

Abstract
Despite its central importance as a regulator of cellular physiology, identification and precise mapping of O-linked N-acetylglucosamine (O-GlcNAc) post-translational modification (PTM) sites in proteins by mass spectrometry (MS) remains a considerable technical challenge. This is due in part to cleavage of the glycosidic bond occurring prior to the peptide backbone during collisionally activated dissociation (CAD), which leads to generation of characteristic oxocarbenium ions and impairs glycosite localization. Herein, we leverage CAD-induced oxocarbenium ion generation to trigger ultraviolet photodissociation (UVPD), an alternate high-energy deposition method that offers extensive fragmentation of peptides while leaving the glycosite intact. Upon activation using UV laser pulses, efficient photodissociation of glycopeptides is achieved with production of multiple sequence ions that enable robust and precise localization of O-GlcNAc sites. Application of this method to tryptic peptides originating from O-GlcNAcylated proteins TAB1 and Polyhomeotic confirmed previously reported O-GlcNAc sites in TAB1 (S395 and S396) and uncovered new sites within both proteins. We expect this strategy will complement existing MS/MS methods and be broadly useful for mapping O-GlcNAcylated residues of both proteins and proteomes.
Graphical Abstract
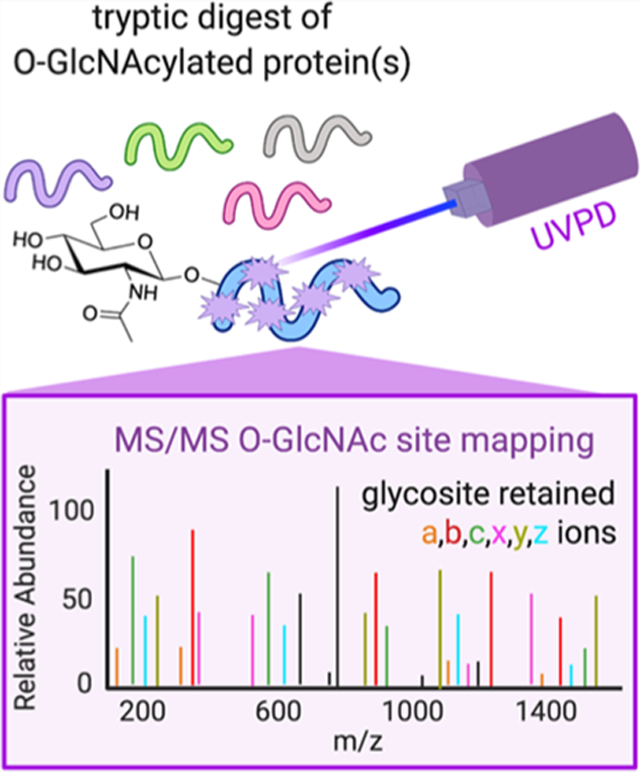
INTRODUCTION
The ubiquitous modification of protein serine and threonine residues by a single O-GlcNAc monosaccharide occurs on hundreds of nucleocytosolic proteins throughout multicellular eukaryotes.1 Enzymatic installation and removal of O-GlcNAc is mediated by just two enzymes; O-GlcNAc transferase (OGT) installs the sugar on target proteins, and O-GlcNAc hydrolase (OGA) removes it. The reversible nature of this modification enables it to serve as a nutrient- and stress-responsive regulator of diverse cellular processes including epigenetic regulation of gene expression, proteostasis, and stress response.1–3 Dysregulation of O-GlcNAc is emerging as a potential contributor to several diseases including cancer, diabetes mellitus, and neurodegeneration.2–5 Despite its importance, however, the cellular and molecular mechanisms underlying how O-GlcNAc regulates these processes remain largely unknown. Accordingly, identification of modified proteins6–8 and mapping of protein O-GlcNAcylation sites9–16 have emerged as topics of intense current interest. Nevertheless, accurate and precise MS-based mapping of O-GlcNAc sites using traditional bottom-up glycoproteomics remains a central limiting challenge for the field for three main reasons: (i) the stoichiometric abundances of O-GlcNAc-modified proteins are often low, (ii) O-GlcNAc sites lack a consensus motif and are often found in serine/threonine-rich stretches in proteins, making their precise localization challenging, and (iii) the high lability of the glycosidic linkage results in liberation of O-GlcNAc during CAD, where fragmentation is dominated by lower energy pathways.6 Although this fragmentation occurs for other O-linked glycans,17 the glycosidic bond in O-GlcNAc is particularly fragile because this fragmentation pathway is facilitated by the trans stereochemical relationship between the glycosidic oxygen and the neighboring N-acetyl group. The N-acetyl group can participate to facilitate fragmentation, and the resulting oxocarbenium ion is stabilized through broader delocalization of the positive charge.
In an attempt to address these challenges, an approach based on β-elimination of O-GlcNAc followed by Michael addition (BEMAD) to install MS-stable reporters and affinity tags for enrichment was developed.18,19 However, a factor limiting the application of BEMAD in complex biological samples is its cross reactivity with O-phosphate and other O-linked glycans, which can lead to false positives in the absence of carefully executed and validated control experiments.20 Therefore, significant effort has gone into improving analytical methods for direct MS analysis of native O-GlcNAcylated peptides. In particular, alternative ion activation methods that preserve labile protein modifications, such as electron capture dissociation (ECD)21 and electron transfer dissociation (ETD),22,23 have significantly facilitated mapping of O-GlcNAc. Furthermore, these methods have been integrated with collisional activation to enhance the ability to characterize O-GlcNAcylation sites. For example, a new hybrid MS/MS method, electron-transfer higher energy collisional dissociation (EThcD),24 has proven beneficial for O-GlcNAcylated peptide analysis by enhancing the conversion of nondissociated ions generated by ETD into meaningful fragment ions via application of supplementary collisional activation.25 Clever targeted methods have been implemented to take advantage of the abundant glycan-specific oxocarbenium ions produced by HCD to trigger subsequent ETD on the same precursor ions.26–28 Oxocarbenium product ion-triggered methods filter the peptides subjected to ETD analysis, thus reducing the duty cycle for collection of sequential HCD and ETD spectra, and more efficiently allocate data acquisition time for glycopeptide analysis.29–31 However, electron-based dissociation methods generally do not cause fragmentation N-terminal to proline,21 which is commonly observed at the −2 and −3 positions upstream of O-GlcNAc sites,32 and more generally within disordered regions in which O-GlcNAc is predominantly found.33 In addition, the rather long periods used for electron activation methods, often tens of milliseconds, are less efficient for high-throughput LCMS analysis of complex mixtures. Therefore, development of additional and complementary fragmentation methods that overcome some of these short-comings is essential to advancing the field.
Recognizing that a key challenge for mapping O-GlcNAc sites relates to the ability to characterize the peptide sequence and localize modifications, we applied UVPD, which uses high-energy laser photons for activation.34 UVPD unlocks fragmentation pathways with significantly higher activation energies, enabling formation of a wide array of diagnostic ions while preserving labile bonds within product ions, leading to its broad utility in PTM mapping including for characterization of glycosylated peptides.35–37 Here, we detail a generally applicable proof of concept approach using a two-part MS/ MS strategy based on HCD-oxocarbenium ion-triggered UVPD to characterize both O-GlcNAcylated peptides and proteins (Figure 1).
