

Acinetobacter baumannii is an ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) opportunistic pathogen, with poorly defined natural habitats/reservoirs outside the clinical setting. A. baumannii arose from the Acinetobacter calcoaceticus-A. baumannii complex as the result of a population bottleneck, followed by a recent population expansion from a few clinically relevant clones endowed with an arsenal of resistance and virulence genes. Still, the identification of virulence traits and the evolutionary paths leading to a pathogenic lifestyle has remained elusive, and thus, the study of nonclinical (“environmental”) A. baumannii isolates is necessary. We conducted here comparative genomic and virulence studies on A. baumannii NCMBI8209 isolated in 1943 from the microbiota responsible for the decomposition of guayule, and therefore well differentiated both temporally and epidemiologically from the multidrug-resistant strains that are predominant nowadays. Our work provides insights on the adaptive strategies used by A. baumannii to escape from host defenses and may help the adoption of measures aimed to limit its further dissemination.
KEYWORDS: environmental Acinetobacter baumannii, preantibiotic-era Acinetobacter baumannii, environmental reservoirs, comparative genomics, insertion sequences, virulence factors
ABSTRACT
Acinetobacter baumannii represents nowadays an important nosocomial pathogen of poorly defined reservoirs outside the clinical setting. Here, we conducted whole-genome sequencing analysis of the Acinetobacter sp. NCIMB8209 collection strain, isolated in 1943 from the aerobic degradation (retting) of desert guayule shrubs. Strain NCIMB8209 contained a 3.75-Mb chromosome and a plasmid of 134 kb. Phylogenetic analysis based on core genes indicated NCIMB8209 affiliation to A. baumannii, a result supported by the identification of a chromosomal blaOXA-51-like gene. Seven genomic islands lacking antimicrobial resistance determinants, 5 regions encompassing phage-related genes, and notably, 93 insertion sequences (IS) were found in this genome. NCIMB8209 harbors most genes linked to persistence and virulence described in contemporary A. baumannii clinical strains, but many of the genes encoding components of surface structures are interrupted by IS. Moreover, defense genetic islands against biological aggressors such as type 6 secretion systems or CRISPR-cas are absent from this genome. These findings correlate with a low capacity of NCIMB8209 to form biofilm and pellicle, low motility on semisolid medium, and low virulence toward Galleria mellonella and Caenorhabditis elegans. Searching for catabolic genes and concomitant metabolic assays revealed the ability of NCIMB8209 to grow on a wide range of substances produced by plants, including aromatic acids and defense compounds against external aggressors. All the above features strongly suggest that NCIMB8209 has evolved specific adaptive features to a particular environmental niche. Moreover, they also revealed that the remarkable genetic plasticity identified in contemporary A. baumannii clinical strains represents an intrinsic characteristic of the species.
IMPORTANCE Acinetobacter baumannii is an ESKAPE (Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, and Enterobacter species) opportunistic pathogen, with poorly defined natural habitats/reservoirs outside the clinical setting. A. baumannii arose from the Acinetobacter calcoaceticus-A. baumannii complex as the result of a population bottleneck, followed by a recent population expansion from a few clinically relevant clones endowed with an arsenal of resistance and virulence genes. Still, the identification of virulence traits and the evolutionary paths leading to a pathogenic lifestyle has remained elusive, and thus, the study of nonclinical (“environmental”) A. baumannii isolates is necessary. We conducted here comparative genomic and virulence studies on A. baumannii NCMBI8209 isolated in 1943 from the microbiota responsible for the decomposition of guayule, and therefore well differentiated both temporally and epidemiologically from the multidrug-resistant strains that are predominant nowadays. Our work provides insights on the adaptive strategies used by A. baumannii to escape from host defenses and may help the adoption of measures aimed to limit its further dissemination.
INTRODUCTION
The genus Acinetobacter, family Moraxellaceae, class Gammaproteobacteria, is characterized by Gram-negative aerobic coccobacilli of ubiquitous environmental distribution and large metabolic capabilities (1). In the Acinetobacter genus, the phylogenetically closely related species composing the A. calcoaceticus-A. baumannii complex represent opportunistic pathogens that are important nowadays (2). Infections due to A. baumannii in particular, rarely reported in health care settings before the 1970s, rapidly increased in importance with the global spread of a limited number of epidemic clonal complexes (CCs) possessing multidrug resistance (MDR) phenotypes (2, 3). Strains composing the CCs generally contain a variety of genetic material obtained by horizontal gene transfer such as plasmids and chromosomally located genomic islands (GIs), including resistance islands (RIs) encompassing different transposons and integrons which play pivotal roles in both antimicrobial and heavy metal resistances (2, 4–6). CC strains also carry a large repertoire of insertion sequences (ISs) capable of mediating gene inactivation as well as genome rearrangements, including deletions and inversions with strong adaptive significances (2, 6–8). The combination of the above factors, added to the intrinsic resistance of A. baumannii to desiccation and fever-associated temperatures (9), are considered main factors of persistence of this pathogen in the nosocomial environment (2, 7).
Whole-genome sequence (WGS) comparisons have become commonplace in examining strain-to-strain variability and in comparing pathogenic strains with environmental relatives of a given species, in efforts to identify underlying genetic determinants and mechanisms responsible for phenotypic dissimilarities. When applied to A. baumannii, these approaches provided valuable information on the clinical population structure of this species, its potential virulence factors, and the origins and acquisition of antimicrobial resistance determinants (2, 4, 6, 7, 10, 11). However, the identification of virulence traits both within and between CC members has remained elusive, suggesting a complex and even multifactorial nature of mechanisms involved (2, 3, 7). The importance of a deeper genomic study of nonclinical (“environmental”) isolates has therefore recently been emphasized to clarify both the virulence potential of the aboriginal A. baumannii population and the evolutionary paths that led toward a pathogenic lifestyle (2). Nevertheless, the natural habitats and potential reservoirs of A. baumannii outside the clinical setting have remained poorly defined, and they are only beginning to be elucidated (2, 11–13).
We have recently traced the origins of the collection strain A. baumannii DSM30011 (11) to an isolate originally classified as Achromobacter lacticum, which was obtained prior to 1944 from the enriched microbiota responsible for the aerobic decomposition of the resinous desert shrub guayule (14, 15). WGS of this strain and subsequent comparative analysis with 32 other complete clinical A. baumannii genomes revealed the presence of 12 unique accessory chromosomal regions in strain DSM30011, including five regions containing phage-related genes, five regions encoding toxins related to the type 6 secretion system, and one region encompassing a novel CRISPR-cas cluster (11). As expected from an environmental isolate obtained before the massive introduction of antimicrobial therapy in the 1950s (2), DSM30011 showed a general antimicrobial susceptibility phenotype and no RI in its genome (11). Notably, the genome of DSM30011 lacked complete IS elements, although some remnants could be predicted on it. Remarkably still, most genes and regulatory mechanisms linked to persistence and virulence in pathogenic Acinetobacter species were identified in the DSM30011 genome. Moreover, several gene clusters encoding components of catabolic pathways related to the degradation of plant defenses were found, thus suggesting that plants may provide an effective niche for A. baumannii (11), as reported for other species of the genus (11). In turn, it also opens the possibility that phytophagous insects feeding on these plants (16–22) may represent both reservoirs and vectors for the dissemination of A. baumannii in the environment.
During the original isolation of DSM30011, two companion isolates also classified as A. lacticum displaying similar phenotypic characteristics were also described (14). Only one of the isolates was saved in collections, and we obtained a replica of this isolate (see Table S1; all supplemental files for this paper can be found at https://www.researchgate.net/publication/342762639_Acinetobacter_baumannii_NCIMB8209_a_rare_environmental_strain_displaying_extensive_insertion_sequence-mediated_genome_remodeling_resulting_in_the_loss_of_exposed_cell_structures_and_defensive_mechanis) which had been deposited on the National Collection of Industrial Food and Marine Bacteria, Aberdeen, Scotland under the strain designation NCIMB8209, aiming to perform WGS comparative studies with other completely sequenced Acinetobacter genomes. We report here the results of these analyses as well as some phenotypic characteristics of this strain, which may provide clues into the environmental reservoirs, genomic diversity, and virulence potential of the preantibiotic era A. baumannii population.
RESULTS AND DISCUSSION
Phylogenetic analysis assigned strain NCIMB8209 to A. baumannii, albeit to a separate clonal lineage compared to its companion strain DSM30011. (i) NCIMB8209 origins.
As described in our previous work (11), strain NCIMB8209 and its companion strain DSM30011 were isolated prior to 1944 from the natural microbiota enriched during the aerobic decomposition of guayule, an industrial procedure (retting) designed to reduce the resinous content of the shrub processed material for the subsequent production of natural latex (14, 15). Our maximum likelihood (ML) phylogenetic analyses based on core gene sequence comparisons derived from the WGS data (see below) allowed us to confidently assign this strain to A. baumannii as a species. In concordance with this assignment, this strain was capable of growing at 44°C (11) in what represents a typical phenotype associated with A. baumannii (1, 23). Still, random amplification PCR (11), phylogenetic, comparative genome analysis, and metabolic studies (see below) indicated significant differences between this strain and its companion A. baumannii strain DSM30011. This evidence suggests that, even though these two A. baumannii strains might have similar environmental niches, they still belong to separate clonal lineages. NCIMB8209 thus provided us with a different A. baumannii strain isolated from a nonclinical source (14) before the massive introduction of antibiotics to treat infections (2).
(ii) NCIMB8209 genomic features.
Genome sequencing indicated that the NCIMB8209 chromosome consisted of 3,751,581 bp in length with a G+C content of 39.1% (Table 1). These values match the average values reported for the genomes of the species composing the Acinetobacter genus (3,870 kbp and 39.6%, respectively) (6). It is noteworthy that 223 of the coding DNA sequences (CDSs) (i.e., around 6% of the total) predicted in the NCIMB8209 genome were pseudogenes. It is worth noting that a similarly high number of nonfunctional genes (272; 9% of the total genes) has been reported for the A. baumannii strain SDF, which was isolated from a human body louse and whose genome is riddled with numerous prophages and ISs (24).
TABLE 1.
General features obtained from NCIMB8209 genomic sequencing
| Genomic parametera | Value for parameter |
|---|---|
| Chromosome | |
| Accession no. | CP028138 |
| Estimated size (bp) | 3,751,581 |
| Average GC content (%) | 39.1 |
| No. of CDSs | 3,596 |
| No. of tRNA genes | 73 |
| No. of rRNA genes | 18 |
| No. of ncRNA genes | 4 |
| No. of phage regions | 5 |
| No. of IS copies | 79 |
| Plasmid | |
| Accession no. | CP028139 |
| Estimated size (bp) | 133,709 |
| Average GC content (%) | 40.1 |
| No. of CDSs | 156 |
| No. of tRNA genes | 1 |
| No. of rRNA genes | |
| No. of ncRNA genes | |
| No. of phage regions | 2 |
| No. of IS copies | 14 |
Based on NCBI Prokaryotic genomic annotation pipeline. ncRNA, noncoding RNA.
Our analysis also showed the presence of a large plasmid of 133,709 bp with a G+C content of 40.1%, hereafter designated pAbNCIMB8209_134 (Table 1). Comparison of pAbNCIMB8209_134 with other plasmids deposited in databases indicated extensive sequence identity with a group of A. baumannii plasmids with a length of more than 100 kb, including pABTJ2 (25). All of these plasmids share a previously undescribed Rep-3 superfamily (pfam0151) replication initiation protein gene (C4X49_18465). The presence of both partition (C4X49_18550) and toxin/antitoxin genes (C4X49_18715 to C4X49_18720) related to plasmid stability were detected in pAbNCIMB8209_134. In contrast, genes involved in mobilization, conjugation, antimicrobial resistance or virulence functions could not be identified in pAbNCIMB8209_134. Still, this plasmid encodes functions which may provide some adaptive advantages to their Acinetobacter hosts, such as a putative glutathione-dependent pathway of formaldehyde detoxification (C4X49_18640 to -18650).
(iii) Comparisons of the chromosomal architectures of A. baumannii environmental strains NCIMB8209 and DSM30011.
Comparison of the overall chromosome structures of the A. baumannii NCIMB8209 and DSM30011 strains showed that the former is 198 kbp smaller than that of DSM30011 (Fig. 1). Furthermore, a number of GIs and prophages distinguished these two chromosomes as will be described in greater detail below. Despite these differences, a general shared synteny was observed between the two chromosomes, with the notable exception of an inversion of a 53.1-kb region located between the first and sixth rRNA operons (Fig. 1). A similar situation was found in the community-acquired strain A. baumannii D1279779 (26), in which the same region was inverted compared to DSM30011 and to most other clinical strains, including the type strain ATCC 17978 (see Fig. S1 at the URL given above). This rearrangement was probably mediated by homologous recombination between two oppositely oriented rRNA operons bordering this region (26). Still, and although this inversion reverses the orientation of a number of critical housekeeping genes as well as the origin of chromosomal replication (oriC), it has no substantial effects on the growth rate of NCIMB8209 compared to DSM30011 in either rich or minimal medium (data not shown).
