

Abstract
The aim of this paper is to summarize ultrastructural evidence for glutamatergic dysregulation in several linked regions in postmortem schizophrenia brain. Following a brief summary of glutamate circuitry and how synapses are identified at the electron microscopic (EM) level, we will review EM pathology in the cortex and basal ganglia. We will include the effects of antipsychotic drugs and the relation of treatment response. We will discuss how these findings support or confirm other postmortem findings as well as imaging results. Briefly, synaptic and mitochondrial density in anterior cingulate cortex was decreased in schizophrenia, versus normal controls (NCs), in a selective layer specific pattern. In dorsal striatum, increases in excitatory synaptic density were detected in caudate matrix, a compartment associated with cognitive and motor function, and in the putamen patches, a region associated with limbic function and in the core of the nucleus accumbens. Patients who were treatment resistant or untreated had significantly elevated numbers of excitatory synapses in limbic striatal areas in comparison to NCs and responders. Protein levels of vGLUT2, found in subcortical glutamatergic neurons, were increased in the nucleus accumbens in schizophrenia. At the EM level, schizophrenia subjects had an increase in density of excitatory synapses in several areas of the basal ganglia. In the substantia nigra, the protein levels of vGLUT2 were elevated in untreated patients compared to NCs. The density of inhibitory synapses was decreased in schizophrenia versus NCs. In schizophrenia, glutamatergic synapses are differentially affected depending on the brain region, treatment status, and treatment response.
1. Introduction
Evidence from in vivo imaging, postmortem studies and animal models of schizophrenia implicate the glutamatergic system in schizophrenia (Coyle, 2006; Goff and Coyle, 2001; Javitt, 2004; Krystal, 2008). The aim of this paper is to provide ultrastructural evidence of glutamatergic dysregulation in schizophrenia and discuss how these findings relate to other postmortem findings and imaging studies. It is noteworthy that quantitative ultrastructural studies of postmortem brain in schizophrenia are rarely conducted outside our lab (Aganova and Uranova, 1992; Uranova and Levitt, 1987; Kolomeets and Uranova, 1999; Kolomeets et al.,2005, 2007). Although cohort size is small in these ultrastructural studies, the results are consistent with those of other studies using methodologies that permit larger cohorts. Psychotomimetic agents, acting on glutamate receptors, produce psychosis in healthy controls and exacerbate psychosis in people with schizophrenia (Itil et al., 1967; Lahti et al., 1995). Glutamate levels in the basal ganglia are elevated in both in vivo and postmortem studies of schizophrenia. Moreover, in experimental animals, psychotomimetics increase striatal neuronal activity, which is reversed by antipsychotic drugs (APDs) (White et al., 1995).
1.1. Glutamate circuitry in the basal ganglia and connecting structures
A simplified version of pertinent connections of the basal ganglia is shown in Figure 1A. Diverse regions of the cerebral cortex including limbic areas (such as orbitofrontal and anterior cingulate cortices), associative and cognitive areas (such as dorsolateral prefrontal cortex, DLPFC), and motor and premotor cortex project to the caudate nucleus and putamen (Alexander et al., 1986; Chikama et al., 1997; Eblen and Graybiel, 1995; Goldman-Rakic, 1994; Goldman-Rakic and Selemon, 1986; Haber et al., 1995, 2000; Kunishio and Haber, 1994; Parent and Hazrati, 1995; Smith et al., 1998) in a topographical pattern (Haber, 2000). The vast majority of striatal synapses are glutamatergic and form with the dendritic spines of medium spiny neurons (the major striatal cell type) (Kemp and Powell, 1971). The outputs of the striatum and substantia nigra pars reticulata are all GABAergic. However, the ventral tegmental area contains a population of neurons that are glutamatergic (Root et al., 2016). The subthalamic nucleus (Benarroch 2008) and the pedunculopontine nucleus (Charara et al. 1996) send glutamatergic projections to the substantia nigra/ ventral tegmental area.
Figure 1.

A) Simplified drawing illustrating glutamatergic connectivity between cortex, basal ganglia and thalamus. The cortex receives glutamatergic inputs from the thalamus; illustrated here is the connection between medial dorsal thalamus and layer III of granular cortex. Glutamatergic neurons in the cortex project to the caudate nucleus, the putamen and the nucleus accumbens. Layers III, V,VI project to the dorsal striatum (caudate, putamen). Specifically, layer III projects to the matrix and layers V,VI project to the patches. The subthalamic nucleus (STN) and the pedunculopontine nucleus (PPN) send glutamate projections to the substantia nigra, ventral tegmental area. B) Schematic drawing illustrating synaptic morphology. Several spines emerge from the dendrite and one is shown enlarged. Synaptic types are defined by the morphology of the postsynaptic density (PSD) and their target (dendrite, spine, axon initial segment, or soma). Type I synapses are mostly excitatory, and have been classically called “asymmetric”, based on a thick PSD in comparison to a nonexistent density on the presynaptic side. Type II synapses are mostly inhibitory, and have been classically called “symmetric” based on a thin PSD more comparable in size to the non-existent density on the presynaptic side. Axospinous synapses are depicted. AT, axon terminal.
1.2. Electron microscopy in human postmortem brain
Electron microscopy in postmortem human brain is difficult due to issues with obtaining tissue with very short postmortem intervals. Our data presented in this review have been previously published and details of each cohort of subjects are found in our articles (Kung et al., 1998; Roberts and Knickman, 2002; Roberts et al., 2005a,b, 2012, 2015; McCollum et al., 2015; Mabry et al., 2019. Human postmortem brain tissue was obtained from the Maryland and Alabama Brain Collections, with family permission and with IRB approved protocols. Briefly, tissue was dissected from the fresh brain and stored until used in a fixative solution of 1% glutaraldehyde and 4% paraformaldehyde in 0.1M phosphate buffer (pH 7.4). Subjects with schizophrenia were matched to the control subjects for age, gender, postmortem interval and race when possible. Controls had no history of central nervous system or neurological diseases. Antipsychotic drug treatment, duration of illness, predominant symptomology, treatment response, and other medical details were obtained from hospital charts, autopsy reports and family interviews. The diagnosis of schizophrenia was made by two research psychiatrists according to the DSM criteria (DSM-III-DSM-V) at the time of the diagnosis.
1.3. Synaptic ultrastructure
Synaptic types are categorized by the morphology of the postsynaptic density and their target (dendrite, spine, axon initial segment, or soma) (Figure 1B). Synapses are defined by having three criteria: an axon terminal with vesicles at the synapse, a postsynaptic density, and parallel pre and postsynaptic membranes (Figure 1B). Traditionally, excitatory and inhibitory synapses can be identified by the morphology of the postsynaptic density (Gray, 1969). Synapses with thick postsynaptic densities are called “asymmetric” and usually identify excitatory synapses, while synapses with thin postsynaptic densities are called “symmetric” and identify inhibitory synapses. These terms assume no presynaptic density. The vast majority of glutamatergic axon terminals form synapses with dendritic spines (Harris and Kater, 1994; Arellano et al., 2007). Glutamate receptors and other molecules needed for glutamate function (Krueger-Burg et al. 2017) are localized to synapses with thick postsynaptic densities. For example, PSD95 is a scaffolding protein located in the postsynaptic density of excitatory synapses (Cho et al., 1992; Sheng & Hoogenraad, 2007). Gephyrin is a postsynaptic scaffolding protein found exclusively at GABAergic and glycinergic synapses (Craig et al., 1996).
Tract tracing, alone or in combination with immunohistochemistry, is used in animal models to identify projections and circuitry within the brain. Thus, particular projections can be identified by the combination of the postsynaptic target and symmetry of the synapse. For example, parvalbumin containing chandelier cells form symmetric synapses on the axon initial segment of cortical pyramidal cells (Lewis et al., 2002). Glutamatergic pyramidal cells from layer V cortex project to the striatum and form asymmetric axospinous synapses on medium spiny neurons (Kemp and Powell, 1971). Anatomical studies in human brain use the results of circuitry from the animal literature to make educated predictions as to the origin of synapses based on morphology.
1.3.1. Methods for counting synapses
Synapses are counted using two or three-dimensional techniques. Two dimensional techniques measure the density of synapses per unit area and are obtained from a method called simple profile counting. Three-dimensional reconstruction or stereological methods measure the density of synapses per unit volume (Geinisman et al., 1996). Stereological techniques were used to count synapses in the cortex (Figure 2A) and striatum (Figures 3&4), while simple profile counts were used in the substantia nigra. Examples of synapses are shown for each region in corresponding figures.
Figure 2.

Anterior Cingulate Cortex. Electron micrographs and graphs are modified from Roberts et al., 2015 as indicated. A,A’) Representative working images (magnification=15,000) used for synapse counting. Each is a montage stitched together from eight micrographs; A and A’ are taken from serial sections. Arrows indicate synapses (identified at higher magnification). Modified from Figure 2. B) An excitatory axospinous synapse (arrow) forms on a spine (sp) emerging from a dendrite (den) from a subject with schizophrenia. m, mitochondria. C) An inhibitory axospinous synapse (arrow) forms on a spine from a subject with schizophrenia. Modified from Fig 4D. D) A dendrite is postsynaptic to two excitatory synapses from a NC case. Modified from Fig 4B. E) Representative electron micrograph of a pyramidal cell, outlined in black, from a subject with schizophrenia. Modified from Fig 8B. F) Total synaptic density is decreased in layers III-VI combined in schizophrenia, due to a selective loss of axospinous synapses. Modified from Fig 5A. G) The number of mitochondria per neuron is decreased in layers III-VI combined in schizophrenia, due to a selective loss in the deep layers. Modified from Fig 3B. H) Mitochondria are reduced in number in axon terminals forming asymmetric (excitatory) synapses in layer III and in axon terminals forming symmetric (inhibitory) synapses in layers V,VI. Modified from Fig7B. Scale bars: A, E=5μm; B-D=500nm. Significance: *, p<0.05; **, p<0.01; ***, p<0.005.
Figure 3.