Figure 1.

MS/MS mapping of O-GlcNAcylated peptides. Schematic diagram of the higher energy collisional dissociation (HCD) oxocarbenium ion-triggered UVPD of O-GlcNAcylated peptides. Primary sequence fragmentation schematic with color code used in figures.
RESULTS AND DISCUSSION
To test our approach, we synthesized three peptides bearing known O-GlcNAcylated residues by in vitro OGT-catalyzed glycosylation: TAB1-O-GlcNAc (biotin-PVSVPYS(g)SAQSTS), CKII-O-GlcNAc (YPGGSTPVS(g)SANMM), and HCF-O-GlcNAc(YVRVCSNPPCS(g)THETGTTNTATTATS) (see Figure S1 for HPLC traces and MS1 spectra). HCD experiments using the TAB1-O-GlcNAc glycopeptide revealed the majority of the dominant ions produced are b/y-type ions as well as oxocarbenium ions released by cleavage of the glycosidic bond (Figure 2A, Table S2, expanded in Figure S2). Owing to loss of O-GlcNAc, the b/y ions are valuable for peptide sequencing but not for glycosite localization. Importantly, the array of low-mass oxocarbenium ions (m/z 126.06, 138.06, 144.07, 168.07, 186.08, and 204.09) generated upon HCD are characteristic of N-acetylglucosamine (GlcNAc)38 and can be used as a reporter ion filter to trigger UVPD of the same precursor ion that was selected for HCD. Therefore, we implemented a two-part HCD/UVPD method, whereby observation of these oxocarbenium ions within the top 20 product ions in an initial HCD scan triggers UVPD of the same precursor ion in the subsequent scan. The high-speed duty cycle of HCD (<1 ms per scan) allows many peptides to be screened with high efficiency, and UVPD can be triggered selectively for exclusive characterization of targeted glycopeptides in complex mixtures.
Figure 2.

HCD oxocarbenium ion-triggered UVPD of O-GlcNAcylated peptides generates a rich array of glycosylated sequence ions for confident site mapping. (A) HCD (NCE 28) mass spectrum of TAB1-O-GlcNAc peptide (2+, m/z 869.90), showing a distinctive oxocarbenium ion pattern, labeled with red asterisks (m/z 126.06, 138.06, 144.07, 168.07, 186.08, and 204.09). PCS score: 8.6e-29. (B) 193 nm UVPD mass spectrum of TAB1-O-GlcNAc peptide (2+, m/z 869.90). PCS score: 2.6e-50. (C) 193 nm UVPD mass spectrum of HCF-O-GlcNAc peptide (3+, m/z 967.76. PCS score: 5.8e-27). In B and C, two laser pulses of 1.5 mJ were used for UVPD. (D) EThcD mass spectrum of HCF-O-GlcNAc peptide (3+, m/z 967.76) using an optimized 50 ms reaction time for ETD with 25% supplemental collisional energy. PCS score: 3.4e-16. In each spectrum, the double-headed arrow and ΔGlcNAc indicate the mass shift representing loss of the O-GlcNAc-modified amino acid between key fragment ions (labeled in gray at each end of the double-headed arrow) that bracket the site of modification. Oxonium ions are labeled with red asterisks. Triangles denote fragment ions that have lost the GlcNAc modification. Backbone cleavages leading to N-terminal and C-terminal fragment ions are marked in blue and red, respectively, on the peptide sequence shown in each spectrum. Complete lists of identified fragment ions are shown in Tables S2, S3, S5, and S7.
Application of this combined method to the TAB1-O-GlcNAc glycopeptide provides extensive fragmentation and preservation of O-GlcNAc-containing fragments. Owing to its high-energy deposition that leads to fast ion dissociation, UVPD can induce fragmentation pathways that retain the O-GlcNAc (i.e., backbone cleavages occur prior to loss of labile modifications). In particular, many of the b and y ion series retain O-GlcNAc for UVPD, whereas conventional collisional activation (like HCD) more frequently produces b/y ions that have lost O-GlcNAc. Particularly notable is that the UVPD mass spectrum showcases a larger array of useful ion types for sequencing the peptide as compared to HCD, including those that retain O-GlcNAc (a7, a9, a10, a11, b9, b10, x8, x11, y9, z7, z8, z12) and allow it to be confidently localized to Ser7 (Figure 2B, Figure S3, Table S3). For comparison, because EThcD is currently considered a gold-standard method for O-GlcNAc mapping, the same TAB1-O-GlcNAc glycopeptide was also analyzed using an analogous HCD-triggered EThcD strategy that was optimized to enable direct comparison of these methods of analyses.10 Even after optimization of both ETD reaction time and added supplemental activation energy, we found that although the GlcNAc modification is retained in some of the product ions (b6, b12, c6, c7, c8, c9, y6, y9, y10, y11, z10, z11, z12), EThcD does not induce bidirectional fragmentation of the middle region of the peptide beyond the N-terminal c-ion series (Figure S4, Table S4). These initial experiments confirmed the ability of UVPD to map O-GlcNAc sites and provide an initial indication that UVPD can expand upon the current arsenal of MS fragmentation methods for mapping O-GlcNAc.
After this initial proof of concept, three glycopeptides (TAB1-O-GlcNAc, CKII-O-GlcNAc, and HCF-O-GlcNAc) were used for systematic optimization of UVPD parameters. In particular, the number of laser pulses and the laser power were varied to evaluate their impact on sequence coverage and retention of O-GlcNAc based on the average number of matching fragment ions (Figure S5). UVPD parameters that consistently yielded the most informative spectra with optimum sequence coverage and glycosite retention were the use of two 5 ns pulses of 1.5 mJ; 193 nm UVPD is capable of producing high levels of peptide sequence coverage with several important differences from electron-activation techniques. In UVPD experiments, experimental parameters are optimized to minimize excessive energy deposition which can lead to secondary dissociation of fragment ions and conversion into small, less informative product ions. In addition, UVPD results in a greater array of fragmentation channels, thus leading to spectra that are data rich. This combination of optimizing the energy deposition and extensive fragmentation means that the resulting spectra are dense with low-abundance fragment ions which still have excellent signal-to-noise ratios that permit reliable assignments. These spectra also often show a large portion of nondissociated precursor ions. Unlike electron-activation methods, UVPD does not generate significant abundances of charge-reduced products. Similar to EThcD, however, production of oxonium ions as well as fragment ions that have lost the O-GlcNAc modification are observed owing to the highly labile nature of the O-glycosidic bond. Importantly, however, the O-GlcNAc is predominantly retained on a, c, x, and z fragment ion series (Tables S3, S5, and S6), which is what enables confident assignment of the sites of modification. However, the high-energy activation of UVPD allows for modifications to be retained even for collisional activation-type ions. Some neutral loss of O-GlcNAc is observed during UVPD, particularly for b-and y-type ions. While the exact mechanism for UVPD fragmentation has yet to be elucidated, this is likely due to the redistribution of excess vibrational energy, causing cleavage of the labile glycosidic bond, similar to collisional dissociation.
