects caused by systemic treatments.
The ESMO clinical recommendations for diagnosis, treatment, and follow‐up of MF (Dummer 2008) also suggest a wait‐and‐see policy or a skin‐directed therapeutic approach for early‐stage disease, followed by PUVA and IFN‐α or extracorporeal photopheresis for stages II to IV as first‐line treatments. TSEB, oral bexarotene, denileukin diftitox and chemotherapy are suggested as second‐line therapy according to the stage of the disease.
For the treatment of MF by stem cell transplantation, this review could not identify a single RCT. This is in accordance with the findings of two other reviews addressing this issue by Wu 2009 for mycosis fungoides and Sézary syndrome and more thoroughly investigated by Schlaak 2013 for cutaneous T‐cell lymphoma.
Authors' conclusions
Implications for practice.
At this point, no exact cause for the disease has been found and no curative treatment for mycosis fungoides (MF) has been established.
Our review found the following key findings.
There may be little to no difference between psoralen plus ultraviolet A (PUVA) alone versus PUVA plus interferon‐α (IFN‐α) in complete response (low‐certainty evidence). This comparison did not report the objective response rate (ORR).
Very low‐certainty evidence means that we are uncertain of the effects of extracorporeal photopheresis (ECP) compared with PUVA on the reported outcomes of complete response (CR) and ORR with PUVA.
There may be little to no difference in CR and ORR in participants given bexarotene in combination with PUVA compared to PUVA alone (low‐certainty‐evidence).
Low‐certainty evidence indicates that IFN‐α + acitretin may lead to a lower complete response rate than IFN‐α + PUVA. This comparison did not report the ORR.
One study compared PUVA maintenance versus no maintenance in patients with CR, but our key outcomes were not assessable or data were not provided.
Mild nausea was reported by some participants after PUVA, and one participant withdrew from the ECP group due to hypotension (very‐low certainty evidence). In the bexarotene plus PUVA group, one participant reported photosensitivity compared to no participants in the PUVA‐alone group (low‐certainty evidence). There may be little to no difference between IFN‐α combined with either PUVA or acitretin in the occurrence of flu‐like symptoms (low‐certainty evidence). Common adverse events were not measured in the comparison of PUVA alone versus PUVA plus IFN‐α (low‐certainty evidence).
The interventions in our review showed a wide range of adverse effects, some of them potentially life‐threatening. Generally, the studies suggested that more aggressive therapeutic approaches led to substantially increased, and more severe, adverse effects.
When treating MF, the clinician should be aware of the limited evidence supporting the different types of treatments. Despite a wide variety of existing treatments for MF, the data we extracted from the 20 randomised controlled trials (RCTs) in this systematic review did not provide us with much useful evidence. The studies included only, on average, 68 participants and showed heterogeneous designs, making it difficult to draw firm conclusions. Only five trials assessed PUVA treatment, either given alone or combined.
In the early stages of this disease, skin‐directed treatment approaches are more often recommended than systemic or combination therapies because progression of the disease is slow, and in the very early stages life expectancy is similar to age‐matched control groups. In this review, we were not able to include RCTs investigating the effect of topical corticosteroids versus placebo. Despite its widespread use, no RCTs could be included in this review investigating the effect of UVB or localised radiotherapy.
Even if the majority of the reviewed trials compared an intervention to an active comparator, clinicians and people with early stage MF should consider what is called 'watchful waiting' or 'expectant policy' as one of their treatment options. Treating patients at early stages of disease might temporary improve their condition. Nevertheless, potential adverse effects can negatively affect quality of life resulting in lower therapeutic value.
Clinicians should motivate people with MF to enter multicentre RCTs, especially for advanced stages of MF as they represent an unmet medical need, in order to facilitate evidence‐based recommendations.
Implications for research.
Research should be based on commonly‐accepted diagnostic and staging criteria. With the publication of the joint consensus statement of the ISCL, EORTC and the United States Cutaneous Lymphoma Consortium (USCLC) by Olsen 2011, commonly‐accepted criteria have been established regarding the conduct of MF studies and their outcome measurements. The consensus statement emphasised the RCT as the ideal and a goal that may be reached by co‐operative studies.
For all studies, the stage of the disease and its activity are needed in order to refer treatments under investigation to a suitable group of participants. The inclusion of subtypes of MF should be clearly stated, and it is recommended that the results are stratified according to the different MF entities. A joint commitment of the specialised centres to high‐quality multicentre RCTs would be helpful to gain sufficient sample sizes.
Regarding the two primary outcome measures chosen for this review, it is surprising that while quality of life measurements are paid attention to in an extra chapter in the consensus statement, no end points were proposed on how to measure or address adverse effects. This might be due to the fact that many clinicians do not consider adverse effects to be a relevant end point for studies at all, but still we remain of the opinion that they should be reported in all further trials regarding the treatment of MF.
The first‐line treatment options already recommended for early stages (i.e. topical application of corticosteroids, nitrogen mustard, hypericin, intralesional injection of IFN‐α, or PUVA) should be a primary target for further research, since they apply to the largest group of people with MF. Treatment options often used as concomitant therapy, such as topical corticosteroids, should be investigated for their inherent efficacy.
Comparable, clearly‐defined, and standardised end points have been proposed by international societies, and they should be consistently used in further research in order to gain comparability (Olsen 2011). Furthermore, involvement of patients at the stage of study design would lead to adequate evaluations of their desires and needs. In addition to efficacy measures, toxicities should be reported not only by physicians but also by patients in order to facilitate the decision for treatment allocation. Quality of life should be addressed based on different treatment arms and not according to response as has been done in the included studies. A measure of the quality of life was often not implemented in the RCTs under investigation, but is essential in order to balance the risks and benefits of treatments. This is particularly applicable to early stage disease where life expectancy is not likely to be affected.
Validated patient‐reported outcome measures are crucial for the comparison of different interventions.
Feedback
Confidence intervals tend to be unreliable when event numbers are very small,
Summary
In response to a letter sent to the author team by Peggy Wu (Beth Israel Deaconess Medical Center) we would like to highlight the issue that in nine instances the Fisher test showed significant results whereas the 95% confidence interval (CI) computed by the Cochrane standard software indicated no significant difference for the relative risk.
Reply
For these instances we computed additionally the 95% CIs by the use of the method described by Miettinen 1985 (CI‐M) which are closer to the exact confidence intervals and provide the results together with the standard CI here:
Duvic 2001a, Analysis 1.1 (rash): RR 7.24, 95% CI 0.92 to 56.76, 95% CI‐M 1.22 to 45.1, Fisher test P = 0.041.
Vonderheid 1987, Analysis 3.1 (mild erythema): RR 11.00, 95% CI 0.74 to 163.49, 95% CI‐M 1.83 to not estimable, Fisher test P = 0.016.
Wolff 1985, Analysis 3.1 (mild fever): RR 11.00, 95% CI 0.70 to 173.66, 95% CI‐M 1.59 to not estimable, Fisher test P =0.03.
Whittaker 2012, Analysis 8.1 (liver toxicities): RR 11.62, 95% CI 0.7 to 200.07, 95% CI‐M 1.46 to not estimable, Fisher test P = 0.03.
Whittaker 2012, Analysis 8.1 (Increased fasting cholesterol): RR 11.62, 95% CI 0.7 to 200.07, 95% CI‐M 1.46 to not estimable, Fisher test P = 0.03.
Prince 2017, Analysis 10.1 (complete response): RR 11.22, 95% CI 0.64 to 197.60, 95% CI‐M 1.36 to not estimable, Fisher test P = 0.03.
Child 2004, Analysis 11.2 (Objective response rate): RR 0.07, 95% CI 0.00 to 1.00, 95% CI‐M 0.00 to 0.40, Fisher test P = 0.002.
Stadler 1998, Analysis 13.1 (adverse effects requiring discontinuation): RR 4.29, 95% CI 0.99 to 18.63, 95% CI‐M 1.13 to 17.1, Fisher test P = 0.049.
Thestrup‐Pedersen 1982, Analysis 14.1 (complete response): RR 0.09, 95% CI 0.01 to 1.41, 95% CI‐M 0.00 to 0.61, Fisher test P = 0.03.
Contributors
Our Statistical Editors Jo Leonardi‐Bee and Matthew Grainge, our Co‐ordinating Editor Hywel Williams, our Feedback Editor Urbà González and the lead author Tobias Weberschock.
What's new
| Date | Event | Description |
|---|---|---|
| 3 July 2020 | New citation required but conclusions have not changed | This update included new therapeutic options, particularly for advanced stages of the disease. The conclusions remain the same. |
| 3 July 2020 | New search has been performed | We added six new studies with 694 participants in total. We used GRADE methodology to assess evidence quality and draw conclusions about our certainty in the review findings. |
History
Protocol first published: Issue 1, 2011 Review first published: Issue 9, 2012
| Date | Event | Description |
|---|---|---|
| 16 May 2013 | Amended | Computation error in feedback corrected |
| 28 March 2013 | Feedback has been incorporated | Methodological discussion about statistical misinterpretation within the review |
| 28 March 2013 | Amended | Feedback added |
Acknowledgements
Special thanks to Dr Finola Delamere, Elizabeth Doney, Emma Axon, Laura Prescott, and Helen Scott for their continuous and patient help in the whole process and Philipp Rehberger for the help in the planning phase.
The Cochrane Skin editorial base wishes to thank Dr Sam Gibbs who was the Key Editor for this updated review; Dr Ben Carter who was the Statistical Editor; Dr Rubeta Matin and Prof Sean Whittaker who were the clinical referees; the consumer referee, Ann Fonfa; Heather Maxwell who copy‐edited the review; and Nicole Pitcher and Elizabeth Royle who wrote the plain language summary.
Appendices
Appendix 1. Cochrane Skin Specialised Register/CRS search strategy
"mycosis fungoide?" or "cutaneous T‐cell lymphoma" or "Alibert Bazin" or "Granuloma fungoide?" or "Granuloma sarcomatode?*" or "facies leon*" or Wucherflechte or parapsoriasis
Appendix 2. CENTRAL (Cochrane Library) search strategy
#1 Granuloma sarcomatode*:ti,ab,kw #2 Granuloma fungoide*:ti,ab,kw #3 facies leon*:ti,ab,kw #4 mycosis fungoide*:ti,ab,kw #5 MeSH descriptor: [Mycosis Fungoides] explode all trees #6 Alibert Bazin:ti,ab,kw #7 Wucherflechte:ti,ab,kw #8 cutaneous T cell lymphoma:ti,ab,kw #9 MeSH descriptor: [Lymphoma, T‐Cell, Cutaneous] explode all trees #10 parapsoriasis:ti,ab,kw #11 MeSH descriptor: [Parapsoriasis] explode all trees #12 {or #1‐#11}
Appendix 3. MEDLINE (Ovid) search strategy
1. mycosis fungoide$.mp. 2. exp Mycosis Fungoides/ 3. cutaneous T‐cell lymphoma.mp. 4. exp Lymphoma, T‐Cell, Cutaneous/ 5. Alibert Bazin.mp. 6. Granuloma fungoide$.mp. 7. Wucherflechte.mp. 8. Granuloma sarcomatode$.mp. 9. facies leon$.mp. 10. parapsoriasis.mp. 11. exp Parapsoriasis/ 12. or/1‐11 13. randomized controlled trial.pt. 14. controlled clinical trial.pt. 15. randomized.ab. 16. placebo.ab. 17. clinical trials as topic.sh. 18. randomly.ab. 19. trial.ti. 20. 13 or 14 or 15 or 16 or 17 or 18 or 19 21. exp animals/ not humans.sh. 22. 20 not 21 23. 12 and 22
[Lines 13‐22: Cochrane Highly Sensitive Search Strategy for identifying randomized trials in MEDLINE: sensitivity‐ and precision‐maximizing version (2008 revision); Ovid format, from section 3.6.1 in Lefebvre C, Glanville J, Briscoe S, Littlewood A, Marshall C, Metzendorf M‐I, Noel‐Storr A, Rader T, Shokraneh F, Thomas J, Wieland LS. Technical Supplement to Chapter 4: Searching for and selecting studies. In: Higgins JPT, Thomas J, Chandler J, Cumpston MS, Li T, Page MJ, Welch VA (eds). Cochrane Handbook for Systematic Reviews of Interventions Version 6. Cochrane, 2019. Available from: www.training.cochrane.org/handbook]
Appendix 4. EMBASE (Ovid) search strategy
1. mycosis fungoide$.tw. 2. exp mycosis fungoides/ 3. cutaneous T‐cell lymphoma.tw. 4. exp cutaneous T cell lymphoma/ 5. parapsoriasis.tw. 6. exp PARAPSORIASIS/ 7. Alibert Bazin.tw. 8. Granuloma fungoide$.tw. 9. Granuloma sarcomatode$.tw. 10. facies leon$.tw. 11. Wucherflechte.tw. 12. or/1‐11 13. crossover procedure.sh. 14. double‐blind procedure.sh. 15. single‐blind procedure.sh. 16. (crossover$ or cross over$).tw. 17. placebo$.tw. 18. (doubl$ adj blind$).tw. 19. allocat$.tw. 20. trial.ti. 21. randomized controlled trial.sh. 22. random$.tw. 23. or/13‐22 24. exp animal/ or exp invertebrate/ or animal experiment/ or animal model/ or animal tissue/ or animal cell/ or nonhuman/ 25. human/ or normal human/ 26. 24 and 25 27. 24 not 26 28. 23 not 27 29. 12 and 28
[Lines 13‐23: Based on terms suggested for identifying RCTs in Embase (section 3.6.2) in Lefebvre C, Glanville J, Briscoe S, Littlewood A, Marshall C, Metzendorf M‐I, Noel‐Storr A, Rader T, Shokraneh F, Thomas J, Wieland LS. Technical Supplement to Chapter 4: Searching for and selecting studies. In: Higgins JPT, Thomas J, Chandler J, Cumpston MS, Li T, Page MJ, Welch VA (eds). Cochrane Handbook for Systematic Reviews of Interventions Version 6. Cochrane, 2019. Available from: www.training.cochrane.org/handbook]
Appendix 5. LILACS search strategy
(((micosis or mycosis) and fungoide$) or (Alibert Bazin) or (Granuloma fungoide$) or (Granuloma sarcomatode$) or (facies leon$) or parapsoriasis)
These terms were combined with the LILACS Controlled clinical trials topic‐specific query filter.
Appendix 6. MEDLINE Adverse Effects search strategy (OVID)
1. exp PUVA Therapy/ 2. exp Photopheresis/ 3. imiquimod.mp. 4. exp Whole‐Body Irradiation/ 5. exp Cyclophosphamide/ 6. exp Doxorubicin/ 7. exp Etoposide/ 8. exp Vincristine/ 9. exp Transfer Factor/ 10. exp Bleomycin/a<y 11. exp Methotrexate/ 12. exp Interferon‐alpha/ 13. bexarotene.mp. 14. peldesine.mp. 15. denileukin diftitox.mp. 16. exp Mechlorethamine/ 17. or/1‐16 18. exp product surveillance, postmarketing/ or exp adverse drug reaction reporting systems/ or exp clinical trials, phase iv/ 19. adverse events.mp. 20. adverse effects.mp. 21. exp hypersensitivity/ or exp drug hypersensitivity/ or exp drug eruptions/ or exp hypersensitivity, delayed/ or exp hypersensitivity, immediate/ 22. exp hypersensitivity, immediate/ or exp anaphylaxis/ or exp conjunctivitis, allergic/ or exp dermatitis, atopic/ or exp food hypersensitivity/ or exp respiratory hypersensitivity/ or exp urticaria/ 23. side effect$.mp. 24. exp Poisoning/ 25. exp hepatitis, toxic/ or exp hepatitis, chronic, drug‐induced/ 26. exp Substance‐Related Disorders/ 27. exp Drug Toxicity/ 28. exp Abnormalities, Drug‐Induced/ 29. exp Teratogens/ 30. exp Mutagens/ 31. exp Carcinogens/ 32. metabolites.mp. 33. exp dermatitis, contact/ or exp dermatitis, allergic contact/ or exp dermatitis, irritant/ or exp dermatitis, phototoxic/ 34. photoallergic reactions.mp. 35. exp dermatitis, allergic contact/ or exp dermatitis, photoallergic/ 36. phototoxicity.mp. 37. sensitization.mp. 38. exp Burning Mouth Syndrome/ 39. stinging.mp. 40. burning.mp. 41. fetal abnormalities.mp. 42. exp Drug Monitoring/ 43. harm$ effects.mp. 44. (toxic effects or drug effects).mp. 45. Sleep Apnea, Obstructive/ 46. ARRHYTHMIA/ 47. undesirable effect$.mp. 48. (safe or safety).mp. [mp=protocol supplementary concept, rare disease supplementary concept, title, original title, abstract, name of substance word, subject heading word, unique identifier] 49. toxicity.mp. 50. noxious.mp. 51. serious reaction$.mp. 52. complication$.mp. 53. treatment emergent.mp. 54. tolerability.mp. 55. (adverse adj3 (effect$ or reaction$ or event$ or outcome$)).mp. [mp=protocol supplementary concept, rare disease supplementary concept, title, original title, abstract, name of substance word, subject heading word, unique identifier] 56. rebound.mp. 57. Hypercalcemia/ci [Chemically Induced] 58. Urinary Calculi/ci [Chemically Induced] 59. Tachyphylaxis/ci, de [Chemically Induced, Drug Effects] 60. Substance Withdrawal Syndrome/ci, de [Chemically Induced, Drug Effects] 61. ATROPHY/ci [Chemically Induced] 62. TELANGIECTASIS/ci [Chemically Induced] 63. skin thinning.mp. 64. Liver Diseases/ci [Chemically Induced] 65. Kidney Diseases/ci [Chemically Induced] 66. Disseminated Intravascular Coagulation/ci [Chemically Induced] 67. Multiple Organ Failure/ci [Chemically Induced] 68. Stevens‐Johnson Syndrome/ci [Chemically Induced] 69. Epidermal Necrolysis, Toxic/ci [Chemically Induced] 70. Heart Block/ci [Chemically Induced] 71. COMA/ci [Chemically Induced] 72. PARALYSIS/ci [Chemically Induced] 73. exp Nausea/dt [Drug Therapy] 74. exp Vomiting/dt [Drug Therapy] 75. 18 or 19 or 20 or 21 or 22 or 23 or 24 or 25 or 26 or 27 or 28 or 29 or 30 or 31 or 32 or 33 or 34 or 35 or 36 or 37 or 38 or 39 or 40 or 41 or 42 or 43 or 44 or 45 or 46 or 47 or 48 or 49 or 50 or 51 or 52 or 53 or 54 or 55 or 56 or 57 or 58 or 59 or 60 or 61 or 62 or 63 or 64 or 65 or 66 or 67 or 68 or 69 or 70 or 71 or 72 or 73 or 74 76. mycosis fungoides.mp. or exp Mycosis Fungoides/ 77. cutaneous T‐cell lymphoma.mp. or exp Lymphoma, T‐Cell, Cutaneous/ 78. Alibert‐Bazin.mp. 79. Granuloma fungoides.mp. 80. Granuloma sarcomatodes.mp. 81. facies leontina.mp. 82. Wucherflechte.mp. 83. parapsoriasis.mp. or exp Parapsoriasis/ 84. 76 or 77 or 78 or 79 or 80 or 81 or 82 or 83 85. 17 and 75 and 84
Appendix 7. Assessment of external validity
In order to assess external quality, we discussed the following questions based on Dekkers 2010.
