

Abstract
Background and Purpose
Although diabetes mellitus (DM) is an important risk factor for Alzheimer's disease (AD), the detailed mechanism(s) by which DM regulates amyloid β (Aβ) processing is still unclear. The longer residence time of amyloid precursor protein (APP) in endosomes is critical for Aβ production and DM is known to cause endosomal dysregulation. Here we have examined the effects of high glucose on APP‐producing endosomes and related signaling pathways.
Experimental Approach
To identify the underlying mechanisms, we investigated the effects of high glucose on abnormalities in early endosomes and related signalling pathways in human neuroblastoma cells. In vivo, diabetic mice treated with pharmacological inhibitors were used to examine endosomal dysfunction.
Key Results
The hippocampus of diabetic animals presented endosomal abnormalities and Aβ up‐regulation. High glucose increased Aβ production through early endosomal enlargement achieved by increased lipid raft‐mediated APP endocytosis. High glucose induced ROS‐stimulated Sp1 activation, up‐regulating phosphatidylinositol binding clathrin assembly protein (PICALM), clathrin heavy chain, and adaptor‐related protein complex 2 alpha 1. PICALM facilitated clathrin‐mediated APP endocytosis resulting in early endosomal enlargement. Meanwhile, AMPK/mTORC1‐mediated autophagy defect and ROS‐ and mTORC1‐mediated lysosomal dysfunction aggravated early endosomal enlargement under high glucose. Moreover, the increased Aβ production and cognitive deficits in diabetic mice were reversed by inhibition of early endosomal enlargement.
Conclusion and Implications
High glucose induces early endosomal abnormalities through PICALM‐induced APP endocytosis and mTORC1‐inhibited endosomal clearance, up‐regulating Aβ production. Thus, targeting PICALM and mTORC1 to prevent endosomal disorders is a promising strategy for managing diabetes‐induced AD.
Abbreviations
- AP2A1
adaptor‐related protein complex 2 alpha 1
- AD
Alzheimer's disease
- Aβ
Amyloid β
- APP
amyloid precursor protein
- BACE1
β‐secretase 1
- CTSB
cathepsin B
- CAV1
caveolin‐1
- CHC
clathrin heavy chain
- CME
clathrin‐mediated endocytosis
- DM
diabetes mellitus
- FLOT1
flotillin‐1
- LMP
lysosomal membrane permeabilization
- LAMP1
lysosomal‐associated membrane protein 1
- mTOR
mammalian target of rapamycin
- MβCD
methyl‐β‐cyclodextrin
- NAC
N‐acetylcysteine
- PICALM
phosphatidylinositol binding clathrin assembly protein
- STZ
streptozotocin
- VAMP
vesicle‐associated membrane protein
- ZDF
Zucker diabetic fatty
- ZLC
Zucker lean control
What is already known
Diabetes increases the likelihood of developing Alzheimer's disease and upregulates Aβ, an important causative agent.
Early endosomal enlargement is the earliest manifestations of Alzheimer's disease.
What this study adds
Early endosomal abnormalities facilitate Aβ production and cognitive impairment in diabetes.
PICALM and mTORC1‐induced endo‐lysosomal dysfunction modulates APP processing under high glucose conditions.
What is the clinical significance
Targeting endosomal disorders and related factors allow early diagnosis and prevention of diabetes‐mediated Alzheimer's disease .
1. INTRODUCTION
Epidemiological and neuropathological studies have suggested that diabetes mellitus (DM) can contribute substantially to the onset or progression of Alzheimer's disease (AD) (Biessels & Despa, 2018; Lacy et al., 2018). Although there are many potential causal factors (Moreno‐Gonzalez et al., 2017), the precise molecular mechanisms by which DM contributes to the development of AD are still unknown. Regarding the dysregulation of endosomal trafficking, a focus has been placed on amyloid β (Aβ) generation (Small, Simoes‐Spassov, Mayeux, & Petsko, 2017). In particular, the enlargement of early endosomes with Aβ immunoreactivity is the earliest pathological change in both diagnosed and undiagnosed AD patients (Cataldo et al., 2004). Furthermore, AD models using human‐induced pluripotent stem cell‐derived neurons commonly have endosomal abnormalities (Kwart et al., 2019). Early diagnosis of disease is important because Aβ is already saturated before the onset of symptoms (Masters et al., 2015). Therefore, investigating what causes endosomal disorders may provide a high potential for disease prevention. A previous report showed that DM up‐regulates Aβ production by aggravating endosomal dysfunction (Okabayashi, Shimozawa, Yasutomi, Yanagisawa, & Kimura, 2015). Although the precise role and regulation of endosomes in Aβ generation have not yet been clarified, these results investigations suggested that abnormalities of early endosomes could be a risk factor for DM‐induced AD pathology at an early stage.
Abnormalities in the early endosome can be the result of numerous factors, including the up‐regulation of endocytic genes, increased endocytosis, and the overactivation of Rab5, which is a small GTPase that is a marker of the early endosome (Nixon, 2017). According to a genome‐wide association study, endocytic genes are risk factors for AD via mechanisms related to the impairment of trafficking (Guimas Almeida, Sadat Mirfakhar, Perdigao, & Burrinha, 2018). The allelic variants of those factors aggravate the effects of DM on cognitive decline (McFall, Wiebe, Vergote, Anstey, & Dixon, 2015). Among them, the PICALM gene, which encodes the phosphatidylinositol binding clathrin assembly protein (PICALM), which plays an important role in clathrin‐mediated endocytosis (CME) (Xu, Tan, & Yu, 2015) and is associated with an increased risk of both AD and gestational diabetes (Vacinova et al., 2017). Given that endocytic disturbance is an important pathogenic mechanism in diabetes (Teng et al., 2016), PICALM is likely to play a role in the endosomal abnormalities associated with the disease. However, PICALM's role and regulation in the development of diabetes remain poorly understood. Thus, investigating PICALM and related signalling pathways in DM may help identify targets to alleviate endosomal dysfunction. As endosomes are degraded through the autophagy pathway or through direct fusion with intact lysosomes (Colacurcio, Pensalfini, Jiang, & Nixon, 2018), the impairment of these pathways contributes to the dysregulation of early endosomes. Previous studies have shown that endocytic materials, such as amyloid precursor protein (APP) substrates and secretases, are enriched in AD‐induced dysfunctional autophagic vacuoles (Yu et al., 2005), which increases the generation of Aβ. Furthermore, such impairments functionally slow the turnover rate of endosomal trafficking, resulting in defective endosomal clearance (Lee et al., 2015). Although it is debated whether DM‐mediated oxidative stress or intracellular signalling induces the impairment (Gonzalez et al., 2014) or overactivation (Moruno, Perez‐Jimenez, & Knecht, 2012) of intracellular clearance systems, an altered clearance system may influence early endosomal degradation.
In the present study, we used a STZ‐induced mouse model (Furman, 2015) and a Zucker diabetic fatty (ZDF) rat (Srinivasan & Ramarao, 2007) model of Type 1 and Type 2 DM, respectively, to determine whether early endosomal enlargement is a common risk factor for AD in different types of diabetes. To investigate the precise molecular mechanisms involved, we used a human neuroblastoma cell line, SK‐N‐MC, treated with high glucose to create hyperglycaemic conditions, which is the main pathophysiological feature of both types of DM. Using these experimental models, we hypothesized that DM is a metabolic risk factor for AD through inducing early endosomal abnormalities leading to the up‐regulation of Aβ production. To address this hypothesis, we investigated the role of enlarged early endosomes in APP processing and assessed the associated regulatory mechanisms. We also measured the effects of regulating early endosomes as a potential strategy for treating diabetes‐associated AD.
2. METHODS
2.1. Animals
Handling and care of animal were conducted in compliance with the guidelines established by the international laws and policies (NIH Guide for the Care and Use of Laboratory Animals, NIH Publication No. 85‐23, 1985, revised 2011). The experimental protocols were approved by the Institutional Animal Care and Use Committee of Seoul National University (Approvals No.: SNU‐140219‐1 and No. SNU‐190122‐1). Animal studies are reported in compliance with the ARRIVE guidelines (Kilkenny et al., 2010) and with the recommendations made by the British Journal of Pharmacology.
Male and female heterozygous types (Leprfa/+) of ZDF rats were obtained from Genetic Models Co. (Indianapolis, USA) and were mated with each other. Based on PCR genotyping, homozygous ZDF (Leprfa/fa, leptin receptor deficiency) and Zucker lean control (ZLC) (Lepr+/+) rats were used for Type 2 DM and its control group, respectively. The rats were housed in a standard environmental state which sets adequate temperature (20–25°C) and humidity (under 60%) with a 12‐h light/12‐h dark cycle. They were allowed ad libitum access to proper diet (Purina 5008, Purina Korea, Korea; recommended by Genetic Models Co.) and tap water.
Male ICR mice (9 weeks of age) were purchased from Han Lim Experimental Animal (Suwon, Korea) and housed in a conventional environmental conditions (20–25°C, 60% humidity, and 12‐h light/12‐h dark cycle). The mice were allowed free access to chow and drinking solution.
2.2. Experimental designs of animal study
Although sex is an important consideration for animal experiments (Docherty et al., 2019), we determined that using male animals in these experiments was reasonable, as the aim of our study is to determine how DM modulates the processing of APP, which induces the up‐regulation of Aβ and cognitive impairment. A total of 16 rats and 60 mice were used in these experiments. Experiments were designed to generate groups of equal size using randomization and blinded analysis. Group size estimation was conducted in compliance with guidelines established by the Institutional Animal Care and Use Committee of Seoul National University (expected effect size: 12% and 15%, SD: 6%, number of groups: 2 and 4, power (1‐β): 0.8, α: 0.05 for both rats and mice). The number of animals used was increased to account for potential decreases in animal numbers due to death during the experiments. Randomization was also applied to the drug treatments. In order to investigate the effects of Type 2 DM on early endosomal disturbance, male rats were divided into two groups: ZLC and ZDF groups (n = 6 in each group). For chronic Type 2 DM, we kept rats until 33 weeks of age. Blood glucose and body weight were measured using a portable glucose monitor (ACCU‐CHEK GO; Roche, Mannheim, Germany) and a scale, respectively, at 6 and 33 weeks of age (Table S1). Rats were killed at 33 weeks of age. To investigate the effects of Type 1 DM on endosomal dysregulation, STZ‐induced DM was performed in mice, as described previously (Furman, 2015). Briefly, mice were randomly chosen for i.p. injection of STZ (180 mg·kg−1) in 200 μl of sodium citrate buffer (0.1 M, pH 4.5, vehicle) and 200 μl of vehicle for either induction of type 1 DM and its control group, respectively. Blood glucose levels were measured using a portable glucose monitor (ACCU‐CHEK GO; Roche, Mannheim, Germany) every 2 days, starting at 72 h after injection of STZ. Mice with blood glucose levels exceeding 300 mg·dl−1 were considered as having severe diabetes (Furman, 2015). Seven days after injection of STZ, enough diabetic mice were obtained for experimentation. Animal experiments consisted of two parts: In experiment 1, mice were randomly divided into four groups (n = 6 per each group)—vehicle, STZ, STZ + Dynasore, and Dyansore alone. Mice were anaesthetized by i.p. injection of a mixture of alfaxan (50 mg·kg−1) and xylazine (20 mg·kg−1) before drug administration. A warm mat was used to maintain their body temperature while mice were under anaesthesia. Adequate anaesthesia was confirmed by assessing the foot‐pinch response. Dyansore (80 μM) (Hansen et al., 2011) in 4 μl of DMSO (1:1,000 with PBS, vehicle) and 4 μl of vehicle were delivered via i.c.v. injection (Kim, Lee, Chung, Kim, & Kim, 2016). Drugs were injected twice, at 5 and 21 days after DM induction. In experiment 2, mice were randomly divided into four groups (n = 6 per each group)—vehicle, STZ, STZ + Rapamycin, and Rapamycin alone. Rapamycin (8.5 mg·kg−1) (Zhou et al., 2009) in 200 μl of the vehicle solution (99% corn oil and 1% DMSO) and 200 μl of the vehicle were delivered via i.p. injection. Drugs were injected once a day for five consecutive days, starting at 5 days after DM induction. Blood glucose levels and body weight were measured at 9 and 18 weeks of age (Table S2). Mice were subjected to behavioural testing at 18 weeks of age and killed for further biochemical experiments.
2.3. Cell culture experiments
Cells from the human neuroblastoma cell line SK‐N‐MC (KCLB, #30010, RRID:CVCL_0530) were obtained from Korean Cell Line Bank (Seoul, South Korea). The SK‐N‐MC cells were cultured with low glucose DMEM (Hyclone, #SH30021FS), 10% FBS, and 1% antibiotic‐antimycotic solution at 37°C with 5% CO2. After cells had grown to 70% confluency, the medium was replaced with DMEM with 2% Knockout™ serum replacement (SR; Gibco, #10828028) (Ferlemann, Menon, Condurat, Rossler, & Pruszak, 2017) which did not alter cell characteristics and kept the cells in good condition during drug treatment period and 1% antibiotic–antimycotic mixture for 12 h prior to experiments. In order to create the hyperglycaemic condition, cells were treated with 25 mM D‐glucose (Haythorne et al., 2019). The effects of the drugs were tested, and the appropriate concentrations were used for each drug. The dose of each drug did not have a toxic effect on cell viability (Figure S1a).
2.4. Y‐maze spontaneous alternation test
Y‐maze behaviour test is used for assessing the cognitive dysfunction which is dependent on the hippocampus. Rodents instinctively prefer to challenge a new arm of the Y‐maze. Before the test, the animals were placed at the testing room for 2 h to minimize the effects of stress on behaviour. The mice were placed in the randomly selected arm of Y‐shaped maze acquired from Sam‐Jung Company (Seoul, Korea). Each mouse was allowed to explore freely through the open field for 8 min. The total number of arm entries and sequence were recorded. Only when all four limbs were placed in the arm was an entry considered to be completed. Percentage alteration is the number of triads which was divided by the maximum alterations (total entries‐2) × 100. When the animals show lower alteration percentage, the animals show impaired memory function.