Application of the optimized HCD/UVPD method to the 2.9 kDa HCF-O-GlcNAc glycopeptide revealed consistent production of the same three oxocarbenium ion fragments, [C8 H10 O3N]+ (m/z 168.07), [C8 H12 O4N]+ (m/z 186.08), and [GlcNAc]+ (m/z 204.09) (Figure S6) upon HCD of the glycopeptide in three different charge states (2+, 3+, 4+). This was an important observation as this two-step HCD triggering strategy relies on production of these oxocarbenium ions across different peptides and charge states. The corresponding UVPD spectrum for this large HCF-O-GlcNAc glycopeptide (3+) yielded 92% sequence coverage and numerous diagnostic sequence ions, including a10, a11, b11, x15, x16, and y15, all of which retain O-GlcNAc and allow simple and unambiguous localization to Ser11 (Figure 2C, Table S5). Similarly, a third glycopeptide CKII-O-GlcNAc shows 100% sequence coverage, and UVPD affords confident localization of O-GlcNAc to Ser9 as expected (Figure S7A, Table S6). For optimization of EThcD for comparative analyses, ETD activation time (ms) and the amount of supplemental collisional energy during HCD was varied to evaluate its impact on sequence coverage and retention of O-GlcNAc based on the average number of matching fragment ions for each standard glycopeptide (Figure S8). This optimization led to the use of an ETD activation time of 50 ms and supplemental collisional energy of 25 NCE for the EThcD experiments. Applying these optimized EThcD parameters for analysis of HCF-O-GlcNAc and CKII-OGlcNAc resulted in 52% and 85% sequence coverage, respectively. For HCF-O-GlcNAc, the O-GlcNAc was localized to the N-terminal side of HCF; however, EThcD did not allow confident localization to Ser11 (Figure 2D, Table S7). Similar to the outcome for analysis of TAB1-O-GlcNAc, EThcD of CKII-O-GlcNAc did not produce bidirectional diagnostic ions but did afford N-terminal b-type fragment ions that successfully pinpointed O-GlcNAc to Ser9 (Figure S7B, Table S8). These results highlight the enhanced ability of UVPD compared to EThcD to generate a larger array of information-rich fragment ions that retain O-GlcNAc, enabling precise mapping of O-GlcNAc sites within glycopeptides, especially those with high numbers of serine/threonine residues.
To evaluate the performance of HCD-triggered UVPD for O-GlcNAc site mapping in more complex mixtures, two O-GlcNAc-modified recombinant proteins were generated using a previously described OGT coexpression system.39,40 The first protein was Homo sapiens transforming growth factor (TGF)-β-activated protein kinase 1 (TAK1) binding protein 1 (TAB1, aa 1–504), involved in regulating innate immunity and inflammation through promoting pro-inflammatory cytokine signaling. O-GlcNAcylation of TAB1 boosts activation of TAK1, and in separate studies, O-GlcNAc was mapped to S39541 and S396,13 located within its disordered C-terminal region. The second protein corresponds to Drosophila melanogaster Polyhomeotic (Ph, aa 1397–1589), a component of the polycomb repressive complex 1 (PRC1), a central player in the regulation of development within Drosophila through modulation of HOX gene expression.43 This construct consists of an N-terminal region of predicted disorder followed by a C-terminal sterile α-motif (SAM) domain. O-GlcNAcylation of Ph at unknown site(s) was previously demonstrated to influence its aggregation and be required for repression of a subset of polycomb target genes in Drosophila.43 Prior to MS analysis, the presence of O-GlcNAc was confirmed by incubation of the OGT-coexpressed proteins with either WT Bacteroides thetaiotaomicron GH84 (WT BtGH84) to selectively remove O-GlcNAc or a catalytically dead mutant (D242A BtGH84),39 followed by immunoblot analyses using a panspecific anti-O-GlcNAc antibody (CTD110.6, Figure S9).
Following confirmation of O-GlcNAcylation, we employed the tandem HCD/UVPD approach for LCMS/MS analysis of tryptic digests of these two O-GlcNAcylated model proteins (chromatographic traces, Figure S10). The resulting HCD and UVPD mass spectra were searched against a human or Drosophila proteome reference database with addition of each recombinant protein sequence appended to each respective FASTA file. Upon HCD, few of the sequence ions retained the labile GlcNAc modification, preventing assignment of OGlcNAcylation sites. However, despite the lack of O-GlcNAc-specific sequence ions upon HCD, the abundant oxocarbenium ions corresponding to the GlcNAc signature38 (m/z 168, 186, 204) were reliably produced for every observed tryptic glycopeptide. At the same time, UVPD enabled confident identification of peptide sequences as well as definitive localization of glycosylation sites for every peptide that generated the oxocarbenium ions upon HCD. For TAB1, 97% protein sequence coverage was observed using the HCD-triggered UVPD strategy based on detection of 67 unique peptides. In total, UVPD identified seven unique glycopeptides corresponding to seven different O-GlcNAc sites across each of the three replicate experiments (Figure 3A, Table 1) with greater than 94% average sequence coverage of each peptide at a false discovery rate (FDR) of <1%.
Figure 3.
