

Abstract
Cancer cells survive and adapt to many types of stress including hypoxia, nutrient deprivation, metabolic and oxidative stress. These stresses are sensed by diverse cellular signaling processes leading to either degradation of mitochondria or alleviation of mitochondrial stress. This review will discuss signaling during sensing and mitigation of stress involving mitochondrial communication with the endoplasmic reticulum, and how retrograde signaling upregulates the mitochondrial stress response to maintain mitochondrial integrity. The importance of the mitochondrial unfolded protein response, an emerging pathway that alleviates cellular stress, will be elaborated with respect to cancer. Detailed understanding of cellular pathways will establish mitochondrial stress response as a key mechanism for cancer cell survival leading to cancer progression and resistance, and provide a potential therapeutic target in cancer.
Keywords: cancer cell survival, heat shock protein 60 (HSP60), mitochondrial unfolded protein response, mitochondrial stress response, cancer progression and therapeutic resistance
Cancer Cell Mitochondria and Stress Response Pathways
Cancer cells rely on the mitochondrial oxidative phosphorylation (OXPHOS) system to sustain high proliferative capacities, metastatic spread, chemotherapy resistance, and stemness [1–4]. Mitochondria generate other macromolecules besides ATP that are essential for cellular function. Mitochondria synthesize amino acids, fatty acids, cholesterol, heme, Fe-S clusters, nucleotides and a myriad of metabolic intermediates that can act as signaling molecules (reviewed extensively in [5]). The mitochondrion is home to other major metabolic pathways such as the tricarboxylic acid (TCA) cycle and fatty acid oxidation (FAO), which occur in the mitochondrial matrix. The TCA intermediates fuel OXPHOS Complexes and are used for biosynthesis of amino acids as well as redox balance while FAO is used as an alternate energy source for ATP [6–9]. Fatty acid and cholesterol synthesis can be upregulated in cancer cell mitochondria to be used as building blocks of the lipid bilayer membrane, an essential requirement for dividing cells. Mitochondrial cholesterol is the sole precursor for steroids including testosterone and estrogen, which drive prostate and breast cancer progression, respectively [10–12]. Therefore, cancer cells often induce mitochondrial biogenesis and metabolic reprogramming to keep up with the energy demands and to produce essential macromolecular building blocks required for constant cell division and oncogenic signaling. Since evasion of apoptosis is one of the hallmarks of cancer [13], cancer cells may increase the apoptotic threshold of mitochondria by activating mitochondrial maintenance programs.
Mitochondria play important roles in cellular physiology by affecting cell viability and function. Therefore, cellular stresses that impact mitochondria need to be managed to avoid any deleterious effects to cells. Cancer cells employ multiple stress response pathways to counteract exogenous or endogenous stressors and to enhance their survival and proliferation. These pathways include the integrated stress response (ISR), the cytosolic heat shock response (HSR), and the unfolded protein response (UPR) mediated by organelles such as the endoplasmic reticulum (ER) and mitochondrion [14–16]. Although the cytoprotective ISR pathway, mediated by the phosphorylation of eukaryotic translation initiation factor 2 alpha, is interlinked with other stress response pathways [14, 15], the activation of the endoplasmic reticulum UPR (UPRER) and the mitochondrial UPR (UPRmt) maintains protein homeostasis specifically in these two organelles in response to exogenous or endogenous stressors [16, 17]. Mitochondria have evolved several maintenance and preservation pathways to attenuate various stresses, such as UPRmt, mitochondrial fission and fusion, and mitophagy [15, 18–21]. In cancer cells, these mechanisms are deregulated due to altered signaling leading to long-term proliferative and survival advantages, which are intrinsically linked to tumor progression and therapeutic resistance [22–24]. This review describes current updates on mitigation of persistent cellular stresses mediated by the mitochondrial stress response (Figure 1) and highlights some outstanding questions that require immediate attention to efficiently target stress signaling to block tumor growth and to prevent development of therapeutic resistance in cancer.
Figure 1. Key Figure. The activation of mitochondrial stress response and cellular signaling.
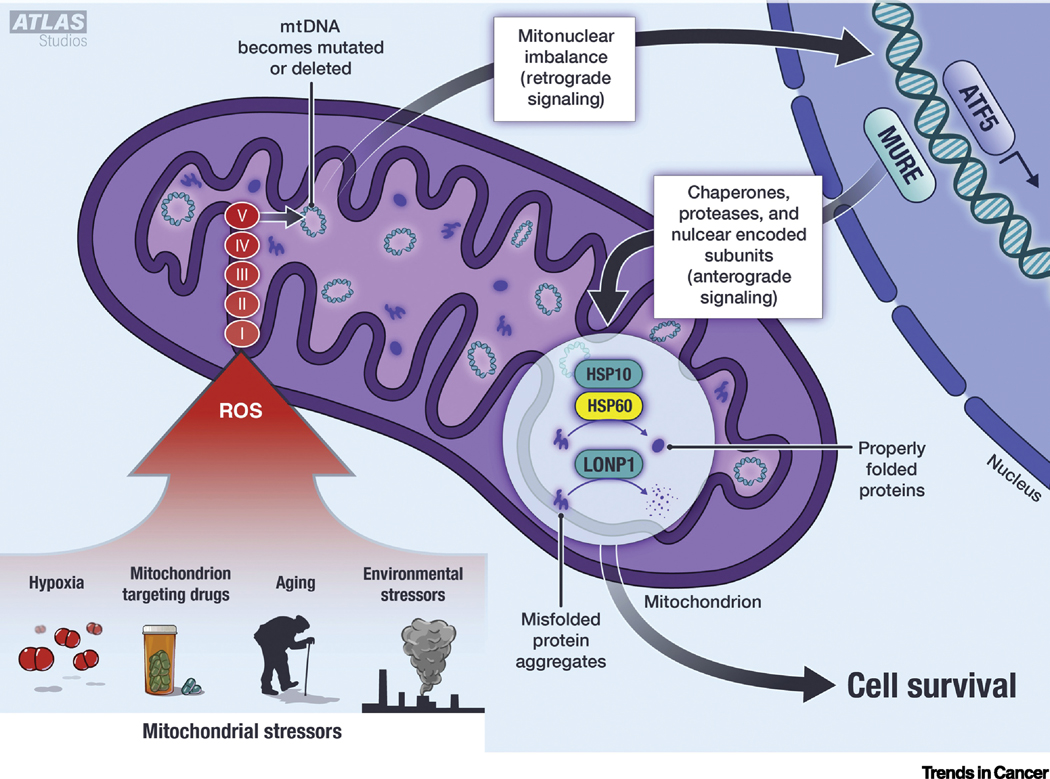
Mitochondrial stresses associated with aging, environmental toxins, hypoxia and mitotoxic drugs promote ROS production. ROS damages mitochondrial proteins including the OXPHOS complexes, which may further enhance ROS production. The continued ROS production damages mtDNA. The damaged mtDNA and OXPHOS complexes create an imbalance of the mitochondria-nuclear proteins and induce retrograde signaling. This includes nuclear translocation of the UPRmt transcription factor ATF5. ATF5 binds to Mitochondrial Unfolded Protein Response Element (MURE) to initiate transcription of UPRmt genes including HSP60, HSP10 and LONP1. These transcripts are translated and imported into the mitochondria (anterograde signaling). Once within the mitochondria, HSP60 and HSP10 work together to properly fold damaged proteins, and LONP1 cleaves and degrades those proteins that are damaged beyond repair. The UPRmt proteins promote cell survival by maintaining mitochondrial integrity, thereby preventing cytochrome c release and inhibiting the initiation of intrinsic apoptosis. Abbreviations: HSP = heat shock protein, LONP1 = Lon Protease, ROS = reactive oxygen species, mtDNA = mitochondrial DNA, OXPHOS = oxidative phosphorylation, ATF5 = activating transcription factor 5, MURE = mitochondrial unfolded protein response elements.
Cellular Stress and Mitochondrial Dysfunction in Cancer
Mitochondria are interconnected with other cellular organelles and perform various functions including bioenergetics, macromolecular synthesis, cellular life and death decisions. These mitochondrial functions are deregulated in cancer rendering long-term survival and proliferative advantages to cancer cells leading to aggressive disease and therapeutic resistance [13, 25]. Key reasons for impaired mitochondrial function (i.e., dysfunction) in cancer cells include endogenous stresses such as hypoxia, and metabolic and proliferative stresses [26]. Mitochondrial dysfunction in cancer is adapted in a way that help cancer cells survive, proliferate, migrate, and develop plasticity [26, 27]. The adaptation of cancer cells under persistent stress is mediated by robust stress sensing and mitigation systems in mitochondria and other cellular compartments [28].