Does the proportion of eligible participants actually included in the study indicate selectivity?
Is there a run‐in period that is likely to exclude non‐compliant participants or participants with a high risk of having side‐effects?
Does the selection of participating centres lead to a selection of participants likely to limit the treatment effects to a narrow subgroup?
Have important changes in medical practice occurred since the original study was performed that will influence treatment effects?
Is ethnicity likely to interact with the treatment effect?
Are there geographical or socioeconomic aspects that are likely to interact with the treatment effect?
Does the study exclude participants of certain ages, thus, limiting the treatment effects to a narrow subgroup of participants?
Does the study exclude participants with certain comorbidities, thus, limiting the treatment effects to a narrow subgroup of participants?
Does extraordinary training for study physicians exist that is likely to limit the treatment effects to a narrow subgroup of participants?
Does the treatment setting (e.g. study nurse, interval of assessments) differ from daily practice, and is this likely to limit the treatment effects to study participants?
Do administrative policies (e.g. immediate access to treatment) differ from daily practice, thus, in all likelihood, limiting the treatment effects to study participants?
Data and analyses
Comparison 1. Topical peldesine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1.1 Common adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1.1 Pruritus | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.1.2 Rash | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 1.2 Complete response | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.3 Objective response rate | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 2. Topical hypericin versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 2.1 Objective response rate | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 3. IFN‐α versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 3.1 Common adverse effects | 2 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1.1 Erythema | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1.2 Fever | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1.3 Myalgia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1.4 Chills or weakness | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.1.5 Nausea, arthralgia, and malaise | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3.2 Complete response | 2 | 36 | Risk Ratio (M‐H, Fixed, 95% CI) | 7.00 [1.56, 31.47] |
Comparison 4. Mechlorethamine gel vs mechlorethamine ointment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 4.1 Common adverse event | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.1 Skin irritation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.2 Pruritus | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.3 Erythema | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.4 Contact dermatitis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.5 Skin hyperpigmentation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.1.6 Folliculitis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 4.2 Complete response | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 4.3 Objective response rate | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 5. IFN‐α + PUVA versus PUVA alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 5.1 Complete response | 2 | 122 | Risk Ratio (M‐H, Random, 95% CI) | 1.07 [0.87, 1.31] |
Comparison 6. Denileukin diftitox high versus low dose.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 6.1 Common adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.1 Constitutional symptoms | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.2 Grade 3 to 4 constitutional symptoms | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.3 Infections | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.4 Grade 3 to 4 infections | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.5 Gastrointestinal syndromes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.6 Grade 3 to 4 gastrointestinal syndromes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.7 CNS syndromes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.8 Grade 3 to 4 CNS syndromes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.9 Rash | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.10 Grade 3 to 4 rash | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.11 Vascular leak syndrome | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.12 Grade 3 to 4 vascular leak syndrome | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.13 Thrombotic events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.14 Grade 3 to 4 thrombotic events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.15 Cardiopulmonary events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.16 Grade 3 to 4 cardiopulmonary events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.17 Acute infusion related events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.18 Grade 3 to 4 acute infusion related events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.1.19 Grade 3 to 4 laboratory abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 6.2 Complete response | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 6.3 Objective response rate | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 7. Bexarotene high versus low dose.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 7.1 Common adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1.1 Photosensitivity | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1.2 Nausea | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1.3 Constipation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1.4 Fatigue | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1.5 Pruritus | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1.6 Arthralgia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1.7 Nasopharyngitis | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1.8 Headache | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1.9 Insomnia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1.10 Free T4 abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1.11 Cholesterol abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1.12 Triglycerid abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.1.13 SGOT/SGPT abnormalities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 7.2 Complete response | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.3 Relapse | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 7.4 Objective response rate | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 8. Bexarotene + PUVA vs PUVA alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 8.1 Common adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.1.1 Liver toxicities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.1.2 Renal toxicity | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.1.3 Haematological toxicities | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.1.4 Increased fasting cholesterol | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.1.5 Photosensitivity | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.1.6 Pruritus | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.1.7 Rash | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.1.8 Hypertriglyceridaemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 8.2 Complete response | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 8.3 Objective response rate | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 9. Lenalidomide maintenance versus observation after debulking therapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 9.1 Common adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1.1 Neutropenia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1.2 Hyperbilirubinaemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1.3 Hypercalcaemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1.4 Hypokalaemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1.5 Hypophosphataemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1.6 Erythema multiforme | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1.7 Periorbital oedema | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1.8 Pruritus | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1.9 Other AE | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected |
Comparison 10. Brentuximab vedotin vs. physician's choice (MTX or bexarotene).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 10.1 Complete response | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 10.2 Objective response rate | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 11. Extracorporeal photopheresis versus PUVA.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 11.1 Complete response | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 11.2 Objective response rate | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 12. Combined therapy versus conservative therapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 12.1 Common adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.1.1 Hospitalization | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.1.2 Fatal myocardial infarction | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.1.3 Cutaneous toxicity from electron beam therapy | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.1.4 Acute non‐lymphocytic leukaemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.1.5 Non‐melanoma skin cancer | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.1.6 Unspecified | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 12.2 Complete response | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.3 Relapse | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.4 Overall survival | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 12.5 Objective response rate | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 13. IFN‐α + acitretin versus IFN‐α + PUVA.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 13.1 Common adverse effects | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.1 Grade I to II adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.2 Grade III adverse events | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.3 Adverse events requiring treatment discontinuation | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.4 Flu‐like symptoms | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.5 Dryness/redness of skin or hair loss | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.6 Neurological disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.7 Psychiatric disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.8 Gastrointestinal disorders | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.9 Elevated liver or biliary tract enzymes | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.10 Elevated triglycerides | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.11 Anemia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.12 Leukopenia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.13 Impotentia | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.1.14 Redness and infiltration at application site | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 13.2 Complete response | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 14. Topical nitrogen mustard with active transfer factor versus topical nitrogen mustard with inactivated transfer factor.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 14.1 Complete response | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.2 Overall survival | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 14.3 Objective response rate | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 15. Mogamulizumab vs. Vorinostat.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 15.1 Objective response rate | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only |
Comparison 16. PUVA maintenance vs. no maintenance.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 16.1 Disease‐free survival | 1 | Hazard Ratio (IV, Random, 95% CI) | Totals not selected |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bagot 2017.
| Study characteristics | ||
| Methods | This study was an open‐label, multicentre, randomised phase III clinical trial in patients with advanced‐stage MF or Sézary syndrome (SS), which was initially designed for 560 days. | |
| Participants | After having received a debulking therapy with either gemcitabine hydrochloride or pegylated liposomal doxorubicin hydrochloride IV (with or without radiotherapy), patients were either randomised into an observation‐only arm or lenalidomide maintenance therapy. Debulking took place before study inclusion. This study recruited 30 patients with MF or SS, 9 of those could not be randomised. Of the remaining 21 patients, 12 were randomised to observation only. Another 9 patients were allocated to the lenalidomide arm. Demographics of the included participants
Relevant exclusion criteria of the trial
|
|
| Interventions |
|
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | No efficacy end points could be included in our review due to low sample size and premature termination of study. Only the adverse effects could be extracted. This study was supported by a research grant from Celgene Inc. and the EORTC Cancer Research Fund. No conflict of interest stated. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The study did not provide information about this. We sought information and had an email response: "Randomisation was performed at EORTC headquarters via a web based application developed at EORTC." |
| Allocation concealment (selection bias) | Low risk | The study did not provide information about this. We sought information and had an email response: "Allocation to treatment arm was concealed." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was an open‐label study. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | The study did not provide information about this. We sought information and had an email response: "The outcome assessors were not blinded." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The study was terminated prematurely due to lack of financial support. |
| Selective reporting (reporting bias) | Low risk | There were several outcome measures missing in the publication compared to the information on clinicaltrials.gov. This might be explained by the premature termination of the study. We sought information and had an email response: "No additional outcome data available.". We therefore decided this study was at low risk of selective reporting. |
| Other bias | High risk | As the authors concluded themselves: "Due to the premature closure of this trial by the provider of the research grant and due to the severely underpowered trial, a meaningful statistical analysis was not possible." |
Child 2004.
| Study characteristics | ||
| Methods | This was a randomised, open‐label, cross‐over trial, which lasted 12 months. | |
| Participants | The study recruited 16 participants (10 were in the PUVA‐first group; 6 were in the ECP‐first group) with plaque‐stage (1B ⁄ T2, Bunn Lamberg 1B) MF and a peripheral blood T‐cell clone (detected by polymerase chain reaction (PCR)‐single‐strand conformational polymorphism (SSCP) methodology), but with no evidence of lymph node involvement. Demographics of the included participants
Exclusion criteria of the trial
|
|
| Interventions |
|
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | No information for funding and conflict of interests given. This study was conducted at Skin Tumour Clinic, St John's Institute of Dermatology, United Kingdom. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "envelopes, numbered from 1 to 20, randomly allocating patients to Group 1 or Group 2, were generated by the statistician." Comment: we judged this to be of low risk. |
| Allocation concealment (selection bias) | Unclear risk | It was unclear whether the used envelopes were sealed and opaque. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | This was not possible because of different types of interventions, so we judged this domain as unclear risk. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There was insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 8/16 participants (50%) were lost to follow‐up: 3/10 participants in the PUVA‐first group and 5/6 participants in the ECP‐first group. |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | High risk | It was unclear if concomitant medication was permitted. |
Chong 2004.
| Study characteristics | ||
| Methods | This was a randomised, double‐blind, parallel‐group trial, which lasted 4 months. | |
| Participants | The study recruited 4 participants (3 in the intervention group and 1 in the control group) with histologically‐proven MF plaque stage 1B MF (T2N0M0). Demographics of the included participants
Exclusion criteria of the trial These were not reported. |
|
| Interventions |
|
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | The funding body was 3M Health Care Limited supplied Aldara. Conflict of interest not reported. This study was conducted in the United Kindom (1 centre). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was described as double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | There were insufficient information to permit judgement. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts were reported. |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | Low risk | None were found. |
Duvic 2001.
| Study characteristics | ||
| Methods | This was a randomised, open‐label, parallel‐group trial, which lasted 16 weeks. | |
| Participants | The study recruited 58 participants (15 in the low‐dose group with 6.5 mg/m² daily versus 43 in the high‐dose group, which consisted of 28 participants who had 300 mg/m² daily and 15 participants who had 650 mg/m² daily) with histologically‐confirmed mycosis fungoides:
Demographics of the included participants
Exclusion criteria of the trial
|
|
| Interventions |
|
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | The high‐dose was reduced twice (from 650 mg/m²/day to > 500 mg/m²/day, and from 500 mg/m²/day to > 300 mg/m²/day) during the trial; there was separate assessments of the high‐dose and "optimal" dose groups. 11/15 participants in the low‐dose group crossed over to high‐dose therapy after 8 weeks of treatment. Randomisation discontinued during the trial after interim analysis and was reinstalled after consideration by the U.S. Food and Drug Administration (FDA). The dropout rate for withdrawals was 72.4%. Dr Duvic was funded by research grants from Ligand Pharmaceuticals, San Diego California, USA (R21‐CA74117); from the National Institutes of Health, Bethesda, Maryland; and from the MD Anderson Cancer Centre (CA16672‐22). This study was conducted in 18 CTCL clinics at academic referral centres in the USA, Canada, Australia, and Europe. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information, but received no response. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information, but received no response. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Blinding was not possible because of the number of capsules given." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The physician was blinded to CA response because it was calculated from the case report form." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The dropout rate for withdrawals was 72.4%. Reasons for dropout were withdrawal due to adverse effect, progressive disease, withdrawal of consent or patients being lost to follow‐up. |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | High risk | The initial dose in the intervention group was reduced from 650 mg/m²/day to 500 mg/m²/day to 300 mg/m²/day due to adverse reactions. The study discontinued randomisation. |
Duvic 2001a.
| Study characteristics | ||
| Methods | This was a randomised, double‐blind, parallel‐group trial, which lasted 24 weeks. | |
| Participants | The study recruited 89 participants (43 in the intervention group and 46 in the control) with histologically‐confirmed MF manifested as patches with or without plaques (stage I), but without enlarged nodes, visceral involvement, or generalised erythroderma. Demographics of the included participants
Exclusion criteria of the trial
|
|
| Interventions |
|
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | Funding came from BioCryst Pharmaceuticals, Inc (Birmingham, Alabama, USA). According to the publication none of the authors had a commercial association with this study that posed a conflict of interest. This study was conducted in 10 tertiary care centres in the USA. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No specific information was given, other than that the data were managed by a third party (Quintiles Inc). |
| Allocation concealment (selection bias) | Low risk | The data were managed by a third party (Quintiles Inc), so it was considered likely that allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Only the sponsor was able to un‐blind in case of withdrawal. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Only the sponsor was able to un‐blind in case of withdrawal. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | ITT analysis was carried out and last observation carried forward. 24/89 participants were lost to follow‐up: 14/43 (33%) in the intervention group and 11/46 (24%) in the placebo group. |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | Low risk | None were found. |
Guitart 2002.
| Study characteristics | ||
| Methods | This was a randomised, open‐label, parallel‐group trial, which lasted 24 weeks. | |
| Participants | The study recruited 43 participants (20 in the high‐dose group and 23 in the low‐dose group) with histologically‐proven mycosis fungoides stages IB and IIA, with lymph node biopsies negative for MF involvement. Demographics of the included participants
Exclusion criteria of the trial These were not reported. |
|
| Interventions |
|
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | Data were abstracted from the manuscript sent by the corresponding author; the sample size was smaller than planned according to the author. Dose reduction was necessary in 14/39 participants because of hyperlipidaemic side‐effects, although antilipidaemic therapy was prescribed for each participant. This study was conducted in 12 tertiary care centres in the USA. This study was funded by Ligan Pharmaceutical (San Diego, CA). Several authors have participated in the speakers bureau and/or received research grants from Ligand Pharmaceuticals. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was an open‐label trial. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No separate outcome assessor was mentioned. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis was carried out: 4 participants (10%) dropped out after randomisation without receiving a single treatment; 1 person (5%) in the 300 mg/day bexarotene group dropped out for "other" reason. |
| Selective reporting (reporting bias) | Low risk | We contacted the corresponding author for additional outcome data, and we had an email response. We were sent a manuscript of unpublished data, and we had further confirmation by email that all outcomes were reported. |
| Other bias | High risk | The study had a smaller sample size than planned; dose reduction was necessary in 14/39 participants due to hypertriglyceridaemia, although preventive antilipidaemic therapy was prescribed for each participant. |
Kaye 1989.
| Study characteristics | ||
| Methods | This was a randomised, open‐label, parallel‐group trial. | |
| Participants | The study recruited 103 participants (52 in the combined‐therapy group and 51 in the conservative‐therapy group) with histologically‐proven MF of all stages. Demographics of the included participants
Exclusion criteria of the trial
|
|
| Interventions |
a) oral methotrexate (20 mg/m² orally twice weekly for stage IVB participants) b) PUVA (oral methoxsalen 0.6 mg/kg body weight followed by UVA light therapy 3 x/week) c) electron‐beam therapy (as described in the combined‐therapy group) combined with methotrexate (as described above) d) systemic chemotherapy (as described in the combined‐therapy group) |
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | The funding body was not declared. No conflicts of interests reported. This study was conducted in 7 secondary/tertiary care centres in the USA. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Stratified block randomisation was undertaken. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | This was not possible because of different interventions used. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | ITT analysis and last observation carried forward was carried out. There were 6/52 (12%) dropouts in the combined‐therapy group (2 refused to receive treatment; 1 withdrew because of congestive heart failure; 1 withdrew because of residual cutaneous disease; and 2 refused treatment after clinical response) and 2/51 (4%) in conservative‐treatment group (no reasons were stated). |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, and the author requested original data from their former employer, but the data were not available so far. |
| Other bias | Unclear risk | It was unclear if previous treatment was stopped. |
Kim 2018.
| Study characteristics | ||
| Methods | This study is a randomised, controlled, open‐label trial on mogamulizumab versus vorinostat in patients with histologically‐confirmed diagnosis of mycosis fungoides (MF) or Sézary syndrome (SS). | |
| Participants | The study recruited 372 participants (186 in the mogamulizumab group and 186 in the vorinostat group) with histologically‐proven MF or Sezary Syndrome in stages IB ‐ IVB. Inclusion criteria
Exclusion Criteria
|
|
| Interventions |
Arm I
Mogamulizumab 1.0 mg/kg weekly x 4 in cycle 1 then every other week until progression Arm II Vorinostat 400 mg once daily |
|
| Outcomes |
Primary outcome of the trial
Progression‐free survival Secondary outcomes of the trial
|
|
| Notes | Funding was provided by Kyowa Kirin. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Patients were randomised using an interactive voice web response system (IVRS). |
| Allocation concealment (selection bias) | Low risk | Low risk due to IVRS. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was an open‐label study, therefore high risk of performance bias. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Low risk due to blinded assessor of outcome data. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis was carried out (analysis 362 of 362): Out of 372 randomised patients, 2 patients did not receive a single treatment, 1 participant was lost to follow‐up. |
| Selective reporting (reporting bias) | High risk | Initial trial registration contained less secondary outcomes than the final publication. However, several patient‐reported outcomes are missing in this publication, which according to the authors will be published in a secondary article. |
| Other bias | High risk | Distribution of concomitant topical or systematic steroids not reported, might lead to imbalance between treatment arms and overestimation of treatment effect. |
Lessin 2013.
| Study characteristics | ||
| Methods | This was a phase II, randomised, double‐blind, parallel‐group safety/efficacy study. | |
| Participants | This study recruited 260 patients with mycosis fungoides stage IA to IIA, who were randomised to receive a topical chemotherapy (mechlorethamine) in different base formulations. Demographics of the included participants
Exclusion criteria
|
|
| Interventions |
|
|
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
|
| Notes | Dr Lessin serves as a consultant to Ceptaris Therapeutics, Inc. This study was partially supported by the Food and Drug Administration and by Ceptaris. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Since blinding of participants was hardly possible, we judged lack of blinding as an unclear risk. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Tumor response an AEs were assessed [...] blinded to treatment type" Comment: Outcome assessor was blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis was carried out (analysis 260 of 260): Out of 260 randomised patients, 5 patients did not receive a single treatment, 7 participants (2.7%) were lost to follow‐up, 81 dropped out after randomisation due to various reasons. |
| Selective reporting (reporting bias) | High risk | Initial trial registration contained more secondary outcomes than the final publication. We did not receive a reply after contacting the author. |
| Other bias | Low risk | None were found. |
Olsen 2001.