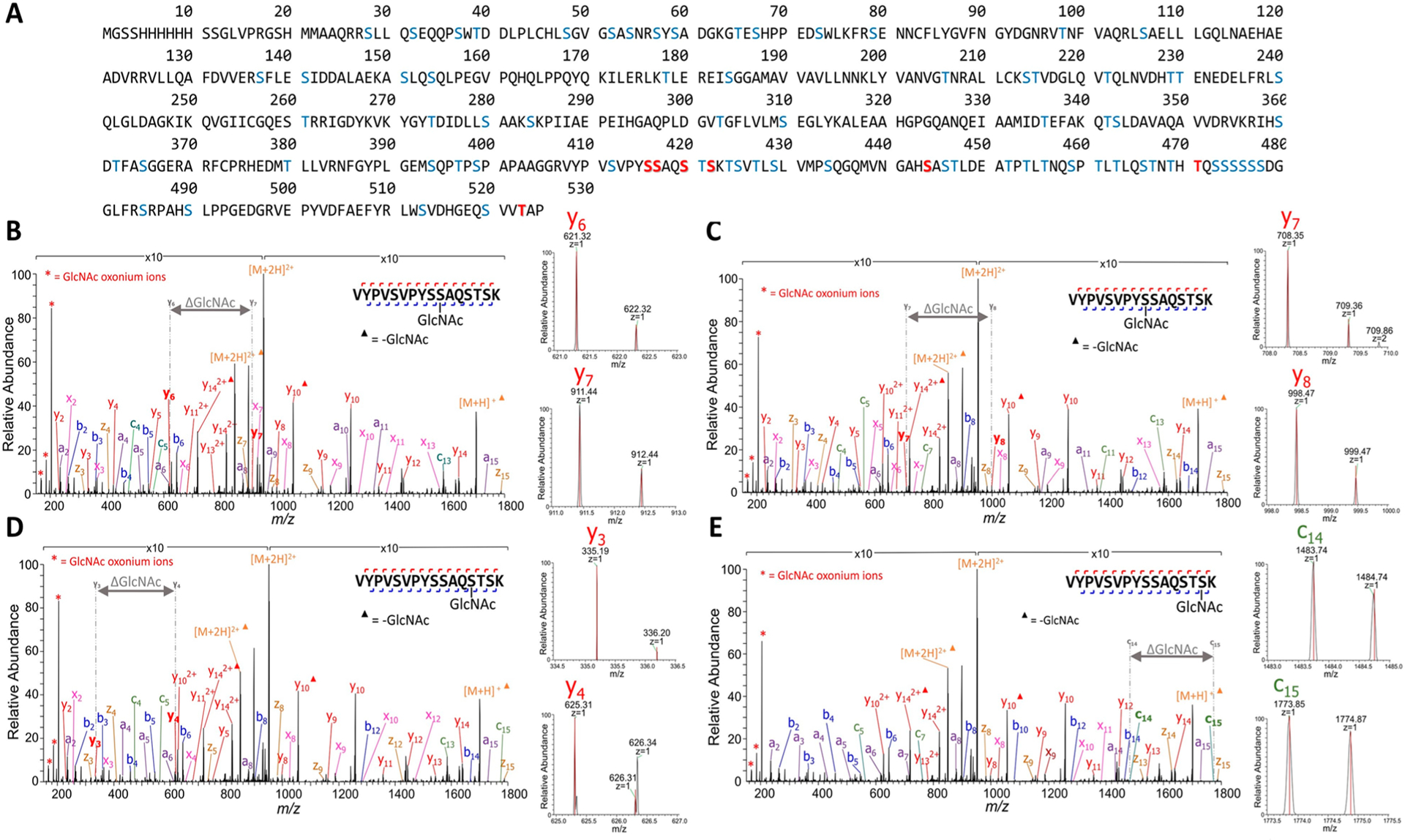
HCD oxocarbenium ion-triggered UVPD enables robust glycosite mapping of tryptic peptides originating from O-GlcNAc-modified TAB1. (A) TAB1 sequence with possible O-GlcNAc sites indicated in blue font and identified sites in red font. (B–E) Representative UVPD mass spectra of four differentially O-GlcNAc-modified peptide isoforms originating from a tryptic digest of glycosylated recombinant TAB1. Shown are (B) VYPVSVPYS[+203]SAQSTSK (Ser395 site), (C) VYPVSVPYSS[+203]AQSTSK (Ser396 site), (D) VYPVSVPYSSAQS[+203]TSK (Ser399 site), and (E) VYPVSVPYSSAQSTS[+203]K (Ser401 site), each (2+, m/z 951.97). Double-headed arrows and ΔGlcNAc indicate the mass shifts representing loss of the O-GlcNAc-modified amino acid between fragment ions (labeled in gray at each end of the double-headed arrow) that bracket the site of modification. Oxonium ions are labeled with red asterisks. Triangles denote fragment ions that exhibit a GlcNAc neutral loss. Backbone cleavages leading to N-terminal and C-terminal fragment ions are marked in blue and red, respectively, on the peptide sequence shown in each spectrum. Expansions of the m/z regions corresponding to selected pairs of diagnostic fragment ions for assignment of the O-GlcNAc sites are shown to the right of each panel, with the theoretical m/z for each fragment marked in red. Two laser pulses of 1.5 mJ were used for UVPD. Complete lists of identified fragment ions are shown in Tables S9–S12.
Table 1.
Summary of O-GlcNAc-Containing Peptides Identified Using HCD-Triggered UVPD for Analysis of Tryptic Digests of O-GlcNAc-Modified TAB1 and Ph Proteins
| protein | glycopeptide | charge state (+) | Detected ion (m/z) | peptide mass (Da) | Mass error (ppm) | PEP2Db | Delta Mod score | Sequence coverage | Modified resc/known site |
|---|---|---|---|---|---|---|---|---|---|
| TAB1 | VYPVSVPYSa(g1)SAQSTSK | 2 | 951.97 | 1902.93 | 1.2 | 1.5e-13 | 766 | 93% | S395/Y41 |
| TAB1 | VYPVSVPYSS(g2)AQSTSK | 2 | 951.97 | 1902.93 | 1.3 | 6.1e-13 | 771 | 100% | S396/Y13 |
| TAB1 | VYPVSVPYSSAQS(g3)TSK | 2 | 951.97 | 1902.93 | 0.3 | 1.5e-12 | 761 | 100% | S399/N |
| TAB1 | VYPVSVPYSSAQSTS(g4)K | 2 | 951.97 | 1902.93 | 1.1 | 1.5e-12 | 659 | 100% | S401/N |
| TAB1 | TSVTLSLVMPSQGQMVNGAHS(g5)ASTLDEATPTLTNQSPTLTLQSTNTHTQSSSSSSDGGLFR | 4 | 1617.77 | 6467.07 | −0.8 | 1.4e-13 | 1120 | 92% | S423/N |
| TAB1 | TSVTLSLVMPSQGQMVNGAHSASTLDEATPTLTNQSPTLTLQSTNTHT(g6)QSSSSSSDGGLFR | 4 | 1617.77 | 6467.07 | −1.1 | 8.2e-15 | 1035 | 73% | T450/N |
| TAB1 | LWSVDHGEQSVVT(g7)AP | 2 | 914.44 | 1826.87 | −0.2 | 3.9e-9 | 697 | 100% | T502/N |
| ph1397–1589 | LSESFPILGASTEVPPMS(g2)LPVQAAISAPSPLAMPLGSPLSVALPTLAPLSVVTSGAAPK | 5 | 118.24 | 5937.19 | −0.7 | 7.2e-11 | 345 | 91% | S1457/N |
| ph1397–1589 | LSESFPILGASTEVPPMSLPVQAAIS(g3)APSPLAMPLGSPLSVALPTLAPLSVVTSGAAPK | 5 | 1188.24 | 5937.19 | 1.5 | 7.2e-16 | 537 | 95% | S1465/N |
| ph1397–1589 | SSEVNGTDRPPIS(g4)S WSVDDVSNFIR | 3 | 989.80 | 2967.40 | 1.6 | 1.3e-16 | 730 | 100% | S1511/N |
(g) denotes an identified O-GlcNAc modification site
Scoring criteria used for assessing the quality of PTM site localization based on a previously established two-dimensional target decoy strategy (PEP 2D scores < 10−5),42 and the Delta Mod metric yields a measure of site localization of each identified O-GlcNAc modification (Delta Mod scores > 40) using Byonic.
Modified amino acid position in the full-length protein.
Data are based on triplicate runs.
UVPD offers the unprecedented identification of four different monoglycosylated isoforms of the same TAB1 tryptic peptide (VYPVSVPS395(g1)S396(g2)AQS399(g3)TS401(g4)K, Figure 3B–E, Table S9–S12). For example, the HCD-triggered UVPD mass spectra for the Ser396-modified isoform yielded 100% sequence coverage and numerous diagnostic sequence ions, including a10, b11, c13, x7, y7, and z7, all which retain the O-GlcNAc modification and allow confident glycosite localization (Figure 3B). The UVPD mass spectra for the other three monoglycosylated isoforms of VYPVSVPS395(g1)S396(g2)AQS399(g3)T-S401(g4)K similarly enabled confident O-GlcNAc site mapping through chromatographic separation from the most abundant isoform (Table 1, Figure 3C–E, Figure S11A). For the S395 and S396 positional isomers, we estimate that approximately 93% of the theoretical fragment ions will be shared owing to the small difference in the position of O-GlcNAc. Moreover, approximately 86% of the canonical fragment ions will be shared among the S399 and S401 positional isomers. This means that reliable glycosite assignments depend on uncovering just a few unique fragment ions.
