

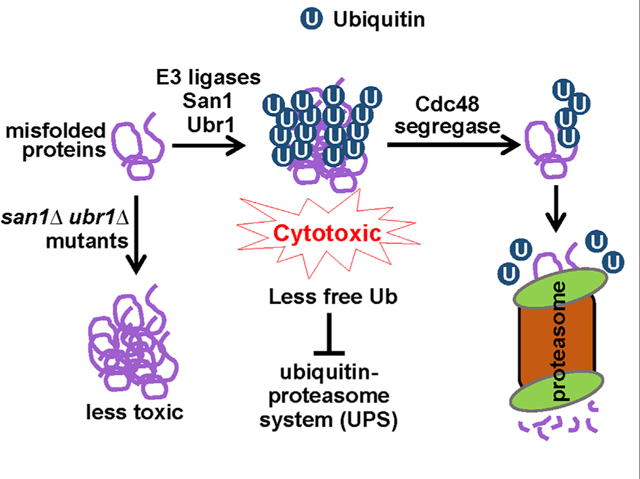
SUMMARY
The accumulation of misfolded proteins is associated with multiple neurodegenerative disorders, but it remains poorly defined how this accumulation causes cytotoxicity. Here, we demonstrate that the Cdc48/p97 segregase machinery drives the clearance of ubiquitinated model misfolded protein Huntingtin (Htt103QP) and limits its aggregation. Nuclear ubiquitin ligase San1 acts upstream of Cdc48 to ubiquitinate Htt103QP. Unexpectedly, deletion of SAN1 and/or its cytosolic counterpart UBR1 rescues the toxicity associated with Cdc48 deficiency, suggesting that ubiquitin depletion, rather than compromised proteolysis of misfolded proteins, causes the growth defect in cells with Cdc48 deficiency. Indeed, Cdc48 deficiency leads to elevated protein ubiquitination levels and decreased free ubiquitin, which depends on San1/Ubr1. Furthermore, enhancing free ubiquitin levels rescues the toxicity in various Cdc48 pathway mutants and restores normal turnover of a known Cdc48-independent substrate. Our work highlights a previously unappreciated function for Cdc48 in ensuring the regeneration of monoubiquitin that is critical for normal cellular function.
Graphical Abstract
In Brief
Misfolded protein accumulation causes cytotoxicity, but the mechanism remains poorly understood. Using budding yeast as a model organism, Higgins et al. show that ubiquitination of misfolded proteins depletes free ubiquitin, which compromises ubiquitin-dependent cellular functions and causes cytotoxicity. The Cdc48/p97 segregase antagonizes this cytotoxicity by promoting ubiquitin recycling from misfolded proteins.
INTRODUCTION
Correct folding of proteins is essential for their function. Although protein folding is a tightly regulated process, misfolded proteins are still generated within cells for various reasons. Low levels of protein misfolding occur spontaneously, but gene mutations, translational errors, aging, and various chemical stressors escalate protein misfolding (Tyedmers et al., 2010). The accumulation of misfolded proteins is associated with multiple neurodegenerative disorders, including Alzheimer’s and Huntington diseases (Knowles et al., 2014; Soto, 2003). In addition, protein misfolding has been implicated in diabetes and cancer (de Oliveira et al., 2015; Mukherjee et al., 2015). Unfortunately, why misfolded proteins are cytotoxic and how cells counteract this toxicity remain poorly understood.
Misfolded proteins are prone to aggregation due to exposed hydrophobic surfaces. The association between hydrophobic domains results in amorphous aggregates. These amorphous aggregates are oligomeric and mostly below the detection limit by microscopy (nm size) (Mogk et al., 2018). An increased concentration of misfolded proteins leads to the formation of microscopically detectable inclusion bodies (μm size) (Hipp et al., 2012; Mogk et al., 2018). Oligomeric aggregates (hereafter, aggregates) are potentially cytotoxic and may contribute to cytotoxicity by sequestrating transcription factors, RNA, and chaperones or by causing endoplasmic reticulum stress (Hartl et al., 2011; Leitman et al., 2013; Ogen-Shtern et al., 2016; Yang and Hu, 2016). However, a more general mechanism likely contributes to the toxicity of misfolded protein aggregates.
Misfolded proteins can be refolded with the assistance of molecular chaperones. They can also be degraded by the ubiquitin-proteasome system (UPS) or by the autophagy pathway. The sequential actions of E1 ubiquitin-activating, E2 ubiquitin-conjugating, and E3 ubiquitin-ligase enzymes covalently attach a polyubiquitin chain to misfolded substrates, which targets them to the proteasome for degradation (Finley et al., 2012). Among the dozens of ubiquitin ligases in budding yeast, San1 and Ubr1 are responsible for the ubiquitination and degradation of misfolded proteins. San1 is the predominant ubiquitin ligase involved in nuclear substrate ubiquitination (Dasgupta et al., 2004; Gardner et al., 2005), whereas cytosolic misfolded proteins are mainly ubiquitinated by Ubr1 (Eisele and Wolf, 2008; Samant et al., 2018). The E3 ligase Ubr1 relies on chaperones to detect and bind misfolded substrates, but San1 appears to bind directly to a wide variety of misfolded proteins (Heck et al., 2010; Rosenbaum et al., 2011). Misfolded protein aggregates require enzymes for disaggregation and the subsequent refolding or degradation. Yeast cells utilize the AAA+ (ATPase associated with various cellular activities) chaperone Hsp104 as a powerful disaggregase (Miller et al., 2015). Hsp104 contains a hydrophobic pocket in its N-terminal domain that interacts with substrates (Aguado et al., 2015).
Yeast cells also utilize a conserved AAA ATPase, Cdc48 (p97/VCP in metazoans), to separate proteins from one another and thus has been termed a segregase. Cdc48 is composed of an N-terminal domain, two centrally located ATPase domains, and a C-terminal tail. Six Cdc48 monomers form a double-ring structure surrounding a central pore (Bodnar and Rapoport, 2017b). This homohexameric structure, along with the help of cofactors, extracts polyubiquitinated substrates from membranes and macromolecular complexes, which facilitates protein relocalization or proteasomal degradation (Bodnar and Rapoport, 2017a). Recent in vitro evidence indicates that the Cdc48 complex acts as an unfoldase to generate unstructured ubiquitin or segments for its substrates (Olszewski et al., 2019; Twomey et al., 2019). The prominent cofactors for Cdc48, Npl4 and Ufd1, contain ubiquitin binding domains (Bodnar et al., 2018). The Cdc48Ufd1/Npl4 complex is involved in chromatin remodeling, DNA replication, endoplasmic-reticulum-associated degradation (ERAD), selective autophagy, and membrane fusion (Ye et al., 2017). However, the segregase function of Cdc48 in response to proteotoxic stress remains poorly defined.
Ubiquitin exists in the cells as monomers (free ubiquitin) or as chains, most of which are covalently attached to other proteins. The balance between these two pools (ubiquitin homeostasis) is tightly regulated by the antagonistic actions of ubiquitin ligases, which assemble chains, and deubiquitinating enzymes, which disassemble them (Komander et al., 2009; Reyes-Turcu et al., 2009). In yeast, deubiquitinase Doa4 controls the free ubiquitin level by recycling polyubiquitin, and doa4Δ cells exhibit free ubiquitin depletion and hypersensitivity to proteotoxic stressors (Swaminathan et al., 1999). Rfu1, a negative regulator of Doa4, was identified as a high-copy suppressor of the cdc48–3 mutant, indicating that Cdc48 might be involved in ubiquitin homeostasis (Kimura et al., 2009). However, the function of Cdc48 in ubiquitin homeostasis and the response to proteotoxic stress remain unclear. Here, we present data suggesting that the ubiquitination of misfolded proteins causes cytotoxicity by draining free ubiquitin and compromising UPS function. The Cdc48 complex combats this toxic effect by disaggregating ubiquitinated protein aggregates, which facilitates ubiquitin recycling and UPS-mediated protein degradation. These observations shed light on the molecular basis for the cytotoxicity of misfolded proteins, as well as the critical role of the Cdc48Ufd1/Npl4 complex in alleviating this toxicity.
RESULTS
The Cdc48Ufd1/Npl4 Complex Is Essential for Proteasomal Degradation of Mutant Huntingtin
The Cdc48Ufd1/Npl4 complex extracts polyubiquitinated proteins from membranes or macromolecular complexes (Bodnar and Rapoport, 2017b). In yeast cells, Cdc48 has been shown to associate with mutated Huntingtin proteins containing a polyQ expansion and a proline-rich region (Htt103QP) (Wang et al., 2009). More recently, p97, the human homolog of Cdc48, was shown to colocalize with mutated Huntingtin in mammalian cells and exhibit segregase activity (Ghosh et al., 2018). Because our previous work shows that Htt103QP is readily degraded and proteasome inhibitor MG132 blocks this degradation (Chuang et al., 2016; Higgins et al., 2018), we tested if the Cdc48Ufd1/Npl4 complex promotes proteasomal degradation of Htt103QP. For this purpose, we induced FLAG-Htt103QP-GFP expression in wild-type (WT) and cdc48–3 cells from a galactose-inducible promoter (GAL) for 1 h before shutoff by adding glucose. At the semi-permissive temperature 34°C, we observed significant stabilization of Htt103QP in cdc48–3 mutant cells, but WT cells showed very efficient Htt103QP degradation. Mutation of the Cdc48 cofactors Npl4 and Ufd1 also caused a significant delay in Htt103QP degradation (Figure 1A). We further compared Htt103QP stability in WT and cdc48–3 mutant cells after protein synthesis inhibition by cycloheximide. Impaired Htt103QP degradation was also observed in cdc48–3 cells by using the cycloheximide chase method (Figure 1B). The expression of mutated Huntingtin lacking the flanking proline-rich region (Htt103QΔP) in yeast cells alters the shape and number of inclusions and causes cytotoxicity (Dehay and Bertolotti, 2006). Compared to Htt103QP, the degradation of Htt103QΔP was much less efficient, and Htt103QΔP overexpression led to the formation of numerous inclusion bodies in each yeast cell (Figure S1). These results indicate an essential role for the Cdc48Ufd1/Npl4 complex in proteasomal degradation of Htt103QP and that the flanking proline-rich region is critical for Htt103QP degradation.
Figure 1. The Cdc48Npl4/Ufd1 Complex Is Required for Htt103QP Degradation but Is Dispensable for Its Ubiquitination.