Caudate Nucleus and Putamen. A) Synaptic density is shown for the caudate nucleus and putamen in normal controls (NC) and subjects with schizophrenia (SZ). All synapses combined (total), asymmetric axospinous (AS) and all asymmetric synapses (Asym) are increased in density in the caudate nucleus in schizophrenia. The putamen shows a similar pattern, but without reaching statistical significance. Modified from Figures 4A and 5A, Roberts et al., 2005a. B) A light microscopic image of a calbindin immunolabeled section from the striatum. Calbindin immunolabeling, which is much more robust in the matrix (M) than in the patches (P), was used to distinguish the two compartments. The trapezoids indicate regions within each compartment that were sampled for synaptic density. Small arrows indicate immunolabeled neurons. Modified from Figure 2, Roberts and Knickman, 2002. C) Total synaptic density in subjects with schizophrenia was significantly higher in the caudate matrix (CM all) and the putamen patches (PP all) than in controls. The density of asymmetric axospinous (AS) synapses was significantly higher in the putamen patches (PP AS) in schizophrenia; the same pattern, albeit only at a trend level, was found in the caudate matrix (CM AS). Modified from Figures 3A and 4A from Roberts et al., 2005b. D) The schizophrenia cohort was subdivided into treatment responders and treatment resistant/or off APDs. In the striatal patches, the treatment resistant/off APD cases had higher synaptic densities of total synapses, asymmetric axospinous synapses and all asymmetric synapses than controls and treatment responders; treatment responders had a higher density of asymmetric axodendritic synapses than controls and treatment resistant/off APD cases. Total Modified from Figure 6, Roberts et al., 2012. Significance: *, p<0.05; **, p<0.01.
Figure 4.

Nucleus Accumbens. A) Western blot analysis of vGLUT2 in control (NC), schizophrenia as a group (SZ) and divided by APD status at the time of death: on-drug (SZ-ON), and off-drug (SZ-OFF). B) Graphs showing optical density of vGLUT2 where the schizophrenia group has significantly higher protein levels of vGLUT2; A and B were modified from Figure 1C in McCollum and Roberts 2015. Electron micrographs and synapse quantification are modified from McCollum et al., 2015 as indicated. C) An axon terminal (AT) forms an asymmetric synapse (arrow) with a spine (sp); modified from Figure 1 D). An AT forms an asymmetric synapse (arrow) with a dendrite (DEN). E) A spine (SP) receives convergent input from axon terminals (AT) making asymmetric (black arrows) and symmetric (white arrow) synapses; modified from Figure 1B. F) An axon terminal (AT) forms a symmetric synapse (arrow) with a dendrite (DEN); modified from Figure 1C. G-I) Quantification of synaptic density in the core and shell. G,H) Density of all synapses (Total), total asymmetric (Total Asym) and total symmetric (Total Sym) synapses, total axospinous (Total Sp) and total axodendritic (Total Den) synapses. I-J) Density of synapse types: asymmetric axospinous (AS), asymmetric axodendritic (AD), symmetric axospinous (SS), and symmetric axodendritic (SD). K,L) Serial sections of an axon terminal forming an elaborate asymmetric multi-perforated synapse with a spine. Scale bars (B-D, K,L) = 500 nm.
2. Synaptic abnormalities in schizophrenia
2.1. Anterior Cingulate Cortex
The anterior cingulate cortex (ACC) is part of prefrontal cortex and is composed of distinct anatomical subregions, each with different functional properties (Peterson et al., 1999; Vogt et al., 1992). Human imaging studies indicate that the ACC is involved in mediating attention, executive functions, cognitive tasks (Koski and Petrides, 2001), response conflict (Barch et al., 2001; Kerns et al., 2004), response inhibition, error commission (Braver et al., 2001; Mathalon et al., 2002), and emotional processing (Bush et al., 2000).
In an ultrastructural study of the ACC (Figure 2A–E), we found that all synapses combined were decreased in density by 28% in the schizophrenia cases compared to the NCs (Figure 2F) (Roberts et al., 2015). Asymmetric axospinous synapses (Figure 2B) were reduced in density by approximately 30% in the patients vs. NCs (Figure 2F). Symmetric axospinous synapses (Figure 2C) were reduced in density by 46% in the schizophrenia group vs. NCs. Axodendritic synapses (Figure 2D) showed no difference in density between the schizophrenia group and NCs. In the combined layers of the ACC, the number of mitochondria per neuronal somata (Figure 2E) in the schizophrenia cases was decreased by 43% in comparison to NCs (Figure 2G). This decrease was due to a selective loss of mitochondria from neurons in layers V/VI. In layer III, the density of mitochondria in axon terminals forming any type of synapse was lower in schizophrenia by 34% as compared to NCs (Figure 2H). This finding was due to a selective loss of mitochondria in terminals forming asymmetric axospinous synapses. In layer V,VI, the density of mitochondria in terminals forming symmetric synapses, both with spines and dendrites was lower in the schizophrenia group than NCs (Figure 2H).
Our findings showed that the ACC in schizophrenia has a decrease in number of excitatory synaptic connections, possibly arising from multiple sources, which may have effects on several downstream pathways (Roberts et al., 2015). Analysis of the vesicular glutamate transmitters (vGLUTs) yields important information about the location of the cell bodies of origin (Bellocchio et al., 2000; Vigneault et al., 2015). The amount of the vGLUTs is directly related to quantal release of glutamate and thus is a good measure of glutamate levels (Wilson et al., 2005). vGLUT1 mRNA is found in the cell bodies of origin of cortical glutamatergic neurons, and the protein is found in the terminals, while vGLUT2 mRNA is found in the cell bodies of origin of subcortical glutamatergic neurons, and the protein is found in the terminals (Vigneault et al., 2015). In the ACC, protein analysis of the vGLUTs (Oni-Orisan et al., 2007) demonstrated that levels of vGLUT1, but not vGLUT2, were reduced in schizophrenia, and not a cause of medication. Taken together, these data suggest that cortical-cortical connections are decreased in the ACC in schizophrenia.
Pyramidal neurons in layers V,VI project to the striatum, brainstem, or thalamus (Goldman and Nauta, 1977). The reduction in number of mitochondria in neurons in these layers suggests that neurons that project to these subcortical regions may have a compromised metabolism. Another downstream target is the hippocampus, where decreased numbers of excitatory axospinous synapses have been reported in schizophrenia cases (Kolomeets et al., 2005, 2007). Dopaminergic terminals form symmetric axospinous synapses (Kung et al., 1998), and this type of synapse is reduced in density in prefrontal cortex in schizophrenia, possibly reflecting a loss of dopamine input. In support of this finding are previous studies showing a decrease in density of dopaminergic fibers in deep cortical layers in the ACC (Benes et al., 1997), as well as other regions of prefrontal cortex (Akil et al., 1999).
Although a number of magnetic resonance spectroscopy (MRS) studies of the ACC have been conducted in schizophrenia, inconsistent results have been reported (Merritt et al., 2016). Such varying results are likely to be due to differences in stage of illness and medication status, magnet strength, sequence acquisitions and data post processing. However, consistent with postmortem findings, two recent studies conducted in first episode psychosis subjects using ultra-high field magnets [7 Tesla (T)] to obtain better spectral resolution reported a decrease in ACC glutamate (Reid et al., 2019, Wang et al., 2019). Fewer studies have evaluated ACC GABA levels in schizophrenia (Egerton et al., 2017). Wang and colleagues (Wang et al., 2019) reported decreases in both ACC glutamate and GABA levels in first episode psychosis subjects.
MRS studies in schizophrenia have also consistently reported decreased N-acetyl-aspartate levels in the ACC (Jessen et al., 2013; Reid et al., 2010, 2019). N-acetyl-aspartate is an amino acid that is primarily synthesized in neuronal mitochondria (Bates et al., 1996) and is metabolized in oligodendrocytes to acetate, a precursor of myelin lipid synthesis (Chakraborty et al., 2001). Thus, the amount of NAA detected in imaging studies may reflect neuronal integrity, the number of axon terminals and synapses, myelin integrity, and/or mitochondrial function (Bates et al., 1996; Chakraborty et al., 2001; Ledeen et al., 2006; Lentz et al., 2005; Moffett et al., 2007; Stork and Renshaw, 2005). Therefore, the results of the present study and those of Aganova and Uranova (1992) support that any of these mechanisms could underlie decreased NAA as seen in in vivo imaging studies. The decrease in density of axospinous synapses together with a lower proportion of remaining terminals that contain mitochondria and lower somatic mitochondrial counts, suggests a decrease in cortical synaptic efficiency. Overall, our changes suggest alterations in multiple cortical connections that may impact on cognitive functions subserved by the ACC. Cognitive tasks of inhibitory control are known to activate a network of regions that includes the ACC as its core region (Kouneiher et al., 2009). Again, consistent with postmortem findings, functional MRI (fMRI) studies in schizophrenia have repeatedly demonstrated altered ACC functional activation during cognitive control tasks (Kerns et al., 2005; Weiss et al., 2007; Minzenberg et al., 2009). In addition, an abnormal relationship between ACC glutamate and the blood oxygen level dependent signal have been reported in schizophrenia (Flakenberg et al., 2014; Cadena et al., 2018; Overbeek et al., 2019).
MRS can also be used to measure neuromatabolites linked to mitochrondial function and brain bioenergetics, such as glutathione, an antioxidant, and lactate, a byproduct of glycosis. In schizophrenia, reduced ACC glutathione has been reported by several (Kumar et al., 2018; Das et al., 2019), including one study at 7 T (Wang et al., 2019) suggesting a decreased protective buffer from oxidative stress. Interestingly, elevated lactate levels have been reported in several brain areas in schizophrenia (Hagihara et al., 2017; Sullivan et al., 2019), including the ACC (Rowland et al., 2016). Increased lactate levels, as well as decreased pH, are features of schizophrenia as well as other psychiatric illnesses (Hagihara et al., 2017). Importantly, these findings are also present in animal models of several psychiatric disorders and are thought to be an undelying pathology of illness rather than an artifact of postmortem interval (Hagihara et al., 2017; Sullivan et al., 2019). The finding of elevated lactate in schizophrenia suggests that, in the presence of impaired mitochondrial function, a shift to greater glycosis results in the accumulation of lactate. In addition, phosphorus MRS (31P MRS) allows the quantification of phosphorus metabolites, including energy related metabolites, such as adenosine triphosphate (ATP), phosphocreatine (PCr), inorganic phosphate (Pi), and nicotinamide adenine dinucleotide (NAD) signals. In the respiratory chain of the mitochondria, ATP is formed from adenosine diphosphate (ADP) and Pi, a process coupled to the reaction transferring the energy moiety of ATP to Cr to generate PCr. The reversible reaction can then generate ATP from ADP and PCr. Not only can these metabolites be measured, but also the rates of these chemical exchanges can be estimated using 31P MRS. In schizophrenia, a number of studies have reported abnormalities of these metabolites, as well as altered chemical exchange rates (Yuksel et al., 2015). Together, these imaging studies strongly implicate impaired mitochondrial function in schizophrenia.