For these isoforms, three differing C-terminal fragments were found for the S395/S396 glycoforms using high-resolution Orbitrap detection (Figure S11B), and five differing N-terminal fragments are found for the S399/S401 glycopeptide positional isomers (Figure S11C). In contrast, a prior peptide-based study using HCD revealed only a single site corresponding to S395 in the full-length protein.41
The HCD-triggered-UVPD method was able to confidently assign additional O-GlcNAc modification sites on Thr502 and, remarkably, two additional sites (Ser423 and Thr450) arising from distinct monoglycosylated isoforms of a large 6.5 kDa peptide in the C-terminal region of TAB1 (Table 1, Figures S12A and S13, Tables S13–15).44 LCMS/MS analysis of the same TAB1 tryptic digest using the optimized HCD/EThcD method confirmed three of the four glycopeptide isoforms [VYPVSVPS395(g1)S396(g2)AQ-S399(g3)TSK] (Table S1, Figure S14, Tables S16–S18) as well as O-GlcNAc localization on the C-terminal end of TAB1 (LWSVDHGEQSVVT523(g7)AP) (Table S1, Figures S12B, Tables S19). However, the HCD-triggered-EThcD approach failed to identify the various monoglycoslylated isoforms of the 6.5 kDa peptide from the C-terminal region of TAB1. Taken together, not only did the new HCD/UVPD method validate the previously reported O-GlcNAc sites of TAB1 (S39541 and S39613), it also identified five new sites mapping to its C-terminal disordered region (Figure 3A). Though analyzed on recombinant protein, these sites may also be involved in O-GlcNAc-dependent TAK1 activation or other cellular roles. In particular, p38-mediated phosphorylation of TAB1 Ser423 was previously shown to negatively regulate TAK1 kinase. Therefore, bearing in mind that both PTMs are most often substoichiometric, it would be interesting to carefully monitor whether reciprocal regulation of phosphorylation and O-GlcNAcylation occurs at this site and whether it might impact TAK1 signaling in cells.
With regard to Ph, application of HCD-triggered-UVPD enabled identification and localization of three O-GlcNAc sites within the Ph1397–1589 sequence (Table 1, Figure S15) and additional sites located just N-terminally to the N-terminal hexahistidine affinity purification tag (Figures S16 and S17A, Tables S20 and S21). Most notably, two different monoglycosylated isoforms of the same 5.9 kDa Ph tryptic peptide (LSESFPILGASTEVPPMS(g2)LPVQAAIS(g3)-APSPLAMPLGSPLSVALPTLAPLSVVTSGAAPK, Figures S18A and S19, Tables S22–S23) were observed with >90% sequence coverage, allowing confident characterization of each isoform. O-GlcNAc was confidently localized to Ser1511 via a series of bidirectional bracketing fragments, b12, b13, c12, c13, y12, and y13, allowing confident assignment of O-GlcNAc modification to this site (Figure S20A, Table S24). Using the optimized HCD-triggered-EThcD method for Ph, three O-GlcNAc sites were localized in comparison to the five sites located using the HCD-triggered UVPD approach (Table S1, Figures S17B, S18B, and S20B, Tables S25–S27).
CONCLUSION
Collectively, these proof of concept data validate the use of triggered HCD/UVPD as a powerful method to identify and precisely map protein O-GlcNAcylation sites. The rich mixture of informative fragment ions that retain O-GlcNAc should prove complementary to existing HCD-and ETD-based methods that are the current standard approaches used for O-GlcNAc mapping. Moreover, the demonstrated utility of this method for mapping multiple O-GlcNAc sites within large peptides highlights the potential for UVPD to be leveraged for top-down O-GlcNAc mapping of multiple proteoforms, as has been done for other PTMs,45 to enable targeted site discovery. Finally, we expect that combining this method with recent direct capture and chemoenzymatic enrichment strategies10,46 will prove valuable for high-throughput mapping of O-GlcNAc sites in complex proteomic samples.
EXPERIMENTAL SECTION
Preparation of O-GlcNAc Modified Peptides.
Glycopeptides used in this study were generated chemoenzymatically using recombinantly expressed O-GlcNAc transferase (OGT). All unmodified peptides were obtained commercially (Biomatik Corp., Cambridge, ON). Generally, 1–2 mg of oligopeptide at a final concentration of 1 mM were incubated with UDP-GlcNAc (5 mM), OGT (1 μM, prepared as previously described39) and 2U shrimp alkaline phosphatase (New England Biolabs, Whitby, ON) in PBS at pH 7.2 containing 12.5 mM MgCl2. Reactions were incubated at 37 °C for 4 h or overnight. Prior to purification, the reactions were terminated by heating at 95 °C for 10 min and then centrifuged at 13 000g for 2 min. The supernatants were recovered and purified on an Agilent 1200 series HPLC equipped with an Agilent XDB-C18 Eclipse reversed-phase column (9.4 × 250 mm, 5 μ particle size). Glycopeptides were eluted using a mobile phase consisting of H2O and CH3CN with 0.1% trifluoroacetic acid over a gradient of 10–50% acetonitrile as appropriate. Fractions containing the product were lyophilized to yield up to 1 mg of glycopeptide as white powders. All glycopeptides were purified to >95% purity and analyzed by high-resolution mass spectrometry performed using a Bruker maXis Impact UltraHigh-Resolution Quadrupole Time-of-Flight (UHR-QTOF) mass spectrometer (mobile phase water:acetonitrile 1:1 + 0.1% formic acid, flow rate of 0.3 mL/min).
Protein Expression and Purification.
The Homo sapiens TAB1 gene (NCBI accession number Q15750.1) corresponding to resides 1–504 was cloned into the pET28a expression vector containing a C-terminal thrombin-cleavable 6XHis tag as previously described.39 A construct corresponding to residues 1397–1589 from the Drosophila melanogaster Ph gene (NCBI accession number CAA45211) was codon optimized, synthesized, and subcloned into pET28a using NdeI and BamHI restriction sites by Genscript. The full-length Homo sapiens OGT gene (NCBI accession O15294.3, residues 1–1046) was cloned into the multiple cloning site of pMal-c2X vector (New England Biolabs) as previously described.40
TAB1 and Ph1397–1589 plasmids were cotransformed with OGT-pMAL into One Shot chemically competent Escherichia coli BL21-DE3 cells (Invitrogen C600003). TAB1 and Ph1397–1589 were grown at 37 °C until OD600 = 0.7 was reached, and expression was induced by addition of 1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) followed by overnight incubation at 25 °C. Harvested cells (20 g) were resuspended in lysis buffer (20 mM sodium phosphate pH 7.5, 500 mM NaCl, 1 mg/mL lysozyme, 1 EDTA free protease inhibitor tablet from Roche), lysed by sonication, and centrifuged (45 000g) for 1 h. Supernatant was passed through a 1 mM HiTrap HP His column pre-equilibrated in lysis buffer. The column was then washed using 10 column volumes of either TAB1 wash buffer (50 mM Tris pH 7.5, 300 mM NaCl, 2.0 mM imidazole) or Ph wash buffer (50 mM Tris pH 7.5, 200 mM NaCl, 10 mM imidazole). Elution buffer (TAB1; TAB1 wash buffer + 1 M imidazole, Ph; Ph wash buffer + 1 M imidazole) was used to elute the His-tagged proteins with a gradient of imidazole from 10 to 50 mM over 50 min. Fractions containing purified proteins were pooled and dialyzed overnight into either TAB1 final buffer (10 mM sodium phosphate pH7.2) or Ph final buffer (20 mM Tris pH 8.5, 200 mM NaCl) and concentrated to ~1 mg/mL.