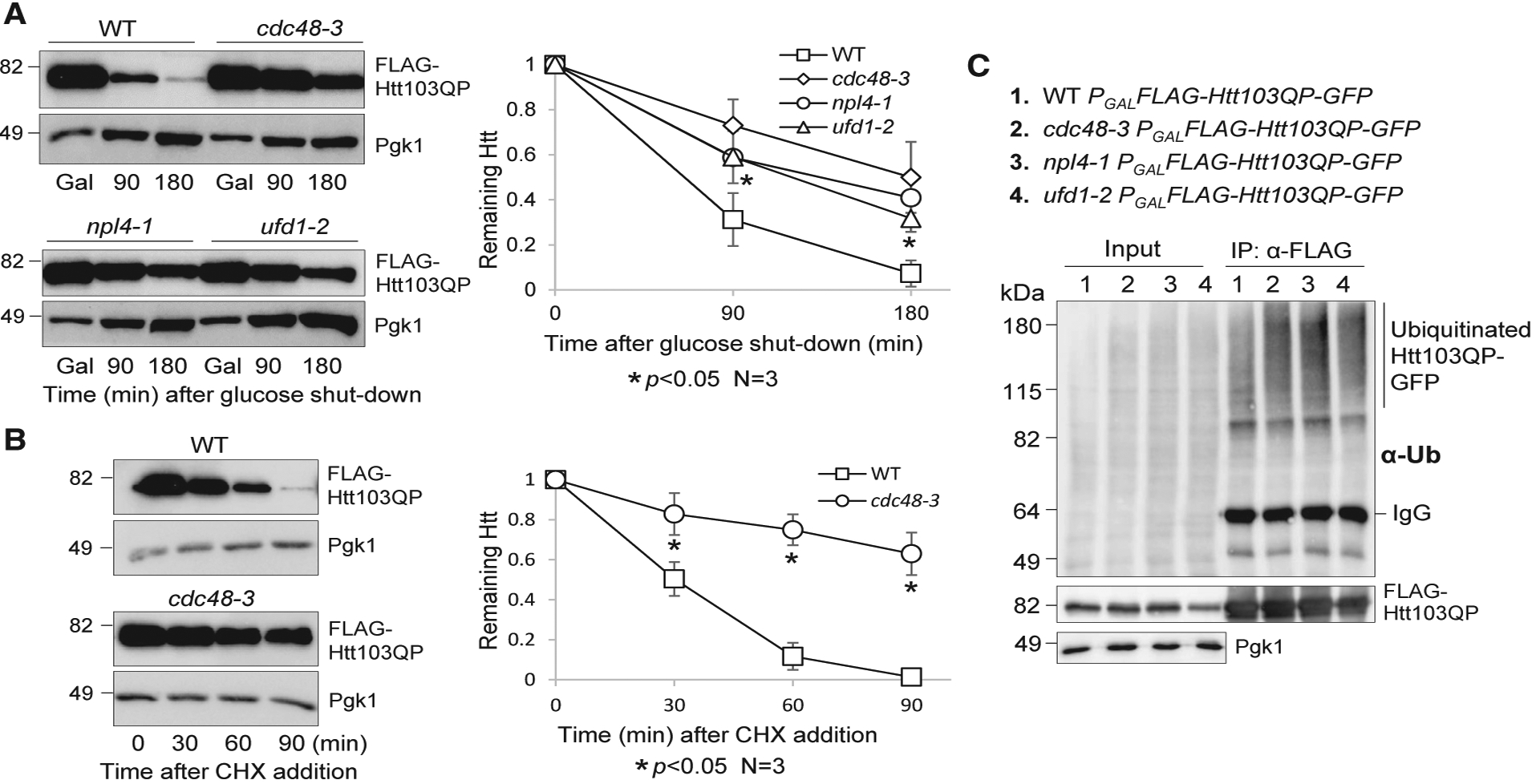
(A) The degradation of Htt103QP depends on the Cdc48Npl4/Ufd1 complex. WT (3419–1-1), cdc48–3 (3598–2-3), npl4–1 (3387–3-4), and ufd1–2 (3385–4-4) mutants containing an integrating plasmid, PGALFLAG-Htt103QP-GFP, were grown in non-inducing raffinose medium (YPR) to mid-log phase at 25°C. Galactose was added to the medium (2% final concentration) for 50 min to induce FLAG-Htt103QP-GFP expression at 25°C. Cells were then shifted to 34°C for 10 additional min, and glucose was added to shut off the expression. FLAG-Htt103QP-GFP protein levels were detected using anti-FLAG antibody. Pgk1, loading control. The levels of Htt103QP and Pgk1 are shown on the left. The intensity of protein bands and the Htt103QP/Pgk1 ratio were analyzed using ImageJ. The remaining Htt103QP after glucose shutoff was calculated based on the results from three independent experiments. The results are represented as mean ± SD (standard deviation). Wilcoxon rank-sum test was used to calculate the p values. The statistical difference is significant (*) when p < 0.05.
(B) Impaired Htt103QP degradation was estimated by cycloheximide (CHX) chase. WT (3419–1-1) and cdc48–3 (3598–2-3) strains with PGALFLAG-Htt103QP-GFP were grown in YPR at 25°C to mid-log phase. Galactose was added to the medium for 30 min, followed by temperature shift to 34°C for 30 min. Then, CHX (200 μg/mL) was added, and the protein levels of Htt103QP and Pgk1 were determined over time. The experiment was repeated three times. Quantification of the Htt103QP/Pgk1 ratio and statistical analysis were performed as described above, and the relative levels of Htt103QP to Pgk1 are represented as mean ± SD. *p < 0.05.
(C) Cdc48 is not required for Htt103QP ubiquitination. WT (3419–1-1), cdc48–3 (3598–2-3), npl4–1 (3387–3-4), and ufd1–2 (3385–4-4) mutant cells containing PGALFLAG-Htt103QP-GFP were grown in YPR to mid-log phase at 25°C. Galactose was then added, and cells were shifted to 34°C for 3 h. The protein extracts were prepared as described in the STAR Methods. FLAG-Htt103QP-GFP was immunoprecipitated using M2 anti-FLAG beads. Htt103QP protein levels were detected using an anti-FLAG antibody, and protein ubiquitination was detected using anti-Ub antibody. Pgk1, loading control.
The impaired degradation of Htt103QP in cdc48 mutants could be a result of defective Htt103QP ubiquitination. To test this possibility, FLAG-Htt103QP-GFP was immunoprecipitated from protein extracts prepared using WT, cdc48–3, npl4–1, and ufd1–2 mutant cells after a 3-h galactose induction of Htt103QP at 34°C. Htt103QP ubiquitination was then examined with an anti-ubiquitin antibody. We observed a much stronger accumulation of ubiquitinated Htt103QP in all the mutants compared to WT cells (Figure 1C), suggesting that the functional Cdc48 complex is essential for the degradation of mutated Huntingtin but is dispensable for its ubiquitination.
San1 Ubiquitinates Htt103QP for Proteasomal Degradation
We next sought to determine which E3 ubiquitin ligase(s) is responsible for Htt103QP ubiquitination and degradation. E3 ligase Ltn1 promotes the ubiquitination of mutant Huntingtin in yeast cells (Yang et al., 2016). However, no significant difference was detected for Htt103QP degradation efficiency between WT and ltn1Δ cells (Figure 2A), indicating that a different or additional E3 ligase catalyzes Htt103QP ubiquitination. To identify the ligase(s), we assessed Htt103QP degradation in 71 yeast strains that each carried a deletion of a verified or putative ubiquitin ligase gene (Table S1; Fang et al., 2011). As described previously (Chuang et al., 2016), a haploid strain containing PGALFLAG-Htt103QP-GFP was crossed with these 71 deletion mutants. Diploids were selected and sporulated and then haploid strains containing PGALFLAG-Htt103QP-GFP and a ubiquitin ligase deletion were isolated. We examined Htt103QP protein stability in these mutants after galactose induction of Htt103QP for 1 h and followed by expression shutoff with glucose for 3 h. Of the 71 tested mutants, only deletion of SAN1, a nuclear ubiquitin ligase that promotes the ubiquitination of misfolded proteins (Heck et al., 2010; Samant et al., 2018), resulted in obvious Htt103QP stabilization (Figure 2A). Ubr1 ubiquitin ligase localizes in the cytoplasm to ubiquitinate misfolded proteins and thereby acts as the cytosolic equivalent of San1. Unexpectedly, no significant Htt103QP stabilization was detected in ubr1Δ mutant cells. In addition, the absence of Ufd2, a ligase that synthesizes branched ubiquitin chains (Liu et al., 2017), had a much less severe but statistically significant defect in Htt103QP degradation that was evident at longer time points (Figure 2A). Therefore, these results support the conclusion that San1 E3 ligase plays a critical role for Htt103QP degradation.
Figure 2. E3 Ubiquitin Ligase San1 Ubiquitinates Htt103QP for Degradation.
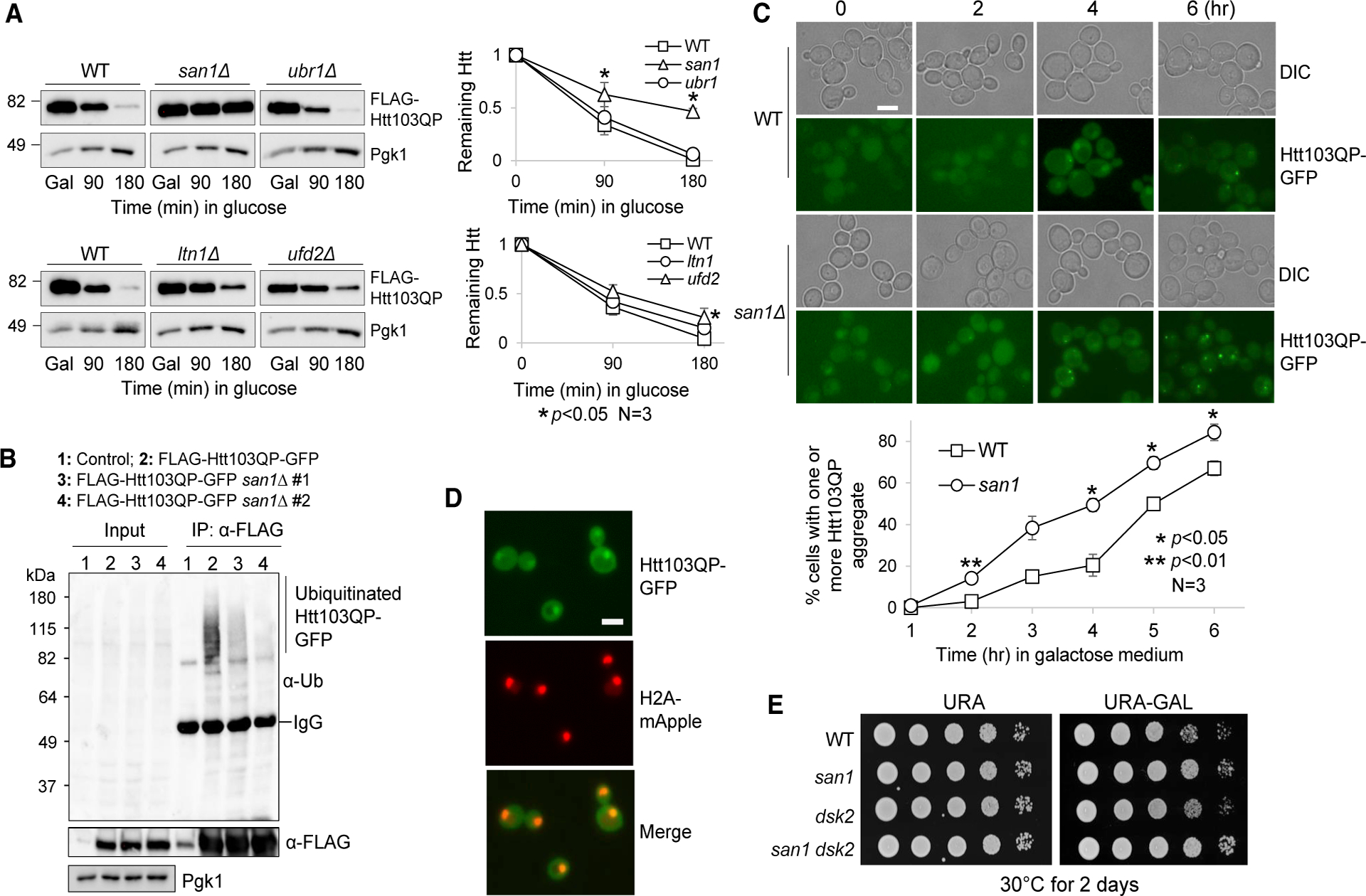
(A) San1 is required for Htt103QP degradation. WT (3419–1-1), san1Δ (RH142), ubr1Δ (3522–4-4), ltn1Δ (3287–1-1), and ufd2Δ (3288–1-3) cells containing PGALFLAG-Htt103QP-GFP were grown in YPR to mid-log phase at 30°C. Galactose was then added to induce Htt103QP expression for 1 h. Glucose was then added to shut off galactose-induced expression. Htt103QP protein levels were detected using an anti-FLAG antibody. The experiment was repeated three times. The western blotting result for Htt103QP level is shown on the left panel. Pgk1, loading control. Protein band intensity and Htt103QP/Pgk1 ratio were analyzed using ImageJ. Quantification results are represented as mean ± SD (right panel). * indicates statistical significant (p < 0.05).
(B) San1 promotes ubiquitination of Htt103QP. WT (3419–1-1) and two san1Δ mutant strains (RH142 and 3301–2-2) containing PGALFLAG-Htt103QP-GFP were grown in YPR to mid-log phase at 30°C. Galactose was then added to induce Htt103QP overexpression for 3 h. After preparation of cell lysates, Htt103QP was immunoprecipitated using M2 anti-FLAG beads. Htt103QP protein levels were detected using anti-FLAG antibody, and the ubiquitination level was detected using anti-Ub antibody. Pgk1, loading control.