2.2. Caudate nucleus and putamen
The dorsal striatum, including the caudate nucleus and putamen, processes motor, cognitive and limbic functions. It is also the region of the brain with the highest density of dopamine D2 receptors, the target of antipsychotic drugs (Carlsson, 1988, 2006; Creese et al., 1976). The striatum receives robust projections from most regions of cortex, including the ACC (Kunishio and Haber, 1994). The majority of neurons in the striatum are medium spiny neurons and they receive the majority of inputs onto their spines; these inputs from cortex and thalamus form glutamatergic synapses (Kemp and Powell, 1971). We found higher synaptic density in the caudate nucleus in schizophrenia with similar trends in the putamen (Figure 3A) (Roberts et al., 2005a). This increase is due to a selective increase in asymmetric synapses onto spines, suggesting the presence of more spines receiving glutamatergic synapses.
The caudate nucleus and putamen are organized in a striosomal pattern (Graybiel and Ragsdale, 1978), which contains patches (or striosomes) imbedded in a larger matrix. These two anatomically distinct striatal compartments differ from each other in several ways including neurochemical composition (Gerfen et al.,1985; Graybiel and Ragsdale, 1978; Holt et al., 1997; Prensa et al., 1999), neuronal organization (Hirsch et al., 1989; Holt et al., 1997; Kubota and Kawaguchi, 1993; Penny et al., 1988), connectivity (Gerfen, 1989, 1992; Kincaid and Wilson, 1996; Ragsdale and Graybiel, 1991), and response to antipsychotic drugs (Bubser and Deutch, 2002). Layer III glutamatergic neurons of the cortex project to the matrix, while the glutamate neurons in layers V/VI project to the patch compartments (Gerfen, 1989).
Patches and matrix are easily distinguished by calbindin immunolabeling, which is present more abundantly in the matrix (Gerfen et al., 1985) (Figure 3B). The matrix compartment in associative regions preferentially receives inputs from the dorsolateral prefrontal cortex (Eblen and Graybiel, 1995), which processes higher cognitive functions, such as working memory (Goldman-Rakic, 1999). Motor and somatosensory cortices also project to the matrix in a topographical fashion (Flaherty and Graybiel, 1993). When considering the patch and matrix separately, total synaptic density was elevated in the caudate matrix and the putamen patches, with asymmetric axospinous synapses driving this increase (Figure 3C) (Roberts et al., 2005b). Thus, it would be expected that increased glutamatergic synaptic density in the striatal matrix could impact cognition, working memory, motor, and somatosensory functions, while increased glutamatergic inputs to the patches would impact limbic function.
When considering treatment response and treatment resistance, the increase in total and asymmetric synapses in the patches was confined to the treatment resistant group (Figure 3D) (Roberts et al., 2012). Treatment responders had normal levels of synapses with the exception of an increase in asymmetric axodendritic synapses, typical of thalamic afferents (Sadikot et al., 1992a,b) in both the patch and matrix. This could be due to an increase in input of thalamic inputs or retraction of spines, but not the synapses onto them. The source of these extra axodendritic synapses could be determined by localizing vGLUT1 and vGLUT2 to identify cortical and thalamic inputs, respectively (Fremeau et al., 2001, 2004).
Although our cohort of treatment resistant and responsive patients was small, in vivo imaging studies have confirmed our results. Demjaha and colleagues (Demjaha et al., 2014) found that treatment resistance is associated with higher striatal glutamate levels and normal dopamine levels. MRS studies have shown increased glutamate in the striatum of individuals at high risk to develop schizophrenia, in patients with schizophrenia who were off medication and floridly psychotic, or drug naive at their first episode of psychosis (de la Fuente-Sandoval, 2009, 2011, 2012, 2013). Moreover, elevated levels of glutamate, located in the associative striatum in medication naive first episode patients, return to normal following antipsychotic treatment (de la Fuente-Sandoval, 2013). The associative region of the striatum is also the location of increased dopamine release in schizophrenia (Kegeles et al., 2010). These data suggest that increases in striatal glutamate predate the illness, are not a medication effect, and occur in the same region where increased dopamine release is found.
Disturbance in striatal functional connectivity is more pervasive in treatment-resistant patients (White et al., 2016). Prior to treatment, striatal functional connectivity patterns, as well as changes in striatal functional connectivity with treatment were associated with better treatment response (Cadena et al., 2019; Sarpal et al., 2015). In addition, caudate volumes measured prior to treatment were also associated with subsequent treatment response (Hutcherson et al., 2014). Thus, like in postmortem studies, imaging studies in schizophrenia found evidence of altered glutamatergic activity, as well as associations between striatal function, structure, and treatment response.
2.3. Nucleus Accumbens.
The nucleus accumbens integrates signaling from multiple regions of the brain, receiving input from areas including the prefrontal cortex, hippocampus, amygdala, thalamus, and midbrain. (Groenewegen and Trimble, 2007). Importantly, all of these areas have been associated with schizophrenia, making the nucleus accumbens a prime region for integrating multiple disrupted areas (Grace, 2000). In a recent study, we found that vGLUT2 protein levels in the nucleus accumbens were significantly elevated in the schizophrenia group (Figure 4A,B) (McCollum and Roberts, 2015). Protein levels of vGLUT2 did not differ between schizophrenia subjects on medication (SZ-ON) and schizophrenia subjects off medication (SZ-OFF) subjects. Protein levels of vGLUT1 were similar in control and schizophrenia subjects, with no difference between SZ-ON and SZ-OFF subjects (data not shown). Similar values in schizophrenia both on and off medication suggest that there is no effect of APDs. The selective increase in vGLUT2 protein implicates subcortical afferents. mRNA expression studies in other brain areas such as the temporal cortex (Uezato et al., 2009) and thalamus (Smith et al., 2001) also support an increase in glutamate expression in neurons in subcortical locations.
The nucleus accumbens is divided into a core and shell, the former being more similar to the dorsal striatum in neurochemical composition and connectivity. Using calbindin immunolabeling as a template, we were able to quantify synapses selectively in the core versus the shell (McCollum et al., 2015). Most synaptic morphologies (Figure 4C–F) were typical of dorsal striatum in both the core and shell. Within the core, there was an increased density of total synapses in the schizophrenia group (Figure 4G). This increased density was specific to asymmetric synapses, while symmetric synapses had a similar density between the two groups. The density of all axospinous synapses was increased at a trend level (p = 0.054). Further, when analyzing synapse subtypes, the increased density was found exclusively in asymmetric axospinous synapses (Figure 4I). These differences were unique to the core region; the shell had similar densities of all synaptic types between the two groups (Figure 4H–J).
Large, elaborate multi-perforated synapses (Figure 4K,L) were found in both groups with the same frequency. These enormous synapses have not been reported in other species to our knowledge or in other regions of the human brain. Increased spine volume, synapse length, and perforations are all associated with increased synaptic activity (Calverley et al., 1990; Hering and Sheng, 2001; Yuste and Bonhoeffer, 2001). Thus, these synapses could be a manifestation of the complex interconnectivity of the nucleus accumbens with many other brain regions. Since they were present in both groups, they may be a unique characteristic of the human nucleus accumbens, albeit independent of the disease state in schizophrenia.
The increase in density of asymmetric axospinous synapses indicates an increase in glutamatergic-type inputs to the core. This is consistent with our vGLUT2 findings and in the synaptic data in the dorsal striatum described above. In agreement with this finding is evidence for an increased expression of excitatory postsynaptic density proteins after ketamine stimulation and in the NAcc in postmortem schizophrenia (Kajimoto et al., 2003; Iasevoli et al., 2007). Rodent models of schizophrenia also exhibit increased indices of glutamatergic-type input in the NAcc. Rats treated with psychotomimetics have increased mRNA expression of excitatory postsynaptic density genes (Das et al., 2019) and increased dendritic spine density in the NAcc (Flores et al., 2007; Robinson and Kolb 1997, 1999), both of which are tightly correlated with the density of asymmetric axospinous synapses (Li et al., 2003). A likely source of this elevated input in schizophrenia and its functional implications are discussed below.
One of the major targets of the cortex is the striatum. Psychotomimetics increase striatal spine density (Li et al., 2003; Flores et al., 2007; Robinson & Kolb, 1996, 1997) and increase striatal neuron firing (Dietz et al., 2009; White et al., 2005), linking psychosis to excessive striatal glutamate levels and spine number. In adult rodents, intermittent chronic exposure to PCP results in elaboration of the dendritic arbor and increased dendritic spine density in striatum (Robinson & Kolb, 1997, 1999). These changes are persistent, as they remain one month after treatment termination, and appear to be specific to brain regions that receive dopamine innervation because no differences in spine density between PCP and saline-treated groups are observed elsewhere (Field et al., 2011). These agents given to animals increase striatal glutamate levels and spine density on striatal neurons, linking psychosis with increased striatal glutamatergic synapses (Flores et al., 2007). Spine number is highly coupled to glutamatergic synapse number and vice versa.
The ventral striatum/ nucleus accumbens is a region which is typically activated during task tapping into reward processing (Daniel and Pollmann, 2014), and a large body of reward studies in schizophrenia have reported abnormal engagement of this region (Chase et al., 2018; Radua et al., 2015). In schizophrenia, abnormal prediction error signal in this region was correlated with glutamate levels measured in the substantia nigra, suggesting that glutamatergic dysfunction might contribute to abnormal reward activation (White et al., 2015).