Stress sensing and mitigation signaling may require inter-organellar communications, mitochondrial biogenesis, unfolded protein response, mitochondrial dynamics and downstream signaling [15, 18–20, 29]. Mitochondrial biogenesis is controlled by both nuclear DNA (nDNA) and mitochondrial DNA (mtDNA). MtDNA encodes 13 structural proteins of the OXPHOS system, 22 tRNAs, and 2 rRNAs [30]. Thus, mitochondrial biogenesis and function relies on import of the nDNA-encoded proteins, which are synthesized in the cytosol [31] and synthesis of the mtDNA-encoded proteins within mitochondria. Mutations of both nDNA and mtDNA compromise mitochondrial function and create mitochondrial stress leading to deregulation of cellular signaling and tumorigenesis. Warburg’s original hypothesis suggests a causative role of the respiratory chain injury and enhanced aerobic glycolysis in cancer cells [32, 33]. However, current evidences also support that tumorigenesis requires both glycolysis and OXPHOS for energy production and the synthesis of macromolecules [13, 25, 34, 35]. Therefore, it is essential to understand the role of glycolysis and OXPHOS in tumor growth and progression in the broader context of mitochondrial stress response.
Mitochondrial Stress and Inter-organellar Networking
Mitochondria experience stresses in many forms including generation of mitochondrial reactive oxygen species (mtROS) due to hypoxia, metabolic stress, nutrient depletion, defects in protein folding within mitochondria, and defects in protein import [26, 31, 36]. Sensing and mitigation of these stresses require instant communication between various cellular compartments such as mitochondria, ER, lysosomes, and nucleus [15, 18–20, 28, 36]. In response to stress, mitochondria undergo dynamic changes and regulate their biogenesis as well as biogenesis of other cellular organelles such as lysosomes and ER. For example, in response to stress, a decline in ATP generation leads to the upregulation of AMP-activated protein kinase (AMPK), which enhances lysosomal biogenesis to degrade dysfunctional mitochondria via mitophagy and maintain mitochondrial homeostasis [37]. Inter-organellar crosstalk between ER and mitochondria via mitochondria-associated ER membranes (MAMs) ensures that mitochondrial stresses are shared between organelles [38–40].
Under normal condition, the ER stress transducer inositol-requiring enzyme 1α (IRE1α) maintains the composition and functions of MAMs, and mediates ER-to-mitochondria communication in order to fine-tune mitochondrial respiration via facilitating local Ca2+ transfer from the ER to the mitochondrial matrix [41]. Increased mitochondrial respiration and expression of the mitochondrial protease LONP1 upon ER stress, including increased ER stress induced by generation of mtROS, suggest a unique coordination and stress sensing mechanism between these two organelles [42–45]. However, persistent accumulation of Ca2+ in the mitochondrial matrix via MAMs induces the mitochondrial permeability transition pore (mPTP), which triggers apoptotic cell death [46, 47]. Additionally, MAMs regulate other tightly controlled cellular processes such as autophagy and mitochondrial dynamics. Given that MAMs are critical for various cellular functions, their deregulation may impact cancer progression and therapeutic resistance. Upregulation of mitochondrial fusion components such as mitochondrial GTPases mitofusin 1 (MFN-1) and mitofusin 2 (MFN-2) promote prostate cancer progression [48]. It is also interesting to note that MFN-1 and MFN-2 are important for physical tethering between mitochondria and ER at the MAMs [49]. By contrast, increased mitochondrial division and expression of GTPase dynamin related protein 1 (Drp1), an essential component of the mitochondrial division (mitochondrial fission) machinery, as well as decreased mitochondrial fusion promote tumorigenesis [50, 51]. Therefore, there is a context-dependent role of mitochondrial dynamics, ER, and MAMs to alleviate cellular stress and promote cancer progression. Notably, mitochondrial networking with other organelles are important to maintain cellular functioning, for example, crosstalk between mitochondria and nucleus is important for both normal and cancer cells.
The UPRmt Alleviates Stress and Promotes Cancer Cell Survival
Cancer cells employ multiple stress response pathways to counteract endogenous and exogenous or environmental stresses. The cytosolic heat shock response (HSR) is regulated by the transcription factor heat shock factor 1 (HSF1) [52]. The HSR is induced by unfolded protein accumulation within the cytosol, which results in HSF1 trafficking to the nucleus to induce cytosolic chaperones, such as heat shock protein 27 (HSP27) and heat shock protein 90 (HSP90), which are involved in protein folding [53–55]. Stressors such as temperature, oxidative stress, and calcium depletion can cause protein denaturation and misfolding resulting in the accumulation of aggregates. The cellular response to accumulation of unfolded proteins is referred to as UPR, which is activated in different cellular compartments.
Mitochondrial-specific stress signaling was first reported in 1996 by Martinus et al. [56]. They observed that depletion of the mtDNA led to an increase in the expression of nuclear-encoded mitochondrial chaperones, HSP10 and HSP60 [56]. However, no change in the expression of HSP70, an ER chaperone, and HSP72, a cytosolic chaperone, were noted. Thus, it was concluded that mtDNA depletion resulted in mitochondria-specific stress response marked by the induction of mitochondrial chaperones [56]. Apart from mtDNA depletion, the accumulation of unfolded protein within the mitochondrial matrix can also result in the induction of mitochondria-specific stress proteins, such as HSP10 and HSP60 [57]. The knockdown of paraplegin (or SPG7), a mitochondrially localized protease, also induces mitochondrial stress response by impairing the assemblies of multiprotein complexes [58]. Therefore, in analogy to UPRER, the transcriptional induction of mitochondrial stress proteins in response to the accumulation of unfolded proteins within mitochondria was termed as the UPRmt. Together, these findings support the notion that mitochondrial protein misfolding and accumulation of insoluble mitochondrial proteins activates UPRmt [57, 59–61].
Destabilizing genetic mutations and oxidizing conditions can cause accumulation of insoluble aggregates of unfolded proteins in cells, which are not cleared efficiently by mitochondrial proteases [60, 61]. Both aging and hypoxia induce the formation of insoluble mitochondrial proteins (aggregates) that can be resolved after enhanced activation of UPRmt [60]. Aging results in accumulation of mutations in both the mitochondrial and nuclear genomes. However, compared to nDNA, the mtDNA is more susceptible to mutations due to the lack of protective histones and fewer DNA repair mechanisms within mitochondria [62–64]. The oxidative environment and lack of protective histones within mitochondria renders mtDNA more susceptible to damage by ROS. ROS can also directly oxidize mitochondrial proteins leading to their misfolding [65–68]. Thus, both mtDNA mutations and mitochondrial protein oxidation can induce UPRmt.
The mitochondrial proteome is comprised of 1200–1500 proteins of mitochondrial and nuclear origins [69]. Only 13 of these, which are components of the OXPHOS system, are encoded by mtDNA [30, 70]. The remaining mitochondrial proteins including OXPHOS proteins are encoded by nDNA. The 13 mtDNA-encoded proteins must exist in a state of balanced stoichiometry with the rest of the nuclear-encoded OXPHOS proteins. Therefore, mtDNA depletion and mutations result in a “mitochondrial-nuclear imbalance” and trigger activation of UPRmt [17, 71]. The status of UPRmt activation ultimately determines the cell fate. The cancer cells may utilize UPRmt to evade apoptosis and ensure their survival similar to cardiomyocytes [72].
The key proteins involved in UPRmt are chaperones HSP10, HSP60, and mtHSP70 and proteases ClpP and LONP1. Among these, HSP60 is a key component of UPRmt activation process across different species (Box 1). HSP10, HSP60, and mtHSP70 work together to properly fold denatured and nascent polypeptides [73]. The ClpP and LONP1 proteases cleave the irreversibly damaged proteins and mark them for degradation [74, 75]. Collectively, the chaperones and proteases restore proteostasis within mitochondria, and thus promote cell survival. Apart from the chaperones and proteases that resolve misfolded mitochondrial proteins, UPRmt activation also induces expression of gene sets including those involved in metabolic adaptation such as glycolytic genes, OXPHOS recovery, and antioxidants such as MnSOD2 [76–78]. When the level of proteotoxic stress is insurmountable, then mitochondria release cytochrome c into the cytosol and initiate the intrinsic apoptotic pathway [79].
Box 1. HSP60 oligomerization, protein folding, proteotoxic stress in mitochondria.