2.5. Immunohistochemistry
Mice and rats were fully anaesthetized and were perfused transcardially with PBS followed by 4% PFA in 0.1‐M phosphate buffer (pH 7.4). The brains were removed and underwent post‐fixation with 4% PFA. Then, brains were placed in 30% sucrose in PBS for 1–2 days. Coronal sections (40 μm thick) were obtained by cutting serially using a cryostat (Leica Biosystems, Nussloch, Germany). Free‐floating hippocampus sections were processed carefully. The immuno‐related procedures used comply with the recommendations made by the British Journal of Pharmacology (Alexander et al., 2018). Sections were blocked with 5% normal goat serum (NGS; Sigma‐Aldrich, #566380) at room temperature for 1 h. Samples were incubated with primary antibody (1:100) for overnight at room temperature. Sections were washed three times with PBS and incubated with secondary antibody (1:200 dilution) for 2 h. Immunostained slides were visualized by Eclipse Ts2™ fluorescence microscopy (Nikon, Tokyo, Japan). Immunohistochemistry (IHC) images were analysed with the Fiji software (RRID:SCR_002285). Signal intensities were measured after applying the same threshold and normalized by each area.
2.6. siRNA transfections for gene silencing
When the SK‐N‐MC cells had grown to 60% confluence, cells were incubated with low glucose DMEM containing 25 nM of the indicated siRNAs, transfection reagent TurboFect™ (Thermo Fisher, #R0531), and 2% SR for 12 h. The medium was replaced with the medium with 1% antibiotics prior to experiments. NT siRNA was used as the negative control. The efficacies of siRNAs were confirmed in Figure S1b,c.
2.7. Western blot analysis
Harvested cells or tissues were incubated with the appropriate lysis buffer and protease and phosphatase inhibitors cocktail (100X) (Thermo Fisher, #78440). Then, samples were homogenized by using sonicator and vortexer for 30 min on ice. The lysates were cleared by centrifugation (13,000 g, 4°C, 20 min). Protein concentrations were determined by using the bicinchoninic acid (BCA) quantification assay (Thermo Fisher, #23227) with two technical replicates each. Equal amount of samples (5–10 μg) were loaded into 8%–12% SDS‐PAGE and transferred to a PVDF membrane. Tris‐buffered saline containing 0.2% Tween‐20 (TBST; 150 mM NaCl, 10 mM Tris–HCl [pH 7.6]) was used for washing and incubating membrane. The membrane was blocked with 5% skim milk (Gibco, #232100) for 1 h. The blocked membrane was washed three times and incubated with a primary antibody (1:1,000 dilution) for overnight at 4°C. Then, the membrane was washed and incubated with HRP‐conjugated secondary antibody (1:10,000 dilution) at room temperature for 2 h. Blotting bands were detected by using chemiluminescence detection kit (Advansta Inc., #K‐12045‐D50). Protein bands quantification were carried out by using the Image J software (developed by Wayne Rasband, National Institutes of Health, Bethesda, MD, USA, RRID:SCR_003070). Protein expression levels were normalized by β‐actin expression levels. The EzSubcell™ subcellular fractionation kit (Atto, #WSE‐7421) was applied to the preparation of the cytosolic and nuclear‐fractionized samples. Cytosolic and nuclear samples were acquired according to the manufacturer's instructions. Protein expression levels of cytosolic and nuclear samples were normalized by β‐tubulin and lamin A/C expression levels, respectively.
2.8. Sucrose density gradient fractionation
The SK‐N‐MC cells were washed with cold PBS (Hyclone, #SH30256) and scraped into 1 ml of lysis buffer (10 mM EDTA, 500 mM Na2CO3, pH 11 in distilled water) with the proteinase and phosphatase inhibitor cocktail (100X). Lysates were homogenized by using sonicator (Branson Sonicator 250, Branson Ultrasonic Corp., Danbury, CT, USA) and incubated with vortexing for 30 min on ice. To form 5%–45% discontinuous gradient, 4 ml of 45% sucrose and 4 ml of 5% sucrose dissolved in MES‐buffered solution (MBS: 25 mM MES [pH 6.5], 0.15 M NaCl) were sequentially placed in an ultracentrifuge tube (Beckman Coulter, Fullerton, CA, USA). Then, equal amounts of protein were prepared in 4 ml of 5% sucrose and added to the ultracentrifuge tube. Sample tubes were centrifuged at 200,000 x g for 24 h in an SW41 rotor (Beckman Coulter). Fractionated samples were acquired and analysed by western blotting.
2.9. Immunofluorescence analysis
The SK‐N‐MC cells were washed twice with PBS and fixed with 4% paraformaldehyde (PFA; Lugen Sci, Seoul, Korea, #LGB‐1175) for 10 min. To permeabilize the cell membrane, cells were incubated in 0.2% TBST or 0.1% Triton X‐100 (Sigma, T8787) for 10 min. Cells were blocked with 5% NGS for 40 min and incubated with primary antibodies (1:100 dilution) for overnight in 4°C. Then, cells were washed three times with PBS and incubated with Alexa Fluor™ 488 or 555‐conjugated secondary antibodies (1:200 dilution) at room temperature for 2 h. Immunostained samples were visualized by a super‐resolution radial fluctuations (SRRF) imaging system (Andor Technology, Belfast, UK) (Gustafsson et al., 2016). Fiji software was used to quantify the fluorescent intensities. The area of Rab5+ endosomes and acidic lysosomes was measured after applying the same threshold. The Manders coefficients were used to analyse co‐localization. The Pitstop 2 could detect transient cargo‐adaptor interactions by binding to the terminal domain of CHC and perturbing clathrin‐coated pit dynamics without affecting pit assembly or the sequestration of the cargoes (von Kleist et al., 2011).
2.10. Measurements of intracellular pH and ROS
The cell permeable pH‐sensitive fluorescent probe BCECF‐AM [2′,7′‐bis‐(2‐carboxyethyl)‐5‐(and‐6)‐carboxyfluorescein, acetoxymethyl ester; Thermo Fisher, #B1150] and CM‐H2DCFDA (Thermo Fisher, #C6821) were used for measuring the intracellular pH and ROS, respectively. After drug treatment, the cells were incubated with 2 μM BCECF‐AM or 10 μM DCFDA in medium and kept at 37°C for 30 min. Then, cells were washed three times with PBS. The signals of BCECF‐AM‐ and DCFDA‐stained cells were measured via flow cytometry (Beckman Coulter, Atlanta, USA).
2.11. Marking and staining lysosomes
Lysotracker deep red (Thermo Fisher, #L12492) which has a high affinity for acidic organelles was used for staining the intact lysosomes. After drug treatment, the cells were incubated with 2 μM Lysotracker red in medium and kept at 37°C for 30 min. Then, cells were washed three times with PBS. The signals of Lysotracker‐stained cells were measured via flow cytometry (Beckman Coulter, Atlanta, USA), or cells were further experimented with ICC. LAMP1‐positive organelles are also referred to as intact lysosomes. However, a significant portion of LAMP1‐positive organelles does not contain lysosomal hydrolases (Cheng et al., 2018). Thus, aberrant increase in LAMP1 may suggest the accumulation of dysfunctional lysosomes.
2.12. Quantitative real‐time PCR
RNA samples were extracted by using RNA extraction kit (TaKaRa, #9767), according to the manufacturer's instructions. Then, the cDNA was made by using RT‐PCR premix (iNtRON Biotechnology, #25081). Reverse transcription was performed for 1 h at 45°C followed by 5 min at 95°C. The cDNA samples with two technical replicates were amplified with the mRNA primers listed in Table S3 and a TB™ Green Premix Ex Taq™ (TaKaRa, #RR420A) by using Rotor‐Gene 6000 real‐time thermal cycling system (Corbett Research, NSW, Australia). The qPCR was performed as follows: 15 min at 95°C for DNA polymerase activation and 60 cycles of 20 s at 94°C, 20 s at 55°C, and 30 s at 72°C. The specificity and identity of the amplified product were validated by the analysis of melting curve. The quantification of mRNA expression levels of the target genes was performed with double δCt analysis, and the data were normalized with those of ACTB gene.
2.13. Measurement of Aβ in in vitro and in vivo samples
In vitro, the culture medium of SK‐N‐MC cells was collected and stored at −70°C with phosphatase inhibitors cocktail (100X). In vivo, equal amount of the hippocampus samples (200 μg) obtained from animals were prepared with proper lysis buffer and BCA assay with two technical replicates each. According to the manufacturer's instructions, the OD of Aβ (1–42) in culture medium and animal samples were acquired by using human Aβ (1–42) ELISA kit (Thermo Fisher, #KHB3544) and mouse and rat Aβ (1–42) ELISA kit (Thermo Fisher, #KMB3441), respectively. The mean OD values were converted into the concentration by using the standard curve.
2.14. GTP‐Rab5 activation assay
Measurement of Rab5 activation was performed by using Rab5 activation assay kit (Neweast Bioscience, #83701). All procedures were done in accordance with the manufacturer's instructions. Briefly, cells were incubated with the provided lysis buffer containing anti‐active Rab5 monoclonal antibody (NewEast Biosciences, #26911, RRID:AB_2617182). Then, by using protein A/G agarose, the bound active Rab5 was pulled down. The precipitated active Rab5 was detected by western blot using Rab5‐specific polyclonal antibody. The ratio of expression levels of active Rab5 and total Rab5 was analysed.
2.15. APP internalization assay
The SK‐N‐MC cells were washed with cold PBS and incubated with Aβ antibody (Xiao et al., 2012) (1:100 dilution) at 4°C for 1 h to label surface APP. Cells were washed with cold PBS twice and incubated at 37°C for 0 and 15 min to permit internalization. Internalization was stopped by rapid cooling on ice. Cells were fixed with 4% PFA for 10 min at room temperature, permeabilized with 0.1% Triton X‐100 for 10 min, and blocked with 5% NGS for 40 min. Cells were incubated with primary antibodies (1:100 dilution) for overnight in 4°C. Then, cells were washed three times with PBS and incubated with Alexa Fluor™ 488 or 555‐conjugated secondary antibodies (1:200 dilution) at room temperature for 2 h. Immunostained samples were visualized by an SRRF imaging system (Andor Technology, Belfast, UK).
2.16. Cell surface biotinylation assay and internalization assay
In order to prevent proteins from shedding at the cell surface, cells were exposed to GM6001 (an MMP inhibitor; 20 μM) for 30 min prior to drug treatments (Kanatsu et al., 2014). Surface protein isolation was conducted by using cell surface protein isolation kit (Biovision, #K295‐10). All procedures were done in accordance with the manufacturer's instructions. Briefly, cells were washed with cold PBS and labelled with Sulfo‐NHS‐SS‐Biotin, a non‐membrane‐permeable, thiol‐cleavable, biotinylation reagent. For the internalization assay, biotinylated cells were incubated at 37°C for 0 and 15 min to permit internalization. Internalization was stopped by rapid cooling on ice. To remove remained biotin at the cell surface, cells were incubated with MesNA (50 mM 2‐mercaptoethanesulfonic acid in 50 mM Tris–HCl, 100 mM NaCl, 2.5 mM CaCl2, and pH 8.7) three times for 20 min at 4°C. After quenching the biotin reagent with 0.1 M glycine, cells were lysed, and labelled surface proteins were isolated by using streptavidin beads. Then, cells were incubated with DTT solution to release attached beads. Biotin‐labelled proteins were subjected to western blot.
2.17. Data and statistical analysis
The data and statistical analysis comply with the recommendations of the British Journal of Pharmacology on experimental design and analysis in pharmacology (Curtis et al., 2018). The group size represents the number of independent values, and statistical analyses were performed using these independent values. Statistical analysis was performed only for studies where each group size was at least n = 5. The exploratory data with large effects and accuracy have a small sample size but may have significant implications. Outliers were assessed in compliance with the exclusion criteria beyond an outer fence (Q1–3 * IQ or Q3 + 3 * IQ) in a box plot construction, and we did not observe any outliers. All the data achieved normality (using the Shapiro–Wilk test) and were analysed by parametric statistics. Imaging experiments and animal tests were conducted and assessed in a blinded fashion. The unpaired Student's t‐test was used to compare the means of the treatment groups with that of the control group. One‐way ANOVA (with Dunnett's multiple comparison test) or two‐way ANOVA (with Tukey's multiple comparison test) was used for analysing the differences among multiple groups. The homogeneity of sample variance was confirmed (with Levene's test), and logarithmic transformation was performed if the variance was not satisfied. The units of a variable were determined by percentage‐matched control values following data transformation. Quantitative data are expressed as the mean ± SEM and analysed with the Prism 6 software (Graphpad, CA, USA, RRID:SCR_000306) and SigmaPlot 12.0 (Systat software Inc., CA, USA, RRID:SCR_003210). The level of probability (P) constitutes the threshold for statistical significance for determining whether groups differ and is not varied later in results. A P value <0.05 was considered statistically significant.
2.18. Materials
FBS and antibiotics were purchased from Hyclone (Logan, UT, USA) and Gibco (Grand Island, NY, USA), respectively. The antibodies to β‐actin (sc‐47778, RRID:AB_626632), caveolin‐1 (CAV1; sc‐53564, RRID:AB_628859), Flotillin‐1 (FLOT1; sc‐74566, RRID:AB_2106563), clathrin heavy chain (CHC) (sc‐12734), PICALM (sc‐271224), lamin A/C (sc‐2068), lysosomal‐associated membrane protein 1 (LAMP1) (sc‐20011, RRID:AB_626853), p‐AMPKα (Thr 172) (sc‐33524), BACE1 (sc‐33711, RRID:AB_626716), VAMP3 (sc‐514843), VAMP8 (sc‐166820, RRID:AB_2212959), Beclin1 (sc‐11427), and CTSB (sc‐365558, RRID:AB_10842446) were acquired from Santa Cruz Biotechnology (Santa Cruz, CA, USA). The antibodies to β‐tubulin (CSB‐PA03874A0Rb) and AP2A1 (610502) were purchased from Cusabio (Wuhan, Hubei, China) and BD Biosciences (San Jose, CA, USA), respectively. The antibodies to Rab5 (NB120‐13253), P62 (NBP1‐48320), and LC3 (NB100‐2220) were obtained from Novus Biologicals (Centennial, CO, USA). The antibodies to p‐mTOR (Ser 2448) (2971S), mTOR (2983S, RRID:AB_2105622), and AMPK (2532S) were acquired from Cell Signaling Technology (Beverly, MA, USA). The antibodies to APP (ab32136), Sp1 (ab227383), and Aβ (ab2539) were purchased from Abcam (Cambridge, England). The antibody to C99 (802801) was obtained from Biolegend (San Diego, CA, USA). VAMP2 (GTX133241) antibody was purchased from Genetex (Irvine, CA, USA). Methyl‐β‐cyclodextrin (MβCD) (lipid raft disruptor), Dynasore hydrate (dynamin inhibitor, CME inhibitor), Rapamycin (mTOR inhibitor), N‐acetylcysteine (NAC) (ROS scavenger), PF‐4708671 (S6K1 inhibitor), DAPI, D‐glucose, L‐glucose, leupeptin (inhibitor of serine and thiol proteases), and STZ were purchased from Sigma Chemical Company (St. Louis, MO, USA). GM6001 and Mithramycin A (Sp1 inhibitor) were acquired from Cayman Chemical (Ann Arbor, MI, USA) and Tocris Bioscience (Bristol, UK), respectively. mRNA primers for PICALM, AP2A1, RAB5, EEA1, RABGEF1, RABEP1, APPL1, ATG14, SNAP29, VTI1B, STX17, VAMP7, and ACTB were purchased from Cosmo Genetech (Seoul, Korea). Primers for CHC and siRNAs for RAB5 and PICALM were acquired from Bioneer (Daejeon, Korea). Non‐targeting (NT) siRNA was purchased from Dharmacon (Lafayette, CO, USA).