2.5. Substantia Nigra/Ventral Tegmental Area
The substantia nigra/ventral tegmental area (SN/VTA) houses one of the largest clusters of dopaminergic cells in the brain (Hökfelt et al. 1973; Fallon et al. 1978).The SN/VTA is composed of distinct anatomical subregions, each with different connections and neurotransmitter phenotypes (McRitchie et al., 1996; Haber, 2000; Gorelova et al., 2012; Root et al.,2016). Elevated striatal dopamine via the nigrostriatal pathway is associated with psychosis (see review by Howes et al. 2009). The preponderance of evidence indicates that dopamine abnormalities are pathway specific and underlie different symptoms (for review see Weinstein et al. 2017). The SN/VTA in schizophrenia displays abnormal glutamate receptor density (Mueller et al. 2004), hyperplasia of mitochondria in axon terminals (Kolomeets and Uranova 1999), myelin pathology and cytoskeletal abnormalities (Walker et al. 2017). Moreover, dopaminergic neurons have larger nucleoli, increased transcriptional and synaptic activity (Williams et al. 2013. A number of studies indicate increased dopamine activity, protein, and/or mRNA levels (Toru et al. 1988; Howes et al. 2013; Williams et al. 2013; Schoonover et al. 2017), while others do not (Ichinose et al. 1994; Perez-Costas et al. 2012; Rice et al., 2016). The reason for these discrepancies could be methodology, location of the SN sampled or differences in patient populations.
We recently examined the SN/VTA in postmortem tissue using protein analysis (Schoonover et al., 2017), immunohistochemistry and electron microscopy (Mabry et al., 2018). We found that protein levels of vGLUT2 were different in a comparison of NCs, and patients on or off APDs (Figure 5A), (Schoonover et al., 2017). Specifically vGLUT2 levels were increased in off drug patients compared to NCs. While vGLUT1 levels were not altered in the schizophrenia group as a whole, the relationship of vGLUT1 with GAD67 was opposite in in the central region (on a mediolateral axis) of NCs versus the schizophrenia group (Figure 5B) (Mabry et al., 2018). The central region of the SN projects to the central, associative region of the striatum (Haber et al. 2000; Haber 2014) and this region exhibits an increase in dopamine release in schizophrenia (Kegeles et al. 2010). Thus, it appears that there is an abnormal relationship between glutamate and GABA in schizophrenia.
Figure 5.

The Substantia Nigra. A) Sample western blots of vGLUT2 protein in controls (NC) and the schizophrenia group divided by APD status at the time of death: on-drug (SZ-on), and off-drug (SZ-OFF). Graph showing between group differences in the optical density of vGLUT2; posthoc analysis showed that the SZ-off group has significantly higher protein levels of vGLUT2 than did the NCs. Blots and graphs were modified from Figures 1A and 2E, respectively, in Schoonover et al., 2016. B) Regional analysis of correlations between vGLUT1 and GAD67 for NCs and schizophrenia patients. A two correlation coefficient test showed a significant difference between groups (two-tailed, p<0.023); modified from Figure 5B in Mabry et al., 2018. C,D) Electron micrographs from the substantia nigra. D) modified from Figure 7C in Mabry et al., 2018. C,D Scale bars=500nm. E) Graph comparing the synaptic density per 100μm2 of all synapses, asymmetric synapses and symmetric synapses; modified from Figure 8A in Mabry et al., 2018. Patients with schizophrenia had a trend toward fewer total synapses and had significantly fewer inhibitory synapses than controls.
Interestingly, we observed a wide variety of different synaptic morphologies that had postsynaptic densities that were characteristic of excitatory synapses, inhibitory synapses, or had PSDs of intermediate thickness (Figure 5C,D) (Mabry et al., 2018). Moreover, many synapses had presynaptic densities (Figure 5D). Quantification of synaptic density revealed a decrease in symmetric, or inhibitory synapses in patients, and a trend toward more synapses overall (Figure 5E). The neurochemical organization of the SN/VTA region is more complicated than other regions of the brain. Recent studies have shown that neurons in the SN/VTA contain multiple transmitters, and use more than one transmitter in a given synapse (Barker et al. 2016; Root et al. 2016), which could lead to the variability in synaptic morphologies. Immunohistochemical localization of glutamate and GABA at the electron microscopic level would be required to identify and parse out the function of the synapses with unusual morphologies. Our data shows an increase in glutamate from subcortical sources, an imbalance of excitatory and inhibitory relationships, and fewer inhibitory synapses in patients.
The investigation of the SN in schizophrenia patients using MRI and MRS spectroscopy imaging techniques has only begun recently (Bolding et al. 2013). Functional MRI reports show disturbed functional connectivity (Hadley et al 2014), which is correlated with increased dopamine synthesis capacity (Howes et al. 2013; Watanabe et al. 2014). MRS spectroscopy indicates that Glx, a combined measure of glutamate and glutamine, is elevated in schizophrenia, suggesting excessive glutamate release (White et al., 2015). Schizophrenia patients have a higher maximum signalling of neuromelanin, suggesting increased dopaminergic neuronal activity (Graham, 1779; Shibata et al. 2008; Watanabe et al. 2014). Moreover, there is a different distribution of neuromelanin density in the SN, suggesting heterogeneity in dopamine turnover within the SN. (Mabry et al., 2018). The SN and other areas of the basal ganglia display hyperactivity during cognitive tasks (Yoon et al. 2013), which is correlated with symptom severity (Yoon et al. 2014). Increased excitatory neurotransmission and/or decreased inhibitory neurotransmission in the SN could underlie these imaging findings.
3. Summary:
Glutamate release, as measured by the vGLUTs, and glutamatergic synapses, as measured by morphologically defined synapses, are differentially affected in different brain regions in schizophrenia. Overall decreases in synaptic density in the schizophrenia group in the ACC are consistent with the reduced neuropil hypothesis, which posits fewer cortical synaptic connections (Selemon and Goldman-Rakic, 1999). Fewer axospinous synapses in the ACC in schizophrenia are consistent with previously reported spine loss (Glantz and Lewis, 1999). Fewer mitochondria in specific populations of axon terminals and in the somata of neurons suggest abnormal energy levels in those structures. However, what happens in the cortex, appears to stay in the cortex, because there are multiple lines of evidence of increased glutamatergic release and density of excitatory synapses in all regions of the basal ganglia that have been studied. This evidence comes from ultrastructural studies, other postmortem methodologies and is confirmed with in vivo imaging studies.
Figure 6.
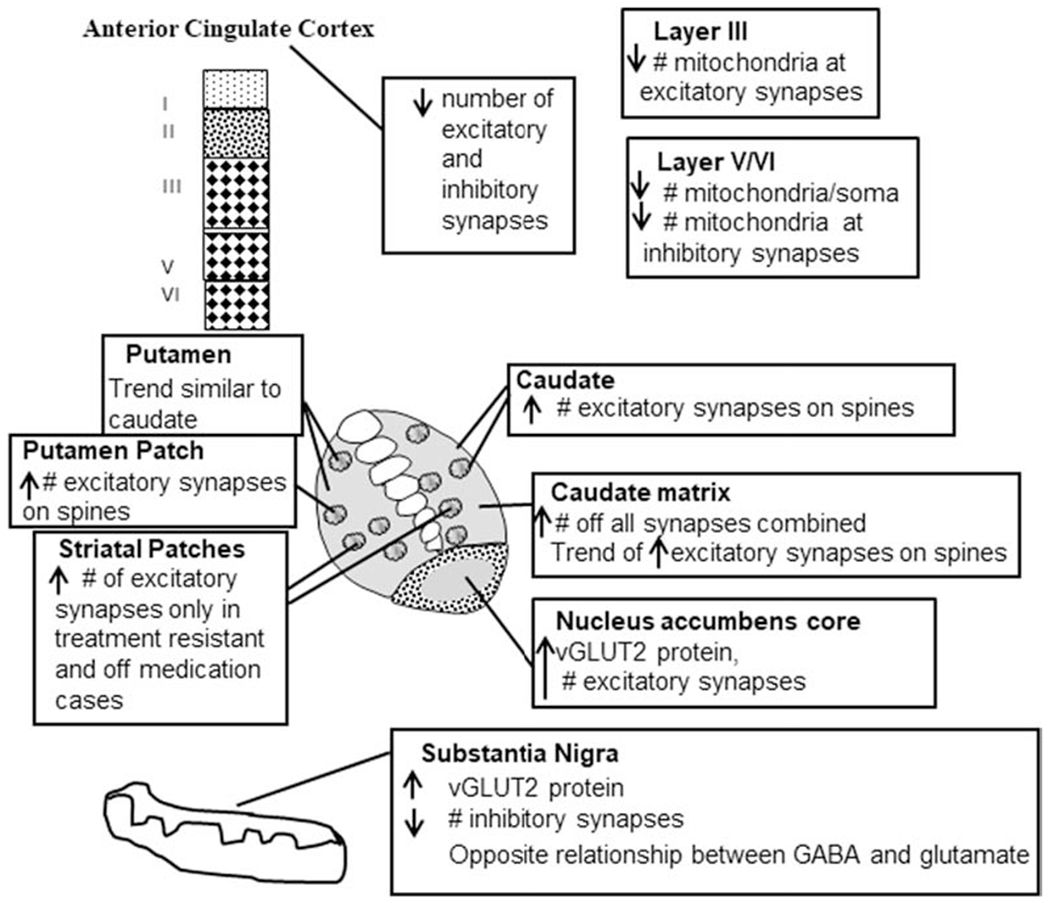
Summary diagram synthesizing the findings. Results from our ultrastructural, light microscopic and protein studies have shown very discrete changes in schizophrenia. In the anterior cingulate, decreases in excitatory and inhibitory synapses, and layer specific changes in mitochondrial density occur. In the caudate, putamen and nucleus accumbens increases in the number of excitatory synapses as well as levels of vGLUT2 were present in specific compartments. The substantia nigra showed an increase in vGLUT2 protein, a decrease in number of inhibitory synapses and an altered relationship between GABA and glutamate.
Acknowledgments
We would like to thank the staff of the Maryland and Alabama Brain Collections and the donors and their families for the brain donations.