FIG 1.

Linear comparison of the genomes of A. baumannii strains NCIMB8209 and DSM30011 inferred using Mauve. Each block corresponds to a DNA fragment of the chromosome distinctively colored for clarity. The degree of conservation is indicated by the vertical bars inside the blocks. Their position relative to the genome line denotes colinear and inverted regions. For a better appreciation of genome synteny, DSM30011 chromosomal DNA shown corresponds to the reverse complementary strand. Putative prophage (PhX-N for NCIMB8209 and PhX-D for DSM30011) and genomic island (GI) insertion sites are indicated (see Tables 2 and 3 for details). Two out of the six rRNA-encoding operons in strain NCIMB8209 are also represented (rRNA1 and rRNA6). Regions encoding interbacterial competition islands (T6-1 to T6-5 and Tps), heavy metals (dusA-GI and Tn6018-like) and antimicrobial (ade-GI) resistance islands and a CRISPR-cas cluster are indicated for DSM30011 genome.
(iv) Phylogenomic and MLST analyses.
A phylogenetic study based on the comparisons of the concatenated sequences of 383 core genes of the environmental strains NCIMB8209 and DSM30011 and a number of Acinetobacter genomes that encompassed 99 other A. baumannii strains as well as 26 non-A. baumannii representatives (26 strains; Table S1) reinforced the affiliation of NCIMB8209 to A. baumannii as a species (Fig. 2 and Fig. S2). Different authors have noted the lack of a defined phylogenetic structure for the general A. baumannii population on phylogenetic trees based on core gene comparisons, with the exception of different terminal clusters each corresponding to an epidemic CC (2, 3, 11, 27). The incorporation of NCIMB8209 core genome sequences to this phylogenetic study did not change this general picture (Fig. 2), but some observations derived from this analysis are worth noting. First, strains NCIMB8209 and DSM30011, which were isolated more than 70 years ago, neither emerged close to the root nor formed a separate “environmental” cluster in the A. baumannii subtree. In contrast, they appeared intermixed between more contemporary clinical strains (Fig. 2). Second, NCIMB8209 emerged in a compact cluster (100% bootstrap support) with a number of recently isolated A. baumannii strains that included PR07 (isolated in 2012 in Malaysia from the blood of a hospitalized patient; NCBI BioProject accession no. PRJNA185400, direct submission) and ABNIH28, a MDR strain isolated in 2016 in the United States from a hospital closet drain (28). Further analyses aiming to compare strain NCIMB8209 with the phylogenetically closest strains ABNIH28 and PR07 (Table S1, ABNIH28-PR07) were performed. We additionally included representative strains of the CCI (ACICU), CCII (AYE), and the NCIMB8209 environmental companion strain DSM30011. Different calculations reflecting evolutionary distance, such as average nucleotide identity (ANI) values, genomic distance, and core-genomic distance shown in this table, reinforced the inferred phylogenetic proximity among the strains included in the subclade under study (Fig. 2). In silico DNA-DNA hybridization values were also in line with the calculated values obtained for evolutionary distances. Regarding common gene families, the observed percentages ranged from 76 to 84% for all strains included in the analysis, whereas for additional and absent gene families, these values ranged between 16 and 24% and 18 and 21%, respectively. These nonshared genes, which are part of the accessory genome in each strain, may contribute to the adaption of each particular strain to a particular niche.
FIG 2.

Maximum likelihood phylogenetic analysis of A. baumannii strains. (A) The ML phylogeny was computed based on 383 concatenated core gene sequences (the full tree is shown in Fig. S2). Numbers at nodes correspond to bootstrap values (BV) (100 replicates of the original data set). For simplicity, subnodes for which internal branches showed BV = 100 were collapsed. The scale bar below corresponds to evolutionary distance (average number of the substitutions per site). CC1 and CC2 correspond to subclusters formed by A. baumannii species assigned to epidemic clonal complexes CC1 and CC2, respectively.
Strain NCIMB8209 was assigned to a novel sequence type (ST) (1197 [Table S1]) in the Pasteur scheme of A. baumannii multilocus sequence type (MLST) classification (3). Notably, and in sharp contrast to strain DSM30011, which shared only four of the seven alleles with its closest matching isolate (11), NCIMB8209 shared six of the seven alleles with its closest matching isolates with all differences being restricted to the rplB allele (Table S1). Of note, the gdhB gene used in the A. baumannii MLST Oxford classification scheme (29) is missing from the NCIMB8209 genome (Table S1), an observation experimentally corroborated by a specific PCR assay (see Materials and Methods for details). A deeper comparative analysis with other A. baumannii genomes indicated that NCIMB8209 lacks a 4.8-kbp region between genes C4X49_10220 and C4X49_10225 which, in other genomes (DSM30011 included), harbors the gdhB gene. In the DSM30011 genome, the gdhB gene is flanked by a hypothetical gene (DSM30011_07590) and a gene encoding a universal stress protein (DSM30011_07600), and a similar situation occurs in other A. baumannii genomes. This observation indicated that gdhB is nonessential for A. baumannii, and in this context, its interruption by IS elements has been previously noted in a number of A. baumannii isolates of clinical origin (30). Moreover, a recent analysis performed with 730 Acinetobacter genomes revealed in 76% of them a paralogous gdhB gene (gdhB2) in a different locus, the overall observations indicating that the Pasteur MLST scheme presents some advantages for the characterization of epidemiologically related A. baumannii isolates (31).
(v) NCIM8209 comparative genomics.
A comparative genomic analysis that included the previously mentioned 127 different Acinetobacter sp. strains (Table S1) indicated that 510 representatives of the protein families detected in the NCIMB8209 genome (see Materials and Methods) are present in all of these genomes (Acinetobacter core genome), and 1,149 of them are present in all 101 A. baumannii genomes analyzed (A. baumannii core genome). These numbers differ from estimations obtained by other authors (6) of the core genome content of the Acinetobacter genus (950 genes; 133 genomes analyzed) and A. baumannii in particular (1,590 genes; 34 genomes analyzed). It is worth remarking that even when similar numbers of total genomes were used in both studies, Touchon and collaborators (6) used mostly non-baumannii strains, whereas in our study most of the strains corresponded to A. baumannii. This difference and the inclusion of two bona fide A. baumannii environmental strains in our calculations might explain why the A. baumannii core genome estimation was reduced by 27.7% in our study. In this context, recent estimations conducted by other authors (32) with 78 A. baumannii genomes, which included the environmental strain DS002, inferred a core genome of 1,344 genes for this species. The inclusion of more environmental strains in these calculations will certainly contribute to obtain a more accurate value for the core genes repertoire of the species.
NCIMB8209 antimicrobial resistance.
Conventional antimicrobial susceptibility assays indicated that strain NCIMB8209 showed susceptibility to most clinically employed antimicrobials tested except nitrofurantoin and, among β-lactams, to ampicillin at MIC values just above the CLSI recommended breakpoints (Table S2). In full concordance (Table 2 and Table S2), this strain lacks AbaR resistance islands. The marginal ampicillin resistance of this strain (see above) most likely reflects the presence of a number of β-lactamase genes (Table S2). Of these genes, it is worth mentioning C4X49_08235 (blaOXA-78) encoding an OXA-51-type carbapenemase with 100% identity to the class D β-lactamase OXA-78 (NCBI WP_005139262.1), and C4X49_12575 (blaADC-154) encoding an enzyme identical to the class C ADC-154 β-lactamase (WP_005138362). The presence of these genes in the chromosome of NCIMB8209 reinforces proposals that they provided for the intrinsic β-lactamase gene repertoire of A. baumannii (7, 33–35). NCIMB8209 also contains a carO gene (C4X49_13345) encoding a carO variant II allele described so far only among the A. baumannii clinical population (36), and also an oprD/occAB1 homolog (C4X49_01130). These genes encode different outer membrane proteins proposed to participate in the permeation of carbapenems into the periplasm (37). NCIMB8209 also carries genes encoding enzymes providing resistance to aminoglycosides and chloramphenicol including ant(3”)-II and catB, respectively (Table S2). This indicates both the potentiality to evolve such resistances under selective pressure and an environmental reservoir of these resistance genes.
TABLE 2.
Genomic islands identified in the chromosome of strain NCIMB8209a
| Region | Length (kbp) | Location | Integrase gene | Flanking genes (accession no.) | Description |
|---|---|---|---|---|---|
| GI1 | 39.7 | 116596–156278 | C4X49_00725 | LysR-family transcriptional regulator (C4X49_00535)/tRNA-Gly-GCC (C4X49_00740) |
Type III restriction modification system. TA systems (HypBA, YefM-YoeB). ISs |
| GI2 | 30.5 | 569359–599853 | C4X49_02720 | Hypothetical protein (C4X49_02715)/ peptide chain release factor 3 (C4X49_02900) |
Phage proteins. Type I restriction modification system |
| GI3 | 19.1 | 613054–632163 | C4X49_02950 |
dusA (C4X49_02945)/MFS transporter (C4X49_03050) |
Arsenic and heavy metal resistance island |
| GI4 | 12.6 | 1165713–1178263 | C4X49_05820 | tRNA-Ser-TGA (C4X49_05765)/ hypothetical protein (C4X49_05860) |
Hypothetical proteins of unknown function |
| GI5 | 19.1 | 1927268–1946350 | C4X49_09525 | Hypothetical protein (C4X49_09520)/ TetR-AcrR family transcriptional regulator (C4X49_09610) |
Fatty acid catabolic pathway |
| GI6 | 29.3 | 2818151–2847423 | C4X49_13865 | Peptide synthetase (C4X49_13745)/ tRNA-Ser-TGA (C4X49_13870) |
Heavy metal resistance island |
| GI7 | 13.3 | 3133811–3147160 | C4X49_15320 | Sensor histidine kinase (C4X49_15265)/ tRNA-Leu-CAA (C4X49_15325) |
Type I restriction modification system |
According to NCBI and RAST annotations.
Strain NCIMB8209 shares with DSM30011 (11) susceptibility to folate pathway inhibitors such as sulfamethoxazole-trimethoprim (Table S2). These susceptibilities correlate with the absence of sul or dfrA resistance genes in the genome (Table S2) and represent a notable exception compared to most clinical strains of A. baumannii, including strains more contemporary to NCIMB8209 such as ATCC 19606 and ATCC 17978 (4, 7, 38–41). This characteristic is compatible with the original isolation of these two strains in an environment still free of the selection pressure derived from the use of sulfonamides common to the clinical setting at that time (11).
Genes encoding components of the RND (resistance-nodulation-cell division), DMT (drug/metabolite transporter), MATE (multidrug and toxic efflux), MFS (major facilitator superfamily), and SMR (small multidrug resistance) efflux systems involved in clinical Acinetobacter strains in the extrusion of toxic compounds, including some antimicrobials (7) were also found in the NCIMB8209 genome (Table S2). Remarkably, a region present in most A. baumannii genomes encoding phage-related proteins and the AdeRS two-component regulatory system and associated AdeABC efflux components (Fig. 2) was missing in the NCIMB8209 genome. A genomic island (GI5) was instead found in an equivalent genomic location (Table 2). The loss of this gene cluster has also been documented in a number of other A. baumannii strains, irrespective of their clinical or environmental origins (10, 31).
A. baumannii NCIMB8209 shows reduced virulence toward Galleria mellonella and Caenorhabditis elegans.
G. mellonella moth larvae and the nematode C. elegans provide reliable models to study the virulence of numerous human pathogens, among them Acinetobacter genus species (42, 43). We thus decided to use these two models to evaluate the virulence of strain NCIMB8209 compared to those of strain DSM30011 and the soil organism Acinetobacter baylyi ADP1 (44). In the G. mellonella model, DSM30011 showed high virulence (45, 46), whereas a low-virulence capacity has been demonstrated for strain ADP1 (47). In contrast, virulence in the C. elegans model has not been assessed before for this group of strains. As observed in Fig. 3, NCIMB8209 was less virulent than DSM30011 in either of these models, with the latter environmental strain showing in particular much higher capacity to kill C. elegans. In contrast, NCIMB8209 killing capacity was close to that observed for A. baylyi ADP1 in either assay (Fig. 3). The observed differences in virulence between NCIMB8209 and DSM30011 indicate relevant phenotypic differences between them (see also following sections), regardless of their isolation as companion strains from a similar environmental origin following similar enrichment and culture protocols (14).
FIG 3.
