

Abstract
Recent advances in sequencing and informatic technologies have led to a deluge of publicly available genomic data. While it is now relatively easy to sequence, assemble, and identify genic regions in diploid plant genomes, functional annotation of these genes is still a challenge. Over the past decade, there has been a steady increase in studies utilizing machine learning algorithms for various aspects of functional prediction, because these algorithms are able to integrate large amounts of heterogeneous data and detect patterns inconspicuous through rule‐based approaches. The goal of this review is to introduce experimental plant biologists to machine learning, by describing how it is currently being used in gene function prediction to gain novel biological insights. In this review, we discuss specific applications of machine learning in identifying structural features in sequenced genomes, predicting interactions between different cellular components, and predicting gene function and organismal phenotypes. Finally, we also propose strategies for stimulating functional discovery using machine learning–based approaches in plants.
Keywords: big data, bioinformatics, gene function, machine learning, predictive genomics
Recent advances in computing power, bioinformatics algorithms, and sequencing technologies have made assembling a genome and annotating genic features relatively easy and commonplace. However, the process of determining the functions of the annotated genes is still a laborious task. Even in Arabidopsis thaliana (L.) Heynh., the workhorse of plant research, 10%, 17%, and 18% of genes have no Gene Ontology (GO) cellular component, molecular function, and biological process annotations, respectively, and only 28%, 16%, and 24% of genes in these categories have experimentally determined functions (TAIR, 2019). The sheer volume of data produced via sequencing itself presents a challenge for deriving biological meaning from sequences. In this review, we discuss how machine learning is being used in different model systems to integrate a variety of pieces of high‐ and low‐throughput data and address the problem of gene function discovery.
In reviewing this topic, it becomes necessary to define gene function. Historically, a “gene” was considered to be a locus in which sequence alterations led to inactivation of a trait of interest (Gerstein et al., 2007), resulting in function being associated with an assayable phenotype. In contrast, the more recent ENCODE project defined a genomic sequence as having function if it “participated in at least one biochemical RNA and/or chromatin associated event in at least one cell type” (The ENCODE Project Consortium, 2012). This relaxed definition of function, which led to the authors declaring 80.4% of the human genome as functional, was heavily criticized (Graur et al., 2013). Nonetheless, with ever‐increasing genomics data and projects such as ENCODE, we now have a better idea of signatures associated with known functional sequences, which machine learning methods exploit for identifying genes and defining their functions. Functional definitions can be encoded at different organizational levels, such as (i) the structural type of a given sequence feature (e.g., gene, non‐coding RNA, pseudogene, transposon, intergenic sequence), (ii) interaction of a gene product with other cellular entities (e.g., microRNA–target interactions, protein–protein interactions [PPIs], subcellular localization, enzyme–substrate interactions), and (iii) phenotypic influence of the feature. The widespread availability of genomic and post‐genomic data in the past decade has created novel opportunities to understand and predict these functions.
In recent years, machine learning has been successfully used in many biological contexts. Machine learning–based algorithms are efficient enough to handle massive data sets that exhibit high amounts of noise, dimensionality, and/or incompleteness, and make minimal assumptions about the data’s underlying probability distributions and generation methods. The primary focus of machine learning methods is prediction, which is different from the inferential focus of traditional statistical approaches (Bzdok et al., 2018), although in practical terms the distinction between machine learning and statistics is rather blurry (Fig. 1A). Machine learning algorithms can be broadly divided into two types—supervised and unsupervised (Fig. 1A, Fig. 2). Supervised algorithms such as random forests (RF), support vector machine (SVM), and k‐nearest neighbors (kNN) are frequently used for the purposes of binary/multi‐class classification of test instances or for numerical prediction of the trait values (regression) and require explicit definitions of labels, while unsupervised methods such as principal components analysis, k‐means clustering, and self‐organizing maps are label‐free and are primarily used for clustering and feature extraction. Machine learning algorithms can further be classified into feature‐based and artificial neural network (ANN)–based methods, depending on the inherent algorithm development process. Whereas feature‐based methods such as RF and SVM require explicit specification of various features, ANN methods such as convolutional neural networks (CNNs) and recurrent neural networks (RNNs) can extract features from the training data by themselves. Furthermore, while a few dozen to a few hundred input training data points can be sufficient for feature‐based methods, ANN algorithms typically require thousands to millions of input data points for accurate model development. The use of ANNs comprising multiple layers of neurons (Fig. 1A, Fig. 2) is referred to as deep learning.