| Study characteristics | ||
| Methods | This was a randomised, parallel‐group trial, which lasted 6 months. | |
| Participants | The study recruited 71 participants (35 in the low‐dose group and 36 in the high‐dose group) with histologically‐proven mycosis fungoides type with ≥ 20% of lymphocytes within the skin biopsy stain positively for CD25 by immunohistochemistry. Further inclusion criteria was as follows: stage Ib‐III CTCL (CTCL Cooperative Group staging) recurred or persisted after ≥ 4 previous treatments for CTCL (excluding topical or systemic corticosteroids) or stage IVa CTCL participants who failed at least 1 previous therapy study consideration, and Eastern Cooperative Oncology Group Performance Status of 0, 1, or 2. Lymph node involvement was no greater than LN₂, and no CTCL involvement of bone marrow. Demographics of the included participants
Exclusion criteria of the trial
|
|
| Interventions |
|
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | Only 42% of all randomised participants received 8 courses of treatment as planned. There was no comparison reported between both treatment groups regarding QoL from baseline to the end of the study (only subgroup analyses of responders vs non‐responders). The funding body was Seragen, Inc (a wholly‐owned subsidiary of Ligand Pharmaceuticals Inc, San Diego, CA). Two of the authors had equity interests in Seragen. This study was conducted in 20 centres across the USA. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was stratified by stage of CTCL for multicentre trial. It was likely to have been carried out by a third party and concealed, although this was not formally stated. |
| Allocation concealment (selection bias) | Unclear risk | Randomisation was stratified by stage of CTCL for multicentre trial. However, it was unclear whether randomisation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was unlikely to be blinded, since the drug was diluted to a certain minimum concentration in both arms and administered by a pump device for 15 to 60 minutes. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All responses were verified by an independent panel of physicians [the Data End Point Review Committee]." Comment: outcome assessment was blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 41/71 (58%) participants dropped out. Discontinuation was due to adverse events (11/35 (31%) participants in the 9 µg/kg/day group vs 15/36 (42%) in the 18 µg/kg/day group) and treatment failure (6/35 (17%) in the 9 µg/kg/day group vs 2/36 (6%) in the 18 µg/kg/day group). |
| Selective reporting (reporting bias) | High risk | The quality of life assessment was compared between responders and non‐responders instead of comparing treatment groups. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | Unclear risk | There were insufficient information to permit judgement. |
Prince 2017.
| Study characteristics | ||
| Methods | This was a randomised, open‐label, phase 3, multicentre study of brentuximab vedotin versus conventional therapy for previous treated patients with CD30‐positive cutaneous T‐cell lymphoma. | |
| Participants | This study recruited 97 patients with mycosis fungoides stage IA to IVB, who were randomised to receive either brentuximab vedotin or physician's choice (MTX or bexarotene). A histologically‐confirmed CD30+ disease by central laboratory assessment and pathology review was required to enrol in this study. Demographics of the included participants
Exclusion criteria
|
|
| Interventions |
|
|
| Outcomes |
Primary outcome of the trial
Secondary outcome of the trial
|
|
| Notes | This study was funded by Millennium Pharmaceuticals Inc. The authors stated several conflicts of interest, for details see original article. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The randomisation list was generated by the Takeda statistician who was not involved in the remainder of the trial." Comment: We judged this to be of low risk of selection bias. |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were randomly assigned (1:1) by an interactive voice and web response system [...]." Comment: We judged this to be of low risk. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | This was an open‐label trial. However, blinding participants and personnel was hardly possible because of different types of interventions, therefore we rated this trial to be of unclear risk, according to our prespecified criteria. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The study did not provide information about this. We sought information and had an email response: "The outcome assessors were blinded to the agent being administered." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis was carried out. |
| Selective reporting (reporting bias) | High risk | We contacted the author about additional outcome data. The author replied that some of the outcomes are not published yet. The author states that they were planning to publish the remaining data. |
| Other bias | Low risk | Slight imbalance in treatment arms (no stage IVB in physician's choice arm). |
Rook 2010.
| Study characteristics | ||
| Methods | This was a randomised, double‐blind (verified by author contact), within‐participant trial, which lasted 6 weeks. | |
| Participants | The study recruited 12 participants (with 1 lesion per treatment): men or non‐pregnant women aged 18 to 70 with stable patch or plaque phase MF of at least 4 months' duration. Demographics of the included participants
Exclusion criteria of the trial These were not reported. |
|
| Interventions |
|
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | The study was supported in part by the USA's Department of Energy Merit Review. Funding came from the Department of Veterans Affairs (Dr Wood) and Vimrx Inc. Disclosure: Dr Rook has been a consultant to Hy BioPharma Inc. This study was conducted in 4 tertiary care centres in the USA. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information and had an email response: "The corresponding author no longer had access to data from the former employer." |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information and had an email response: "The corresponding author no longer had access to data from the former employer." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The study was described in the publication as "double‐blind" and "open‐label". The email response from the author confirmed that the study was double‐blind. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study did not provide information about this. We sought information and had an email response: "The corresponding author no longer had access to data from the former employer." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | There were no dropouts. |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We sought information and had an email response: "The corresponding author no longer had access to data from the former employer." |
| Other bias | Unclear risk | It was unclear if previous treatment was stopped and if concomitant medication was permitted. |
Stadler 1998.
| Study characteristics | ||
| Methods | This was a randomised, open‐label, parallel‐group trial, which lasted 48 weeks. | |
| Participants | The study recruited 82 participants (40 in the IFN‐α + PUVA group and 42 in the IFN‐α + acitretin group) with small‐ to medium‐sized pleomorphic T‐cell lymphoma or mycosis fungoides stage I or II. The principle investigator (Stadler) stated on author contact that all participants had histologically‐proven mycosis fungoides. Demographics of the included participants
Exclusion criteria of the trial These were not reported. |
|
| Interventions |
|
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | Funding body and conflict of interests not declared. This study was conducted in 21 tertiary care centres in Germany, Austria, and Switzerland. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was by a central institution/third party (Estimate GmbH, Augsburg/Germany) and stratified by pretreatment. |
| Allocation concealment (selection bias) | Low risk | Randomisation was by central institution/third party (Estimate GmbH, Augsburg/Germany) and stratified by pretreatment. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | This was not possible because of different interventions (PUVA vs capsules). |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The primary analysis was per‐protocol. ITT analysis was also carried out for comparison between study groups regarding complete remission: 16/98 (16%) participants dropped out (6 participants did not receive any treatment; 6 participants had insufficient data monitored; and 4 participants had wrong staging at enrolment); there was no distribution between groups reported. 40/49 participants in the PUVA group and 42/49 participants in the acitretin group were evaluable. |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | Unclear risk | It was unclear if previous treatment was stopped and if concomitant medication was permitted. |
Stadler 2006.
| Study characteristics | ||
| Methods | This was a randomised parallel‐group trial, which lasted 52 weeks. | |
| Participants | The study recruited 124 participants with cutaneous T‐cell lymphoma (stages lA to IIA) ‐ type mycosis fungoides or small to medium cellular pleomorphic type. The principle investigator (Stadler) stated on author contact that all participants had histologically‐proven mycosis fungoides. Demographics of the included participants
Exclusion criteria of the trial These were not reported. |
|
| Interventions |
|
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | The funding body was not declared. The disclosure in Stadler 2006 stated: "No significant financial relationships to disclose." This study was conducted in 26 tertiary care centres in Germany and Switzerland. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding was unlikely since no placebo injections were described. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 31/124 (25%) randomised participants were not evaluable, and no reasons for this were stated. |
| Selective reporting (reporting bias) | High risk | The only outcome reported was complete remission; there was no report on adverse effects. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | Unclear risk | It was unclear if previous treatment was stopped and if concomitant medication was permitted. |
Thestrup‐Pedersen 1982.
| Study characteristics | ||
| Methods | This was a randomised, double‐blind, parallel‐group trial, which lasted 12 months. | |
| Participants | The study recruited 16 participants (8 in the intervention group and 8 in the control group) with histologically‐proven MF van Scott stage II to IV. Demographics of the included participants
Exclusion criteria of the trial These were not reported. |
|
| Interventions |
|
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | The funding body was Landsforeningen til kraeftens bekaempelse (a grant came from the National Institution for Cancer Prevention of Danish Cancer Society). Conflicts of interest were not declared. This study was conducted in the Department of Dermatology, University of Aarhus, Denmark. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. |
| Allocation concealment (selection bias) | High risk | The corresponding trial author confirmed in an email response that the randomisation list was open. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This was a double‐blind study. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | The corresponding trial author confirmed in an email response that the outcome assessors were blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No losses to follow‐up were reported, but the number of participants randomised was not stated. |
| Selective reporting (reporting bias) | Low risk | This was unknown. We contacted the corresponding author for additional outcome data, and the author responded with a completed data extraction form. |
| Other bias | High risk | Concomitant treatment was permitted. |
Vieyra‐Garcia 2019.
| Study characteristics | ||
| Methods | A multi‐centre, randomised study on oral 8‐methoxypsoralen plus UVA with or without maintenance therapy in Mycosis Fungoides EORTC/ISCL Stage IA to IIA | |
| Participants | This study recruited 27 participants with cutaneous T‐cell lymphoma stages IA‐IIA, who received treatment with oral 8‐methoxypsoralen followed by UV‐A exposure 2 times per week for 12 to 24 weeks until CR. Then, patients with CR were randomised to PUVA maintenance for 9 months (14 total exposures) or no maintenance. Demographics of the included participants
Exclusion criteria of the trial
|
|
| Interventions |
Arm I
PUVA maintenance for 9 months (14 total exposures) after complete response or Arm II No maintenance therapy after complete response |
|
| Outcomes |
Primary outcomes of the trial
Secondary outcomes of the trial
|
|
| Notes | This study was supported by several research grants:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Patients were randomised by a computer‐generated list. |
| Allocation concealment (selection bias) | Low risk | Allocation was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Since blinding of participants was hardly possible, we judged lack of blinding as an unclear risk. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study did not provide information about this. The authors stated that blinding was hardly possible due to tanning of the skin. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis was carried out. There was one dropout due to adverse effects. However, this occurred before randomisation. Seven out of 27 participants were excluded because they did not reach CR, which was a prerequisite for randomisation. |
| Selective reporting (reporting bias) | High risk | Authors initially planned to report several secondary outcomes such as quality of life or the hospital anxiety depression score. However, these were not reported in the final publication. |
| Other bias | High risk | The calculated sample size for statistical significance was not met (82 participants and an assumed 10% dropout rate) |
Vonderheid 1987.
| Study characteristics | ||
| Methods | This was a randomised, double‐blind, within‐participant trial, which lasted 4 weeks. | |
| Participants | The study recruited 6 participants (2 lesions per treatment) with plaque phase MF, MFCG stage nomenclature of 1979 stage IA (T1, Nx, T0, M0), stage IB (T2, Nx, T0, M0), or stage IIA (T2, N1, T0, M0) Demographics of the included participants
Exclusion criteria of the trial
|
|
| Interventions |
|
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | The funding body and conflicts of interest were not declared. This study was conducted in a tertiary care centre in the USA. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Lesions were allocated by a random code; no further information was given; information was sought, but we received no response. |
| Allocation concealment (selection bias) | Unclear risk | Information was sought, but we received no response. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | The trial was described as double‐blind for the first part, which we data extracted. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No separate outcome assessor was described. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis was carried out. There were no dropouts. |
| Selective reporting (reporting bias) | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | Unclear risk | There were insufficient information to permit judgement. |
Whittaker 2012.
| Study characteristics | ||
| Methods | This was a randomised, open‐label, multicentre study which lasted 16 weeks. | |
| Participants | This study recruited 93 patients with mycosis fungoides stage IB to IIA, who were randomised to receive bexarotene in combination with PUVA vs. PUVA alone. A histologically‐confirmed mycosis fungoides stage IB or IIA, confirmed by current or prior diagnostic lesion biopsy, was required to enrol in this study. Demographics of the included participants
Exclusion criteria of the trial
|
|
| Interventions |
|
|
| Outcomes |
Primary outcomes of the trial
Secondary outcomes of the trial
|
|
| Notes | This study was funded by educational grants from Ligand Pharmaceuticals Inc./Eisai Co., Ltd. and by a donation from Cancer Research
U.K. through the EORTC Charitable Trust. The authors stated no conflicts of interest. This study was conducted in tertiary care hospitals in 11 participating countries. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The authors used a minimisation method for randomising patients into treatment arms. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information but got no response. We judged this to be of unclear risk. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Study personnel were not blinded to study groups. |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | This was unknown. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis was carried out. 6 of 93 patients did not start treatment after randomisation. For 5 of those 6, reasons for not starting therapy were not stated. The other patient was ineligible because of prior treatment. Additionally 1 patient lost to follow‐up and 1 drop out for "other" reasons. |
| Selective reporting (reporting bias) | Low risk | Although the primary outcome measure was changed during the conduct of the trial, the authors clearly stated the reason for this change (low accrual and overestimation of CR). The primary end point was changed from cumulative dose of UVA necessary to achieve a CR to cumulative dose of UVA to achieve an ORR. |
| Other bias | High risk | The primary end point was changed after realising that the a priori expected rate of complete responses was overestimated in trial design. |
Wolff 1985.
| Study characteristics | ||
| Methods | This was a randomised, double‐blind, parallel‐group trial, which lasted 8 weeks. | |
| Participants | The study recruited 12 participants (9 from the intervention group and 3 from the control group) with early plaque or patch stage MF (stage IA or IB), with no evidence of physical examination on lymphadenopathy or organomegaly. Demographics of the included participants
Exclusion criteria of the trial
|
|
| Interventions |
a) IFN‐α 2MU in superficial dermis 3 times weekly; b) betamethasone dipropionate ointment 0.05% twice daily; or c) no treatment.
a) placebo (buffered glycine serum human albumin) in superficial dermis 3 times weekly; b) betamethasone dipropionate ointment 0.05% twice daily; or c) no treatment. |
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | Objective response was reported as mean difference in size of lesions without the possibility to identify participants' objective response rate according to our defined secondary outcome. Lesions in the IFN‐α group generally improved better than in the placebo group, possibly due to a systemic effect of IFN‐α as discussed by the authors. The IFN‐α was supplied by Schering Corp. This study was conducted in a tertiary care centre in Pittsburgh, USA. Conflicts of interest were not declared. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information, but received no response. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information, but received no response. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | This trial had a double‐blind setting. |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Histopathological features of biopsies were assessed without knowledge of the treatment or group. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis was carried out. There were no dropouts. |
| Selective reporting (reporting bias) | High risk | Only the mean difference in the decrease of the lesions were reported; no incidence of partial remission (i.e. > 50% reduction of disease) was reported. The corresponding author was contacted for additional outcome data but did not respond within 4 weeks. |
| Other bias | High risk | The groups were unequal: There was a higher proportion of stage 1B, more men, longer duration of skin disease, and longer time since diagnosis in intervention group. |
Wozniak 2008.
| Study characteristics | ||
| Methods | This was a randomised, open‐label, parallel‐group trial, which lasted 24 weeks. | |
| Participants | The study recruited 29 participants (12 in the intervention group and 17 in the control group) with mycosis fungoides stage IA to IIA. Demographics of the included participants
Exclusion criteria of the trial
|
|
| Interventions |
|
|
| Outcomes |
Outcomes of the trial
|
|
| Notes | The funding body was Ministerio de Ciencia y Tecnologia (BIO2000‐0275‐C02 ⁄01‐⁄02, SAF2001‐0060, SAF2005‐00221), Comunidad Autonoma de Madrid (CAM 08.1 ⁄0011 ⁄2001.1), and the Ministerio de Sanidad y Consumo (FISP05 ⁄1710, FIS 01‐0035, G03 ⁄179, PI051623) RETICS, Spain. The author, MBW, was supported by FISP05 ⁄1710, and LT was supported by grants from the CNIO and the Higher Education Authority of Ireland, St James Hospital, Dublin. Participants were categorised to responders and non‐responders instead of treatment groups. Some information was taken from the clinicaltrials.gov website (NCT00630903). The main primary aim of the study was to examine the gene expression profiles of primary skin biopsies from these participants. This study was conducted in 9 tertiary care hospitals in Madrid, Spain. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | The study did not provide information about this. We sought information, but received no response. |
| Allocation concealment (selection bias) | Unclear risk | The study did not provide information about this. We sought information, but received no response. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | This was taken from the previous version of the NCT00630903 protocol: "The study was described as open‐label." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | The study did not provide information about this. We sought information, but received no response. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analysis was carried out. There were no dropouts. |
| Selective reporting (reporting bias) | High risk | Participants were characterised and divided into responders and non‐responders instead of treatment groups. We contacted the corresponding author for additional outcome data, but we received no reply within 4 weeks. |
| Other bias | High risk | Some information was taken from protocol NCT00630903 (www.clinicaltrials.gov). The study was described as terminated due to insufficient accrual. |
AEs: adverse effects; CR: complete response; CTCL: cutaneous T‐cell lymphomas; ITT: intention‐to‐treat; LCT: large cell trnsformation; MF: Mycosis fungoides; MFCG: Mycosis Fungoides Cooperative Group; mSWAT: modified severity weighted assessment tool; PR: partial response; PUVA: psoralen plus ultraviolet A; QoL: quality of life;SS: Sézary syndrome.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Anonymous 1982 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. |
| Anonymous 2000 | This study was not a randomised controlled trial. |
| Argyropoulos 1979 | There was no relevant end point according to the protocol report. |
| Aviles 2015 | This study was not a randomised controlled trial. |
| Bazex 1975 | This study was not a randomised controlled trial. |
| Breneman 1991 | There was no relevant end point according to the protocol report. |
| Cooper 1994 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. |
| Currie 1980 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. |
| Dang 2007 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. |
| Doan 1958 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. |
| Dueck 2010 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. |
| Duvic 2010 | This study was not a randomised controlled trial. |
| Fawzi 2010 | There was not enough information to confirm inclusion criteria and we had no reaction when we attempted to contact the corresponding author. |
| Fisher 1993 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. |
| Foss 2011 | This study was not a randomised controlled trial. |
| Groth 1979 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. |
| Heald 2003 | This study was not a randomised controlled trial. |
| JapicCTI‐050041 | There was not enough information to confirm inclusion criteria and we had no reply when we attempted to contact the corresponding author. |
| Kaung 1969 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. |
| Kujawska 2003 | There was not enough information to confirm inclusion criteria and we had no reply when we attempted to contact the corresponding author. |
| Kuzel 2010 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. |
| Lambert 1986 | This study was not a randomised controlled trial. |
| Lansigan 2010 | There was not enough information to abstract data from the publication. |
| Loescher 1984 | This study was not a randomised controlled trial. |
| Marsden 1968 | This study was not a randomised controlled trial. |
| Moog 2008 | This study was not a randomised controlled trial. |
| NCT00054171 | An email response confirmed that the study was not completed. (The Principal Investigator passed away). |
| NCT00091208 | This study was not a randomised controlled trial. |
| NCT01007448 | There was not enough information to confirm inclusion criteria and we had no reaction when we attempted to contact the corresponding author. |
| NCT01187446 | Study terminated due to "business decision". We contacted the corresponding author for results but did not receive a reply. |
| NCT01386398 | This study was withdrawn prior to enrolment according to www.clinicaltrials.gov. |
| NCT01625455 | Study terminated due to difficult recruiting according to www.clinicaltrials.gov. |
| Neering 1972 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. |
| Negro‐Vilar 2007 | There was not enough information to confirm inclusion criteria and we had no reply when we attempted to contact the corresponding author. |
| O'Neill 2013 | This study was not a randomised controlled trial. |
| Olsen 1986 | This study was not a randomised controlled trial. |
| Pan 2007 | There was not enough information to confirm inclusion criteria and we had no reply when we attempted to contact the corresponding author. |
| Peugeot 1995 | This was scientific fraud (see Grant 2009). |
| Plettenberg 2001 | This was a report of an ongoing trial; there was not enough information to confirm inclusion criteria and we had no reply when we attempted to contact the corresponding author. |
| Prince 2010 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. |
| Schrag 1997 | There was no relevant end point according to the protocol report. |
| Serri 1990 | This study was not a randomised controlled trial. |
| Shi 2015 | This study was not a randomised controlled trial. |
| Simon 2010 | This study explicitly excluded MF. |
| Thomsen 1977 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. This was identified by Molin 1979 and retrieved as a reference in the adverse event search. |
| Thomsen 1979 | This study was not a randomised controlled trial. |
| Thomsen 1989 | This study was not a randomised controlled trial. |
| Touraine 1978 | This study was not a randomised controlled trial. |
| Wain 2005 | There was not enough information to confirm inclusion criteria and we had no reply when we attempted to contact the corresponding author. |
| Wiernik 1998 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. |
| Wilson 1995 | This study was not a randomised controlled trial. |
| Zubrod 1960 | Less than 90% of enrolled participants had Alibert‐Bazin type MF and no subgroup analysis was available. |
Characteristics of studies awaiting classification [ordered by study ID]
Bashey 2014.
| Methods | This is a multicentre, open‐label, randomised, phase I/II study evaluating the safety and efficacy of low‐dose (12 Gy) total skin electron beam therapy (TSEBT) combined with vorinostat versus low‐dose TSEBT monotherapy in patients with mycosis fungoides. |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Arm I:
Arm II:
|
| Outcomes |
Primary outcome
Secondary outcome
|
| Notes | Additional information was sought but we received no answer. |
Kim 2014.
| Methods | This is a phase 1b multicentre, double‐blind, placebo‐controlled, randomised trial in stage IA‐IIA CTCL to assess safety, pharmacokinetic, pharmacodynamic, and preliminary efficacy with SHP‐141 applied twice daily to index lesions (maximum 5% of body surface area) for 28 days. |
| Participants |
Inclusion criteria
Exclusion Criteria
|
| Interventions | This study had 4 treatment arms.
|
| Outcomes |
Outcomes of the trial
|
| Notes | This was a meeting abstract. Additional information was sought but we received no response. This study is NCT01433731. |
CTCL: cutaneous T‐cell lymphomas.