Immunoblots.
To confirm O-GlcNAc modification, glycosylated proteins were treated with a bacterial O-GlcNAcase Bacteroides thetaiotaomicron GH84 in either the wild-type (WT) or the catalytic mutant form D242A for 4 h at 25 °C. Following incubation, 1 μg of the samples was loaded on a 10% SDS-PAGE gel followed by electrophoresis at 70 V for 1.5 h and proteins were transferred onto 0.45 μm nitrocellulose membranes using the TransBlot Turbo (Bio-Rad) semidry transfer system according to the manufacturer’s instructions. Blots were incubated in blocking buffer (PBS, 4% BSA) for 1 h prior to addition of primary antibody [mouse anti O-GlcNAc antibody (BioLegend, CTD110.6) and rabbit monoclonal Anti-His antibody (Invitrogen; 21HCLC)]. The blots were incubated with primary antibody overnight and imaged using the LI-COR fluorescent secondary antibodies and gel imager according to the manufacturer’s instructions.
LC-MS Analysis Using an Oxonium Ion-Triggered MS/MS Approach.
SDS-PAGE-separated O-glycosylated proteins were reduced using 10 mM dithiothreitol and alkylated in 55 mM iodoacetamide (Sigma-Aldrich, St. Louis, MO) prior to in-gel proteolytic digestion with trypsin (1:50, enzyme: protein ratio) for 18 h (Pierce, Rockford, IL). Following heat inactivation for 10 min at 80 °C to quench the digestion, tryptic peptides were extracted from the polyacrylamide gel using 0.1% formic acid in 70% acetonitrile and dried under medium heat to concentrate sample. Samples were reconstituted in 0.1% formic acid, 2% acetonitrile in water.
Each aliquot was separated on a Dionex RSLC 3000 nano-LC system (Thermo Fisher Scientific, San Jose, CA, USA). Approximately 450 ng of digested protein was injected onto an in-house packed 5 cm C-18 trapping column (3 μm, 300 Å pore size, 75 μm i.d.). Peptides were then eluted onto a 20 cm fritted C-18 (1.8 μm, 300 Å pore size, 75 um ID, packed in-house) analytical column (New Objective, Woburn, MA, USA). Separation was performed using mobile phase A consisting of 0.1% formic acid in water and mobile phase B consisting of 0.1% formic acid in acetonitrile. A 125 min linear gradient was applied from 3% to 32% mobile phase B at a flow rate of 300 nL/min. The nanoLC system was interfaced to a Thermo Scientific Instruments Lumos Orbitrap mass spectrometer (San Jose, CA, USA) equipped with a 193 nm excimer laser for UVPD as described previously.37 MS1 spectra were collected in a top-speed data-dependent fashion with a dynamic exclusion of the precursor for 20 s after two repeated activation events. All MS2 spectra were acquired in the Orbitrap (30 000k resolution at 200 m/z). HCD mass spectra were collected using 28 NCE, and UVPD was performed in the high-pressure linear ion trap using two 5 ns pulses (1.5 mJ per pulse) from a 193 nm, 500 Hz excimer laser (Coherent Excistar, Santa Clara, CA). EThcD was performed in the high-pressure linear ion trap with an optimized 50 ms reaction time for ETD (2 × 105 reagent AGC) with 25% supplemental collisional energy.
Database Searching of O-GlcNAcylated Peptides.
Byonic (Protein Metrics, Cupertino, CA, USA) proteomic database search was used for large-scale identification and site localization of O-linked glycopeptides with precursor and fragment mass tolerance of 10 ppm. The main scoring metrics used to gauge the confidence of identified glycopeptides are the Delta Mod Score and the PEP 2D score. Byonic provides a site-localization statistic called “Delta Mod score”, which is the decrease in certainty from the top-scoring glycopeptide to the second-best possible glycopeptide and so on. A Delta Mod score below about 20.0 means that the glycosite identification is uncertain, usually in the site localization of an O-GlcNAc. Delta Mod above ~40.0 means that the identification should be correct in every detail. Manual validation was performed for each identified glycopeptide.47 The “posterior error probability” or PEP score is a statistic ranking of a peptide spectral match (PSM), giving the probability that a PSM came from a decoy or false distribution rather than the top protein hit.47 The PEP 2D statistic gives a bonus score to a PSM scored from the top protein emanating from the human protein database used in this analysis, giving further confidence that this PSM corresponds to our glycoproteins of interest. In addition, each identified glycopeptide was validated by deconvolution using the Xtract algorithm (Thermo) and analysis using Prosight Lite using a fragment tolerance of 10 ppm. The “protein characterization score” (PCS) is used in the analysis of infused glycopeptides. The PCS uses shuffled amino acid sequences during analysis to create a decoy distribution to which the original (or forward) result is compared.48
Supplementary Material
ACKNOWLEDGMENTS
D.J.V. is a Tier I Canada Research Chair in Chemical Biology. D.T.K. was supported by a postdoctoral fellowship from CIHR and a Trainee Award from the Michael Smith Foundation for Health Research. Funding from the UT System for support of the UT System Proteomics Core Facility Network is gratefully acknowledged.
Funding
This work was supported by grants from the Canadian Institutes for Health Research (CIHR) (D.J.V., PJT-148732, PJT-156202), the Natural Sciences and Engineering Council of Canada (Discovery-RGPIN298406), the National Science Foundation (CHE-1402753, J.S.B.), the Welch Foundation (F-1155, J.S.B.), and the NIH (R01 GM121714, J.S.B.).
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.0c04710.
Proteins and DNA sequences, detailed experimental methods, immunoblot data, mass spectrometry data (PDF)
Notes
The authors declare no competing financial interest.
Complete contact information is available at: https://pubs.acs.org/10.1021/jacs.0c04710
Contributor Information
Edwin E. Escobar, Department of Chemistry, The University of Texas at Austin, Austin, Texas 78712, United States;.
Dustin T. King, Department of Biochemistry, Simon Fraser University, Burnaby, British Columbia V5A 1S6, Canada.
Jesús E. Serrano-Negroń, Department of Biochemistry, Simon Fraser University, Burnaby, British Columbia V5A 1S6, Canada
Matthew G. Alteen, Department of Chemistry, Simon Fraser University, Burnaby, British Columbia V5A 1S6, Canada
David J. Vocadlo, Department of Chemistry and Department of Molecular Biology and Biochemistry, Simon Fraser University, Burnaby, British Columbia V5A 1S6, Canada.
