

Abstract
Oxygen is both vital and toxic to life. Molecular oxygen is the most used substrate in the human body and is required for several hundred diverse biochemical reactions. The discovery of the PHD-HIF-pVHL system revolutionized our fundamental understanding of oxygen sensing and cellular adaptations to hypoxia. It deepened our knowledge of the biochemical underpinnings of numerous diseases, ranging from anemia to cancer. Cellular dysfunction and tissue pathology can result from a mismatch of oxygen supply and demand. Recent work has shown that mitochondrial disease models display tissue hyperoxia and that disease pathology can be reversed by normalization of excess oxygen, suggesting that certain disease states can potentially be treated by modulating oxygen levels. In this review, we describe cellular and organismal mechanisms of oxygen sensing and adaptation. We provide a revitalized framework for understanding pathologies of too little or too much oxygen.
Reflections on the Early Adventures of Oxygen Research
Oxygen was introduced into Earth’s atmosphere nearly 2.5 billion years ago, marking a new era in the evolution of complex life forms. The appearance of photosynthetic cyanobacteria led to the Great Oxidation Event and caused a mass extinction of anaerobic species [1]. Eventually, this selective pressure gave rise to the first oxygen-sensing and detoxifying pathways, while simultaneously allowing for hundreds of oxygen-dependent biochemical reactions and highly efficient ATP production through oxidative phosphorylation (Box 1). The apparent trade-off between the pros and cons of an oxygen-rich environment are a constant theme in human development, health, and disease. Here, we discuss several key concepts central to this theme: (i) How do cells and organisms sense changes in oxygen levels? (ii) How do cells and organisms adapt to variations in oxygen levels? (iii) What happens when oxygen supply and demand are mismatched? (iv) Which diseases and pathologies arise from such an imbalance? (v) How can we modulate oxygen levels for therapeutic benefit?
Box 1. Oxygen-Dependent Reactions.
Oxygen is the most used substrate in the human body and is required for several hundred different biochemical reactions. Yet, little is known about which reactions are affected as a function of O2 tension. Oxygen-dependent enzymes include over 60 different α-ketoglutarate (2-oxoglutarate)-dependent dioxygenases [6]. Such enzymes are obligate steps in a diverse set of pathways, including collagen maturation (e.g., LOX), carnitine synthesis (e.g., TMLHE), histone demethylation (e.g., KDM and JMJ enzymes), DNA methylation (e.g., TET enzymes), protein hydroxylation [e.g., prolyl hydroxylase domain (PHD) hydroxylases], and ribosome modifications (e.g., OGFOD1), to name a few [7–11].
Oxygen is also central to amino acid catabolism and lipid metabolism. For example, tryptophan dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO1) shunt tryptophan towards de novo NAD+ synthesis and separately, tryptophan hydroxylase (TPH) produces serotonin from tryptophan. Tyrosine hydroxylase (TH) is O2-dependent and responsible for the synthesis of catecholamines such as dopamine, epinephrine, and norepinephrine [12]. Additionally, aerobic conditions are needed for fatty acid and sterol metabolism. The cytochrome P450 hydroxylases use molecular oxygen to introduce hydroxyl groups at multiple steps in the mevalonate pathway for steroid and cholesterol synthesis (e.g., Cyp51A). Cholesterol synthesis is a highly O2-consuming process, requiring 11 molecules of O2 for every cholesterol molecule in eukaryotes. Furthermore, fatty acid desaturation is O2-dependent (e.g., SCD1) and is needed to maintain an optimal ratio of saturated to unsaturated fatty acids, thereby regulating cell membrane fluidity and preventing lipotoxicity [13].
And perhaps most well-known is the role of O2 as the final electron acceptor in the mitochondrial electron transport chain. Electron transport chain dysfunction can have secondary metabolic effects, including a collapse in membrane potential, impaired NADH oxidation, and secondary stress responses. As is evident from this discussion, O2 enabled a great diversification of biochemistries during the transition from anaerobic to aerobic conditions. In fact, oxygen plays a key role in nearly every aspect of mammalian metabolism. As a result, hundreds of oxygen-dependent reactions can fail during states of ischemia. The challenge ahead lies in more comprehensively studying such reactions and related adaptations as a function of PO2.
Before delving into the biochemistry of oxygen metabolism, we look to the past and draw inspiration from the rich history of the oxygen research field. Oxygen was first discovered in 1774 by the British experimental chemist, Joseph Priestley, who described ‘dephlogisticated air’ as a highly reactive, colorless gas that supported combustion. Soon after, Antoine Lavoisier named this element ‘oxygen’ from the Greek term for ‘acid-former’ [2]. The decades that followed were characterized by colorful stories of high-altitude expeditions and hot air balloon rides to study the physiological effects of low oxygen, or hypoxia (see Glossary). In the mid-19th century, the aeronaut and French chemist Gaston Tissandier experienced the life-threatening effects of acute hypobaric hypoxia when he survived the rise to an elevation of 8600 meters. His colleagues, Joseph Crocé-Spinelli and Théodore Sivel, were less fortunate and perished [3]. In a more controlled environment, Paul Bert, the French physiologist, demonstrated the acute central nervous system (CNS) effects of oxygen toxicity on larks [4]. Recently, researchers have focused on problems related to oxygen sensing and adaptation, from the carotid body at the whole-body level to the hypoxia transcriptional factor (HIF) program at the molecular scale.
The Goldilocks Oxygen Principle
Here, we review the recent advances in the field and propose a revitalized framework for oxygen metabolism: ‘The Goldilocks Oxygen Principle’. Every tissue has an ideal oxygen tension (PO2) that allows it to maintain metabolic homeostasis, which is regulated by extrinsic variables (e.g., temperature, barometric pressure) and intrinsic factors (e.g., metabolic rate, fuel preferences, physiological reserve). Tissue PO2 is regulated by oxygen delivery, oxygen consumption, and blood flow, which are controlled by complex interactions of the cardiovascular, respiratory, and nervous systems. Under normal conditions, the partial pressure of oxygen (PO2), varies widely in the human body, ranging from 100 mmHg (13.2% O2) in arterial blood, 34 mmHg (4.4% O2) in the brain, and 30 mmHg (3.8% O2) in skeletal muscle [5]. Certain organs, such as the brain and heart, are highly aerobic and sensitive to hypoxia, whereas others, such as the skeletal muscle, are more tolerant of prolonged hypoxia. Not only do different tissues have different ‘set-points’ for oxygen, we believe that different individuals and disease states also have an optimal oxygen set-point. By understanding such states of hypoxia or hyperoxia, we can better understand the interplay between homeostasis and disease pathology, allowing us to achieve the ideal set-point by ‘turning the oxygen dial’ as a therapy.
Cellular Oxygen Sensors
At the molecular level, oxygen sensors can either directly sense oxygen or respond to redox reactions (e.g., cysteine oxidation, Fe-S cluster oxidation). Here, we define oxygen sensors as: (i) having a physiologically relevant KM for O2, (ii) transducing O2-dependent reactions into a signaling cascade, and (iii) resulting in adaptive or stress responses in hypoxic conditions. The canonical PHD-HIF-pVHL pathway is conserved across all metazoans and its components have been extensively characterized as master regulators of oxygen homeostasis (Figure 1). Gregg L. Semenza, William G. Kaelin, Jr., and Sir Peter J. Ratcliffe were awarded the Nobel Prize in Physiology or Medicine in 2019 for delineating the biochemical details of this elegant stress response pathway. The three HIF-α isoforms (1α, 2α, and 3α) are basic helix-loop-helix, PAS domain-containing, oxygen labile proteins that form heterodimers with the constitutively expressed HIF-1β (ARNT) [14]. Three PHD proteins (PHD 1,2,3), which are also known as egg-laying defective nine (EglN 2, 1, and 3, respectively) isoenzymes, regulate HIF-α abundance via prolyl hydroxylation [15–17]. Of these, PHD2 is the main oxygen sensor and has the lowest O2 affinity. Under normoxic conditions, HIF-α undergoes oxygen-dependent hydroxylation on two highly conserved prolyl residues. This hydroxylated form of HIF is recognized by the E3 ligase and tumor suppressor protein, pVHL, which marks the protein for degradation [18,19]. Factor inhibiting HIF (FIH-1) is an additional regulator that serves as a fine-tuner of the HIF response. More specifically, FIH-1 impairs HIF-1 α transcriptional activity by hydroxylating the C terminal transactivation domain of HIF-1α, thus inhibiting its interaction with its transcriptional coactivators, p300 and CREP binding protein (CBP) [20–22]. For comprehensive reviews of the PHD-HIF pathway, refer to [23–25].
Figure 1. Cellular Oxygen Sensors.
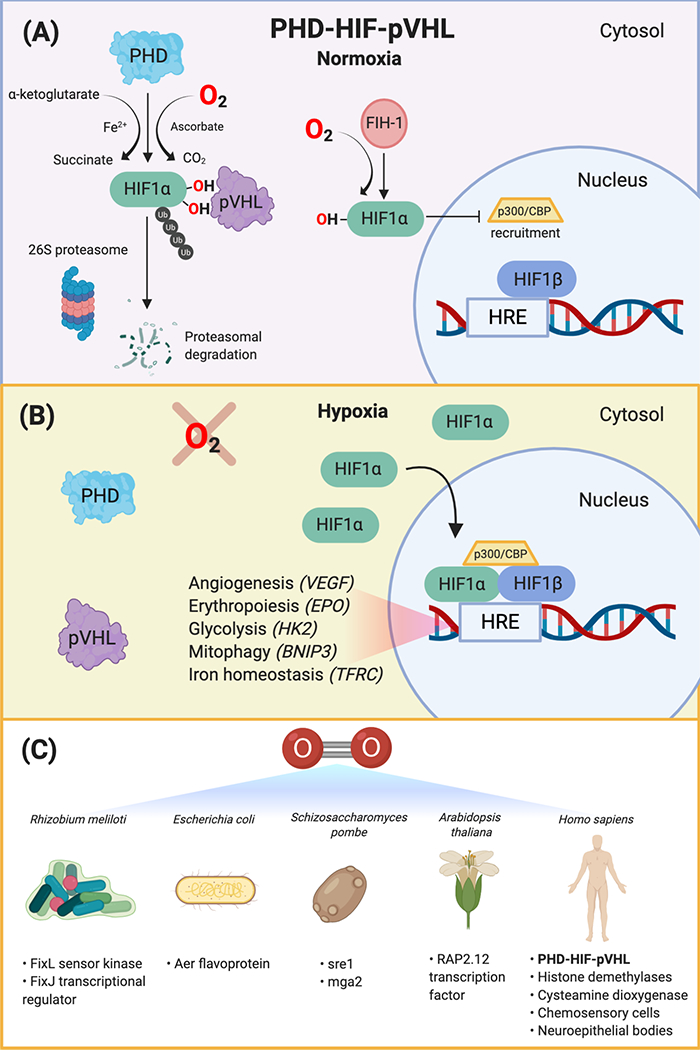
(A)The PHD-HIF-pVHL oxygen sensing pathway is conserved across all metazoans. Under normoxic conditions, PHD proteins hydroxylate the HIF-α transcription factor at two highly-conserved prolyl residues on the oxygen-dependent degradation domain, leading to its recognition by the tumor suppressor, pVHL. pVHL is the recognition component of a ubiquitin E3 ligase complex that polyubiquitylates HIF-α, marking it for proteasomal degradation by the 26S proteasome. Factor inhibiting HIF1 (FIH-1) is an asparagine hydroxylase that hydroxylates HIF in normoxia and prevents recruitment of the transcription coactivators, p300 and CBP. (B) Under hypoxic conditions, HIF-α is not degraded. HIF-α accumulates in the cytosol and translocates to the nucleus, where it binds to the conserved HRE sequence on DNA, its constitutively active and oxygen-insensitive partner, HIF-1β (ARNT), and the transcription coactivators, p300 and CBP. This complex transcriptionally activates hundreds of genes that allow cells to adapt to hypoxic environments, including VEGF, EPO, HK2, BNIP3, and TFRC. (C) There are various oxygen sensors across organisms, including Gram-negative bacteria (Rhizobium meliloti and Escherichia coli), fission yeast (Schizosaccharomyces pombe), plants (Arabidopsis thaliana), and metazoans (e.g., Homo sapiens). These systems, including the FixL sensor kinase and FixJ transcriptional response regulator, Aer flavoprotein, sre1 and mga2 transcriptional regulators, RAP2.12 transcription factor, histone demethylases, chemosensory cells in the carotid body, and intrapulmonary neuroepithelial bodies, transduce oxygen-dependent reactions into a signaling cascade that result in adaptive responses in hypoxic conditions. Abbreviations: BNIP3, BCL2 interacting protein 3; CBP, cyclic-AMP response element binding protein (CREB) binding protein; EPO, erythropoietin; TFRC, transferrin receptor; HIF, hypoxia-inducible factor; HK2, hexokinase-2, HRE, hypoxia response element; PHD, prolyl hydroxylase domain; pVHL, von-Hippel-Lindau tumor suppressor protein; VEGF, vascular endothelial growth factor.
Recently, researchers have discovered several additional oxygen-sensing pathways that appear to be HIF-independent. As mentioned above, there are 60+ enzymes that belong to the same enzyme family as PHDs: the -ketoglutarate-dependent dioxygenases. Of these, several histone demethylase enzymes have been shown to sense oxygen and induce changes in histone methylation, thereby affecting gene expression. KDM5A and KDM6A are O2-sensitive Jumonji C domain histone lysine demethylases that directly control chromatin and cell fate by regulating histone methylation homeostasis [26,27]. Other members of this family are also likely to be oxygen sensors that coordinate cellular adaptations to hypoxia.