(C) Htt103QP inclusion bodies form at an accelerated rate in san1Δ mutant. WT (3419–1-1) and san1Δ (RH142) cells containing PGALFLAG-Htt103QP-GFP were grown in YPR to mid-log phase at 30°C. Galactose was then added to induce Htt103QP expression. Differential interference contrast (DIC) and GFP fluorescence images were obtained every h for 6 h. Scale bar, 5 μm. The quantitative result is the average from three independent experiments. Data are represented as mean ± SD. Two-way ANOVA with Tukey’s multiple comparisons test was performed for each dataset. *p < 0.05.
(D) Htt103QP shows nuclear localization. Yeast cells with H2A-mApple and PGALFLAG-Htt103QP-GFP (2925–3-2) were grown in YPR, and then galactose was added to induce Htt103QP expression. Images were acquired after incubation in galactose for 1 h. Scale bar, 5 μm.
(E) Cells lacking SAN1 are not sensitive to Htt103QP overexpression. WT (3419–1-1), san1Δ (3301–2-2), dsk2Δ (3222–1-1), and dsk2Δ san1Δ (3514–1-2) strains with PGALFLAG-Htt103QP-GFP were grown to saturation in YPR, and then 10-fold serially diluted and spotted onto URA (uracil) dropout plates containing glucose or galactose. The plates were incubated at 30°C for 2 days.
We further determined the role of San1 in the ubiquitination of Htt103QP. We found that san1Δ mutant cells exhibited a dramatic decrease in Htt103QP ubiquitination (Figure 2B). Of note, some ubiquitination of Htt103QP was detected in san1Δ cells, which is likely attributable to other E3 ligases. To the point, a previous study shows a defect in Htt103QP ubiquitination in yeast mutants lacking E3 ligase Ltn1 (Yang et al., 2016). Moreover, ubiquitin ligase Rsp5 has been shown to catalyze the ubiquitination of Htt96Q (mutated Htt with 96 polyQ repeats) in yeast cells (Lu et al., 2014). Unlike the Htt103QP protein, Htt96Q lacks the proline-rich region. Regardless, the dramatic defect in Htt103QP ubiquitination and degradation in san1Δ cells suggest that San1 plays a major role in the ubiquitination of mutated Huntingtin in yeast cells.
Htt103QP forms inclusion bodies in yeast cells when it is over-expressed (Chuang et al., 2016; Higgins et al., 2018; Wang et al., 2009). We found that in san1Δ cells, Htt103QP inclusion bodies formed at an accelerated rate compared to WT cells after Htt103QP induction in galactose medium (Figure 2C). In addition, many san1Δ cells had two or more inclusion bodies after Htt103QP induction, but very few WT cells showed more than one inclusion body. These results suggest that ubiquitination of Htt103QP by San1 is not necessary for its recruitment to inclusion bodies, which is consistent with previous observations in mammalian cells (Bersuker et al., 2016). Because the E3 ligase San1 localizes in the nucleus, the nuclear localization of Htt103QP is likely required for its ubiquitination by San1. After Htt103QP-GFP induction for 1 h, before inclusion bodies had formed, we detected colocalization of Htt103QP-GFP with histone H2A-mApple, which marks the nucleus (Figure 2D). In spite of the stabilization of Htt103QP in san1Δ mutants, san1Δ cells grew slightly better than WT cells on galactose plates that induced Htt103QP overexpression. We found that deletion of the ubiquilin ortholog DSK2 in yeast cells resulted in sensitivity to Htt103QP overexpression (Chuang et al., 2016), but this sensitivity was abolished in dsk2Δ san1Δ cells (Figure 2E), indicating that high levels of un-modified Htt103QP is not cytotoxic. Taken together, these results suggest that E3 ubiquitin ligase San1 ubiquitinates Htt103QP-GFP and targets it for proteasomal degradation, whereas defective ubiquitination of Htt103QP leads to accelerated inclusion body formation with no accompanying cytotoxicity.
San1- and Ubr1-Dependent Ubiquitination Causes Cytotoxicity in cdc48 Mutant Cells
Considering that both San1 and the Cdc48Ufd1/Npl4 complex promote Htt103QP degradation and that San1 is the major E3 ligase that ubiquitinates Htt103QP, we speculated that San1-dependent ubiquitination of Htt103QP and other misfolded proteins in the nucleus enables Cdc48-mediated dissolution of protein aggregates. Therefore, the combination of SAN1 deletion and cdc48 mutation may exacerbate protein aggregation and cause proteotoxicity. Surprisingly, the growth defect of cdc48–3 at elevated temperatures (32°C) was partially suppressed by san1Δ (Figure 3A). E3 ligase Ubr1 is the cytosolic counterpart of San1. Compared to the san1Δ mutant, ubr1Δ had a more profound suppression of the cdc48–3 growth defects at 32°C, and the suppression was even noticeable at 37°C. Similarly, the suppression of the temperature sensitivity of ufd1–2 mutants by ubr1Δ was more dramatic than san1Δ. Strikingly, the growth defect of ufd1–2 at 37°C was near-completely suppressed by ubr1Δ and san1Δ ubr1Δ. We also observed the suppression of the temperature sensitivity of the npl4–1 mutant by san1Δ ubr1Δ at 32°C and 34°C (Figure 3B). Therefore, San1/Ubr1-dependent ubiquitination of misfolded proteins likely contributes to the growth defect in cells with Cdc48Ufd1/Npl4 deficiency, and Ubr1-dependent ubiquitination of misfolded proteins in the cytoplasm seems to play a more important role.
Figure 3. The Absence of San1 and Ubr1 Partially Suppresses the Temperature Sensitivity of cdc48–3, npl4–1, and ufd1–2 Mutants.
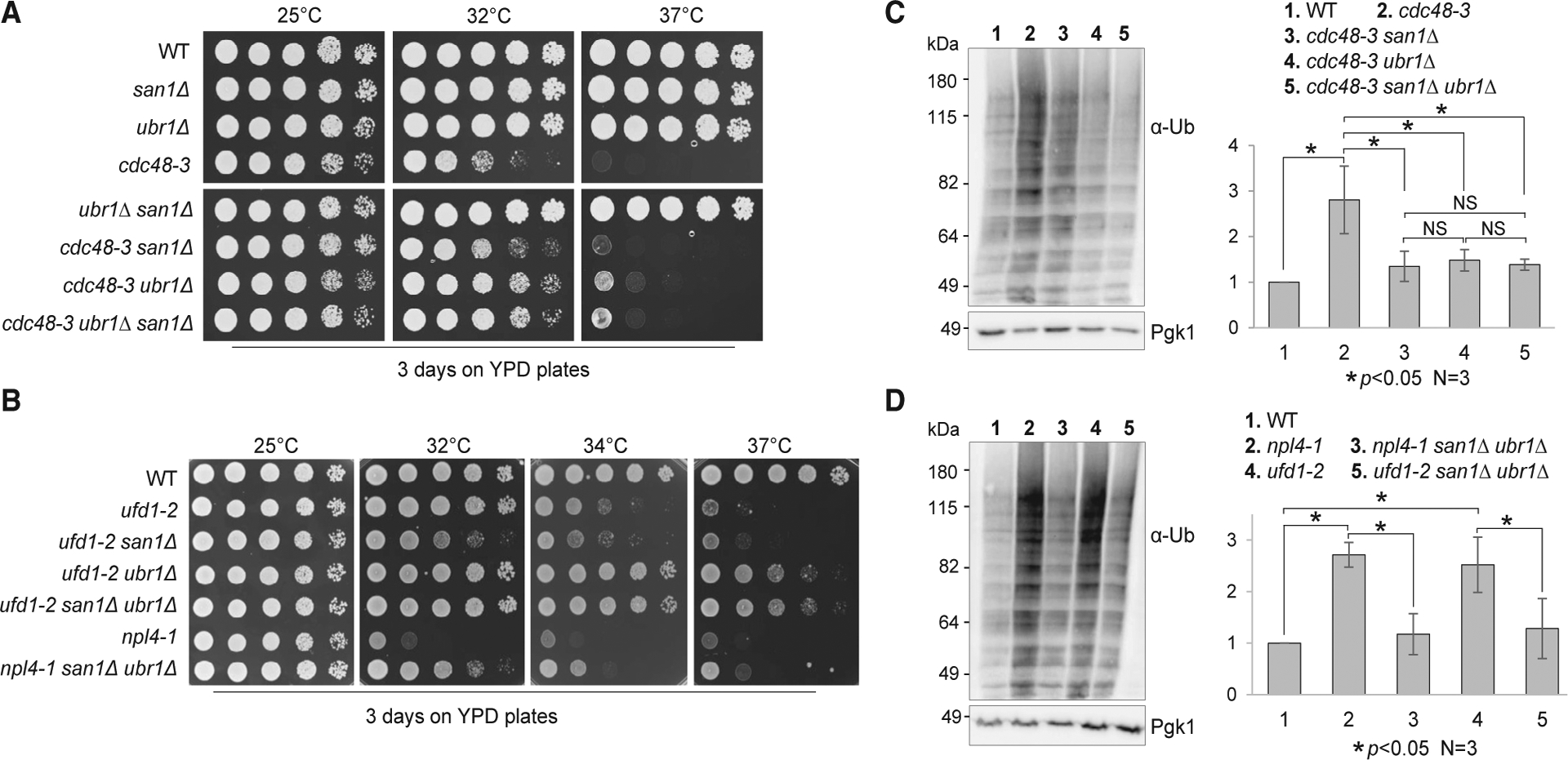
(A) The suppression of the temperature sensitivity of cdc48–3 mutants by san1Δ, ubr1Δ, and san1Δ ubr1Δ mutants. Cells with the indicated genotypes were grown in YPD to saturation and then 10-fold serially diluted onto YPD plates. The plates were incubated at 25°C, 32°C, and 37°C for 2 days. Strains used in this experiment were WT (Y300), cdc48–3 (MHY3512), cdcc48–3 san1Δ (3550–5-3), cdc48–3 ubr1Δ (3550–6-3), and cdc48–3 san1Δ ubr1Δ (3550–2-1).
(B) The growth defect of npl4–1 and ufd1–2 is partially suppressed by san1Δ ubr1Δ mutants. The strains used were WT (Y300), ufd1–2 (1122), ufd1–2 san1Δ (3556–1-1), ufd1–2 ubr1Δ (3556–2-3), ufd1–2 san1Δ ubr1Δ (3556–3-3), npl4–1 (1126), and npl4–1 san1Δ ubr1Δ (3555–5-1). The plates were incubated at 25°C, 32°C, 34°C, and 37°C for 2 days.
(C) The accumulation of ubiquitinated proteins in cdc48–3 mutant cells is suppressed by san1Δ, ubr1Δ, and san1Δ ubr1Δ mutants. Cells with the indicated genotypes were grown in YPD at 25°C and then shifted to 34°C for 5 h. Protein samples were prepared, and ubiquitinated protein species were detected using an anti-Ub antibody. Pgk1, loading control. The experiment was repeated three times. The band intensity of ubiquitinated protein species and Pgk1 was analyzed using ImageJ. The relative ubiquitination level over Pgk1 is shown as mean ± SD. *p < 0.05. NS, not statistically significant. Strains used in this experiment were WT (Y300), cdc48–3 (MHY3512), cdc48–3 san1Δ (3550–5-3), cdc48–3 ubr1Δ (3550–6-3), and cdc48–3 san1Δ ubr1Δ (3550–2-1).
(D) The accumulation of ubiquitinated proteins in npl4 and ufd1 mutants is suppressed by san1Δ ubr1Δ. Quantification of relative level of protein ubiquitination and statistical analysis were performed as described above. The strains used in this experiment were WT (Y300), npl4–1 (1126), npl4–1 san1Δ ubr1Δ (3555–5-1), ufd1–2 (1122), and ufd1–2 san1Δ ubr1Δ (3556–3-3).
Downregulation of Cdc48 results in significant accumulation of polyubiquitinated proteins in Drosophila, zebrafish, and human cells (Imamura et al., 2012). We tested if yeast cdc48 mutants accumulate more ubiquitinated proteins and if this accumulation depends on San1 and Ubr1. When grown at 34°C, cdc48–3 mutants exhibited a much higher level of ubiquitinated protein species than did WT cells. Strikingly, deletion of SAN1, UBR1, or SAN1 UBR1 strongly suppressed this accumulation, and the suppression was statistically significant. Although the suppression of the temperature sensitivity by ubr1Δ was more profound than san1Δ, we did not observe stronger suppression of the accumulation of ubiquitinated protein species in cdc48–3 by ubr1Δ or san1Δ ubr1Δ than san1Δ (Figure 3C). One explanation is that the ubiquitinated misfolded proteins in the nucleus or cytoplasm exhibit different toxicity. Similarly, a dramatic increase in ubiquitinated protein species was detected in npl4–1 and ufd1–2 mutants grown at 34°C, and this accumulation was abolished by san1Δ ubr1Δ (Figure 3D). These results suggest that most Cdc48Ufd1/Npl4 substrates are ubiquitinated by San1 and Ubr1 E3 ligases.