Jennifer S. Brodbelt, Department of Chemistry, The University of Texas at Austin, Austin, Texas 78712, United States;.
REFERENCES
- (1).Yang X; Qian K Protein O-GlcNAcylation: Emerging Mechanisms and Functions. Nat. Rev. Mol. Cell Biol 2017, 18 (7), 452–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Banerjee PS; Lagerlöf O; Hart GW Roles of O-GlcNAc in Chronic Diseases of Aging. Mol. Aspects Med 2016, 51, 1–15. [DOI] [PubMed] [Google Scholar]
- (3).Hanover JA; Chen W; Bond MR O-GlcNAc in Cancer: An Oncometabolism-Fueled Vicious Cycle. J. Bioenerg. Biomembr 2018, 50 (3), 155–173. [DOI] [PubMed] [Google Scholar]
- (4).Yabing Chen; Xinyang Zhao; Hui Wu Metabolic Stress and Cardiovascular Disease in Diabetes Mellitus. Arterioscler. Thromb. Vasc. Biol 2019, 39 (10), 1911–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Ryan P; Xu M; Davey AK; Danon JJ; Mellick GD; Kassiou M; Rudrawar S O-GlcNAc Modification Protects against Protein Misfolding and Aggregation in Neurodegenerative Disease. ACS Chem. Neurosci 2019, 10 (5), 2209–2221. [DOI] [PubMed] [Google Scholar]
- (6).Vercoutter-Edouart A-S; Yazidi-Belkoura IE; Guinez C; Baldini S; Leturcq M; Mortuaire M; Mir A-M; Steenackers A; Dehennaut V; Pierce A; Lefebvre T Detection and Identification of O-GlcNAcylated Proteins by Proteomic Approaches. Proteomics 2015, 15 (5–6), 1039–1050. [DOI] [PubMed] [Google Scholar]
- (7).Guo J; Zhang G; Ma J; Zhao C; Xue Q; Wang J; Liu W; Liu K; Wang H; Liu N; Song Q; Li J Detection and Identification of O-GlcNAc-Modified Proteins Using 6-Azido-6-Deoxy-N-Acetyl-Galactosamine. Org. Biomol. Chem 2019, 17 (17), 4326–4334. [DOI] [PubMed] [Google Scholar]
- (8).Darabedian N; Pratt MR Chapter Thirteen - Identifying Potentially O-GlcNAcylated Proteins Using Metabolic Labeling, Bioorthogonal Enrichment, and Western Blotting. In Methods in Enzymology; Shukla AK, Ed.; Chemical and Synthetic Biology Approaches To Understand Cellular Functions – Part B; Academic Press, 2019; Vol. 622, pp 293–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Griffin ME; Jensen EH; Mason DE; Jenkins CL; Stone SE; Peters EC; Hsieh-Wilson LC Comprehensive Mapping of O-GlcNAc Modification Sites Using a Chemically Cleavable Tag. Mol. BioSyst 2016, 12 (6), 1756–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Ma J; Wang W-H; Li Z; Shabanowitz J; Hunt DF; Hart GW O-GlcNAc Site Mapping by Using a Combination of Chemoenzymatic Labeling, Copper-Free Click Chemistry, Reductive Cleavage, and Electron-Transfer Dissociation Mass Spectrometry. Anal. Chem 2019, 91 (4), 2620–2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Woo CM; Lund PJ; Huang AC; Davis MM; Bertozzi CR; Pitteri SJ Mapping and Quantification of Over 2000 O-Linked Glycopeptides in Activated Human T Cells with Isotope-Targeted Glycoproteomics (Isotag). Mol. Cell. Proteomics 2018, 17 (4), 764–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Vosseller K; Trinidad JC; Chalkley RJ; Specht CG; Thalhammer A; Lynn AJ; Snedecor JO; Guan S; Medzihradszky KF; Maltby DA; Schoepfer R; Burlingame AL O-Linked N-Acetylglucosamine Proteomics of Postsynaptic Density Preparations Using Lectin Weak Affinity Chromatography and Mass Spectrometry. Mol. Cell. Proteomics 2006, 5 (5), 923–934. [DOI] [PubMed] [Google Scholar]
- (13).Trinidad JC; Barkan DT; Gulledge BF; Thalhammer A; Sali A; Schoepfer R; Burlingame AL Global Identification and Characterization of Both O-GlcNAcylation and Phosphorylation at the Murine Synapse. Mol. Cell. Proteomics 2012, 11 (8), 215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Vosseller K; Hansen KC; Chalkley RJ; Trinidad JC; Wells L; Hart GW; Burlingame AL Quantitative Analysis of Both Protein Expression and Serine/Threonine Post-Translational Modifications through Stable Isotope Labeling with Dithiothreitol. Proteomics 2005, 5 (2), 388–398. [DOI] [PubMed] [Google Scholar]
- (15).Leney AC; El Atmioui D; Wu W; Ovaa H; Heck AJR Elucidating Crosstalk Mechanisms between Phosphorylation and OGlcNAcylation. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (35), E7255–E7261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Worth M; Li H; Jiang J Deciphering the Functions of Protein O-GlcNAcylation with Chemistry. ACS Chem. Biol 2017, 12 (2), 326–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Thaysen-Andersen M; Packer NH Advances in LC−MS/MS-Based Glycoproteomics: Getting Closer to System-Wide Site-Specific Mapping of the N-and O-Glycoproteome. Biochim. Biophys. Acta, Proteins Proteomics 2014, 1844 (9), 1437–1452. [DOI] [PubMed] [Google Scholar]
- (18).Wells L; Vosseller K; Cole RN; Cronshaw JM; Matunis MJ; Hart GW Mapping Sites of O-GlcNAc Modification Using Affinity Tags for Serine and Threonine Post-Translational Modifications. Mol. Cell. Proteomics 2002, 1 (10), 791–804. [DOI] [PubMed] [Google Scholar]
- (19).Ma J; Banerjee P; Whelan SA; Liu T; Wei A-C; Ramirez-Correa G; McComb ME; Costello CE; O’Rourke B; Murphy A; Hart GW Comparative Proteomics Reveals Dysregulated Mitochondrial O-GlcNAcylation in Diabetic Hearts. J. Proteome Res 2016, 15 (7), 2254–2264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Khidekel N; Ficarro SB; Peters EC; Hsieh-Wilson LC Exploring the O-GlcNAc Proteome: Direct Identification of OGlcNAc-Modified Proteins from the Brain. Proc. Natl. Acad. Sci. U. S. A 2004, 101 (36), 13132–13137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Zhurov KO; Fornelli L; Wodrich MD; Laskay ÜA; Tsybin YO Principles of Electron Capture and Transfer Dissociation Mass Spectrometry Applied to Peptide and Protein Structure Analysis. Chem. Soc. Rev 2013, 42 (12), 5014–5030. [DOI] [PubMed] [Google Scholar]
- (22).Riley NM; Coon JJ The Role of Electron Transfer Dissociation in Modern Proteomics. Anal. Chem 2018, 90 (1), 40–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Myers SA; Daou S; Affar EB; Burlingame A Electron Transfer Dissociation (ETD): The Mass Spectrometric Breakthrough Essential for O-GlcNAc Protein Site Assignments – A Study of the OGlcNAcylated Protein Host Cell Factor C1. Proteomics 2013, 13 (6), 982–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Zhang Y; Xie X; Zhao X; Tian F; Lv J; Ying W; Qian X Systems Analysis of Singly and Multiply O-Glycosylated Peptides in the Human Serum Glycoproteome via EThcD and HCD Mass Spectrometry. J. Proteomics 2018, 170, 14–27. [DOI] [PubMed] [Google Scholar]