Cysteamine dioxygenase (ADO) is a cysteine dioxygenase that transduces responses to hypoxia in both animals and plants via an O2-dependent pathway for protein degradation [28]. More specifically, in normoxia, an O2-dependent modification of an N terminal cysteine residue triggers degradation of a subset of proteins. In Arabidopsis thaliana, a conserved N terminal amino acid sequence of the ethylene response factor transcription factor, RAP2.12, can undergo post-translational modification in response to oxygen; in hypoxic conditions, RAP2.12 translocates to the nucleus and activates anaerobic gene expression. In normoxia, this transcription factor is degraded via the N-end rule pathway [29]. In this manner, hypoxia can be sensed on a shorter timescale, directly affecting the protein stability of targets. This oxygen-sensing pathway may serve as a bridge to HIF-dependent adaptations that require an orchestrated transcriptional response over a longer timescale (Figure 1).
Lower organisms that lack PHD-HIF enzymes have other unique methods of sensing and adapting to hypoxia. The gram-negative nitrogen-fixing bacterium, Rhizobium meliloti, utilizes a two-component system oxygen sensor, FixL (sensor kinase)/FixJ (transcriptional activator), to control expression of nitrogen fixation genes, nifA and fixK, in response to hypoxia via a heme-binding region [30,31]. Escherichia coli can sense oxygen levels using the Aer redox flavoprotein, enabling migration towards or away from oxygen gradients [32]. This behavior allows E. coli to respond to changes in PO2 and optimize energy utilization [33]. The fission yeast, Schizosaccharomyces pombe, uses homologs of the sterol regulatory SREBP pathway, mga2 (functionally analogous to SREBP-1) and sre1 (SREBP-2), to stimulate transcription of sterol synthesis genes and other O2-dependent enzymes that are required for anaerobic growth (Figure 1) [34–36].
Whole-Body Oxygen Sensors
In addition to cellular oxygen sensing, there are whole-body mechanisms of oxygen sensing and adaptation. A few notable examples exist during developmental transition from the hypoxic womb to the relatively O2-rich atmosphere after birth. Placental trophoblasts are cells that provide nutrients to the early embryo and first-trimester placenta, which is characterized by cellular hypoxia (PO2 <20 mmHg). In the second trimester, PO2 levels rise, driving trophoblast differentiation and placental maturation [37]. As another example, fetal blood circulation bypasses the nonfunctional pulmonary circulation via the foramen ovale and the ductus arteriosus [38]. Upon the first few breaths of the newborn, PO2 rapidly rises and pulmonary vascular resistance decreases, leading to the anatomical closure of the ductus. Failure of this O2-dependent closure can lead to pulmonary hypertension (PH).
Postdevelopment, mammals can acutely sense and adapt to oxygen levels, largely via cardiorespiratory responses. The carotid body is a neural crest-derived organ located at the bifurcation of the common carotid artery that senses arterial blood O2 tension and lactate by type I (glomus) cells and acts as the principal arterial chemoreceptor. In hypoxia, hyperventilation is triggered via inhibition of O2-sensitive K+ channels in glomus cells and subsequent afferent nerve activation to the brainstem respiratory center [39,40]. Hypoxia-induced hyperventilation increases the alveolar ventilation rate, thereby causing respiratory alkalosis and a leftward shift of the hemoglobin saturation curve. Additional hypoxia sensors include intrapulmonary neuroepithelial bodies (NEB), which respond to changes in airway oxygenation. NEB are composed of innervated clusters of cells that regulate NADPH oxidase-O2 sensitive K+ channels and downstream intracellular signaling pathways in response to hypoxia [41]. It is likely that additional tissues sense and respond to O2. Future work is needed to discover additional oxygen sensors that integrate the cellular and whole-body oxygen-sensing mechanisms.
Cellular Oxygen Adaptations
Hypoxia-induced HIF activity initiates a robust transcriptional program of more than 200 genes. Broadly, many of these adaptations are aimed at either increasing oxygen delivery or decreasing oxygen consumption, in order to meet metabolic demands and normalize local PO2. For example, HIF induction triggers erythropoiesis [erythropoietin (EPO)] and iron homeostasis [e.g., transferrin (TF)] to increase circulating red blood cell (RBC) levels [42,43]. Additional adaptations are aimed at shifting carbon flux from oxidative phosphorylation to anaerobic glycolysis (e.g., induction of nearly all glycolytic enzymes, glucose transporters, and lactate transporters). Hypoxia results in phosphorylation and inactivation of pyruvate dehydrogenase (via HIF-dependent upregulation of PDK1), to downregulate glucose-derived oxidative phosphorylation and tricarboxylic acid (TCA) cycle flux [44]. Hypoxia-induced reductive carboxylation is a process by which glutamine-derived carbons are used to run the TCA cycle in reverse, in order to generate citrate for de novo fatty acid synthesis [45,46]. In Caenorhabditis elegans, hypoxia leads to H2S accumulation and promotes interaction of PHD and CYSL-1, a member of the cysteine synthase/sulfhydrylase gene family, thus stabilizing HIF-1 transcriptional activity [47]. Additionally, several isoform/subunit switches have been reported in the electron transport chain that regulate more efficient electron flux during hypoxia, including the HIF-dependent induction of NDUFA4L2 and COX4I2 subunits [48,49]. Together, such adaptations redirect fuel sources for biosynthetic and energy-producing reactions, in a manner that minimizes dependence on the O2-dependent electron transport chain (Figure 2) [50].
Figure 2. Cellular Hypoxia Adaptations.
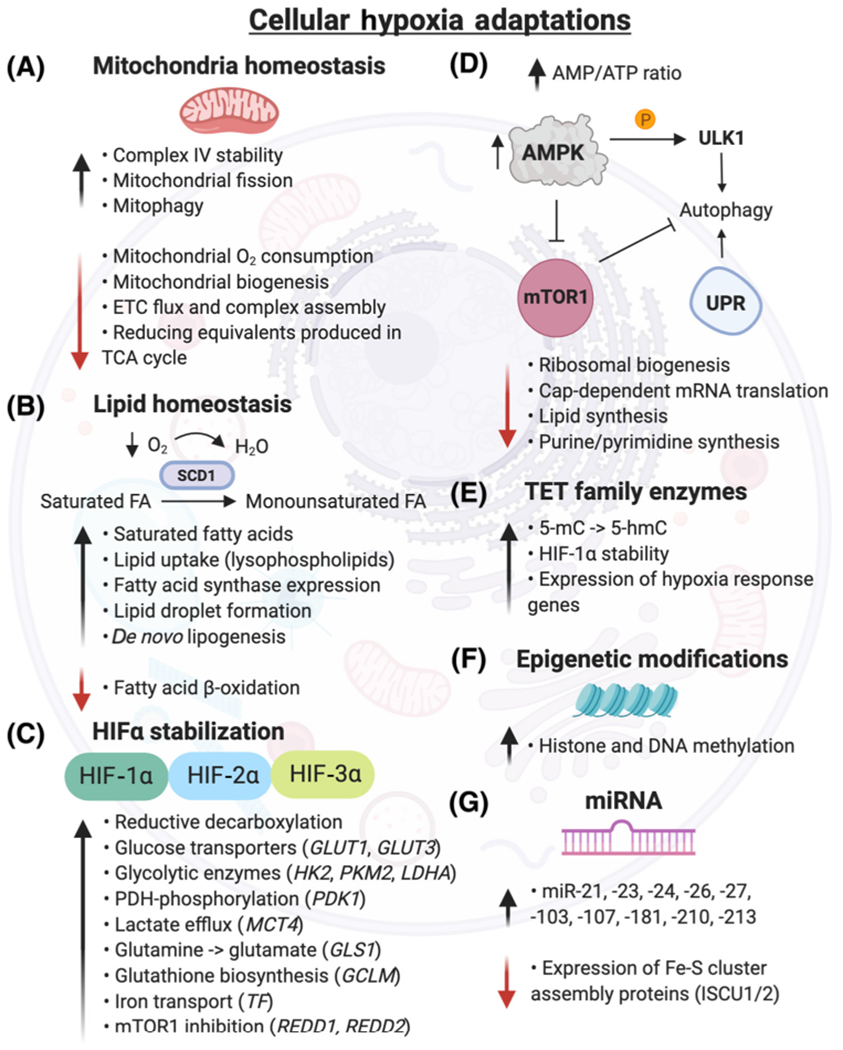
(A) Mitochondria adapt to hypoxia by increasing cytochrome c oxidase (complex IV) stability, which facilitates electron transport to O2. Mitochondria biogenesis, ETC flux, and respiration are downregulated, reducing O2 consumption and reactive oxygen species production [223]. Hypoxia induces mitochondrial fission by increasing the activity of the E3 ubiquitin ligase, SIAH2, which degrades AKAP121, subsequently increasing Drp1/Fis1 interaction [224]. (B) Under hypoxia, cells become deficient in unsaturated fatty acids by reduced O2-dependent desaturation of saturated fatty acids by stearoyl-CoA desaturases (e.g., SCD1). Hypoxic cells increase uptake of lysophospholipids and formation of lipid droplets, which serve as extra supply stores for lipids and help buffer against saturated lipid species [225]. Hypoxia downregulates fatty acid β-oxidation [226]. (C) Gene expression of several HIF targets are highlighted, though hundreds of genes are regulated by HIF stabilization [24]. (D) Hypoxia andthe subsequent energy deficiency (elevated AMP/ATP ratio) inhibit mTOR1 through AMP-activated protein kinase (AMPK). The downstream pathways regulated by mTOR1, including ribosomal and lipid synthesis, are downregulated in hypoxia, while apoptosis is increased. Unc-51-like kinase 1 (ULK1), an autophagy initiation serine/threonine protein kinase, is directly phosphorylated by AMPK in hypoxia, initiating a proautophagy signal [227]. Severe hypoxia (<0.01% O2) promotes the transcriptional response of essential autophagy genes and activates the UPR in a HIF-independent manner [228]. (E) Ten-eleven translocation methylcytosine dioxygenases (TET) catalyze the conversion of 5-methylcytosine (5-mC) to 5-hydroxymethylcytosine (5-hmC), increasing DNA methylation and gene expression of hypoxia responsive genes [8]. (F) Hypoxia induces changes in histone and DNA methylation via KDM and TET enzymes, respectively, which regulate chromatin and methylation homeostasis. (G) Numerous microRNAs are induced by hypoxia. miR-210 is increased in a HIF-1α-dependent manner and represses iron-sulfur cluster assembly proteins (ISCU1/2), resulting in decreased integrity of Fe-S cluster proteins [53]. Abbreviations: ETC, electron transport chain; FA, fatty acid; GCLM, glutamate-cysteine ligase modifier subunit; GLS1, glutaminase; GLUT, glucose transporter; HIF, hypoxia-inducible factor; HK2, hexokinase 2; KDM, lysine demethylase; LDHA, lactate dehydrogenase A; MCT4, monocarboxylate transporter 4; mTOR, mammalian target of rapamycin; PDK1, pyruvate dehydrogenase kinase; PKM2, pyruvate kinase muscle isozyme 2; REDD1/2, DNA damage response; TCA, tricarboxylic acid cycle; TF, transferrin; UPR, unfolded protein response.
Additional cellular adaptations are HIF-independent. These include chromatin remodeling, epigenetic changes (TETs and DNA methylation) [51], miRNA expression (also known as ‘hypoxamirs’) [52,53], post-translational processes (e.g., cysteine thiol redox status) [54], and induction of additional stress responses (e.g., NF-κβ, CEBP, and AP-1 stress responses) [55]. For example, hypoxia can cause a secondary ATP crisis, resulting in a high AMP/ATP ratio. This is sensed by AMP-activated protein kinase (AMPK), which results in mammalian target of rapamycin (mTOR) inhibition and downregulation of energy-intensive processes, such as translation and lipid synthesis, and upregulation of autophagy. Hypoxia also inhibits mTOR1 independently of AMPK by induction of the HIF-1α target genes, DNA damage response 1 and 2 (REDD1, REDD2) [56]. Lipid homeostasis is also tightly regulated by hypoxia since sterol synthesis and fatty acid desaturation are oxygen-requiring processes (e.g., formation of monounsaturated fatty acids by SCD1) and are important to maintain proper cell integrity and membrane fluidity in hypoxia [57] Figure 2). We recently found that genes involved in lipid metabolism (e.g., AMFR, SREBF1), peroxisomal biogenesis (e.g., PEX10, ACSL4), and ether phospholipid biosynthesis (e.g., FAR1, AGPS) can be selectively essential in low oxygen, likely in part to protect against saturated fatty acid toxicity in hypoxia [113]. Focused discussions of cellular adaptations to hypoxia are reviewed elsewhere [58].