2.19. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY (Harding et al., 2018), and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20 (Alexander et al., 2019).
3. RESULTS
3.1. High glucose up‐regulates Aβ production through early endosomal abnormalities
To elucidate the effects of DM on APP‐processing early endosomes, we first measured hippocampal C99 (APP‐CTFβ) and Rab5, located in early endosomes and used as an early endosomal marker. Compared with the levels in ZLC rats, both proteins increased in ZDF rats (Figure 1a). In addition, ZDF rats had higher Aβ levels and Rab5 staining intensities (Law et al., 2017) at the hippocampus than ZLC rats (Figure 1b,c). Moreover, STZ‐diabetic mice had increased C99, Rab5 (Figure 1d) expressions, and Rab5 staining intensities at the hippocampus (Figure 1e), compared with vehicle‐treated mice. In in vitro experiments involving treatment of cells with either d‐ or l‐glucose, only d‐glucose increased C99, Rab5 (Figure 1f), and the levels of Aβ (Figure 1g), implying that osmotic effects were not involved in amyloidogenesis. In addition to increasing expression of Rab5 protein, high glucose also increased early endosomal activation, which was measured by determining the level of the GTP‐Rab5 form (Figure 1h). To investigate that high glucose increased APP processing in early endosomes, along with our previous report demonstrating up‐regulation of the protein levels and activity of BACE1, but not γ‐secretase (presenilin 1), under high glucose conditions (Lee et al., 2016), we found that high glucose induced early endosomal enlargement (Kwart et al., 2019) and increased co‐localization with the C‐terminus of APP and BACE1, suggesting an increase in APP‐processing in enlarged early endosomes (Figure 1i,j). Furthermore, high glucose‐induced increase of C99 and Aβ were reversed by RAB5 knockdown (Figure 1k,l). These findings suggest that early endosomal abnormalities are critically involved in the increase of Aβ in high glucose (DM) conditions.
FIGURE 1.
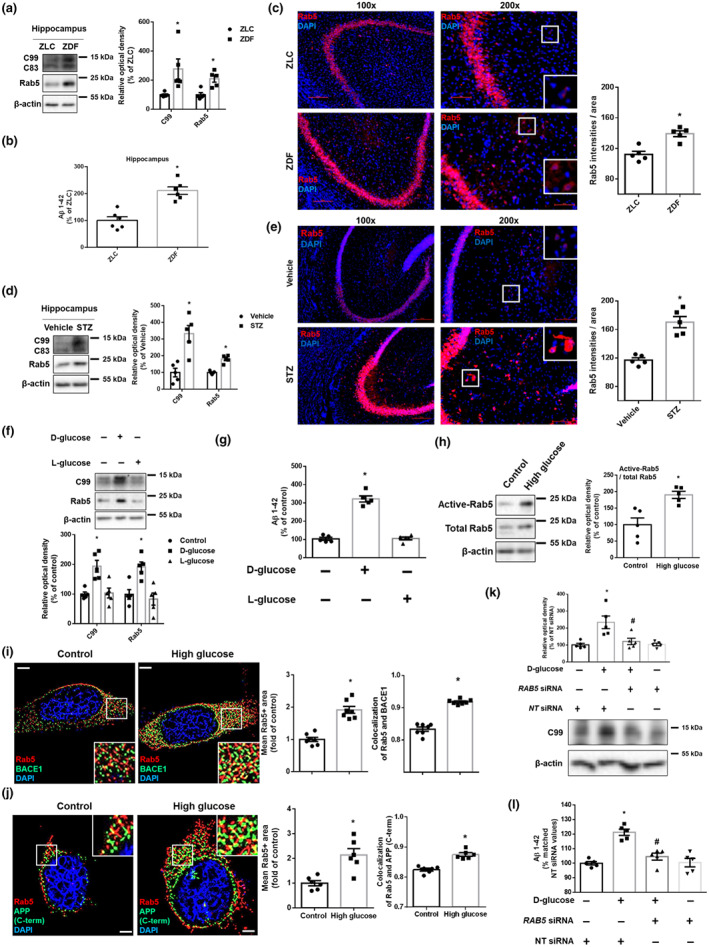
High glucose up‐regulates Aβ production through early endosomal enlargement. (a–c) Samples were obtained from ZLC and ZDF rat hippocampus. (a) C99, Rab5, and β‐actin were detected by western blot. n = 5 in each group. (b) Aβ 1–42 were measured by rat and mouse Aβ 1–42 specific ELISA assay. n = 6 in each group with two technical replicates each. (c) Tissue slides for IHC were immunostained with Rab5‐specific antibody and counterstained with DAPI. Scale bars, 50 μm (magnification, ×200). n = 5 in each group. * P < .05, significantly different from ZLC rats. (d,e) Samples were obtained from vehicle‐ and STZ‐treated mice hippocampus. (d) C99, Rab5, and β‐actin were detected by western blot. n = 5 in each group. (e) Tissue slides for IHC were immunostained with Rab5‐specific antibody and counterstained with DAPI. Scale bars, 200 μm (magnification, ×200). n = 5 in each group. *P < .05, significantly different from vehicle‐treated mice. (f) The SK‐N‐MC cells were treated with d‐glucose (25 mM) or l‐glucose (25 mM) for 24 h. Then, C99, Rab5, and β‐actin were detected by western blot. n = 5 from independent experiments. (g) d‐glucose (25 mM) or l‐glucose (25 mM) was treated for 48 h in the cells. Then, Aβ 1–42 from cell culture medium were measured by human Aβ 1–42 specific ELISA assay. n = 5 from independent experiments with two technical replicates each. (h) The cells were treated with high glucose (25 mM) for 24 h. Active Rab5 (GTP‐bound Rab5) was immunoprecipitated with Rab5‐specific antibody (upper panels). Expression of Rab5 and β‐actin in total lysate is shown in the middle and lower panels. n = 5 from independent experiments. (i) The cells were treated with high glucose (25 mM) for 24 h which were immunostained with Rab5 and BACE1‐specific antibodies and counterstained with DAPI. Scale bars, 8 μm (magnification, ×1,000). n = 7 from independent experiments. (j) Immunostaining of cells treated with high glucose (25 mM) for 24 h was visualized. Rab5 and APP (C‐terminus)‐specific antibodies were used and counterstained with DAPI. Scale bars, 8 μm (magnification, ×1,000). n = 6 from independent experiments. *P < .05, significantly different from control. (k,l) The cells were transfected with NT siRNA or RAB5 siRNA for 12 h prior to high glucose (25 mM) treatment for 24 and 48 h, respectively. (k) C99 and β‐actin were detected by western blot. n = 5 from independent experiments. (l) Aβ 1–42 from cell culture medium were measured by human Aβ 1–42 specific ELISA assay. n = 5 from independent experiments with two technical replicates each. Logarithmic transformations were performed for homogeneity of the sample variance. * P < .05, significantly different from NT siRNA transfection, # P < .05, significantly different from high glucose with NT siRNA transfection. Quantitative data are presented as a mean ± SEM. All blots and immunofluorescence images are representative
3.2. High glucose induces early endosomal enlargement by increasing APP endocytosis at lipid rafts
To explore how high glucose induces early endosomal over‐activation along with increased expression of Rab5 protein (Figure 1h), we measured mRNA for Rab5 and its effector proteins. However, we did not identify any significant changes (Figure S2a). Next, we hypothesized that high glucose increased APP endocytosis resulting in early endosomal enlargement. We confirmed that high glucose increased protein expression levels of APP (Figure S2b) and its endocytosis (Figure 2a). Given that high glucose facilitated lipid raft reorganization (Lee et al., 2016), exploratory data showed high glucose markedly moved APP to the lipid raft part as indicated by CAV1 and FLOT1 (Figure 2b). The expression levels of surface APP in cells treated with high glucose and MβCD were higher than those of cells treated with high glucose (Figure 2c). In addition, high glucose increased internalization of APP, which was reversed by MβCD (Figure 2d). Furthermore, pretreatment with MβCD decreased high glucose‐induced early endosomal enlargement and increase of Aβ, which means that APP endocytosis at lipid rafts contributed to early endosomal enlargement (Figure 2e,f). These results suggest that early endosomal enlargement under high glucose conditions is mediated by APP endocytosis at lipid rafts.
FIGURE 2.
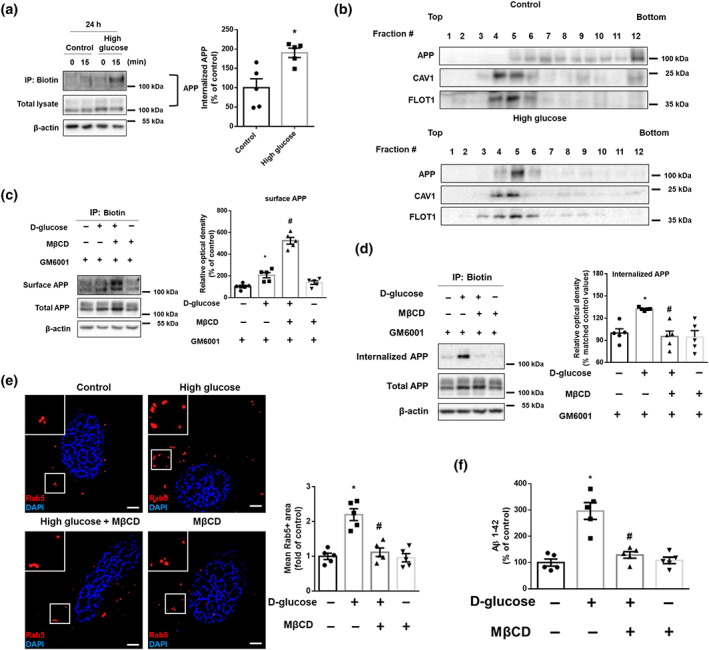
High glucose facilitates lipid raft‐mediated APP endocytosis resulting in early endosomal enlargement. (a) Biotin surface labelling and internalization assay from the SK‐N‐MCs treated with high glucose (25 mM) for 24 h were carried out. 0 and 15 min indicate time points of internalization. In the bottom, total lysates were subjected to western blot. APP and β‐actin were detected. Percent of internalized APP was measured by normalizing with total APP at time 15 in each conditions. n = 5 from independent experiments. (b) High glucose (25 mM) was treated for 24 h in the cells. Sucrose gradient‐fractionized lysates were subjected to western blot. APP, CAV1, and FLOT1 were detected. Exploratory data. (c,d) The cells were treated with GM6001 (20 μM) for 30 min and then with MβCD (1 mM) for 30 min and high glucose (25 mM) for 24 h. (c) Cell surface was biotinylated, and labelled proteins were pulled down by streptavidin beads. Surface APP, total APP, and β‐actin were detected by western blot. n = 5 from independent experiments. (d) Biotin surface labelling and internalization assay including 15 min for internalization were carried out. Internalized APP, total APP, and β‐actin were detected by western blot. n = 5 from independent experiments. Logarithmic transformations were performed for homogeneity of the sample variance. (e) The cells were incubated with MβCD (1 mM) for 30 min prior to high glucose treatment (25 mM) for 24 h. The cells were immunostained with Rab5‐specific antibody and counterstained with DAPI. Scale bars, 8 μm (magnification, ×1,000). n = 5 from independent experiments. (f) The cells were pretreated with MβCD (1 mM) for 30 min before high glucose treatment (25 mM) for 48 h. Aβ 1–42 from cell culture medium were measured by human Aβ 1–42 specific ELISA assay. n = 5 from independent experiments with two technical replicates each. *P < .05, significantly different from control, # P < .05, significantly different from high glucose. Quantitative data are presented as a mean ± SEM. All blots and immunofluorescence images are representative
3.3. High glucose‐induced ROS increase the expression of endocytic proteins by facilitating Sp1 nuclear translocation
Next, we investigated how high glucose facilitates APP endocytosis and hypothesized that it regulates endocytosis‐related proteins. Given that the main cause of pathological situations under high glucose conditions is ROS, we found that high glucose significantly increased the amount of ROS after 24 h (Figure 3a). However, there were no significant changes over time in the control conditions. Furthermore, among the kinases controlled by ROS, we found that Sp1 is related to endocytic protein expression (Bai et al., 2017). Indeed, high glucose‐stimulated Sp1 nuclear translocation was blocked by pretreatment of NAC (Figure 3b,c). As shown in Figure 3d,e, mRNA and protein expression levels of PICALM, AP2A1, and CHC were increased by high glucose but inhibited by pretreatment with Mithramycin A. Moreover, compared with the levels in control animals, ZDF rats and STZ‐treated mice had high expression levels of the hippocampal PICALM, AP2A1, and CHC (Figure 3f,g). These findings suggest that high glucose up‐regulates the expression of endocytic proteins by ROS‐stimulated Sp1 activation.
FIGURE 3.