Given that San1 and Ubr1 ubiquitinate misfolded proteins that are prone to aggregation, cells with Cdc48 deficiency might accumulate more misfolded protein aggregates. To test this idea, we used C-terminally GFP-tagged Hsp104, a disaggregase chaperone, to mark endogenous protein aggregates (Higgins et al., 2018; Jacobson et al., 2012). After shifting the temperature from 25°C to 34°C for 1 h, both WT and cdc48–3 cells exhibited an initial increase in the number of Hsp104-GFP foci, presumably due to heat shock. The average number of GFP foci in WT cells was approximately one focus per cell, but the average number was over three in cdc48–3 cells. After 3 h at 34°C, the number of Hsp104-GFP foci in WT cells decreased but remained relatively constant in cdc48–3 cells (Figure S2A). Similarly, an increase in Hsp104-GFP foci was also observed in the npl4–1 and ufd1–2 mutant cells growing at 34°C (Figure S2B). Because the foci appear similar in size in WT and mutant cells, these results suggest that compromised Cdc48Ufd1/Npl4 function results in the accumulation of more protein aggregates due to a reduced efficiency of clearance.
Impairment of the UPS in cdc48–3 Mutants
Our results suggest that San1/Ubr1-dependent ubiquitination of misfolded proteins partially contributes to the growth defect in cells with Cdc48 deficiency. One further question is how the ubiquitination of misfolded proteins contributes to the growth defect in cdc48 mutant cells. One possibility is that the ubiquitination of large amounts of misfolded proteins compromises UPS function by occupying and potentially inhibiting proteasomes. Alternatively, this ubiquitination results in the depletion of free ubiquitin necessary for UPS function and/or other cellular processes. To test this idea, we first used genetic methods to examine the UPS function in the cdc48–3 mutant. Rpn4 is a transcription factor, and RPN4 deletion compromises UPS function by downregulating the expression of proteasome subunits (Xie and Varshavsky, 2001). We crossed rpn4Δ with cdc48–3, but after tetrad dissection, no rpn4Δ cdc48–3 double mutants were viable, indicating their synthetic lethality (Figure 4A, left). Rpn10 is a proteasome regulatory particle (RP) subunit that maintains the structural integrity of the RP while also acting as a polyubiquitin receptor (Glickman et al., 1998; Tomko and Hochstrasser, 2011). Cells lacking RPN10 are viable, but rpn10Δ cdc48–3 double mutants were also synthetically lethal (Figure 4A, right). The synthetic lethality between cdc48–3 and two mutants (rpn4Δ and rpn10Δ) that show compromised UPS function suggests that the cdc48–3 mutation likely impairs UPS function as well. We next analyzed the genetic interaction between cdc48–3 and ubr2Δ mutants. Ubr2 is an E3 ubiquitin ligase that targets Rpn4 for degradation; thus, ubr2Δ cells accumulate high levels of the Rpn4 protein, which increases the expression of proteasome subunits (Wang et al., 2004; Xie and Varshavsky, 2001). In clear contrast, we found that UBR2 deletion partially rescued the growth defects of cdc48–3, npl4–1, and ufd1–2 mutants at 34°C (Figure 4B). This is consistent with a previous finding that Rpn4 overexpression suppresses the growth defect of cdc48–3 function (Chien and Chen, 2013). Together, these results indicate impaired UPS function in cdc48 mutants.
Figure 4. cdc48 Mutants Show Compromised UPS Function.
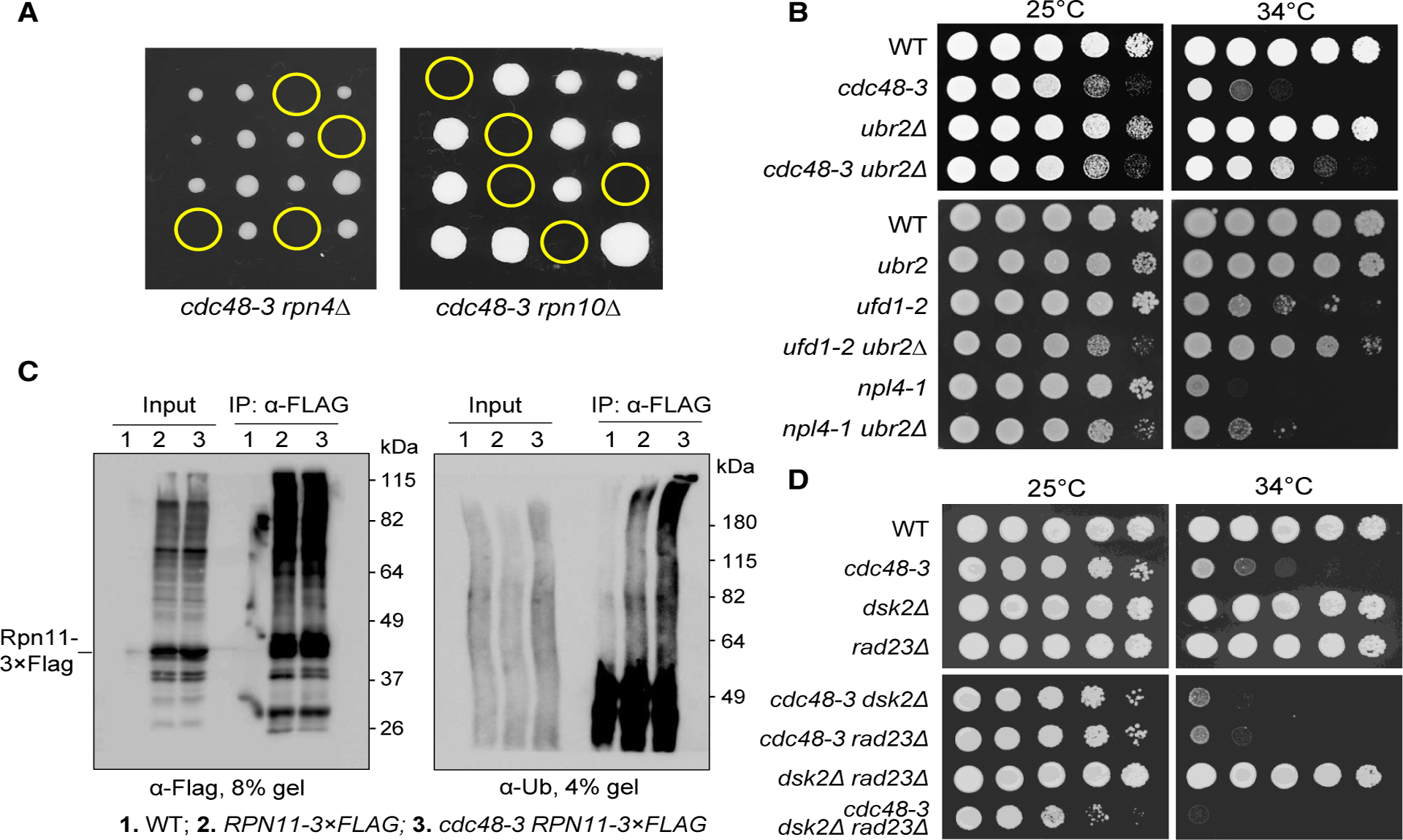
(A) The cdc48–3 mutation is synthetically lethal with rpn4Δ and rpn10Δ. The cdc48–3 mutant was crossed to rpn4Δ and rpn10Δ mutants to obtain diploid cells. The growth of the resultant spores at 25°C after tetrad dissection is shown. Yellow circles indicate spores of cdc48–3 rpn4Δ or cdc48–3 rpn10Δ double mutants.
(B) The temperature sensitivity of cdc48–3, npl4–1, and ufd1–2 mutants is partially suppressed by ubr2Δ. Cells with the indicated genotypes were grown to saturation in YPD, 10-fold serially diluted, and spotted onto YPD plates. Cells were grown at 25°C and 34°C for 2 days. The yeast strains used in this experiment were WT (Y300), cdc48–3 (MHY3512), ubr2Δ (3969–4-4), cdc48–3 ubr2Δ (3655–2-4), npl4–1 (1126), ufd1–2 (1122), npl4–1 ubr2Δ (3658–1-4), and ufd1–2 ubr2Δ (3659–1-2).
(C) The cdc48–3 mutant cells show increased accumulation of ubiquitinated substrates on proteasomes. Log-phase cells of pdr5Δ (3589–1-4), pdr5Δ RPN11–3 × FLAG (3592–4-4), and pdr5Δ cdc48–3 RPN11–3 × FLAG (3592–5-2) grown in 25°C YPD were shifted to 34°C for 3 h. MG-132 was then added at 50 mM for 1 h to inhibit proteasome activity. The cells were then lysed, the Rpn11–3 × FLAG protein was immunoprecipitated using M2 anti-FLAG beads, and ubiquitinated proteins were detected using anti-Ub antibody. We used 4% SDS-PAGE to visualize high-molecular-weight ubiquitinated species (right panel).
(D) dsk2Δ rad23Δ mutation does not rescue the growth defect in cdc48 mutants. Cells with indicated genotypes were grown in YPD to saturation, then 10-fold serially diluted, and spotted onto YPD plates. Cells were grown at 25°C or 34°C for 2 days. The strains used were WT (Y300), cdc48–3 (MHY3512), dsk2Δ (YYW14), rad23Δ (3553–2-4), cdc48–3 dsk2Δ (3553–7-2), cdc48–3 rad23Δ (3553–10-3), dsk2Δ rad23Δ (3553–5-3), and cdc48–3 dsk2Δ rad23Δ (3553–3-2).
The Accumulation of Ubiquitinated Proteins on Proteasomes Plays an Insignificant Role in the Growth Defect of cdc48 Mutants
Previous studies showed increased accumulation of ubiquitinated proteins on proteasomes in cdc48 mutants (Tsuchiya et al., 2017; Verma et al., 2011). This enhanced proteasome occupancy may compromise normal cellular protein turnover. Thus, we first confirmed the accumulation of ubiquitinated substrates on proteasomes by using a previously described protocol (Tsuchiya et al., 2017). WT and cdc48–3 mutant cells expressing 3× FLAG-tagged Rpn11, a proteasome subunit, were grown at a semi-permissive temperature (34°C), and proteasomes were isolated by immunoprecipitating Rpn11. We noticed that the accumulation of ubiquitinated substrates on proteasomes in cdc48–3 mutant cells was greater than that in WT cells (Figure 4C). Here, we used anti-ubiquitin immunoblotting of a 4% SDS-PAGE gel to detect ubiquitinated high-molecular-weight species. Using Htt103QP as a model misfolded protein, we also detected an increased association of Htt103QP with proteasomes in cdc48–3 mutant cells (Figure S3), suggesting that Cdc48 deficiency leads to increased association of misfolded proteins with proteasomes. Because San1 and Ubr1 are primarily responsible for the ubiquitination of misfolded proteins, we examined whether their absence would decrease the association of misfolded proteins with proteasomes in the cdc48–3 mutant. Not surprisingly, we found that the accumulation of ubiquitinated proteins on proteasomes in cdc48–3 mutants was partially suppressed by san1Δ ubr1Δ (Figure S4). These results support the notion that Cdc48 deficiency increases the accumulation of ubiquitinated misfolded proteins on proteasomes, which is consistent with the previous observation (Tsuchiya et al., 2017).
It has been shown that the proteasome shuttling factors Dsk2 and Rad23 are responsible for the increased accumulation of ubiquitinated substrates on proteasomes in cdc48–3 mutant cells (Tsuchiya et al., 2017). Our results confirmed Dsk2/Rad23-dependent proteasome accumulation of ubiquitinated proteins in cdc48–3 mutant cells (Figure S5). Surprisingly, the dsk2Δ rad23Δ mutation did not suppress the temperature sensitivity of cdc48–3 mutants but rather resulted in a more severe growth defect at both 25°C and 34°C (Figure 4D). Although more ubiquitinated substrates accumulate on proteasomes in cdc48–3 mutant cells, this result indicates that this accumulation may play a minor role in the growth defect caused by Cdc48 deficiency. The synthetic growth defect of cdc48–3 dsk2Δ rad23Δ could be explained by the roles of Dsk2, Rad23, and Cdc48 in UPS-dependent protein degradation and suggests the source of misfolded protein toxicity lies upstream of the segregation of aggregates and delivery to the proteasome.