Characteristics of ongoing studies [ordered by study ID]
NCT01738594.
| Study name | A randomized phase I dose‐escalation trial of carfilzomib with and without romidepsin in cutaneous T‐cell lymphoma |
| Methods | This is a randomised, controlled trial on carfilzomib with and without romidepsin in patients with cutaneous T‐cell lymphoma |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Arm I
Arm II
|
| Outcomes |
Primary outcomes of the trial
Secondary outcomes of the trial
|
| Starting date | January 2013 |
| Contact information |
Sponsors and collaborators
Investigators
|
| Notes | ‐ |
NCT02213861.
| Study name | A randomized phase 2 study to evaluate three treatment regimens of SHAPE, a histone deacetylase inhibitor, in patients with stage IA, IB or IIA cutaneous T‐cell lymphoma |
| Methods | This is a randomised, controlled trial comparing three treatment regimens of SHAPE, a histone deacetylase inhibitor, in patients with mycosis fungoides |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Arm I
Arm II
Arm III
|
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Starting date | November 2014 |
| Contact information |
Sponsors and collaborators
|
| Notes | ‐ |
NCT02301494.
| Study name | Feasibility study to determine effectiveness of 3.75% topical imiquimod cream and topical Vanos (fluocinonide) cream 0.1% in the treatment of early stage cutaneous T‐cell lymphoma |
| Methods | This is a randomised, controlled trial comparing topical 3.75% Imiquimod cream vs. 0.1% Fluocinonide cream in patients with mycosis fungoides |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Arm I
Arm II
|
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Starting date | November 2014 |
| Contact information |
Sponsors and Collaborators
Investigators
|
| Notes | ‐ |
NCT02323659.
| Study name | Comparison of methotrexate versus interferon‐alfa 2b on efficacy, safety and quality of life in patients with primary cutaneous T‐cell lymphomas |
| Methods | This is a randomised, controlled, open‐label trial comparing methotrexate vs. interferon alfa‐2b on efficacy, safety and quality of life in patients with primary cutaneous T‐cell lymphomas after failure of topical or phototherapy treatment |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Arm I
Arm II
|
| Outcomes |
Primary outcome of the trial
Secondary outcomes of the trial
|
| Starting date | June 2014 |
| Contact information |
Sponsors and collaborators
Investigators
|
| Notes | ‐ |
NCT02448381.
| Study name | A phase 3 multicenter, randomised, double‐blind, placebo controlled study to determine the efficacy of topical SGX301 (synthetic hypericin) and fluorescent Bulb‐Light Irradiation for the treatment of cutaneous T‐cell lymphoma |
| Methods | This study is a randomised, controlled trial comparing topical SGC301, a topical photosensitising agent, vs. placebo to treat patients with patch/plaque phase cutaneous T‐cell lymphoma (mycosis fungoides) |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Arm I
Arm II
|
| Outcomes |
Primary outcomes of the trial
Secondary outcomes of the trial
|
| Starting date | December 2015 |
| Contact information |
Sponsors and collaborators
|
| Notes | ‐ |
NCT02811783.
| Study name | A double blind randomized vehicle controlled crossover study to evaluate the safety and efficacy of topical Naloxone Hydrochloride Lotion 0.5% for the relief of pruritus in patients With the MF Form of Cutaneous T‐Cell Lymphoma (CTCL) |
| Methods | This is a randomised, controlled trial comparing naloxone hydrochloride lotion vs. placebo for the relief of pruritus in patients with CTCL |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Arm I
Arm II
|
| Outcomes |
Primary outcome of the trial:
Secondary outcomes of the trial
|
| Starting date | January 2017 |
| Contact information |
Sponsors and collaborators
Investigators
|
| Notes | ‐ |
NCT02943642.
| Study name | Safety and effectiveness of A‐dmDT390‐bisFv(UCHT1) fusion protein (Resimmune®) in subjects With Mycosis Fungoides: a phase II multi‐center randomized clinical trial |
| Methods | This is a randomised, controlled trial comparing A‐dmDT390‐bisFv(UCHT1) vs Vorinostat in patients with mycosis fungoides |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Arm I
Arm II
|
| Outcomes |
Primary outcomes of the study
Secondary outcomes of the study
|
| Starting date | January 2017 |
| Contact information |
Sponsors and collaborators
|
| Notes | ‐ |
NCT02953301.
| Study name | A multicentre, double blind, randomised, placebo‐controlled, Phase II trial to evaluate resminostat for maintenance treatment of patients with advanced stage (Stage IIB‐IVB) Mycosis Fungoides (MF) or Sézary Syndrome (SS) that have achieved disease control with systemic Tterapy ‐ the RESMAIN study |
| Methods | This is a randomised, controlled trial comparing resminostat vs. placebo in patients with advanced stage mycosis fungoides (MF) or Sézary syndrome (SS) |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Arm I
Arm II
|
| Outcomes |
Primary outcomes of the study
Secondary outcomes of the study
|
| Starting date | November 2016 |
| Contact information |
Sponsors and collaborators
Investigators
|
| Notes | ‐ |
NCT03011814.
| Study name | A Phase 1/2 Trial of durvalumab (MEDI4736) when given as a single agent or in combination With lenalidomide in patients with relapsed/ refractory peripheral T‐cell lymphoma, including cutaneous T‐cell lymphoma |
| Methods | This is a randomised, controlled, open‐label trial on durvalumab (MEDI4736) with or without lenalidomide in patients with cutaneous or peripheral T‐cell lymphoma |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions |
Arm I
Arm II
|
| Outcomes |
Primary outcomes of the trial
Secondary outcomes of the trial
|
| Starting date | February 2017 |
| Contact information |
Sponsors and collaborators
Investigators
|
| Notes | ‐ |
NCT03292406.
| Study name | A safety, efficacy and pharmacokinetics study of CD11301 for the treatment of Cutaneous T‐Cell Lymphoma (CTCL) |
| Methods | This is a randomised, controlled trial comparing topical treatment with CD11301 or placebo in patients with mycosis fungoides |
| Participants |
Inclusion Criteria
Exclusion Criteria
|
| Interventions |
Arm I
Arm II
Arm III
|
| Outcomes |
Primary outcomes of the study
Secondary outcomes of the study
|
| Starting date | December 2017 |
| Contact information |
Sponsors and Collaborators
Investigators
|
| Notes | ‐ |
NCT03454945.
| Study name | Efficacy of doxycycline in the treatment of early stages of mycosis fungoides: a randomizedcControlled yrial |
| Methods | This is a randomised controlled trial comparing vibramycin vs. UVA + psoralen in patients with mycosis fungoides. |
| Participants |
Inclusion Criteria
Exclusion Criteria
|
| Interventions |
Arm I
Oral vibramycin antibiotic100 mg capsule every 12 hours for 3 months Arm II UVA + psoralen 3 sessions per week for 3 months |
| Outcomes |
Primary outcomes of the study
Secondary outcomes of the study
|
| Starting date | 2017 |
| Contact information |
Sponsors and collaborators
None Investigators Principal Investigator: Hagar El Sayed |
| Notes | ‐ |
NCT03713320.
| Study name | SOLAR: A Phase 2, randomized, open‐label, parallel‐group, active comparator, ,ulti‐center study to investigate the efficacy and aafety of cobomarsen (MRG‐106) in subjects with cutaneous T‐cell lymphoma (CTCL), Mycosis Fungoides (MF) Subtype |
| Methods | This is a randomised controlled trial comparing cobomarsen vs. vorinostat in patients with mycosis fungoides. |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions | Arm I At least weekly doses of cobomarsen throughout study treatment period. Arm II Daily doses of vorinostat throughout study treatment period. |
| Outcomes |
Primary outcomes of the study Proportion of participants achieving an objective response of at least 4 months duration (ORR4) based on composite global response criteria including radiological imaging, flow cytometry, and the modified Severity Weighted Assessment Tool (mSWAT). Secondary outcomes of the study Progression‐free survival Pruritus Numerical Rating Scale Skindex‐29 Dermatological Survey Pain Numerical Rating Scale Difference in drug tolerability by weekly patient impression of treatment side effects Duration of composite global response for responding subjects Complete response rate Skin disease severity based on modified Severity‐weighted Assessment Tool (mSWAT) Time to progression Overall survival Number of participants with treatment‐related adverse events as assessed by CTCAE v5.0 Plasma concentration of cobomarsen |
| Starting date | 2018 |
| Contact information |
Sponsors and collaborators
miRagen Therapeutics, Inc. Investigators Principal Investigator: Christiane Querfeld |
| Notes | ‐ |
UMIN000029537.
| Study name | Efficacy and safety of Targretin capsule 75‐mg alone or in combination with phototherapy in Japanese patients with cutaneous T‐cell lymphomas |
| Methods | This is a randomised controlled trial comparing bexarotene alone vs. bexarotene plus phototherapy for cutaneous T‐cell lymphoma (CTCL) patients. |
| Participants |
Inclusion criteria
Exclusion criteria
|
| Interventions | Arm I Patients receive are administrated a 300 mg/m2 dose of bexarotene orally once daily for 8 weeks. Arm II Patients are administrated a 300 mg/m2 dose of bexarotene orally once daily for 8 weeks. Patients are treated with psoralen baths preceding treatment with UVA radiation 5 times weekly. The initial dose of UVA was 0.5 J/cm2, dose increment of 0.5 J/cm2 each radiation. The maximum dose was 4.0 J/cm2. The initial dose of narrowband UVB administered is 50% to 70% of the MPD or 0.5‐0.7 J/cm2. The dose of NB‐UVB for the subsequent NB‐UVB sessions is elevated 20% increments with each successive treatment session. The maximum dose is 2.0 J/cm2. |
| Outcomes |
Primary outcomes of the study
The primary efficacy end points evaluated though the 8 weeks of treatment were follows: Modified Severity‐weighted Assessment Tool (mSWAT), Physician's Global Assessment (PGA). Secondary outcomes of the study Efficacy: time to cutaneous tumour response, time to cutaneous tumour progression, amount of irradiation and UV dose, amount of bexarotene, capsules, and compliance rate, LDH, sIL‐2R, TARC, T‐cell receptor repertoire analysis Safety: adverse events, haematology, blood chemistry |
| Starting date | 2017 |
| Contact information |
Sponsors and collaborators
Minophagen Pharmaceutical Co., Ltd. Investigators Principal Investigator: Akimichi Morita |
| Notes | ‐ |
Differences between protocol and review
We added an assessment of external validity as suggested by an external peer review board of the German Federal Ministry of Education and Research.
For the search of clinical trials databases, we focused on the two most commonly used aggregator databases: ClinicalTrials.gov and the World Health Organization International Clinical Trials Registry Platform (ICTRP). This is in contrast to the published protocol, where we stated we would search:
The meta Register of Controlled Trials (www.controlledtrials.com)
The US National Institutes of Health Ongoing Trials Register (www.clinicaltrials.gov)
The Australian New Zealand Clinical Trials Registry (www.anzctr.org.au)
The World Health Organization International Clinical Trials Registry (www.who.int/trialsearch)
The Register of the European Organization of Research and Treatment of Cancer (www.eortc.be)
Trials Central (www.trialscentral.org/index).
Since the Cochrane Skin Group Ongoing Trials Register no longer exists, we were not able to search that resource for this update.
We replaced survival data from van Doorn 2000 with data from the more recently‐published study, Agar 2010.
In order to gain more accurate analyses for smaller sample sizes, we added Fisher tests, which is a deviation from the review protocol. This change was made in order to avoid spurious (non‐)significance in studies with small sample sizes or low numbers of events. P values ≤ 0.05 were considered to be significant. In response to a letter sent to the author team by Peggy Wu (Beth Israel Deaconess Medical Center), we would like to highlight the issue that in nine instances the Fisher test showed significant results, whereas the 95% confidence interval (CI) computed by the Cochrane standard software indicated no significant difference for the relative risk. For these instances we additionally computed the 95% CIs by the use of the method described by Miettinen 1985 (CI‐M), which are closer to the exact confidence intervals and provided the results together with the standard CI.
We added the section Unit of analysis issues.
We made minor changes and clarifications in the 'Measures of treatment effects' and 'Data synthesis' sections to allow for the evolution of methods since the protocol and first version of this review were published.
The application of the rule of three by Hanley 1983 was removed, since it is not applicable for missing data.
Asssessment of reporting bias, subgroup analysis, and investigation of heterogeneity as well as sensitivity analyses were not performed because of the low number of comparable trials.
We added "Summary of findings tables" and implemented GRADE (GRADE pro GDT 2015) according to the methodological expectations of Cochrane Intervention Reviews (MECIR).
In contrast to the protocol we included studies with different investigated diseases (e.g. mycosis fungoides and other lymphomas) but separate outcome data for the mycosis fungoides cohort meeting our inclusion criteria had to be available.
The protocol for the initial review was designed and published before the publication of the consensus statement by Olsen 2011. Accepting the importance of this consensus statement and considering the need for standardised end points to gain comparability, slight changes of nomenclature and definition of our outcomes were necessary. Therefore, we adjusted our end points to the outcomes proposed by Olsen 2011. More particularly, clearance and improvement have been changed to complete response and objective response rate. Survival rate and disease‐free interval have been changed to overall survival and disease‐free survival. Only two of 14 of the initially included studies had to be updated according to the new definition of outcomes (Child 2004; Stadler 1998).
For our primary outcome 1: Improvement in health‐related quality of life as defined by participant questionnaires we added that the questionnaires were "all self‐completed".
For "Types of studies", we clarified that we would only include non‐RCT evidence for our outcome of "rare adverse effects".
Contributions of authors
AV was the contact person with the editorial base and co‐ordinated contributions from the co‐authors.
MJ and AV wrote the final draft of the review, TW, PW, JS and RS revised it. MJ and AV screened papers against eligibility criteria. MJ and AV obtained data on ongoing and unpublished studies. MJ, AV and TW appraised the quality of papers. MJ, AV and PW extracted data for the review and sought additional information about papers. MJ and AV entered data into RevMan. MJ and AV entered data into Grade. MJ and AV analysed and interpreted data. MJ, AV and TW worked on the methods sections. TW drafted the initial clinical sections of the background, which were updated by AV and MJ. AV, MJ and TW responded to the clinical comments of the referees. AV and TW responded to the methodology and statistics comments of the referees. CB was the consumer co‐author and checked the review for readability and clarity, as well as ensuring outcomes are relevant to consumers. MJ, AV and PW are the guarantors of the update.
Disclaimer
This project was supported by the National Institute for Health Research, via Cochrane Infrastructure funding to the Cochrane Skin Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Sources of support
Internal sources
Evidence‐based Medicine Working Group, Institute for General Practice, Goethe‐University Frankfurt, Germany
External sources
German Federal Ministry of Education and Research (FKZ: 01KG1011), Germany
-
The National Institute for Health Research (NIHR), UK
The NIHR, UK, is the largest single funder of the Cochrane Skin Group.
Declarations of interest
Arash Valipour: I received travel expenses and accommodation for the annual German Dermatology Society Meeting 2019 from UCB Pharma, but this company is not involved in the development of drugs for mycosis fungoides. I attended a meeting called 'Inflammation campus' (a dermatology/rheumatology‐oriented medical meeting) in July 2015, which was organised by Pfizer. Pfizer paid for my travel expenses and accommodation. Manuel Jäger: nothing to declare. Peggy Wu: nothing to declare. Jochen Schmitt: I acted as a consultant for Lilly and Sanofi and received institutional grants for investigator‐initiated trials outside the submitted work from Novartis, Sanofi, ALK, and Pfizer. Charles Bunch: nothing to declare. Tobias Weberschock: nothing to declare.
Clinical referee, Prof Sean Whittaker: "I led and co‐authored the updated U.K. Cutaneous Lymphoma Group guidelines for the management of primary cutaneous lymphomas, which were published in 2018 (Gilson 2019)."