HSP60 is a 60-kDa protein encoded by the nuclear HSPD1 gene [144] and translated in the cytosol. The newly translated peptide contains a mitochondrial import signal (MIS) [145] at the N-terminus [146]. The MIS is a sequence of 26 amino acids, the majority of which are hydroxylated, and contributes to mitochondrial import in cells [146]. The MIS is cleaved upon mitochondrial import and HSP60 reaches its final conformation [146, 147]. Each HSP60 monomer has three domains: the apical domain, intermediate domain, and equatorial domain. The equatorial domain contains the ATP-binding site, the apical domain binds unfolded proteins, and the intermediate domain acts as a hinge by connecting the apical and the equatorial domain. Once in the mitochondria, HSP60 monomers self-assemble into ring-shaped heptameric oligomers, two of which associate to form a barrel-shaped tetradecamer [73]. In the inactive state (ATP unbound), the exposed residues of the apical domain in the core of the barrel-like structure are mostly hydrophobic and bind unfolded, non-native proteins. In the active state, ATP binds the equatorial domain and the co-chaperonin HSP10 covers the activated complex. The culmination of these two events induce a conformational shift and causes hydrophilic residues of the apical domain to become exposed in the core of the barrel. This charge turnover from hydrophobic to hydrophilic drives the folding of the encapsulated substrate proteins and their subsequent release from the HSP60-HSP10 complex [73, 148–150]. Reduced expression of mitochondrial chaperones causes a buildup of misfolded protein aggregates resulting in a proteotoxic stress [78, 151]. The proteotoxic stress triggers ATF5 to translocate to the nucleus and activate UPRmt.
Retrograde Signaling and Mitochondrial Stress Relieving Response Pathways
To sense and mitigate mitochondrial stress, mitochondria-to-nuclear (retrograde) signaling is activated. The signals from mitochondria are relayed to the cytosol and nucleus by mitochondrial metabolites, such as TCA cycle intermediates, ATP, and ROS, which initiate key protein modifications (e.g. histone acetylation) and activate transcriptional regulatory elements in nuclear genes [80–82]. Several transcription factors are involved in UPRmt activation. For example, the transcription factor CCAAT-enhancer-binding protein homologous protein (CHOP) is known to activate UPRmt. AKT and AMPK are proposed to serve as sensors, which detect proteotoxic stress and elevations of ROS in mitochondria, leading to activation of CCAAT-enhancer-binding protein beta (C/EBPβ), followed by induction of CHOP [57, 83, 84]. Stress in the inter-membrane space of mitochondria, and not in the mitochondrial matrix, induces elevated levels of CHOP [85]. However, C/EBPβ and CHOP are better known to activate UPRER and are not specific to the activation of UPRmt [86]. Unlike CHOP, a UPRmt-specific transcription factor, ATFS-1 has been discovered in C. elegans [87]. Under normal conditions, ATFS-1 accumulates in the mitochondria where it is constantly degraded by the LONP1 protease [88]. Under stress conditions, mitochondrial protein import becomes impaired and results in cytosolic accumulation of ATFS-1 [88]. Because ATFS-1 has both mitochondrial and nuclear localization signals, its cytosolic accumulation results in nuclear translocation where it facilitates the transcription of genes involved in UPRmt and OXPHOS [77, 88]. ATFS-1 targets include HSP60, mtHSP70 and nuclear-encoded OXPHOS subunits. Thereby, ATFS-1 ultimately promotes OXPHOS recovery during mitochondrial stress [77]. It is interesting to note that UPRmt activation limits pathogen infection as ATFS-1 induces innate immune genes [89, 90], which may modulate host cell signaling to promote cellular proliferation and cancer [91].
The mammalian homolog of ATFS-1 is activating transcription factor 5 (ATF5), which can be activated by heat shock [92], amino acid depletion [93], inhibition of proteasome function [94], increases in ROS [67, 94], and ER stress [94, 95]. ATF5 complements the genetic ATFS-1 deficiency in C. elegans by rescuing UPRmt [67]. ATF5 may induce UPRmt using a similar mechanism to ATFS-1 because both localize to mitochondria and the nucleus. ATF5 is required for transcriptional induction of HSP60, mtHSP70, and LONP1 in response to paraquat, a mitotoxic agent [67]. Like ATFS-1, the knockdown of ATF5 reduces mitochondrial respiration. ATF5 promotes OXPHOS recovery after mitochondrial stress [67]. In addition to transactivating UPRmt components, ATF-5 is also reported to regulate expression levels of Egr-1, BCL-2, and MCL1 to mediate proliferation and survival in cancer [96–98]. ATF5 has been upregulated in many types of cancer such as colorectal, breast and pancreatic cancer, and contribute to tumor cell cells survival and growth [82, 99, 100]. Furthermore, high ATF5 levels in lung cancer and malignant glioma are correlated with reduced survival in patients [101, 102]. Therefore, pharmacological inhibition of ATF5 may provide therapeutic benefits to patients with cancer.
The activation of retrograde signaling ensures that mitochondrial stress is relieved. However, the presence of persistent stress may lead to dysfunctional mitochondria, which either leads to mitophagy or apoptosis. Mitophagy is initiated by identification of severely damaged mitochondria, which are tagged by PTEN-induced putative kinase 1 (PINK1)-dependent phosphorylation of ubiquitin ligase Parkin. The activated Parkin leads to poly-ubiquitination of multiple outer mitochondrial proteins, which target mitochondria for degradation upon their fusion with lysosomes [103, 104].
In the presence of a persistent internal stress, the mitochondrial outer membrane is permeabilized to release cytochrome c into the cytosol. Cytochrome c binds with the apoptotic protease-activating factor 1 (Apaf-1), which recruits caspase 9 to form the apoptosome, a large protein complex. The apoptosome activates caspase 9, which subsequently leads to the activation of executioner caspases such as caspase-3 and −7. This cascade of activating caspases initiates the process of cellular degradation via apoptosis [105–110]. Apoptosis can be inhibited by the prevention of cytochrome c release from mitochondria into the cytosol or prevention of cytochrome-c interaction with Apaf-1 by many factors including intracellular nucleotide pool [111–114]. Thus, mitochondrial dysfunction in the form of cytochrome c release plays a key role in initiating apoptosis. Each cell type has an apoptotic threshold, which is determined by mitochondrial homeostasis in the presence of stress. When a stress disturbs mitochondrial homeostasis beyond the threshold, then cells initiate apoptosis. Compared to normal cells, cancer cells raise their apoptotic threshold by overexpressing prosurvival factors as well as by the activation of UPRmt (Box 2) [79, 115].
Box 2. Constitutive activation of the UPRmt enhances apoptosis threshold in cancer cells.
Elevated HSP60 expression has been observed in many cancers such as acute myeloid leukemia, breast ductal invasive carcinoma, pancreatic ductal adenocarcinoma, ovarian carcinoma, prostate adenocarcinoma, and others [135, 152]. Protective function of HSP60 and UPRmt is based on hypothesis that it is a “chaperonopathy by mistake” [153]. Thus, instead of protecting the host, HSP60 facilitates cancer cell growth and proliferation by increasing the apoptotic threshold of the cell via folding and refolding of oncoproteins and denatured/partially misfolded proteins within mitochondria. An increase in the ratio of mutated and misfolded proteins to chaperones can trigger UPRmt. The UPRmt enhances the protein-folding capacity of the mitochondria by activating the transcription factor ATF5 and CHOP, which promotes transcription of genes involved in mitochondria quality control such as the protease LONP1, and chaperonins HSP10 and HSP60 [67, 154]. Therefore, the elevated HSP60 expression within cancer cells may be a sign of constitutively activated UPRmt, which would increase the apoptotic threshold. Thus, increased HSP60 works against the host by aiding tumor progression via constitutive activation of the UPRmt [153].
Regulation of UPRmt by Mitochondrial Bioenergetics
The key role of UPRmt is to minimize the impact of mitochondrial stress and protect cells. Cells may experience mitochondrial stress by different ways, e.g. by OXPHOS defects and ROS production. A typical strategy used to induce UPRmt in cell culture is the treatment of cells with paraquat. Paraquat causes superoxide production (O2.−) by receiving electrons from the respiratory chain Complex III [67, 116]. The electrons from Complex III are trapped by paraquat before they reach Complex IV. Thus, paraquat disrupts electron flow along the respiratory chain and impairs OXPHOS, resulting in UPRmt activation [116]. The knockdown of proteases affecting OXPHOS biogenesis, such as spg-7 also induces UPRmt in C. elegans [77]. Partial loss-of-function mutation in NDUFS7, a subunit of the respiratory chain Complex I, also induces mitochondrial stress response [117]. Thus, OXPHOS defects may play a regulatory role in UPRmt activation. However, whether specific defects of the OXPHOS Complexes, e.g. isolated deficiencies of Complexes I-V, can induce UPRmt needs further confirmation.