San1/Ubr1 Ubiquitin Ligases and the Cdc48 Complex Control Ubiquitin Homeostasis
We showed San1/Ubr1-dependent accumulation of ubiquitinated proteins in cells with dysfunctional Cdc48Ufd1/Npl4 (Figures 3C and 3D). This accumulation may decrease free ubiquitin levels. If so, deletion of SAN1 and UBR1 may abolish this decrease. Thus, we incubated WT, cdc48–3, npl4–1, and ufd1–2 mutant cells at 34°C for 5 h and then examined their free ubiquitin levels. Indeed, all three mutants exhibited a significant decrease of free ubiquitin compared to WT cells, and the difference was statistically significant. Remarkably, a san1Δ ubr1Δ mutation restored free ubiquitin levels in all of these mutants (Figure 5A). These results suggest that the accumulation of ubiquitinated protein species in Cdc48Ufd1/Npl4 mutants decreases the free ubiquitin pool and that blocking misfolded protein ubiquitination by deleting SAN1 and UBR1 restores free ubiquitin levels.
Figure 5. Disrupted Ubiquitin Homeostasis and the Growth Defect of cdc48 Mutants.
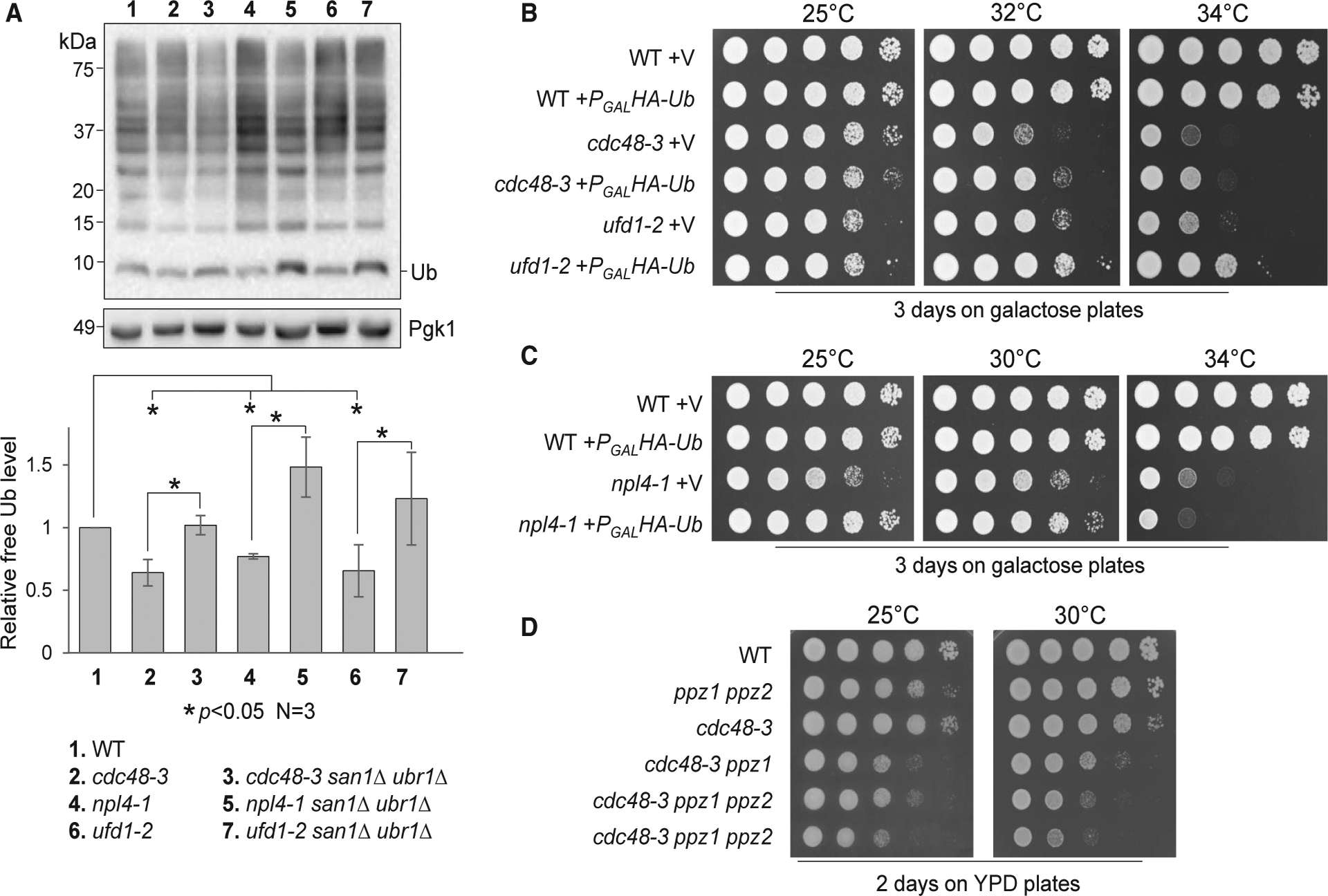
(A) Cdc48Ufd1/Npl4 mutants show decreased free ubiquitin. Cells with the indicated genotypes were first grown in YPD at 25°C to mid-log phase and then shifted to 34°C for 5 h. Samples were prepared using Laemmli buffer (no lysis method used). Samples were resolved using Tricine-SDS-PAGE. Unconjugated (free) ubiquitin levels were detected using anti-Ub antibody. Pgk1, loading control. ImageJ was used to measure the intensity of mono-ubiquitin and Pgk1 bands. The ubiquitin/Pgk1 ratio represents the relative free ubiquitin level. The quantified result (mean ± SD) is from three independent experiments. *p < 0.05. Strains used in this experiment were WT (Y300), cdc48–3 (MHY3512), cdc48–3 san1Δ ubr1Δ (3550–2-1), npl4–1 (1126), npl4–1 san1Δ ubr1Δ (3555–5-1), ufd1–2 (1122), and ufd1–2 san1Δ ubr1Δ (3556–3-3).
(B) Overexpression of ubiquitin partially rescues the temperature sensitivity of cdc48–3 and ufd1–2 mutants. WT (Y300), cdc48–3 (MHY3512), and ufd1–2 (1122) cells containing either CEN-TRP1 control vector p1217 (V) or PGALHA-Ub (Ub) were grown to saturation in TRP (tryptophan) dropout medium containing raffinose, then 10-fold serially diluted, and spotted onto TRP dropout plates containing galactose. Cells were grown at 25°C, 32°C, and 34°C for 3 days.
(C) Overexpression of ubiquitin partially rescues the temperature sensitivity of npl4–1. WT (Y300) and npl4–1 (1126) with vector and PGALHA-Ub plasmid were used for this experiment. The plates were incubated at 25°C, 30°C, and 34°C for 3 days.
(D) Deletion of ubiquitin kinases exacerbates the growth defect of cdc48–3 mutants. Saturated WT, ppz1Δ ppz2Δ (PHY648), cdc48–3 (MHY3512), cdc48–3 ppz1Δ (4023–1-1), and cdc48–3 ppz1Δ ppz2Δ (4023–2-4, 4023–8-4) cells were serially 10-fold diluted and spotted onto YPD plates. The plates were imaged after a 3-day incubation at 25°C and 30°C.
We showed that san1Δ ubr1Δ mutants suppress the growth defect of Cdc48Ufd1/Npl4 mutants at elevated temperatures (Figure 3). If this suppression is attributable to the restoration of free ubiquitin levels, high levels of ubiquitin expression should also suppress the growth defect in these mutants. Therefore, we introduced an empty vector or PGALHA-Ub (ubiquitin) plasmid into WT, cdc48–3, npl4–1, and ufd1–2 cells. Ubiquitin overexpression partially rescued the temperature sensitivity of cdc48–3 and ufd1–2 mutants at 32°C and 34°C (Figure 5B). Although ubiquitin overexpression did not suppress the growth defect of the npl4–1 mutant at 34°C, the overexpression did improve its growth at 25°C and 30°C (Figure 5C). We confirmed the induction of hemagglutinin (HA)-tagged ubiquitin in galactose medium (Figure S6A). In contrast to this result, a previous study showed that expression of monomeric ubiquitin from a high copy number plasmid (2μ) under a GAP1 promoter was toxic to a cdc48–3 mutant (Kimura et al., 2009). To clarify this discrepancy, WT and cdc48–3 mutant cells were transformed with an empty vector or PADH1RPS31(UBI3) plasmid, which expresses a high level of yeast ubiquitin (UBI3) from a strong ADH1 promotor (Lee et al., 2017). Although the suppression of the temperature sensitivity of cdc48–3 by PADH1RPS31 was not as clear as PGALHA-Ub, ubiquitin overexpression using this plasmid did not show toxicity to cdc48–3 (Figure S6B).
If the function of Cdc48 is to promote ubiquitin homeostasis, decreased free ubiquitin levels would be expected to exacerbate the growth defect of cdc48 mutants. However, deleting one of the ubiquitin encoding genes, UBI4, in cdc48–3 mutants failed to cause a synthetic growth defect at the temperatures tested. The observation that the expression of the UBI4 gene is only induced by stresses may explain this result (Finley et al., 1987). A recent study shows that Ppz1 and Ppz2 phosphorylate ubiquitin protein at Ser57, which stabilizes ubiquitin. Ubiquitin levels decrease in yeast cells lacking Ppz1 and Ppz2 (Lee et al., 2017). Strikingly, we found that deletion of either PPZ1 alone or both PPZ1 and PPZ2 aggravated the growth defect of cdc48–3 cells at both 25°C and 30°C (Figure 5D), indicating that cdc48 mutant cells are sensitive to a reduced free ubiquitin pool. This result argues against the proteasome occupancy model, as decreased ubiquitin levels would decrease the delivery of ubiquitinated protein aggregates to the proteasome and thus would be expected to “unclog” the proteasome. Together, these results suggest that the Cdc48Ufd1/Npl4 complex may drive ubiquitin recycling by disaggregating ubiquitinated protein aggregates. Furthermore, it suggests that a reduced free ubiquitin pool likely contributes to the growth defect in mutants with impaired Cdc48Ufd1/Npl4 function.
Compromised UPS Function in cdc48 Mutants and Ubiquitin Homeostasis
Our preceding results indicate compromised UPS activity and a ubiquitin homeostasis defect in cdc48 mutant cells. The reduced free ubiquitin level in cdc48 mutants may lead to compromised UPS function. To test this idea, we further verified compromised UPS activity in cdc48–3 cells by measuring the degradation kinetics of Clb5, an S-phase cyclin. Clb5 shows ubiquitin-dependent proteasomal degradation (Shirayama et al., 1999; Wang et al., 2001). We used a glucose shutoff assay to compare Clb5 stability in WT and cdc48–3 mutant cells incubated at 34°C, as described for Htt103QP. To rule out any cell-cycle-related effects on protein degradation, the cells were arrested in G1 phase for the entire experiment using the α-factor. After HA-Clb5 expression shutoff, cdc48–3 cells showed a slower rate for Clb5 degradation than WT cells, as indicated by the significantly higher level of Clb5 in cdc48–3 cells than WT cells after Clb5 expression was turned off for 120 and 150 min (Figure 6A), suggesting that functional Cdc48 is required for the efficient destruction of proteasomal substrates. We have shown that the temperature sensitivity of cdc48–3 is suppressed either by a ubr2Δ mutant, which increases the expression of proteasome subunits, or by san1Δ ubr1Δ mutation, which decreases the ubiquitination of misfolded proteins. Strikingly, both ubr2Δ and san1Δ ubr1Δ mutations significantly suppressed the Clb5 degradation defect in cdc48–3 cells (Figure 6A). As a segregase, Cdc48 may facilitate Clb5 degradation by separating cyclin Clb5 from cyclin-dependent kinase, but the suppression of the Clb5 degradation defect in cdc48–3 cells by deletion of San1 and Ubr1 argues against this possibility. In addition, we used the cycloheximide chase method to assess the degradation kinetics of endogenous Clb5 in WT, cdc48–3, and cdc48–3 san1Δ ubr1Δ strains expressing HA-CLB5. Compromised Clb5 degradation was also observed in cdc48–3 mutant cells after cycloheximide addition. A san1Δ ubr1Δ mutation suppressed the Clb5 degradation defect in cdc48–3 mutant cells (Figure 6B). These results suggest that Cdc48 deficiency compromises proteasome-mediated protein degradation, and this UPS defect is likely caused by the accumulation of misfolded protein aggregates, which decreases the free ubiquitin pool.