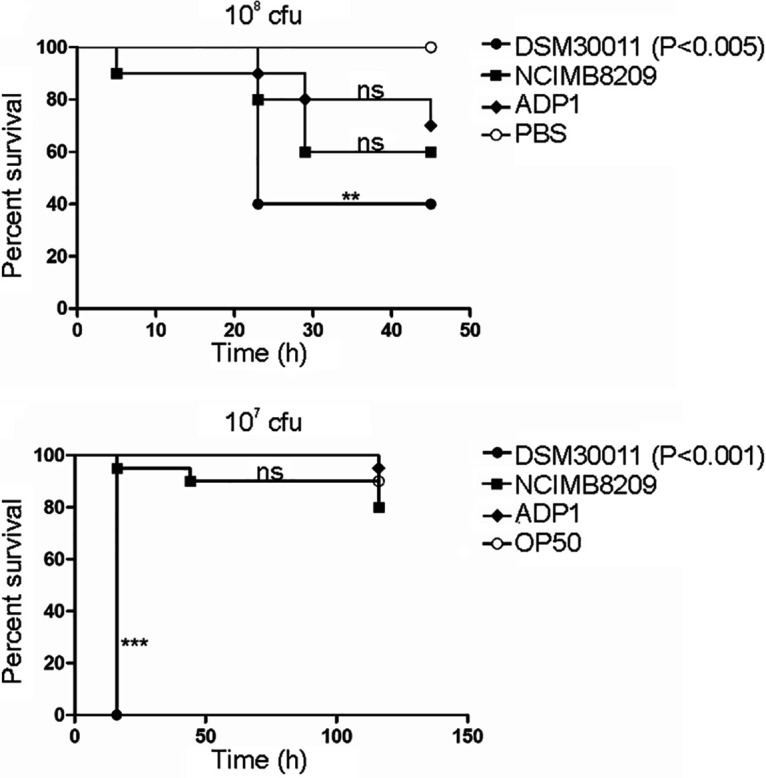
G. mellonella (top) and C. elegans (bottom) lethality curves. Comparative survival analysis between A. baumannii DSM30011 and NCIMB8209 and A. baylyi ADP1 strains. PBS was used as control. (Top) 108 CFU were used. (Bottom) 107 CFU were used. Data are representative of three separate survival experiments, each performed with 20 larvae/worms. Survival curves were constructed by the Kaplan-Meier method and compared by log rank analysis. ns, not significant. **, P < 0.01; ***, P < 0.005.
ISs and prophages have extensively modified the NCIMB8209 genome.
The lower virulence displayed by strain NCIMB8209 compared to that of DSM30011 (see above) led us to analyze in more detail their genomes, aiming to find genetic differences that might explain these distinct phenotypes. When comparing the accessory genomes of DSM30011 and NCIMB8209, some differences worth noting were observed in the latter (Fig. 1) such as: (i) the presence of seven GIs (Table 2), which will be described throughout the text; (ii) the presence of five regions harboring putative prophages (Table 1 and Table S3), which will also be described in detail below; (iii) the notorious absence of a CRISPR-cas gene cluster common to DSM30011 and other A. baumannii strains (11, 48); (iv) the lack of interbacterial competition islands (ICI), such as those encoding the type 6 secretion system components and/or its associated toxins (45); (v) significant differences regarding the number of ISs and composition between these two strains (see below).
(i) Prophage content.
Recent bioinformatic analyses uncovered a wide distribution and diversity of prophage contents in A. baumannii (49), with some of the prophages contributing to horizontal gene transfer between strains by generalized transduction (50). Our bioinformatic analysis of phage sequences in the NCIMB8209 genome indicated five regions (Ph1-N to Ph5-N) encompassing phage-related genes (Table 3). According to PHASTER integrity predictions, two “questionable” prophages (Ph1-N and Ph2-N) and three “incomplete” prophages (Ph3-5-N; Table S3) were identified among them. The integration sites for Ph4-N and Ph5-N in the NCIMB8209 genome were the same hot spots found in other A. baumannii genomes (10), whereas those of Ph1-N to Ph3-N represent novel integration sites (Table 3). Of note, both Ph1-N and Ph5-N sequences share significant identity with the podoviral lytic bacteriophage YMC/09/02/B1251 ABA BP (GenBank NC_019541.1) (51), while Ph2-N, Ph3-N, and Ph4-N showed homology with the Acinetobacter phage vB_AbasS_TRS1 (NC_031098) (52). It is also worth noting that the prophages found in NCBIM8209 shared only limited homology with those predicted for DSM30011 (11).
TABLE 3.
Prophages inserted into the chromosome of strain NCIMB8209a
| Region | Length (kbp) |
No. of predicted proteins |
Genome position | GC (%) | Flanking genes (accession no.) |
|---|---|---|---|---|---|
| Ph1-N | 25.8 | 35 | 860213–886091 | 40.3 |
smpB (C4X49_04195)/phosphopantetheine adenylyltransferase (C4X49_04375) |
| Ph2-N | 28.4 | 41 | 955316– 983747 | 37.7 |
ssrA (tmRNA; C4X49_04705)/DUF927 domain- containing protein (C4X49_04925) |
| Ph3-N | 25.2 | 29 | 1240267–1265538 | 41.5 | Aspartate kinase (C4X49_06155)/alanine-tRNA ligase (C4X49_06310) |
| Ph4-N | 39.1 | 32 | 2140199–2179329 | 40.5 | MFS transporter (C4X49_10600)/hypothetical protein (C4X49_10635) |
| Ph5-N | 13.9 | 11 | 2414964–2428883 | 37.7 |
gspL (C4X49_11920)/phosphohistidine phosphatase SixA (C4X49_12010) |
According to PHASTER predictions. tmRNA, transfer-messenger RNA.
(ii) IS content.
IS elements are important drivers of genome evolution in A. baumannii and other pathogens, modulating antibiotic resistance gene expression, mediating genome rearrangements, including deletions of substantial chromosomal regions, promoting insertional gene inactivation, etc. (53). Notably, and in sharp contrast to A. baumannii DSM30011 (11), a high representation of transposable genetic elements such as ISs or transposons was found in the genome of strain NCIMB8209. ISSaga predictions (54) (Table S3) followed by manual examination corroborated the presence of 12 different ISs (a total of 79 IS copies) in the NCIMB8209 chromosome and 9 different ISs (a total of 14 IS copies) in the plasmid that this strain harbors (Table 4). These values are significantly higher than the estimated average of 33 IS copies per A. baumannii genome determined by Adams and collaborators (8). Moreover, the diversity of IS families (16 total) found in the NCIMB8209 genome is also remarkable when considering that only 7 out of 976 A. baumannii genomes were found to carry 10 or more different IS elements (8). Although most of the IS elements identified in NCIMB8209 were previously reported in different species of the Acinetobacter genus, this strain contains a very particular IS profile for an A. baumannii strain, with two of the most frequent IS elements (ISAha2 and ISAha3) originally detected in A. haemolyticus (Table 4). Furthermore, as also seen in Table 4, NCIMB8209 harbors two of the five most represented ISs in A. baumannii (8), namely, ISAba26 (15 copies) and ISAba13 (14 copies). The absence of ISAba1 copies in NCIMB8209 is also remarkable, considering the large impact of this IS among clinical A. baumannii genomes (8). Of note, the three most represented chromosomal ISs (ISAha3, ISAba26, and ISAba13) are also carried by plasmid pAbNCIMB8209, suggesting that this plasmid was the vehicle enabling their acquisition.
TABLE 4.
IS contenta
| IS name | Origin | No. of DNA molecules |
|
|---|---|---|---|
| Chromosomeb | pAbNCIMB8209 | ||
| ISAha3 | A. haemolyticus | 18 (2) | 1 |
| ISAba26 | A. baumannii | 15 (4) | 2 |
| ISAba13 | A. baumannii | 14 (7) | 3 |
| ISAba44 | A. baumannii | 11 (2) | |
| ISAha2 | A. haemolyticus | 7 (2) | |
| IS1006 | A. junnii | 4 | |
| ISAba45 | A. baumannii | 3 | |
| IS17 | A. haemolyticus | 2 (1) | |
| IS1008 | A. calcoaceticus | 2 (1) | |
| ISAba22 | A. baumannii | 1 (1) | 2 |
| ISAba4 | A. baumannii | 1 | |
| ISAba31 | A. baumannii | 1 | 2 |
| ISAba2 | A. baumannii | 1 | |
| ISAba46 | A. baumannii | 1 | |
| IS1007 | Acinetobacter sp. | 1 | |
| ISAcsp3 | Acinetobacter sp. | 1 | |
According to ISSaga and ISFinder predictions.
The numbers of gene-interrupting IS copies are indicated in parentheses.
From the detected ISs in the NCIMB8209 genome, three represented novel elements that were deposited in the ISSaga database (54) under the names ISAba44 (IS630 family), ISAba45 (IS3 family), and ISAba46 (IS66 family). From these elements, ISAba44 is present in 11 copies in the NCIMB8209 chromosome (Table 4). This particular mobile element was also found in the chromosomes of three other A. baumannii strains (ABNIH28, B8300, and B8342) and in A. johnsonnii XBB1 (75% identity, query coverage of 100%). No homologs to this IS were found in other organisms outside the Acinetobacter genus. ISAba45 (three copies) was also detected in four other A. baumannii strains (PR07, ABNIH28, A1296, and IOMTU433, the former two strains closely linked to NCIMB8209, Fig. 2) and in A. soli GFJ2 (87% identity, query coverage of 99%). Moreover, ISAba45 displays significant nucleotide identity with similar ISs present in other Acinetobacter strains (76%) and in Moraxella osloensis plasmids (70%). The ISAba46-only copy is carried by plasmid pAbNCIMB8209, and it was also found in the chromosomes and plasmids of numerous strains of A. pittii, A. baumannii, A. lwoffii, A. junii, A. johnsonnii, and A. haemolyticus (>86% identity, query coverage of 100%). Concerning the locations of the above IS elements, we found that 17 of them are interrupting an equivalent number of CDSs, very likely precluding their expression (Table S3). Seven of these 17 CDSs are interrupted by ISAba13 copies (Table 4). Remarkably, a similar situation was reported previously for strain D1279779, which harbors 18 ISAba13 copies (26). We also detected that some IS elements were positioned on one of the borders of several genomic islands (GIs), including GI1, GI2, GI4, and GI6 (Table 2). It is tempting to speculate that these insertions probably interfere with the excision mechanism of the corresponding GIs and that they were thus selected due to the subsequent retention of these GIs in the chromosome (55).
In summary, the plethora of IS elements found in strain NCIMB8209 have significantly helped remodel the genome of this strain.
A genome devoid of interbacterial competition islands.
As mentioned above, the genome of NCIMB8209 carries neither a type 6 secretion system (T6SS) main gene cluster nor any T6SS-associated locus encoding VgrG-like proteins or their cognate toxins (45, 56). Searching for other competition mechanisms such as the two-partner systems (Tps) already identified in strain DSM30011 (Fig. 2) related to contact-dependent inhibition (CDI) (57) also resulted in negative results. These observations led us to hypothesize that the ability of strain NCIMB8209 to outcompete other bacteria was severely compromised. To test this prediction, bacterial competition assays were performed essentially following previously described procedures (45). Results shown in Fig. S3 demonstrate that strain NCIMB8209 was not capable of outcompeting Escherichia coli when the former strain was used as the attacker and E. coli DH5α was the prey. In contrast, the DSM30011 strain was clearly capable of outcompeting E. coli DH5α in a similar assay, an effect that specifically depended on the T6SS system as shown by the lack of effect observed in a DSM30011 ΔtssM mutant (Fig. S3A). In correlation with these results, NCIMB8209, similarly to DSM30011 ΔtssM, did not secrete detectable amounts of Hcp (a marker of a functional T6SS) into the growth medium (Fig. S3B). We additionally tested the capacity of NCIMB8209 and DSM30011 to outcompete each other. While DSM30011 completely eliminated NCIMB8209 (Fig. S3C) when coincubated in a 10:1 attacker/prey ratio, the latter strain was not capable of outcompeting the former under similar experimental conditions (Fig. S3D).
Persistence and virulence.
To analyze the presence of genes potentially involved in persistence and virulence in strain NCIMB8209, we performed a BLASTN homology search using as the query a list of potential candidates described in Acinetobacter strains (11). This list included genes coding for the synthesis of the capsule and other exopolysaccharides, appendages, outer membrane (OM) proteins, and the T2SS, genes encoding phospholipases and proteases, and genes involved in traits such as motility and iron scavenging. Our searching indicated that 131 out of the 146 genes (sequence identity of ≥76%) encoding potential virulence factors analyzed were present in NCIMB8209 (Table S2). Among the 15 putative virulence factors included in the search and not detected in the NCIMB8209 genome, most of them encode or are involved in the synthesis of surface-exposed molecules (Table S2). These included the Prp pilus (58), the MFS transporter Pmt (probably involved in DNA transport necessary for biofilm formation [59]), and a surface motility-associated molecule (60). Moreover, the gene encoding the pilus 3 fimbrial adhesion precursor (C4X49_08175), and the pilus 3 fimbria-anchoring protein (C4X49_08180) are both incomplete, and a gene coding for a polymorphic toxin of the RTX family (61) was interrupted by an ISAba44 copy (C4X49_17180 to C4X49_17185). We also noticed that several genes whose products are directly or indirectly involved in functions related to adhesion or biofilm formation in clinical strains were absent or interrupted by ISs. For instance, the gene encoding the Bap protein (62) could not be detected (Table S2), and instead an ISAba13 copy was identified in the locus encompassing the bap gene in other A. baumannii strains. Other examples are the gene coding for the Ata adhesin (63) which was interrupted by an ISAba26 copy, and a gene encoding the O-antigen ligase TfpO normally involved in glycan decoration of capsule and proteins, which was interrupted by an ISAba13 copy in NCIMB8209 (64, 65). We then hypothesized that the lack of this group of genes could have impacted traits such as biofilm and pellicle formation as well as motility. To evaluate this hypothesis, the capacity of NCIMB8209 to form pellicle and biofilm when grown overnight in rich medium was compared to that of DSM30011, which was found to represent a high-biofilm/biopellicle producer (45). This assay indicated that NCIMB8209 was neither capable of forming pellicle (Fig. S4A) nor attaching to glass surfaces (Fig. S4B), supporting the above prediction that the ability to form biofilm/biopellicles is severely compromised in this strain. Furthermore, motility assays on semisolid medium showed that the ability of NCIMB8209 to perform surface-associated motility was highly reduced (Fig. S4B), again in sharp contrast to DSM30011 (46).