The sirtuins (Sirt 1–7), a family of NAD-dependent enzymes, detect cellular energy perturbations and alter the metabolic state of cells [118]. NAD+ is used as a substrate by sirtuins such as Sirt1 and Sirt3 [119]. Sirt1 deacetylates PGC-1α, a master transcription factor controlling mitochondrial biogenesis [120]. The administration of NAD+ and overexpression of Sirt1 increases transcription and translation of HSP60, ClpP and superoxide dismutase (SOD2) genes, which are involved in UPRmt [121]. Sirt3 has been implicated in maintaining mitochondrial stability by activating antioxidant machinery during periods of mitochondrial proteotoxic stress [78]. The members of poly ADP-ribose polymerase (PARP) family also uses NAD+ as a substrate, and compete with sirtuins for the use of NAD+ [122]. An acute activation of PARP can deplete NAD+ pool and initiate cell death. In C. elegans, the inhibition of PARP increases lifespan by boosting mtDNA content and ATP production. In mammalian cells, PARP inhibition also elevates HSP60, ClpP and SOD2 protein levels in a Sirt1-dependent manner [121]. Sirt1 is a key nuclear transcription factor, whose activity is highly responsive to NAD+ levels. Therefore, PARP inhibition promotes UPRmt by making NAD+ available for Sirt1 [121].
The mitochondrial maintenance transcription factor nuclear respiratory factor 1 (NRF1) recruits Sirt7 to the promoters of mitochondrial ribosomal proteins and translational factors. In doing so, NRF1 represses mitochondrial metabolic activity and reduces protein-folding stress to promote stem cell quiescence [123]. Loss of Sirt7 increases transcription of HSP10, HSP60, ClpP, mtDNA content, and cell proliferation. These phenotypes are reversible when Sirt7 was reintroduced to the system [123].
Cancer cells rely on functional mitochondria to generate macromolecules required for unchecked proliferative capacity. To meet the increased demand for proliferation, cancer cells reprogram their metabolism to enhance macromolecule biosynthesis, readjust bioenergetics, and redox status [124]. They also take up macromolecules from their microenvironment. Because of the increased demand for amino acids and protein synthesis, some of the non-essential amino acids may become essential for cancer growth. The TCA cycle is the hub of anabolic and catabolic metabolism. It is dynamically linked with the OXPHOS. The function of the respiratory chain is dependent on an adequate supply of NADH and FADH2. Severe deficiency of the respiratory chain causes accumulation of NADH resulting in reductive stress, particularly inside mitochondria. The build-up of NADH blocks the TCA cycle by the feedback inhibition of the NAD+-dependent dehydrogenases. This results in oxaloacetate deficiency, which is required to generate the amino acids such as aspartate. Aspartate is a nonessential amino acid synthesized inside mitochondria. Severe respiratory chain deficiency makes aspartate essential for cell proliferation [125]. The role of the respiratory chain in cellular physiology has been rediscovered after about 40 years. In late 1970s, the Scheffler group at University of California San Diego demonstrated that genetic respiratory chain deficiency could block the TCA cycle. The respiration-deficient lung fibroblasts were unable to grow in a bicarbonate-free medium. In addition, the respiration-deficient mutants also became dependent on aspartate and asparagine for their growth [126–128]. Aspartate is used in the urea cycle and released as fumarate, which links the urea cycle with the TCA cycle. The most important role of urea cycle is to clear toxic ammonia generated from the protein metabolism. The overexpression of a mutant ornithine decarboxylase (OTC), a urea cycle enzyme, in mitochondria led to the discovery of mitochondria-specific stress response or the UPRmt [57]. Therefore, mitochondrial metabolism controls multiple aspects of cellular physiology. The major ones being the TCA and urea cycles, and Fe-S cluster biosynthesis. Alterations in the TCA cycle and urea cycles are implicated in cancer development [129, 130]. Without the UPRmt activation, the mitochondria in cancer cells may not meet the demand for macromolecules biosynthesis required for cell growth and proliferation. Thus, UPRmt is essential to maintain proper mitochondrial function to sustain unchecked proliferation during tumorigenesis (Box 2).
Concluding Remarks
The UPRmt is a mitochondrial stress specific response. Like UPRER [131], the UPRmt can play a pivotal role in evading apoptosis by alleviating mitochondrial stress in cancer cells. Cancer cells experience mitochondrial stress as they undergo unchecked cellular proliferation and generate ROS, which damage mtDNA and crucial mitochondrial proteins, such as components of the OXPHOS family causing mitochondrial dysfunction. Cancer cells rely on functional mitochondria to generate macromolecules such as amino acids, nucleotides and cholesterol to maintain their high proliferative capacity. Thus, cancer cells activate the mitochondrial stress response to alleviate mitochondrial dysfunction and protein aggregation, which subsequently promotes tumor growth and progression (Figure 2). These notions have been validated by recent findings indicating that persistent activation of UPRmt provides survival advantage to cancer cells leading to tumor progression [22], suggesting that a mediator of UPRmt such as superoxide dismutase 1 (SOD1) may represent a therapeutic target in cancer [132]. Mitochondrial stress relieving response requires inter-organellar communication, retrograde signaling, mitophagy, mitochondrial bioenergetics, mitochondrial dynamics, and apoptosis. These cellular and biochemical signaling pathways provide multiple targets to develop efficacious therapeutics in various types of cancer (Figure 3). HSP60, a key player of the UPRmt, is overexpressed in many cancer types, and its expression has been correlated with the metastatic potential of cancers and overall survival of cancer patients [133–137]. HSP60 upregulation within mitochondria raises cancer cells threshold for surviving stressful conditions. The extramitochondrial HSP60 may also promote tumor growth, apoptosis resistance, and metastatic spread (Box 3). An upstream regulator of UPRmt, ATF5 functions as a pro-survival protein and promotes tumor progression and therapeutic resistance [98, 99, 138]. Increased mitochondrial fission mediated by Drp1 promotes tumor growth [50, 51] and pharmacological inhibition of mitochondrial fission improve survival in mice [139]. Importantly, targeting the components of mitochondrial stress response, such as mitochondrial ClpP protease and other mitochondrial peptidase, have shown selective therapeutic efficacy in cancer cells [140–143]. Although inhibiting UPRmt offers a unique therapeutic strategy for targeting cancer cells, important challenges need to be addressed experimentally to exploit mitochondrial stress response pathways in cancer (see Outstanding Questions).
Figure 2. Mitochondrial stress response promotes cancer growth, progression and therapeutic resistance in cancer.

A schematic describing how different stresses induce mitochondrial dysfunction leading to the activation of the mitochondrial stress response, restoration of mitochondrial integrity, cancer cell survival, which subsequently promotes tumor growth and progression.
Figure 3. Mitochondrial stress response (UPRmt) regulates various cellular signaling and functions.

A graphical representation depicting the overarching reach of UPRmt signaling in various cellular functions including bioenergetics, mitochondrial homeostasis, ROS detoxification, cell survival and proliferation.
Box 3. The extramitochondrial roles of HSP60 in cancer.
In addition to its established localization in mitochondria, HSP60 also localizes to other cellular compartments such as cytosol and may contribute to malignant phenotypes (Figure I). Cytosolic HSP60 promotes cancer cell survival and contributes to malignant phenotypes function [155]. Cytosolic HSP60 interacts with the IKK complex and enhances the activation of IKK [155]. This interaction directly increases the expression of NF-κB targets such as Bfl-1/A-1 and MnSOD, and promotes cancer cell survival under stressful conditions [155]. Cytosolic HSP60 enhances IKK activation independent of its chaperone activity, suggesting alternative functions of HSP60 beyond protein folding (Figure I) [155].
Cytosolic HSP60 also interacts with proapoptotic proteins Bax and Bak but not with antiapoptotic Bcl-2 in adult cardiac myocytes [156]. HSP60 interacts with the tumor suppressor p53 and restrain its transcriptional function, which reduces Bax expression [134]. HSP60 upregulation also sequesters Bax in the cytosol, and thus inhibits onset of apoptosis in cancer cells (Figure I).