These authors contributed equally to this work
These authors contributed equally to this work
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Bagot 2017 {published and unpublished data}
- Bagot M, Hasan B, Whittaker S, Beylot-Barry M, Knobler R, Shah E, et al. A phase III study of lenalidomide maintenance after debulking therapy in patients with advanced cutaneous T-cell lymphoma - EORTC 21081 (NCT01098656): results and lessons learned for future trial designs. European Journal of Dermatology 2017;27(3):286–94. [CENTRAL: CN-01395486] [PMID: ] [DOI] [PubMed] [Google Scholar]
- NCT01098656. A phase III study of lenalidomide maintenance after debulking therapy in patients with advanced cutaneous T-Cell lymphoma. clinicaltrials.gov/ct2/show/NCT01098656 (first received 5 April 2010). [NCT01098656]
Child 2004 {published data only (unpublished sought but not used)}
- Child FJ, Mitchell TJ, Whittaker SJ, Scarisbrick JJ, Seed PT, Russel-Jones R. A randomized cross-over study to compare PUVA and extracorporeal photopheresis in the treatment of plaque stage (T2) mycosis fungoides. Clinical & Experimental Dermatology 2004;29(3):231-6. [CENTRAL: CN-00468455] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Child FJ, Mitchell TJ, Whittaker SJ, Watkins P, Seed P, Russell-Jones R. A randomised cross-over study to compare Puva and extracorporeal photopheresis (ECP) in the treatment of plaque stage (T2) mycosis fungoides. British Journal of Dermatology 2001;145(Suppl 59):16. [CENTRAL: CN-00430854] [DOI] [PubMed] [Google Scholar]
Chong 2004 {published data only (unpublished sought but not used)}
- Chong A, Loo WJ, Banney L, Grant JW, Norris PG. Imiquimod 5% cream in the treatment of mycosis fungoides - a pilot study. Journal of Dermatological Treatment 2004;15(2):118-9. [CENTRAL: CN-00467258] [PMID: ] [DOI] [PubMed] [Google Scholar]
Duvic 2001 {published data only}
- Duvic M, Martin AG, Kim Y, Olsen E, Wood GS, Crowley CA, et al. Phase 2 and 3 clinical trial of oral bexarotene (Targretin capsules) for the treatment of refractory or persistent early-stage cutaneous T-cell lymphoma. Archives of Dermatology 2001;137(5):581-93. [CENTRAL: CN-00347816] [PMID: ] [PubMed] [Google Scholar]
- Prince HM, McCormack C, Ryan G, Baker C, Rotstein H, Davison J, et al. Bexarotene capsules and gel for previously treated patients with cutaneous T-cell lymphoma: results of the Australian patients treated on phase II trials. Australasian Journal of Dermatology 2001;42(2):91-7. [CENTRAL: CN-00442169] [PMID: ] [DOI] [PubMed] [Google Scholar]
Duvic 2001a {published data only}
- Duvic M, Olsen EA, Omura GA, Maize JC, Vonderheid EC, et al. A phase III, randomized, double-blind, placebo-controlled study of peldesine (BCX-34) cream as topical therapy for cutaneous T-cell lymphoma. Journal of the American Academy of Dermatology 2001;44(6):940-7. [CENTRAL: CN-00348073] [PMID: ] [DOI] [PubMed] [Google Scholar]
Guitart 2002 {published and unpublished data}
- Guitart J, Tucker R, Stevens V. Low dose bexarotene (Targretin®) capsules and phototherapy for early stage cutaneous T-Cell lymphoma. Journal of Investigative Dermatology 2002;119(1):241. [CENTRAL: CN-00794450] [Google Scholar]
- NCT00030589. A muliticenter, dose-randomized evaluation of Targretin capsules plus PUVA in patients with stage IB - IIA cutaneous T-Cell lymphoma. clinicaltrials.gov/ct/show/NCT00030589 (first received 27 Jaunry 2003).
Kaye 1989 {published data only}
- Kaye F, Eddy J, Ihde DC, Steinberg S, Fischmann AB, Glatstein E, et al. Conservative vs. aggressive therapy in mycosis fungoides. Proceedings of the American Society of Clinical Oncology 1989;8:257. [CENTRAL: CN-00694960] [Google Scholar]
- Kaye F, Ihde D, Fischmann A, Glatstein E, Minna J, Schechter G, et al. A randomized trial comparing conservative and aggressive therapy in mycosis fungoides (MF). Proceedings of the American Society of Clinical Oncology 1986;5:195. [CENTRAL: CN-00695166] [Google Scholar]
- Kaye FJ, Bunn PA, Steinberg SM, Stocker JL, Ihde DC, Fischmann AB, et al. A randomized trial comparing combination electron-beam radiation and chemotherapy with topical therapy in the initial treatment of mycosis fungoides. New England Journal of Medicine 1989;321(26):1784-90. [CENTRAL: CN-00064211] [PMID: ] [DOI] [PubMed] [Google Scholar]
Kim 2018 {published data only}
- Bagot M, Dalle S, Sokol L, Tsianakas A, Musiek A, Ortiz-Romero P, et al. Long-term clinical benefit to anti-CCR4 mogamulizumab: results from the phase 3 mavoric study in previously treated cutaneous t-cell lymphoma (CTCL). Blood 2018;132(Supp 1):2901. [DOI: 10.1182/blood-2018-99-118473] [DOI] [Google Scholar]
- Cowan R, Scarisbrick J, Dwyer K, Leoni M, Grebennik D, Morris S. Mogamulizumab demonstrates significant improvement in PFS compared to vorinostat in patients with previously treated cutaneous T-cell lymphoma (CTCL). British Journal of Haematology 2018;181(S1):77–8. [DOI: 10.1111/bjh.15226] [DOI] [Google Scholar]
- Kim YH, Bagot M, Pinter-Brown L, Rook AH, Porcu P, Horwitz SM, et al. Mogamulizumab versus vorinostat in previously treated cutaneous T-cell lymphoma (MAVORIC): an international, open-label, randomised, controlled phase 3 trial. Lancet Oncology 2018;19(9):1192-1204. [PMID: ] [DOI] [PubMed] [Google Scholar]
- NCT01728805. Study of KW-0761 versus vorinostat in relapsed/refractory CTCL. clinicaltrials.gov/ct2/show/NCT01728805 (first received 20 November 2012).
- Porcu P, Hudgens S, Quaglino P, Cowan R, Floden L, Leoni M, et al. Quality of life in cutaneous T-cell lymphoma subjects treated with anti-CCR4 monoclonal antibody mogamulizumab versus vorinostat: results from the phase 3 MAVORIC trial. Journal of Clinical Oncology 2018;36(15):-. [DOI: 10.1097/HS9.0000000000000060] [DOI] [Google Scholar]
- Quaglino P, Hudgens S, Porcu P, Cowan R, Floden L, Tsianakas A, et al. Quality of life in cutaneous t-cell lymphoma subjects treated with the anti-ccr4 monoclonal antibody mogamulizumab versus vorinostat: results from the phase 3 mavoric trial. Hemasphere 2018;2(S1):85. [Google Scholar]
- Scarisbrick J, Zinzani PL, Horwitz S, Kim YH, Moskowitz AJ, Porcu P, et al. Efficacy of mogamulizumab by prior systemic therapy in patients with previously treated cutaneous T-cell lymphoma: Post hoc analysis from the phase 3 MAVORIC study. British Journal of Haematology 2019;185(Suppl 1):94. [Google Scholar]
- Tsianakas A, Quaglino P, Hudgens S, Floden L, Leoni M, Dale S, et al. Quality of life in cutaneous T-cell lymphoma subjects treated with the anti-CCR4 monoclonal antibody mogamulizumab versus vorinostat: Results from the phase 3 MAVORIC trial. Oncology Research and Treatment 2018;41(Suppl 4):272. [Google Scholar]
- Zinzani PL, Horwitz SM, Kim YH, Moskowitz AJ, Porcu P, Scarisbrick J, et al. Efficacy of mogamulizumab by prior systemic therapy in patients with previously treated cutaneous T-cell lymphoma: post hoc analysis from the phase 3 mavoric study. Blood 2018;132(Suppl 1):1619. [DOI: 10.1182/blood-2018-99-114819] [DOI] [Google Scholar]
Lessin 2013 {published data only}
- Lessin S, Duvic M, Guitart J, Pandya A, Strober B, Olsen E, et al. Positive results of a pivotal phase ii trial testing a manufactured 0.02% Mechlorethamine gel in mycosis fungoides (mf). Journal of Investigative Dermatology 2011;131(Suppl 1):S84. [CENTRAL: CN-00843792] [Google Scholar]
- Lessin SR, Duvic M, Guitart J, Pandya AG, Strober BE, Olsen EA, et al. Topical chemotherapy in cutaneous T-cell lymphoma: positive results of a randomized, controlled, multicenter trial testing the efficacy and safety of a novel mechlorethamine, 0.02%, gel in mycosis fungoides. JAMA Dermatology 2013;149(1):25-32. [CENTRAL: CN-00859487] [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCT00168064. Safety and efficacy of nitrogen mustard in treatment of mycosis fungoides. clinicaltrials.gov/ct2/show/NCT00168064 (first received 14 September 2005).
Olsen 2001 {published and unpublished data}
- Duvic M, Kuzel TM, Olsen EA, Martin AG, Foss FM, Kim YH, et al. Quality-of-life improvements in cutaneous T-cell lymphoma patients treated with denileukin diftitox (ONTAK). Clinical Lymphoma 2002;2(4):222-8. [CENTRAL: CN-00514479] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Kuzel T, Olsen E, Martin A, Kim Y, Duvic M, Frankel A, et al. Pivotal phase III trial of two dose levels of DAB389IL-2 (Ontak®) for the treatment of mycosis fungoides (MF). Blood 1997;90(10 Suppl 1 (Pt 1)):586a. [CENTRAL: CN-00518069] [Google Scholar]
- NTC00050999. Study of ONTAK (Denileukin Diftitox) in cutaneous T-Cell lymphoma (CTCL) patients. clinicaltrials.gov/ct2/show/NCT00050999 (first received 3 January 2003).
- Olsen E, Duvic M, Frankel A, Kim Y, Martin A, Vonderheid E, et al. Pivotal phase III trial of two dose levels of denileukin diftitox for the treatment of cutaneous T-cell lymphoma. Journal of Clinical Oncology 2001;19(2):376-88. [CENTRAL: CN-00328506] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Olsen E, Duvic M, Martin A. Pivotal phase III trial of two dose levels of DAB 389 IL-2 (ONTAK) for the treatment of cutaneous T-cell lymphoma (CTCL). Journal of Investigative Dermatology 1998;110(4):678. [CENTRAL: CN-00353452] [Google Scholar]
Prince 2017 {published data only}
- Horwitz S, Whittaker S, Duvic M, Dummer R, Kim YH, Scarisbrick J, et al. Response by stage in CD30-positive (CD30+) cutaneous T cell lymphoma (CTCL) patients receiving brentuximab vedotin (BV) vs physician's choice (PC) in the phase 3 ALCANZA study. In: Hematological oncology. Conference: 14th international conference on malignant lymphoma palazzo dei congressi. Switzerland. Vol. 35. 2017:245–7. [CENTRAL: CN-01408605] [DOI: 10.1002/hon.2438] [DOI]
- Horwitz SM, Akilov OE, Fisher D, Kuzel TM, Wang Y, Palanca-Wessels MC, et al. Brentuximab vedotin or physician’s choice in CD30-positivecutaneous T-cell lymphoma (ALCANZA): an international,open-label, randomised, phase 3, multicentre trial. Lancet 2017;390(10094):555-66. [CENTRAL: CN-01403731] [DOI] [PubMed] [Google Scholar]
- Horwitz SM, Scarisbrick JJ, Dummer R, Duvic M, Kim YH, Walewski J, et al. Updated analyses of the international, open-label, randomized, phase 3 alcanza study: longer-term evidence for superiority of brentuximab vedotin versus methotrexate or bexarotene for CD30-positive cutaneous T-cell lymphoma (CTCL). In: Blood. Conference: 59th annual meeting of the american society of hematology, ASH 2017. United states. Vol. 130. 2017. [CENTRAL: CN-01450312]
- Kim YH, Prince HM, Whittaker S, Horwitz SM, Duvic M, Scarisbrick J, et al. Brentuximab vedotin vs physician's choice in CTCL patients from the phase 3 ALCANZA study: analysis of outcomes by CD30 expression. In: Hematological Oncology. Conference: 14th international conference on malignant lymphoma palazzo dei congressi. Switzerland. Vol. 35. 2017:77–8. [CENTRAL: CN-01408482] [DOI: 10.1002/hon.2437] [DOI]
- Kim YH, Prince HM, Whittaker S, Horwitz SM, Duvic M, Scarisbrick J, et al. Outcomes by CD30 expression in patients with CTCL receiving brentuximab vedotin (BV) vs physician's choice (PC) in the Phase 3 ALCANZA study. Journal of Clinical Oncology 2017;35(15 Supplement 1):7517. [EMBASE: 617388795] [Google Scholar]
- Kim YH, Whittaker S, Dummer R, Geskin LJ, Gauthier P, Little M, et al. Phase 3 study of brentuximab vedotin versus physician's choice of methotrexate or bexarotene in patients (PTS) with CD30-positive (CD30+) cutaneous T-cell lymphoma (CTCL). The alcanza study. Hematological Oncology 2013;31(Suppl 1):278. [CENTRAL: CN-01531598] [EMBASE: 71147875] [Google Scholar]
- Kim YH, Whittaker S, Horwitz SM, Duvic M, Dummer R, Scarisbrick J, et al. Brentuximab vedotin demonstrates superior activity to standard therapy in CD30-expressing (CD30+) cutaneous T-cell lymphoma (CTCL) in the randomized phase 3 ALCANZA study. Journal of Investigative Dermatology 2017;137(5 Suppl 1):S45. [CENTRAL: CN-01375469] [EMBASE: 616393777] [Google Scholar]
- Kim YH, Whittaker S, Horwitz SM, Duvic M, Dummer R, Scarisbrick JJ, et al. Brentuximab vedotin demonstrates significantly superior clinical outcomes in patients with CD30-expressing cutaneous T cell lymphoma versus physician's choice (Methotrexate or Bexarotene): the phase 3 alcanza study. In: Blood. Conference: 58th annual meeting of the american society of hematology, ASH 2016. United states. Conference start: 20161203. Conference end: 20161206. Vol. 128. 2016. [CENTRAL: CN-01303081] [EMBASE: 614225065]
- NCT01578499. A randomised, open-label, phase 3 trial of brentuximab vedotin (SGN-35) versus physician's choice (methotrexate or bexarotene) in patients with CD30-positive cutaneous T-cell lymphoma. clinicaltrials.gov/ct2/show/NCT01578499 (first received 17 April 2012).
- Prince HM, Dummer R, Whittaker S, Horwitz S, Duvic M, Scarisbrick J, et al. Patient-reported outcomes and quality of life in patients with cutaneous T cell lymphoma: results from the phase 3 ALCANZA study. In: Hematological Oncology. Conference: 14th international conference on malignant lymphoma palazzo dei congressi. Switzerland. Vol. 35. 2017:247–8. [CENTRAL: CN-01408603] [DOI: 10.1002/hon.2438] [DOI]
- Zagadailov E, Prince HM, Whittaker S, Horwitz S, Duvic M, Kim Y, et al. Phase 3 alcanza study of brentuximab vedotin (BV) or physician's choice (PC) of methotrexate (MTX) or bexarotene (BEX) in CD30-positive cutaneous T-cell lymphoma (CTCL): number needed to treat analysis. In: Haematologica. Conference: 22th congress of the european hematology association. Spain. Vol. 102. 2017:251. [CENTRAL: CN-01399195]
Rook 2010 {published and unpublished data}
- Rook AH, Wood GS, Duvic M, Vonderheid EC, Tobia A, Cabana B. A phase II placebo-controlled study of photodynamic therapy with topical hypericin and visible light irradiation in the treatment of cutaneous T-cell lymphoma and psoriasis. Journal of the American Academy of Dermatology 2010;63(6):984-90. [CENTRAL: CN-00772559] [PMID: ] [DOI] [PubMed] [Google Scholar]
Stadler 1998 {published and unpublished data}
- Otte HG, Kuhl S, Stadler R, Luger T, Henz B, Sterry W. Treatment of cutaneous T-cell lymphoma with Interferon alpha and PUVA versus Interferon alpha and Acitretin - results of a prospective randomised multi-centre study [Behandlung des kutanen T-Zell-Lymphoms mit Interferon alpha und PUVA versus Interferon alpha und Acirentin - Ergebnisse einer prospektiv-randomisierten Multicenter-Studie]. Der Hautarzt 1997;48(Suppl):S97. [CENTRAL: CN-00495120] [Google Scholar]
- Otte HG, Stadier R, Luger T, Henz B, Sterry W. Open controlled clinical trial on the use of interferon plus acitretin versus interferon plus PUVA in patients with cutaneous T-cell lymphoma (CTCL). Annals of Oncology 1996;7(Suppl 3):66. [CENTRAL: CN-00308243] [PubMed] [Google Scholar]
- Stadler R, Otte HG, Luger T, Henz BM, Kuhl P, Zwingers T, et al. Prospective randomized multicenter clinical trial on the use of interferon -2a plus acitretin versus interferon -2a plus PUVA in patients with cutaneous T-cell lymphoma stages I and II. Blood 1998;92(10):3578-81. [CENTRAL: CN-00156682] [PMID: ] [PubMed] [Google Scholar]
Stadler 2006 {published and unpublished data}
- Bohmeyer J, Otte HG, Stadler R, Luger T, Sterry W. Therapy of cutaneous T-cell lymphoma with PUVA versus interferon alfa plus PUVA: first results. Journal of the European Academy of Dermatology & Venereology 1999;12(Suppl 2):S268. [CENTRAL: CN-00478472] [Google Scholar]
- Bohmeyer J, Stadler R, Luger T, Sterry W. Prospective randomized multicentre therapy optimizing protocol for therapy of cutaneous t-cell-lymphoma with PUVA versus Interferon α + PUVA [Prospectiv randomisiertes, multizentrisches Therapieoptimierungsprotokoll zur Therapie kutaner T-Zell-Lymphome mit PUVA versus Interferon α + PUVA]. Zeitschrift für Hautkrankheiten 2001;76(Suppl 1):S55. [CENTRAL: CN-01446038] [Google Scholar]
- Kremer A, Bohmeyer J, Stadler R, Luger T, Sterry W. Prospective randomized multicentre therapy optimizing protocol for therapy of cutaneous t-cell-lymphoma with PUVA versus Interferon α + PUVA [Prospectiv randomisiertes, multizentrisches Therapieoptimierungsprotokoll zur Therapie kutaner T-Zell-Lymphome mit PUVA versus Interferon α + PUVA]. Journal der Deutschen Dermatologischen Gesellschaft 2003;1(Suppl 1):S99. [CENTRAL: CN-01446037] [Google Scholar]
- Otte HG, Stadler R, Luger T, Sterry W. Therapy of cutaneous t-cell-lymphoma with PUVA versus Interferon α plus PUVA: first results of a prospective randomized multicentre therapy optimizing protocol [Therapie kutaner T-Zell-Lymphome mit PUVA versus Interferon α plus PUVA: Erste Ergebnisse des prospektiven randomisierten multizentrischen Therapieoptimierungsprotokolls]. Der Hautarzt 1999;50(Suppl 1):S2. [CENTRAL: CN-01457423] [Google Scholar]
- Otte HG, Stadler R. Combination therapy of cutaneous t-cell-lymphoma with interferon alfa 2a and PUVA [Kombinationstherapie kutaner T-Zell-Lymphome mit Interferon Alfa 2a und PUVA]. Zentralblatt Haut- und Geschlechtskrankheiten 1993;162(Suppl):P2.03.08. [CENTRAL: CN-01457422] [Google Scholar]
- Stadler M, Kremer A, Luger T, Sterry W. Prospective, randomized, multicentre clinical trial on the use of interferon a 2a plus PUVA versus PUVA monotherapy in patients with cutaneous T-cell lymphoma, stages I and II [Abstract 7541]. ASCO annual meeting proceedings. Journal of Clinical Oncology 2006;24(18S Pt 1):432. [CENTRAL: CN-00625001] [Google Scholar]
Thestrup‐Pedersen 1982 {published and unpublished data}
- Thestrup-Pedersen K, Grunnet E, Zachariae H. Transfer factor therapy in mycosis fungoides: a double-blind study. Acta Dermato-Venereologica 1982;62(1):47-53. [CENTRAL: CN-00027377] [PMID: ] [PubMed] [Google Scholar]
Vieyra‐Garcia 2019 {published data only}
- NCT01686594. A multi-center, randomized study on oral 8-methoxypsoralen plus UVA with or without maintenance therapy in mycosis fungoides EORTC/ISCL stage IA to IIB. clinicaltrials.gov/ct2/show/NCT01686594 (first received 18 September 2012).