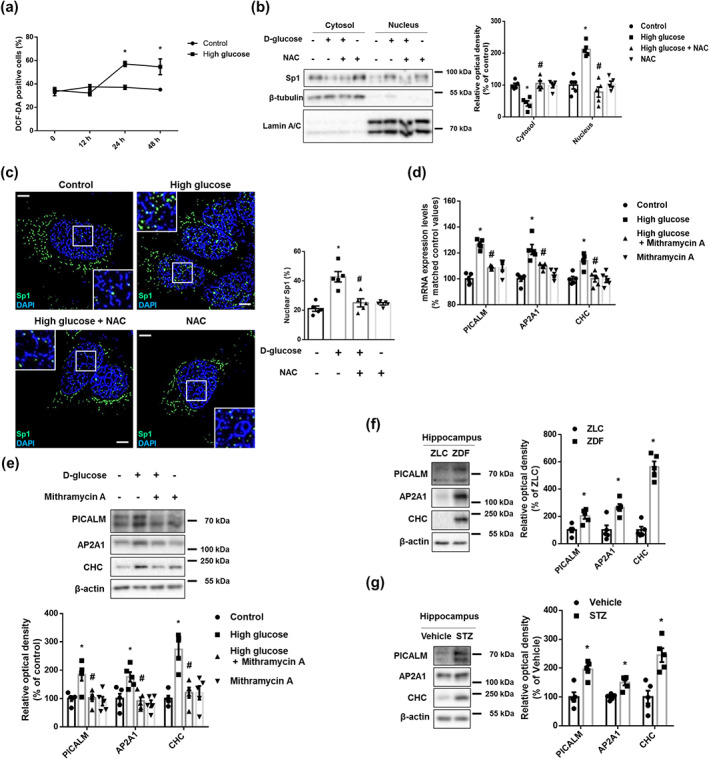
High glucose increased PICALM, AP2A1, and CHC expression through ROS‐stimulated Sp1 nuclear translocation. (a) The SK‐N‐MC cells were treated with distilled water (DW) or 25 mM high glucose in a time response. Intracellular ROS was measured by DCF‐DA staining and analysed by flow cytometry. n = 5 from independent experiments. (b,c) The cells were incubated with NAC (4 mM) for 30 min prior to high glucose treatment (25 mM) for 6 h. (b) Sp1, β‐tubulin, and Lamin A/C in cytosolic and nuclear fraction samples were detected by western blot. n = 5 from independent experiments. (c) The cells were immunostained with Sp1‐specific antibody and counterstained with DAPI. Scale bars, 8 μm (magnification, ×1,000). n = 5 from independent experiments. (d) The cells were pretreated with Mithramycin A (25 nM) for 30 min before high glucose treatment (25 mM) for 12 h. mRNA expression levels of PICALM, AP2A1, and CHC were analysed by quantitative real‐time PCR. n = 5 from independent experiments with two technical replicates each. Logarithmic transformations were performed for homogeneity of the sample variance. (e) The cells were incubated with Mithramycin A (25 nM) for 30 min prior to high glucose treatment (25 mM) for 24 h. PICALM, AP2A1, CHC, and β‐actin were subjected to western blot. n = 5 from independent experiments. *P < .05, significantly different from control, # P < .05, significantly different from high glucose. (f) The hippocampal samples were obtained from ZLC and ZDF rats. PICALM, AP2A1, CHC, and β‐actin were detected by western blot. n = 5 in each group. *P < .05, significantly different from ZLC rats. (g) The hippocampal samples were obtained from vehicle‐ and STZ‐treated mice. PICALM, AP2A1, CHC, and β‐actin were detected by western blot. n = 5 in each group. *P < .05, significantly different from vehicle‐treated mice. Quantitative data are presented as a mean ± SEM. All blots and immunofluorescence images are representative
3.4. PICALM regulates clathrin‐dependent APP endocytosis, which up‐regulates Aβ production
Given that PICALM is considered to be a common risk factor for both AD and DM (McFall et al.2015) and recruits CHC and AP‐2 complex (Xu et al., 2015), we studied the role of PICALM in the endocytosis under high glucose and AD pathology. First, exploratory data showed that PICALM was located in lipid rafts under both normal and high glucose conditions (Figure S3a). We further found that the levels of co‐localization of APP with AP2A1 and CHC under high glucose in the cell transfected with a NT siRNA were higher than those of the PICALM siRNA transfected cell treated with high glucose (Figure 4a,b). Moreover, prior to endocytosis (0 min), surface‐labelled APP and PICALM were observed exclusively on the cell surface. After endocytosis (15 min), surface‐labelled APP and PICALM were observed in intracellular vesicles and were co‐localized. This indicates that high glucose caused APP co‐localization with PICALM during endocytosis (Figure 4c). Similarly, the expression levels of surface APP in cells transfected with PICALM siRNA under high glucose conditions were higher than those of NT siRNA‐transfected cells treated with high glucose (Figure 4d). Additionally, high glucose increased the internalization of APP, which was reversed by PICALM RNAi (Figure 4e). As we aimed to measure APP internalization and not the recruitment ability of PICALM, we used GM6001, which inhibited APP shedding in Figures 4d,e. Thus, compared with total and surface APP levels of Figures 4a,b, those of Figures 4d,e were not reduced by PICALM siRNA. High glucose‐induced increases in Aβ were reversed by PICALM knockdown (Figure 4f). These results indicate that up‐regulation of PICALM by high glucose induces clathrin‐mediated APP endocytosis. To confirm the effects of CME on APP endocytosis, we used a Dynasore. The expression levels of surface APP in the samples treated with high glucose and dynasore were higher than those of samples treated with high glucose alone (Figure 4g). Furthermore, Dynasore reversed high glucose‐induced early endosomal enlargement and increment of Aβ (Figure 4h,i). These results demonstrated that increased PICALM facilitated clathrin‐dependent APP endocytosis, resulting in Aβ production.
FIGURE 4.
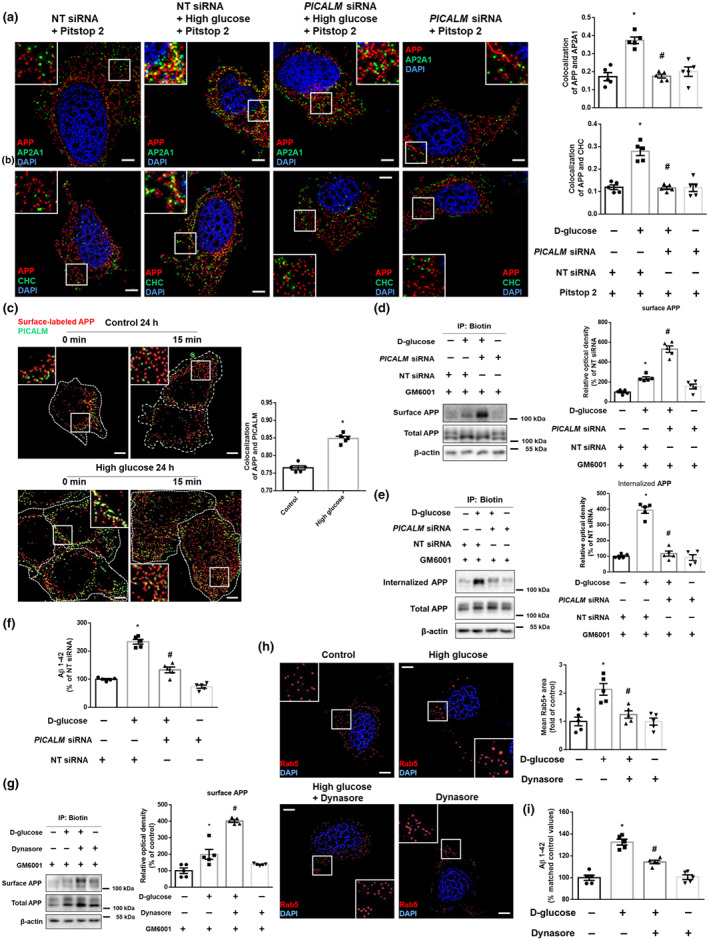
PICALM facilitates clathrin‐dependent APP endocytosis under high glucose conditions. (a,b) The SK‐N‐MC cells were transfected with NT siRNA or PICALM siRNA for 12 h and then treated with Pitstop 2 (30 μM) for 30 min prior to high glucose treatment (25 mM) for 24 h. (a) The cells were immunostained with APP‐ and AP2A1‐specific antibodies and counterstained with DAPI. Scale bars, 8 μm (magnification, ×1,000). n = 5 from independent experiments. (b) The cells were immunostained with APP‐ and CHC‐specific antibodies and counterstained with DAPI. Scale bars, 8 μm (magnification, ×1,000). n = 5 from independent experiments. *P < .05, significantly different from NT siRNA transfection, # P < .05, significantly different from high glucose with NT siRNA transfection. (c) Internalization assay from the cells treated with high glucose (25 mM) for 24 h was done by immunostaining with Aβ for surface‐labelled APP and PICALM specific antibodies. 0 and 15 min indicate time points of internalization. Scale bars, 8 μm (magnification, ×1,000). n = 5 from independent experiments. (d,e) The SK‐N‐MC cells were transfected with NT siRNA or PICALM siRNA for 12 h and then treated with GM6001 (20 μM) for 30 min prior to high glucose treatment (25 mM) for 24 h. (d) Cell surface was biotinylated, and labelled proteins were pulled down by streptavidin beads. Surface APP, total APP, and β‐actin were detected by western blot. n = 5 from independent experiments. (e) Biotin surface labelling and internalization assay including 15 min for internalization were done. Internalized APP, total APP, and β‐actin were detected by western blot. n = 5 from independent experiments. (f) The cells were transfected with NT siRNA or PICALM siRNA for 12 h prior to high glucose treatment (25 mM) for 48 h. Aβ 1–42 from cell culture medium were measured by human Aβ 1–42 specific ELISA assay. n = 5 from independent experiments with two technical replicates each. *P < .05, significantly different from NT siRNA transfection, # P < .05, significantly different from high glucose with NT siRNA transfection. (g) The cells were pretreated with GM6001 (20 μM) for 30 min and then treated with Dynasore (25 μM) for 30 min before high glucose treatment (25 mM) for 24 h. Cell surface was biotinylated, and labelled proteins were pulled down by streptavidin beads. Surface APP, total APP, and β‐actin were detected by western blot. n = 5 from independent experiments. (h) The cells were pretreated with Dynasore (25 μM) for 30 min immunostained with Rab5‐specific antibody and counterstained with DAPI. Scale bars, 8 μm (magnification, ×1,000). n = 5 from independent experiments. (i) The cells were incubated with Dynasore (25 μM) for 30 min prior to high glucose treatment (25 mM) for 48 h. Aβ 1–42 from cell culture medium were measured by human Aβ 1–42 specific ELISA assay. n = 5 from independent experiments with two technical replicates each. Logarithmic transformations were performed for homogeneity of the sample variance. *P < .05, significantly different from control, # P < .05, significantly different from high glucose . Quantitative data are presented as a mean ± SEM. All blots and immunofluorescence images are representative
3.5. Defects in AMPK/mTORC1‐mediated autophagy impairs early endosomal clearance under high glucose conditions
We hypothesized that the block of the degradative trafficking pathway also causes early endosomal enlargement. Compared with the levels in control animals, ZDF rats and STZ‐treated mice had increased hippocampal P62 and a decreased LC3II/LC3I ratio (Figure 5a,b). In in vitro models, high glucose decreased the LC3II/LC3I ratio and increased P62 after 12 h (Figure S4a). Compared with l‐glucose, only d‐glucose decreased the LC3II/LC3I ratio and increased P62 (Figure S4b). This suggests that autophagic dysfunction is induced under DM. As PICALM is known to induce autophagy, we further investigated whether PICALM regulates autophagy under high glucose conditions. The interaction between LC3 and PICALM (Figure S5a) (Tian, Chang, Fan, Flajolet, & Greengard, 2013) and endocytosis of VAMP3 (Figure S5b) (Moreau et al., 2014) were increased under high glucose conditions. PICALM siRNA transfection further reduced the LC3‐II/LC3‐I ratio compared to high glucose, with NT siRNA transfection (Figure S5c). These data suggest that increased PICALM under high glucose conditions facilitates autophagy but is insufficient to fully reverse the reduction in autophagy. Furthermore, ROS promote autophagy through increasing the expression of Beclin1 through MAPK signalling (Moruno et al., 2012). However, we did not identify any significant changes (Figure S6a). The ROS/Beclin1‐mediated pathway was not dominant in autophagy under our experimental conditions. Given that mTORC1 is a master regulator of autophagy, we found that high glucose down‐regulated AMPKα phosphorylation at Thr172 and up‐regulated mTORC1 phosphorylation at Ser2448 in a time‐dependent manner (Figure 5c). The decrease in the LC3II/LC3I ratio, co‐localization of lysosomes with LC3, and the increase in P62 that is induced by high glucose were reversed by pretreatment with Rapamycin (Figure 5d,e). To investigate the relationship between mTORC1‐mediated autophagic impairment with the increase in Rab5 protein levels, we used PF‐4708671 and Rapamycin. Under high glucose conditions, Rab5 protein expression levels were decreased after pretreatment with Rapamycin, but not upon pretreatment with PF‐4708671. This suggests that increased Rab5 is not due to increased protein synthesis by S6k1, but rather, due to a deficiency in autophagy (Figure 5f). Indeed, we confirmed that Rapamycin attenuated the high glucose‐induced increment of Rab5+ endosomal size and Aβ (Figure 5g,h). These results revealed that high glucose impairs early endosomal clearance through AMPK/mTORC1‐mediated defect in autophagy.
FIGURE 5.