Whole-Body Oxygen Adaptations
Several non-human animals are particularly tolerant of hypoxic environments. We highlight a few of these organisms that have developed unique adaptive mechanisms of extreme hypoxia tolerance (Figure 3). The naked mole rat (Heterocephalus glaber) lives in subterranean, unventilated environments with extremely low O2 levels [59]. It is also the longest-living rodent with a similar longevity quotient to humans [60]. It has several interesting adaptations, such as unique O2-binding globins in the brain, enhanced ammonia detoxication, and expression of genes associated with DNA damage repair that allow it to be relatively insensitive to low oxygen levels [61]. More recently, it was found to uptake large amounts of fructose via increased expression of ketohexokinase and the fructose transporter GLUT5 as an alternative entry point into glycolysis [62]. High-altitude birds (e.g., Gyps rueppelli) have four hemoglobins with a wide range of O2 affinities, permitting oxygen loading along a broad spectrum of O2 tensions [63]. Turtles (e.g., Chrysemys picta) use their mineralized shells to buffer against increased lactate levels from hypoxic tissues by releasing calcium and magnesium carbonates [64]. Diving mammals, such as Weddell seals (Leptonychotes weddellii), are able to acutely increase tissue oxygen delivery by triggering contractions of their spleen during deep dives, which release stored RBCs into circulation [65]. In these animals, the circulating hemoglobin concentration can increase 25–60% during dives due to splenic contraction. Interestingly, human elite apneic-divers (e.g., professional Korean diving women) are able to adapt to prolonged breath-holds by splenic contractions during deep dives to transiently increase circulating RBCs [66,67]. It is likely that future research will uncover even more creative adaptations that exist in nature across extreme organisms. Perhaps, remnants of these adaptations exist in humans or can serve as inspiration for future therapeutic approaches for states of ischemia.
Figure 3. Organismal Hypoxia Adaptations in Extreme Animals and Homo sapiens.
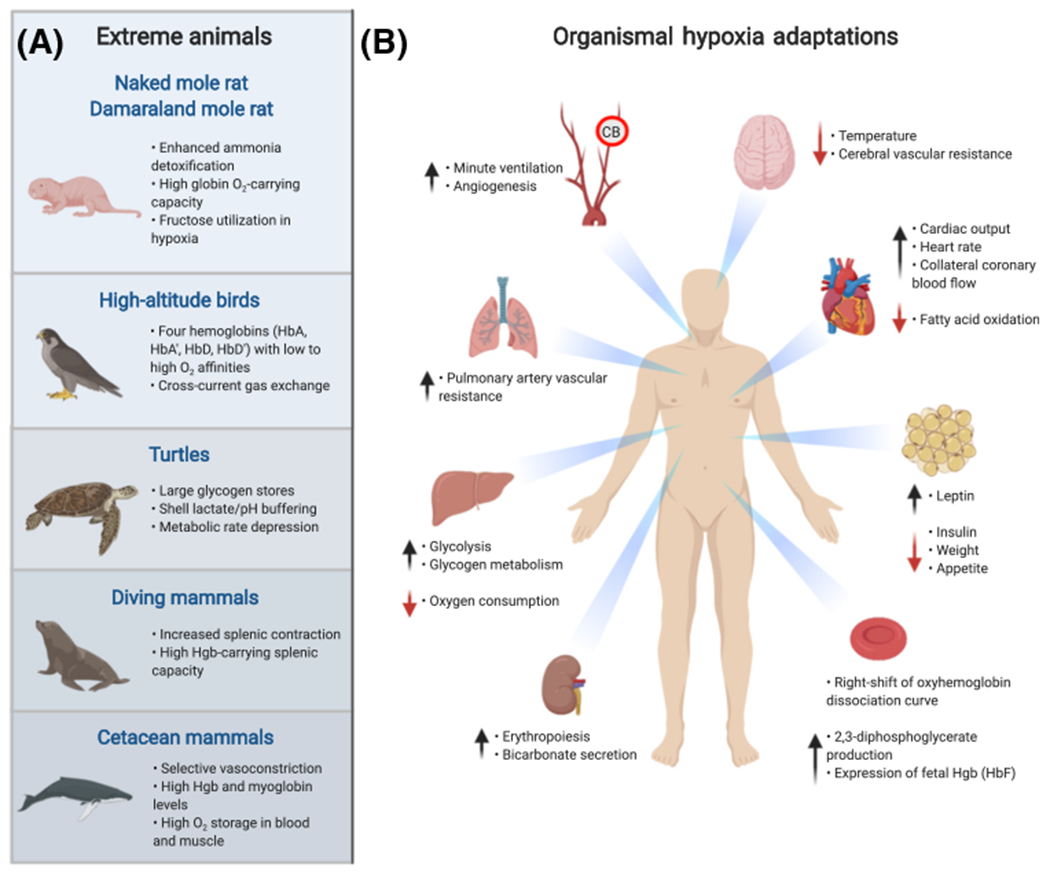
(A) The naked mole rat (Heterocephalus glaber), high-altitude birds (e.g., Gyps rueppelli), turtles (e.g., Chrysemys picta), diving mammals (e.g., Leptonychotes weddellii), and cetacean mammals (e.g., Tursiops truncatus) [229], have developed unique metabolic and physiologic adaptations to extreme hypoxic environments. (B) Hypoxia induces whole-body physiologic and tissue metabolic adaptations in Homo sapiens involving the brain, heart, adipose tissue, kidney, liver, pulmonary and systemic vasculature, and carotid body (CB). Hypoxia induces generalized vasodilation with the exception of HIF-2α-mediated vasoconstriction of pulmonary arteries, leading to pulmonary hypertension. Metabolic changes include increased circulating leptin levels, decreased insulin levels, and increased glycogen metabolism [230]. Cardiac adaptations include increased heart rate, cardiac output, and coronary artery blood flow, and decreased fatty acid oxidation rate [231]. Chemosensory type I glomus cells in the CB sense hypoxia and increase the ventilation rate via activation of action potentials in the glossopharyngeal nerve that excite central chemoreceptors in the brain. Kidneys adapt by increasing bicarbonate secretion and erythropoiesis via HIF-2α-dependent induction of erythropoietin, augmenting oxygen-carrying capacity to tissues. The oxyhemoglobin dissociation curve shifts to the right, facilitating unloading of oxygen into tissues. Hypoxia promotes erythroid expansion of mature red blood cell (RBC) precursors containing fetal hemoglobin (HbF) [232]. Abbreviation: Hgb, hemoglobin.
Human whole-body hypoxia adaptions lead to an increase of oxygen supply and decrease of oxygen demand at the tissue level (Figure 3). Acute physiologic changes that augment oxygen supply include hyperventilation mediated by the chemosensory cells in the carotid body, systemic vasodilation, and increased cardiac output. The pulmonary arteries are unique in their ability to vasoconstrict in response to hypoxia, allowing blood to be shunted away from hypoxic alveoli and subsequently correcting ventilation-perfusion mismatch. More chronic adaptations include erythropoiesis mediated by HIF-2α stabilization, angiogenesis, suppression of whole-body O2 consumption, decreased food intake, and decreased body temperature [68,69]. Chronic hypoxia also increases 2,3-diphosphoglycerate, which is a glycolytic intermediate that shifts the oxygen-hemoglobin dissociation curve to the right and improves oxygen off-loading in peripheral tissues at lower O2 tensions [70].
Toxicity of Low Oxygen
Hypoxia can have deleterious effects on the acute, chronic, and intermittent timescales. Acute, severe hypoxia exposure, which occurs when healthy individuals ascend rapidly to high altitudes (>2500 m), can cause acute mountain sickness, high-altitude cerebral edema (HACE), and high-altitude pulmonary edema (HAPE) (Table 1). In general, these adverse effects subside with acclimatization and are reversed upon return to normoxia [71]. The metabolic and physiologic susceptibility risk factors for HAPE and HACE are unknown.
Table 1.
Toxicity of Low Oxygen or the Hypoxia Response
| Type | System/organ | Exposure/condition | Effect | Refs |
|---|---|---|---|---|
| Acute | Systemic | - High altitude - Carbon monoxide poisoning |
- Acute mountain sickness - High altitude pulmonary edema (HAPE) - Pulmonary hypertension, pulmonary edema, hypoxic respiratory failure - High altitude cerebral edema (HACE) - Cerebellar ataxia, retinal hemorrhages, unconsciousness, death |
[158,159] |
| Chronic | Systemic | - High altitude - Hypoxic diseases - Chronic obstructive pulmonary disease - Emphysema - Diffuse parenchymal lung disease - VHL loss of function mutation (e.g., 598C->T Chuvash polycythemia) - Germline VHL deficiency (homozygous synonymous mutation C.222C->A, p.V74V) - PHD2 loss of function mutations - HIF-2α, EPOR gain of function mutations |
- Chronic mountain sickness - Decreased fertility - Low birth weight - Erythrocytosis - World Health Organization Group III pulmonary hypertension - Right-sided heart failure - Hypotension - Persistent hypoglycemia |
[160–168] |
| Intermittent | Systemic | - Obstructive sleep apnea - Central sleep apnea - Cheyne-Stokes respiration - Intermittent hypoxia |
- Hypertension - Obesity - Non-alcoholic fatty liver disease (NAFLD) and steatohepatitis - Insulin resistance - Dyslipidemia - Atrial fibrillation - Ventricular arrhythmias - Sudden cardiac death |
[169–175] |
| Ischemia | Myocardium Brain Acute limb ischemia Renal infarction |
- Non-ST segment elevation myocardial
infarction (MI), ST-elevation MI - Acute coronary syndrome - Ischemic stroke - Thromboembolic event |
- MI - Ischemic stroke - Kidney infarction - Peripheral arterial thrombosis - Pulmonary embolus |
[176–179] |
| Local | White adipose tissue | - Overexpression of constitutively active form
of Hif-1α - Adipocyte-specific Vhl KO - High-fat diet-induced obesity and adipose hypoxia |
- Increased local inflammation and
fibrosis - Increased proinflammatory cytokines, pathologic cardiac hypertrophy - Increased expression of inflammatory genes - Decreased expression of adiponectin - Increased adipose tissue inflammation and uncoupled respiratory state |
[180–184] |
| Intestine | - Distal ileum HIF-2α
expression in obese subjects - Intestinal epithelium-specific Vhl disruption |
- Increased obesity, NAFLD, hepatic
steatosis - Increased colon tumor multiplicity and progression from adenomas to carcinomas |
[185,186] | |
| Myocardium | - Chronic cardiac-specific
Phd inactivation - Cardiac-specific Hif-1α and Vhl KO mouse model |
- Dilated cardiomyopathy - Increased cardiac steatosis, fatty acid uptake, and eccentric hypertrophy |
[187,188] | |
| Kidney | - Conditional inactivation of
Vhl in Pepck-Cre mutants - Kidney-specific Vhl KO - Pkd1 and Hif-1α co-deletion in kidney epithelium - Podocyte-specific Vhl gene loss |
- Increased renal cyst development - Polycythemia - Increased interstitial fibrosis and kidney cyst growth - Glomerulomegaly and glomerulosclerosis |
[189–192] | |
| Brain | - Controlled cortical impact traumatic brain
injury mouse model - Mouse cerebral hypoxia-ischemia model |
- Induction of LRRK2, exacerbation of neuronal
cell death - Increased expression of TIM-3 and inflammatory immune cells |
[193,194] | |
| Liver | - Temporal liver-specific Vhl
disruption - Vhl-Hif-1α mutant mice |
- Increased hepatic lipid accumulation,
steatosis, and inflammation - Severe steatohepatitis, impaired fatty acid β-oxidation - Increased circulating cholesterol levels |
[117,195,196] | |
| Lung | - Alveolar type 2 cells-specific Hif-1α conditional KO with closed-chest unilateral lung contusion | - Increased acute proinflammatory cytokines (IL-1β, IL-6, macrophage inflammatory protein-2) | [197] | |
| Malignancy | Tumor | - Inactivation of VHL tumor
suppressor gene - Hypoxia in solid tumors |
- Clear cell renal cell carcinoma - Sporadic hemangioblastoma - Paraganglioma - Pheochromocytoma - Increased chemoresistance, metastatic potential, and treatment resistance - Poor prognosis marker for malignancies, including cervical cancer, breastcancer, colorectal cancer, lymphoma, glioblastoma multiforme |
[81,198–202] |
Ischemia, defined as the lack of oxygen coupled with nutrient deprivation (e.g., glucose, glutamine, fatty acids) from restriction of blood flow, is characteristic of myocardial infarction and stroke, which are leading causes of morbidity and mortality worldwide [72]. While the upstream pathology may differ, organ dysfunction and tissue necrosis ultimately result from a lack of oxygen and nutrients. The critical thresholds and responses to ischemia, including tissue metabolic rate, fuel use, oxygen extraction, inflammation, and antioxidant responses, in different organs need to be better studied to elucidate mechanisms of intrinsic ischemia tolerability.
Chronic hypoxia is linked to both systemic and tissue-level adaptation and pathology. Approximately 140 million people live at 2500 meters above sea level or higher. At moderate altitudes, children are born significantly underweight and at extreme altitudes, childbirth is no longer possible [73,74]. The causes of developmental issues and infertility are unknown. The adaptive effects of chronic hypoxia can also result in PH from endothelial HIF-2α upregulation and right-sided heart failure [75,76]. A proportion of individuals living at high altitude suffer from chronic mountain sickness, which is a disease secondary to polycythemia and hypoxia [77]. However, many individuals are disease-free and others (e.g., Tibetan high-dwellers), have acquired genetic adaptations in the HIF2a (EPAS1) and the PHD2 genes, thereby decreasing erythrocytosis and PH [78,79].