- Vieyra-Garcia P, Fink-Puches R, Porkert S, Lang R, Pöchlauer S, Ratzinger G, et al. Evaluation of low-dose, low-frequency oral psoralen-UV-A treatment with or without maintenance on early-stage mycosis fungoides: A randomized clinical trial. JAMA Dermatology 2019;155(5):538-47. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Vonderheid 1987 {published data only (unpublished sought but not used)}
- Vonderheid EC, Thompson R, Smiles KA, Lattanand A. Recombinant interferon alfa-2b in plaque-phase mycosis fungoides. Intralesional and low-dose intramuscular therapy. Archives of Dermatology 1987;123(6):757-63. [CENTRAL: CN-00047990] [PMID: ] [PubMed] [Google Scholar]
Whittaker 2012 {published data only}
- NCT00056056. Ultraviolet light therapy using methoxsalen with or without Bexarotene in treating patients with mycosis fungoides. clinicaltrials.gov/ct2/show/NCT00056056 (first received 7 March 2003).
- Whittaker S, Ortiz P, Dummer R, Ranki A, Hasan B, Meulemans B, et al. Efficacy and safety of bexarotene combined with psoralen-ultraviolet A (PUVA) compared with PUVA treatment alone in stage IB-IIA mycosis fungoides: final results from the EORTC Cutaneous Lymphoma Task Force phase III randomized clinical trial 21011 (NCT00056056). British Journal of Dermatology 2012;167(3):678-87. [CENTRAL: CN-00856432] [PMID: ] [DOI] [PubMed] [Google Scholar]
- Whittaker S, Ortiz-Romero PL, Dummer R, Ranki A, Hasan B, Meulemans B, et al. Efficacy and safety of bexarotene combined with psoralen/ultraviolet A light (PUVA) compared to PUVA treatment alone in stage IB-IIa mycosis fungoides (MF): Final results from EORTC cutaneous lymphoma task force (CLTF) phase III clinical trial 21011. Journal of Clinical Oncology 2012;30(15 Suppl 1):8076. [DOI] [PubMed] [Google Scholar]
Wolff 1985 {published data only (unpublished sought but not used)}
- Wolff JM, Zitelli JA, Rabin BS, Smiles KA, Abell E. Intralesional interferon in the treatment of early mycosis fungoides. Journal of the American Academy of Dermatology 1985;13(4):604-12. [CENTRAL: CN-00040943] [PMID: ] [DOI] [PubMed] [Google Scholar]
Wozniak 2008 {published data only (unpublished sought but not used)}
- Wozniak MB, Tracey L, Ortiz-Romero PL, Montes S, Alvarez M, Fraga J, et al. Psoralen plus ultraviolet A +/- interferon-alpha treatment resistance in mycosis fungoides: the role of tumour microenvironment, nuclear transcription factor-kappaB and T-cell receptor pathways. British Journal of Dermatology 2008;160(1):92-102. [CENTRAL: CN-00668828] [PMID: ] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Anonymous 1982 {published data only}
- No authors listed. Comparison of the use of teniposide and vincristine in combination chemotherapy for non-Hodgkin's lymphoma. Cancer Treatment Reports 1982;66(1):49-55. [PubMed] [Google Scholar]
Anonymous 2000 {published data only}
- Anonymous. Bexarotene (Targretin) for cutaneous T-cell lymphoma. Medical Letter on Drugs and Therapeutics 2000;42(1075):31-2. [PMID: ] [PubMed] [Google Scholar]
Argyropoulos 1979 {published data only}
- Argyropoulos CL, Lamberg SI, Clendenning WE, Fischmann AB, Jegasothy B, Zackheim H, et al. Preliminary evaluation of 15 chemotherapeutic agents applied topically in the treatment of mycosis fungoides. Cancer Treatment Reports 1979;63(4):619-21. [PubMed] [Google Scholar]
Aviles 2015 {published data only}
- Aviles A, Neri N, Fernandez-Diez J, Silva L, Nambo MJ. Interferon and low doses of methotrexate versus interferon and retinoids in the treatment of refractory/relapsed cutaneous T-cell lymphoma. Hematology 2015;20(9):538–42. [PMID: 25592781] [DOI] [PubMed] [Google Scholar]
Bazex 1975 {published data only}
- Bazex A, De Mouzon J, Bazex J. Not available [Expérimentation du Dipropionate de Bétaméthazone en application locale]. Revue de Medecine de Toulouse 1975;11:1137-47. [Google Scholar]
Breneman 1991 {published data only}
- Breneman DL, Nartker AL, Ballman EA, Pruemer JM, Blumsack RF, Davis M, et al. Topical mechlorethamine in the treatment of mycosis fungoides. Uniformity of application and potential for environmental contamination. Journal of the American Academy of Dermatology 1991;25(6 Pt 1):1059-64. [DOI] [PubMed] [Google Scholar]
Cooper 1994 {published data only}
- Cooper IA, Wolf MM, Robertson TI, Fox RM, Matthews JP, Stone JM, et al. Randomized comparison of MACOP-B with CHOP in patients with intermediate-grade non-Hodgkin's lymphoma. The Australian and New Zealand Lymphoma Group. Journal of Clinical Oncology 1994;12(4):769-78. [DOI] [PubMed] [Google Scholar]
Currie 1980 {published data only}
- Currie VE, Kempin SJ, Young CW. Phase I trial of metoprine in patients with advanced cancer. Cancer Treatment Reports 1980;64(8-9):951-6. [PubMed] [Google Scholar]
Dang 2007 {published data only}
- Dang NH, Pro B, Hagemeister FB, Samaniego F, Jones D, Samuels BI, et al. Phase II study of denileukin diftitox for relapsed/refractory B-Cell non-Hodgkin's lymphoma. British Journal of Haematology 2007;136(3):439-47. [PMID: ] [DOI] [PubMed] [Google Scholar]
Doan 1958 {published data only}
- Doan CA, Wiseman BK, Bouroncle BA. Clinical evaluation of CB 1348 in leukemias and lymphomas. Annals of the New York Academy of Sciences 1958;68(3):979-95. [DOI] [PubMed] [Google Scholar]
Dueck 2010 {published data only}
- Dueck G, Chua N, Prasad A, Finch D, Stewart D, White D, et al. Interim report of a phase 2 clinical trial of lenalidomide for T-cell non-Hodgkin lymphoma. Cancer 2010;116(19):4541-8. [DOI] [PubMed] [Google Scholar]
Duvic 2010 {published data only}
- Duvic M, Martin A, Olsen EA, Fivenson D, Prince M. Efficacy of denileukin diftitox retreatment in patients with cutaneous T-cell lymphoma who relapsed after initial response. Blood 2010;116(21):2863. [Google Scholar]
Fawzi 2010 {published data only}
- Fawzi M, El Mofty M, Ramadan S, Hegazy R, Sayed S. Broadband UVA versus PUVA in the treatment of early stage mycosis fungoides: a comparative study. Melanoma Research 2010;20:e85. [DOI: 10.1097/01.cmr.0000382933.41435.dc] [DOI] [Google Scholar]
Fisher 1993 {published data only}
- Fisher RI, Gaynor ER, Dahlberg S, Oken MM, Grogan TM, Mize EM, et al. Comparison of a standard regimen (CHOP) with three intensive chemotherapy regimens for advanced non-Hodgkin's lymphoma. New England Journal of Medicine 1993;328(14):1002-6. [DOI] [PubMed] [Google Scholar]
Foss 2011 {published data only}
- Foss F, Duvic M, Olsen EA. Predictors of complete responses with denileukin diftitox in cutaneous T-cell lymphoma. American Journal of Hematology 2011;86(7):627-30. [DOI] [PubMed] [Google Scholar]
Groth 1979 {published data only}
- Groth O, Molin L, Thomsen K. Tumour stage of mycosis fungoides treated with bleomycin and methotrexate: report from the Scandinavian mycosis fungoides study group. Acta Dermato-Venereologica 1979;59(1):59–63. [PMID: 84469] [PubMed] [Google Scholar]
Heald 2003 {published data only}
- Heald P, Mehlmauer M, Martin AG, Crowley CA, Yocum RC, Reich SD, et al. Topical bexarotene therapy for patients with refractory or persistent early-stage cutaneous T-cell lymphoma: Results of the phase III clinical trial. Journal of the American Academy of Dermatology 2003;49(5):801–15. [DOI: 10.1067/S0190-9622(3)01475-0] [PMID: ] [DOI] [PubMed] [Google Scholar]
JapicCTI‐050041 {published data only}
- JapicCTI-050041. Post-marketing clinical study of Ogamma 100 in patients with mycosis fungoides. www.clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicCTI-050041 (first received 12 September 2005).
Kaung 1969 {published data only}
- Kaung DT, Wittington RM, Spencer H, Patno ME. Comparison of chlorambucil and streptonigrin (NSC-45383) in the treatment of malignant lymphomas. Cancer 1969;23(6):1280-3. [DOI] [PubMed] [Google Scholar]
Kujawska 2003 {published data only}
- Kujawska A, Abrou A, Fivenson DP, Lim HW. PUVA vs. PUVA and interferon alpha for treatment of mycosis fungoides. Journal of Investigative Dermatology 2003;121(1):1185. [Google Scholar]
Kuzel 2010 {published data only}
- Kuzel T. Correlates of capillary leak syndrome (CLS) in patients with cutaneous T-cell lymphoma (CTCL) during treatment with denileukin diftitox (DD) in a placebo (PBO)-controlled phase III trial. Journal of Clinical Oncology 2010;28(15 Suppl):e18509. [Google Scholar]
Lambert 1986 {published data only}
- Lambert D, Bousser AM, Dalac S, Godard W, Lepinoy D, Horiot JC. Mycosis fungoides and radiotherapy. Value of combined total skin electron therapy and whole-body photon therapy. Annales de Dermatologie et de Venereologie 1986;113(6-7):541–7. [PMID: ] [PubMed] [Google Scholar]
Lansigan 2010 {published data only}
- Lansigan F, Foss FM. Rate of infection and immunosuppression in two phase III studies of denileukin diftitox (DD) in cutaneous T-cell lymphoma (CTCL). Journal of Clinical Oncology 2010;28(15 Suppl):e18515.
Loescher 1984 {published data only}
- Loescher L, Kessler J, Jones S, Meyskens F, Levine N, Sauer K. 13-Cis retinoic acid as treatment for mycosis fungoides: Clinical results of a phase II trial. Federation Proceedings 1984;43(4):3896. [Google Scholar]
Marsden 1968 {published data only}
- Marsden CW. Fluocinolone acetonide 0.2 per cent cream--a co-operative clinical trial. British Journal of Dermatology 1968;80(9):614-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Moog 2008 {published data only}
- Moog R. Therapeutic apheresis - Many ways to go. Transfusion Medicine and Hemotherapy 2008;35(1):5-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT00054171 {published data only}
- NCT00054171. Photodynamic therapy in treating patients with lymphoma or chronic lymphocytic leukemia. clinicaltrials.gov/ct2/show/NCT00054171 (first received 6 February 2003).
NCT00091208 {unpublished data only}
- NCT00091208. A phase I/II open label, multi-center study for the evaluation of Pf-3512676 (CPG 7909) in patients with stage Ib to Iva cutaneous T-Cell lymphoma. clinicaltrials.gov/ct2/show/NCT00091208 (first received 12 August 2002). [NCT00091208]
NCT01007448 {unpublished data only}
- NCT01007448. Study evaluating two dose levels of Targretin capsules in patients with refractory cutaneous T-cell lymphoma (CTCL). clinicaltrials.gov/ct2/show/NCT01007448 (first received 4 November 2009).
NCT01187446 {unpublished data only}
- NCT01187446. Low-dose (12 Gy) TSEBT+Vorinostat versus low-dose TSEBT monotherapy in mycosis fungoides. clinicaltrials.gov/ct2/show/NCT01187446 (first received 24 August 2010).
NCT01386398 {unpublished data only}
- NCT01386398. Vorinostat with or without Bortezomib in treating patients with refractory or recurrent stage IIB, stage III, or stage IV cutaneous T-Cell lymphoma. clinicaltrials.gov/ct2/show/NCT01386398 (first received 1 July 2011).
NCT01625455 {unpublished data only}
- NCT01625455. Effect of Neurokinin-1 Receptor (NK1R) antagonism on pruritus in patients with Sezary syndrome. clinicaltrials.gov/ct2/show/NCT01625455 (first received 21 June 2012).
Neering 1972 {published data only}
- Neering H, Kroon HV, Roeleveld CG. Treatment of localized skin lesions with betamethasone 17-valerate and triamcinolone acetonide in alcoholic solution under occlusive dressing. A double blind comparative study. Dermatologica 1972;145(6):395-9. [DOI] [PubMed] [Google Scholar]
Negro‐Vilar 2007 {published data only}
- Negro-Vilar A, Dziewanowska Z, Groves ES, Stevens V, Zhang JK, Prince M, et al. Efficacy and safety of denileukin diftitox (Dd) in a phase III, double-blind, placebo-controlled study of CD25+ patients with cutaneous T-cell lymphoma (CTCL). Journal of Clinical Oncology 2007;25(18 Suppl):8026. [Google Scholar]
O'Neill 2013 {published data only}
- O'Neill B, Geskin L, Story S. Novel combination therapy for the treatment of cutaneous T-cell lymphoma. Journal of the American Academy of Dermatology 2013;68(4 Suppl 1):AB147.
Olsen 1986 {published and unpublished data}
- Olsen EA, Diab NG. Interferon alfa-2A in the treatment of cutaneous T cell lymphoma. Journal of Investigative Dermatology 1986;86(4):498. [DOI] [PubMed] [Google Scholar]
Pan 2007 {published data only}
- Pan Y, Prince HM, Ellis L, Culver K. LBH589, a novel deacetylase inhibitor (DACi), in the treatment of cutaneous T-cell lymphoma (CTCL): changes in tumor gene expression profiles related to clinical response after therapy. In: 11th World Congress on Cancers of the Skin. 2007 June 8-11. Amsterdam, The Netherlands. 2007:Abstract 00172.
Peugeot 1995 {published data only}
- Peugeot RL. Clinical efficacy of a novel purine nucleoside phosphorylase inhibitor (BCX-34) in the treatment of cutaneous T-cell lymphoma. Journal of Investigative Dermatology 1995;104(4):563. [Google Scholar]
Plettenberg 2001 {published data only}
- Plettenberg H, Stege H, Megahed T, Ruzicka T, Hosokawa Y, Tsuji T, et al. UVA1-Phototherapie versus PUVA-Photochemotherapie in der Behandlung von Patienten mit kutanem T-Zell-Lymphom (CTCL). Zeitschrift für Hautkrankheiten 2001;76(Suppl 1):S96. [Google Scholar]
Prince 2010 {published data only}
- Prince HM, Duvic M, Martin A, Sterry W, Assaf C, Sun Y, et al. Phase III placebo-controlled trial of denileukin diftitox for patients with cutaneous T-cell lymphoma. Journal of Clinical Oncology 2010;28(11):1870-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Schrag 1997 {published data only}
- Schrag H-J, Dippel E, Goerdt S, Orfanos CE. Extracorporal photophoresis (ECP) + Interferon-alpha 2a vs. ECP in patients with cutaneous T-cell lymphoma. Evaluation using a new score (CTCL-SI). Der Hautarzt 1997;48(Suppl):S96. [Google Scholar]
Serri 1990 {published data only}
- Serri F, De Simone C, Venier A, Rusciani L, Marchetti F. Combination of retinoids and PUVA (Re-PUVA) in the treatment of cutaneous T cell lymphomas. Current Problems in Dermatology 1990;19:252-7. [PMID: ] [DOI] [PubMed] [Google Scholar]
Shi 2015 {published data only}
- Shi Y, Dong M, Zhu J, Zhou D, Huang H, Tu P, et al. Phase II study of chidamide, a new subtype-selective oral histone deacetylase inhibitor, in patients with relapsed or refractory cutaneous T-cell lymphoma. Blood 2015;126(23):1513. [Google Scholar]
Simon 2010 {published data only}
- Simon A, Peoch M, Casassus P, Deconinck E, Colombat P, Desablens B, et al. Upfront VIP-reinforced-ABVD (VIP-rABVD) is not superior to CHOP/21 in newly diagnosed peripheral T cell lymphoma. Results of the randomized phase III trial GOELAMS-LTP95. British Journal of Haematology 2010;151(2):159-66. [PMID: ] [DOI] [PubMed] [Google Scholar]
Thomsen 1977 {published data only (unpublished sought but not used)}
- Thomsen K. Scandinavian mycosis fungoides study group. Bulletin du Cancer 1977;64(2):287-90. [PMID: ] [PubMed] [Google Scholar]
Thomsen 1979 {published data only}
- Thomsen K. Scandinavian mycosis fungoides trial. Cancer Treatment Reports 1979;63(4):709–11. [PMID: ] [PubMed] [Google Scholar]
Thomsen 1989 {published data only}
- Thomsen K, Hammar H, Molin L, Volden G. Retinoids plus PUVA (RePUVA) and PUVA in mycosis fungoides, plaque stage. Acta Dermato-Venereologica 1989;69(6):536-8. [PubMed] [Google Scholar]
Touraine 1978 {published data only}
- Touraine R, Revuz J. Topical chemotherapy in mycosis fungoides [Chimiotherapie locale du mycosis fongoide: Dermatologica]. Dermatologica 1978;157(6):397-406. [PubMed] [Google Scholar]
Wain 2005 {published data only}
- Wain EM, Whittaker SJ, Russell-Jones R. A randomized, open, crossover study to compare the efficacy of extracorporeal photopheresis with methotrexate in the treatment of erythrodermic primary cutaneous T-cell lymphoma. British Journal of Dermatology 2005;153(Suppl 1):10. [Google Scholar]
Wiernik 1998 {published data only}
- Wiernik PH, Moore DF, Bennett JM, Vogl SE, Harris JE, Luger S, et al. Phase II study of mitoguazone, cyclophosphamide, doxorubicin, vincristine and prednisone for patients with diffuse histologic subtypes of non-Hodgkin's lymphoma: an Eastern Cooperative Oncology Group Study (PE481). Leukemia & Lymphoma 1998;30(5-6):601-7. [DOI] [PubMed] [Google Scholar]
Wilson 1995 {published data only}
- Wilson LD, Licata AL, Braverman IM, Edelson RL, Heald PW, Feldman AM, et al. Systemic chemotherapy and extracorporeal photochemotherapy for T3 and T4 cutaneous T-cell lymphoma patients who have achieved a complete response to total skin electron beam therapy. International Journal of Radiation Oncology, Biology, Physics 1995;32(4):987–95. [PMID: ] [DOI] [PubMed] [Google Scholar]
Zubrod 1960 {published data only}
- Zubrod CG, Schneiderman M, Frei E, Brindley C, Gold GL, Shnider B, et al. Appraisal of methods for the study of chemotherapy of cancer in man: Comparative therapeutic trial of nitrogen mustard and triethylene thiophosphoramide. Journal of Chronic Diseases 1960;11(1):7-33. [Google Scholar]
References to studies awaiting assessment
Bashey 2014 {published data only}
- Bashey S, Tavallaee M, Duvic M, Dabaja B, Talpur R, Wilson L, et al. A multicenter, open-label, randomized, phase I/II study evaluating the safety and efficacy of low-dose (12 Gy) total skin electron beam therapy (TSEBT) combined with vorinostat versus low-dose TSEBT monotherapy in patients with mycosis fungoides. Journal of Investigative Dermatology 2014;134(Suppl 1):S104. [DOI: ] [Google Scholar]
Kim 2014 {published data only}
- Kim YH, Krathen M, Duvic M, Wong H, Porcu P, Tacastacas J, et al. A phase 1b study in cutaneous T-cell lymphoma (CTCL) with the novel topically applied skin-restricted histone deacteylase inhibitor (HDAC-i) SHP-141. Journal of Clinical Oncology 2014;32(15 Suppl):8525. [Google Scholar]
- Kim YH, Krathen MS, Duvic M, Wong H, Porcu P, Tacastacas J, et al. Tolerability and encouraging clinical activity of SHP-141, a topical skin-restricted HDAC inhibitor, in a phase 1B study in cutaneous t-cell lymphoma. Journal of Investigative Dermatology 2014;134(Suppl 1):S93. [Google Scholar]
- NCT01433731. Safety, pharmacodynamics (PD), pharmacokinetics (PK) study of SHP141 in 1A, 1B, or 2A cutaneous T-cell lymphoma (CTCL). clinicaltrials.gov/ct/show/NCT01433731 (first received 14 September 2011).