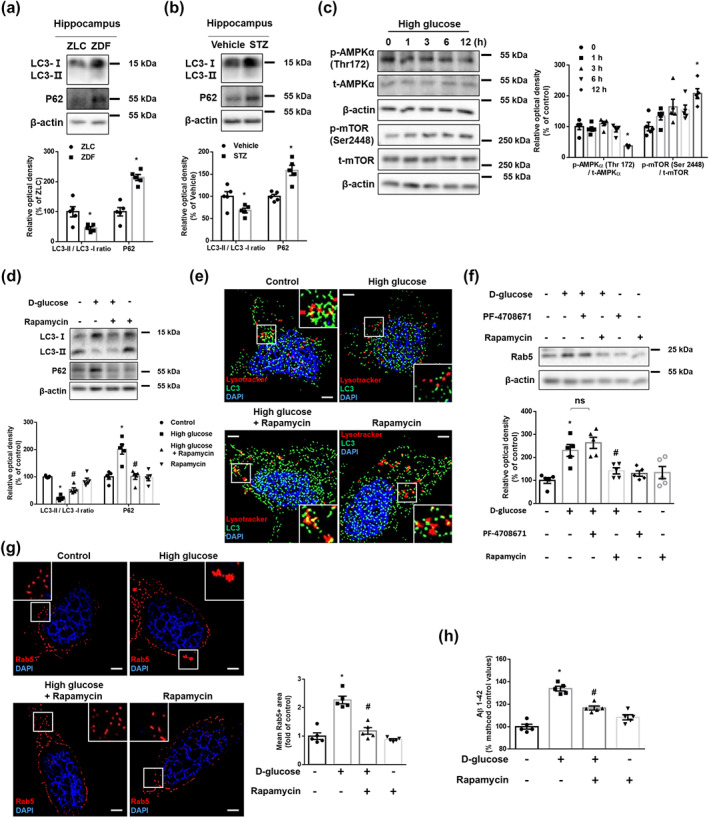
High glucose impairs endosomal clearance via AMPK/mTORC1‐mediated autophagy defect. (a) The hippocampal samples were obtained from ZLC and ZDF rats. LC3, P62, and β‐actin were detected by western blot. n = 5 in each group. *P < .05, significantly different from ZLC rats. (b) The hippocampal samples were obtained from vehicle‐ and STZ‐treated mice. LC3, P62, and β‐actin were subjected to western blot. n = 5 in each group. *P < .05, significantly different from vehicle‐treated mice. (c) The SK‐N‐MC cells were treated with high glucose (25 mM) in a time response. p‐AMPKα (Thr172), t‐AMPKα, p‐mTOR (Ser2448), t‐mTOR, and β‐actin were detected by western blot. n = 5 from independent experiments. (d,e,g) The cells were incubated with Rapamycin (200 nM) for 30 min before high glucose treatment (25 mM) for 24 h. (d) LC3, P62, and β‐actin were subjected to western blot. n = 5 from independent experiments. (e) The cells were immunostained with LC3‐specific antibody and stained with Lysotracker red and DAPI. Scale bars, 8 μm (magnification, ×1,000). Exploratory data. (f) The cells were pretreated with PF‐4708671 (10 μM) or Rapamycin (200 nM) for 30 min prior to high glucose treatment (25 mM) for 24 h. Rab5 and β‐actin were detected by western blot. n = 5 from independent experiments. (g) The cells were immunostained with Rab5‐specific antibody and counterstained with DAPI. Scale bars, 8 μm (magnification, ×1,000). n = 5 from independent experiments. (h) The cells were pretreated with Rapamycin (200 nM) for 30 min before high glucose treatment (25 mM) for 48 h. Aβ 1–42 from cell culture medium were measured by human Aβ 1–42 specific ELISA assay. n = 5 from independent experiments with two technical replicates each. Logarithmic transformations were performed for homogeneity of the sample variance. *P < .05, significantly different from control, # P < .05, significantly different from high glucose. Quantitative data are presented as a mean ± SEM. All blots and immunofluorescence images are representative
3.6. Lysosomal dysfunction induced by ROS and mTORC1 up‐regulates Aβ production through impairment of endosomal clearance
To further investigate endosomal degradative trafficking, we tested the relationship between early endosomes with lysosomes. High glucose increased Rab5 co‐localization with lysosomes (Figure 6a). As there were no significant alterations of mRNA expression levels of endolysosomal fusion‐related genes (Figure S7a), we hypothesized that lysosomal dysfunction impairs the degradation of enlarged endosomes. Indeed, high glucose decreased the mean size of acidic lysosomes (Usenovic, Tresse, Mazzulli, Taylor, & Krainc, 2012) (Figure 6a) and the percentage of lysotracker‐positive cells after 12 h. However, there were no significant changes over time in the control conditions (Figure 6b), which meant that high glucose reduced the number of intact lysosomes. Hippocampal LAMP1 increased in ZDF rats and STZ‐treated mice compared with that of control groups (Figure 6c,d). In in vitro models, LAMP1 also increased after 12 h under high glucose conditions (Figure 6e) indicating that dysfunctional lysosomes were increased under high glucose. Given that lysosomal dysfunction is caused by ROS‐mediated lysosomal membrane permeabilization (LMP) (Serrano‐Puebla & Boya, 2016) and impairment of lysosomal quality control (Papadopoulos & Meyer, 2017), we demonstrated that pretreatment with NAC or Rapamycin reversed the elevated LAMP1 expression levels induced by high glucose (Figure 6f,g). Furthermore, the attenuation of the signals of both lysotracker and BCECF‐AM by high glucose was recovered by pretreatment with NAC and Rapamycin, respectively (Figure 6h,i). In addition to the impairment of early endosomal degradation, we found that co‐localization of CTSB, an Aβ‐degradative lysosomal enzyme (Mueller‐Steiner et al., 2006), was decreased by high glucose (Figure 6j). Moreover, pretreatment with leupeptin further increased the amount of Aβ up‐regulated by high glucose (Figure 6k). These findings suggest that ROS‐ and mTORC1‐mediated lysosomal dysfunction is critical for the blockage of endosomal clearance and increment of Aβ under high glucose conditions.
FIGURE 6.
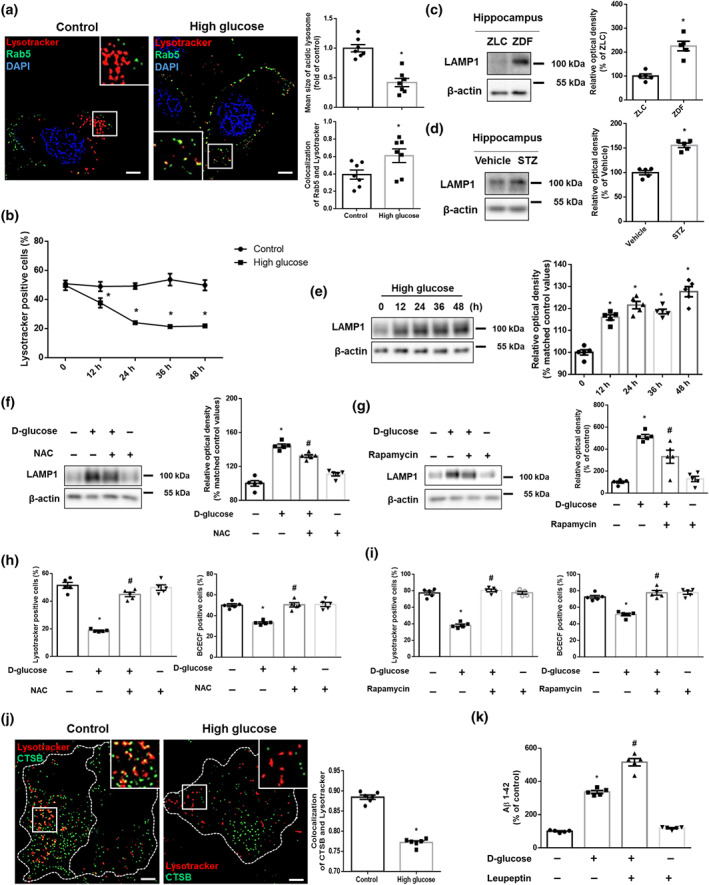
ROS‐ and mTORC1‐mediated lysosomal dysfunction aggravates endosomal clearance and up‐regulates Aβ production. (a,j) The SK‐N‐MC cells were treated with high glucose (25 mM) for 24 h. (a) The cells were immunostained with Rab5‐specific antibody and stained with Lysotracker red and DAPI. n = 7 from independent experiments. (b) The cells treated with DW or high glucose (25 mM) were labelled with Lysotracker red and analysed by flow cytometry in a time response. n = 5 from independent experiments. *P < .05, significantly different from control. (c) The hippocampal samples were obtained from ZLC and ZDF rats. LAMP1 and β‐actin were detected by western blot. n = 5. *P < .05, significantly different from ZLC rats in each group. (d) The hippocampal samples were obtained from vehicle‐ and STZ‐treated mice. LAMP1 and β‐actin were subjected to western blot. n = 5 in each group. *P < .05, significantly different from vehicle‐treated mice. (e) The cells in a time response with 25 mM high glucose were subjected to western blot. LAMP1 and β‐actin were detected. n = 5 from independent experiments. Logarithmic transformations were performed for homogeneity of the sample variance. (f,h) The cells were pretreated with NAC (4 mM) for 30 min before high glucose treatment (25 mM) for 24 h. (f) LAMP1 and β‐actin were detected by western blot. n = 5 from independent experiments. Logarithmic transformations were performed for homogeneity of the sample variance. (g,i) The cells were incubated with Rapamycin (200 nM) for 30 min prior to high glucose treatment (25 mM) for 24 h. (g) LAMP1 and β‐actin were detected by western blot. n = 5 from independent experiments. (h,i) Intact lysosomes and intracellular pH were measured by Lysotracker red and BCECF‐AM staining, respectively. n = 5 from independent experiments. (j) The cells were immunostained with CTSB‐specific antibody and stained with Lysotracker red. (k) The cells were pretreated with leupeptin (100 nM) for 30 min before high glucose treatment (25 mM) for 48 h. Aβ 1–42 from cell culture medium were measured by human Aβ 1–42 specific ELISA assay. n = 5 from independent experiments with two technical replicates each. *P < .05, significantly different from control, # P < .05, significantly different from high glucose. Quantitative data are presented as a mean ± SEM. All blots and immunofluorescence images are representative
3.7. Increased early endosomes, Aβ increment, and cognitive impairment are recovered by Dynasore and Rapamycin in STZ‐induced DM mice model
To show clinical relevance, we tested alterations of signature proteins and the clinical symptoms in STZ‐treated mice. As described in Section 2, animal experiments consisted of two parts (Figure 7a). In the histological data, signal intensities of hippocampal early endosomes of STZ‐treated mice were higher than both those of vehicle‐treated mice and those of STZ + Dynasore‐treated mice (Figure 7b). With increased early endosomes, STZ up‐regulated hippocampal Aβ levels, which were inhibited by Dynasore treatment (Figure 7c). Furthermore, the Y‐maze results showed that STZ‐treated mice showed cognitive impairment, whereas STZ‐treated mice with Dynasore treatment showed recovery of cognition (Figure 7d). Meanwhile, in STZ‐treated mice, phosphorylation of mTORC1 at Ser2448 was increased, which was down‐regulated by treatment with Rapamycin (Figure S8a). In the histological data, signal intensities of hippocampal early endosomes of STZ‐treated mice were higher than both those of vehicle‐treated mice and those of STZ + Rapamycin‐treated mice (Figure 7e). Increased protein expression levels of hippocampal Rab5 in STZ‐treated mice were reversed in STZ + Rapamycin‐treated mice (Figure 7f). In addition, STZ increased hippocampal Aβ levels and induced cognitive impairment, which were recovered by Rapamycin treatment (Figure 7g,h). These results demonstrate that DM induces cognitive impairment and Aβ increments through endocytosis‐ and autophagy‐mediated early endosomal dysregulation.
FIGURE 7.
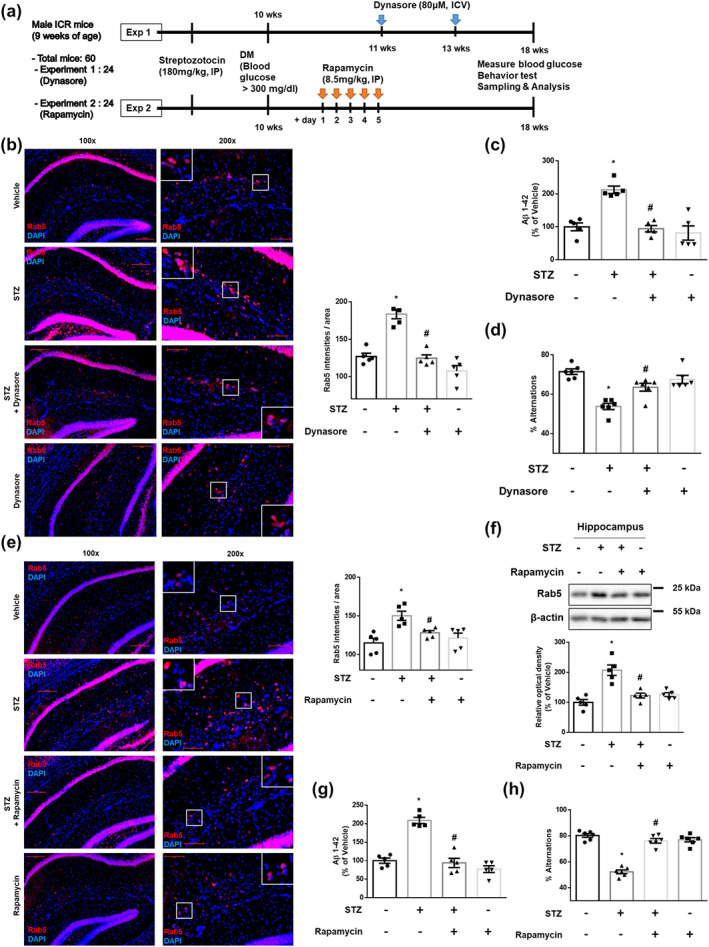
Increases in early endosomes, Aβ and cognitive impairment are reversed by Dynasore and Rapamycin, respectively, in STZ‐induced DM mouse model. (a) Scheme of the experimental protocols in vivo. (b–h) The experimental mice were treated with vehicle, STZ, Dynasore, or Rapamycin, as described in Section 2. (b,e) Hippocampal slides for IHC were immunostained with Rab5‐specific antibody and counterstained with DAPI. Scale bars, 50 μm (magnification, ×200). n = 5 in each group. (c,g) Hippocampal Aβ 1–42 were measured by rat and mouse Aβ 1–42 specific ELISA assay. n = 5 in each group with two technical replicates each. (d,h) Mice were subjected to Y‐maze test to evaluate cognitive function. n = 6 in each group. (f) Hippocampal Rab5 and β‐actin were subjected to western blot. n = 5 in each group. *P < .05, significantly different from vehicle‐treated mice, # P < .05, significantly different from STZ‐treated mice. Quantitative data are presented as a mean ± SEM. All blots and immunofluorescence images are representative
4. DISCUSSION
The present study has revealed that high glucose facilitates early endosomal abnormalities through increasing APP endocytosis and impairment of endosomal clearance, which leads to the up‐regulation of Aβ production. A previous paper had demonstrated that membrane cholesterol and lipid rafts are important factors for amyloidogenic APP endocytosis (Cheng et al., 2007). High glucose‐induced lipid raft reorganization has also been shown to facilitate BACE1 localization on lipid rafts, leading to increased Aβ production (Lee et al., 2016). Consistent with these findings, we found that lipid rafts mediate APP endocytosis under high glucose conditions. In addition to lipid rafts, proteins involved in endocytosis are associated with the pathogenesis of AD (Alsaqati, Thomas, & Kidd, 2018) and DM (Teng et al., 2016). Our data showed that high glucose contributed to increases in PICALM, CHC, and AP2A1, through ROS‐stimulated Sp1 activation. Levels of the long‐form of PICALM (upper band) appear to be decreased compared to the short form, in Mithramycin‐treated groups, and these levels were lower than those of the control. As Sp1 is a transcription factor that regulates PICALM expression, it would not have a significant effect on PICALM isoforms made via alternative splicing. Thus, differences in mRNA stability between the isoforms may be the reason for these findings. As all the isoforms increased under high glucose conditions, the differences in the isoform expression should not significantly affect overall outcome. Therefore, we suggest that excessive production of endocytic proteins due to ROS‐activated Sp1 are crucial molecular factors for APP endocytosis under high glucose conditions, leading to functional abnormalities. PICALM mediated APP endocytosis and Aβ generation in neural cell and PICALM knockdown decreased plaque deposition in APP transgenic mice (Xiao et al., 2012). PICALM up‐regulated Aβ generation by regulating CME of γ‐secretase (Kanatsu et al., 2014) and β‐secretase (Thomas et al., 2016) in neural cell and Aβ was reduced in the Picalm+/− mouse brain (Kanatsu et al., 2016). In addition, our data showed that PICALM facilitated APP endocytosis through the recruitment of CHC and AP2A1 to APP. On the other hand, PICALM‐induced trans‐endocytosis of Aβ in endothelial cells, which plays a role in Aβ clearance (Zhao et al., 2015). These distinct functions of PICALM are probably achieved by specific signalling in different cell types but require further study. However, considering our finding that high glucose increased both Aβ and PICALM and the knockdown of PICALM decreased Aβ production, we suggest that targeting PICALM is an efficient strategy for the down‐regulation of Aβ production.