Overexpression of HSP60 significantly increases cellular migration and invasion in vitro as well as increased tumor volume and metastasis in vivo [157]. HSP60 directly interacts with β-catenin via its apical domain. β-catenin expression is critical for HSP60-mediated in vitro and in vivo metastatic activity [157] implying that HSP60 overexpression represents an alternate route for β-catenin activation. Intriguingly, the reverse process occurs in neuronal tissue, whereby canonical Wnt signaling (i.e., β-catenin -dependent) induces UPRmt [158]. HSP60 is highly expressed on the surface of pancreatic metastatic cells. In these cancer cells, HSP60 seems to play a critical role in metastasis [159]. Surface HSP60 interacts with proteins involved in metastasis e.g. integrin α3β1 [160]. Activated integrin α3β1 induces motility and cell adhesion property in breast cancer cells [161] via preferential association with HSP60 located on the cell surface. Exogenous addition of HSP60 also activates integrin α3β1 and increases cell motility in breast cancer cells [160]. Conversely, mizoribine, the HSP60-binding drug, inhibits HSP60 association with integrin α3β1 [160]. Thus, activation of integrin α3β1 by HSP60 is independent of its chaperone activity (Figure I).
Outstanding questions.
-
Cellular stresses are sensed by various organelles with diverse mechanisms of action.
How is the specificity of mitochondrial stress response determined?
-
How does ATF5 translocate from mitochondria to the nucleus during stress response?
This mechanism was elucidated in the homolog ATFS-1 in C.elegans but has yet to be determined in cancer cells.
How does the mitochondria relay stress signal to other cellular organelles such as endoplasmic reticulum and nucleus in cancer cells?
What are the underlying mechanisms of activation of ATF5 and retrograde signaling during mitochondrial stress response in cancer?
HSP60, a key component of mitochondrial stress response, contains mitochondrial localization sequence and is traditionally located in the mitochondrial matrix. How does HSP60 regulate tumor promoting factors localized in the cytosol and nucleus?
Figure I. Extramitochondrial roles of HSP60.
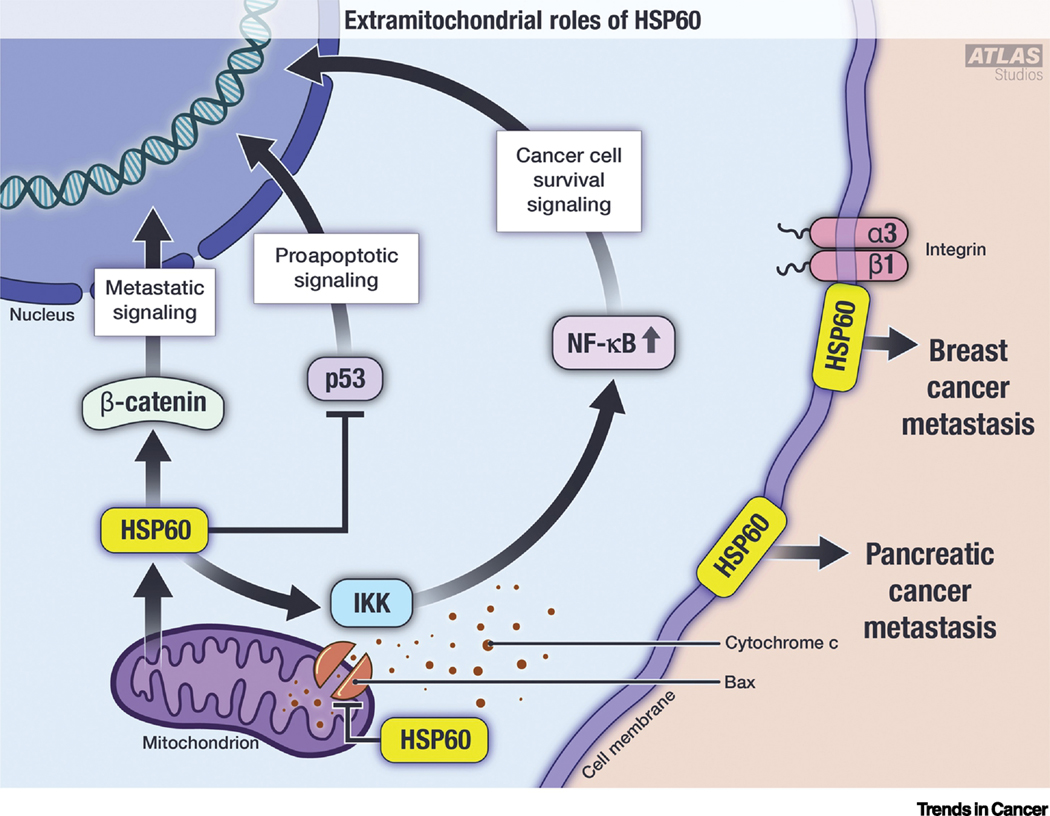
Graphical representation of nontraditional roles of HSP60. Cytosolic HSP60 independently interacts with β-catenin, IKK and p53 to induce metastatic signaling, cell survival, and inhibit pro-apoptotic signaling, respectively. Cytosolic HSP60 also binds with Bax at the mitochondrial membrane to inhibit cytochrome c release and inhibit intrinsic apoptosis. On the cell surface, HSP60 interacts with integrin α3β1 to promote breast cancer metastasis. Cell surface HSP60 is also implicated in pancreatic cancer metastasis.
Highlights.
Cancer cell mitochondria are susceptible to oxidative stress due to inherent production of reactive oxygen species and a lack of robust protective mechanisms causing persistent mitochondrial stress.
Cancer cells activate mitochondrial stress response to alleviate endogenous stresses in order to prolong survival, enhance proliferation and metastatic potential.
Mitochondrial unfolded protein response (UPRmt) comprising chaperones and proteases
remove proteotoxic stress to maintain cellular homoeostasis.
Elevated expression of HSP60 and its upstream regulator ATF5 enhances apoptotic threshold in cancer cells leading to therapeutic resistance in various types of cancer.
Targeting of mitochondrial stress response by pharmacologic inhibition of its components provides novel therapeutic targets in cancer.
Acknowledgements
This work was supported by the National Cancer Institute of the National Institutes of Health under Award Number RO1-CA160685 and the American Cancer Society Research Scholar Grant RSG-12-214-01 - CCG to DC, and in part by the National Cancer Institute Center Support Grant P30-CA016056 to the Roswell Park Comprehensive Cancer Center. We apologize to those colleagues whose publications inadvertently could not be cited.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Davis RT et al. (2020) Transcriptional diversity and bioenergetic shift in human breast cancer metastasis revealed by single-cell RNA sequencing. Nat Cell Biol 22 (3), 310–320. [DOI] [PubMed] [Google Scholar]
- 2.Lee KM et al. (2017) MYC and MCL1 Cooperatively Promote Chemotherapy-Resistant Breast Cancer Stem Cells via Regulation of Mitochondrial Oxidative Phosphorylation. Cell Metab 26 (4), 633–647 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Molina JR et al. (2018) An inhibitor of oxidative phosphorylation exploits cancer vulnerability. Nat Med 24 (7), 1036–1046. [DOI] [PubMed] [Google Scholar]
- 4.Weinberg SE and Chandel NS (2015) Targeting mitochondria metabolism for cancertherapy. Nat Chem Biol 11 (1), 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spinelli JB and Haigis MC (2018) The multifaceted contributions of mitochondria tocellular metabolism. Nat Cell Biol 20 (7), 745–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson NM et al. (2018) The emerging role and targetability of the TCA cycle in cancer metabolism. Protein Cell 9 (2), 216–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Carracedo A et al. (2013) Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer 13 (4), 227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma Y et al. (2018) Fatty acid oxidation: An emerging facet of metabolic transformation in cancer. Cancer Lett 435, 92–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez-Outschoorn UE et al. (2017) Cancer metabolism: a therapeutic perspective. NatRev Clin Oncol 14 (1), 11–31. [DOI] [PubMed] [Google Scholar]
- 10.Miller WL (2007) Steroidogenic acute regulatory protein (StAR), a novel mitochondrial cholesterol transporter. Biochim Biophys Acta 1771 (6), 663–76. [DOI] [PubMed] [Google Scholar]
- 11.Twiddy AL et al. (2011) Cholesterol as a potential target for castration-resistant prostate cancer. Pharm Res 28 (3), 423–37. [DOI] [PubMed] [Google Scholar]
- 12.Bose HS et al. (2002) Rapid regulation of steroidogenesis by mitochondrial protein import. Nature 417 (6884), 87–91. [DOI] [PubMed] [Google Scholar]
- 13.Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144 (5), 646–74. [DOI] [PubMed] [Google Scholar]
- 14.Quiros PM et al. (2016) Mitonuclear communication in homeostasis and stress. Nat RevMol Cell Biol 17 (4), 213–26. [DOI] [PubMed] [Google Scholar]
- 15.Pakos-Zebrucka K et al. (2016) The integrated stress response. EMBO Rep 17 (10), 1374–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hetz C (2012) The unfolded protein response: controlling cell fate decisions under ER stress and beyond. Nat Rev Mol Cell Biol 13 (2), 89–102. [DOI] [PubMed] [Google Scholar]
- 17.Jovaisaite V et al. (2014) The mitochondrial unfolded protein response, a conserved stress response pathway with implications in health and disease. J Exp Biol 217 (Pt 1), 137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chevet E et al. (2015) Endoplasmic reticulum stress-activated cell reprogramming in oncogenesis. Cancer Discov 5 (6), 586–97. [DOI] [PubMed] [Google Scholar]
- 19.Fiorese CJ and Haynes CM (2017) Integrating the UPR(mt) into the mitochondrial maintenance network. Crit Rev Biochem Mol Biol 52 (3), 304–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qureshi MA et al. (2017) The mitochondrial unfolded protein response: Signaling from the powerhouse. J Biol Chem 292 (33), 13500–13506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Melber A and Haynes CM (2018) UPR(mt) regulation and output: a stress response mediated by mitochondrial-nuclear communication. Cell Res 28 (3), 281–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kenny TC et al. (2019) Mitohormesis Primes Tumor Invasion and Metastasis. Cell Rep 27 (8), 2292–2303 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carrasco DR et al. (2007) The differentiation and stress response factor XBP-1 drives multiple myeloma pathogenesis. Cancer Cell 11 (4), 349–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Oakes SA (2017) Endoplasmic reticulum proteostasis: a key checkpoint in cancer. Am J Physiol Cell Physiol 312 (2), C93–C102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ward PS and Thompson CB (2012) Metabolic reprogramming: a cancer hallmark even warburg did not anticipate. Cancer Cell 21 (3), 297–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kreuzaler P et al. (2020) Adapt and conquer: Metabolic flexibility in cancer growth, invasion and evasion. Mol Metab 33, 83–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Guerra F et al. (2017) Mitochondria and cancer chemoresistance. Biochim Biophys Acta Bioenerg 1858 (8), 686–699. [DOI] [PubMed] [Google Scholar]