At the tissue level, tissue-specific genetic activation of the HIF transcriptional response can have pathologic effects (Table 1). HIF activation and hypoxia are not synonymous and artificially activating HIF in normoxia can paradoxically lead to increased oxygen delivery, thereby potentially causing tissue hyperoxia. Furthermore, chronic hypoxia in solid tumors and the tumor microenvironment has been linked to aberrant angiogenic signaling, tumor proliferation and survival, resistance to chemotherapy, increased metastatic potential, and poor prognosis [80,81]. The majority of cases of clear cell renal cell carcinoma (ccRCC), the most common type of kidney cancer, are caused by VHL gene inactivation, leading to aberrant HIF accumulation. Recently, studies have shown that two pVHL targets, the transcription factors zinc fingers and homeoboxes 2 (ZHX2) and Scm-like with four malignant brain tumor domains 1 (SFMBT1), contribute to ccRCC carcinogenesis by promoting NF-κβ activation and sphingosine kinase 1 transcription, respectively [82,83]. Both of these transcription factors undergo prolyl hydroxylation (SFMBT1 by PHD2), demonstrating that the pro-oncogenic phenotype driven by solid tumor hypoxia is regulated by both HIF-dependent and HIF-independent downstream pathways. For recent reviews on hypoxia and tumor metabolism, refer to [84–86].
Toxicity of High Oxygen
At the other extreme, hyperoxia (>21% O2) has harmful effects at the cellular, tissue, and whole-body levels (Table 2). The mechanisms of hyperoxic injury are not completely understood; increased O2 levels cause superoxide radical formation, followed by dismutation to H2O2 by superoxide dismutase. In the presence of Fe2+, H2O2 is catalyzed into the highly reactive hydroxyl free radical via the Fenton reaction and serves as a membrane-permeable signaling molecule. Oxidative stress can lead to lipid peroxidation of cellular membranes, protein dysfunction, and inactivation of critical cellular enzymes, all of which contribute to cellular dysfunction, autophagy, and cell death [87,88]. However, it is largely unknown how cells adapt to hyperoxia and the pathways affected by excess oxygen independent of signaling related to reactive oxygen species (ROS).
Table 2.
Toxicity of High Oxygen
| Type | System/organ | Exposure/cond ition | Effect | Refs |
|---|---|---|---|---|
| Acute | Systemic Lungs Brain Heart Ocular |
- Supplemental oxygen in excess - Deep diving with supplemental nitrox |
- Irritability, disorientation, seizures,
ataxia, coma, death (Paul Bert effect) - Pulmonary edema, alveolar and bronchial damage - Activation of lung inflammasome - Decreased cardiac output - Higher systemic vascular resistance - Decreased coronary artery blood flow - Increased coronary vascular resistance - Increased cardiac biomarkers and larger infarct size on cardiac magnetic resonance imaging following myocardial infarction - Worse short-term functional outcomes after traumatic brain injury - Higher in-hospital mortality |
[203–207] |
| Subacute | Systemic Brain |
- Supplemental oxygen | - Increased intensive care unit
mortality - Worse functional outcomes after mechanical thrombectomy - Neurologic disability |
[208–210] |
| Chronic | Brain | - Ndufs4 KO mouse model of complex I deficiency | - Increased tissue hyperoxia leading to neurologic disease and decreased lifespan | [109] |
| Ischemia-reperfusion | Systemic Heart Brain Lung Kidney Gut Skeletal muscle |
- Ischemia followed by reoxygenation - Organ-specific ischemia-reperfusion injury - Organ allogeneic transplant |
- Induction of cytochrome p450 and increased
oxidation of polyunsaturated fatty acids into eicosanoids - Increased production of reactive oxygen species (e.g., superoxide) - Leukocyte and neutrophil infiltration - Opening of the mitochondrial permeability transition pore and subsequent apoptotic cell death - ATP depletion - Increased MCT4 expression and lactate extrusion - Decreased nitric oxide bioavailability - Succinate accumulation |
[210–218] |
| Infants | Retina Lungs |
- Supplemental oxygen | - Retinopathy of prematurity - Bronchopulmonary dysplasia Airway injury and inflammation Airway smooth muscle hypertrophy - Lower alveolar density, enlargement of parenchymal air spaces |
[219–222] |
In premature infants with immature lung development, supplemental oxygen is often delivered to support normal gas exchange. However, excess oxygen can cause retinopathy of prematurity, a pathological vasoproliferative disorder of the retina that can lead to blindness [89]. In adults, hyperoxia causes vasoconstriction, reduction in coronary blood flow, and decreased cardiac output. Chronic hyperoxia can cause adverse effects involving the CNS (e.g., altered mental status, seizures), eyes (e.g., cataracts), and lungs (e.g., pulmonary interstitial fibrosis) (Table 2). Relative hyperoxia from oxygen influx in the setting of restoration of blood flow after ischemia, known as ischemia/reperfusion (I/R), causes cell death and tissue injury through various mechanisms, including generation of ROS, lactate, and succinate accumulation, release of proinflammatory eicosanoids and cytokines, and dysregulation of nitric oxide signaling (Table 2).
Supplemental oxygen is one of the most common therapies provided in hospitalized patients, often without clear evidence of hypoxia or in excess of what is needed by the human body [i.e., arterial partial pressure of oxygen (PaO2) >100 mmHg] [90]. There is increasing clinical evidence that this therapy is not benign. In critically ill patients, excess oxygen is associated with increased intensive care unit (ICU) mortality, decrease in ventilator-free days, and neurologic disability in patients with traumatic brain injury and following cardiac resuscitation (Table 2). More rigorous prospective clinical trials are needed to delineate the toxicities associated with hyperoxia.
Hypoxia as a Therapy
The imbalance between oxygen supply and demand, coupled with organ-specific set points for (low or high) oxygen tolerance, results in negative effects from hypoxia and hyperoxia, as described. Understanding the pathologies of low or high oxygen provides us with a unique therapeutic opportunity. There is emerging preclinical evidence that oxygen can be modulated for therapeutic benefit and that hypoxia can ameliorate certain disease phenotypes, decrease the risk of cardiovascular disease, and even significantly improve survival in rare genetic disorders such as mitochondrial disease.
Hypoxia and Aging
The potential benefits of hypoxia are apparent across species from yeast to C. elegans to humans. In C. elegans, the hypoxia response via loss of VHL-1 increases lifespan, enhances resistance to β-amyloid toxicity, and improves protein homeostasis [91–93]. Hypoxia improves replicative lifespan in yeast and delays senescence in patient fibroblasts [94,95]. Indeed, almost all the hallmarks of aging are known to be ameliorated by low oxygen in cell culture settings: stem cell exhaustion, senescence, telomere attrition, and mitochondrial dysfunction [96–99]. The in vivo and organismal relevance of hypoxia on aging and age-associated pathologies remains to be determined.
Moreover, there is epidemiological data suggesting that systemic hypoxia might improve longevity in humans, as well. For example, the number of centenarians is significantly higher in Tibetans who have lived at altitude for centuries compared with the Han Chinese who ascended in more recent times [100]. The mechanism of improved longevity is unclear and might be secondary to a range of factors, including lower temperature, decreased basal metabolic rates, and adaptations of aging-associated genes in hypoxia. Furthermore, there is extensive epidemiologic data that demonstrate decreased cardiovascular disease and age-related risk factors, including diabetes, hypertension, ischemic heart disease, and obesity, in populations that live at or have been adapted to high altitude [101–104]. Interestingly, patients with cyanotic heart disease (e.g., tetralogy of Fallot) also seem to be protected from atherosclerosis and dyslipidemia [105,106]. More rigorous epidemiological studies will be needed to determine correlations between high altitude, chronic hypoxia, and disease incidence.
Hypoxia and Mitochondrial Disease
We have several lines of evidence that support using hypoxia or the hypoxia response as a potential therapeutic strategy for specific metabolic disorders. Recently, we showed that hypoxia can prevent and even reverse disease pathologies associated with mitochondrial dysfunction in rodent models of disease. In a mouse model of the pediatric mitochondrial disease, Leigh syndrome, exposure to chronic normobaric hypoxia (11% O2) rescued body weight, behavior, and neuropathology [107]. Moreover, the lifespan of this complex 1 disease mouse model was extended by fivefold. Amazingly, hypoxia exposure was even able to reverse neuropathology at a very late stage of neurologic disease. These benefits were not observed with a more conservative degree of continuous normobaric hypoxia (17% O2) or intermittent hypoxia and quickly reversed upon return to normoxia [108].
While cellular studies suggested that HIF activation could rescue the effects of electron transport chain deficiency, this did not translate in vivo. Though hypoxia exposure itself rescued disease in the Leigh syndrome mouse model, activating the HIF response was insufficient to rescue disease [109]. Instead, it appears that a broken electron transport chain leads to decreased tissue oxygen consumption, thereby causing brain hyperoxia. Indeed, others have previously observed decreased oxygen extraction and venous hyperoxia in mitochondrial disease patients upon exercise [110]. Exactly how elevated oxygen leads to tissue damage remains to be determined. We believe we have identified some of the first metabolic disorders of ‘excess oxygen’. It will be interesting to characterize which additional genetic disorders result from tissue hyperoxia.
And so the question remains: Will this therapeutic approach generalize to additional mitochondrial disorders, unrelated metabolic disorders, and common conditions? More recently, researchers showed that an additional genetic model of Leigh syndrome caused by a mutation in the complex 2 subunit succinate dehydrogenase (SDHC) could be rescued by hypoxia [111]. Additionally, hypoxia delayed symptoms in a disease model of Friederich’s ataxia [112]. In order to prioritize additional diseases for hypoxia therapy, we recently performed a genome-wide CRISPR knockout (KO) screen comparing cell growth and survival at 21% O2 versus 1% O2 [113]. This revealed over 75 monogenic disorders whose gene KO could be rescued by hypoxia. Potential disease candidates for hypoxia therapy include pyruvate dehydrogenase (PDH) deficiency, sideroblastic anemia (GLRX5), autosomal dominant optic atrophy (OPA1), and monogenic forms of Parkinson’s disease (HTRA2), among others. Future preclinical work is needed to determine whether these cellular findings extend to in vivo models of disease. In addition to mitochondrial diseases, there are preclinical studies that suggest therapeutic benefit of hypoxia or the hypoxia response in fatty liver disease [114], insulin sensitivity [115], obesity [116,117], kidney injury [118,119], chemotherapy-related toxicities [120], multiple sclerosis [121], colitis [122], and transplant allograft vasculopathy [123,124].
Ischemic Preconditioning
Due to potential long-term complications associated with chronic hypoxia, shorter-duration therapy could be safer in specific contexts. Local and remote ischemic preconditioning (RIPC) are two techniques that utilize hypoxia to protect against ischemic injury. Numerous preclinical studies demonstrate that ischemic preconditioning can attenuate subsequent ischemic injury in animal models of focal brain ischemia [125], ischemic and fibrotic kidney injury [126,127], neurodegenerative diseases (e.g., Parkinson’s disease) [128], and myocardial infarction [129]. Patients who experience stable angina prior to myocardial infarction have smaller infarctions and fewer cardiac complications [130]. This protection is partially conferred by hypoxia-induced angiogenesis and arteriogenesis, with development of collateral arteries [131].
In RIPC, brief episodes of hypoxia to an organ, for example, the skeletal muscle, can provide ischemic protection to a distant organ (e.g., heart from ischemia-reperfusion injury). The mechanisms are not fully understood, though an increase of protective secreted factors, including interleukin (IL)-10 and kynurenic acid, and reduction of proinflammatory cytokines, such as IL-6, have been implicated [132–135]. Clinical applications of RIPC in patients undergoing elective coronary artery bypass grafting surgery have largely been negative, possibly due to interaction of protective factors with hepatically metabolized anesthetic agents [136,137]. Future trials should assess the role of RIPC in acute and chronic conditions (e.g., stroke, myocardial infarction, neurodegenerative disease).
Regenerative Medicine
Most stem cells exist in hypoxic niches within the body [138]. Severe hypoxia has also been shown to cause proliferation of fetal cardiomyocytes and allow adult mammalian cardiomyocytes to exit cell cycle arrest [139,140]. Hypoxic cardiomyocytes display characteristics of proliferative neonatal cardiomyocytes and are less sensitive to oxidative DNA damage. In a preclinical adult mouse model, hypoxia can induce heart regeneration and decrease infarct size after I/R injury through mechanisms that are not fully understood [141]. PHD2 inactivation in skeletal progenitor cells supports postimplantation bone repair and survival by preserving redox balance via HIF-1α mediated conversion of glutamine to glutathione [142]. The mechanisms by which hypoxia activates stem cell niches or quiescent cells need further investigation and can potentially be widely applicable to human diseases related to aging, senescence, and tissue injury.
HIF Response as a Therapy
Recent drug developments have taken advantage of the HIF response for therapeutic benefit [143]. PHD inhibitors, which partially stabilize HIF levels and upregulate expression of HIF target genes, have shown promise in patients with chronic kidney disease (CKD)-related anemia (Figure 4). EPO is a glycoprotein hormone and specific HIF-2α target that is produced by renal interstitial fibroblasts. In patients with CKD, treatment with the PHD inhibitor, FG-4592, induces EPO as a therapeutic strategy for anemia and also decreases cholesterol levels through unclear mechanisms [144,145]. In a preclinical mouse model, PHD inhibition increases hepatic cholesterol elimination and ceramide catabolism [146]. It remains unknown what other diseases could be treated with PHD inhibition.
Figure 4. Hypoxia Response Therapy and Candidate Diseases Amenable to Hypoxia/HIF Activation or HIF Inhibition.