A previous study reported that overexpression of the dominant inhibitory form of Rab5 in human adipocytes could mitigate pathophysiological changes in diabetes (Tessneer, Jackson, Griesel, & Olson, 2014). On the other hand, a study showed that Rab5 knockdown in murine fibroblasts aggravated DM (Su, Lodhi, Saltiel, & Stahl, 2006). Even if the effects of early endosomes on DM are still debatable, we showed that abnormalities in early endosomes were induced in high glucose conditions. Consistent with our results, early endosomal enlargement was reported in diabetic monkey brain, which up‐regulated Aβ production (Okabayashi et al., 2015). These findings indicate that the overactivation of early endosomes induced by high glucose may have a detrimental effect in AD. Persistent activation of an adaptor protein localized at early endosomes induced the activation of Rab5 (Kim, Sato, et al., 2016) and the mRNA (Ginsberg, Alldred, et al., 2010) and protein levels (Ginsberg, Mufson, et al., 2010) of Rab5 were up‐regulated in AD patients. Our finding that increased APP endocytosis is the cause of endosomal disorders differs from previous studies, and this finding may be specific to diabetes. Although further studies on the expression and activity of other secretases associated with Aβ production in diabetes and its association with endosomal disorders are needed, our data show that high glucose increased Rab5 co‐localization with both BACE1 and APP‐CTFβ, which produced excess Aβ. Similarly, a previous study described that the overexpression of Rab5 increased the coincidence of APP‐CTFβ and early endosomal compartments and Aβ generation (Grbovic et al., 2003). Taking these findings together, we demonstrate that high glucose up‐regulates Aβ production through inducing abnormalities in early endosomes.
Early endosomes are degraded by the auto‐lysosomal pathway. However, there is controversy over the changes in this degradative pathway in diabetes. DM increased autophagy in a STZ‐induced mouse model and primary cultured mouse hippocampal neurons through promotion of the JNK pathway (Kong et al., 2018) and in mouse osteoblasts through inhibition of the mTORC1 pathway (Wang, Feng, Li, Chen, & Tang, 2016). On the other hand, autophagy was impaired in diabetic beta‐cells (Bartolome et al., 2014) and in high glucose‐treated human glomerular endothelial cells (Lim et al., 2018) through hyper‐activation of mTORC1. Our results demonstrate that the impairment of autophagy was induced by the AMPK/mTORC1 signalling pathway under high glucose conditions. These findings indicate the different regulation of autophagy may depend on the cell type and preferentially involved mechanisms. Indeed, a previous study suggested that decreased activity of AMPK is a common characteristic for both DM and AD (Steinberg & Kemp, 2009). In addition, activation of AMPK protected hippocampal neurons against Aβ toxicity (Culmsee, Monnig, Kemp, & Mattson, 2001). The post mortem brains from adults with AD‐like pathology have high levels of mTORC1 and low levels of DEPTOR, the inhibitory protein of mTOR (Crino, 2016). We showed that the inhibition of mTORC1 down‐regulated high glucose‐induced Rab5 protein expression and Aβ production. These results suggest that dysfunction of the mTORC1‐mediated autophagy is a critical cause of AD pathology by causing defects in the clearance of early endosomes, in high glucose conditions. Meanwhile, destabilization of lysosomes was demonstrated in the hippocampus of DM mice (Sims‐Robinson, Bakeman, Rosko, Glasser, & Feldman, 2016). In accordance with these results, our data also indicated that high glucose facilitated lysosomal dysfunction. Previous reports demonstrated that high glucose facilitated ROS‐mediated increase of LMP (Abuarab, Munsey, Jiang, Li, & Sivaprasadarao, 2017). Furthermore, severe DM accompanying mTORC1‐mediated impairment of autophagy induced the defective clearance of lysosomes, lysophagy, which aggravated lysosomal dysfunction (Takahashi et al., 2017). Indeed, our data showed that lysosomal dysfunction was induced by both ROS and mTORC1. Although there is a need for more precise study of the molecular mechanisms behind lysosomal dysfunction, our findings reveal that ROS and mTORC1‐mediated lysosomal dysfunction aggravate the impairment of early endosomal clearance and up‐regulate Aβ production. Overall, we suggest that targeting the auto‐lysosomal pathway is a promising strategy for reducing the amount of Aβ by enhancing the clearance of APP‐producing early endosomes.
Our data showed that DM animal models also presented increased levels of early endosomes, which induced increased Aβ production and cognitive impairment. Although previous studies reported that treatment with an endocytosis blocker (Wang et al., 2018) and Rapamycin (Zhou et al., 2009) restored cognitive impairment, the mechanisms underlying these processes had not been previously identified. We tested the relevance of APP‐producing endosomes by applying pharmacological treatments to the diabetic STZ‐treated mice. As the drugs did not act as insulin, they may have little effect on controlling hyperglycaemia induced by STZ. Thus, reducing Aβ without decreasing blood glucose may be used in combination with conventional anti‐diabetic drugs, providing synergistic effects on reducing levels of Aβ. The potential limitations to this study include that the behaviour and rescue experiments were only performed in mice. We modified the experimental design as it was difficult to apply drugs that could have effects on the brain during the entire breeding period of ZLC and ZDF rats. However, our results showed that the expression levels of endo‐lysosomal proteins were up‐regulated in both DM models. Based on the above findings, we suggest that the dysfunction of endocytosis and the auto‐lysosomal pathway have detrimental effects on Aβ‐producing endosomes in diabetes. Additionally, it is possible that early endosomal abnormalities are a common feature that facilitates AD pathology.
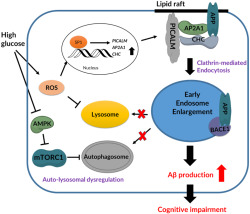
In conclusion, the present study demonstrated that high glucose conditions up‐regulated Aβ production through early endosomal abnormalities induced by PICALM‐mediated increases of APP endocytosis and ROS‐ and mTORC1‐mediated impairment of the auto‐lysosomal pathway (Figure 8). Indeed, our investigation is the first to identify early endosomal dysregulation and their precise mechanisms in Aβ pathology in diabetes. These findings shed light into novel diabetes‐related varieties of aberrant molecular and cellular processes leading to AD. Thus, the present study provides compelling evidence that targeting PICALM and mTORC1 to prevent endosomal abnormalities is an attractive and promising strategy for managing diabetes‐induced AD.
FIGURE 8.
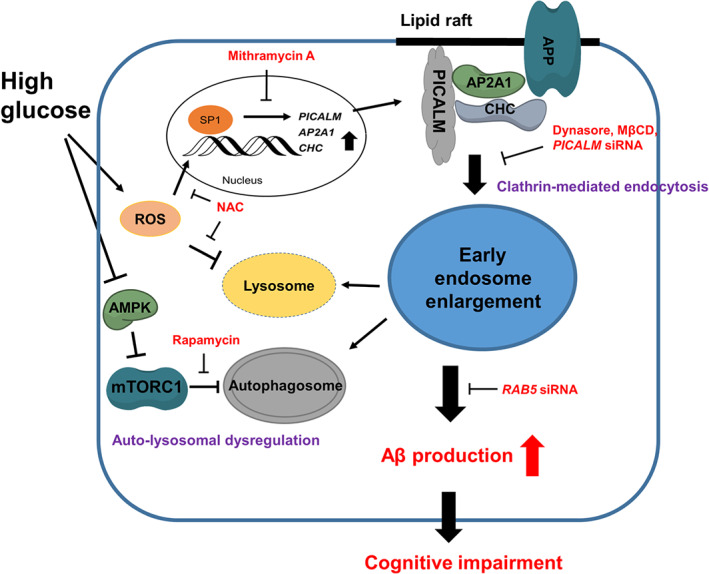
Diagram of the pathways underlying the up‐regulation of Aβ production by abnormalities in early endosomes, under high glucose conditions. High glucose increased PICALM, AP2A1, and CHC expression through ROS‐stimulated Sp1 nuclear translocation. PICALM facilitates clathrin‐mediated APP endocytosis at lipid rafts, which induces early endosomal enlargement. On the one hand, high glucose impairs endosomal clearance via AMPK/mTORC1‐ and ROS‐mediated auto‐lysosomal dysregulation. The consequences of abnormalities in early endosomes are overproduction of Aβ and cognitive impairment
AUTHOR CONTRIBUTIONS
C.W.C, H.J.L, G.E.C, and H.J.H. conceptualized and designed the paper. C.W.C., G.E.C., J.S.K., J.R.L, and S.Y.K. did the data acquisition. C.W.C, I.K.H., J.K.S., and H.J.H. interpreted and analyzed the data. C.W.C, H.J.L, Y.H.J, and H.J.H. wrote the manuscript. All authors reviewed and revised the final version of this manuscript.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
DECLARATION OF TRANSPARENCY AND SCIENTIFIC RIGOUR
This declaration acknowledges that this paper adheres to the principles for transparent reporting and scientific rigour of preclinical research as stated in the BJP guidelines for Natural Product Research, Design and Analysis, Immunoblotting and Immunochemistry, and Animal Experimentation, and as recommended by funding agencies, publishers, and other organizations engaged with supporting research.
Supporting information
Figure S1. The efficacy of siRNAs and toxic effects of drugs used in experiments. a The cells were treated with vehicle or drugs, Dynasore (25 μM), Mithramycin A (25 nM), Leupeptin (100 nM), Rapamycin (200 nM), GM6001 (20 μM), NAC (4 mM), MβCD (1 mM), and Pitstop 2 (30 μM), respectively for 24 h. Cell viability was measured by trypan blue exclusion cell viability assay. n = 5 from independent experiments. b The SK‐N‐MCs were transfected with NT siRNA or RAB5 siRNA for 12 h and incubated in medium for 24 h. Rab5 were detected by western blot. n = 5 from independent experiments. c The cells were transfected with NT siRNA or PICALM siRNA for 12 h and incubated in medium for 24 h. PICALM were detected by western blot. n = 5 from independent experiments. *p < 0.05 vs. NT siRNA transfection. Quantitative data are presented as a mean ± S.E.M. All blots are representative.
Figure S2. High glucose has no significant alterations on the mRNA expression of Rab5 and its effector proteins. a The SK‐N‐MCs were treated with high glucose (25 mM) in a time dependent manner. The mRNA expression levels of RAB5, EEA1, RABGEF1, RABEP1, and APPL1 were analyzed by quantitative real‐time PCR. n = 5 from independent experiments with 2 technical replicates each. b The cells were treated with D‐glucose (25 mM) or L‐glucose (25 mM) for 24 h. Then, APP and β‐actin were detected by western blot. n = 7 from independent experiments. *p < 0.05 vs. control, #p < 0.05 vs. D‐glucose. Quantitative data are presented as a mean ± S.E.M. All blots are representative.
Figure S3. PICALM is located in lipid rafts under both normal‐ and high glucose‐conditions. a High glucose (25 mM) were treated for 24 h in the cells. Sucrose gradient‐fractionized lysates were subjected to western blot. PICALM, CAV1, and FLOT1 were detected. Exploratory data.
Figure S4. High glucose impairs autophagy in a time dependent manner. a The SK‐N‐MCs were treated with high glucose (25 mM) in a time dependent manner. LC3, P62, and β‐actin were subjected to western blot. n = 5 from independent experiments. b The cells were treated with D‐glucose (25 mM) or L‐glucose (25 mM) for 24 h. Then, LC3, P62, and β‐actin were detected by western blot. n = 5 from independent experiments. *p < 0.05 vs. control. Quantitative data are presented as a mean ± S.E.M. All blots are representative.
Figure S5. PICALM induces autophagy under high glucose conditions. a, b The SK‐N‐MCs were treated with high glucose (25 mM) for 24 h. a Co‐immunoprecipitation of PICALM with IgG and LC3 antibodies were shown in left panel. Total protein expressions in lysate were shown in right panel. n = 5 from independent experiments. *p < 0.05 vs. Control. b The cells were immunostained with VAMP2 or VAMP8 or VAMP3‐specific antibodies and counterstained with DAPI. Scale bars, 8 μm (magnification, ×1,000). n = 5 from independent experiments. Exploratory data. c The cells were transfected with NT siRNA or PICALM siRNA for 12 h prior to high glucose (25 mM) treatment for 24 h. LC3 and β‐actin were detected by western blot. n = 5 from independent experiments. Logarithmic transformations were performed for homogeneity of the sample variance. *p < 0.05 vs. NT siRNA transfection, #p < 0.05 vs. NT siRNA transfection + high glucose. Quantitative data are presented as a mean ± S.E.M. All blots and immunofluorescence images are representative.
Figure S6. High glucose has no significant alterations on the protein expression of Beclin1. The SK‐N‐MCs were treated with high glucose (25 mM) in a time dependent manner. The Beclin1 were detected by western blot. n = 5 from independent experiments. Quantitative data are presented as a mean ± S.E.M. All blots are representative.
Figure S7. High glucose has no significant alterations on the mRNA expression of endo‐lysosomal fusion related proteins. a The SK‐N‐MCs were treated with high glucose (25 mM) in a time dependent manner. The mRNA expression levels of ATG14, SNAP29, VTI1B, STX17, and VAMP7 were analyzed by quantitative real‐time PCR. n = 5 from independent experiments with 2 technical replicates each. Quantitative data are presented as a mean ± S.E.M.