Role of funding sources
This research was supported by R01MH66123 (RCR), R01MH60744 (RCR), R01MH081014 (ACL), F31MH098566 (LAM) and the UAB President’s Summer Research Scholarship (SJM). The funding sources paid for salary, supplies, and travel to meetings. The funding sources had no role in study design, data collection, analysis and interpretation of data, writing the manuscript, or in the decision to submit the paper for publication.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors have no conflicts of interest to declare.
8. References
- Alexander GE, DeLong MR, Strick PL, 1986. Parallel organization of functionally segregated circuits linking basal ganglia and cortex. Ann. Rev. Neurosci 9, 357–381. [DOI] [PubMed] [Google Scholar]
- Aganova EA, Uranova NA, 1992. Morphometric analysis of synaptic contacts in the anterior limbic cortex in the endogenous psychoses. Neurosci. Behav. Physiol 22 (1) 59–65. [DOI] [PubMed] [Google Scholar]
- Akil M, Pierri JN, Whitehead RE, Edgar CL, Mohila C, Sampson AR, Lewis DA, 1999. Lamina-specific alterations in the dopamine innervation of the prefrontal cortex in schizophrenic subjects. Am. J. Psych 156 (10) 1580–1589. [DOI] [PubMed] [Google Scholar]
- Arellano JI, Benavides-Piccione R, Defelipe J, Yuste R, 2007. Ultrastructure of dendritic spines: correlation between synaptic and spine morphologies. Front Neurosci. 1 (1): 131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barch DM, Braver TS, Akbudak E, Conturo T., Ollinger J., Snyder A., 2001. Anterior cingulate cortex and response conflict: effects of response modality and processing domain. Cereb Cortex 11 (9) 837–848. [DOI] [PubMed] [Google Scholar]
- Barker DJ, Root DH, Zhang S, Morales M, 2016. Multiplexed neurochemical signaling by neurons of the ventral tegmental area. J Chem Neuroanat. 73:33–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bates TE, Strangward M, Keelan J, Davey GP, Munro PM, Clark JB, 1996. Inhibition of N-acetylaspartate production: implications for 1H MRS studies in vivo. NeuroReport. 7 (8) 1397–1400. [PubMed] [Google Scholar]
- Bellocchio E, Reimer R, Fremeau R Jr., Edwards RH, 2000. Uptake of glutamate into synaptic vesicles by an inorganic phosphate transporter. Science 289(5481): 957–960. [DOI] [PubMed] [Google Scholar]
- Benes FM, Todtenkopf MS, Taylor JB, 1997. Differential distribution of tyrosine hydroxylase fibers on small and large neurons in layer II of anterior cingulate cortex of schizophrenic brain. Synapse 25 (1) 80–92. [DOI] [PubMed] [Google Scholar]
- Benes FM, Vincent SL, Todtenkopf M, 2001. The density of pyramidal and nonpyramidal neurons in anterior cingulate cortex of schizophrenic and bipolar subjects. Biol Psychiatry 50 (6) 395–406. [DOI] [PubMed] [Google Scholar]
- Benarroch EE, 2008. Subthalamic nucleus and its connections: Anatomic substrate for the network effects of deep brain stimulation. Neurology 70(21): 1991–1995. [DOI] [PubMed] [Google Scholar]
- Bolding MS, Reid MA, Avsar KB, Roberts RC, Gamlin PD, Gawne TJ, White DM, den Hollander JA, Lahti AC, 2013. Magnetic transfer contrast accurately localizes substantia nigra confirmed by histology. Biol Psychiatry 73(3):289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braver TS, Barch DM, Gray JR, Molfese DL, Snyder A, 2001. Anterior cingulate cortex and response conflict: effects of frequency, inhibition and errors. Cereb Cortex 11 (9) 825–836. [DOI] [PubMed] [Google Scholar]
- Bubser M, Deutch AY, 2002. Differential effects of typical and atypical antipsychotic drugs on striosome and matrix compartments of the striatum. Eur J Neurosci. 15, 713–720. [DOI] [PubMed] [Google Scholar]
- Bush G, Luu P, Posner MI, 2000. Cognitive and emotional influences in anterior cingulate cortex. Trends Cog Sci. 4 (6) 215–222. [DOI] [PubMed] [Google Scholar]
- Cadena EJ, White DM, Kraguljac NV, Redi MA, Maximo JO, Nelson EA, Gawronski BA, Lahti AC, 2018. A Longitudinal Multimodal Neuroimaging Study to Examine Relationships Between Resting State Glutamate and Task Related BOLD Response in Schizophrenia. Front Psychiatry 9, 632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadena EJ, White DM, Kraguljac NV, Reid MA, Jindal R, Pixley RM, Lahti AC, 2019. Cognitive control network dysconnectivity and response to antipsychotic treatment in schizophrenia. Schizophr Res. 204, 262–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calverley RK, Jones DG, 1990. Contributions of dendritic spines and perforated synapses to synaptic plasticity. Brain Res Brain Res Rev. 15(3):215–249. [DOI] [PubMed] [Google Scholar]
- Carlsson A, 1988. The current status of the dopamine hypothesis of schizophrenia. Neuropsychopharmacology 1, 179–186. [DOI] [PubMed] [Google Scholar]
- Carlsson A, 2006. The neurochemical circuitry of schizophrenia. Pharmacopsychiatry 39 Suppl. 1, S10–14. [DOI] [PubMed] [Google Scholar]
- Chakraborty G, Mekala P, Yahya D, Wu G, Ledeen RW, 2001. Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipid synthesis: evidence for myelin-associated aspartoacylase. J Neurochem. 78(4): 736–745. [DOI] [PubMed] [Google Scholar]
- Chase HW, Loriemi P, Wensing T, Eickhoff SB, Nickl-Jockschat T, 2018. Meta-analytic evidence for altered mesolimbic responses to reward in schizophrenia. Hum Brain Mapp. 39(7), 2917–2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chikama M, McFarland N, Amaral DG, Haber SN, 1997. Insular cortical projections to functional regions of the striatum correlate with cortical cytoarchitectonic organization in the primate. J Neurosci. 17, 9686–9705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KO, Hunt CA, Kennedy MB, 1992. The rat brain postsynaptic density fraction contains a homolog of the Drosophila discs-large tumor suppressor protein. Neuron 9(5):929–942. [DOI] [PubMed] [Google Scholar]
- Coyle JT, 2006. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol. 26(4-6):365–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AM, Banker G, Chang W, McGrath ME, Serpinskaya AS, 1996. Clustering of gephyrin at GABAergic but not glutamatergic synapses in cultured rat hippocampal neurons. J Neurosci. 16:3166–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creese I, Burt DR, Snyder SH, 1976. Dopamine receptor binding predicts clinical and pharmacological potencies of antischizophrenic drugs. Science 192(4238):481–483. [DOI] [PubMed] [Google Scholar]
- Daniel R and Pollmann S, 2014. A universal role of the ventral striatum in reward-based learning: evidence from human studies. Neurobiol Learn Mem. 114: p. 90–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das TK, Javadzadeh A, Dey A, Sabesan P, Théberge J, Radua J, Palaniyappan L, 2019. Antioxidant defense in schizophrenia and bipolar disorder: A meta-analysis of MRS studies of anterior cingulate glutathione. Prog Neuropsychopharmacol Biol Psychiatry 91: p. 94–102. [DOI] [PubMed] [Google Scholar]
- de la Fuente-Sandoval C, Favila R, Alvarado P, León-Ortiz P, Díaz-Galvis L, Amezcua C, García-Muñoz E, Graff-Guerrero A, 2009. Glutamate increase in the associative striatum in schizophrenia: a longitudinal magnetic resonance spectroscopy preliminary study. Gac Med Mex. 145(2):109–113. [PubMed] [Google Scholar]
- de la Fuente-Sandoval C, Leon-Ortiz P, Azcarraga M, Favila R, Stephano S, Graff-Guerrero A, 2012. Striatal glutamate and the conversion to psychosis: a prospective 1H-MRS imaging study. Int J Neuropsychopharmacology 1–5. [DOI] [PubMed] [Google Scholar]
- de la Fuente-Sandoval C, León-Ortiz P, Azcárraga M, Stephano S, Favila R, Díaz-Galvis L, Alvarado-Alanis P, Ramírez-Bermúdez J, Graff-Guerrero A, 2013. Glutamate levels in the associative striatum before and after 4 weeks of antipsychotic treatment in first-episode psychosis: a longitudinal proton magnetic resonance spectroscopy study. JAMA Psychiatry 70(10):1057–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente-Sandoval C, Leon-Ortiz P, Favila R, Stephano S, Mamo D, Ramirez-Bermudez J, Graff-Guerrero A, 2011. Higher levels of glutamate in the associative-striatum of subjects with prodromal symptoms of schizophrenia and patients with first-episode psychosis. Neuropsychopharmacology, 36(9):1781–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demjaha A, Egerton A, Murray RM, Kapur S, Howes OD, Stone JM, McGuire PK, 2014. Antipsychotic treatment resistance in schizophrenia associated with elevated glutamate levels but normal dopamine function. Biol Psychiatry 75(5): e11–3. [DOI] [PubMed] [Google Scholar]
- Dietz D, Dietz K, Nestler E, Russo S, 2009. Molecular mechanisms of psychostimulant-induced structural plasticity. Pharmacopsychiatry 42 (Suppl 1):S69–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eblen F, Graybiel AM, 1995. Highly restricted origin of prefrontal cortical inputs to striosomes in the macaque monkey. J Neurosci. 15 (9) 5999–6013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerton A, Modinos G, Ferrera D, McGuire P, 2017. Neuroimaging studies of GABA in schizophrenia: a systematic review with meta-analysis. Transl Psychiatry 7(6): p. e1147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallon J, Moore R, 1978. Catecholamine innervation of the basal forebrain IV: Topography of the dopamine projection to the basal forebrain and neostriatum. J Comp Neurol. 180(3): 545–80. [DOI] [PubMed] [Google Scholar]