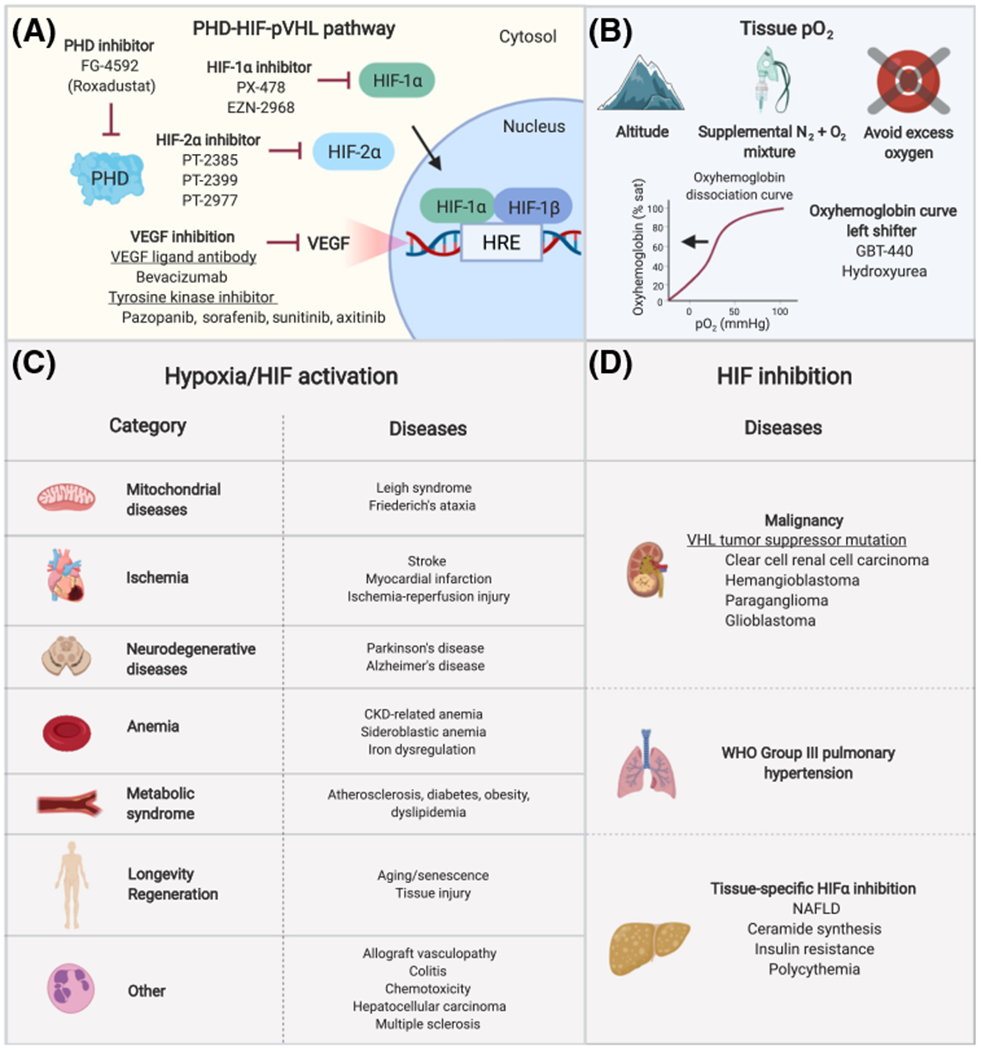
(A) Medications and novel small-molecule compounds that inhibit prolyl hydroxylase domain (PHD) proteins, HIF-1α, HIF-2α, and VEGF have shown clinical benefit in preclinical models and clinical trials [153,154,183,185,233]. PHD inhibition increases the HIF transcriptional response by preventing HIF hydroxylation, whereas HIF-1α and HIF-2α inhibition downregulate the HIF response. (B) Hypobaric hypoxia can be achieved by ascent to altitude or use of hypobaric hypoxia chambers, which simulate hypoxia by reducing atmospheric pressure. Normobaric hypoxia can be achieved by delivery of a mixture of nitrogen and oxygen gases and pressure swing adsorption systems. Tissue hypoxia can be mimicked by administration of oxyhemoglobin curve left shifters. Certain mitochondrial diseases are associated with tissue hyperoxia and preclinical studies show that pathologic phenotypes can be ameliorated by hypoxia exposure. In these conditions, excess oxygen should be avoided unless clinically indicated. (C) Category and examples of diseases that can potentially be treated with hypoxia or HIF activation (e.g., PHD inhibition). (D) Candidate diseases that can be treated with inhibition of the HIF response (e.g., HIF or VEGF inhibition). Abbreviations: CKD, chronic kidney disease; HIF, hypoxia-inducible factor; HRE, hypoxia response element; NAFLD, non-alcoholic fatty liver disease; PHD, prolyl hydroxylase domain protein; pO2, oxygen tension; VEGF, vascular endothelial growth factor; VHL, von Hippel-Lindau tumor suppressor protein; WHO, World Health Organization.
Furthermore, the downregulation of the HIF response using selective and orally active HIF-1 and HIF-2 antagonists has demonstrated therapeutic potential in preclinical studies and human Phase I and II clinical trials for various neoplasms (Figure 4). A subset of patients with ccRCC, which is driven by pVHL inactivation and constitutive overexpression of HIF-2α have shown clinical benefit with oral HIF-2α inhibitors. VEGF inhibitors, including VEGF ligand antibodies and tyrosine kinase inhibitors, are FDA approved systemic therapies for advanced renal cell carcinoma [147]. Preclinical models of PHD inhibition or HIF activation suggest therapeutic potential for neuroprotection following brain ischemia [148], allograft dysfunction [149,150], chemotherapy-related adverse effects [120], cardiac dysfunction [151], inflammatory bowel diseases [122]. Alternatively, HIF inhibition could be a therapeutic strategy for chronic hypoxia-induced WHO group III PH [152,153] and certain malignancies characterized by abnormal hypoxia signaling [154]. FIH inhibitors might be helpful for amelioration of metabolic syndromes, including diabetes and obesity [155]. Future research should further delineate which diseases can be treated by targeted manipulation of the hypoxia response, such as HIF stabilization, versus hypoxia itself, which involves numerous interrelated regulatory pathways.
Implementation of Hypoxia Therapy
Hypobaric hypoxia can be achieved by living at altitude or by gradual reduction of atmospheric pressure in a hypobaric chamber. As a reference, there is 15% O2 at 2400 meters above sea level (Aspen, CO, USA), 14% O2 at 3400 meters (Cusco, Peru), and 7% O2 at 8840 meters (Mount Everest). Commercially available at-home systems mimic altitude training by decreasing the oxygen percentage of air using pressure swing adsorption. Hypoxic air can be created by mixing nitrogen and oxygen gases at different ratios and it is feasible to make this system portable, comparable with portable oxygen tanks with delivery via nasal cannula. We can also imagine small-molecule approaches to causing tissue hypoxia. For example, left shifters of the oxyhemoglobin dissociation curve are already being developed for sickle cell anemia and can cause tissue hypoxia when given at high doses (Figure 4) [156,157].
The hypoxia response can also be targeted by medications or small-molecule inhibitors, such as PHD inhibitors and pVHL inhibitors (Figure 4). Drugs that target the HIF pathway have already shown great potential for treating anemia and renal cell carcinoma and future drug development should aim to discover druggable HIF and non-HIF targets involving the hypoxia response [83]. Prior to implementation of hypoxia therapy, future studies will need to determine which organs and diseases are most amenable to hypoxia therapy, as well as the risks of chronic use that could lead to prohibitive treatment-related adverse events or treatment resistance. Therefore, additional mechanistic studies might allow us to develop more specific and targeted small-molecule therapies.
Concluding Remarks
Oxygen is both vital and toxic to life. Hypoxia is a double-edged sword that can have detrimental and beneficial effects at the cellular, tissue, and systemic levels. Every tissue, disease state, and individual has an optimal oxygen set-point. A mismatch in oxygen supply and demand disrupts this set-point and can result in pathology. Several important questions remain unanswered about the mechanisms of how hypoxia and the hypoxia response can treat certain diseases and how to target the hypoxia pathway in select disease states. For hypoxia therapy, the degree and duration of hypoxia will need to be carefully determined to maximize the benefit to risk ratio. On the opposite end of the spectrum, much remains unknown about cellular responses to hyperoxia. There is mounting evidence that excess oxygen is toxic at the cellular and systemic levels for reasons that are not fully understood. Oxygen delivery should be considered a therapy that should be administered when indicated, titrated to meet tissue oxygen demands, and discontinued when no longer required to minimize toxic systemic side effects. Importantly, supplemental oxygen should be avoided unless absolutely indicated in patients with mitochondrial disease, with evidence indicating that pathology and disease progression are related to tissue and venous hyperoxia and that normalization of hyperoxia can have dramatic phenotypic improvements, including prolongation of life. Future research should aim to unravel how cells and tissues sense, respond, and adapt to hyperoxia, and the mechanisms by which certain organs are more tolerant of hyperoxia than others. The horizon of oxygen research looks bright as it may unlock clues to treatment for neurological and cardiovascular disease, mitochondrial dysfunction, proteotoxic conditions, dopaminergic neurodegeneration, malignancies driven by abnormal hypoxia signaling, and aging. We have come a long way since the days of hot-air balloon rides and high-altitude expeditions. Yet, boundless unchartered territory remains to explore the highs and lows of oxygen metabolism (see Outstanding Questions).
Outstanding Questions.
Which additional α-ketoglutarate-dependent dioxygenases are able to sense physiological changes in oxygen and coordinate stress responses under hypoxic conditions?
What are novel cellular adaptations to hypoxia that are independent of the transcriptional response regulated by the PHD-HIF-pVHL pathway?
Which oxygen-dependent reactions fail during states of ischemia and lead to adaptive metabolic changes that improve cell survival?
Are there metabolic adaptations in animals tolerant of extreme hypoxia that are widely evolutionarily conserved and applicable to human disease?
Which diseases can be treated by modulating oxygen tensions or the related stress responses, such as HIF stabilization or HIF inhibition?
How do cells and organisms sense and adapt to hyperoxia? Are there human diseases that are defined by tissue hyperoxia that can be corrected by normalization of tissue pO2 by hypoxia?
Highlights.
Molecular oxygen is an essential substrate in mammalian metabolism. Imbalances in oxygen levels are associated with a wide range of conditions in human health and disease.
The Jumonji C domain histone lysine demethylases, KDM5A and KDM6A, are recently discovered oxygen sensors that regulate histone demethylation and cellular differentiation.
Some animals are tolerant of extreme hypoxia and have developed unique adaptations that might have relevance to oxygen sensing and adaptation in humans.
Hypoxia can have both beneficial and toxic effects depending on the severity and duration of hypoxia, in combination with cell- and tissue-specific metabolic demands.
In a mouse model of pediatric mitochondrial disease, hypoxia itself, but not activation of the hypoxia transcriptional factor (HIF) response, is sufficient to rescue disease. Decreased tissue oxygen consumption and resulting excess oxygen contributes to pathology, which can be reversed with normalization of oxygen levels by hypoxia.
Preclinical and early clinical studies demonstrate that hypoxia or manipulation of the hypoxia response can potentially be used to treat various diseases, including mitochondrial diseases, neurodegenerative and cardiovascular diseases, anemia, and malignancies, among others.
Acknowledgments
We thank Dengke Ma, Ethan Weiss, and members of the Jain lab for their critical review of this paper. Figures created with Biorender.com. This work was supported by T32 HL007731 (A.H.B.) and DP5 OD026398 (I.H.J.).
Glossary
- Hypoxia
in the atmosphere, defined as less than 21% oxygen. Hypobaric hypoxia results from decreased barometric pressure (e.g., high altitude). Normobaric hypoxia results from decreased inspired fraction of oxygen (FiO2).
- Intermittent hypoxia
alternating episodes of normoxia and hypoxia, leading to cyclical bursts of deoxygenation and reoxygenation. Intermittent hypoxia is a feature of obstructive sleep apnea and central sleep apnea.
- Ischemia
severe hypoxia or anoxia (complete lack of oxygen), coupled with reduced availability of nutrients, including glucose, fatty acids, amino acids, and vitamins. In tissues, ischemia results from inadequate blood flow due to arterial blood flow restriction, leading to accumulation of metabolic waste products, cellular dysfunction, and tissue damage.
- KM
the Michaelis constant value at which the substrate concentration permits a reaction rate that is half of Vmax, the maximum rate ofan enzymatic reaction when saturated by the substrate. Enzymes with high KM have low affinity for their substrates. As an example, PHD proteins have low oxygen affinities (high KM), enabling them to sense oxygen in physiological ranges.
- Oxygen tension
the partial pressure of oxygen (PO2). The partial pressure of oxygen at sea level is 21% of the standard atmospheric pressure of 760 mmHg, equivalent to 160 mmHg. At sea level, Earth’s atmosphere is composed of 78% nitrogen, 21% oxygen, 0.9% argon, 0.03% carbon dioxide, and trace amounts of other gases.
Footnotes
Disclaimer Statement
A.H.B. has no conflicts of interest to declare. I.H.J. is listed on patents related to the use of hypoxia therapy for metabolic disorders.