Another gene absent in strain NCIMB8209 is cpaA (Table S2), which encodes the surface-exposed metallopeptidase Cpa endowed with the ability to cleave fibrinogen and coagulation factor XII, and thus proposed to deregulate blood coagulation (66, 67). CpaA represents a substrate of the type II secretion system (68), and it has been proposed as a bona fide A. baumannii virulence factor (69). Therefore, its conspicuous absence in NCIMB8209 (Table S2) and also in its companion DSM30011, as well as in the clinical strains ATCC 17978 and ATCC 19606 isolated by the middle of last century (11) supports previous proposals that this gene was recently acquired by A. baumannii by horizontal gene transfer (68).
Genetic features contributing to NCIMB8209 environmental adaptation. (i) Gene acquisition by lateral gene transfer.
Our genomic comparative analysis and subsequent BLASTN-based homology search (see Materials and Methods) indicated that 77 CDSs (59 CDSs located in the chromosome and the extra 18 in the plasmid) are unique to strain NCIMB8209 (Table S4). From the 59 unique chromosomal genes, 38 were located within different prophage and GI regions and showed no significant hits in databases. A BLAST search against the NCBI Protein database using as the query the amino acid sequences of the remaining 21 chromosomal CDSs revealed that 20 of them had significant best hits with proteins found in the database, 16 of them (i.e., 80%) located in species of the Acinetobacter genus (12 of them in A. baumannii) with potential roles in transport, motility, transcriptional regulation, and lipopolysaccharide synthesis (see Table S4 for details). Notably, four CDSs related to capsule synthesis (see below) were best affiliated with homologs located either in species of different orders among the class Gammaproteobacteria to which Acinetobacter belongs, including the Alteromonodales (Alteromonas sp.) and the Enterobacteriales (Yersinia sp.), and also to a different class (Azoarcus sp., Betaproteobacteria) or even to a different phylum (Vitellibacter sp., Bacteroidetes/Chlorobi) (Table S4). This indicated a remarkable ability of A. baumannii NCIMB8209 to co-opt genes from both phylogenetically related and distant species as the result of horizontal gene transfer.
(ii) NCIMB8209 carries a novel K locus.
Among idiosyncratic features of strain NCIMB8209 worth noting, we found differences in content and organization of the genes linked to the production of the K capsule. The K locus identified in the NCIMB8209 genome (Table S2) displayed a gene organization that resembles the polysaccharide gene cluster PSgc6 reported for other clinical A. baumannii strains (70), with the exception of the wafQRST cluster which is missing in NCIMB8209. Furthermore, three extra genes were found in this novel K locus, namely, C4X49_00290 encoding an O-acetylase (absent in other members of the Acinetobacter genus), C4X49_00300 encoding a glycosyltransferase, and C4X49_00305 encoding a pyruvyl transferase (Table S2). Pyruvyl-capped N-acetyl-d-galactosamine (d-GalpNAcA) branches constitute rare structures described so far only in A. baumannii D78, a strain assigned to CC1 (71). The K-locus arrangement described above for strain NCIMB8209 was not found in other Acinetobacter strains by database searching, therefore revealing a previously unreported PSgc locus in A. baumannii. Of note, the weeK gene coding for an UDP-N-acetylglucosamine 4,6-dehydratase (involved in the biosynthesis of UDP-linked sugar precursors used for capsule synthesis) is also interrupted by an ISAba44 copy in NCIMB8209 (C4X49_00335-C4X49_00350, Table S2).
Strain NCIMB8209 shares with DSM30011 a similar gene locus involved in the synthesis of outer core polysaccharides (OC) of the lipid A core moiety, with the exception of an additional gene encoding a glycosyltransferase (C4X49_15205). However, this gene is annotated as a pseudogene and is probably nonfunctional (Table S2). As previously described for strain DSM30011 (11), this cluster includes the rmlBDAC genes (Table S2) responsible for the biosynthesis of dTDP-l-rhamnose (70, 72). The observed genetic organization of the OC locus in NCIMB8209 corresponds to OCL6 according to the classification proposed by Kenyon and collaborators (73).
(iii) Mechanisms of resistance to toxic compounds.
Strain NCIMB8209 also contains many gene clusters encoding systems involved in the resistance to toxic compounds. Some of these clusters are scattered throughout the genome, while others are concentrated in three regions. One of these regions is constituted by GI3 (Table 2) integrated next to the dusA gene (C4X49_02945). The dusA locus has been found to represent a common integration site for this kind of genetic island in A. baumannii (74, 75). Although GI3 (19.1 kb long) is shorter than homologous GIs carried by other A. baumannii strains (for instance, in strain DSM30011, this GI is 33 kb long), this island still includes genes for putative arsenate and heavy metal ion detoxification systems (ars and czc genes) and other genes involved in Fe ion transport (feoAB). Another case is GI6, which carries a cluster of genes encoding a putative copper ion detoxification system (C4X49_13800 to C4X49_13840, Table 2). More interestingly, GI6 also harbors a mobA gene (C4X49_13855) encoding a protein with a relaxase domain which might be responsible for its mobilization after excision from the genome (Table 2). This may suggest a plasmid origin for this GI, and it also opens the possibility that this gene could even mediate its mobilization by horizontal gene transfer after excision from the genome. The third cluster (C4X49_16225 to C4X49_16400) contains a merR-merTPCAD gene cluster (C4X49_16305 to C4X49_16300 to C4X49_16280) coding for a complete Hg ion detoxification system (76). Since there is no gene coding for an integrase nearby, this region was not considered a GI. Remarkably, however, it is flanked by several IS copies which might have contributed to its mobilization and integration in this locus.
Inspection of the NCIMB8209 genome also evidenced the presence of three putative catalase genes (C4X49_07525, C4X49_02120, and C4X49_17825). Catalases represent one of the main strategies evolved by cells to cope with the accumulation of reactive oxygen species (77). It is then noteworthy that the number of putative catalase proteins encoded by this strain is higher than that of the environmental strains DSM30011 (two proteins, NCBI:protein PNH13446.1 and PNH14300.1) and A. baylyi ADP1 (one protein, CAG67388.1), and similar to that found in the A. baumannii clinical strain ATCC 17978 (three proteins, ABO11814.2, ABO10867.2, and ABO13771.2). Moreover, a comparative analysis of the tolerance of the above strains to strong oxidants, as judged by their survival when exposed to H2O2 (77), indicated a strong correlation between their catalase gene content and oxidative stress resistances (Fig. S6).
(iv) Catabolic abilities.
Previous WGS analysis of the environmental A. baumannii DSM30011 strain predicted the presence in its genome of 28 gene clusters encoding many metabolic pathways involved in the utilization of a large variety of plant substances (11). The presence and organization of similar catabolic genes were also investigated in strain NCIMB8209, and despite some differences in the organization of catabolic loci between these two strains, 27 out of the 28 catabolic loci found in DSM30011 (11) were also present in this strain. The only exception was the salicylate/gentisate (sal2/gen) cluster, which was totally missing in NCIMB8209. In addition, the betABI locus present in both strains (previously thought to be a catabolic gene cluster [11, 44]) has been shown in A. baylyi to be involved in the synthesis rather than in the degradation of glycine betaine (78). In agreement, none of the A. baumannii strains tested including DSM30011, NCIMB8209, and ATCC 17978, nor A. baylyi, were capable of utilizing glycine betaine as the only carbon source for growth (Table S5).
From the 27 predicted catabolic loci shared between strains NCIMB8209 and DSM30011 mentioned above, 17 equivalent clusters are also found in the genome of A. baylyi and are involved in the degradation of plant substances and the recycling of plant material (44). These include loci such as pca, qui, pob, hca, van, and ben, involved in the degradation of aromatic acids and hydroxylated aromatic acids such as hydroxycinnamic acids constituting the building blocks of plant protective heteropolymers such as suberin (44, 79–81). These aromatic compounds are ultimately catabolized through the beta-ketoadipate pathway yielding Krebs cycle intermediary substrates, therefore allowing bacterial growth when used as the substrates (44). Our analysis of the ability of A. baumannii strains NCIMB8209 and DSM30011 to employ different compounds as the substrates for growth (Table S5) indicated that these two strains share with A. baylyi the ability to utilize many aromatic acids found in plants, including benzoate, 4-hydroxy-benzoate, 4-hydroxy-cinnamate, and shikimate, as the sole carbon sources. Moreover, the activity of the mdc pathway involved in the catabolism of dicarboxylic malonic acid, another plant-synthesized compound (44), was inferred from the growth observed by all A. baumannii strains tested in malonate as the only carbon source (Table S5). These observations are compatible with the isolation of A. baumannii strains NCIMB8209 and DSM30011 from an enriched consortium specialized in recycling resinous plant material (14). Remarkably still, all of the above-mentioned catabolic capabilities are also shared by A. baumannii clinical strains such as ATCC 17978 (Table S5 and data not shown).
Besides the above-described similarities with A. baylyi, strains NCIMB8209 and DSM30011 are endowed with some idiosyncratic catabolic clusters also related to the degradation of particular plant compounds. Among them we could mention the paa (phenylacetic acid [PAA]) and liu (leucine/isovalerate) clusters (11). The presence of a paa cluster in both NCIMB8209 and DSM30011, but not in A. baylyi, correlates with the capability of these A. baumannii strains to grow on PAA as the sole carbon source (Table S5). PAA is a plant auxin derived from the catabolism of phenylalanine (82, 83) endowed with substantial antimicrobial activity (84). PAA degradation by A. baumannii clinical strains has already been noted (82) (see also Table S5) and found to play an important role during A. baumannii infection by reducing the levels of this powerful phagocyte chemoattractant (83). Concerning the liu (leucine/isovalerate) catabolic cluster, evidence of its activity was obtained by the growth observed for NCIMB8209 and DSM30011 on l-leucine or isovalerate as the sole carbon sources (Table S5). In P. aeruginosa, the liu pathway complements the atu pathway responsible for the degradation of acyclic terpenes produced by plants in response to phytopathogens (85), and a similar situation may also occur in NCIMB8209 and DSM30011 (11). It follows that these A. baumannii strains have the capability not only to participate in the degradation of many plant aromatic compounds, but they are also endowed with the additional ability to degrade compounds produced by plants in response to stress situations, including the attack of phytopathogens and phytophagous insects (79–81).
Besides the above-described similarities at both the genomic level and metabolic capabilities between strains DSM30011 and NCIMB8209, a differential capacity for the utilization of the basic amino acids arginine and ornithine was observed between these two strains (Table S5). This may be explained by the differential presence in DSM30011 of a 10.5-kb fragment containing a gdh-ascC-astA-astD-astB-astE gene cluster encoding a complete arginine succinyltransferase (AST) pathway responsible for the catabolism of these basic amino acids (86), which was missing in NCIMB8209 (Fig. 1). Instead, a region of 27.6 kb bordered by IS1008/ISOur remnants and encompassing putative ion mercury detoxification genes was found in NCIMB8209, which has in turn been heavily impacted by several ISs of different types (Table S3). It is worth noting in the above context that the chromosomal region adjacent to astA in A. baumannii CC2 strains is also an integration site for different mobile elements such as AbGRI2-type resistance islands among others, which have provoked different rearrangements in their vicinity including various deletions (10, 53). This has resulted in some CC2 strains in which a AbGRI2-type element is found adjacent to a complete ast gene cluster, and other strains in which the ast genes have been completely deleted (10, 53; also data not shown). It has been shown in P. aeruginosa that the N-succinyl transferase AstA, the first enzyme of the AST pathway, is able to use both l-arginine and l-ornithine as the substrates (87), and a similar substrate specificity may also occur for A. baumannii AstA. Furthermore, genes coding for a putrescine importer (puuP) and a gamma-aminobutyraldehyde dehydrogenase (patD), the latter part of the transaminase pathway of putrescine degradation (86), were identified in DSM30011 (51% and 48% identity with the corresponding proteins from Escherichia coli strain K-12 substrain MG1655; accession numbers AAC74378.2 and AAC74526.1, respectively) but not in NCIMB8209. The latter observations might also explain the inability of NCIMB8209 to grow on putrescine, compared to the other A. baumannii strains tested (Table S12). In any case, the observed phenotypic profiles suggest a narrowing of NCIMB8209 substrate utilization capabilities when basic amino acids and polyamines in particular are considered, which suggests an adaptation of this strain to a specific niche.