References to ongoing studies
NCT01738594 {published data only}
- NCT01738594. Dose-escalation trial of carfilzomib with and without Romidepsin in cutaneous T-cell lymphoma. clinicaltrials.gov/ct2/show/NCT01738594 (first received 30 November 2012).
NCT02213861 {published data only}
- Duvic M, Kim YH, LeBoeuf NR, Porcu P, Hastings J, Bassuner J, et al. A phase 2 randomized study of SHAPE Gel (SHP-141) in patients with early-stage cutaneous T-cell lymphoma: Interim results. Journal of Clinical Oncology 2016;34(15 Suppl):7562. [DOI: 10.1200/JCO.2016.34] [DOI] [Google Scholar]
- NCT02213861. Efficacy, safety and tolerability study of SHAPE in IA, IB or IIA cutaneous T-cell lymphoma. clinicaltrials.gov/ct2/show/NCT02213861 (first received 12 August 2014).
NCT02301494 {published data only}
- NCT02301494. Effectiveness of imiquimod topical cream in early stage cutaneous T-cell lymphoma (CTCL). clinicaltrials.gov/ct2/show/NCT02301494 (first received 26 November 2014).
NCT02323659 {published data only}
- NCT02323659. Comparison of methotrexate versus interferon-alfa 2b in patients with primary cutaneous T-cell lymphomas. clinicaltrials.gov/ct2/show/NCT02323659 (first received 23 December 2014).
NCT02448381 {published data only}
- NCT02448381. FLASH [Fluorescent Light Activated Synthetic Hypericin] clinical study: topical SGX301 (synthetic hypericin) for the treatment of cutaneous T-cell lymphoma (mycosis fungoides). clinicaltrials.gov/ct2/show/NCT02448381 (first received 19 May 2015).
NCT02811783 {published data only}
- NCT02811783. Naloxone hydrochloride study for relief of pruritus in patients with MF or SS forms of CTCL. clinicaltrials.gov/ct2/show/NCT02811783 (first received 23 June 2016).
NCT02943642 {published data only}
- NCT02943642. Safety and effectiveness of A-dmDT390-bisFv(UCHT1) fusion protein in subjects with mycosis fungoides (Resimmune®). clinicaltrials.gov/ct2/show/NCT02943642 (first received 25 October 2016).
NCT02953301 {published data only}
- NCT02953301. Resminostat for maintenance treatment of patients with advanced stage mycosis fungoides (MF) or Sézary syndrome (SS) (RESMAIN). clinicaltrials.gov/ct2/show/NCT02953301 (first received 2 November 2016).
- Stadler R, Scarisbrick J, Knobler R, Quaglino P, Borgmann M, Orlovius M, et al. A multicentre, double blind, randomised, placebo controlled, Phase II trial to evaluate Resminostat for maintenance treatment of patients with advanced stage (Stage IIB-IVB) mycosis fungoides (MF) or Sezary Syndrome (SS) that have achieved disease control with systemic therapy-the RESMAIN Study. Journal of the European Academy of Dermatology and Venereology 2017;31(Suppl 3):50. [Google Scholar]
- Stadler R, Scarisbrick J, Knobler R, Quaglino P, Borgmann M, Orlovius M, et al. A multicentre, double blind, randomised, placebo-controlled, Phase II trial to evaluate Resminostat for maintenance treatment of patients with advanced stage (Stage IIB-IVB) mycosis fungoides (MF) or Sezary Syndrome (SS) that have achieved disease control with systemic therapy: The RESMAIN Study. Journal der Deutschen Dermatologischen Gesellschaft [Journal of the German Society of Dermatology] 2017;15(Suppl 3):74–5. [DOI: 10.1111/ddg.13300] [DOI] [Google Scholar]
NCT03011814 {published data only}
- NCT03011814. Durvalumab with or without lenalidomide in treating patients with relapsed or refractory cutaneous or peripheral T cell lymphoma. clinicaltrials.gov/ct2/show/NCT03011814 (first received 5 January 2017).
NCT03292406 {published data only}
- NCT03292406. A safety, efficacy and pharmacokinetics study of CD11301 for the treatment of cutaneous T-cell lymphoma (CTCL) (CTCL). clinicaltrials.gov/ct2/show/NCT03292406 (first received 25 September 2017).
NCT03454945 {published data only}
- NCT03454945. Efficacy of doxycycline in the treatment of early stages of mycosis fungoides: A randomized controlled trial. clinicaltrials.gov/ct2/show/NCT03454945 (first received 6 March 2018).
NCT03713320 {published data only}
- NCT03713320. SOLAR: A phase 2, randomized, open-label, parallel-group, active comparator, multi-center study to investigate the efficacy and safety of cobomarsen (MRG-106) in subjects with cutaneous T-cell lymphoma (CTCL), mycosis fungoides (MF) subtype. clinicaltrials.gov/ct2/show/NCT03713320 (first received 17 October 2018).
UMIN000029537 {published data only}
- UMIN000029537. Efficacy and safety of Targretin capsule 75-mg alone or in combination with phototherapy in Japanese patients with cutaneous T-cell lymphomas. upload.umin.ac.jp/cgi-open-bin/ctr_e/ctr_view.cgi?recptno=R000033748 (date first received 16 October 2017).
Additional references
Abbott 2009
- Abbott RA, Whittaker SJ, Morris SL, Russell-Jones R, Hung T, Bashir SJ, et al. Bexarotene therapy for mycosis fungoides and Sézary syndrome. British Journal of Dermatology 2009;160(6):1299-307. [DOI] [PubMed] [Google Scholar]
Agar 2010
- Agar NS, Wedgeworth E, Crichton S, Mitchell TJ, Cox M, Ferreira S, et al. Survival outcomes and prognostic factors in mycosis fungoides/Sezary syndrome: validation of the revised International Society for Cutaneous Lymphomas/European Organisation for Research and Treatment of Cancer staging proposal. Journal of Clinical Oncology 2010;28(31):4730-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Akpek 1999
- Akpek G, Koh HK, Bogen S, O'Hara C, Foss FM. Chemotherapy with etoposide, vincristine, doxorubicin, bolus cyclophosphamide, and oral prednisone in patients with refractory cutaneous T-cell lymphoma. Cancer 1999;86(7):1368-76. [PubMed] [Google Scholar]
Aronsson 1982
- Aronsson A, Jonsson N, Tegner E. Transient lymphomatoid papulosis in mycosis fungoides. Acta Dermato-Venereologica 1982;62(6):529-31. [PubMed] [Google Scholar]
Avarbock 2008
- Avarbock AB, Loren AW, Park JY, Junkins-Hopkins JM, Choi J, Litzky LA, et al. Lethal vascular leak syndrome after denileukin diftitox administration to a patient with cutaneous gamma/delta T-cell lymphoma and occult cirrhosis. American Journal of Hematology 2008;83(7):593-5. [DOI] [PubMed] [Google Scholar]
Bernstein 1989
- Bernstein L, Deapen D, Ross RK. Mycosis fungoides. JAMA 1989;261(13):1882. [PMID: ] [PubMed] [Google Scholar]
Bohmeyer 2003
- Bohmeyer J, Stadler R, Kremer A, Nashan D, Muche M, Gellrich S, et al. Bexarotene--an alternative therapy for progressive cutaneous T-cell lymphoma? First experiences. Journal der Deutschen Dermatologischen Gesellschaft [Journal of the German Society of Dermatology] 2003;1(10):785-9. [DOI] [PubMed] [Google Scholar]
Bradford 2009
- Bradford PT, Devesa SS, Anderson WF, Toro JR. Cutaneous lymphoma incidence patterns in the United States: a population-based study of 3884 cases. Blood 2009;113(21):5064-73. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Braverman 1987
- Braverman IM, Yager NB, Chen M, Cadman EC, Hait WN, Maynard T. Combined total body electron beam irradiation and chemotherapy for mycosis fungoides. Journal of the American Academy of Dermatology 1987;16(1 Pt 1):45-60. [DOI] [PubMed] [Google Scholar]
Bunn 1979
- Bunn PA Jr, Lamberg SI. Report of the committee on staging and classification of cutaneous T-cell lymphomas. Cancer Treatment Reports 1979;63(4):725-8. [PMID: ] [PubMed] [Google Scholar]
Campo‐Voegeli 1998
- Campo-Voegeli A, Estrach T, Marti RM, Corominas N, Tuset M, Mascaró JM. Acrocyanosis induced by interferon alpha(2a). Dermatology 1998;196(3):361-3. [DOI] [PubMed] [Google Scholar]
Carson 2014
- Carson KR, Newsome SD, Kim EJ, Wagner-Johnston ND, Geldern G, Moskowitz CH, et al. Progressive multifocal leukoencephalopathy associated with brentuximab vedotin therapy: a report of 5 cases from the Southern Network on Adverse Reactions (SONAR) project. Cancer 2014;120(16):2464-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cella 1993
- Cella DF, Tulsky DS, Gray G, Sarafian B, Linn E, Bonomi A, et al. The Functional Assessment of Cancer Therapy scale: development and validation of the general measure. Journal of Clinical Oncology 1993;11(3):570-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Cerroni 2018
- Cerroni L. Mycosis fungoides - clinical and histopathologic features, differential diagnosis, and treatment. Seminars in Cutaneous Medicine and Surgery 2018;37(1):2-10. [DOI: 10.12788/j.sder.2018.002] [DOI] [PubMed] [Google Scholar]
Chan 2004
- Chan AW, Hróbjartsson A, Haahr MT, Gøtzsche PC, Altman DG. Empirical evidence for selective reporting of outcomes in randomized trials: comparison of protocols to published articles. JAMA 2004;291(20):2457-65. [DOI] [PubMed] [Google Scholar]
Corbin 2017
- Corbin ZA, Nguyen-Lin A, Li S, Rahbar Z, Tavallaee M, Vogel H, et al. Characterization of the peripheral neuropathy associated with brentuximab vedotin treatment of Mycosis Fungoides and Sézary Syndrome. Journal of Neuro-oncology 2017;132(3):439-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Criscione 2007
- Criscione VD, Weinstock MA. Incidence of cutaneous T-cell lymphoma in the United States, 1973-2002. Archives of Dermatology 2007;143(7):854-9. [PMID: ] [DOI] [PubMed] [Google Scholar]
Daughters 1973
- Daughters D, Zackheim H, Maibach H. Urticaria and anaphylactoid reactions after topical application of mechlorethamine. Archives of Dermatology 1973;107(3):429-30. [PubMed] [Google Scholar]
Dekkers 2010
- Dekkers OM, Elm E, Algra A, Romijn JA, Vandenbroucke JP. How to assess the external validity of therapeutic trials: a conceptual approach. International Journal of Epidemiology 2010;39(1):89-94. [PMID: ] [DOI] [PubMed] [Google Scholar]
Desai 1988
- Desai KR, Pezner RD, Lipsett JA, Vora NL, Luk KH, Wong JY, et al. Total skin electron irradiation for mycosis fungoides: relationship between acute toxicities and measured dose at different anatomic sites. International Journal of Radiation Oncology, Biology, Physics 1988;15(3):641-5. [DOI] [PubMed] [Google Scholar]
Duarte 2010
- Duarte R, Canals C, Onida F, Gabriel I, Arranz R, Arcese W, et al. Allogeneic hematopoietic cell transplantation for patients with mycosis fungoides and Sézary syndrome: a retrospective analysis of the Lymphoma Working Party of the European Group for Blood and Marrow Transplantation.. Journal of Clinical Oncology 2010;28(29):4492-9. [PMID: 20697072] [DOI] [PubMed] [Google Scholar]
Dueck 2010a
- Dueck G, Chua N, Prasad A, Finch D, Stewart D, White D, et al. Interim report of a phase 2 clinical trial of lenalidomide for T-cell non-Hodgkin lymphoma. Cancer 2010;116(19):4541-8. [DOI] [PubMed] [Google Scholar]
Dummer 2008
- Dummer R, Dreyling M. Primary cutaneous lymphoma: ESMO clinical recommendations for diagnosis, treatment and follow-up. Annals of Oncology 2008;19(Suppl 2):ii72-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
Dummer 2012
- Dummer R, Quaglino P, Becker JC, Hasan B, Karrasch M, Whittaker S, et al. Prospective international multicenter phase II trial of intravenous pegylated liposomal doxorubicin monochemotherapy in patients with stage IIB, IVA, or IVB advanced mycosis fungoides: final results from EORTC 21012. Journal of Clinical Oncology 2012;30(33):4091-7. [DOI] [PubMed] [Google Scholar]
Duvic 2001b
- Duvic M, Hymes K, Heald P, Breneman D, Martin AG, Myskowski P, et al. Bexarotene is effective and safe for treatment of refractory advanced-stage cutaneous T-cell lymphoma: multinational phase II-III trial results. Journal of Clinical Oncology 2001;19(9):2456-71. [DOI] [PubMed] [Google Scholar]
Duvic 2002
- Duvic M, Kuzel TM, Olsen EA, Martin AG, Foss FM, Kim YH, et al. Quality-of-life improvements in cutaneous T-cell lymphoma patients treated with denileukin diftitox (ONTAK). Clinical Lymphoma 2002;2(4):222-8. [DOI] [PubMed] [Google Scholar]
Duvic 2013
- Duvic M, Martin AG, Olsen EA, Fivenson DP, Prince HM. Efficacy and safety of denileukin diftitox retreatment in patients with relapsed cutaneous T-cell lymphoma. Leukemia & Lymphoma 2013;54(3):514-9. [DOI] [PubMed] [Google Scholar]
Duvic 2015
- Duvic M, Tetzlaff MT, Gangar P, Clos AL, Sui D, Talpur R. Results of a phase II trial of brentuximab vedotin for CD30+ cutaneous T-cell lymphoma and lymphomatoid papulosis. Journal of Clinical Oncology 2015;33(32):3759-65. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elder 2018
- Elder DE, Massi D, Scolyer RA, Willemze R. WHO Classification of Skin Tumours (4th edition). IARC Press, 2018. [ISBN-13 9789283224402] [Google Scholar]
Foss 2001
- Foss FM, Bacha P, Osann KE, Demierre MF, Bell T, Kuzel T. Biological correlates of acute hypersensitivity events with DAB(389)IL-2 (denileukin diftitox, ONTAK) in cutaneous T-cell lymphoma: decreased frequency and severity with steroid premedication. Clinical Lymphoma 2001;1(4):298-302. [DOI] [PubMed] [Google Scholar]
Gilson 2019
- Gilson D, Whittaker SJ, Child FJ, Scarisbrick JJ, Illidge TM, Parry EJ, et al. British Association of Dermatologists and U.K. Cutaneous Lymphoma Group guidelines for the management of primary cutaneous lymphomas 2018. British Journal of Dermatology 2019;180(3):496-526. [PMID: ] [DOI] [PubMed] [Google Scholar]
Goday 1990
- Goday JJ, Aguirre A, Ratón JA, Díaz-Pérez JL. Local bullous reaction to topical mechlorethamine (mustine). Contact Dermatitis 1990;22(5):306-7. [DOI] [PubMed] [Google Scholar]
GRADE pro GDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime) GRADEpro GDT. Version accessed prior to 29 March 2019. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015.Available at gradepro.org.
Grant 2009
- Grant B. Biotech Baddies. The Scientist 2009;23(4):48. [Google Scholar]
Grunnet 1976
- Grunnet E. Contact urticaria and anaphylactoid reaction induced by topical application of nitrogen mustard. British Journal of Dermatology 1976;94(1):101-3. [DOI] [PubMed] [Google Scholar]
Guilhou 1980
- Guilhou JJ, Barnéon G, Malbos S, Peyron JL, Michel B, Meynadier J. Perforating follicular mucinosis and immediate hypersensitivity to mechlorethamine in a patient with mycosis fungoides. Annales de Dermatologie et de Venereologie 1980;107(1-2):59-62. [PubMed] [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Vist GE, Kunz R, Falck-Ytter Y, Alonso-Coello P, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 2008;336(7650):924-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hanley 1983
- Hanley JA, Lippman-Hand A. If nothing goes wrong, is everything all right? Interpreting zero numerators. JAMA 1983;249(13):1743-5. [PMID: ] [PubMed] [Google Scholar]
Herrmann 1995
- Herrmann JJ, Roenigk HH Jr, Hurria A, Kuzel TM, Samuelson E, Rademaker AW, et al. Treatment of mycosis fungoides with photochemotherapy (PUVA): long-term follow-up. Journal of the American Academy of Dermatology 1995;33(2 Pt 1):234-42. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane-handbook.org.