Figure 6. The Ubiquitination of Misfolded Proteins Compromises UPS Function in cdc48 Mutants.
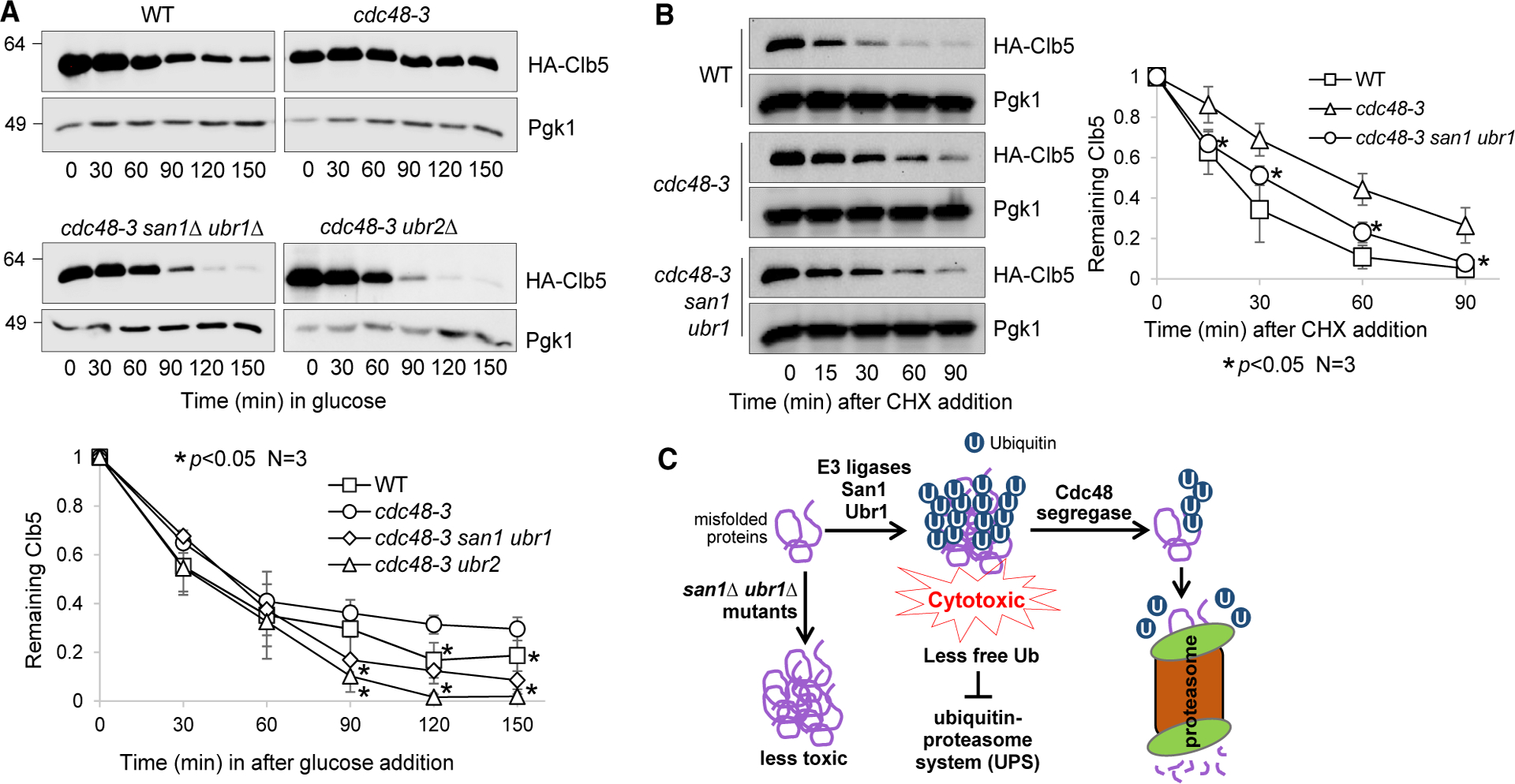
(A) The cdc48–3 mutant shows compromised degradation of S-phase cyclin Clb5, and this defect is suppressed by enhanced proteasome activity or decreased ubiquitination of misfolded proteins. WT (FY-13–1), cdc48–3 (3504–3-2), cdc48–3 san1Δ ubr1Δ (3580–1-3), and cdc48–3 ubr2Δ (3660–1-4) cells containing PGALHA-CLB5 were grown to mid-log phase in YPR (raffinose medium) at 25°C and then treated with α-factor for G1 phase arrest for 2 h. Cells were shifted to 34°C for 30 min, and galactose was added for 1 h to induce HA-Clb5 overexpression. Glucose was then added to shut off HA-Clb5 expression. Cells were collected to determine HA-Clb5 protein levels. G1 arrest was maintained throughout the experiment. Pgk1, loading control. The relative level of HA-Clb5 to Pgk1 was analyzed using ImageJ. The quantification shown at the bottom is the average of three independent experiments (mean ± SD). *p < 0.05.
(B) san1Δ ubr1Δ mutation alleviates the compromised degradation of endogenous Clb5 in cdc48–3 mutant cells using CHX chase. WT (229–3-2), cdc48–3 (3968–5-1), and cdc48–3 san1Δ ubr1Δ (3968–4-3) cells with HA-tagged CLB5 in the chromosome locus were grown to mid-log phase in YPD at 25°C, and then the temperature was shifted to 34°C. After temperature shift for 30 min, CHX (200 μg/mL) was added to the medium, and cells were collected over time to examine the HA-Clb5 protein level using an anti-HA antibody. Pgk1, loading control. The experiment was repeated three times. The quantification results are represented as mean ± SD on the right panel. *p < 0.05.
(C) Working model. San1 and Ubr1 E3 ligases ubiquitinate misfolded proteins, which form aggregates. Cdc48-dependent disaggregation enables the degradation of misfolded proteins and ubiquitin recycling. Deletion of SAN1 and UBR1 overcomes the requirement for the Cdc48 complex in ubiquitin recycling.
DISCUSSION
This research provides novel insight into the nature of the cytotoxicity of misfolded proteins. Our results suggest that ubiquitination of misfolded proteins causes toxicity to cells at least in part by depleting free ubiquitin, thereby compromising UPS function. In response to proteotoxic stress, the Cdc48 complex alleviates the cytotoxicity by promoting the disaggregation of ubiquitinated protein aggregates and driving ubiquitin recycling. Blocking misfolded protein ubiquitination in yeast cells by deleting E3 ligases San1 and Ubr1 decreases the cytotoxicity of misfolded proteins and reduces dependence on the Cdc48Ufd1/Npl4 complex to combat proteotoxicity (Figure 6C).
Although the Cdc48Ufd1/Npl4 complex has been shown to extract ubiquitinated substrates from membranes and macromolecular complexes (Blythe et al., 2017; Bodnar and Rapoport, 2017b; Twomey et al., 2019), it remains largely unknown how Cdc48Ufd1/Npl4 acts in the proteotoxic stress response. Using Htt103QP as a model misfolded protein, we found compromised Htt103QP degradation and elevated accumulation of ubiquitinated Htt103QP in cdc48–3 mutant cells. Furthermore, using an endogenous marker of protein aggregates, Hsp104-GFP, we also observed increased protein aggregation in cells with Cdc48Ufd1/Npl4 deficiency. These results indicate the role of Cdc48 in segregating ubiquitinated protein aggregates, which likely facilitates protein degradation and ubiquitin recycling.
E3 ligases San1 and Ubr1 are responsible for the ubiquitination of misfolded proteins in budding yeast (Samant et al., 2018). One interesting observation is that deletion of San1 and Ubr1 suppresses the growth defect in cells with Cdc48 deficiency. Because deletion of these two ubiquitin ligases impairs ubiquitination of misfolded proteins, the accumulation of ubiquitinated proteins is likely a contributing factor to the growth defect in Cdc48Ufd1/Npl4 mutants. Two avenues were explored for the reason why the accumulation of ubiquitinated proteins is toxic. First, the polyubiquitinated protein aggregates occupy proteasomes and compromise their function. Second, the accumulation of polyubiquitinated proteins decreases free ubiquitin levels, which impairs UPS function indirectly. Our results support the second possibility.
Our findings, combined with previous reports, indicate that dysfunctional Cdc48 results in the accumulation of polyubiquitinated substrates on proteasomes (Tsuchiya et al., 2017; Verma et al., 2011). The absence of proteasome shuttling factors Rad23 and Dsk2 abolishes this accumulation (Tsuchiya et al., 2017) but yields no suppression of the growth defect of cdc48–3 cells. Although we cannot exclude the possibility that the occupancy of proteasomes by misfolded protein aggregates impairs UPS function, this may not be the major contribution to the growth defect in cdc48-3 cells. We have provided several lines of evidence indicating that polyubiquitinated protein aggregates act as a sponge to drain the free ubiquitin pool and impair UPS function. First, the absence of San1 and Ubr1 ubiquitin ligases reduces the accumulation of polyubiquitinated protein species in cdc48-3, npl4–1, and ufd1-2 mutants, while also partially suppressing their growth defects. Second, san1Δ ubr1Δ restores the efficiency of UPS-dependent degradation of cyclin Clb5 in cdc48–3 mutant cells. Third, and importantly, ubiquitin overexpression suppresses the temperature sensitivity of Cdc48Ufd1/Npl4 complex mutants, but a lower free ubiquitin level caused by ppz1Δ ppz2Δ exacerbates the growth defect of cdc48–3 mutants. Together, our data suggest a new mechanism for how misfolded proteins impose cytotoxicity. The ubiquitination of misfolded proteins appears to be a double-edged sword. Cells target misfolded proteins for degradation through their ubiquitination; however, under certain conditions, this drains free ubiquitin and impairs UPS function. Given the high abundance of the Cdc48 protein (Baek et al., 2013), cells are well equipped to combat spontaneous protein misfolding and aggregation, but upon Cdc48 inactivation, the accumulation of polyubiquitinated protein aggregates poses a greater risk to cell health.
Overall, our findings highlight that ubiquitination of misfolded proteins can cause cytotoxicity if not degraded in a timely manner, at least in part due to the decrease of the free ubiquitin pool. Moreover, our results suggest that the Cdc48Ufd1/Npl4 complex not only facilitates proteasomal degradation of misfolded proteins but also indirectly stimulates ubiquitin recycling. In addition, the absence of two ubiquitin ligases, San1 and Ubr1, known to target misfolded proteins results in a decrease in the toxicity of misfolded proteins. Our data support a novel mechanism by which misfolded proteins cause cytotoxicity and highlight the important roles of the Cdc48Ufd1/Npl4 segregase and ubiquitin ligases in ubiquitin homeostasis (Figure 6C).
The Cdc48Ufd1/Npl4 complex is conserved from yeast to humans, indicating that human cells likely use the same mechanism to combat the cytotoxicity of misfolded proteins. Mutations in the human Cdc48 segregase p97/VCP are associated with inclusion body myopathy with Paget disease of bone(Watts et al., 2004).Additionally, aneuploid cancer cells show increased proteotoxic stress due to the synthesis of unnecessary proteins, which results in hypersensitivity to inhibitors of Cdc48/p97 (Chapman et al., 2015; Luo et al., 2009). Furthermore, recent research indicates the anticancer activity of the alcohol-abuse drug disulfiram, which targets Cdc48 cofactor Npl4 (Skrott et al., 2017). Therefore, our research provides a new angle to understand the function of the Cdc48 complex in response to proteotoxic stress.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Yanchang Wang (yanchang.wang@med.fsu.edu).
Materials Availability
All of the yeast strains and plasmids generated by this work will be available upon request.