Conclusions.
A. baumannii NCIMB8209 represents to our knowledge the second reported environmental A. baumannii strain, isolated from a desert plant source at the onset (or even before) the massive introduction of antimicrobials to treat infections (11). In concordance, and similarly to its companion strain DSM30011, NCIMB8209 showed general susceptibility to most clinically employed antimicrobials, including folate pathway inhibitors (this work and reference 11). As expected from their common plant source and retting enrichment before isolation in media containing guayule resins as the substrates for growth (14), both A. baumannii strains share the ability to degrade a number of substances produced by plants, including many hydroxylated aromatic acids constituting the building blocks of plant protective hydrophobic heteropolymers and also repellents against predators (Table S5). However, WGS and subsequent phylogenetic and comparative genome analysis, as well as different biochemical studies conducted in this work, indicated significant differences between these two environmental strains. First, compared to DSM30011, NCIMB8209 has undergone a significant genome reduction and lacks many genetic clusters encoding components involved in defense mechanisms against other biological competitors such as the CRISPR-cas complex, T6SS, and two-partner systems. Moreover, and although NCIMB8209 contains most genes associated with persistence and virulence in A. baumannii clinical strains, many genes encoding components of surface structures are interrupted by IS elements whose relatively high number and variability impacted heavily on the NCIMB8209 genome. Among IS-interrupted genes, we found those encoding pili components, the O-antigen ligase TfpO, a biosynthetic route for surfactants compounds, Bap and Ata adhesins, a RTX-toxin, etc. (Table S3). Comparative biofilm and pellicle production, as well as motility assays, yielded evidence for the fact that NCIM8209 is severely compromised in these pathogenicity-associated traits (Fig. S4). Altogether, these observations can explain the low relative virulence potential observed for NCIM8209 on the G. mellonella and C. elegans infection models (Fig. S3).
Loss of T6SS genes has been observed in a number of A. baumannii clinical strains causing infections, suggesting that this system is not required once A. baumannii invades its host (53, 88, 89). Moreover, the absence of T6SS has been linked with higher chances of evasion of A. baumannii from the host immune system (90, 91). The situation described above for strain NCIMB8209 therefore resembles that reported for A. baumannii SDF isolated from a human body louse (24), which has undergone extensive genome reductions and rearrangements mediated by ISs (24). Also, albeit more moderately, loss of T6SS and IS-mediated inactivation of genes encoding surface structures was also observed in A. baumannii D1279779, a community-acquired strain isolated from the bacteremic infection of an indigenous Australian (26).
It is tempting to speculate that the changes observed in A. baumannii NCIMB8209 are also related to its adaptation to a particular niche. In this context, several reports have shown the profound associations existing between different bacterial groups, including many members of the Acinetobacter genus with a number of insects feeding on plants (16–22). Many of these bacterial species are located in the guts of their insect hosts conducting mutualistic or symbiotic associations based on nutrition/protection relationships (16, 18–20, 92, 93). The bacterial counterpart thus degrades toxic compounds for the insect of the plant diet, and the insect host in return provides a stable environment, supply, or resources, and a vector for the rapid spreading and inoculation into fresh plant tissues. For those bacterial species displaying low pathogenic potential, selective pressure eventually favors more stable relationships with concomitant structural and metabolic changes (93). A general trend thus observed for Gram-negative species with these characteristics is genome reduction with the loss or modification of surface-exposed molecules, thus reducing interactions with the innate immune system of their hosts which may trigger defensive responses and elimination (92, 93). This situation can certainly be applied to NCIMB8209, as extensively detailed above. In this context, we note the relative high tolerance of this strain to pro-oxidants such as H2O2 (Fig. S6), which correlates with the identification of three catalase genes in its genome (94). The innate immune system of insects closely resembles that of vertebrates at both the molecular and cellular levels (92). Thus, while the cellular immune response in vertebrates is mediated by professional phagocytes, in insects, this function is conducted by phagocytic cells known as hemocytes (92). Since phagocytic cells use oxidative burst as a common strategy to counteract pathogens (92, 95), it is then possible that the selection of a higher antioxidant ability in NCIMB8209 led to an increased tolerance to oxidative stress and therefore increased survival in an insect niche. Of note, recent evidence indicates that A. baumannii strains with high catalase production are more resistant against intracellular killing by macrophages (96).
Our observations also provide further clues on the high genomic plasticity of A. baumannii as a species (6), underscoring the fact that this highly advantageous feature in adaptive terms is not exclusive to A. baumannii strains of clinical origin only. In summary, WGS analysis and complementary phenotypic characterization of A. baumannii strains of environmental origin such as DSM30011 and NCIMB8209 provided important clues on the genomic content and diversity of this species before the strong selection pressure associated with the current antibiotic era, and suggested potential niches of this species outside the clinical setting.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Acinetobacter sp. strain NCIMB8209 was obtained from the National Collection of Industrial Food and Marine Bacteria, Aberdeen, Scotland. For details on the original isolation and different denominations assigned to this strain by various collections, see our previous publication (11) under “Tracing DSM30011 origins.”
The use of carbon or carbon and nitrogen sources by the A. baumannii strain NCIMB8209, DSM30011, or ATCC 17978 was conducted on solid BM2 minimal medium [62 mM potassium phosphate (pH 7.0), 7 mM (NH4)2SO4, 0.5 mM MgSO4, 10 μM FeSO4] supplemented with 1.5% Difco bacteriological agar and 0.2% (wt/vol) of the tested substrates (97). The plates were incubated at 30°C for 48 to 96 h before colony growth was inspected. Other bacterial strains such as Acinetobacter baylyi ADP1 (44) and Pseudomonas aeruginosa PAO1 (98) were also included in these tests as controls for the utilization of particular compounds.
Plates containing 0.3% agarose, 10 g/liter tryptone, and 5 g/liter NaCl were used as a tool to detect cell motility on a semisolid surface. The plates were inoculated on the surface with bacteria lifted from overnight LB agar cultures using flat-ended sterile wooden sticks or depositing 10 μl of LB cultures grown to an optical density at 600 nm (OD600) of 0.1. Plates were incubated for 30 h at 30°C in the dark before inspection and recording.
Tolerance to H2O2 was performed as previously described (77). Briefly, bacterial cultures were collected when they reached an OD600 of 0.6 and subjected to serial dilutions. Aliquots of 10 μl were then loaded onto LB agar plates, supplemented with 400 μM H2O2, and incubated for 30 h at 30°C before inspection.
NCIMB8209 genome sequencing and annotation.
NCIMB8209 DNA was isolated using a commercial kit (Wizard Genomic DNA purification kit; Promega), following the manufacturer’s instructions. The genomic sequence was obtained using a hybrid strategy combining PacBio Sequel (MrDNALab, Shallowater, TX, USA) and Illumina MiSeq (single-end reads, University of Chicago genomic facilities) methods. A total of 3,238,476,791 reads were generated with the PacBio Sequel approach (depth of coverage ∼834×), which were subjected to quality assessment prior to de novo genome assembly using Falcon (99) followed by further polishment by the Arrow algorithm. In turn, the sequence data generated by the Illumina strategy were assembled into contigs using Spades version 3.0.11 (100), and used to close the gaps left by the PacBio Sequel strategy. The replication origin (oriC) was predicted with OriFinder (101). The final assembled NCIMB8209 chromosome was annotated using the pipeline available at NCBI and deposited in DDBJ/ENA/GenBank under accession number CP028138. In turn, the plasmid sequences derived from the above sequencing data were deposited in the GenBank nucleotide sequence database under accession number CP028139.
The presence of IS elements in the NCIMB8209 genome was determined using IS Finder version 2.0 (102) (https://www-is.biotoul.fr/). Novel ISs were deposited into the ISSaga database (54). Putative prophage sequences were identified by PHASTER (103, 104). Antimicrobial resistance genes were detected with ResFinder 3.2 (https://cge.cbs.dtu.dk/services/ResFinder/) (105). Catabolic clusters present in the NCIMB8209 genome were searched by a BLASTN approach (106) using as queries the catabolic genes previously described for the DSM30011 strain (11).
Protein family analyses and phylogenomic calculations.
The proteomes of the 126 Acinetobacter strains analyzed in this work, which included 99 A. baumannii strains other than NCIMB8209 and DSM30011, were first retrieved from the NCBI ftp database and gathered into a local database together with the NCIMB8209 proteome. Families of homologous proteins were clustered with PGAP version 1.2.1 by using the gene family (GF) method (107). Briefly, the BLASTALL tool was executed among all mixed protein sequences, and the filtered BLAST result was clustered by the MCL algorithm. For each protein pair of the same cluster, the global match region and identity were set to be equal or higher than 70% of the longer sequence. This analysis allowed us to assemble 27,367 protein families among these genomes, from which 6,383 corresponded to protein families presenting a single representative in all Acinetobacter genomes analyzed. Regarding strain NCIMB8209 (3,596 predicted total CDSs), 3,551 protein families contained at least one representative sequence in this strain, including 79 families that contained more than one.
Maximum likelihood (ML) phylogenetic trees were then constructed using these data. For each protein family, the corresponding nucleotide sequences were retrieved, individually aligned using ClustalW2 (108), and trimmed using GBlock 0.91b (109). The resulting alignments were combined to build a large supermatrix (362,285 nucleotide positions) using the Python3 package AMAS 0.98 (110). Finally, the evolutionary history of the strains was inferred as implemented by Espariz et al. (111) using the RAxML software (112) and the general tree reversible (GTR) substitution model with gamma distribution. The individual parameters for the model were estimated and optimized for each concatenated gene as indicated by Stamatakis (112). Reliability of the inferred tree was tested by bootstrapping with 100 repetitions.
In order to depurate the list of protein families specific for strain NCIMB8209 according to PGAP predictions, their coding genes were used as queries in a BLASTN sequence similarity-based comparison (106) against all 126 Acinetobacter genomes present in our local database. Coverage and identity thresholds of 70% and an E-value cutoff of 1E−30 were used for these calculations. Those CDSs for which a significant hit was found were then deleted from the list.
Sequence typing.
Assignment of sequence types (ST) for strain NCIMB8209 was done using the housekeeping genes cpn60, gltA, gpi, gyrB, recA, and rpoD (Oxford scheme) (29) or cpn60, fusA, gltA, pyrG, recA, rplB, and rpoD (Pasteur scheme) (3). For details, see the A. baumannii MLST Databases website (http://pubmlst.org/abaumannii/) (113).
gdhB gene detection by PCR.
PCRs were carried out following standard protocols, using the primer pair GHDB_1F (GCTACTTTTATGCAACAGAGC)/GHDB775R (GTTGAGTTGGCGTATGTTGTGC) for ghdB detection and the degenerate primer pair 16S RDNAF (AGAGTTTGATCHTGGYTYAGA)/16S_RDNAR (ACGGYTACCTTGTTACGACTTC) for 16S rRNA gene detection.
Antimicrobial susceptibility testing.
The general antimicrobial susceptibility of strain NCIMB8209 was evaluated using the Vitek 2 System (bioMérieux) following criteria recommended by the Clinical and Laboratory Standards Institute (CLSI) (114). Susceptibility tests to tetracycline, chloramphenicol, and macrolides such as azithromycin and erythromycin were done separately by disk assays on Mueller-Hinton agar (MHA) following CLSI protocols. In short, NCIMB8209 cells were grown overnight at 37°C, resuspended in LB broth to a turbidity of 0.5 McFarland units, and spread on the surfaces of MHA-containing petri plates. Antibiotic disks were then carefully deposited at the center of the agar surface, and the plates were incubated at 37°C for 16 h before measuring the diameter of the corresponding growth inhibition zones.
Bacterial competition assays.
The different A. baumannii and E. coli strains used in competition assays were grown overnight in 2 ml of L-broth (10 g/liter tryptone, 5 g/liter yeast extract, and 0.5 g/liter NaCl) medium. Cultures were then diluted in L-broth medium to an absorbance at 600 nm (Abs600) of 0.1. The cells were mixed in a 10:1 ratio (predator/prey). Aliquots of 20 μl of these cell mixtures were laid on the surface of L-broth medium supplemented with 1.5% agar and incubated at 37°C for 4 h. The bacterial spot on the agar surface was subsequently removed and vigorously resuspended in an Eppendorf tube with 500 μl of PBS. The mixtures were serially diluted 1:10 in PBS using a 96-well polystyrene microtiter plate and then plated onto solid selective media for colony counting. For E. coli DH5α, selective medium containing 20 μg/ml nalidixic acid was used. For Acinetobacter strains, selective media contained either 30 μg/ml gentamicin or 75 μg/ml rifampicin were employed as indicated. For all bacterial competition experiments, one representative image of the growth results obtained in selective medium is shown.
Hcp secretion analysis.