References
- 1.Gumsley AP et al. (2017) Timing and tempo of the Great Oxidation Event. Proc. Natl. Acad. Sci. U. S. A 114, 1811–1816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Severinghaus JW (2003) Fire-air and dephlogistication. Revisionisms of oxygen’s discovery. Adv. Exp. Med. Biol 543, 7–19 [PubMed] [Google Scholar]
- 3.Ashcroft FM (2000) Life at the Extremes, University of California Press [Google Scholar]
- 4.Dejours P and Dejours S (1992) The effects of barometric pressure according to Paul Bert: the question today. Int. J. Sports Med 13, S1S–5 [DOI] [PubMed] [Google Scholar]
- 5.Carreau A et al. (2011) Why is the partial oxygen pressure of human tissues a crucial parameter? Small molecules and hypoxia. J. Cell. Mol. Med 15, 1239–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Markolovic S et al. (2015) Protein hydroxylation catalyzed by 2-oxoglutarate-dependent oxygenases. J. Biol. Chem 290, 20712–20722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schneider C et al. (2007) Control of oxygenation in lipoxygenase and cyclooxygenase catalysis. Chem. Biol 14, 473–488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J et al. (2017) Tet1 facilitates hypoxia tolerance by stabilizing the HIF-α proteins independent of its methylcytosine dioxygenase activity. Nucleic Acids Res. 45, 12700–12714 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mariani CJ et al. (2014) TET1-mediated hydroxymethylation facilitates hypoxic gene induction in neuroblastoma. Cell Rep. 7, 1343–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Erler JT et al. (2009) Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell 15, 35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stoehr A et al. (2019) The ribosomal prolyl-hydroxylase OGFOD1 decreases during cardiac differentiation and modulates translation and splicing. JCI Insight 5, 128496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Daubner SC et al. (2011) Tyrosine hydroxylase and regulation of dopamine synthesis. Arch. Biochem. Biophys 508, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang D et al. (2006) Saturated fatty acids promote endoplasmic reticulum stress and liver injury in rats with hepatic steatosis. Endocrinology 147, 943–951 [DOI] [PubMed] [Google Scholar]
- 14.Hu C-J et al. (2003) Differential roles of hypoxia-inducible factor 1alpha (HIF-1alpha) and HIF-2alpha in hypoxic gene regulation. Mol. Cell. Biol 23, 9361–9374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jaakkola P et al. (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292, 468–472 [DOI] [PubMed] [Google Scholar]
- 16.Bruick RK and McKnight SL (2001) A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294, 1337–1340 [DOI] [PubMed] [Google Scholar]
- 17.Epstein AC et al. (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell, 107 43–54 [DOI] [PubMed] [Google Scholar]
- 18.Maxwell PH et al. (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271−275 [DOI] [PubMed] [Google Scholar]
- 19.Ivan M et al. (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292, 464–468 [DOI] [PubMed] [Google Scholar]
- 20.Lando D et al. (2002) FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-induciblefactor. Genes Dev. 16, 1466–1471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hewitson KS et al. (2002) Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J. Biol. Chem 277, 26351–26355 [DOI] [PubMed] [Google Scholar]
- 22.Masson N et al. (2012) The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep. 13, 251–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kaelin WG and Ratcliffe PJ (2008) Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell 30, 393–402 [DOI] [PubMed] [Google Scholar]
- 24.Ivan M and Kaelin WG (2017) The EGLN-HIF O2-sensing system: multiple inputs and feedbacks. Mol. Cell66, 772–779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schito L and Semenza GL (2016) Hypoxia-inducible factors: master regulators of cancer progression. Trends Cancer 2, 758–770 [DOI] [PubMed] [Google Scholar]
- 26.Batie M et al. (2019) Hypoxia induces rapid changes to histone methylation and reprograms chromatin. Science 363, 1222–1226 [DOI] [PubMed] [Google Scholar]
- 27.Chakraborty AA et al. (2019) Histone demethylase KDM6A directly senses oxygen to control chromatin and cell fate. Science 363, 1217–1222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Masson N et al. (2019) Conserved N-terminal cysteine dioxygenases transduce responses to hypoxia in animals and plants. Science 365, 65–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Licausi F et al. (2011) Oxygen sensing in plants is mediated by an N-end rule pathway for protein destabilization. Nature 479, 419–422 [DOI] [PubMed] [Google Scholar]
- 30.Agron PG et al. (1994) Oxygen regulation of expression of nitrogen fixation genes in Rhizobium meliloti. Res. Microbiol 145, 454–459 [DOI] [PubMed] [Google Scholar]
- 31.Monson EK et al. (1995) The oxygen sensor protein, FixL, of Rhizobium meliloti. Role of histidine residues in heme binding, phosphorylation, and signal transduction. J. Biol. Chem 270, 5243–5250 [DOI] [PubMed] [Google Scholar]
- 32.Taylor BL et al. (1999) Aerotaxis and other energy-sensing behavior in bacteria. Annu. Rev. Microbiol 53, 103–128 [DOI] [PubMed] [Google Scholar]
- 33.Bibikov SI et al. (1997) A signal transducer for aerotaxis in Escherichia coli. J. Bacteriol 179, 4075–4079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hughes AL et al. (2005) SREBP pathway responds to sterols and functions as an oxygen sensor in fission yeast. Cell 120, 831−842 [DOI] [PubMed] [Google Scholar]
- 35.Burr R et al. (2016) Mga2 transcription factor regulates an oxygen-responsive lipid homeostasis pathway in fission yeast. J. Biol. Chem 291, 12171–12183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burr R et al. (2017) Coordinate regulation of yeast sterol regulatory element-binding protein (SREBP) and Mga2 transcription factors. J. Biol. Chem 292, 5311–5324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huppertz B et al. (2009) Oxygen as modulator of trophoblast invasion. J. Anat 215, 14–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gournay V (2011) The ductus arteriosus: physiology, regulation, and functional and congenital anomalies. Arch. Cardiovasc. Dis 104, 578–585 [DOI] [PubMed] [Google Scholar]
- 39.López-Barneo J et al. (2009) Oxygen sensing in the carotid body. Ann. N. Y. Acad. Sci 1177,119–131 [DOI] [PubMed] [Google Scholar]
- 40.Chang AJ et al. (2015) Oxygen regulation of breathing through an olfactory receptor activated by lactate. Nature 527,240–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cutz E and Jackson A (1999) Neuroepithelial bodies as airway oxygen sensors. Respir. Physiol 115, 201–214 [DOI] [PubMed] [Google Scholar]
- 42.Shah YM and Xie L (2014) Hypoxia-inducible factors link iron homeostasis and erythropoiesis. Gastroenterology 146, 630–642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang F et al. (2019) Inhibition of suicidal erythrocyte death by chronic hypoxia. High Ait. Med. Biol 20, 112–119 [DOI] [PubMed] [Google Scholar]
- 44.Kim J et al. (2006) HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 3, 177–185 [DOI] [PubMed] [Google Scholar]
- 45.Metallo CM et al. (2011) Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wise DR et al. (2011) Hypoxia promotes isocitrate dehydrogenase-dependent carboxylation of α-ketoglutarate to citrate to support cell growth and viability. Proc. Natl. Acad. Sci. U. S. A 108, 19611–19616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ma DK et al. (2012) CYSL-1 interactswith the O2-sensing hydroxylase EGL-9 to promote H2S-modulated hypoxia-induced behavioral plasticity in C. elegans. Neuron 73, 925–940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tello D et al. (2011) Induction ofthe mitochondrial NDUFA4L2 protein by HIF-1α decreases oxygen consumption by inhibiting Complex I activity. Cell Metab. 14, 768–779 [DOI] [PubMed] [Google Scholar]
- 49.Moreno-Dominguez A et al. (2020) Acute O2 sensing through HIF2α-dependent expression of atypical cytochrome oxidase subunits in arterial chemoreceptors. Sci. Signal 13, eaay9452. [DOI] [PubMed] [Google Scholar]
- 50.Papandreou I et al. (2006) HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 3, 187–197 [DOI] [PubMed] [Google Scholar]
- 51.Wu H and Zhang Y (2011) Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 25, 2436–2452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kulshreshtha R et al. (2007) A microRNA signature of hypoxia. Mol. Cell. Biol 27, 1859–1867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chan SY et al. (2009) MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab. 10, 273–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Izquierdo-Alvarez A et al. (2012) Differential redox proteomics allows identification of proteins reversibly oxidized at cysteine residues in endothelial cells in response to acute hypoxia. J. Proteome 75, 5449–5462 [DOI] [PubMed] [Google Scholar]
- 55.D’Ignazio L and Rocha S (2016) Hypoxia induced NF-kB. Cells 5, E10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu L et al. (2006) Hypoxia-induced energy stress regulates mRNA translation and cell growth. Mol. Cell 21, 521–531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bensaad K et al. (2014) Fatty acid uptake and lipid storage induced by HIF-1 α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 9, 349–365 [DOI] [PubMed] [Google Scholar]
- 58.Lee P et al. (2020) Cellular adaptation to hypoxia through hypoxia inducible factors and beyond. Nat. Rev. Mol. Cell Biol. 21, 268–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fang X et al. (2014) Adaptations to a subterranean environment and longevity revealed by the analysis of mole rat genomes. Cell Rep. 8, 1354–1364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kim EB et al. (2011) Genome sequencing reveals insights into physiology and longevity of the naked mole rat. Nature 479, 223–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lee BP et al. (2020) Negligible senescence in naked mole rats may be a consequence of well-maintained splicing regulation. Geroscience Published online January 11, 2020 10.1007/s11357-019-00150-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Park TJ et al. (2017) Fructose-driven glycolysis supports anoxia resistance in the naked mole-rat. Science 356, 307–311 [DOI] [PubMed] [Google Scholar]
- 63.Scott GR (2011) Elevated performance: the unique physiology of birds that fly at high altitudes. J. Exp. Biol 214, 2455–2462 [DOI] [PubMed] [Google Scholar]
- 64.Jackson DC (2000) How a turtle’s shell helps it survive prolonged anoxic acidosis. News Physiol. Sci 15, 181–185 [DOI] [PubMed] [Google Scholar]
- 65.Hurford WE et al. (1996) Splenic contraction, catecholamine release, and blood volume redistribution during diving in the Weddell seal. J. Appl. Physiol 80, 298–306 [DOI] [PubMed] [Google Scholar]
- 66.Schagatay E et al. (2012) Size matters: spleen and lung volumes predict performance in human apneic divers. Front. Physiol 3, 173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hurford WE et al. (1990) Splenic contraction during breath-hold diving in the Korean ama. J. Appl. Physiol 69, 932–936 [DOI] [PubMed] [Google Scholar]
- 68.Tschop M et al. (1998) Raised leptin concentrations at high altitude associated with loss of appetite. Lancet 352,1119–1120 [DOI] [PubMed] [Google Scholar]
- 69.DiPasquale DM et al. (2015) Acute normobaric hypoxia reduces body temperature in humans. High Alt. Med. Biol 16, 61–66 [DOI] [PubMed] [Google Scholar]
- 70.MacDonald R (1977) Red cell 2,3-diphosphoglycerate and oxygen affinity. Anaesthesia 32, 544–553 [DOI] [PubMed] [Google Scholar]
- 71.Peacock AJ (1998) ABC of oxygen: oxygen at high altitude. BMJ 317, 1063–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Benjamin EJ et al. (2017) Heart disease and stroke statistics-2017 update: a report from the American Heart Association. Circulation 135, e146–e603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bailey BA et al. (2019) High altitude continues to reduce birth weights in Colorado. Matern. Child Health J 23, 1573–1580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Verratti V et al. (2008) Evidence that chronic hypoxia causes reversible impairment on male fertility. Asian J. Androl 10, 602–606 [DOI] [PubMed] [Google Scholar]
- 75.Tang H et al. (2018) Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition. Am. J. Physiol. Lung Cell Mol. Physiol 314, L256–L275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Galiè N et al. (2016)2015 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension: The Joint Task Force for the Diagnosis and Treatment of Pulmonary Hypertension of the European Society of Cardiology (ESC) and the European Respiratory Society (ERS): endorsed by: Association for European Paediatric and Congenital Cardiology (AEPC), International Society for Heart and Lung Transplantation (ISHLT). Eur. Heart J 37, 67–119 [DOI] [PubMed] [Google Scholar]
- 77.Zhou D et al. (2013) Whole-genome sequencing uncovers the genetic basis of chronic mountain sickness in Andean highlanders. Am. J. Hum. Genet 93, 452–462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lorenzo FR et al. (2014) A genetic mechanism for Tibetan high-altitude adaptation. Nat. Genet 46, 951–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Bigham AW and Lee FS (2014) Human high-altitude adaptation: forward genetics meets the HIF pathway. Genes Dev. 28, 2189–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mandriota SJ et al. (2002) HIF activation identifies early lesions in VHL kidneys: evidence for site-specific tumor suppressor function in the nephron. Cancer Cell 1,459–468 [DOI] [PubMed] [Google Scholar]
- 81.Lewis CA et al. (2015) SREBP maintains lipid biosynthesis and viability of cancer cells under lipid- and oxygen-deprived conditions and defines a gene signature associated with poor survival in glioblastoma multiforme. Oncogene 34, 5128–5140 [DOI] [PubMed] [Google Scholar]