Figure S8. The treatment of rapamycin attenuated STZ‐induced phosphorylation of mTORC1. a Hippocampal sample was obtained from vehicle‐, STZ‐, STZ + rapamycin‐, or rapamycin‐treated mice. Then, p‐mTOR (Ser 2,448), t‐mTOR, and β‐actin were subjected to western blot. n = 5 in each animal. *p < 0.05 vs. vehicle‐treated mice, #p < 0.05 vs. STZ‐treated mice. Quantitative data are presented as a mean ± S.E.M. All blots are representative.
Table S1. Body weight and blood glucose levels in ZLC and ZDF rats. Blood glucose and body weight were determined with a portable glucose monitor at 6 and 33 weeks of age. Data are presented as a mean ± S.E.M.
Table S2. Body weight and blood glucose levels in experimental mice. Blood glucose and body weight were determined with a portable glucose monitor at 9 and 18 weeks of age. Data are presented as a mean ± S.E.M.
Table S3. Human gene primers for real‐time qPCR.
ACKNOWLEDGEMENT
This research was supported by National R&D Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (NRF‐2020R1A2B5B02002442) and BK21 PLUS Program for Creative Veterinary Science Research.
Chae CW, Lee HJ, Choi GE, et al. High glucose‐mediated PICALM and mTORC1 modulate processing of amyloid precursor protein via endosomal abnormalities. Br J Pharmacol. 2020;177:3828–3847. 10.1111/bph.15131
REFERENCES
- Abuarab, N. , Munsey, T. S. , Jiang, L. H. , Li, J. , & Sivaprasadarao, A. (2017). High glucose‐induced ROS activates TRPM2 to trigger lysosomal membrane permeabilization and Zn2+‐mediated mitochondrial fission. Science Signaling, 10, eaal4161 10.1126/scisignal.aal4161 [DOI] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Fabbro, D. , Kelly, E. , Mathie, A. , Peters, J. A. , Veale, E. L. , … Collaborators, C. G. T. P. (2019). THE CONCISE GUIDE TO PHARMACOLOGY 2019/20: Enzymes. British Journal of Pharmacology, 176, S297–S396. 10.1111/bph.14752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, S. P. H. , Roberts, R. E. , Broughton, B. R. S. , Sobey, C. G. , George, C. H. , Stanford, S. C. , … Ahluwalia, A. (2018). Goals and practicalities of immunoblotting and immunohistochemistry: A guide for submission to the British Journal of Pharmacology. British Journal of Pharmacology, 175, 407–411. 10.1111/bph.14112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alsaqati, M. , Thomas, R. S. , & Kidd, E. J. (2018). Proteins involved in endocytosis are upregulated by ageing in the normal human brain: Implications for the development of Alzheimer's Disease. The Journals of Gerontology. Series a, Biological Sciences and Medical Sciences, 73, 289–298. 10.1093/gerona/glx135 [DOI] [PubMed] [Google Scholar]
- Bai, X. L. , Yang, X. Y. , Li, J. Y. , Ye, L. , Jia, X. , Xiong, Z. F. , … Jin, S. (2017). Cavin‐1 regulates caveolae‐mediated LDL transcytosis: Crosstalk in an AMPK/eNOS/NF‐κB/Sp1 loop. Oncotarget, 8, 103985–103995. 10.18632/oncotarget.21944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolome, A. , Kimura‐Koyanagi, M. , Asahara, S. , Guillen, C. , Inoue, H. , Teruyama, K. , … Uchiyama, Y. (2014). Pancreatic β‐cell failure mediated by mTORC1 hyperactivity and autophagic impairment. Diabetes, 63, 2996–3008. 10.2337/db13-0970 [DOI] [PubMed] [Google Scholar]
- Biessels, G. J. , & Despa, F. (2018). Cognitive decline and dementia in diabetes mellitus: Mechanisms and clinical implications. Nature Reviews. Endocrinology, 14, 591–604. 10.1038/s41574-018-0048-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cataldo, A. M. , Petanceska, S. , Terio, N. B. , Peterhoff, C. M. , Durham, R. , Mercken, M. , … Nixon, R. A. (2004). Aβ localization in abnormal endosomes: Association with earliest Aβ elevations in AD and Down syndrome. Neurobiology of Aging, 25, 1263–1272. 10.1016/j.neurobiolaging.2004.02.027 [DOI] [PubMed] [Google Scholar]
- Cheng, H. , Vetrivel, K. S. , Gong, P. , Meckler, X. , Parent, A. , & Thinakaran, G. (2007). Mechanisms of disease: new therapeutic strategies for Alzheimer's disease‐targeting APP processing in lipid rafts. Nature Clinical Practice. Neurology, 3, 374–382. 10.1038/ncpneuro0549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, X. T. , Xie, Y. X. , Zhou, B. , Huang, N. , Farfel‐Becker, T. , & Sheng, Z. H. (2018). Characterization of LAMP1‐labeled nondegradative lysosomal and endocytic compartments in neurons. The Journal of Cell Biology, 217, 3127–3139. 10.1083/jcb.201711083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colacurcio, D. J. , Pensalfini, A. , Jiang, Y. , & Nixon, R. A. (2018). Dysfunction of autophagy and endosomal‐lysosomal pathways: Roles in pathogenesis of Down syndrome and Alzheimer's Disease. Free Radical Biology & Medicine, 114, 40–51. 10.1016/j.freeradbiomed.2017.10.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crino, P. B. (2016). The mTOR signalling cascade: Paving new roads to cure neurological disease. Nature Reviews. Neurology, 12, 379–392. 10.1038/nrneurol.2016.81 [DOI] [PubMed] [Google Scholar]
- Culmsee, C. , Monnig, J. , Kemp, B. E. , & Mattson, M. P. (2001). AMP‐activated protein kinase is highly expressed in neurons in the developing rat brain and promotes neuronal survival following glucose deprivation. Journal of Molecular Neuroscience, 17, 45–58. 10.1385/JMN:17:1:45 [DOI] [PubMed] [Google Scholar]
- Curtis, M. J. , Alexander, S. , Cirino, G. , Docherty, J. R. , George, C. H. , Giembycz, M. A. , … Ahluwalia, A. (2018). Experimental design and analysis and their reporting II: Updated and simplified guidance for authors and peer reviewers. British Journal of Pharmacology, 175, 987–993. 10.1111/bph.14153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Docherty, J. R. , Stanford, S. C. , Panattieri, R. A. , Alexander, S. P. H. , Cirino, G. , George, C. H. , … Ahluwalia, A. (2019). Sex: A change in our guidelines to authors to ensure that this is no longer an ignored experimental variable. British Journal of Pharmacology, 176, 4081–4086. 10.1111/bph.14761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlemann, F. C. , Menon, V. , Condurat, A. L. , Rossler, J. , & Pruszak, J. (2017). Surface marker profiling of SH‐SY5Y cells enables small molecule screens identifying BMP4 as a modulator of neuroblastoma differentiation. Scientific Reports, 7, 13612 10.1038/s41598-017-13497-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman, B. L. (2015). Streptozotocin‐induced diabetic models in mice and rats. Current Protocols in Pharmacology, 70, 5–47. 41‐20 [DOI] [PubMed] [Google Scholar]
- Ginsberg, S. D. , Alldred, M. J. , Counts, S. E. , Cataldo, A. M. , Neve, R. L. , Jiang, Y. , … Che, S. (2010). Microarray analysis of hippocampal CA1 neurons implicates early endosomal dysfunction during Alzheimer's disease progression. Biological Psychiatry, 68, 885–893. 10.1016/j.biopsych.2010.05.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg, S. D. , Mufson, E. J. , Counts, S. E. , Wuu, J. , Alldred, M. J. , Nixon, R. A. , & Che, S. (2010). Regional selectivity of rab5 and rab7 protein upregulation in mild cognitive impairment and Alzheimer's disease. Journal of Alzheimer's Disease, 22, 631–639. 10.3233/JAD-2010-101080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez, C. D. , Lee, M.‐S. , Marchetti, P. , Pietropaolo, M. , Towns, R. , Vaccaro, M. I. , … Wiley, J. W. (2014). The emerging role of autophagy in the pathophysiology of diabetes mellitus. Autophagy, 7, 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grbovic, O. M. , Mathews, P. M. , Jiang, Y. , Schmidt, S. D. , Dinakar, R. , Summers‐Terio, N. B. , … Cataldo, A. M. (2003). Rab5‐stimulated up‐regulation of the endocytic pathway increases intracellular β‐cleaved amyloid precursor protein carboxyl‐terminal fragment levels and Aβ production. The Journal of Biological Chemistry, 278, 31261–31268. 10.1074/jbc.M304122200 [DOI] [PubMed] [Google Scholar]
- Guimas Almeida, C. , Sadat Mirfakhar, F. , Perdigao, C. , & Burrinha, T. (2018). Impact of late‐onset Alzheimer's genetic risk factors on β‐amyloid endocytic production. Cellular and Molecular Life Sciences, 75, 2577–2589. 10.1007/s00018-018-2825-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson, N. , Culley, S. , Ashdown, G. , Owen, D. M. , Pereira, P. M. , & Henriques, R. (2016). Fast live‐cell conventional fluorophore nanoscopy with ImageJ through super‐resolution radial fluctuations. Nature Communications, 7(1), 12471 10.1038/ncomms12471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, C. , Angot, E. , Bergstrom, A. L. , Steiner, J. A. , Pieri, L. , Paul, G. , … Li, J. Y. (2011). α‐Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. The Journal of Clinical Investigation, 121, 715–725. 10.1172/JCI43366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding, S. D. , Sharman, J. L. , Faccenda, E. , Southan, C. , Pawson, A. J. , Ireland, S. , … NC‐IUPHAR . (2018). The IUPHAR/BPS Guide to PHARMACOLOGY in 2018: updates and expansion to encompass the new guide to IMMUNOPHARMACOLOGY. Nucleic Acids Research, 46, D1091–D1106. 10.1093/nar/gkx1121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haythorne, E. , Rohm, M. , van de Bunt, M. , Brereton, M. F. , Tarasov, A. I. , Blacker, T. S. , … Ashcroft, F. M. (2019). Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic β‐cells. Nature Communications, 10(1), 2474 10.1038/s41467-019-10189-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatsu, K. , Hori, Y. , Takatori, S. , Watanabe, T. , Iwatsubo, T. , & Tomita, T. (2016). Partial loss of CALM function reduces Aβ42 production and amyloid deposition in vivo. Human Molecular Genetics, 25, 3988–3997. 10.1093/hmg/ddw239 [DOI] [PubMed] [Google Scholar]
- Kanatsu, K. , Morohashi, Y. , Suzuki, M. , Kuroda, H. , Watanabe, T. , Tomita, T. , & Iwatsubo, T. (2014). Decreased CALM expression reduces Aβ42 to total Aβ ratio through clathrin‐mediated endocytosis of γ‐secretase. Nature Communications, 5(1), 3386 10.1038/ncomms4386 [DOI] [PubMed] [Google Scholar]
- Kilkenny, C. , Browne, W. , Cuthill, I. C. , Emerson, M. , Altman, D. G. , & Group NCRRGW . (2010). Animal research: Reporting in vivo experiments: the ARRIVE guidelines. British Journal of Pharmacology, 160, 1577–1579. 10.1111/j.1476-5381.2010.00872.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H. Y. , Lee, D. K. , Chung, B. R. , Kim, H. V. , & Kim, Y. (2016). Intracerebroventricular injection of amyloid‐β peptides in normal mice to acutely induce Alzheimer‐like cognitive deficits. Journal of Visualized Experiments, 109, e53308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, S. , Sato, Y. , Mohan, P. S. , Peterhoff, C. , Pensalfini, A. , Rigoglioso, A. , … Nixon, R. A. (2016). Evidence that the rab5 effector APPL1 mediates APP‐βCTF‐induced dysfunction of endosomes in Down syndrome and Alzheimer's disease. Molecular Psychiatry, 21, 707–716. 10.1038/mp.2015.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong, F. J. , Ma, L. L. , Guo, J. J. , Xu, L. H. , Li, Y. , & Qu, S. (2018). Endoplasmic reticulum stress/autophagy pathway is involved in diabetes‐induced neuronal apoptosis and cognitive decline in mice. Clinical Science (London, England), 132, 111–125. 10.1042/CS20171432 [DOI] [PubMed] [Google Scholar]
- Kwart, D. , Gregg, A. , Scheckel, C. , Murphy, E. , Paquet, D. , Duffield, M. , … Tessier‐Lavigne, M. (2019). A large panel of isogenic APP and PSEN1 mutant human iPSC neurons reveals shared endosomal abnormalities mediated by APP β‐CTFs, not Aβ. Neuron, 140(2), 256–270. [DOI] [PubMed] [Google Scholar]
- Lacy, M. E. , Gilsanz, P. , Karter, A. J. , Quesenberry, C. P. , Pletcher, M. J. , & Whitmer, R. A. (2018). Long‐term glycemic control and dementia risk in Type 1 diabetes. Diabetes Care, 41, 2339–2345. 10.2337/dc18-0073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law, F. , Seo, J. H. , Wang, Z. , DeLeon, J. L. , Bolis, Y. , Brown, A. , … Rocheleau, C. E. (2017). The VPS34 PI3K negatively regulates RAB‐5 during endosome maturation. Journal of Cell Science, 130, 2007–2017. 10.1242/jcs.194746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, H. J. , Ryu, J. M. , Jung, Y. H. , Lee, S. J. , Kim, J. Y. , Lee, S. H. , … Han, H. J. (2016). High glucose upregulates BACE1‐mediated Aβ production through ROS‐dependent HIF‐1α and LXRα/ABCA1‐regulated lipid raft reorganization in SK‐N‐MC cells. Scientific Reports, 6(1), 36746 10.1038/srep36746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, J. H. , McBrayer, M. K. , Wolfe, D. M. , Haslett, L. J. , Kumar, A. , Sato, Y. , … Mitchell, C. H. (2015). Presenilin 1 maintains lysosomal Ca2+ homeostasis via TRPML1 by regulating vATPase‐mediated lysosome acidification. Cell Reports, 12, 1430–1444. 10.1016/j.celrep.2015.07.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, J. H. , Kim, H. W. , Kim, M. Y. , Kim, T. W. , Kim, E. N. , Kim, Y. , … Park, C. W. (2018). Cinacalcet‐mediated activation of the CaMKKβ‐LKB1‐AMPK pathway attenuates diabetic nephropathy in db/db mice by modulation of apoptosis and autophagy. Cell Death & Disease, 9(3), 270 10.1038/s41419-018-0324-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters, C. L. , Bateman, R. , Blennow, K. , Rowe, C. C. , Sperling, R. A. , & Cummings, J. L. (2015). Alzheimer's disease. Nature Reviews. Disease Primers, 1, 15056 10.1038/nrdp.2015.56 [DOI] [PubMed] [Google Scholar]