Data and Code Availability
This study did not generate any unique datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains used in this study are of the W303 background. The relevant genotypes of the strains used in this study are listed in Table S2. Gene deletions and GFP tagging of HSP104 were performed using a PCR-based method (Longtine et al., 1998). The PGAL-FLAG-Htt103QP-GFP plasmid was originally from the Lindquist laboratory (Krobitsch and Lindquist, 2000). Manipulation of this plasmid and the construction of the p1217/PGALHA-Ub plasmid were previously described (Higgins et al., 2018). Yeast extract/peptone media supplied with raffinose (YPR), galactose (YPG), or glucose (YPD) were used for the growth of yeast strains, except for those carrying centromeric plasmids, in which case synthetic dropout medium was used.
METHOD DETAILS
Western Blotting
Unless otherwise noted, protein samples were prepared using an alkaline method and resolved by 8% SDS–PAGE. The resources of the antibodies used in this study are listed in the above table. After ECL, the western blot membranes were imaged using Bio-Rad ChemiDoc.
Ubiquitination of Htt103QP
The protocol was adapted from a previous publication (Higgins et al., 2018). Briefly, cells containing PGALFLAG-Htt103QP-GFP were grown in YPR (raffinose medium) to OD600 = 0.4. Galactose was then added to the medium to the final concentration of 2% to induce FLAG-Htt103QP-GFP expression for 3 hours. Ten mL of cells were harvested by centrifugation at 10,000 rpm for 5 minutes. Cells were washed once with water and resuspended in RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM EDTA, 0.05% Tween-20) along with sodium azide and protease inhibitor cocktail. Samples were frozen in liquid nitrogen and subsequently crushed using a freezer mill. Samples were then thawed on ice in the presence of 20 mM deubiquitinase inhibitor N-ethylmaleimide. After centrifugation at 15,000 rpm for 20 minutes at 4°C, the supernatant was collected and centrifuged again at 15,000 rpm for 20 minutes at 4°C. Input sample was taken prior to the addition of M2 FLAG agarose beads to immunoprecipitate FLAG-Htt103QP-GFP. Beads were washed three times, resuspended in Laemmli buffer, and boiled for 5 minutes. Western blotting was performed using anti-FLAG, anti-Ub, and anti-Pgk1 antibodies.
The interaction of ubiquitinated substrates with proteasomes
We used a previous protocol with minor modifications to detect the interaction of ubiquitinated substrates with proteasomes (Tsuchiya et al., 2017). All strains used in these experiments were in the pdr5Δ background, which allows efficient proteasome inhibition by the proteasome inhibitor MG-132 (Ravid and Hochstrasser, 2007). Briefly, cells were grown in 50 mL YPD (glucose medium) at 25°C to OD600 = 0.4, followed by a temperature shift to 34°C for 3 hours. MG-132 (50 μM) was added for one additional hour at 34°C. Cells were then harvested and resuspended in 10 mL YPD containing 1% of paraformaldehyde for 10 minutes. Glycine was then added at 250 mM to quench the paraformaldehyde. Cells were harvested, resuspended in 200 μl lysis buffer, and lysed by a bead beater. To the lysate, 200 μl of lysis buffer containing 2% Triton X-100 was added and samples were incubated on ice for 30 minutes. The samples were centrifuged at 15,000 rpm for 10 minutes. We removed 25 μl of supernatant as an input, and M2 anti-FLAG agarose beads were added to the remaining supernatant to immunoprecipitate Rpn11–3 × FLAG. The beads were washed three times with lysis buffer containing 1% Triton X-100. Beads were resuspended in NuPAGE 1 × SDS loading buffer (Invitrogen) and incubated at 65°C for 10 minutes. Samples were resolved using SDS-PAGE.
Monitoring degradation of Htt103QP
The degradation of Htt103QP was performed using a shut-off assay as described previously (Chuang et al., 2016; Higgins et al., 2018). Briefly, the expression of FLAG-Htt103QP-GFP was induced in YPG (galactose medium) for 1 hour, then glucose was added and the protein levels of Htt103QP were determined by western blotting using an anti-FLAG antibody.
The degradation of Htt103QP was also performed with cycloheximide chase assay. Cells with indicated genotypes were grown to mid-log phase. After Htt103QP expression was induced in YPG for 30 minutes, cycloheximide was added to the medium at the final concentration of 200 μg/mL. Cells were collected over time to measure Htt103QP protein levels.
Clb5 degradation kinetics
Cells containing an integrated PGALHA-CLB5 plasmid were grown in YPR (raffinose medium) to mid-log phase at 25°C. To arrest cells in G1 phase, we added α-factor (1 μg/mL) to cell cultures for 2 hours at 25°C, followed by a temperature shift to 34°C for 30 minutes. Galactose (2%) was added to induce HA-Clb5 overexpression for 1 hour, and then glucose (2%) was added to shut off expression. G1 arrest was maintained throughout the entire experiment by adding α-factor at 1-hour intervals. Samples were prepared using the alkaline method, resuspended in Laemmli buffer, and boiled for 5 minutes. HA-Clb5 was detected using an anti-HA antibody.
Cycloheximide chase was also used to determine the degradation kinetics of endogenous Clb5 protein. Yeast strains expressing CLB5-HA from the endogenous promotor were grown to mid-log phase and cycloheximide was added to the medium to the final concentration of 200 μg/mL. Cells were collected over time to prepare protein samples as above. The protein level of Clb5 was determined by western blotting with anti-HA antibody.
Free ubiquitin detection
Yeast cells were inoculated into 5 mL of YPD (glucose medium) and incubated overnight at 25°C. The cultures were diluted to an OD600 of 0.1 in 5 mL of fresh YPD and incubated at 25°C for 90 minutes, then transferred to 34°C for 5 hours. The cells were harvested by centrifugation at 10,000 rpm for 1 minute. The resulting pellets were resuspended in 1 × Laemmli loading buffer and boiled at 95°C for 5 minutes. Samples were separated using 12% SDS-PAGE with Tris-Tricine running buffer. The gel was electrophoresed at 80 V for 15 minutes and then switched to 100 V until the dye front escaped. Gels were transferred to PVDF membranes (EMD Millipore) at 100 V for 1 hour at 4°C and immunoblotted with antibodies against ubiquitin and Pgk1. Band intensities were quantified using ImageJ, and the ubiquitin/Pgk1 ratio represents ubiquitin level.
Fluorescence imaging
Fluorescence imaging analysis was performed using an EVOS microscope (Thermo Fisher Scientific, Waltham, MA). For all imaging analysis, yeast cells were fixed in 4% paraformaldehyde for 5 minutes and then resuspended in 1 × PBS buffer.
QUANTIFICATION AND STATISTICAL ANALYSIS
To determine differences in protein degradation kinetics and the level of protein ubiquitination in different yeast strains, we used ImageJ to acquire the intensity of each protein band from western blotting images. Then the protein levels were normalized by determining the ratio to loading control, Pgk1. Normalized protein levels from three repeats were used to calculate the mean and standard deviation (SD). The Wilcoxon rank sum test was used to determine p values. Statistically significance was inferred when p < 0.05 (*).
To compare cluster formation of Hsp104-GFP (Figure 2C) or Htt103QP-GFP (Figure S2) clusters in WT and mutant cells, more than 100 cells were counted for each sample at a given time point. Cells with or without visible GFP clusters (Figure 2C) and the number of GFP foci (Figure S2) were counted and the experiments were repeated three times. We performed a two-way ANOVA on each data-set, and calculated the statistical significance of the differences between genotypes at each time point. The significance was corrected for multiple comparisons using Tukey’s multiple comparisons test. We considered values significantly different if p < 0.05.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Flag | Sigma-Aldrich | Cat# F3165; RRID:AB_259529 |
| Mouse monoclonal anti-Ubiquitin (P4D1) | Santa Cruz | Cat# Sc-8017; RRID:AB_2762364 |
| Mouse monoclonal anti-Pgk1 | Invitrogen | Cat# 459250; RRID:AB_2532235 |
| Mouse monoclonal anti-HA | Biolegend | Cat# 901515; RRID:AB_2565334 |
| Mouse anti-Flag M2 (agarose beads) affinity gel | Sigma | Cat#A2220; RRID:AB_10063035 |
| Mouse anti-GFP antibody | Santa Cruz | Cat# Sc-9966; RRID:AB_627235 |
| Secondary anti-mouse IgG HRP-linked antibody | Cell signaling | Cat# 7076; RRID:AB_330924 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| MG132 | Abeam | Ab141003 |
| Protease inhibitor cocktail set III | Millipore-Calbiochem | 539136 |
| Deubiquitinase inhibitor N-ethylmaleimide | Sigma | E3876 |
| Cycloheximide | Enzo Life Sciences | ALX380–269 G001 |
| ECL | PerkinElmer | NEL 104001 |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae: strain background W303; see Table S1 | This paper | N/A |
| Oligonucleotides | ||
|
SAN1 deletion forward primer ACTATAGATAGAAATTATTTAGCATTTCAGG ATAGTTCGTCGGATCCCCGGGTTAATTAA |
This paper | N/A |
|
SAN1 deletion reverse primer TGGATGACTGCCAATAGGACATATTTTCA TATTAACATACGAATTCGAGCTCGTTTAAAC |
This paper | N/A |
|
UBR1 deletion forward primer GTCCCTAATCTTTACAGGTCACACAAATT ACATAGAACATCGGATCCCCGGGTTAATTAA |
This paper | N/A |
|
UBR1 deletion reverse primer GTCAATCGACGCGCAAATGTTTAATAATGTAT AAGIIIIIGAATTCGAGCTCGTTTAAAC |
This paper | N/A |
|
HTA1-mApple forward primer AAAGAAGTCTGCCAAGGCTACCAAGGCTTC TCAAGAATTAGCGGCCGCTCTAGAACTAGTGG |
This paper | N/A |
|
HTA1-mApple reverse primer TTTAGTTCCTTCCGCCTTCTTTAAAATACCTGAA CCGATCCCCCCTCGAGGTCGACGGTATCG |
This paper | N/A |
| Software and Algorithms | ||
| ImageJ | National Institutes of Health | https://imagej.nih.gov/ij/ |
Highlights.
Cdc48 segregase is required for the degradation of misfolded proteins in yeast
Cdc48 deficiency leads to a decreased pool of free ubiquitin that compromises the UPS
San1 and Ubr1 ubiquitinate misfolded proteins, reducing the free ubiquitin pool
Restoring free ubiquitin suppresses the toxicity associated with Cdc48 deficiency
ACKNOWLEDGMENTS
We sincerely thank Drs. Timothy Megraw, Yi Zhou, and Hong-Guo Yu and the yeast community at Florida State University (FSU) for comments and suggestions for this project. We thank Drs. Rey-Huei Chen, Mark Hochstrasser, and Jason MacGurn for yeast strains and plasmids. We highly appreciate the careful editing of the manuscript by Dr. Terra Bradley. We thank Dr. Wen Li from Department of Psychology, at FSU, for her advice on statistical analysis. We thank the FSU Biology Core Facility for DNA sequencing. This work was supported by a CRC planning grant from FSU, partly by R01GM121786 from National Institute of Health (USA) to Y.W. and partly by R01GM118600 to R.J.T.
Footnotes
DECLARATION OF INTERESTS
There are no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.107898.