Experiments were performed by the method of Repizo et al. (45). Briefly, the A. baumannii strains analyzed for Hcp secretion were inoculated in 50 ml L-broth at an Abs600 of 0.08, and incubated at 37°C until the Abs600 reached a value of ∼1.0. Culture supernatants were obtained by centrifugation at 4,000 × g, followed by filtration using 0.22-μm filters. Secreted proteins present in the supernatants were then concentrated 50-fold using Amicon Ultracel 3K centrifuge filters following the instructions of the manufacturer and subjected to a wash step with 40 mM Tris (pH 8), 200 mM NaCl, and 5% glycerol before being analyzed by 18% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE).
Biofilm assays.
Biofilm formation was qualitatively determined by measuring the adhesion of bacteria to the surfaces of glass tubes. A. baumannii strains were statically grown overnight in 2 ml of L-broth medium. The spent liquid medium was discarded, and the tubes were rinsed twice with water before adding a solution of 1% crystal violet. After 15 min, the dye solution was discarded, and the tubes were rinsed twice with water before inspection. All assays were done at least three times using fresh samples each time.
A. baumannii virulence assays using Caenorhabditis elegans nematode and Galleria mellonella larvae model systems.
Virulence assays of the different A. baumannii strains tested here were evaluated by using two different model systems: G. mellonella moth larvae (42) and the nematode C. elegans (43). G. mellonella larvae were purchased from Knutson’s Live Bait (Brooklyn, MI) and were used the day after arrival. Groups of 20 randomly picked larvae were used for each assay condition. The different A. baumannii strains tested were grown overnight in LB and then diluted with phosphate-buffered saline (PBS) to obtain the CFU titers indicated in the corresponding figure legends, which were verified by colony counts on L-broth agar (LBA) for all inocula. A Hamilton microliter syringe was used to inject 10 μl of the bacterial suspensions into the hemolymph of each larva via the second to the last left proleg. For a control, one group of G. mellonella larvae was injected with 10 μl of PBS. After injection, the larvae were incubated in plastic plates at 37°C, and the numbers of dead individuals were scored regularly.
For C. elegans survival experiments, the N2 Bristol (wild type) was used. Gravid hermaphrodites were bleached, and the resulting eggs were washed using standard protocols (115). Nematodes arrested in larval stage 1 (L1) were then transferred onto NGM plates seeded with fresh E. coli OP50 and grown at 20°C until nematodes reached larval stage 4 (L4). For virulence assays, A. baumannii and E. coli OP50 (control) were grown overnight at 37°C in L-broth medium. The stationary culture was then diluted to 107 total CFU, and 50-μl portions were seeded onto 60-mm nematode growth medium (NGM) plates. Groups of L4 worms were then transferred to the plates seeded with A. baumannii and deposited on the center of the bacterial spot. Assays were performed in duplicates. The nematodes were maintained at 20°C, transferred to fresh plates every 48 h, and monitored daily. Worms that did not respond to stimulation by touch were scored as dead.
Survival curves were plotted using PRISM software, and comparisons in survival were calculated using the log rank Mantel-Cox test and Gehan-Breslow-Wilcoxon test.
Data availability.
The final assembled NCIMB8209 chromosome was annotated using the pipeline available at NCBI and deposited in DDBJ/ENA/GenBank under accession number CP028138. In turn, the plasmid sequences derived from the above sequencing data were deposited in the GenBank nucleotide sequence database under accession number CP028139.
ACKNOWLEDGMENTS
This work was supported by a grant awarded to A.M.V. from the Agencia Nacional de Promocion Cientifica y Tecnologica (ANPCyT grant PICT-2015-1072), by a grant awarded to H.A.S. (NIH grant AI-115203), and by a Fulbright fellowship awarded to G.D.R.
We are indebted to the curator teams of both the Institut Pasteur MLST system (Paris, France) and Oxford University (Oxford, UK) for their help in incorporating NCIMB8209 MLST alleles and profiles at http://pubmlst.org/abaumannii/. We are indebted to D. de Mendoza (IBR-CONICET, Rosario, Argentina) for the kind gift of the C. elegans N2 strain. We also thank V. Müller (Goethe University Frankfurt, Frankfurt am Main, Germany) for calling our attention to the biosynthetic characteristics of the Acinetobacter bet locus. M.E., A.M.V., and G.D.R. are staff members of CONICET. H.A.S. is a member of the Department of Microbiology at the University of Chicago.
G.D.R., A.M.V., and H.A.S. conceived and designed the work. G.D.R. and M.E. conducted the bioinformatic analysis. G.D.R., J.L.S., J.I.D.M., B.A.S., and A.M.V. performed the experimental part. G.D.R., M.E., and A.M.V. analyzed the data. All authors contributed to the writing of the manuscript and read and approved the final version.
We declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
REFERENCES
- 1.Bouvet PJM, Grimont P. 1986. Taxonomy of the genus Acinetobacter with the recognition of Acinetobacter baumannii sp. nov., Acinetobacter haemolyticus sp. nov., Acinetobacter johnsonii sp. nov., and Acinetobacter junii sp. nov. and emended descriptions of Acinetobacter calcoaceticus and Acinetobacter lwoffii. Int J Syst Bacteriol 36:228–240. doi: 10.1099/00207713-36-2-228. [DOI] [Google Scholar]
- 2.Antunes LCS, Visca P, Towner KJ. 2014. Acinetobacter baumannii: evolution of a global pathogen. Pathog Dis 71:292–301. doi: 10.1111/2049-632X.12125. [DOI] [PubMed] [Google Scholar]
- 3.Diancourt L, Passet V, Nemec A, Dijkshoorn L, Brisse S. 2010. The population structure of Acinetobacter baumannii: expanding multiresistant clones from an ancestral susceptible genetic pool. PLoS One 5:e10034. doi: 10.1371/journal.pone.0010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Di Nocera PP, Rocco F, Giannouli M, Triassi M, Zarrilli R. 2011. Genome organization of epidemic Acinetobacter baumannii strains. BMC Microbiol 11:224. doi: 10.1186/1471-2180-11-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nigro SJ, Post V, Hall RM. 2011. Aminoglycoside resistance in multiply antibiotic-resistant Acinetobacter baumannii belonging to global clone 2 from Australian hospitals. J Antimicrob Chemother 66:1504–1509. doi: 10.1093/jac/dkr163. [DOI] [PubMed] [Google Scholar]
- 6.Touchon M, Cury J, Yoon E-J, Krizova L, Cerqueira GC, Murphy C, Feldgarden M, Wortman J, Clermont D, Lambert T, Grillot-Courvalin C, Nemec A, Courvalin P, Rocha EPC. 2014. The genomic diversification of the whole Acinetobacter genus: origins, mechanisms, and consequences. Genome Biol Evol 6:2866–2882. doi: 10.1093/gbe/evu225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roca I, Espinal P, Vila-Farrés X, Vila J. 2012. The Acinetobacter baumannii oxymoron: commensal hospital dweller turned pan-drug-resistant menace. Front Microbiol 3:148. doi: 10.3389/fmicb.2012.00148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adams MD, Bishop B, Wright MS. 2016. Quantitative assessment of insertion sequence impact on bacterial genome architecture. Microb Genom 2:e000062. doi: 10.1099/mgen.0.000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeidler S, Müller V. 2019. Coping with low water activities and osmotic stress in Acinetobacter baumannii: significance, current status and perspectives. Environ Microbiol 21:2212–2230. doi: 10.1111/1462-2920.14565. [DOI] [PubMed] [Google Scholar]
- 10.Chan AP, Sutton G, DePew J, Krishnakumar R, Choi Y, Huang XZ, Beck E, Harkins DM, Kim M, Lesho EP, Nikolich MP, Fouts DE. 2015. A novel method of consensus pan-chromosome assembly and large-scale comparative analysis reveal the highly flexible pan-genome of Acinetobacter baumannii. Genome Biol 16:143. doi: 10.1186/s13059-015-0701-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Repizo GD, Viale AM, Borges V, Cameranesi MM, Taib N, Espariz M, Brochier-Armanet C, Gomes JP, Salcedo SP. 2017. The environmental Acinetobacter baumannii isolate DSM30011 reveals clues into the preantibiotic era genome diversity, virulence potential, and niche range of a predominant nosocomial pathogen. Genome Biol Evol 9:2292–2307. doi: 10.1093/gbe/evx162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eveillard M, Kempf M, Belmonte O, Pailhoriès H, Joly-Guillou ML. 2013. Reservoirs of Acinetobacter baumannii outside the hospital and potential involvement in emerging human community-acquired infections. Int J Infect Dis 17:e802–e805. doi: 10.1016/j.ijid.2013.03.021. [DOI] [PubMed] [Google Scholar]
- 13.Wilharm G, Skiebe E, Higgins PG, Poppel MT, Blaschke U, Leser S, Heider C, Heindorf M, Brauner P, Jäckel U, Böhland K, Cuny C, Łopińska A, Kaminski P, Kasprzak M, Bochenski M, Ciebiera O, Tobółka M, Żołnierowicz KM, Siekiera J, Seifert H, Gagné S, Salcedo SP, Kaatz M, Layer F, Bender JK, Fuchs S, Semmler T, Pfeifer Y, Jerzak L. 2017. Relatedness of wildlife and livestock avian isolates of the nosocomial pathogen Acinetobacter baumannii to lineages spread in hospitals worldwide. Environ Microbiol 19:4349–4364. doi: 10.1111/1462-2920.13931. [DOI] [PubMed] [Google Scholar]
- 14.Allen PJ, Naghski J, Hoover SR. 1944. Decomposition of guayule resins by microorganisms. J Bacteriol 47:559–572. doi: 10.1128/JB.47.6.559-572.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Naghski J, White JW, Hoover SR. 1944. Aerobic decomposition of guayule shrub (Parthenium argentatum Gray). J Bacteriol 48:159–178. doi: 10.1128/JB.48.2.159-178.1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Morales-Jiménez J, Zúñiga G, Villa-Tanaca L, Hernández-Rodríguez C. 2009. Bacterial community and nitrogen fixation in the red turpentine beetle, Dendroctonus valens LeConte (Coleoptera: Curculionidae: Scolytinae). Microb Ecol 58:879–891. doi: 10.1007/s00248-009-9548-2. [DOI] [PubMed] [Google Scholar]
- 17.Keeling CI, Yuen MM, Liao NY, Docking TR, Chan SK, Taylor GA, Palmquist DL, Jackman SD, Nguyen A, Li M, Henderson H, Janes JK, Zhao Y, Pandoh P, Moore R, Sperling FA, Huber DP, Birol I, Jones SJ, Bohlmann J. 2013. Draft genome of the mountain pine beetle, Dendroctonus ponderosae Hopkins, a major forest pest. Genome Biol 14:R27. doi: 10.1186/gb-2013-14-3-r27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim PS, Shin NR, Kim JY, Yun JH, Hyun DW, Bae JW. 2014. Acinetobacter apis sp. nov., isolated from the intestinal tract of a honey bee, Apis mellifera. J Microbiol 52:639–645. doi: 10.1007/s12275-014-4078-0. [DOI] [PubMed] [Google Scholar]
- 19.Vilanova C, Marín M, Baixeras J, Latorre A, Porcar M. 2014. Selecting microbial strains from pine tree resin: biotechnological applications from a terpene world. PLoS One 9:e100740. doi: 10.1371/journal.pone.0100740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu L, Lu M, Xu D, Chen L, Sun J. 2016. Sexual variation of bacterial microbiota of Dendroctonus valens guts and frass in relation to verbenone production. J Insect Physiol 95:110–117. doi: 10.1016/j.jinsphys.2016.09.014. [DOI] [PubMed] [Google Scholar]
- 21.Liu S, Wang Y, Ruan Z, Ma K, Wu B, Xu Y, Wang J, You Y, He M, Hu G. 2017. Acinetobacter larvae sp. nov., isolated from the larval gut of Omphisa fuscidentalis. Int J Syst Evol Microbiol 67:806–811. doi: 10.1099/ijsem.0.001644. [DOI] [PubMed] [Google Scholar]
- 22.Blankenchip CL, Michels DE, Braker HE, Goffredi SK. 2018. Diet breadth and exploitation of exotic plants shift the core microbiome of Cephaloleia, a group of tropical herbivorous beetles. PeerJ 6:e4793. doi: 10.7717/peerj.4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nemec A, Krizova L, Maixnerova M, van der Reijden TJ, Deschaght P, Passet V, Vaneechoutte M, Brisse S, Dijkshoorn L. 2011. Genotypic and phenotypic characterization of the Acinetobacter calcoaceticus-Acinetobacter baumannii complex with the proposal of Acinetobacter pittii sp. nov. (formerly Acinetobacter genomic species 3) and Acinetobacter nosocomialis sp. nov. (formerly Acinetobacter genomic species 13TU). Res Microbiol 162:393–404. doi: 10.1016/j.resmic.2011.02.006. [DOI] [PubMed] [Google Scholar]