- McFall, G. P. , Wiebe, S. A. , Vergote, D. , Anstey, K. J. , & Dixon, R. A. (2015). Alzheimer's genetic risk intensifies neurocognitive slowing associated with diabetes in non‐demented older adults. Alzheimers Dement (Amst), 1, 395–402. 10.1016/j.dadm.2015.08.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau, K. , Fleming, A. , Imarisio, S. , Lopez Ramirez, A. , Mercer, J. L. , Jimenez‐Sanchez, M. , … Rubinsztein, D. C. (2014). PICALM modulates autophagy activity and tau accumulation. Nature Communications, 5(1), 4998 10.1038/ncomms5998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno‐Gonzalez, I. , Edwards Iii, G. , Salvadores, N. , Shahnawaz, M. , Diaz‐Espinoza, R. , & Soto, C. (2017). Molecular interaction between type 2 diabetes and Alzheimer's disease through cross‐seeding of protein misfolding. Molecular Psychiatry, 22, 1327–1334. 10.1038/mp.2016.230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moruno, F. , Perez‐Jimenez, E. , & Knecht, E. (2012). Regulation of autophagy by glucose in mammalian cells. Cell, 1, 372–395. 10.3390/cells1030372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller‐Steiner, S. , Zhou, Y. , Arai, H. , Roberson, E. D. , Sun, B. , Chen, J. , … Gan, L. (2006). Antiamyloidogenic and neuroprotective functions of cathepsin B: implications for Alzheimer's disease. Neuron, 51, 703–714. 10.1016/j.neuron.2006.07.027 [DOI] [PubMed] [Google Scholar]
- Nixon, R. A. (2017). Amyloid precursor protein and endosomal‐lysosomal dysfunction in Alzheimer's disease: Inseparable partners in a multifactorial disease. The FASEB Journal, 31, 2729–2743. 10.1096/fj.201700359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okabayashi, S. , Shimozawa, N. , Yasutomi, Y. , Yanagisawa, K. , & Kimura, N. (2015). Diabetes mellitus accelerates Aβ pathology in brain accompanied by enhanced GAβ generation in nonhuman primates. PLoS ONE, 10, e0117362 10.1371/journal.pone.0117362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papadopoulos, C. , & Meyer, H. (2017). Detection and clearance of damaged lysosomes by the endo‐lysosomal damage response and lysophagy. Current Biology: CB, 27, R1330–R1341. 10.1016/j.cub.2017.11.012 [DOI] [PubMed] [Google Scholar]
- Serrano‐Puebla, A. , & Boya, P. (2016). Lysosomal membrane permeabilization in cell death: New evidence and implications for health and disease. Annals of the New York Academy of Sciences, 1371, 30–44. 10.1111/nyas.12966 [DOI] [PubMed] [Google Scholar]
- Sims‐Robinson, C. , Bakeman, A. , Rosko, A. , Glasser, R. , & Feldman, E. L. (2016). The role of oxidized cholesterol in diabetes‐induced lysosomal dysfunction in the brain. Molecular Neurobiology, 53, 2287–2296. 10.1007/s12035-015-9207-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Small, S. A. , Simoes‐Spassov, S. , Mayeux, R. , & Petsko, G. A. (2017). Endosomal traffic jams represent a pathogenic hub and therapeutic target in Alzheimer's Disease. Trends in Neurosciences, 40, 592–602. 10.1016/j.tins.2017.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srinivasan, K. , & Ramarao, P. (2007). Animal models in type 2 diabetes research: An overview. Indian Journal of Medical Research, 125, 451–472. [PubMed] [Google Scholar]
- Steinberg, G. R. , & Kemp, B. E. (2009). AMPK in health and disease. Physiological Reviews, 89, 1025–1078. 10.1152/physrev.00011.2008 [DOI] [PubMed] [Google Scholar]
- Su, X. , Lodhi, I. J. , Saltiel, A. R. , & Stahl, P. D. (2006). Insulin‐stimulated interaction between insulin receptor substrate 1 and p85α and activation of protein kinase B/Akt require Rab5. The Journal of Biological Chemistry, 281, 27982–27990. 10.1074/jbc.M602873200 [DOI] [PubMed] [Google Scholar]
- Takahashi, A. , Takabatake, Y. , Kimura, T. , Maejima, I. , Namba, T. , Yamamoto, T. , … Isaka, Y. (2017). Autophagy inhibits the accumulation of advanced glycation end products by promoting lysosomal biogenesis and function in the kidney proximal tubules. Diabetes, 66, 1359–1372. 10.2337/db16-0397 [DOI] [PubMed] [Google Scholar]
- Teng, B. , Schroder, P. , Muller‐Deile, J. , Schenk, H. , Staggs, L. , Tossidou, I. , … Schiffer, M. (2016). CIN85 deficiency prevents nephrin endocytosis and proteinuria in diabetes. Diabetes, 65, 3667–3679. 10.2337/db16-0081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessneer, K. L. , Jackson, R. M. , Griesel, B. A. , & Olson, A. L. (2014). Rab5 activity regulates GLUT4 sorting into insulin‐responsive and non‐insulin‐responsive endosomal compartments: A potential mechanism for development of insulin resistance. Endocrinology, 155, 3315–3328. 10.1210/en.2013-2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas, R. S. , Henson, A. , Gerrish, A. , Jones, L. , Williams, J. , & Kidd, E. J. (2016). Decreasing the expression of PICALM reduces endocytosis and the activity of β‐secretase: Implications for Alzheimer's disease. BMC Neuroscience, 17(1), 50 10.1186/s12868-016-0288-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, Y. , Chang, J. C. , Fan, E. Y. , Flajolet, M. , & Greengard, P. (2013). Adaptor complex AP2/PICALM, through interaction with LC3, targets Alzheimer's APP‐CTF for terminal degradation via autophagy. Proceedings of the National Academy of Sciences of the United States of America, 110, 17071–17076. 10.1073/pnas.1315110110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usenovic, M. , Tresse, E. , Mazzulli, J. R. , Taylor, J. P. , & Krainc, D. (2012). Deficiency of ATP13A2 leads to lysosomal dysfunction, α‐synuclein accumulation, and neurotoxicity. The Journal of Neuroscience, 32, 4240–4246. 10.1523/JNEUROSCI.5575-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacinova, G. , Vejrazkova, D. , Lukasova, P. , Lischkova, O. , Dvorakova, K. , Rusina, R. , … Vaňková, M. (2017). Associations of polymorphisms in the candidate genes for Alzheimer's disease BIN1, CLU, CR1 and PICALM with gestational diabetes and impaired glucose tolerance. Molecular Biology Reports, 44, 227–231. 10.1007/s11033-017-4100-9 [DOI] [PubMed] [Google Scholar]
- von Kleist, L. , Stahlschmidt, W. , Bulut, H. , Gromova, K. , Puchkov, D. , Robertson, M. J. , … Chircop, M. (2011). Role of the clathrin terminal domain in regulating coated pit dynamics revealed by small molecule inhibition. Cell, 146, 471–484. [DOI] [PubMed] [Google Scholar]
- Wang, W. , Duclot, F. , Groveman, B. R. , Carrier, N. , Qiao, H. , Fang, X. Q. , … Yu, X. M. (2018). Hippocampal protein kinase D1 is necessary for DHPG‐induced learning and memory impairments in rats. PLoS ONE, 13, e0195095 10.1371/journal.pone.0195095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Feng, Z. , Li, J. , Chen, L. , & Tang, W. (2016). High glucose induces autophagy of MC3T3‐E1 cells via ROS‐AKT‐mTOR axis. Molecular and Cellular Endocrinology, 429, 62–72. 10.1016/j.mce.2016.03.036 [DOI] [PubMed] [Google Scholar]
- Xiao, Q. , Gil, S. C. , Yan, P. , Wang, Y. , Han, S. , Gonzales, E. , … Lee, J. M. (2012). Role of phosphatidylinositol clathrin assembly lymphoid‐myeloid leukemia (PICALM) in intracellular amyloid precursor protein (APP) processing and amyloid plaque pathogenesis. The Journal of Biological Chemistry, 287, 21279–21289. 10.1074/jbc.M111.338376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W. , Tan, L. , & Yu, J. T. (2015). The role of PICALM in Alzheimer's disease. Molecular Neurobiology, 52, 399–413. 10.1007/s12035-014-8878-3 [DOI] [PubMed] [Google Scholar]
- Yu, W. H. , Cuervo, A. M. , Kumar, A. , Peterhoff, C. M. , Schmidt, S. D. , Lee, J. H. , … Nixon, R. A. (2005). Macroautophagy—A novel β‐amyloid peptide‐generating pathway activated in Alzheimer's disease. The Journal of Cell Biology, 171, 87–98. 10.1083/jcb.200505082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Z. , Sagare, A. P. , Ma, Q. , Halliday, M. R. , Kong, P. , Kisler, K. , … Zlokovic, B. V. (2015). Central role for PICALM in amyloid‐β blood‐brain barrier transcytosis and clearance. Nature Neuroscience, 18, 978–987. 10.1038/nn.4025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J. , Blundell, J. , Ogawa, S. , Kwon, C. H. , Zhang, W. , Sinton, C. , … Parada, L. F. (2009). Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural‐specific Pten knock‐out mice. The Journal of Neuroscience, 29, 1773–1783. 10.1523/JNEUROSCI.5685-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. The efficacy of siRNAs and toxic effects of drugs used in experiments. a The cells were treated with vehicle or drugs, Dynasore (25 μM), Mithramycin A (25 nM), Leupeptin (100 nM), Rapamycin (200 nM), GM6001 (20 μM), NAC (4 mM), MβCD (1 mM), and Pitstop 2 (30 μM), respectively for 24 h. Cell viability was measured by trypan blue exclusion cell viability assay. n = 5 from independent experiments. b The SK‐N‐MCs were transfected with NT siRNA or RAB5 siRNA for 12 h and incubated in medium for 24 h. Rab5 were detected by western blot. n = 5 from independent experiments. c The cells were transfected with NT siRNA or PICALM siRNA for 12 h and incubated in medium for 24 h. PICALM were detected by western blot. n = 5 from independent experiments. *p < 0.05 vs. NT siRNA transfection. Quantitative data are presented as a mean ± S.E.M. All blots are representative.
Figure S2. High glucose has no significant alterations on the mRNA expression of Rab5 and its effector proteins. a The SK‐N‐MCs were treated with high glucose (25 mM) in a time dependent manner. The mRNA expression levels of RAB5, EEA1, RABGEF1, RABEP1, and APPL1 were analyzed by quantitative real‐time PCR. n = 5 from independent experiments with 2 technical replicates each. b The cells were treated with D‐glucose (25 mM) or L‐glucose (25 mM) for 24 h. Then, APP and β‐actin were detected by western blot. n = 7 from independent experiments. *p < 0.05 vs. control, #p < 0.05 vs. D‐glucose. Quantitative data are presented as a mean ± S.E.M. All blots are representative.
Figure S3. PICALM is located in lipid rafts under both normal‐ and high glucose‐conditions. a High glucose (25 mM) were treated for 24 h in the cells. Sucrose gradient‐fractionized lysates were subjected to western blot. PICALM, CAV1, and FLOT1 were detected. Exploratory data.
Figure S4. High glucose impairs autophagy in a time dependent manner. a The SK‐N‐MCs were treated with high glucose (25 mM) in a time dependent manner. LC3, P62, and β‐actin were subjected to western blot. n = 5 from independent experiments. b The cells were treated with D‐glucose (25 mM) or L‐glucose (25 mM) for 24 h. Then, LC3, P62, and β‐actin were detected by western blot. n = 5 from independent experiments. *p < 0.05 vs. control. Quantitative data are presented as a mean ± S.E.M. All blots are representative.
Figure S5. PICALM induces autophagy under high glucose conditions. a, b The SK‐N‐MCs were treated with high glucose (25 mM) for 24 h. a Co‐immunoprecipitation of PICALM with IgG and LC3 antibodies were shown in left panel. Total protein expressions in lysate were shown in right panel. n = 5 from independent experiments. *p < 0.05 vs. Control. b The cells were immunostained with VAMP2 or VAMP8 or VAMP3‐specific antibodies and counterstained with DAPI. Scale bars, 8 μm (magnification, ×1,000). n = 5 from independent experiments. Exploratory data. c The cells were transfected with NT siRNA or PICALM siRNA for 12 h prior to high glucose (25 mM) treatment for 24 h. LC3 and β‐actin were detected by western blot. n = 5 from independent experiments. Logarithmic transformations were performed for homogeneity of the sample variance. *p < 0.05 vs. NT siRNA transfection, #p < 0.05 vs. NT siRNA transfection + high glucose. Quantitative data are presented as a mean ± S.E.M. All blots and immunofluorescence images are representative.
Figure S6. High glucose has no significant alterations on the protein expression of Beclin1. The SK‐N‐MCs were treated with high glucose (25 mM) in a time dependent manner. The Beclin1 were detected by western blot. n = 5 from independent experiments. Quantitative data are presented as a mean ± S.E.M. All blots are representative.
Figure S7. High glucose has no significant alterations on the mRNA expression of endo‐lysosomal fusion related proteins. a The SK‐N‐MCs were treated with high glucose (25 mM) in a time dependent manner. The mRNA expression levels of ATG14, SNAP29, VTI1B, STX17, and VAMP7 were analyzed by quantitative real‐time PCR. n = 5 from independent experiments with 2 technical replicates each. Quantitative data are presented as a mean ± S.E.M.
Figure S8. The treatment of rapamycin attenuated STZ‐induced phosphorylation of mTORC1. a Hippocampal sample was obtained from vehicle‐, STZ‐, STZ + rapamycin‐, or rapamycin‐treated mice. Then, p‐mTOR (Ser 2,448), t‐mTOR, and β‐actin were subjected to western blot. n = 5 in each animal. *p < 0.05 vs. vehicle‐treated mice, #p < 0.05 vs. STZ‐treated mice. Quantitative data are presented as a mean ± S.E.M. All blots are representative.
Table S1. Body weight and blood glucose levels in ZLC and ZDF rats. Blood glucose and body weight were determined with a portable glucose monitor at 6 and 33 weeks of age. Data are presented as a mean ± S.E.M.
Table S2. Body weight and blood glucose levels in experimental mice. Blood glucose and body weight were determined with a portable glucose monitor at 9 and 18 weeks of age. Data are presented as a mean ± S.E.M.
Table S3. Human gene primers for real‐time qPCR.