- (25).Yu Q; Wang B; Chen Z; Urabe G; Glover MS; Shi X; Guo L-W; Kent KC; Li L Electron-Transfer/Higher-Energy Collision Dissociation (EThcD)-Enabled Intact Glycopeptide/Glycoproteome Characterization. J. Am. Soc. Mass Spectrom 2017, 28 (9), 1751–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Saba J; Dutta S; Hemenway E; Viner R Increasing the Productivity of Glycopeptides Analysis by Using Higher-Energy Collision Dissociation-Accurate Mass-Product-Dependent Electron Transfer Dissociation. Int. J. Proteomics 2012, 2012. 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Zhao P; Viner R; Teo CF; Boons G-J; Horn D; Wells L Combining High-Energy C-Trap Dissociation and Electron Transfer Dissociation for Protein O-GlcNAc Modification Site Assignment. J. Proteome Res 2011, 10 (9), 4088–4104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Ma C; Qu J; Li X; Zhao X; Li L; Xiao C; Edmunds G; Gashash E; Song J; Wang PG Improvement of Core-Fucosylated Glycoproteome Coverage via Alternating HCD and ETD Fragmentation. J. Proteomics 2016, 146, 90–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).You X; Qin H; Ye M Recent Advances in Methods for the Analysis of Protein O-Glycosylation at Proteome Level. J. Sep. Sci 2018, 41 (1), 248–261. [DOI] [PubMed] [Google Scholar]
- (30).Singh C; Zampronio CG; Creese AJ; Cooper HJ Higher Energy Collision Dissociation (HCD) Product Ion-Triggered Electron Transfer Dissociation (ETD) Mass Spectrometry for the Analysis of N-Linked Glycoproteins. J. Proteome Res 2012, 11 (9), 4517–4525. [DOI] [PubMed] [Google Scholar]
- (31).Hogan JM; Pitteri SJ; Chrisman PA; McLuckey SA Complementary Structural Information from a Tryptic N-Linked Glycopeptide via Electron Transfer Ion/Ion Reactions and Collision-Induced Dissociation. J. Proteome Res 2005, 4 (2), 628–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Xu S-L; Chalkley RJ; Maynard JC; Wang W; Ni W; Jiang X; Shin K; Cheng L; Savage D; Hühmer AFR; Burlingame AL; Wang Z-Y Proteomic Analysis Reveals O-GlcNAc Modification on Proteins with Key Regulatory Functions in Arabidopsis. Proc. Natl. Acad. Sci. U. S. A 2017, 114 (8), E1536–E1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Nishikawa I; Nakajima Y; Ito M; Fukuchi S; Homma K; Nishikawa K Computational Prediction of O-Linked Glycosylation Sites That Preferentially Map on Intrinsically Disordered Regions of Extracellular Proteins. Int. J. Mol. Sci 2010, 11 (12), 4991–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Brodbelt JS Photodissociation Mass Spectrometry: New Tools for Characterization of Biological Molecules. Chem. Soc. Rev 2014, 43 (8), 2757–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Madsen JA; Ko BJ; Xu H; Iwashkiw JA; Robotham SA; Shaw JB; Feldman MF; Brodbelt JS Concurrent Automated Sequencing of the Glycan and Peptide Portions of O-Linked Glycopeptide Anions by Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem 2013, 85 (19), 9253–9261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Ko BJ; Brodbelt JS Comparison of Glycopeptide Fragmentation by Collision Induced Dissociation and Ultraviolet Photodissociation. Int. J. Mass Spectrom 2015, 377 (1), 385–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Klein DR; Holden DD; Brodbelt JS Shotgun Analysis of Rough-Type Lipopolysaccharides Using Ultraviolet Photodissociation Mass Spectrometry. Anal. Chem 2016, 88 (1), 1044–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Wang X; Yuan Z-F; Fan J; Karch KR; Ball LE; Denu JM; Garcia BA A Novel Quantitative Mass Spectrometry Platform for Determining Protein O-GlcNAcylation Dynamics. Mol. Cell. Proteomics 2016, 15 (7), 2462–2475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Shen DL; Gloster TM; Yuzwa SA; Vocadlo DJ Insights into O-GlcNAc Processing and Dynamics through Kinetic Analysis of O-GlcNAc Transferase and O-GlcNAcase Activity on Protein Substrates. J. Biol. Chem 2012, jbc.M111.310664. 28715395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Yuzwa SA; Yadav AK; Skorobogatko Y; Clark T; Vosseller K; Vocadlo DJ Mapping O-GlcNAc Modification Sites on Tau and Generation of a Site-Specific O-GlcNAc Tau Antibody. Amino Acids 2011, 40 (3), 857–868. [DOI] [PubMed] [Google Scholar]
- (41).Pathak S; Borodkin VS; Albarbarawi O; Campbell DG; Ibrahim A; van Aalten DM O-GlcNAcylation of TAB1Modulates TAK1-Mediated Cytokine Release. EMBO J. 2012, 31 (6), 1394–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Bern MW; Kil YJ Two-Dimensional Target Decoy Strategy for Shotgun Proteomics. J. Proteome Res 2011, 10 (12), 5296–5301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Gambetta MC; Müller J O-GlcNAcylation Prevents Aggregation of the Polycomb Group Repressor Polyhomeotic. Dev. Cell 2014, 31 (5), 629–639. [DOI] [PubMed] [Google Scholar]
- (44).Cheung PCF; Campbell DG; Nebreda AR; Cohen P Feedback Control of the Protein Kinase TAK1 by SAPK2a/P38α. EMBO J. 2003, 22 (21), 5793–5805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Greer SM; Brodbelt JS Top-Down Characterization of Heavily Modified Histones Using 193 nm Ultraviolet Photodissociation Mass Spectrometry. J. Proteome Res 2018, 17 (3), 1138–1145. [DOI] [PubMed] [Google Scholar]
- (46).Kim EJ The Utilities of Chemical Reactions and Molecular Tools for O-GlcNAc Proteomic Studies. ChemBioChem 2015, 16 (10), 1397–1409. [DOI] [PubMed] [Google Scholar]
- (47).Bern M; Kil YJ; Becker C Byonic: Advanced Peptide and Protein Identification Software. Curr. Protoc. Bioinforma. Ed. Board Andreas Baxevanis Al 2012, CHAPTER, Unit13.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Fellers RT; Greer JB; Early BP; Yu X; LeDuc RD; Kelleher NL; Thomas PM ProSight Lite: Graphical Software to Analyze Top-down Mass Spectrometry Data. Proteomics 2015, 15 (7), 1235–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.