- Falkenberg LE, Westerhausen R, Craven AR, Johnsen E, Kroken RA, L Berg EM, Specht K, Hugdahl K, 2014. Impact of glutamate levels on neuronal response and cognitive abilities in schizophrenia. Neuroimage Clin. 4: p. 576–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty AW, Graybiel AM, 1993. Two input systems for body representations in the primate striatal matrix: experimental evidence in the squirrel monkey. J Neurosci. 13 (3) 1120–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field JR, Walker AG, Conn PJ, 2011. Targeting glutamate synapses in schizophrenia. Trends Mol Med. 17(12):689–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores C, Wen X, Labelle-Dumais C, Kolb B, 2007. Chronic phencyclidine treatment increases dendritic spine density in prefrontal cortex and nucleus accumbens neurons. Synapse 61(12):978–984. [DOI] [PubMed] [Google Scholar]
- Fremeau RT Jr., Troyer MD, Pahner I, Nygaard GO, Tran CH, Reimer RJ, Bellocchio EE, Fortin D, Storm-Mathisen J, Edwards RH, 2001. The expression of vesicular glutamate transporters defines two classes of excitatory synapse. Neuron 31(2):247–260. [DOI] [PubMed] [Google Scholar]
- Fremeau RT Jr., Voglmaier S, Seal RP, Edwards RH, 2004. VGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci. 27(2):98–103. [DOI] [PubMed] [Google Scholar]
- Geinisman Y, Gundersen HJ, van der Zee E, West MJ, 1996. Unbiased stereological estimation of the total number of synapses in a brain region. J Neurocytol. 25(12):805–819. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, 1989. The neostriatal mosaic: Striatal patch-matrix organization is related to cortical lamination. Science 246, 385–388. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, 1992. The neostriatal mosaic: multiple levels of compartmental organization. Trends Neurosci. 15(4):133–9. Review. [DOI] [PubMed] [Google Scholar]
- Gerfen CR, Baimbridge KG, Miller JJ, 1985. The neostriatal mosaic: compartmental distribution of calcium-binding protein and parvalbumin in the basal ganglia of the rat and monkey. Proc Natl Acad Sci U S A. 82(24):8780–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz LA, Lewis DA, 2000. Decreased dendritic spine density on prefrontal cortical pyramidal neurons in schizophrenia. Arch Gen Psychiatry 57(1):65–73. [DOI] [PubMed] [Google Scholar]
- Goff DC, Coyle JT, 2001. The emerging role of glutamate in the pathophysiology and treatment of schizophrenia. Am J Psychiatry 158(9):1367–1377. [DOI] [PubMed] [Google Scholar]
- Goldman PS, Nauta WJ, 1977. An intricately patterned prefronto-caudate projection in the rhesus monkey. J Comp Neurol. 72(3):369–86. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, 1994. Working memory dysfunction in schizophrenia. J Neuropsychiatry Clin Neurosci. 6:348–357. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, 1999. The physiological approach: functional architecture of working memory and disordered cognition in schizophrenia. Biol Psychiatry 46(5) 650–661. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS, Selemon LD, 1986. Topography of corticostriatal projections in nonhuman primates and implications for functional parcellation of the neostriatum. In: Jones EG, Peters A (Eds.), Cerebral Cortex. Plenum Press, New York, pp. 447–466. [Google Scholar]
- Gorelova N, Mulholland PJ, Chandler LJ, Seamans JK, 2012. The glutamatergic component of the mesocortical pathway emanating from different subregions of the ventral midbrain. Cereb Cortex 22(2): 327–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grace AA., 2000. Gating of information flow within the limbic system and the pathophysiology of schizophrenia. Brain Res Brain Res Rev. 31(2-3):330–41. [DOI] [PubMed] [Google Scholar]
- Graham DG, 1979. On the origin and significance of neuromelanin. Arch Pathol Lab Med. 103: 359–362. [PubMed] [Google Scholar]
- Groenewegen HJ, Trimble M, 2007. The ventral striatum as an interface between the limbic and motor systems. CNS Spectr. 12(12):887–92. [DOI] [PubMed] [Google Scholar]
- Gray EG, 1969. Electron microscopy of excitatory and inhibitory synapses: a brief review. Prog Brain Res. 31:141–55. Review. [DOI] [PubMed] [Google Scholar]
- Graybiel AM, Ragsdale CW Jr., 1978. Histochemically distinct compartments in the striatum of human, monkeys, and cat demonstrated by acetylthiocholinesterase staining. Proc Natl Acad Sci. 75 (11), 5723–5726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, 2014. The place of dopamine in the cortico-basal ganglia circuit. Neuroscience 282:248–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Fudge JL, McFarland NR, 2000. Striatonigrostriatal pathways in primates form an ascending spiral from the shell to the dorsolateral striatum. J Neurosci. 20(6):2369–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber SN, Kunishio K, Mizobuchi M, Lynd-Balta E, 1995. The orbital and medial prefrontal circuit through the primate basal ganglia. J Neurosci. 15, 4851–4867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley J, Nenert R, Kraguljac NV Bolding MS, White DM, Skidmore FM, Visscher KM, Lahti AC, 2014. Ventral tegmental area/midbrain functional connectivity and response to antipsychotic medication in schizophrenia. Neuropsychopharmacology 39(4): 1020–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagihara H, Catts VS, Katayama Y, Shoji H, Takagi T, Huang FL, Nakao A, Mori Y, Huang KP, Ishii S, Graef IA, Nakayama KI, Shannon Weickert C, Miyakawa T, 2018. Decreased Brain pH as a Shared Endophenotype of Psychiatric Disorders. Neuropsychopharmacology 43(3):459–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris K, Kater S, 1994. Dendritic Spines: Cellular Specializations Imparting Both Stability and Flexibility to Synaptic Function. Annu Rev Neurosci. 17(1):341371. [DOI] [PubMed] [Google Scholar]
- Hering H, Sheng M, 2001. Dendritic spines: structure, dynamics and regulation. Nat Rev Neurosci. 2(12):880–8. [DOI] [PubMed] [Google Scholar]
- Hirsch EC, Graybiel AM, Hersh LB, Duyckaerts C, Agid Y, 1989. Striosomes and extrastriosomal matrix contain different amounts of immunoreactive choline acetyltransferase in the human striatum. Neurosci Lett. 96(2):145–50. [DOI] [PubMed] [Google Scholar]
- Hökfelt T, Fuxe K, Goldstein M, 1973. Immunohistochemical studies on monoamine-containing cell systems. Brain Res. 62(2):461–469. [DOI] [PubMed] [Google Scholar]
- Holt DJ, Graybiel AM, Saper CB, 1997. Neurochemical architecture of the human striatum. J Comp Neurol. 384(1):1–25. [DOI] [PubMed] [Google Scholar]
- Howes O, Kapur S, 2009. The dopamine hypothesis of schizophrenia: version III- the final common pathway. Schizophr Bull. 35(3):549–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howes O, Williams M, Ibrahim K, Leung G, Egerton A, McGuire PK, Turkheimer F 2013. Midbrain dopamine function in schizophrenia and depression: a post-mortem and positron emission tomographic imaging study. Brain 136(Pt 11 ):3242–4251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheson NL, Clark DG, Bolding MS, White DM, Lahti AC, 2014. Basal ganglia volume in unmedicated patients with schizophrenia is associated with treatment response to antipsychotic medication. Psychiatry Res. 221(1): p. 6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iasevoli F, Polese D, Ambesi-lmpiombato A, Muscettola G, de Bartolomeis A., 2007. Ketamine-related expression of glutamatergic postsynaptic density genes: possible implications in psychosis. Neurosci Lett. 416(1): 1–5. [DOI] [PubMed] [Google Scholar]
- Ichinose H, Ohye T, Fujita K, Pantucek F, Lange K, Riderer P, Nagatsu T, 1994. Quantification of mRNA of tyrosine hydroxylase and aromatic L-amino acid decarboxylase in the substantia nigra in Parkinson’s disease and schizophrenia. J Neural Transm Park Dis Dement. Sect 8 (1-2):149–158. [DOI] [PubMed] [Google Scholar]
- Itil T, Keskiner A, Kiremitci N, Holden JM, 1967. Effect of phencyclidine in chronic schizophrenics. Can Psychiatr Assoc J. 12(2):209–212. [DOI] [PubMed] [Google Scholar]
- Javitt DC, 2004. Glutamate as a therapeutic target in psychiatric disorders. Mol Psychiatry 9(11):984–997. [DOI] [PubMed] [Google Scholar]
- Jessen F, Fingerhut N, Sprinkart AM, Kühn KU, Petrovsky N, Maier W, Schild HH, Block W, Wagner M, Träber F, 2013. N-acetylaspartylglutamate (NAAG) and N-acetylaspartate (NAA) in patients with schizophrenia. Schizophren Bull. 39 (1) 197–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kajimoto Y, Shirakawa O, Lin XH, Hashimoto T, Kitamura N, Murakami N, Takumi T, Maeda K, 2003. Synapse-associated protein 90/postsynaptic density-95-associated protein (SAPAP) is expressed differentially in phencyclidine-treated rats and is increased in the nucleus accumbens of patients with schizophrenia. Neuropsychopharmacology 28(10):1831–9. [DOI] [PubMed] [Google Scholar]
- Kegeles LS, Abi-Dargham A, Frankie WG, Gil R, Cooper TB, Slifstein M, Hwang DR, Huang Y, Haber SN, Laruelle M, 2010. Increased synaptic dopamine function in associative regions of the striatum in schizophrenia. Arch Gen Psychiatry 67(3):231–9. [DOI] [PubMed] [Google Scholar]
- Kemp JM, Powell TPS, 1971. The termination of fibres from the cerebral cortex and thalamus upon dendritic spines in the caudate nucleus: A study with the Golgi method. Phil Trans R Soc Lond Biol. 262:429–439. [DOI] [PubMed] [Google Scholar]
- Kerns JG, Cohen JD, MacDonald AW, Cho RY, Stenger VA, Carter CS, 2004. Anterior cingulate conflict monitoring and adjustments in control. Science 303 (5660) 1023–1026. [DOI] [PubMed] [Google Scholar]