- 82.Liu X et al. (2020) Genome-wide screening identifies SFMBT1 as an oncogenic driver in cancer with VHL loss. Mol. Cell 77, 1294–1306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang J et al. (2018) VHL substrate transcription factor ZHX2 as an oncogenic driver in clear cell renal cell carcinoma. Science 361,290–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Nakazawa MS et al. (2016) Oxygen availability and metabolic adaptations. Nat. Rev. Cancer16, 663–673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Petrova V et al. (2018) The hypoxic tumour microenvironment. Oncogenesis 7, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Samanta D and Semenza GL (2018) Metabolic adaptation of cancer and immune cells mediated by hypoxia-inducible factors. Biochim. Biophys. Acta Rev. Cancer 1870, 15–22 [DOI] [PubMed] [Google Scholar]
- 87.Dubreuil MM et al. (2020) Systematic identification of regulators of oxidative stress reveals non-canonical roles for peroxisomal import and the pentose phosphate pathway. Cell Rep. 30, 1417–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Moloney JN and Cotter TG (2018) ROS signalling in the biology of cancer. Semin. Cell Dev. Biol 80, 50–64 [DOI] [PubMed] [Google Scholar]
- 89.Saugstad OD (2006) Oxygen and retinopathy of prematurity. J. Perinatol 26, S46–S50 [DOI] [PubMed] [Google Scholar]
- 90.Long-Term Oxygen Treatment Trial Research Group et al. (2016) A randomized trial of long-term oxygen for COPD with moderate desaturation. N. Engl. J. Med 375, 1617–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mehta R et al. (2009) Proteasomal regulation of the hypoxic response modulates aging in C. e/egans. Science 324, 1196–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen S et al. (2019)VHL-1 inactivation and mitochondrial antioxidants rescue C. elegans dopaminergic neurodegeneration, Protein. Cell 10, 610–614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chen D et al. (2009) HIF-1 modulates dietary restriction-mediated lifespan extension via IRE-1 in Caenorhabditis e/egans. PLoS Genet. 5, e1000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bonawitz ND et al. (2007) Reduced TOR signaling extends chronological life span via increased respiration and upregulation of mitochondrial gene expression. Cell Metab. 5, 265–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Packer L and Fuehr K (1977) Low oxygen concentration extends the lifespan of cultured human diploid cells. Nature 267, 423–425 [DOI] [PubMed] [Google Scholar]
- 96.Minamino T et al. (2001) Hypoxia extends the life span of vascular smooth muscle cells through telomerase activation. Mol. Cell. Biol 21, 3336–3342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Lee SH et al. (2013) Hypoxia inhibits cellular senescence to restore the therapeutic potential of old human endothelial progenitor cells via the hypoxia-inducible factor-1α-TWIST-p21 axis. Arterioscler. Thromb. Vasc. Biol 33, 2407–2414 [DOI] [PubMed] [Google Scholar]
- 98.Korski KI et al. (2019) Hypoxia prevents mitochondrial dysfunction and senescence in human c-Kit+ cardiac progenitor cells. Stem Cells 37, 555–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bell EL et al. (2007) Mitochondrial reactive oxygen species trigger hypoxia-inducible factor-dependent extension of the replicative life span during hypoxia. Mol. Cell. Bio. 27, 5737–5745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li Y et al. (2017) Hypoxia potentially promotes Tibetan longevity. Cell Res. 27, 302–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Faeh D et al. (2009) Lower mortality from coronary heart disease and stroke at higher altitudes in Switzerland. Circulation 120, 495–501 [DOI] [PubMed] [Google Scholar]
- 102.Negi PC et al. (2012) Epidemiological study of hypertension in natives of Spiti Valley in Himalayas and impact of hypobaric hypoxemia; a cross-sectional study. J. Assoc. Physicians India 60, 21–25 [PubMed] [Google Scholar]
- 103.Voss JD et al. (2014) Lower obesity rate during residence at high altitude among a military population with frequent migration: a quasi experimental model for investigating spatial causation. PLoS One 9, e93493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Woolcott OO et al. (2014) Inverse association between diabetes and altitude: a cross-sectional study in the adult population of the United States. Obesity (Silver Spring) 22, 2080–2090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Duffels MGJ et al. (2010) Atherosclerosis in patients with cyanotic congenital heart disease. Circ. J 74, 1436–1441 [DOI] [PubMed] [Google Scholar]
- 106.Fyfe A et al. (2005) Cyanotic congenital heart disease and coronary artery atherogenesis. Am. J. Cardio . 96, 283–290 [DOI] [PubMed] [Google Scholar]
- 107.Jain IH et al. (2016) Hypoxia as a therapy for mitochondrial disease. Science 352, 54–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ferrari M et al. (2017) Hypoxia treatment reverses neurode- generative disease in a mouse model of Leigh syndrome. Proc. Natl. Acad. Sci. U. S. A 114, E4241–E4250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Jain IH et al. (2019) Leigh syndrome mouse model can be rescued by interventions that normalize brain hyperoxia, but not HIF activation. Cell Metab. 30, 824–832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Taivassalo T et al. (2002) Venous oxygen levels during aerobic forearm exercise: an index of impaired oxidative metabolism in mitochondrial myopathy. Ann. Neurol 51,38–44 [DOI] [PubMed] [Google Scholar]
- 111.Al Khazal F et al. (2019) A conditional mouse model of complex II deficiency manifesting as Leigh-like syndrome. FASEBJ. 33, 13189–13201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ast T et al. (2019) Hypoxia rescues frataxin loss by restoring iron sulfur cluster biogenesis. Cell 177, 1507–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jain IH et al. (2020) Genetic screen for cell fitness in high or low oxygen highlights mitochondrial and lipid metabolism. Cell 181, 1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Sweeney NW et al. (2019) Hypoxia suppresses high fat diet-induced steatosis and development of hepatic adenomas. Hypoxia (Auckl) 7, 53–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gorgens SW et al. (2017) Hypoxia in combination with muscle contraction improves insulin action and glucose metabolism in human skeletal muscle via the HIF-1 α pathway. Diabetes 66, 2800–280728811274 [Google Scholar]
- 116.Rahtu-Korpela L et al. (2014) HIF prolyl 4-hydroxylase-2 inhibition improves glucose and lipid metabolism and protects against obesity and metabolic dysfunction. Diabetes 63, 3324–3333 [DOI] [PubMed] [Google Scholar]
- 117.Rankin EB et al. (2009) Hypoxia-inducible factor 2 regulates hepatic lipid metabolism. Mol. Cell. Biol 29, 4527–4538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Ahn J et al. (2012) Hypoxia-inducible factor activation protects the kidney from gentamicin-induced acute injury. PLoS One 7, e48952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kapitsinou PP et al. (2014) Endothelial HIF-2 mediates protection and recovery from ischemic kidney injury. J. Clin. Invest 124, 2396–2409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Leite de Oliveira R et al. (2012) Gene-targeting of Phd2 improves tumor response to chemotherapy and prevents side-toxicity. Cancer Cell 22, 263–277 [DOI] [PubMed] [Google Scholar]
- 121.Navarrete C et al. (2018) Hypoxia mimetic activity of VCE-004.8, a cannabidiol quinone derivative: implications for multiple sclerosis therapy. J. Neuroinflammation 15, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Tambuwala MM et al. (2010) Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology 139, 2093–2101 [DOI] [PubMed] [Google Scholar]
- 123.Oda T et al. (2017) Hypoxia-inducible factor-1α expression in kidney transplant biopsy specimens after reperfusion is associated with early recovery of graft function after cadaveric kidney transplantation. Transplant. Proc 49, 68–72 [DOI] [PubMed] [Google Scholar]
- 124.Keränen MAI et al. (2010) Cardiomyocyte-targeted HIF-1alpha gene therapy inhibits cardiomyocyte apoptosis and cardiac allograft vasculopathy in the rat. J. Heart Lung Transplant 29, 1058–1066 [DOI] [PubMed] [Google Scholar]
- 125.Yang T et al. (2020) Ischemic preconditioning provides long- lasting neuroprotection against ischemic stroke: the role of Nrf2. Exp. Neurol. 325,113142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bernhardt WM et al. (2006) Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J. Am. Soc. Nephrol 17, 1970–1978 [DOI] [PubMed] [Google Scholar]
- 127.Tampe B et al. (2018) Pharmacological induction of hypoxia-inducible transcription factor ARNT attenuates chronic kidney failure. J. Clin. Invest 128, 3053–3070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Stetler RA et al. (2014) Preconditioning provides neuroprotection in models of CNS disease: paradigms and clinical significance. Prog. Neurobio. 114, 58–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Sarkar K et al. (2012) Hypoxia-inducible factor 1 transcriptional activity in endothelial cells is required for acute phase cardioprotection induced by ischemic preconditioning. Proc. Nat. Acad. Sci. U. S. A 109, 10504–10509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Solomon SD et al. (2004) Angina pectoris prior to myocardial infarction protects against subsequent left ventricular remodeling. J. Am. Coll. Cardiol 43, 1511–1514 [DOI] [PubMed] [Google Scholar]
- 131.Choo G-H (2015) Collateral circulation in chronic total occlusions – an interventional perspective. Curr. Cardiol. Rev 11, 277–284 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Cai Z et al. (2013) Hypoxia-inducible factor 1 is required for remote ischemic preconditioning of the heart. Proc. Nat . Acad. Sci. U. S. A 110, 17462–17467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Olenchock BA et al. (2016) EGLN1 inhibition and rerouting of α-ketoglutarate suffice for remote ischemic protection. Cell 164, 884–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Liu C et al. (2020) Remote ischemic conditioning reduced cerebral ischemic injury by modulating inflammatory responses and ERK activity in type 2 diabetic mice. Neurochem. Int 135, 104690. [DOI] [PubMed] [Google Scholar]
- 135.Jachova J et al. (2019) Neuroprotection mediated by remote preconditioning is associated with a decrease in systemic oxidative stress and changes in brain and blood glutamate concentration. Neurochem. Int 129,104461. [DOI] [PubMed] [Google Scholar]
- 136.Benstoem C et al. (2017) Remote ischaemic preconditioning for coronary artery bypass grafting (with or without valve surgery). Cochrane Database Syst. Rev 5, CD011719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hausenloy DJ et al. (2015) Remote ischemic preconditioning and outcomes of cardiac surgery. N. Engl. J. Med 373, 1408–1417 [DOI] [PubMed] [Google Scholar]
- 138.Huang X et al. (2018) Hypoxia signaling pathway in stem cell regulation: good and evil. Curr. Stem Cell Rep 4, 149–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Guimarães-Camboa N et al. (2015) HIF1α represses cell stress pathways to allow proliferation of hypoxic fetal cardiomyocytes. Dev. Cell 33, 507–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kimura W et al. (2015) Hypoxiafate mapping identifies cycling cardiomyocytes in the adult heart. Nature 523, 226–230 [DOI] [PubMed] [Google Scholar]
- 141.Nakada Y et al. (2017) Hypoxia induces heart regeneration in adult mice. Nature 541,222–227 [DOI] [PubMed] [Google Scholar]
- 142.Stegen S et al. (2016) HIF-1α promotes glutamine-mediated redox homeostasis and glycogen-dependent bioenergetics to support postimplantation bone cell survival. Cell Metab. 23, 265–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Cho H et al. (2016) On-target efficacy of a HIF-2α antagonist in preclinical kidney cancer models. Nature 539, 107–111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chen N et al. (2019) Roxadustat treatment for anemia in patients undergoing long-term dialysis. N. Engl. J. Med 381, 1011–1022 [DOI] [PubMed] [Google Scholar]
- 145.Chen N et al. (2019) Roxadustat for anemia in patients with kidney disease not receiving dialysis. N. Engl. J. Med 381, 1001–1010 [DOI] [PubMed] [Google Scholar]
- 146.Zhang X et al. (2019) Adipocyte hypoxia-inducible factor 2α suppresses atherosclerosis by promoting adipose ceramide catabolism. Cell Metab. 30, 937–951 [DOI] [PubMed] [Google Scholar]
- 147.Choueiri TK and Motzer RJ (2017) Systemic therapy for metastatic renal-cell carcinoma. N. Engl. J. Med 376, 354–366 [DOI] [PubMed] [Google Scholar]
- 148.Quaegebeur A et al. (2016) Deletion or inhibition of the oxygen sensor PHD1 protects against ischemic stroke via reprogramming of neuronal metabolism. Cell Metab. 23, 280–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Bernhardt WM et al. (2009) Donor treatment with a PHD-inhibitor activating HIFs prevents graft injury and prolongs survival in an allogenic kidney transplant model. Proc. Natl. Acad. Sci. U. S. A 106, 21276–21281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Heim C et al. (2016) Prolyl-hydroxylase inhibitor activating hypoxia-inducible transcription factors reduce levels of transplant arteriosclerosis in a murine aortic allograft model. Interact. Cardiovasc. Thorac. Surg. 22, 561−570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Hyvarinen J et al. (2010) Hearts of hypoxia-inducible factor prolyl 4-hydroxylase-2 hypomorphic mice show protection against acute ischemia-reperfusion injury. J. Biol. Chem 285, 13646–13657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dai Z et al. (2018) Therapeutic targeting of vascular remodeling and right heart failure in pulmonary arterial hypertension with a HIF-2α inhibitor. Am. J. Respir. Crit. Care Med 198, 1423–1434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hu C-J et al. (2019) Suppression of HIF2 signalling attenuates the initiation of hypoxia-induced pulmonary hypertension. Eur. Respir. J. 54, 1900378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kaelin WG (2018) HIF2 inhibitor joins the kidney cancer armamentarium. J. Clin. Oncol 36, 908–910 [DOI] [PubMed] [Google Scholar]