REFERENCES
- Aguado A, Fernández-Higuero JA, Moro F, and Muga A (2015). Chaperone-assisted protein aggregate reactivation: Different solutions for the same problem. Arch. Biochem. Biophys 580, 121–134. [DOI] [PubMed] [Google Scholar]
- Baek GH, Cheng H, Choe V, Bao X, Shao J, Luo S, and Rao H (2013). Cdc48: a swiss army knife of cell biology. J. Amino Acids 2013, 183421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bersuker K, Brandeis M, and Kopito RR (2016). Protein misfolding specifies recruitment to cytoplasmic inclusion bodies. J. Cell Biol 213, 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blythe EE, Olson KC, Chau V, and Deshaies RJ (2017). Ubiquitin- and ATP-dependent unfoldase activity of P97/VCP$NPLOC4$UFD1L is enhanced by a mutation that causes multisystem proteinopathy. Proc. Natl. Acad. Sci. USA 114, E4380–E4388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar N, and Rapoport T (2017a). Toward an understanding of the Cdc48/p97 ATPase. F1000Res 6, 1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar NO, and Rapoport TA (2017b). Molecular Mechanism of Substrate Processing by the Cdc48 ATPase Complex. Cell 169, 722–735.e729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodnar NO, Kim KH, Ji Z, Wales TE, Svetlov V, Nudler E, Engen JR, Walz T, and Rapoport TA (2018). Structure of the Cdc48 ATPase with its ubiquitin-binding cofactor Ufd1-Npl4. Nat. Struct. Mol. Biol 25, 616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman E, Maksim N, de la Cruz F, and La Clair JJ (2015). Inhibitors of the AAA+ chaperone p97. Molecules 20, 3027–3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien CY, and Chen RH (2013). Cdc48 chaperone and adaptor Ubx4 distribute the proteasome in the nucleus for anaphase proteolysis. J. Biol. Chem 288, 37180–37191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang KH, Liang F, Higgins R, and Wang Y (2016). Ubiquilin/Dsk2 promotes inclusion body formation and vacuole (lysosome)-mediated disposal of mutated huntingtin. Mol. Biol. Cell 27, 2025–2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasgupta A, Ramsey KL, Smith JS, and Auble DT (2004). Sir Antagonist 1 (San1) is a ubiquitin ligase. J. Biol. Chem 279, 26830–26838. [DOI] [PubMed] [Google Scholar]
- de Oliveira GA, Rangel LP, Costa DC, and Silva JL (2015). Misfolding, Aggregation, and Disordered Segments in c-Abl and p53 in Human Cancer. Front. Oncol 5, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehay B, and Bertolotti A (2006). Critical role of the proline-rich region in Huntingtin for aggregation and cytotoxicity in yeast. J. Biol. Chem 281, 35608–35615. [DOI] [PubMed] [Google Scholar]
- Eisele F, and Wolf DH (2008). Degradation of misfolded protein in the cytoplasm is mediated by the ubiquitin ligase Ubr1. FEBS Lett 582, 4143–4146. [DOI] [PubMed] [Google Scholar]
- Fang NN, Ng AH, Measday V, and Mayor T (2011). Hul5 HECT ubiquitin ligase plays a major role in the ubiquitylation and turnover of cytosolic misfolded proteins. Nat. Cell Biol 13, 1344–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley D, Ozkaynak E, and Varshavsky A (1987). The yeast polyubiquitin gene is essential for resistance to high temperatures, starvation, and other stresses. Cell 48, 1035–1046. [DOI] [PubMed] [Google Scholar]
- Finley D, Ulrich HD, Sommer T, and Kaiser P (2012). The ubiquitin-proteasome system of Saccharomyces cerevisiae. Genetics 192, 319–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner RG, Nelson ZW, and Gottschling DE (2005). Degradation-mediated protein quality control in the nucleus. Cell 120, 803–815. [DOI] [PubMed] [Google Scholar]
- Ghosh DK, Roy A, and Ranjan A (2018). The ATPase VCP/p97 functions as a disaggregase against toxic Huntingtin-exon1 aggregates. FEBS Lett 592, 2680–2692. [DOI] [PubMed] [Google Scholar]
- Glickman MH, Rubin DM, Coux O, Wefes I, Pfeifer G, Cjeka Z, Baumeister W, Fried VA, and Finley D (1998). A subcomplex of the proteasome regulatory particle required for ubiquitin-conjugate degradation and related to the COP9-signalosome and eIF3. Cell 94, 615–623. [DOI] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, and Hayer-Hartl M (2011). Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. [DOI] [PubMed] [Google Scholar]
- Heck JW, Cheung SK, and Hampton RY (2010). Cytoplasmic protein quality control degradation mediated by parallel actions of the E3 ubiquitin ligases Ubr1 and San1. Proc. Natl. Acad. Sci. USA 107, 1106–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higgins R, Kabbaj MH, Hatcher A, and Wang Y (2018). The absence of specific yeast heat-shock proteins leads to abnormal aggregation and compromised autophagic clearance of mutant Huntingtin proteins. PLoS One 13, e0191490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipp MS, Patel CN, Bersuker K, Riley BE, Kaiser SE, Shaler TA, Brandeis M, and Kopito RR (2012). Indirect inhibition of 26S proteasome activity in a cellular model of Huntington’s disease. J. Cell Biol 196, 573–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura S, Yabu T, and Yamashita M (2012). Protective role of cell division cycle 48 (CDC48) protein against neurodegeneration via ubiquitin-proteasome system dysfunction during zebrafish development. J. Biol. Chem 287, 23047–23056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson T, Navarrete C, Sharma SK, Sideri TC, Ibstedt S, Priya S, Grant CM, Christen P, Goloubinoff P, and Tamás MJ (2012). Arsenite interferes with protein folding and triggers formation of protein aggregates in yeast. J. Cell Sci 125, 5073–5083. [DOI] [PubMed] [Google Scholar]
- Kimura Y, Yashiroda H, Kudo T, Koitabashi S, Murata S, Kakizuka A, and Tanaka K (2009). An inhibitor of a deubiquitinating enzyme regulates ubiquitin homeostasis. Cell 137, 549–559. [DOI] [PubMed] [Google Scholar]
- Knowles TP, Vendruscolo M, and Dobson CM (2014). The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol 15, 384–396. [DOI] [PubMed] [Google Scholar]
- Komander D, Clague MJ, and Urbé S (2009). Breaking the chains: structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol 10, 550–563. [DOI] [PubMed] [Google Scholar]
- Krobitsch S, and Lindquist S (2000). Aggregation of huntingtin in yeast varies with the length of the polyglutamine expansion and the expression of chaperone proteins. Proc. Natl. Acad. Sci. USA 97, 1589–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Tumolo JM, Ehlinger AC, Jernigan KK, Qualls-Histed SJ, Hsu PC, McDonald WH, Chazin WJ, and MacGurn JA (2017). Ubiquitin turnover and endocytic trafficking in yeast are regulated by Ser57 phosphorylation of ubiquitin. eLife 6, e29176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitman J, Ulrich Hartl F, and Lederkremer GZ (2013). Soluble forms of polyQ-expanded huntingtin rather than large aggregates cause endoplasmic reticulum stress. Nat. Commun 4, 2753. [DOI] [PubMed] [Google Scholar]
- Liu C, Liu W, Ye Y, and Li W (2017). Ufd2p synthesizes branched ubiquitin chains to promote the degradation of substrates modified with atypical chains. Nat. Commun 8, 14274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A III, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, and Pringle JR (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Lu K, Psakhye I, and Jentsch S (2014). Autophagic clearance of polyQ proteins mediated by ubiquitin-Atg8 adaptors of the conserved CUET protein family. Cell 158, 549–563. [DOI] [PubMed] [Google Scholar]
- Luo J, Solimini NL, and Elledge SJ (2009). Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 136, 823–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller SB, Mogk A, and Bukau B (2015). Spatially organized aggregation of misfolded proteins as cellular stress defense strategy. J. Mol. Biol 427, 1564–1574. [DOI] [PubMed] [Google Scholar]
- Mogk A, Bukau B, and Kampinga HH (2018). Cellular Handling of Protein Aggregates by Disaggregation Machines. Mol. Cell 69, 214–226. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Morales-Scheihing D, Butler PC, and Soto C (2015). Type 2 diabetes as a protein misfolding disease. Trends Mol. Med 21, 439–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogen-Shtern N, Ben David T, and Lederkremer GZ (2016). Protein aggregation and ER stress. Brain Res 1648, 658–666. [DOI] [PubMed] [Google Scholar]
- Olszewski MM, Williams C, Dong KC, and Martin A (2019). The Cdc48 unfoldase prepares well-folded protein substrates for degradation by the 26S proteasome. Commun. Biol 2, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravid T, and Hochstrasser M (2007). Autoregulation of an E2 enzyme by ubiquitin-chain assembly on its catalytic residue. Nat. Cell Biol 9, 422–427. [DOI] [PubMed] [Google Scholar]
- Reyes-Turcu FE, Ventii KH, and Wilkinson KD (2009). Regulation and cellular roles of ubiquitin-specific deubiquitinating enzymes. Annu. Rev. Biochem 78, 363–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum JC, Fredrickson EK, Oeser ML, Garrett-Engele CM, Locke MN, Richardson LA, Nelson ZW, Hetrick ED, Milac TI, Gottschling DE, and Gardner RG (2011). Disorder targets misorder in nuclear quality control degradation: a disordered ubiquitin ligase directly recognizes its misfolded substrates. Mol. Cell 41, 93–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samant RS, Livingston CM, Sontag EM, and Frydman J (2018). Distinct proteostasis circuits cooperate in nuclear and cytoplasmic protein quality control. Nature 563, 407–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirayama M, Tóth A, Gálová M, and Nasmyth K (1999). APC(Cdc20) promotes exit from mitosis by destroying the anaphase inhibitor Pds1 and cyclin Clb5. Nature 402, 203–207. [DOI] [PubMed] [Google Scholar]
- Skrott Z, Mistrik M, Andersen KK, Friis S, Majera D, Gursky J, Ozdian T, Bartkova J, Turi Z, Moudry P, et al. (2017). Alcohol-abuse drug disulfiram targets cancer via p97 segregase adaptor NPL4. Nature 552, 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto C (2003). Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci 4, 49–60. [DOI] [PubMed] [Google Scholar]
- Swaminathan S, Amerik AY, and Hochstrasser M (1999). The Doa4 deubiquitinating enzyme is required for ubiquitin homeostasis in yeast. Mol. Biol. Cell 10, 2583–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomko RJ Jr., and Hochstrasser M (2011). Incorporation of the Rpn12 subunit couples completion of proteasome regulatory particle lid assembly to lid-base joining. Mol. Cell 44, 907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchiya H, Ohtake F, Arai N, Kaiho A, Yasuda S, Tanaka K, and Saeki Y (2017). In Vivo Ubiquitin Linkage-type Analysis Reveals that the Cdc48-Rad23/Dsk2 Axis Contributes to K48-Linked Chain Specificity of the Proteasome. Mol. Cell 66, 488–502.e487. [DOI] [PubMed] [Google Scholar]
- Twomey EC, Ji Z, Wales TE, Bodnar NO, Ficarro SB, Marto JA, Engen JR, and Rapoport TA (2019). Substrate processing by the Cdc48 ATPase complex is initiated by ubiquitin unfolding. Science 365, eaax1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyedmers J, Mogk A, and Bukau B (2010). Cellular strategies for controlling protein aggregation. Nat. Rev. Mol. Cell Biol 11, 777–788. [DOI] [PubMed] [Google Scholar]
- Verma R, Oania R, Fang R, Smith GT, and Deshaies RJ (2011). Cdc48/p97 mediates UV-dependent turnover of RNA Pol II. Mol. Cell 41, 82–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Liu D, Wang Y, Qin J, and Elledge SJ (2001). Pds1 phosphorylation in response to DNA damage is essential for its DNA damage checkpoint function. Genes Dev 15, 1361–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Mao X, Ju D, and Xie Y (2004). Rpn4 is a physiological substrate of the Ubr2 ubiquitin ligase. J. Biol. Chem 279, 55218–55223. [DOI] [PubMed] [Google Scholar]
- Wang Y, Meriin AB, Zaarur N, Romanova NV, Chernoff YO, Costello CE, and Sherman MY (2009). Abnormal proteins can form aggresome in yeast: aggresome-targeting signals and components of the machinery. FASEB J 23, 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watts GD, Wymer J, Kovach MJ, Mehta SG, Mumm S, Darvish D, Pestronk A, Whyte MP, and Kimonis VE (2004). Inclusion body myopathy associated with Paget disease of bone and frontotemporal dementia is caused by mutant valosin-containing protein. Nat. Genet 36, 377–381. [DOI] [PubMed] [Google Scholar]
- Xie Y, and Varshavsky A (2001). RPN4 is a ligand, substrate, and transcriptional regulator of the 26S proteasome: a negative feedback circuit. Proc. Natl. Acad. Sci. USA 98, 3056–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, and Hu HY (2016). Sequestration of cellular interacting partners by protein aggregates: implication in a loss-of-function pathology. FEBS J 283, 3705–3717. [DOI] [PubMed] [Google Scholar]
- Yang J, Hao X, Cao X, Liu B, and Nyström T (2016). Spatial sequestration and detoxification of Huntingtin by the ribosome quality control complex. eLife 5, e11792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y, Tang WK, Zhang T, and Xia D (2017). A Mighty “Protein Extractor” of the Cell: Structure and Function of the p97/CDC48 ATPase. Front. Mol. Biosci 4, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets or code.