- Kerns JG, Cohen JD, MacDonald AW 3rd, Johnson MK, Stenger VA, Aizenstein H, Carter CS, 2005. Decreased conflict- and error-related activity in the anterior cingulate cortex in subjects with schizophrenia. Am J Psychiatry 162(10): 1833–9. [DOI] [PubMed] [Google Scholar]
- Kincaid AE, Wilson CJ, 1996. Corticostriatal innervation of the patch and matrix in the rat neostriatum. J Comp Neurol. 374:578–592. [DOI] [PubMed] [Google Scholar]
- Kolomeets NS, Orlovskaya DD, Uranova NA, 2007. Decreased numerical density of CA3 hippocampal mossy fiber synapses in schizophrenia. Synapse 61 (8) 615–621. [DOI] [PubMed] [Google Scholar]
- Kolomeets NS, Orlovskaya DD, Rachmanova VI, Uranova NA, 2005. Ultrastructural alterations in hippocampal mossy fiber synapses in schizophrenia: a postmortem morphometric study. Synapse 57 (1) 47–55. [DOI] [PubMed] [Google Scholar]
- Kolomeets NS, Uranova NA, 1999. Synaptic contacts in schizophrenia: studies using immunocytochemical identification of dopaminergic neurons. Neurosci Behav Physiol. 29(2):217–221. [DOI] [PubMed] [Google Scholar]
- Koski L, Petrides M, 2001. Time-related changes in task performance after lesions restricted to the frontal cortex. Neuropsychol. 39 (3) 268–281. [DOI] [PubMed] [Google Scholar]
- Kouneiher F, Charron S, Koechlin E, 2009. Motivation and cognitive control in the human prefrontal cortex. Nat Neurosci. 12(7): p. 939–45. [DOI] [PubMed] [Google Scholar]
- Krueger-Burg D, Papadopoulos T, Brose N, 2017. Organizers of inhibitory synapses come of age. Curr Opin Neurobiol. 45:66–77. Review. [DOI] [PubMed] [Google Scholar]
- Krystal JH, 2008. Capitalizing on extrasynaptic glutamate neurotransmission to treat antipsychotic-resistant symptoms in schizophrenia. Biol Psychiatry 64(5):358–360. [DOI] [PubMed] [Google Scholar]
- Kubota Y, Kawaguchi Y, 1993. Spatial distributions of chemically identified intrinsic neurons in relation to patch and matrix compartments of rat neostriatum. J Comp Neurol. 332(4):499–513. [DOI] [PubMed] [Google Scholar]
- Kung L, Force M, Chute DJ, Roberts RC, 1998. Immunocytochemical localization of tyrosine hydroxylase in the human striatum: a postmortem ultrastructural study. J Comp Neurol. 390(1):52–62. [DOI] [PubMed] [Google Scholar]
- Kumar J, Liddle EB, Fernandes CC, Palaniyappan L, Hall EL, Robson SE, Simmonite M, Fiesal J, Katshu MZ, Qureshi A, Skelton M, Christodoulou NG, Brookes MJ, Morris PG, Liddle PF, 2018. Glutathione and glutamate in schizophrenia: a 7T MRS study. Mol Psychiatry 10.1038/s41380-018-0104-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunishio K, Haber SN, 1994. Primate cingulostriatal projection: limbic striatal versus sensorimotor striatal input. J Comp Neurol. 350, 337–356. [DOI] [PubMed] [Google Scholar]
- Lahti AC, Koffel B, LaPorte D, Tamminga CA, 1995. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology 13(1):9–19. [DOI] [PubMed] [Google Scholar]
- Ledeen RW, Wang J, Wu G, Lu ZH, Chakraborty G, Meyenhofer M, Tyring SK, Matalon R, 2006. Physiological role of N-acetylaspartate: contribution to myelinogenesis. Adv Exp Med Biol. (76) 131–143; discussion 361-363. [DOI] [PubMed] [Google Scholar]
- Lentz MR, Kim JP, Westmoreland SV, Greco JB, Fuller RA, Ratai EM, He J, Sehgal PK, Halpern EF, Lackner AA, Masliah E, Gonzalez RG, 2005. Quantitative neuropathologic correlates of changes in ratio of N-acetylaspartate to creatine in macaque brain. Radiology 235 (2) 461–468. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Melchitzky DS, Burgos GG, 2002. Specificity in the functional architecture of primate prefrontal cortex. J. Neurocytol 31 (3-5) 265–276. [DOI] [PubMed] [Google Scholar]
- Li Y, Kolb B, Robinson TE, 2003. The location of persistent amphetamine-induced changes in the density of dendritic spines on medium spiny neurons in the nucleus accumbens and caudate-putamen. Neuropsychopharmacology 28(6):1082–1085. [DOI] [PubMed] [Google Scholar]
- Mabry SJ, McCollum LA, Farmer CB, Bloom ES, Roberts RC., 2019. Evidence for altered excitatory and inhibitory tone in the post-mortem substantia nigra in schizophrenia. World Journal of Biological Psychiatry 4:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathalon DH, Fedor M, Faustman WO, Gray M, Askari N, Ford JM, 2002. Response-monitoring dysfunction in schizophrenia: an event-related brain potential study. J. Abnorm. Psych 111 (1) 22–41. [PubMed] [Google Scholar]
- Merritt K, Egerton A, Kempton MJ, Taylor MJ, McGuire PK, 2016. Nature of Glutamate Alterations in Schizophrenia: A Meta-analysis of Proton Magnetic Resonance Spectroscopy Studies. JAMA Psychiatry 73(7) 665–674. [DOI] [PubMed] [Google Scholar]
- McCollum LA, Roberts RC, 2015. Uncovering the role of the nucleus accumbens in schizophrenia: A postmortem analysis of tyrosine hydroxylase and vesicular glutamate transporters. Schizophr Res. 169:369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCollum LA, Walker CK, Roche JK, Roberts RC, 2015. Elevated excitatory input to the nucleus accumbens in schizophrenia: a postmortem ultrastructural study. Schizophr Bull. 41 (5): 1123–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McRitchie DA, Hardman CD, Halliday GM, 1996. Cytoarchitectural distribution of calcium binding proteins in midbrain dopaminergic regions of rats and humans. J Comp Neurol. 364(1):121–150. [DOI] [PubMed] [Google Scholar]
- Minzenberg MJ, Laird AR, Thelen S, Carter CS, Glahn DC, 2009. Meta-analysis of 41 functional neuroimaging studies of executive function in schizophrenia. Arch Gen Psychiatry 66(8) 811–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffett JR, Ross B, Arun P, Madhavarao CN, Namboodiri AM, 2007. N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog Neurobiol. 81 (2) 89–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller T, Haroutunian V, Davis KL, Meador-Woodruff JH, 2004. Expression of the ionotropic glutamate receptor subunits and NMDA receptor-associated intracellular proteins in the substantia nigra in schizophrenia. Mol Brain Res. 121(1-2):60–69. [DOI] [PubMed] [Google Scholar]
- Oni-Orisan A, Kristiansen LV, Haroutunian V, Meador-Woodruff JH, McCullumsmith RE, 2008. Altered vesicular glutamate transporter expression in the anterior cingulate cortex in schizophrenia. Biol Psychiatry 63(8):766–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek G, Gawne TJ, Reid MA, Salibi N, Kraguljac NV, White DM, Lahti AC, 2019. Relationship Between Cortical Excitation and Inhibition and Task-Induced Activation and Deactivation: A Combined Magnetic Resonance Spectroscopy and Functional Magnetic Resonance Imaging Study at 7T in First-Episode Psychosis. Biol Psychiatry Cogn Neurosci Neuroimaging, 4(2): p. 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent A, Hazrati LN, 1995. Functional anatomy of the basal ganglia. I. The cortico-basal ganglia-thalamo-cortical loop. Brain Res Rev. 20, 91–127. [DOI] [PubMed] [Google Scholar]
- Penny GR, Wilson CJ, Kitai ST, 1988. Relationship of the axonal and dendritic geometry of spiny projection neurons to the compartmental organization of the neostriatum. J Comp Neurol. 269(2):275–289. [DOI] [PubMed] [Google Scholar]
- Perez-Costas E, Melendez-Ferro M, Rice MW, Conley RR, Roberts RC, 2012. Dopamine pathology in schizophrenia: analysis of total and phosphorylated tyrosine hydroxylase in the substantia nigra. Front Psychiatry 3:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BS, Skudlarski P, Gatenby JC, Zhang H, Anderson AW, Gore JC, 1999. An fMRI study of Stroop word-color interference: evidence for cingulate subregions subserving multiple distributed attentional systems. Biol. Psych 45 (10) 1237–1258. [DOI] [PubMed] [Google Scholar]
- Prensa L, Gimenenz-Amaya JM, Parent A, 1999. Chemical heterogeneity of the striosomal compartment in the human striatum. J Comp Neurol. 413, 603–618. [PubMed] [Google Scholar]
- Radua J, Schmidt A, Borgwardt S, Heinz A, Schlagenhauf F, McGuire P, Fusar-Poli P, 2015. Ventral Striatal Activation During Reward Processing in Psychosis: A Neurofunctional Meta-Analysis. JAMA Psychiatry 72(12): 1243–51. [DOI] [PubMed] [Google Scholar]
- Ragsdale CW Jr., Graybiel AM, 1991. Compartmental organization of the thalamostriatal connection in the cat. J Comp Neurol. 311(1): 134–67 [DOI] [PubMed] [Google Scholar]
- Reid MA, Salibi N, White DM, Gawne TJ, Denney TS, Lahti AC, 2019. 7T proton magnetic resonance spectroscopy of the anterior cingulate cortex in first-episode schizophrenia. Schizophr Bull. 45(1):180–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MA, Stoeckel LE, White DM, Avsar KB, Bolding MS, Akella NS, Knowlton RC, den Hollander JA, Lahti AC, 2010. Assessments of function and biochemistry of the anterior cingulate cortex in schizophrenia. Biol Psych. 68 (7) 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice M, Roberts RC, Melendez-Ferro M, Perez-Costas E, 2016. Mapping dopaminergic deficiencies in the substantia nigra/ventral tegmental area in schizophrenia. Brain Struct Funct. 221(1):185–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RC, Barksdale KA, Roche JK, Lahti AC, 2015. Decreased synaptic and mitochondrial density in the postmortem anterior cingulate cortex in schizophrenia. Schizophr Res. 168(1-2):543–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RC, Knickman JK, 2002. The ultrastructural organization of the patch matrix compartments in the human striatum. J Comp Neurol. 452(2):128–138. [DOI] [PubMed] [Google Scholar]