- 28.Callegari S and Dennerlein S (2018) Sensing the Stress: A Role for the UPR(mt) and UPR(am) in the Quality Control of Mitochondria. Front Cell Dev Biol 6, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valera-Alberni M and Canto C (2018) Mitochondrial stress management: a dynamic journey. Cell Stress 2 (10), 253–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yadav N and Chandra D (2013) Mitochondrial DNA mutations and breast tumorigenesis. Biochim Biophys Acta 1836 (2), 336–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chacinska A et al. (2009) Importing mitochondrial proteins: machineries and mechanisms. Cell 138 (4), 628–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Warburg O (1956) On the origin of cancer cells. Science 123 (3191), 309–14. [DOI] [PubMed] [Google Scholar]
- 33.Warburg O (1956) On respiratory impairment in cancer cells. Science 124 (3215), 269–70. [PubMed] [Google Scholar]
- 34.Yu L et al. (2017) Modeling the Genetic Regulation of Cancer Metabolism: Interplay between Glycolysis and Oxidative Phosphorylation. Cancer Res 77 (7), 1564–1574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pavlova NN and Thompson CB (2016) The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23 (1), 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weidberg H and Amon A (2018) MitoCPR-A surveillance pathway that protects mitochondria in response to protein import stress. Science 360 (6385). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Deus CM et al. (2020) Mitochondria-Lysosome Crosstalk: From Physiology to Neurodegeneration. Trends Mol Med 26 (1), 71–88. [DOI] [PubMed] [Google Scholar]
- 38.Liu Y and Zhu X (2017) Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl Neurodegener 6, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kerkhofs M et al. (2018) Emerging molecular mechanisms in chemotherapy: Ca(2+) signaling at the mitochondria-associated endoplasmic reticulum membranes. Cell Death Dis 9 (3), 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Morciano G et al. (2018) Role of Mitochondria-Associated ER Membranes in CalciumRegulation in Cancer-Specific Settings. Neoplasia 20 (5), 510–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Carreras-Sureda A et al. (2019) Non-canonical function of IRE1alpha determines mitochondria-associated endoplasmic reticulum composition to control calcium transfer and bioenergetics. Nat Cell Biol 21 (6), 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhandary B et al. (2012) An involvement of oxidative stress in endoplasmic reticulum stress and its associated diseases. Int J Mol Sci 14 (1), 434–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bravo R et al. (2011) Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci 124 (Pt 13), 2143–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hori O et al. (2002) Transmission of cell stress from endoplasmic reticulum to mitochondria: enhanced expression of Lon protease. J Cell Biol 157 (7), 1151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yoboue ED et al. (2018) Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis 9 (3), 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bonora M et al. (2017) Mitochondrial permeability transition involves dissociation of F1FOATP synthase dimers and C-ring conformation. EMBO Rep 18 (7), 1077–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Baumgartner HK et al. (2009) Calcium elevation in mitochondria is the main Ca2+ requirement for mitochondrial permeability transition pore (mPTP) opening. J Biol Chem 284 (31), 20796–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Philley JV et al. (2016) Complex-I Alteration and Enhanced Mitochondrial Fusion Are Associated With Prostate Cancer Progression. J Cell Physiol 231 (6), 1364–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Brito OM and Scorrano L (2008) Mitofusin 2 tethers endoplasmic reticulum tomitochondria. Nature 456 (7222), 605–10. [DOI] [PubMed] [Google Scholar]
- 50.Rehman J et al. (2012) Inhibition of mitochondrial fission prevents cell cycle progression in lung cancer. FASEB J 26 (5), 2175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Serasinghe MN et al. (2015) Mitochondrial division is requisite to RAS-induced transformation and targeted by oncogenic MAPK pathway inhibitors. Mol Cell 57 (3), 521–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Perisic O et al. (1989) Stable binding of Drosophila heat shock factor to head-to-head and tail-to-tail repeats of a conserved 5 bp recognition unit. Cell 59 (5), 797–806. [DOI] [PubMed] [Google Scholar]
- 53.Donnelly N et al. (2014) HSF1 deficiency and impaired HSP90-dependent protein folding are hallmarks of aneuploid human cells. EMBO J 33 (20), 2374–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lellahi SM et al. (2018) The long noncoding RNA NEAT1 and nuclear paraspeckles are up-regulated by the transcription factor HSF1 in the heat shock response. J Biol Chem 293 (49), 18965–18976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schopf FH et al. (2017) The HSP90 chaperone machinery. Nat Rev Mol Cell Biol 18 (6),345–360. [DOI] [PubMed] [Google Scholar]
- 56.Martinus RD et al. (1996) Selective induction of mitochondrial chaperones in response toloss of the mitochondrial genome. Eur J Biochem 240 (1), 98–103. [DOI] [PubMed] [Google Scholar]
- 57.Zhao Q et al. (2002) A mitochondrial specific stress response in mammalian cells. EMBO J21 (17), 4411–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yoneda T et al. (2004) Compartment-specific perturbation of protein handling activates genes encoding mitochondrial chaperones. J Cell Sci 117 (Pt 18), 4055–66. [DOI] [PubMed] [Google Scholar]
- 59.Iosefson O et al. (2012) Reactivation of protein aggregates by mortalin and Tid1—the human mitochondrial Hsp70 chaperone system. Cell Stress Chaperones 17 (1), 57–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kaufman DM et al. (2017) Ageing and hypoxia cause protein aggregation in mitochondria. Cell Death Differ 24 (10), 1730–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ruan L et al. (2017) Cytosolic proteostasis through importing of misfolded proteins into mitochondria. Nature 543 (7645), 443–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Khrapko K et al. (1997) Mitochondrial mutational spectra in human cells and tissues. Proc Natl Acad Sci U S A 94 (25), 13798–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pinz KG et al. (1995) Action of mitochondrial DNA polymerase gamma at sites of base loss or oxidative damage. J Biol Chem 270 (16), 9202–6. [DOI] [PubMed] [Google Scholar]
- 64.Labbadia J et al. (2017) Mitochondrial Stress Restores the Heat Shock Response and Prevents Proteostasis Collapse during Aging. Cell Rep 21 (6), 1481–1494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Muftuoglu M et al. (2014) Formation and repair of oxidative damage in the mitochondrial DNA. Mitochondrion 17, 164–81. [DOI] [PubMed] [Google Scholar]
- 66.Han Y and Chen JZ (2013) Oxidative stress induces mitochondrial DNA damage and cytotoxicity through independent mechanisms in human cancer cells. Biomed Res Int 2013, 825065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fiorese CJ et al. (2016) The Transcription Factor ATF5 Mediates a Mammalian Mitochondrial UPR. Curr Biol 26 (15), 2037–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sena LA and Chandel NS (2012) Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48 (2), 158–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Calvo SE and Mootha VK (2010) The mitochondrial proteome and human disease. Annu Rev Genomics Hum Genet 11, 25–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chinnery PF and Hudson G (2013) Mitochondrial genetics. Br Med Bull 106, 135–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Houtkooper RH et al. (2013) Mitonuclear protein imbalance as a conserved longevity mechanism. Nature 497 (7450), 451–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Smyrnias I et al. (2019) Cardioprotective Effect of the Mitochondrial Unfolded Protein Response During Chronic Pressure Overload. J Am Coll Cardiol 73 (14), 1795–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bukau B and Horwich AL (1998) The Hsp70 and Hsp60 chaperone machines. Cell 92 (3),351–66. [DOI] [PubMed] [Google Scholar]