- 24.Vallenet D, Nordmann P, Barbe V, Poirel L, Mangenot S, Bataille E, Dossat C, Gas S, Kreimeyer A, Lenoble P, Oztas S, Poulain J, Segurens B, Robert C, Abergel C, Claverie J-M, Raoult D, Médigue C, Weissenbach J, Cruveiller S. 2008. Comparative analysis of Acinetobacters: three genomes for three lifestyles. PLoS One 3:e1805. doi: 10.1371/journal.pone.0001805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang H, Dong Y, Yang ZL, Luo H, Zhang X, Gao F. 2014. Complete sequence of pABTJ2, a plasmid from Acinetobacter baumannii MDR-TJ, carrying many phage-like elements. Genomics Proteomics Bioinformatics 12:172–177. doi: 10.1016/j.gpb.2014.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Farrugia DN, Elbourne LD, Hassan KA, Eijkelkamp BA, Tetu SG, Brown MH, Shah BS, Peleg AY, Mabbutt BC, Paulsen IT. 2013. The complete genome and phenome of a community-acquired Acinetobacter baumannii. PLoS One 8:e58628. doi: 10.1371/journal.pone.0058628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holt K, Kenyon JJ, Hamidian M, Schultz MB, Pickard DJ, Dougan G, Hall R. 2016. Five decades of genome evolution in the globally distributed, extensively antibiotic-resistant Acinetobacter baumannii global clone 1. Microb Genom 2:e000052. doi: 10.1099/mgen.0.000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weingarten RA, Johnson RC, Conlan S, Ramsburg AM, Dekker JP, Lau AF, Khil P, Odom RT, Deming C, Park M, Thomas PJ, Henderson DK, Palmore TN, Segre JA, Frank KM. 2018. Genomic analysis of hospital plumbing reveals diverse reservoir of bacterial plasmids conferring carbapenem resistance. mBio 9:e02011-17. doi: 10.1128/mBio.02011-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bartual SG, Seifert H, Hippler C, Luzon MAD, Wisplinghoff H, Rodríguez-Valera F. 2005. Development of a multilocus sequence typing scheme for characterization of clinical isolates of Acinetobacter baumannii. J Clin Microbiol 43:4382–4390. doi: 10.1128/JCM.43.9.4382-4390.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tomaschek F, Higgins PG, Stefanik D, Wisplinghoff H, Seifert H. 2016. Head-to-head comparison of two multi-locus sequence typing (MLST) schemes for characterization of Acinetobacter baumannii outbreak and sporadic isolates. PLoS One 11:e0153014. doi: 10.1371/journal.pone.0153014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gaiarsa S, Batisti Biffignandi G, Esposito EP, Castelli M, Jolley KA, Brisse S, Sassera D, Zarrilli R. 2019. Comparative analysis of the two Acinetobacter baumannii multilocus sequence typing (MLST) schemes. Front Microbiol 10:930. doi: 10.3389/fmicb.2019.00930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yakkala H, Samantarrai D, Gribskov M, Siddavattam D. 2019. Comparative genome analysis reveals niche-specific genome expansion in Acinetobacter baumannii strains. PLoS One 14:e0218204. doi: 10.1371/journal.pone.0218204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Turton JF, Woodford N, Glover J, Yarde S, Kaufmann ME, Pitt TL. 2006. Identification of Acinetobacter baumannii by detection of the blaOXA-51-like carbapenemase gene intrinsic to this species. J Clin Microbiol 44:2974–2976. doi: 10.1128/JCM.01021-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hamouda A, Evans BA, Towner KJ, Amyes SGB. 2010. Characterization of epidemiologically unrelated Acinetobacter baumannii isolates from four continents by use of multilocus sequence typing, pulsed-field gel electrophoresis, and sequence-based typing of blaOXA-51-like genes. J Clin Microbiol 48:2476–2483. doi: 10.1128/JCM.02431-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Evans BA, Amyes SJ. 2014. OXA β-lactamases. Clin Microbiol Rev 27:241–263. doi: 10.1128/CMR.00117-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mussi MA, Limansky AS, Relling V, Ravasi P, Arakaki A, Actis LA, Viale AM. 2011. Horizontal gene transfer and assortative recombination within the Acinetobacter baumannii clinical population provide genetic diversity at the single carO gene, encoding a major outer membrane protein channel. J Bacteriol 193:4736–4648. doi: 10.1128/JB.01533-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morán-Barrio J, Cameranesi MM, Relling V, Limansky AS, Brambilla L, Viale AM. 2017. The Acinetobacter outer membrane contains multiple specific channels for carbapenem β-lactams as revealed by kinetic characterization analyses of imipenem permeation into Acinetobacter baylyi cells. Antimicrob Agents Chemother 61:e01737-16. doi: 10.1128/AAC.01737-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Post V, White PA, Hall RM. 2010. Evolution of AbaR-type genomic resistance islands in multiply antibiotic-resistant Acinetobacter baumannii. J Antimicrob Chemother 65:1162–1170. doi: 10.1093/jac/dkq095. [DOI] [PubMed] [Google Scholar]
- 39.Nigro SJ, Hall RM. 2011. GIsul2, a genomic island carrying the sul2 sulphonamide resistance gene and the small mobile element CR2 found in the Enterobacter cloacae subspecies cloacae type strain ATCC 13047 from 1890, Shigella flexneri ATCC 700930 from 1954 and Acinetobacter baumannii ATCC 17978 from 1951. J Antimicrob Chemother 66:2175–2176. doi: 10.1093/jac/dkr230. [DOI] [PubMed] [Google Scholar]
- 40.Loewen PC, Alsaadi Y, Fernando D, Kumar A. 2014. Genome sequence of a tigecycline-resistant clinical isolate of Acinetobacter baumannii strain AB031 obtained from a bloodstream infection. Genome Announc 2:e01036-14. doi: 10.1128/genomeA.01036-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hamidian M, Hall RM. 2017. Acinetobacter baumannii ATCC 19606 carries GIsul2 in a genomic island located in the chromosome. Antimicrob Agents Chemother 61:e01991-16. doi: 10.1128/AAC.01991-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Peleg AY, Jara S, Monga D, Eliopoulos GM, Moellering RC, Jr, Mylonakis E. 2009. Galleria mellonella as a model system to study Acinetobacter baumannii pathogenesis and therapeutics. Antimicrob Agents Chemother 53:2605–2609. doi: 10.1128/AAC.01533-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vallejo JA, Beceiro A, Rumbo-Feal S, Rodríguez-Palero MJ, Russo TA, Bou G. 2015. Optimisation of the Caenorhabditis elegans model for studying the pathogenesis of opportunistic Acinetobacter baumannii. Int J Antimicrob Agents 2015:S0924-8579(15)00241-1. doi: 10.1016/j.ijantimicag.2015.05.021. [DOI] [PubMed] [Google Scholar]
- 44.Barbe V, Vallenet D, Fonknechten N, Kreimeyer A, Oztas S, Labarre L, Cruveiller S, Robert C, Duprat S, Wincker P, Ornston LN, Weissenbach J, Marliere P, Cohen GN, Medigue C. 2004. Unique features revealed by the genome sequence of Acinetobacter sp. ADP1, a versatile and naturally transformation competent bacterium. Nucleic Acids Res 32:5766–5779. doi: 10.1093/nar/gkh910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Repizo GD, Gagné S, Foucault-Grunenwald M-L, Borges V, Charpentier X, Limansky AS, Gomes JP, Viale AM, Salcedo SP. 2015. Differential role of the T6SS in Acinetobacter baumannii virulence. PLoS One 10:e0138265. doi: 10.1371/journal.pone.0138265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wilharm G, Piesker J, Laue M, Skiebe E. 2013. DNA uptake by the nosocomial pathogen Acinetobacter baumannii occurs during movement along wet surfaces. J Bacteriol 195:4146–4153. doi: 10.1128/JB.00754-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gebhardt MJ, Gallagher LA, Jacobson RK, Usacheva EA, Peterson LR, Zurawski DV, Shuman HA. 2015. Joint transcriptional control of virulence and resistance to antibiotic and environmental stress in Acinetobacter baumannii. mBio 6:e01660-15. doi: 10.1128/mBio.01660-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Karah N, Samuelsen Ø, Zarrilli R, Sahl JW, Wai SN, Uhlin BE. 2015. CRISPR-cas subtype I-Fb in Acinetobacter baumannii: evolution and utilization for strain subtyping. PLoS One 10:e0118205. doi: 10.1371/journal.pone.0118205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Costa AR, Monteiro R, Azeredo J. 2018. Genomic analysis of Acinetobacter baumannii prophages reveals remarkable diversity and suggests profound impact on bacterial virulence and fitness. Sci Rep 8:15346. doi: 10.1038/s41598-018-33800-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wachino J, Jin W, Kimura K, Arakawa Y. 2019. Intercellular transfer of chromosomal antimicrobial resistance genes between Acinetobacter baumannii mediated by prophages. Antimicrob Agents Chemother 63:e00334-19. doi: 10.1128/AAC.00334-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jeon J, Kim J, Yong D, Lee K, Chong Y. 2012. Complete genome sequence of the podoviral bacteriophage YMC/09/02/B1251 ABA BP, which causes the lysis of an OXA-23-producing carbapenem-resistant Acinetobacter baumannii isolate from a septic patient. J Virol 86:12437–12438. doi: 10.1128/JVI.02132-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Turner D, Wand ME, Sutton JM, Centron D, Kropinski AM, Reynolds DM. 2016. Genome sequence of vB_AbaS_TRS1, a viable prophage isolated from Acinetobacter baumannii strain A118. Genome Announc 4:e01051-16. doi: 10.1128/genomeA.01051-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wright MS, Haft DH, Harkins DM, Perez F, Hujer KM, Bajaksouzian S, Benard MF, Jacobs MR, Bonomo RA, Adams MD. 2014. New insights into dissemination and variation of the health care-associated pathogen Acinetobacter baumannii from genomic analysis. mBio 5:e00963-13. doi: 10.1128/mBio.00963-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Varani AM, Siguier P, Gourbeyre E, Charneau V, Chandler M. 2011. ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol 12:R30. doi: 10.1186/gb-2011-12-3-r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Osborn AM, Böltner D. 2002. When phage, plasmids, and transposons collide: genomic islands, and conjugative- and mobilizable-transposons as a mosaic continuum. Plasmid 48:202–212. doi: 10.1016/s0147-619x(02)00117-8. [DOI] [PubMed] [Google Scholar]
- 56.Repizo GD, Espariz M, Seravalle JL, Salcedo SP. 2019. Bioinformatic analysis of the type VI secretion system and its potential toxins in the Acinetobacter genus. Front Microbiol 10:2519. doi: 10.3389/fmicb.2019.02519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roussin M, Rabarioelina S, Cluzeau L, Cayron J, Lesterlin C, Salcedo SP, Bigot S. 2019. Identification of a contact-dependent growth inhibition (CDI) system that reduces biofilm formation and host cell adhesion of Acinetobacter baumannii DSM30011 strain. Front Microbiol 10:2450. doi: 10.3389/fmicb.2019.02450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wood CR, Ohneck EJ, Edelmann RE, Actis LA. 2018. A light-regulated type I pilus contributes to Acinetobacter baumannii biofilm, motility, and virulence functions. Infect Immun 86:e00442-18. doi: 10.1128/IAI.00442-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sahu PK, Iyer PS, Gaikwad MB, Talreja SC, Pardesi KR, Chopade BA. 2012. An MFS transporter-like ORF from MDR Acinetobacter baumannii AIIMS 7 is associated with adherence and biofilm formation on biotic/abiotic surface. Int J Microbiol 2012:490647. doi: 10.1155/2012/490647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Clemmer KM, Bonomo RA, Rather PN. 2011. Genetic analysis of surface motility in Acinetobacter baumannii. Microbiology 157:2534–2544. doi: 10.1099/mic.0.049791-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Harding CM, Pulido MR, Di Venanzio G, Kinsella RL, Webb AI, Scott NE, Pachón J, Feldman MF. 2017. Pathogenic Acinetobacter species have a functional type I secretion system and contact-dependent inhibition systems. J Biol Chem 292:9075–9087. doi: 10.1074/jbc.M117.781575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.De Gregorio E, Del Franco M, Martinucci M, Roscetto E, Zarrilli R, Di Nocera PP. 2015. Biofilm-associated proteins: news from Acinetobacter. BMC Genomics 16:933. doi: 10.1186/s12864-015-2136-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Weidensdorfer M, Ishikawa M, Hori K, Linke D, Djahanschiri B, Iruegas R, Ebersberger I, Riedel-Christ S, Enders G, Leukert L, Kraiczy P, Rothweiler F, Cinatl J, Berger J, Hipp K, Kempf VAJ, Göttig S. 2019. The Acinetobacter trimeric autotransporter adhesin Ata controls key virulence traits of Acinetobacter baumannii. Virulence 10:68–81. doi: 10.1080/21505594.2018.1558693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eijkelkamp BA, Stroeher UH, Hassan KA, Paulsen IT, Brown MH. 2014. Comparative analysis of surface-exposed virulence factors of Acinetobacter baumannii. BMC Genomics 15:1020. doi: 10.1186/1471-2164-15-1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Scott NE, Kinsella RL, Edwards AV, Larsen MR, Dutta S, Saba J, Foster LJ, Feldman MF. 2014. Diversity within the O-linked protein glycosylation systems of Acinetobacter species. Mol Cell Proteomics 13:2354–2370. doi: 10.1074/mcp.M114.038315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tilley D, Law R, Warren S, Samis JA, Kumar A. 2014. CpaA a novel protease from Acinetobacter baumannii clinical isolates deregulates blood coagulation. FEMS Microbiol Lett 356:53–61. doi: 10.1111/1574-6968.12496. [DOI] [PubMed] [Google Scholar]