- Roberts RC, Roche JK, Conley RR, 2005a. Synaptic differences in the postmortem striatum of subjects with schizophrenia: a stereological ultrastructural analysis. Synapse 56:185–97. [DOI] [PubMed] [Google Scholar]
- Roberts RC, Roche JK, Conley RR, 2005b. Synaptic differences in the patch matrix compartments of subjects with schizophrenia: a postmortem ultrastructural study of the striatum. Neurobiol Dis. 20: 324–335. [DOI] [PubMed] [Google Scholar]
- Roberts RC, Roche JK, Somerville SM, Conley RR, 2012. Ultrastructural distinctions between treatment responders and non-responders in schizophrenia: postmortem studies of the striatum, in: Labate L (Ed.), Mental Illnesses - Evaluation, Treatments and Implications. InTech, Croatia, 261–286. [Google Scholar]
- Robinson TE, Kolb B, 1997. Persistent structural modifications in nucleus accumbens and prefrontal cortex neurons produced by previous experience with amphetamine. J Neurosci. 17(21):8491–8497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson TE, Kolb B, 1999. Alterations in the morphology of dendrites and dendritic spines in the nucleus accumbens and prefrontal cortex following repeated treatment with amphetamine or cocaine. Eur J Neurosci. 11(5):1598–1604. [DOI] [PubMed] [Google Scholar]
- Root DH, Wang HL, Liu B, Barker DJ, Mód L, Szocsics P, Silva AC, Maglóczky Z, Morales M, 2016. Glutamate neurons are intermixed with midbrain dopamine neurons in nonhuman primates and humans. Sci Rep. 6:30615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland LM, Pradhan S, Korenic S, Wijtenburg SA, Hong LE, Edden RA, Barker PB, 2016. Elevated brain lactate in schizophrenia: a 7 T magnetic resonance spectroscopy study. Transl Psychiatry 6(11): p. e967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadikot AF, Parent A, Francois C, 1992a. Efferent connections of the centromedian and parafascicular thalamic nuclei in the squirrel monkey: A PHA-L study of subcortical projections. J Comp Neurol. 315, 137–159. [DOI] [PubMed] [Google Scholar]
- Sadikot AF, Parent A, Smith Y, Bolam JP, 1992b. Efferent connections of the centromedian and parafascicular thalamic nuclei in the squirrel monkey: a light and electron microscopic study of the thalamostriatal projection in relation to striatal heterogeneity. J Comp Neurol. 320, 228–242. [DOI] [PubMed] [Google Scholar]
- Sarpal DK, Robinson DG, Lencz T, Argyelan M, Ikuta T, Karlsgodt K, Gallego JA, Kane JM, Szeszko PR, Malhotra AK, 2015. Antipsychotic treatment and functional connectivity of the striatum in first-episode schizophrenia. JAMA Psychiatry 72(1): p. 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoonover KE, McCollum LA, Roberts RC, 2017. Tyrosine hydroxylase, GAD67, vGLUH, and vGLUT2 proteins in the substantia nigra in schizophrenia. Neuropsychopharmacology 42(2):540–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selemon LD, Goldman-Rakic PS, 1999. The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biol Psychiatry 45(1): 17–25. Review. [DOI] [PubMed] [Google Scholar]
- Sheng M, Hoogenraad CC, 2007. The postsynaptic architecture of excitatory synapses: a more quantitative view. Annu Rev Biochem. 76:823–847 [DOI] [PubMed] [Google Scholar]
- Shibata E, Sasaki M, Tohyama K, Otsuka K, Endoh J, Terayama Y, Sakai A, 2008. Use of neuromelanin-sensitive MRI to distinguish schizophrenic and depressive patients and healthy individuals based on signal alterations in the substantia nigra and locus ceruleus. Biol Psychiatry 64:401–406. [DOI] [PubMed] [Google Scholar]
- Smith Y, Bevan MD, Shink E, Bolam JP, 1998. Microcircuitry of the direct and indirect pathways of the basal ganglia. Neuroscience 86, 353–387. [DOI] [PubMed] [Google Scholar]
- Smith RE, Haroutunian V, Davis KL, Meador-Woodruff JH, 2001. Vesicular glutamate transporter transcript expression in the thalamus in schizophrenia. NeuroReport 12(13):2885–7. [DOI] [PubMed] [Google Scholar]
- Stork C, Renshaw PF, 2005. Mitochondrial dysfunction in bipolar disorder: evidence from magnetic resonance spectroscopy research. Mol Psych. 10 (10) 900–919. [DOI] [PubMed] [Google Scholar]
- Sullivan CR, Mielnik CA, Funk A, O’Donovan SM, Bentea E, Pletnikov M, Ramsey AJ, Wen Z, Rowland LM, McCullumsmith RE, 2019. Measurement of lactate levels in postmortem brain, iPSCs, and animal models of schizophrenia. Sci Rep. 9(1):5087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torn M, Watanabe S, Shibuya H, Nishikawa T, Noda K, Mitsushio H, Ichikawa H, Kurumaji A, Takashima M, Mataga N, Ogawa A, 1988. Neurotransmitters, receptors, and neuropeptides in post-mortem brains of chronic schizophrenic patients. Acta Psychiatr Scand. 78(2): 121–137. [DOI] [PubMed] [Google Scholar]
- Uezato A, Meador-Woodruff JH, McCullumsmith RE, 2009. Vesicular glutamate transporter mRNA expression in the medial temporal lobe in major depressive disorder, bipolar disorder, and schizophrenia. Bipolar Disord. 11(7):711–25. [DOI] [PubMed] [Google Scholar]
- Uranova NA, Levite OI, 1987. Ultrastructure of the substantia nigra in schizophrenia. Zh Nevropatol Psikhiatr IM S. S. Korsakova 87:1017–1024. [PubMed] [Google Scholar]
- Vigneault E, Poirel O, Riad M, Prud’homme J, Dumas S, Turecki G, Fasano C, Mechawar N, El Mestikawy S, 2015. Distribution of vesicular glutamate transporters in the human brain. Front Neuroanat. 9: 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt BA, Finch DM, Olson CR, 1992. Functional heterogeneity in cingulate cortex: the anterior executive and posterior evaluative regions. Cereb Cortex 2 (6) 435–443. [DOI] [PubMed] [Google Scholar]
- Walker CK, Roche JK, Sinha V, Roberts RC, 2017. Substantia nigra ultrastructural pathology in schizophrenia. Schizophrenia Research Dec 20. pii: S0920-9964(17)30756-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang AM, Pradhan S, Coughlin JM, Trivedi A, DuBois SL, Crawford JL, Sedlak TW, Nucifora FC Jr, Nestadt G, Nucifora LG, Schretlen DJ, Sawa A, Barker PB. 2019. Assessing Brain Metabolism With 7-T Proton Magnetic Resonance Spectroscopy in Patients With First-Episode Psychosis. JAMA Psychiatry 76(3) 314–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe Y, Tanaka A, Tsukabe A, Kunitomi Y, Nishizawa M, Hashimoto R, Yamamori H, Fujimoto M, Fukunaga M, Tomiyama N, 2014. Neuromelanin magnetic resonance imaging reveals increased dopaminergic neuron activity in the substantia nigra of patients with schizophrenia. PLoS One 9(8):e104619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein JJ, Chohan MO, Slifstein M, Kegeles LS, Moore H, Abi-Dargham A, 2017. Pathway-specific dopamine abnormalities in schizophrenia. Biol Psychiatry 81:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss EM, Siedentopf C, Golaszewski S, Mottaghy FM, Hofer A, Kremser C, Felber S, Fleischhacker WW, 2007. Brain activation patterns during a selective attention test--a functional MRI study in healthy volunteers and unmedicated patients during an acute episode of schizophrenia. Psychiatry Res. 154(1): p. 31–40. [DOI] [PubMed] [Google Scholar]
- White IM, Flory GS., Hooper KC, Speciale J, Banks DA, Rebec GV, 1995. Phencyclidine-induced increases in striatal neuron firing in behaving rats: reversal by haloperidol and clozapine. J Neural Transm Gen Sect. 102(2):99–112. [DOI] [PubMed] [Google Scholar]
- White DM, Kraguljac NV, Reid MA, Lahti AC, 2015. Contribution of substantia nigra glutamate to prediction error signals in schizophrenia: a combined magnetic resonance spectroscopy/functional imaging study. NPJ Schizophrenia 1:14001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TP, Wigton R, Joyce DW, Collier T, Fornito A, Shergill SS, 2016. Dysfunctional Striatal Systems in Treatment-Resistant Schizophrenia. Neuropsychopharmacology 41(5): 1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams M, Galvin K, O’Sullivan B, MacDonald CD, Ching EW, Turkheimer F, 2013. Neuropathological changes in the substantia nigra in schizophrenia but not depression. EurArch Psychiatry Clin Neurosci. 264(4):285–296. [DOI] [PubMed] [Google Scholar]
- Wilson N, Kang J, Hueske EV, Leung T, Varoqui H, Murnick JG, Erickson JD, Liu G, 2005. Presynaptic regulation of quantal size by the vesicular glutamate transporter vGLUT1. J Neurosci. 25(26): 6221–6234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon HJ, Mizenberg MJ, Raouf S, D’Esposito M, Carter CS, 2013. Impaired prefrontostriatonigral functional connectivity and substantia nigra hyperactivity in schizophrenia. Biol Psychiatry 74(2):122–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Westphal AJ, Minzenberg MJ, Niendam T, Ragland JD, Lesh T, Solomon M, Carter CS, 2014. Task-evoked substantia nigra hyperactivity associated with prefrontal hypofunction, prefrontonigral disconnectivity and nigrostriatal connectivity predicted psychosis severity in medication naive first episode schizophrenia. Schizophrenia Res. 159(2-3):521–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuksel C, Tegin C, O’Connor L, Du F, Ahat E, Cohen BM., Ongur D., 2015. Phosphorus magnetic resonance spectroscopy studies in schizophrenia. J Psychiatr Res. 68:157–166. [DOI] [PubMed] [Google Scholar]
- Yuste R, Bonhoeffer T, 2001. Morphological changes in dendritic spines associated with longterm synaptic plasticity. Annu Rev Neurosci. 24:1071–1089. [DOI] [PubMed] [Google Scholar]