- 74.Haynes CM and Ron D (2010) The mitochondrial UPR - protecting organelle protein homeostasis. J Cell Sci 123 (Pt 22), 3849–55. [DOI] [PubMed] [Google Scholar]
- 75.Haynes CM et al. (2007) ClpP mediates activation of a mitochondrial unfolded protein response in C. elegans. Dev Cell 13 (4), 467–80. [DOI] [PubMed] [Google Scholar]
- 76.Lin YF and Haynes CM (2016) Metabolism and the UPR(mt). Mol Cell 61 (5), 677–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Nargund AM et al. (2015) Mitochondrial and nuclear accumulation of the transcription factor ATFS-1 promotes OXPHOS recovery during the UPR(mt). Mol Cell 58 (1), 123–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Papa L and Germain D (2014) SirT3 regulates the mitochondrial unfolded protein response. Mol Cell Biol 34 (4), 699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yadav N and Chandra D (2014) Mitochondrial and post mitochondrial survival signaling in cancer. Mitochondrion 16, 18–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vyas S et al. (2016) Mitochondria and Cancer. Cell 166 (3), 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bohovych I and Khalimonchuk O (2016) Sending Out an SOS: Mitochondria as a Signaling Hub. Front Cell Dev Biol 4, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Deng P and Haynes CM (2017) Mitochondrial dysfunction in cancer: Potential roles ofATF5 and the mitochondrial UPR. Semin Cancer Biol 47, 43–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kenny TC and Germain D (2017) From discovery of the CHOP axis and targeting ClpP to the identification of additional axes of the UPRmt driven by the estrogen receptor and SIRT3. J Bioenerg Biomembr 49 (4), 297–305. [DOI] [PubMed] [Google Scholar]
- 84.Basu SK et al. (2018) A RAS-CaMKKbeta-AMPKalpha2 pathway promotes senescence by licensing post-translational activation of C/EBPbeta through a novel 3’UTR mechanism. Oncogene 37 (26), 3528–3548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Papa L and Germain D (2011) Estrogen receptor mediates a distinct mitochondrial unfolded protein response. J Cell Sci 124 (Pt 9), 1396–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Rozpedek W et al. (2016) The Role of the PERK/eIF2alpha/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress. Curr Mol Med 16 (6), 533–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Haynes CM et al. (2010) The matrix peptide exporter HAF-1 signals a mitochondrial UPR by activating the transcription factor ZC376.7 in C. elegans. Mol Cell 37 (4), 529–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Nargund AM et al. (2012) Mitochondrial import efficiency of ATFS-1 regulates mitochondrial UPR activation. Science 337 (6094), 587–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Pellegrino MW et al. (2014) Mitochondrial UPR-regulated innate immunity provides resistance to pathogen infection. Nature 516 (7531), 414–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Deng P et al. (2019) Mitochondrial UPR repression during Pseudomonas aeruginosa infection requires the bZIP protein ZIP-3. Proc Natl Acad Sci U S A 116 (13), 6146–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.van Elsland D and Neefjes J (2018) Bacterial infections and cancer. EMBO Rep 19 (11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang H et al. (2007) ATF5 promotes cell survival through transcriptional activation ofHsp27 in H9c2 cells. Cell Biol Int 31 (11), 1309–15. [DOI] [PubMed] [Google Scholar]
- 93.Watatani Y et al. (2007) Amino acid limitation induces expression of ATF5 mRNA at the post-transcriptional level. Life Sci 80 (9), 879–85. [DOI] [PubMed] [Google Scholar]
- 94.Zhou D et al. (2008) Phosphorylation of eIF2 directs ATF5 translational control in response to diverse stress conditions. J Biol Chem 283 (11), 7064–73. [DOI] [PubMed] [Google Scholar]
- 95.Torres-Peraza JF et al. (2013) Protective neuronal induction of ATF5 in endoplasmic reticulum stress induced by status epilepticus. Brain 136 (Pt 4), 1161–76. [DOI] [PubMed] [Google Scholar]
- 96.Liu DX et al. (2011) p300-Dependent ATF5 acetylation is essential for Egr-1 gene activation and cell proliferation and survival. Mol Cell Biol 31 (18), 3906–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Dluzen D et al. (2011) BCL-2 is a downstream target of ATF5 that mediates the prosurvival function of ATF5 in a cell type-dependent manner. J Biol Chem 286 (9), 7705–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Karpel-Massler G et al. (2016) A Synthetic Cell-Penetrating Dominant-Negative ATF5 Peptide Exerts Anticancer Activity against a Broad Spectrum of Treatment-Resistant Cancers. Clin Cancer Res 22 (18), 4698–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Angelastro JM (2017) Targeting ATF5 in Cancer. Trends Cancer 3 (7), 471–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sun X et al. (2020) Dominant-Negative ATF5 Compromises Cancer Cell Survival by Targeting CEBPB and CEBPD. Mol Cancer Res 18 (2), 216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sheng Z et al. (2010) A genome-wide RNA interference screen reveals an essential CREB3L2-ATF5-MCL1 survival pathway in malignant glioma with therapeutic implications. Nat Med 16 (6), 671–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ishihara S et al. (2015) Activating transcription factor 5 enhances radio resistance and malignancy in cancer cells. Oncotarget 6 (7), 4602–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lazarou M et al. (2015) The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524 (7565), 309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sarraf SA et al. (2013) Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496 (7445), 372–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Cain K et al. (2002) The Apaf-1 apoptosome: a large caspase-activating complex. Biochimie 84 (2–3), 203–14. [DOI] [PubMed] [Google Scholar]
- 106.Cheng TC et al. (2016) A near atomic structure of the active human apoptosome. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Langlais C et al. (2015) In Vitro Assembly and Analysis of the Apoptosome Complex. Cold Spring Harb Protoc 2015 (12), pdb prot087080. [DOI] [PubMed] [Google Scholar]
- 108.Wu CC et al. (2016) The Apaf-1 apoptosome induces formation of caspase-9 homo- and heterodimers with distinct activities. Nat Commun 7, 13565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhou M et al. (2015) Atomic structure of the apoptosome: mechanism of cytochrome c- and dATP-mediated activation of Apaf-1. Genes Dev 29 (22), 2349–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Zou H et al. (1999) An APAF-1.cytochrome c multimeric complex is a functional apoptosome that activates procaspase-9. J Biol Chem 274 (17), 11549–56. [DOI] [PubMed] [Google Scholar]
- 111.Bao Q et al. (2007) Calcium blocks formation of apoptosome by preventing nucleotide exchange in Apaf-1. Mol Cell 25 (2), 181–92. [DOI] [PubMed] [Google Scholar]
- 112.Cain K et al. (2001) Physiological concentrations of K+ inhibit cytochrome c-dependent formation of the apoptosome. J Biol Chem 276 (45), 41985–90. [DOI] [PubMed] [Google Scholar]
- 113.Chandra D et al. (2006) Intracellular nucleotides act as critical prosurvival factors by binding to cytochrome C and inhibiting apoptosome. Cell 125 (7), 1333–46. [DOI] [PubMed] [Google Scholar]