- 67.Waack U, Warnock M, Yee A, Huttinger Z, Smith S, Kumar A, Deroux A, Ginsburg D, Mobley HLT, Lawrence DA, Sandkvist M. 2018. CpaA is a glycan-specific adamalysin-like protease secreted by Acinetobacter baumannii that inactivates coagulation factor XII. mBio 9:e01606-18. doi: 10.1128/mBio.01606-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Harding CM, Kinsella RL, Palmer LD, Skaar EP, Feldman MF. 2016. Medically relevant Acinetobacter species require a type II secretion system and specific membrane-associated chaperones for the export of multiple substrates and full virulence. PLoS Pathog 12:e1005391. doi: 10.1371/journal.ppat.1005391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Harding CM, Hennon SW, Feldman MF. 2018. Uncovering the mechanisms of Acinetobacter baumannii virulence. Nat Rev Microbiol 16:91–102. doi: 10.1038/nrmicro.2017.148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hu D, Liu B, Dijkshoorn L, Wang L, Reeves PR. 2013. Diversity in the major polysaccharide antigen of Acinetobacter baumannii assessed by DNA sequencing, and development of a molecular serotyping scheme. PLoS One 8:e70329. doi: 10.1371/journal.pone.0070329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kenyon JJ, Speciale I, Hall RM, De Castro C. 2016. Structure of repeating unit of the capsular polysaccharide from Acinetobacter baumannii D78 and assignment of the K4 gene cluster. Carbohydr Res 434:12–17. doi: 10.1016/j.carres.2016.07.016. [DOI] [PubMed] [Google Scholar]
- 72.Russo TA, Luke NR, Beanan JM, Olson R, Sauberan SL, MacDonald U, Schultz LW, Umland TC, Campagnari AA. 2010. The K1 capsular polysaccharide of Acinetobacter baumannii strain 307-0294 is a major virulence factor. Infect Immun 78:3993–4000. doi: 10.1128/IAI.00366-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Kenyon JJ, Nigro SJ, Hall RM. 2014. Variation in the OC locus of Acinetobacter baumannii genomes predicts extensive structural diversity in the lipooligosaccharide. PLoS One 9:e107833. doi: 10.1371/journal.pone.0107833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Farrugia DN, Elbourne LD, Mabbutt BC, Paulsen IT. 2015. A novel family of integrases associated with prophages and genomic islands integrated within the tRNA-dihydrouridine synthase A (dusA) gene. Nucleic Acids Res 43:4547–4557. doi: 10.1093/nar/gkv337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Al-Jabri Z, Zamudio R, Horvath-Papp E, Ralph J, Al-Muharrami Z, Rajakumar K, Oggioni M. 2018. Integrase-controlled excision of metal-resistance genomic islands in Acinetobacter baumannii. Genes (Basel) 9:366. doi: 10.3390/genes9070366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Boyd E, Barkay T. 2012. The mercury resistance operon: from an origin in a geothermal environment to an efficient detoxification machine. Front Microbiol 3:349. doi: 10.3389/fmicb.2012.00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Di Capua C, Bortolotti A, Farías ME, Cortez N. 2011. UV-resistant Acinetobacter sp. isolates from Andean wetlands display high catalase activity. FEMS Microbiol Lett 317:181–189. doi: 10.1111/j.1574-6968.2011.02231.x. [DOI] [PubMed] [Google Scholar]
- 78.Scholz A, Stahl J, de Berardinis V, Müller V, Averhoff B. 2016. Osmotic stress response in Acinetobacter baylyi: identification of a glycine-betaine biosynthesis pathway and regulation of osmoadaptive choline uptake and glycine-betaine synthesis through a choline-responsive BetI repressor. Environ Microbiol Rep 8:316–322. doi: 10.1111/1758-2229.12382. [DOI] [PubMed] [Google Scholar]
- 79.Franceschi VR, Krokene P, Christiansen E, Krekling T. 2005. Anatomical and chemical defenses of conifer bark against bark beetles and other pests. New Phytol 167:353–375. doi: 10.1111/j.1469-8137.2005.01436.x. [DOI] [PubMed] [Google Scholar]
- 80.Graça J. 2015. Suberin: the biopolyester at the frontier of plants. Front Chem 3:62. doi: 10.3389/fchem.2015.00062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Widhalm JR, Dudareva N. 2015. A familiar ring to it: biosynthesis of plant benzoic acids. Mol Plant 8:83–97. doi: 10.1016/j.molp.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 82.Cerqueira GM, Kostoulias X, Khoo C, Aibinu I, Qu Y, Traven A, Peleg AY. 2014. A global virulence regulator in Acinetobacter baumannii and its control of the phenylacetic acid catabolic pathway. J Infect Dis 210:46–55. doi: 10.1093/infdis/jiu024. [DOI] [PubMed] [Google Scholar]
- 83.Bhuiyan MS, Ellett F, Murray GL, Kostoulias X, Cerqueira GM, Schulze KE, Mahamad Maifiah MH, Li J, Creek DJ, Lieschke GJ, Peleg AY. 2016. Acinetobacter baumannii phenylacetic acid metabolism influences infection outcome through a direct effect on neutrophil chemotaxis. Proc Natl Acad Sci U S A 113:9599–9604. doi: 10.1073/pnas.1523116113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Somers E, Ptacek D, Gysegom P, Srinivasan M, Vanderleyden J. 2005. Azospirillum brasilense produces the auxin-like phenylacetic acid by using the key enzyme for indole-3-acetic acid biosynthesis. Appl Environ Microbiol 71:1803–1810. doi: 10.1128/AEM.71.4.1803-1810.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Förster-Fromme K, Höschle B, Mack C, Bott M, Armbruster W, Jendrossek D. 2006. Identification of genes and proteins necessary for catabolism of acyclic terpenes and leucine/isovalerate in Pseudomonas aeruginosa. Appl Environ Microbiol 72:4819–4828. doi: 10.1128/AEM.00853-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Schneider BL, Kiupakis AK, Reitzer LJ. 1998. Arginine catabolism and the arginine succinyltransferase pathway in Escherichia coli. J Bacteriol 180:4278–4286. doi: 10.1128/JB.180.16.4278-4286.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Vander Wauven C, Jann A, Haas D, Leisinger T, Stalon V. 1988. N2-succinylornithine in ornithine catabolism of Pseudomonas aeruginosa. Arch Microbiol 150:400–404. doi: 10.1007/BF00408314. [DOI] [PubMed] [Google Scholar]
- 88.Jones CL, Clancy M, Honnold C, Singh S, Snesrud E, Onmus-Leone F, McGann P, Ong AC, Kwak Y, Waterman P, Zurawski DV, Clifford RJ, Lesho E. 2015. Fatal outbreak of an emerging clone of extensively drug-resistant Acinetobacter baumannii with enhanced virulence. Clin Infect Dis 61:145–154. doi: 10.1093/cid/civ225. [DOI] [PubMed] [Google Scholar]
- 89.Kim J, Lee J-Y, Lee H, Choi JY, Kim DH, Wi YM, Peck KR, Ko KS. 2017. Microbiological features and clinical impact of the type VI secretion system (T6SS) in Acinetobacter baumannii isolates causing bacteremia. Virulence 8:1378–1389. doi: 10.1080/21505594.2017.1323164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Snitkin ES, Zelazny AM, Montero CI, Stock F, Mijares L, NISC Comparative Sequence Program, Murray PR, Segre JA. 2011. Genome-wide recombination drives diversification of epidemic strains of Acinetobacter baumannii. Proc Natl Acad Sci U S A 108:13758–13763. doi: 10.1073/pnas.1104404108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Repizo GD. 2017. Prevalence of Acinetobacter baumannii strains expressing the Type 6 secretion system in patients with bacteremia. Virulence 8:1099–1101. doi: 10.1080/21505594.2017.1346768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Waterfield NR, Wren BW, French-Constant RH. 2004. Invertebrates as a source of emerging human pathogens. Nat Rev Microbiol 2:833–841. doi: 10.1038/nrmicro1008. [DOI] [PubMed] [Google Scholar]
- 93.Latorre A, Manzano-Marín A. 2017. Dissecting genome reduction and trait loss in insect endosymbionts. Ann N Y Acad Sci 1389:52–75. doi: 10.1111/nyas.13222. [DOI] [PubMed] [Google Scholar]
- 94.Sartorio MG, Repizo GD, Cortez N. 2020. Catalases of the polyextremophylic Andean isolate Acinetobacter sp. Ver 3 confer adaptive response to H2O2 and UV radiation. FEBS J doi: 10.1111/febs.15244. [DOI] [PubMed] [Google Scholar]
- 95.Qiu H, KuoLee R, Harris G, Van Rooijen N, Patel GB, Chen W. 2012. Role of macrophages in early host resistance to respiratory Acinetobacter baumannii infection. PLoS One 7:e40019. doi: 10.1371/journal.pone.0040019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sato Y, Unno Y, Miyazaki C, Ubagai T, Ono Y. 2019. Multidrug-resistant Acinetobacter baumannii resists reactive oxygen species and survives in macrophages. Sci Rep 9:17462. doi: 10.1038/s41598-019-53846-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Mussi MA, Relling VM, Limansky AS, Viale AM. 2007. CarO, an Acinetobacter baumannii outer membrane protein involved in carbapenem resistance, is essential for l-ornithine uptake. FEBS Lett 581:5573–5578. doi: 10.1016/j.febslet.2007.10.063. [DOI] [PubMed] [Google Scholar]
- 98.Holloway BW. 1969. Genetics of Pseudomonas. Bacteriol Rev 33:419–443. doi: 10.1128/MMBR.33.3.419-443.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chin C-S, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O’Malley R, Figueroa-Balderas R, Morales-Cruz A, Cramer GR, Delledonne M, Luo C, Ecker JR, Cantu D, Rank DR, Schatz MC. 2016. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13:1050–1054. doi: 10.1038/nmeth.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nurk S, Bankevich A, Antipov D, Gurevich AA, Korobeynikov A, Lapidus A, Prjibelski AD, Pyshkin A, Sirotkin A, Sirotkin Y, Stepanauskas R, Clingenpeel SR, Woyke T, McLean JS, Lasken R, Tesler G, Alekseyev MA, Pevzner PA. 2013. Assembling single-cell genomes and mini-metagenomes from chimeric MDA products. J Comput Biol 20:714–737. doi: 10.1089/cmb.2013.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Gao F, Zhang C-T. 2008. Ori-Finder: a web-based system for finding oriCs in unannotated bacterial genomes. BMC Bioinformatics 9:79. doi: 10.1186/1471-2105-9-79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Siguier P, Perochon J, Lestrade L, Mahillon J, Chandler M. 2006. ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res 34:D32–D36. doi: 10.1093/nar/gkj014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhou Y, Liang Y, Lynch KH, Dennis JJ, Wishart DS. 2011. PHAST: a fast phage search tool. Nucleic Acids Res 39:W347–W352. doi: 10.1093/nar/gkr485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Arndt D, Grant J, Marcu A, Sajed T, Pon A, Liang Y, Wishart DS. 2016. PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res 44:W16–W21. doi: 10.1093/nar/gkw387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, Lund O, Aarestrup FM, Larsen MV. 2012. Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67:2640–2644. doi: 10.1093/jac/dks261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhao Y, Wu J, Yang J, Sun S, Xiao J, Yu J. 2012. PGAP: pan-genomes analysis pipeline. Bioinformatics 28:416–418. doi: 10.1093/bioinformatics/btr655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. 2007. ClustalW and ClustalX version 2. Bioinformatics 23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]
- 109.Talavera G, Castresana J. 2007. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst Biol 56:564–577. doi: 10.1080/10635150701472164. [DOI] [PubMed] [Google Scholar]
- 110.Borowiec ML. 2016. AMAS: a fast tool for alignment manipulation and computing of summary statistics. PeerJ 4:e1660. doi: 10.7717/peerj.1660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Espariz M, Zuljan FA, Esteban L, Magni C. 2016. Taxonomic identity resolution of highly phylogenetically related strains and selection of phylogenetic markers by using genome-scale methods: the Bacillus pumilus group case. PLoS One 11:e0163098. doi: 10.1371/journal.pone.0163098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Jolley KA, Maiden MC. 2010. BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Clinical and Laboratory Standards Institute. 2016. Performance standards for antimicrobial susceptibility testing. Document M100S, 26th ed. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 115.Stiernagle T. 11 February 2006. Maintenance of C. elegans. In The C. elegans Research Community, WormBook (ed), WormBook.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The final assembled NCIMB8209 chromosome was annotated using the pipeline available at NCBI and deposited in DDBJ/ENA/GenBank under accession number CP028138. In turn, the plasmid sequences derived from the above sequencing data were deposited in the GenBank nucleotide sequence database under accession number CP028139.