- 155.Zhang N et al. (2010) The asparaginyl hydroxylase factor inhibiting HIF-1alpha is an essential regulator of metabolism. Cell Metab. 11, 364–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Telfer P et al. (2018) Impact of voxelotor (GBT440) on unconjugated bilirubin and jaundice in sickle cell disease. Hematol. Rep 10, 7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wood JC (2019) Brain O2 reserve in sickle cell disease. Blood 133, 2356–2358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Luks AM et al. (2017) Acute high-altitude sickness. Eur. Respir. Rev 26, 160096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Zhou Y et al. (2017) Hypoxia augments LPS-induced inflammation and triggers high altitude cerebral edema in mice. Brain Behav. Immun 64, 266–275 [DOI] [PubMed] [Google Scholar]
- 160.Cole AM et al. (2014) Genetic variation in SENP1 and ANP32D as predictors of chronic mountain sickness. High Alt Med. Biol 15, 497–499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Tintu AN et al. (2007) Hypoxia disturbs fetal hemodynamics and growth. Endothelium 14, 353–360 [DOI] [PubMed] [Google Scholar]
- 162.Greksa LP (2006) Growth and development of Andean high altitude residents. High Alt. Med. Biol 7, 116–124 [DOI] [PubMed] [Google Scholar]
- 163.Lappin TR and Lee FS (2019) Update on mutations in the HIF: EPO pathway and their role in erythrocytosis. Blood Rev. 37,100590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Tuder RM et al. (2007) Hypoxia and chronic lung disease. J. Mol. Med 85, 1317–1324 [DOI] [PubMed] [Google Scholar]
- 165.Wilson R et al. (2016) Erythrocytosis dueto PHD2 mutations: a review of clinical presentation, diagnosis, and genetics. Case Rep. Hematol 6373706, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Perrotta S et al. (2020) Effects of germline VHL deficiency on growth, metabolism, and mitochondria. N. Engl. J. Med 382, 835–844 [DOI] [PubMed] [Google Scholar]
- 167.Bhagwani AR et al. (2020) Clonally selected primitive endothelial cells promote occlusive pulmonary arteriopathy and severe pulmonary hypertension in rats exposed to chronic hypoxia. Sci. Rep 10, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gordeuk VR et al. (2004) Congenital disorder of oxygen sensing: association of the homozygous Chuvash polycythemia VHL mutation with thrombosis and vascular abnormalities but not tumors. Blood 103, 3924–3932 [DOI] [PubMed] [Google Scholar]
- 169.Musso G et al. (2013) Association of obstructive sleep apnoea with the presence and severity of non-alcoholic fatty liver disease. A systematic review and meta-analysis. Obes. Rev 14, 417–431 [DOI] [PubMed] [Google Scholar]
- 170.Polotsky VY et al. (2009) Obstructive sleep apnea, insulin resistance, and steatohepatitis in severe obesity. Am. J. Respir. Crit Care Med 179, 228–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Ryan S et al. (2019) Adiposetissue as a key player in obstructive sleep apnoea. Eur. Respir. Rev 28, 19000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Tanne F et al. (2005) Chronic liver injury during obstructive sleep apnea. Hepatology 41, 1290–1296 [DOI] [PubMed] [Google Scholar]
- 173.Li J et al. (2005) Intermittent hypoxia induces hyperlipidemia in lean mice. Circ. Res 97, 698–706 [DOI] [PubMed] [Google Scholar]
- 174.Kendzerska T et al. (2018) Sleep apnea increases the risk of new hospitalized atrial fibrillation: a historical cohort study. Chest 154, 1330–1339 [DOI] [PubMed] [Google Scholar]
- 175.Morand J et al. (2018) Chronic intermittent hypoxia promotes myocardial ischemia-related ventricular arrhythmias and sudden cardiac death. Sci. Rep 8, 2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Saver JL (2006) Time is brain-quantified. Stroke 37, 263–266 [DOI] [PubMed] [Google Scholar]
- 177.Anderson JL and Morrow DA (2017) Acute myocardial infarction. N. Engl. J. Med 376, 2053–2064 [DOI] [PubMed] [Google Scholar]
- 178.Sharfuddin AA and Molitoris BA (2011) Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol 7, 189–200 [DOI] [PubMed] [Google Scholar]
- 179.Lyaker MR et al. (2013) Arterial embolism. Int. J. Crit. Illn. Inj. Sci 3, 77–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Halberg N et al. (2009) Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol. Cell. Biol 29, 4467–4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Lin Q et al. (2013) Activation of hypoxia-inducible factor-2 in adipocytes results in pathological cardiac hypertrophy. J. Am. Heart Assoc 2, e000548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ye J et al. (2007) Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am. J. Physiol. Endocrinol. Metab 293, E1118–E1128 [DOI] [PubMed] [Google Scholar]
- 183.Lee YS et al. (2014) Increased adipocyte O2 consumption triggers HIF-1α, causing inflammation and insulin resistance in obesity. Cell 157,1339–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.He Q et al. (2011) Regulation of HIF-1{alpha} activity in adipose tissue by obesity-associated factors: adipogenesis, insulin, and hypoxia. Am. J. Physiol. Endocrinol. Metab 300, E877–E885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Xie C et al. (2017) Activation of intestinal hypoxia-inducible factor 2α during obesity contributes to hepatic steatosis. Nat. Med 23,1298–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Xue X et al. (2012) Hypoxia-inducible factor-2α activation promotes colorectal cancer progression by dysregulating iron homeostasis. Cancer Res. 72, 2285–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Krishnan J et al. (2009) Activation of a HIF1alpha-PPARgamma axis underlies the integration of glycolytic and lipid anabolic pathways in pathologic cardiac hypertrophy. Cell Metab. 9, 512–524 [DOI] [PubMed] [Google Scholar]
- 188.Moslehi J et al. (2010) Loss of hypoxia-inducible factor prolyl hydroxylase activity in cardiomyocytes phenocopies ischemic cardiomyopathy. Circulation 122,1004–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Rankin EB et al. (2006) Renal cyst development in mice with conditional inactivation of the von Hippel-Lindau tumor suppressor. Cancer Res. 66, 2576–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Kimura K et al. (2008) Stable expression of HIF-lalpha in tubular epithelial cells promotes interstitial fibrosis. Am. J. Physiol. Renal Physiol 295, F1023–F1029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kraus A et al. (2018) HIF-1α promotes cyst progression in a mouse model of autosomal dominant polycystic kidney disease. Kidney Int 94, 887–899 [DOI] [PubMed] [Google Scholar]
- 192.Brukamp K et al. (2007) Hypoxia and podocyte-specific Vhlh deletion confer risk of glomerular disease. Am. J. Physiol. Renal Physiol 293, F1397–F1407 [DOI] [PubMed] [Google Scholar]
- 193.Bae HY-H et al. (2018) Brain injury induces HIF-1α-dependent transcriptional activation of LRRK2 that exacerbates brain damage. Cell Death Dis. 9, 1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Koh HS et al. (2015) The HIF-1/glial TIM-3 axis controls inflammation-associated brain damage under hypoxia. Nat. Commun 6, 6340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Qu A et al. (2011) Hypoxia-inducible transcription factor 2α promotes steatohepatitis through augmenting lipid accumulation, inflammation, and fibrosis. Hepatology 54, 472–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Ramakrishnan SK et al. (2014) Loss of von Hippel-Lindau protein (VHL) increases systemic cholesterol levels through targeting hypoxia-inducible factor 2α and regulation of bile acid homeostasis. Mol. Cell. Biol 34, 1208–1220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Suresh MV et al. (2014) Activation of hypoxia-inducible factor-1 α in type 2 alveolar epithelial cell is a major driver of acute inflammation following lung contusion. Crit. Care Med 42, e642–e653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sato Y et al. (2013) Integrated molecular analysis of clear-cell renal cell carcinoma. Nat. Genet 45, 860–867 [DOI] [PubMed] [Google Scholar]
- 199.Bachtiary B et al. (2003) Overexpression of hypoxia-inducible factor 1alpha indicates diminished response to radiotherapy and unfavorable prognosis in patients receiving radical radiotherapy for cervical cancer. Clin. Cancer Res 9, 2234–2240 [PubMed] [Google Scholar]
- 200.Jarman EJ et al. (2019) HER2 regulates HIF-2α and drives an increased hypoxic response in breast cancer. Breast Cancer Res. 21,10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Shiraishi A et al. (2017) Hypoxia promotes the phenotypic change of aldehyde dehydrogenase activity of breast cancer stem cells. Cancer Sci. 108, 362–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Sueoka E et al. (2013) Development of lymphoproliferative diseases by hypoxia inducible factor-1alpha is associated with prolonged lymphocyte survival. PLoS One 8, e57833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Lodato RF (1989) Decreased O2 consumption and cardiac output during normobaric hyperoxia in conscious dogs. J. Appl. Physiol 67, 1551–1559 [DOI] [PubMed] [Google Scholar]
- 204.Brenner M et al. (2012) Association between early hyperoxia and worse outcomes after traumatic brain injury. Arch. Surg 147, 1042–1046 [DOI] [PubMed] [Google Scholar]
- 205.Farquhar H et al. (2009) Systematic review of studies of the effect of hyperoxia on coronary blood flow. Am. Heart J 158, 371–377 [DOI] [PubMed] [Google Scholar]
- 206.de Jonge E et al. (2008) Association between administered oxygen, arterial partial oxygen pressure and mortality in mechanically ventilated intensive care unit patients. Crit. Care 12, R156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Chawla A and Lavania AK (2001) Oxygen toxicity. Med. J. Armed Forces India 57, 131–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Damiani E et al. (2014) Arterial hyperoxia and mortality in critically ill patients: a systematic review and meta-analysis. Crit. Care 18,711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Kilgannon JH et al. (2010) Association between arterial hyperoxia following resuscitation from cardiac arrest and inhospital mortality. JAMA 303, 2165–2171 [DOI] [PubMed] [Google Scholar]
- 210.Page D et al. (2018) Emergency department hyperoxia is associated with increased mortality in mechanically ventilated patients: a cohort study. Crit. Care 22, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Ma DK et al. (2013) Cytochrome P450 drives a HIF-regulated behavioral response to reoxygenation by C. elegans. Science 341,554–558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Yokoyama K et al. (1999) Time course of post-ischemic superoxide generation in venous effluent from reperfused rabbit hindlimbs. J. Reconstr. Microsurg. 15, 215–221 [DOI] [PubMed] [Google Scholar]
- 213.Petzelbauer P et al. (2005) The fibrin-derived peptide Bbeta15-42 protects the myocardium against ischemia-reperfusion injury. Nat. Med 11,298–304 [DOI] [PubMed] [Google Scholar]
- 214.Clarke SJ et al. (2002) Sanglifehrin A acts as a potent inhibitor of the mitochondrial permeability transition and reperfusion injury of the heart by binding to cyclophilin-D at a different site from cyclosporin A. J. Bio . Chem 277, 34793–34799 [DOI] [PubMed] [Google Scholar]
- 215.Jaeschke H and Lemasters JJ (2003) Apoptosis versus oncotic necrosis in hepatic ischemia/reperfusion injury. Gastroenterology 125, 1246–1257 [DOI] [PubMed] [Google Scholar]
- 216.Zhu Y et al. (2013) MCT1 and MCT4 expression during myocardial ischemic-reperfusion injury in the isolated rat heart. Cell. Physiol. Biochem 32, 663–674 [DOI] [PubMed] [Google Scholar]
- 217.Jones SP et al. (1999) Myocardial ischemia-reperfusion injury is exacerbated in absence of endothelial cell nitric oxide synthase. Am. J. Phys 276, H1567–H1573 [DOI] [PubMed] [Google Scholar]
- 218.Chouchani ET et al. (2014) Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515, 431–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Kalikkot Thekkeveedu R et al. (2017) Bronchopulmonary dysplasia: a review of pathogenesis and pathophysiology. Respir. Med 132, 170–177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Bashinsky AL(2017) Retinopathy of prematurity. N. C. Med. J 78, 124–128 [DOI] [PubMed] [Google Scholar]
- 221.Hamad AE et al. (2019) Late-onset retinal findings and complications in untreated retinopathy of prematurity. Ophtha mo . Retina Published online December 24, 2019 10.1016/j.oret.2019.12.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Kindermann A et al. (2019) Severe but not moderate hyperoxia of newborn mice causes an emphysematous lung phenotype in adulthood without persisting oxidative stress and inflammation. BMC Pulm. Med 19, 245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Fuhrmann DC and Brune B (2017) Mitochondrial composition and function under the control of hypoxia. Redox Biol 12, 208–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Kim H et al. (2011) Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol. Cell 44, 532–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kamphorst JJ et al. (2013) Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc. Natl. Acad. Sci. U. S. A 110, 8882–8887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Chen J et al. (2019) Hypoxia exacerbates nonalcoholic fatty liver disease via the HIF-2α/PPARa pathway. Am. J. Physiol. Endocrinol. Metab 317, E710–E722 [DOI] [PubMed] [Google Scholar]
- 227.Egan DF et al. (2011) Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331,456–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Rouschop KMA et al. (2010) The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. invest 120, 127–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Tian R et al. (2016) Evolutionary genetics of hypoxiatolerance in cetaceans during diving. Genome Biol. Evo. 8, 827–839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Lippl FJ et al. (2010) Hypobaric hypoxia causes body weight reduction in obese subjects. Obesity (Silver Spring) 18,675–681 [DOI] [PubMed] [Google Scholar]
- 231.Mansor LS et al. (2016) Increased oxidative metabolism following hypoxia in the type 2 diabetic heart, despite normal hypoxia signalling and metabolic adaptation. J. Physiol. (Lend.) 594, 307–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Risso A et al. (2012) Expression of fetal hemoglobin in adult humans exposed to high altitude hypoxia. Blood Cells Mol. Dis 48, 147–153 [DOI] [PubMed] [Google Scholar]
- 233.Baumeister J et al. (2019) Hypoxia-inducible factor 1 (HIF-1) is a new therapeutictarget in JAK2V617F-positive myeloproliferative neoplasms. Leukemia 34, 1062–1074 [DOI] [PubMed] [Google Scholar]