- 114.Mei Y et al. (2010) tRNA binds to cytochrome c and inhibits caspase activation. Mol Cell 37 (5), 668–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Gogada R et al. (2013) Bim, a proapoptotic protein, up-regulated via transcription factor E2F1-dependent mechanism, functions as a prosurvival molecule in cancer. J Biol Chem 288 (1), 368–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Castello PR et al. (2007) Mitochondria are a major source of paraquat-induced reactive oxygen species production in the brain. J Biol Chem 282 (19), 14186–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rauthan M et al. (2015) A Mutation in Caenorhabditis elegans NDUF-7 Activates the Mitochondrial Stress Response and Prolongs Lifespan via ROS and CED-4. G3 (Bethesda) 5 (8), 1639–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Gonzalez Herrera KN et al. (2015) Intersections between mitochondrial sirtuin signaling and tumor cell metabolism. Crit Rev Biochem Mol Biol 50 (3), 242–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.van de Ven RAH et al. (2017) Mitochondrial Sirtuins and Molecular Mechanisms of Aging. Trends Mol Med 23 (4), 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Khan SA et al. (2015) ATGL-catalyzed lipolysis regulates SIRT1 to control PGC- 1alpha/PPAR-alpha signaling. Diabetes 64 (2), 418–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Mouchiroud L et al. (2013) The NAD(+)/Sirtuin Pathway Modulates Longevity through Activation of Mitochondrial UPR and FOXO Signaling. Cell 154 (2), 430–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gibson BA and Kraus WL (2012) New insights into the molecular and cellular functions of poly(ADP-ribose) and PARPs. Nat Rev Mol Cell Biol 13 (7), 411–24. [DOI] [PubMed] [Google Scholar]
- 123.Mohrin M et al. (2015) Stem cell aging. A mitochondrial UPR-mediated metabolic checkpoint regulates hematopoietic stem cell aging. Science 347 (6228), 1374–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cairns RA et al. (2011) Regulation of cancer cell metabolism. Nat Rev Cancer 11 (2), 85–95. [DOI] [PubMed] [Google Scholar]
- 125.Birsoy K et al. (2015) An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162 (3), 540–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Ditta G et al. (1976) The selection of Chinese hamster cells deficient in oxidative energy metabolism. Somatic Cell Genet 2 (4), 331–44. [DOI] [PubMed] [Google Scholar]
- 127.Breen GA and Scheffler IE (1979) Respiration-deficient Chinese hamster cell mutants: biochemical characterization. Somatic Cell Genet 5 (4), 441–51. [DOI] [PubMed] [Google Scholar]
- 128.DeFrancesco L et al. (1976) A respiration-deficient Chinese hamster cell line with a defect in NADH-coenzyme Q reductase. J Biol Chem 251 (15), 4588–95. [PubMed] [Google Scholar]
- 129.Keshet R et al. (2018) Rewiring urea cycle metabolism in cancer to support anabolism. Nat Rev Cancer 18 (10), 634–645. [DOI] [PubMed] [Google Scholar]
- 130.Ryan DG et al. (2019) Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat Metab 1, 16–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shemorry A et al. (2019) Caspase-mediated cleavage of IRE1 controls apoptotic cell commitment during endoplasmic reticulum stress. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Gomez M and Germain D (2019) Cross talk between SOD1 and the mitochondrial UPR in cancer and neurodegeneration. Mol Cell Neurosci 98, 12–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hjerpe E et al. (2013) HSP60 predicts survival in advanced serous ovarian cancer. Int J Gynecol Cancer 23 (3), 448–55. [DOI] [PubMed] [Google Scholar]
- 134.Ghosh JC et al. (2008) Hsp60 regulation of tumor cell apoptosis. J Biol Chem 283 (8), 5188–94. [DOI] [PubMed] [Google Scholar]
- 135.Cappello F et al. (2008) Hsp60 expression, new locations, functions and perspectives for cancer diagnosis and therapy. Cancer Biol Ther 7 (6), 801–9. [DOI] [PubMed] [Google Scholar]
- 136.Tong WW et al. (2016) The tumor promoting roles of HSP60 and HIF2alpha in gastric cancer cells. Tumour Biol 37 (7), 9849–54. [DOI] [PubMed] [Google Scholar]
- 137.Li XS et al. (2014) Heat shock protein 60 overexpression is associated with the progression and prognosis in gastric cancer. PLoS One 9 (9), e107507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Ishihara S and Haga H (2015) ATF5: development of oncogenic resistance to radiotherapy Aging (Albany NY: ) 7 (7), 453–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yu M et al. (2019) Mitochondrial fusion exploits a therapeutic vulnerability of pancreatic cancer. JCI Insight 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cole A et al. (2015) Inhibition of the Mitochondrial Protease ClpP as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell 27 (6), 864–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Goard CA and Schimmer AD (2014) Mitochondrial matrix proteases as novel therapeutic targets in malignancy. Oncogene 33 (21), 2690–9. [DOI] [PubMed] [Google Scholar]
- 142.Ishizawa J et al. (2019) Mitochondrial ClpP-Mediated Proteolysis Induces Selective Cancer Cell Lethality. Cancer Cell 35 (5), 721–737 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Mirali S et al. (2020) The mitochondrial peptidase, neurolysin, regulates respiratory chain super complex formation and is necessary for AML viability. Sci Transl Med 12 (538). [DOI] [PubMed] [Google Scholar]
- 144.Stelzer G et al. (2016) The GeneCards Suite: From Gene Data Mining to Disease Genome Sequence Analyses. Curr Protoc Bioinformatics 54, 1 30 1–1 30 33. [DOI] [PubMed] [Google Scholar]
- 145.Shin BK et al. (2003) Global profiling of the cell surface proteome of cancer cells uncovers an abundance of proteins with chaperone function. J Biol Chem 278 (9), 7607–16. [DOI] [PubMed] [Google Scholar]
- 146.Singh B et al. (1990) Mitochondrial import of the human chaperonin (HSP60) protein. Biochem Biophys Res Commun 169 (2), 391–6. [DOI] [PubMed] [Google Scholar]
- 147.Cheng MY et al. (1990) The mitochondrial chaperonin hsp60 is required for its own assembly. Nature 348 (6300), 455–8. [DOI] [PubMed] [Google Scholar]
- 148.Fenton WA et al. (1994) Residues in chaperonin GroEL required for polypeptide binding and release. Nature 371 (6498), 614–9. [DOI] [PubMed] [Google Scholar]
- 149.Boisvert DC et al. (1996) The 2.4 A crystal structure of the bacterial chaperonin GroEL complexed with ATP gamma S. Nat Struct Biol 3 (2), 170–7. [DOI] [PubMed] [Google Scholar]
- 150.Braig K et al. (1994) The crystal structure of the bacterial chaperonin GroEL at 2.8 A. Nature 371 (6498), 578–86. [DOI] [PubMed] [Google Scholar]
- 151.Radke S et al. (2008) Mitochondrial protein quality control by the proteasome involves ubiquitination and the protease Omi. J Biol Chem 283 (19), 12681–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Zhou C et al. (2018) Oncogenic HSP60 regulates mitochondrial oxidative phosphorylation to support Erk1/2 activation during pancreatic cancer cell growth. Cell Death Dis 9 (2), 161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Macario AJ and Conway de Macario E (2007) Chaperonopathies by defect, excess, or mistake. Ann N Y Acad Sci 1113, 178–91. [DOI] [PubMed] [Google Scholar]
- 154.Aldridge JE et al. (2007) Discovery of genes activated by the mitochondrial unfolded protein response (mtUPR) and cognate promoter elements. PLoS One 2 (9), e874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Chun JN et al. (2010) Cytosolic Hsp60 is involved in the NF-kappaB-dependent survival of cancer cells via IKK regulation. PLoS One 5 (3), e9422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Kirchhoff SR et al. (2002) Cytosolic heat shock protein 60, apoptosis, and myocardial injury. Circulation 105 (24), 2899–904. [DOI] [PubMed] [Google Scholar]
- 157.Tsai YP et al. (2009) Interaction between HSP60 and beta-catenin promotes metastasis. Carcinogenesis 30 (6), 1049–57. [DOI] [PubMed] [Google Scholar]
- 158.Zhang Q et al. (2018) The Mitochondrial Unfolded Protein Response Is Mediated CellNon-autonomously by Retromer-Dependent Wnt Signaling. Cell 174 (4), 870–883 e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Piselli P et al. (2000) Different expression of CD44, ICAM-1, and HSP60 on primary tumor and metastases of a human pancreatic carcinoma growing in scid mice. Anticancer Res 20 (2A), 825–31. [PubMed] [Google Scholar]
- 160.Barazi HO et al. (2002) Identification of heat shock protein 60 as a molecular mediator of alpha 3 beta 1 integrin activation. Cancer Res 62 (5), 1541–8. [PubMed] [Google Scholar]
- 161.Chandrasekaran S et al. (1999) Pro-adhesive and chemotactic activities of thrombospondin-1 for breast carcinoma cells are mediated by alpha3beta1 integrin and regulated by insulin-like growth factor-1 and CD98. J Biol Chem 274 (16), 11408–16. [DOI] [PubMed] [Google Scholar]