Hollis 1999
- Hollis S, Campbell F. What is meant by intention to treat analysis? Survey of published randomised controlled trials. BMJ 1999;319(7211):670-4. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hoppe 1987
- Hoppe RT, Abel EA, Deneau DG, Price NM. Mycosis fungoides: management with topical nitrogen mustard. Journal of Clinical Oncology 1987;5(11):1796-803. [DOI] [PubMed] [Google Scholar]
Humme 2014
- Humme D, Nast A, Erdmann R, Vandersee S, Beyer M. Systematic review of combination therapies for mycosis fungoides. Cancer Treatment Reviews 2014;40(8):927-33. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hwang 2008
- Hwang ST, Janik JE, Jaffe ES, Wilson WH. Mycosis fungoides and Sézary syndrome. Lancet 2008;371(9616):945-57. [PMID: ] [DOI] [PubMed] [Google Scholar]
Hüsken 2012
- Hüsken AC, Tsianakas A, Hensen P, Nashan D, Loquai C, Beissert S, et al. Comparison of pegylated interferon α-2b plus psoralen PUVA versus standard interferon α-2a plus PUVA in patients with cutaneous T-cell lymphoma. Journal of the European Academy of Dermatology and Venereology : JEADV 2012;26(1):71-8. [DOI] [PubMed] [Google Scholar]
Jaffe 2001
- Jaffe ES, Harris NL, Stein H, Vardiman JW, editors. Pathology and Genetics: Tumours of Hematopoetic and Lymphoid Tissues (World Health Organization Classification of Tumours). Lyon: IARC Press, 2001. [Google Scholar]
Jüni 2001
- Jüni P, Altman DG, Egger M. Systematic reviews in health care: Assessing the quality of controlled clinical trials. BMJ 2001;323(7303):42-6. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kairouani 2012
- Kairouani M, Sekkate S, Ismaili N, Abahssain H, Errihani H. A rare case of nephrotic syndrome revealing mycosis fungoide managed successfully with chemotherapy. Pan African Medical Journal 2012;12:67. [PMC free article] [PubMed] [Google Scholar]
Kim 1996
- Kim YH, Jensen RA, Watanabe GL, Varghese A, Hoppe RT. Clinical stage IA (limited patch and plaque) mycosis fungoides. A long-term outcome analysis. Archives of Dermatology 1996;132(11):1309-13. [PMID: ] [PubMed] [Google Scholar]
Kim 2003
- Kim YH, Liu HL, Mraz-Gernhard S, Varghese A, Hoppe RT. Long-term outcome of 525 patients with mycosis fungoides and Sézary syndrome: Clinical prognostic factors and risk for disease progression. Archives of Dermatology 2003;139(7):857-66. [PMID: ] [DOI] [PubMed] [Google Scholar]
Kim 2015
- Kim YH, Tavallaee M, Sundram U, Salva KA, Wood GS, Li S, et al. Phase II investigator-initiated study of brentuximab vedotin in mycosis fungoides and Sézary syndrome with variable CD30 expression level: a multi-institution collaborative project. Journal of Clinical Oncology 2015;33(32):3750–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Korgavkar 2013
- Korgavkar K, Xiong M, Weinstock M. Changing incidence trends of cutaneous T-cell lymphoma. JAMA Dermatology 2013;149(11):1295-9. [DOI: 10.1001/jamadermatol.2013.5526] [DOI] [PubMed] [Google Scholar]
Korpusik 2007
- Korpusik D, Homey B, Stege H, Bruch-Gerharz D, Schulte KW. Mycosis fungoides: Complications of long-term treatment with PUVA and ECP [Mycosis fungoides: Komplikationen einer Langzeitbehandlung mit PUVA und ECP]. Hautarzt 2007;58(4):298-9. [EMBASE: 46623430] [DOI] [PubMed] [Google Scholar]
Kütting 1997
- Kütting B, Böhm M, Luger TA, Bonsmann G. Oropharyngeal lichen planus associated with interferon-alpha treatment for mycosis fungoides: a rare side-effect in the therapy of cutaneous lymphomas. British Journal of Dermatology 1997;137(5):836-7. [DOI] [PubMed] [Google Scholar]
Legroux‐Crespel 2003
- Legroux-Crespel E, Lafaye S, Mahé E, Picard-Dahan C, Crickx B, Sassolas B, et al. Seizures during interferon alpha therapy: three cases in dermatology. Annales de Dermatologie et de Venereologie 2003;130(2 Pt 1):202-4. [PubMed] [Google Scholar]
Lessin 2011
- Lessin S, Duvic M, Guitart J, Pandya A, Strober B, Olsen E, et al. Positive results of a pivotal phase ii trial testing a manufactured 0.02% Mechlorethamine gel in mycosis fungoides (mf). Journal of Investigative Dermatology 2011;131(Suppl 1):S84. [CENTRAL: CN-00843792] [Google Scholar]
Lorincz 1996
- Lorincz AL. Cutaneous T-cell lymphoma (mycosis fungoides). Lancet 1996;347(9005):871-6. [PMID: ] [DOI] [PubMed] [Google Scholar]
McCann 2012
- McCann S, Akilov OE, Geskin L. Adverse effects of denileukin diftitox and their management in patients with cutaneous T-cell lymphoma. Clinical Journal of Oncology Nursing 2012;16(5):E164-72. [DOI] [PubMed] [Google Scholar]
Micaily 1983
- Micaily B, Vonderheid EC, Brady LW. Combined moderate dose electron beam radiotherapy and topical chemotherapy for cutaneous T-Cell lymphoma. International Journal of Radiation Oncology, Biology, Physics 1983;9(4):475-9. [DOI] [PubMed] [Google Scholar]
Miettinen 1985
- Miettinen O, Nurminen M. Comparative analysis of two rates. Statistics in Medicine 1985;4(2):213-26. [PMID: ] [DOI] [PubMed] [Google Scholar]
Mna 2016
- Mna AB, Souissi A, Halouani S, El Euch D, Zahani A, Kchir N, et al. Methotrexate-induced necrolysis in tumoral-stage mycosis fungoides: a challenging diagnosis. Dermatology Online Journal 2016;22(1):16. [PubMed] [Google Scholar]
Moher 1995
- Moher D, Jadad AR, Nichol G, Penman M, Tugwell P, Walsh S. Assessing the quality of randomized controlled trials: An annotated bibliography of scales and checklists. Controlled Clinical Trials 1995;16(1):62-73. [PMID: ] [DOI] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Journal of Clinical Epidemiology 2009;62(10):1006-12. [PMID: ] [DOI] [PubMed] [Google Scholar]
Molin 1979
- Molin L, Thomsen K, Volden G. Mycosis fungoides plaque stage treated with topical nitrogen mustard with and without attempts at tolerance induction: report from the Scandinavian mycosis fungoides study group. Acta Dermato-Venereologica 1979;59(1):64-8. [PMID: ] [PubMed] [Google Scholar]
Molin 1981
- Molin L, Thomsen K, Volden G, Groth O. Photochemotherapy (PUVA) in the pretumour stage of mycosis fungoides: a report from the Scandinavian Mycosis Fungoides Study Group. Acta Dermatovenereologica (Stockholm) 1981;61(1):47-51. [PubMed] [Google Scholar]
Morales 2000
- Morales Suárez-Varela MM, Llopis González A, Marquina Vila A, Bell J. Mycosis fungoides: Review of epidemiological observations. Dermatology 2000;201(1):21-8. [PMID: ] [DOI] [PubMed] [Google Scholar]
Nath 2014
- Nath SK, Yu JB, Wilson LD. Poorer prognosis of African-American patients with mycosis fungoides: an analysis of the SEER dataset, 1988 to 2008. Clinical Lymphoma, Myeloma & Leukemia 2014;14(5):419-23. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Newman 1997
- Newman JM, Rindler JM, Bergfeld WF, Brydon JK. Stevens-Johnson syndrome associated with topical nitrogen mustard therapy. Journal of the American Academy of Dermatology 1997;36(1):112-4. [DOI] [PubMed] [Google Scholar]
Olsen 2007
- Olsen E, Vonderheid E, Pimpinelli N, Willemze R, Kim Y, Knobler R, et al. Revisions to the staging and classification of mycosis fungoides and Sézary syndrome: a proposal of the International Society for Cutaneous Lymphomas (ISCL) and the cutaneous lymphoma task force of the European Organization of Research and Treatment of Cancer (EORTC). Blood 2007;110(6):1713-22. [PMID: ] [DOI] [PubMed] [Google Scholar]
Olsen 2011
- Olsen EA, Whittaker S, Kim YH, Duvic M, Prince HM, Lessin SR, et al. Clinical end points and response criteria in mycosis fungoides and Sezary syndrome: a consensus statement of the International Society for Cutaneous Lymphomas, the United States Cutaneous Lymphoma Consortium, and the Cutaneous Lymphoma Task Force of the European Organisation for Research and Treatment of Cancer. Journal of Clinical Oncology 2011;29(18):2598-607. [PMID: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Olsen 2016
- Olsen EA, Hodak E, Anderson T, Carter JB, Henderson M, Cooper K, et al. Guidelines for phototherapy of mycosis fungoides and Sézary syndrome: A consensus statement of the United States Cutaneous Lymphoma Consortium. Journal of the American Academy of Dermatology 2015;74(1):27-58. [DOI: 10.1016/j.jaad.2015.09.033] [PMID: ] [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration., 2014.
Rupoli 2005
- Rupoli S, Goteri G, Pulini S, Filosa A, Tassetti A, Offidani M, et al. Long-term experience with low-dose interferon-alpha and PUVA in the management of early mycosis fungoides. European Journal of Haematology 2005;75(2):136-45. [DOI] [PubMed] [Google Scholar]
Sanchez 1977
- Sánchez Yus E, Surárez Martín E. Contact urticaria and anaphylactic reactions induced by topical application of nitrogen mustard. Actas Dermo-Sifiliograficas 1977;68(12):39-44. [PubMed] [Google Scholar]
Sauder 2003
- Sauder DN. Imiquimod: modes of action. British Journal of Dermatology 2003;149(s66):5-8. [DOI] [PubMed] [Google Scholar]
Sausville 1988
- Sausville EA, Eddy JL, Makuch RW, Fischmann AB, Schechter GP, Matthews M, et al. Histopathologic staging at initial diagnosis of mycosis fungoides and the Sézary syndrome. Definition of three distinctive prognostic groups. Annals of Internal Medicine 1988;109(5):372-82. [PMID: ] [DOI] [PubMed] [Google Scholar]
Schlaak 2013
- Schlaak M, Pickenhain J, Theurich S, Skoetz N, Bergwelt-Baildon M, Kurschat P. Allogeneic stem cell transplantation versus conventional therapy for advanced primary cutaneous T-cell lymphoma. Cochrane Database of Systematic Reviews 2013, Issue 8. [DOI: 10.1002/14651858.CD008908.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schwartzman 1994
- Schwartzman R, Cidlowski J. Glucocorticoid-induced apoptosis of lymphoid cells. International Archives of Allergy and Immunology 1994;105(4):347-54. [PMID: ] [DOI] [PubMed] [Google Scholar]
Simoni 1987
- Simoni R, Cavalieri R, Coppola G, Ricciotti L, De Pità O, Criscuolo D, et al. Recombinant leukocyte interferon alfa-2a in the treatment of mycosis fungoides. Journal of Biological Regulators and Homeostatic Agents 1987;1(2):93-9. [PubMed] [Google Scholar]
Sokolowska‐Wojdylo 2016
- Sokolowska-Wojdylo M, Florek A, Zaucha JM, Chmielowska E, Giza A, Knopinska-Posluszny W, et al. Polish lymphoma research group experience with bexarotene in the treatment of cutaneous T-cell lymphoma. American Journal of Therapeutics 2016;23(3):e749-56. [DOI] [PubMed] [Google Scholar]
Spaccarelli 2015
- Spaccarelli N, Rook AH. The use of interferons in the treatment of cutaneous T-cell lymphoma. Dermatologic Clinics 2015;33(4):731-45. [PMID: ] [DOI] [PubMed] [Google Scholar]
Spitzer 1981
- Spitzer WO, Dobson AJ, Hall J, Chesterman E, Levi J, Shepherd R, et al. Measuring the quality of life of cancer patients: a concise QL-index for use by physicians. Journal of Chronic Diseases 1981;34(12):585-97. [PMID: ] [DOI] [PubMed] [Google Scholar]
Talpur 2012
- Talpur R, Duvic M. Pilot study of denileukin diftitox alternate dosing regimen in patients with cutaneous peripheral T-cell lymphomas. Clinical Lymphoma, Myeloma & Leukemia 2012;12(3):180-5. [DOI] [PubMed] [Google Scholar]
Toumishey 2015
- Toumishey E, Prasad A, Dueck G, Chua N, Finch D, Johnston J, et al. Final report of a phase 2 clinical trial of lenalidomide monotherapy for patients with T-cell lymphoma. Cancer 2015;121(5):716-23. [DOI] [PubMed] [Google Scholar]
Trautinger 2006
- Trautinger F, Knobler R, Willemze R, Peris K, Stadler R, Laroche L, et al. EORTC consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome. European Journal of Cancer 2006;42(8):1014-30. [PMID: ] [DOI] [PubMed] [Google Scholar]
Trautinger 2017
- Trautinger F, Eder J, Assaf C, Bagot M, Cozzio A, Dummer R, et al. European organisation for research and treatment of cancer consensus recommendations for the treatment of mycosis fungoides/Sézary syndrome - Update 2017. European Journal of Cancer 2017;77:57-74. [DOI: 10.1016/j.ejca.2017.02.027] [PMID: ] [DOI] [PubMed] [Google Scholar]
van Doorn 2000
- Doorn R, Van Haselen CW, Voorst Vader PC, Geerts ML, Heule F, Rie M, et al. Mycosis fungoides: Disease evolution and prognosis of 309 Dutch patients. Archives of Dermatology 2000;136(4):504-10. [PMID: ] [DOI] [PubMed] [Google Scholar]
van Scott 1973
- Van Scott EJ, Kalmanson JD. Complete remissions of mycosis fungoides lymphoma induced by topical nitrogen mustard (HN2). Control of delayed hypersensitivity to HN2 by desensitization and by induction of specific immunologic tolerance. Cancer 1973;32(1):18-30. [PMID: ] [DOI] [PubMed] [Google Scholar]
Vegna 1990
- Vegna ML, Papa G, Defazio D, Pisani F, Coppola G, De Pità O, et al. Interferon alpha-2a in cutaneous T-cell lymphoma. European Journal of Haematology. Supplementum 1990;52:32-5. [DOI] [PubMed] [Google Scholar]
Verhagen 1998
- Verhagen AP, Vet HC, Bie RA, Kessels AG, Boers M, Bouter LM, et al. The Delphi list: A criteria list for quality assessment of randomized clinical trials for conducting systematic reviews developed by Delphi consensus. Journal of Clinical Epidemiology 1998;51(12):1235-41. [PMID: ] [DOI] [PubMed] [Google Scholar]
Vonderheid 2006
- Vonderheid EC, Pena J, Nowell P. Sézary cell counts in erythrodermic cutaneous T-cell lymphoma: Implications for prognosis and staging. Leukemia & Lymphoma 2006;47(9):1841-56. [PMID: ] [DOI] [PubMed] [Google Scholar]
Wain 2003
- Wain EM, Orchard GE, Whittaker SJ, Spittle MF, Russell-Jones R. Outcome in 34 patients with juvenile-onset mycosis fungoides: A clinical, immunophenotypic, and molecular study. Cancer 2003;98(10):2282-90. [PMID: ] [DOI] [PubMed] [Google Scholar]
Weinstock 1988
- Weinstock MA, Horm JW. Mycosis fungoides in the United States. Increasing incidence and descriptive epidemiology. JAMA 1988;260(1):42-6. [PMID: ] [PubMed] [Google Scholar]
Weinstock 1999
- Weinstock MA, Reynes JF. The changing survival of patients with mycosis fungoides: A population-based assessment of trends in the United States. Cancer 1999;85(1):208-12. [PMID: ] [PubMed] [Google Scholar]
Whittaker 2003
- Whittaker SJ, Marsden JR, Spittle M, Russell Jones R. Joint British Association of Dermatologists and U.K. Cutaneous Lymphoma Group guidelines for the management of primary cutaneous T-cell lymphomas. British Journal of Dermatology 2003;149(6):1095-107. [PMID: ] [DOI] [PubMed] [Google Scholar]
Willemze 1997
- Willemze R, Kerl H, Sterry W, Berti E, Cerroni L, Chimenti S, et al. EORTC classification for primary cutaneous lymphomas: a proposal from the Cutaneous Lymphoma Study Group of the European Organization for Research and Treatment of Cancer. Blood 1997;90(1):354-71. [PMID: ] [PubMed] [Google Scholar]
Willemze 2005
- Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, et al. WHO-EORTC classification for cutaneous lymphomas. Blood 2005;105(10):3768-85. [PMID: ] [DOI] [PubMed] [Google Scholar]
Willemze 2019
- Willemze R, Cerroni L, Kempf W, Berti E, Facchetti F, Swerdlow SH, et al. The 2018 update of the WHO-EORTC classification for primary cutaneous lymphomas. Blood 2019 Jan 11 [Epub ahead of print]. [DOI: 10.1182/blood-2018-11-881268] [DOI] [PMC free article] [PubMed]
Wu 2009
- Wu PA, Kim YH, Lavori PW, Hoppe RT, Stockerl-Goldstein KE. A meta-analysis of patients receiving allogeneic or autologous hematopoietic stem cell transplant in mycosis fungoides and Sezary syndrome. Biology of Blood & Marrow Transplantation 2009;15(8):982-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yoo 1996
- Yoo EK, Rook AH, Elenitsas R, Gasparro FP, Vowels BR. Apoptosis induction of ultraviolet light A and photochemotherapy in cutaneous T-cell Lymphoma: relevance to mechanism of therapeutic action. Journal Investigative Dermatology 1996;107(2):235-42. [PMID: ] [DOI] [PubMed] [Google Scholar]
Youssef 2013
- Youssef M, Mokni S, Belhadjali H, Aouem K, Moussa A, Laatiri A, et al. Cyclophosphamide-induced generalised reticulated skin pigmentation: a rare presentation. International Journal of Clinical Pharmacy 2013;35(3):309-12. [DOI] [PubMed] [Google Scholar]
Zackheim 1996
- Zackheim HS, Kashani-Sabet M, Hwang ST. Low-dose methotrexate to treat erythrodermic cutaneous T-cell lymphoma: results in twenty-nine patients. Journal of the American Academy of Dermatology 1996;34(4):626-31. [DOI] [PubMed] [Google Scholar]
Zackheim 1999
- Zackheim HS, Amin S, Kashani-Sabet M, McMillan A. Prognosis in cutaneous T-cell lymphoma by skin stage: Long-term survival in 489 patients. Journal of the American Academy of Dermatology 1999;40(3):418-25. [PMID: ] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Weberschock 2011
- Weberschock T, Rehberger P, Roellig C, Bunch C, Schmitt J, Bauer A. Interventions for mycosis fungoides. Cochrane Database of Systematic Reviews 2011, Issue 1. [DOI: 10.1002/14651858.CD008946] [DOI] [PubMed] [Google Scholar]
Weberschock 2012
- Weberschock T, Strametz R, Lorenz M, Röllig C, Bunch C, Bauer A, et al. Interventions for mycosis fungoides. Cochrane Database of Systematic Reviews 2012, Issue 9. [DOI: 10.1002/14651858.CD008946.pub2] [DOI] [PubMed] [Google Scholar]