Figure 1.
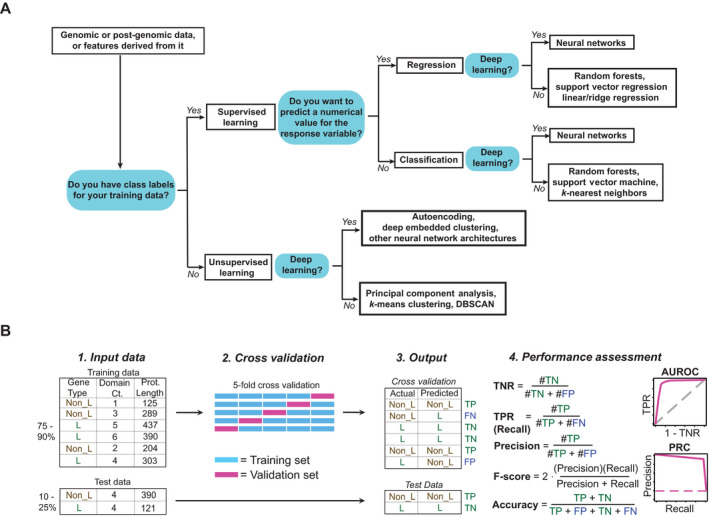
Schematic for choosing and implementing a machine learning (ML) workflow. (A) A simplified decision tree for choosing the type of ML algorithm for a given task. Considerations for choosing deep learning methods over others include whether a large amount of training data and computational power are available, whether features can be extracted readily, and whether the “black box” nature of neural networks is acceptable. (B) A typical workflow for supervised, feature‐based learning. An ML algorithm predicts the value of the response variable for each input data point (instance) based upon its particular feature (dependent variable) values. While we here show the workflow for classification, many ML algorithms are also capable of regression. We present an example with fictional data, based upon the work presented in Lloyd et al. (2015), in which an ML model predicts the response variable of lethality upon gene knockout using the features domain count and protein length. (1) In binary classification, input data consists of positive instances and negative instances (L and Non_L, respectively). Optimally, the number of positive instances should roughly equal the number of negative instances. When there are substantial training data available, the input data set may be split into a Training Set (usually 75–90% of the input data) and a Test Set. When a Test Set cannot be made, cross validation (CV) may be sufficient to estimate the algorithm’s error in classification of test instances. (2) Here, five‐fold CV is shown. In each fold, 20% of the input training data is randomly chosen to be the Validation Set (shown in pink). The model is trained using the training data, and its performance on new instances is determined through the Validation Set. This process is repeated five times. (3) Each instance’s predicted class is compared to its actual class. Here, L is the positive class. TP = true positive, FP = false positive, TN = true negative, FN = false negative. (4) Overall classification performance can be quantified by different metrics: TNR = true negative rate (also specificity), TPR = true positive rate (also sensitivity, recall), precision, accuracy, area under the receiver operating characteristic (AUROC) curve, PRC = precision‐recall curve. For all of the above metrics, values closer to 1 indicate increasingly optimal performance.
Figure 2.

Explanation of four machine learning methods used for various aspects of functional prediction and their variants. Each method has been employed in a study reviewed in this article. For the decision tree (random forest [RF] inlet), k‐nearest neighbor (kNN), and support vector machine (SVM), classification is shown here as based on two features (domain count and protein length); however, many more features are typically used for model development. Using these features, RF and SVM predict the class (L = lethal, N = nonlethal) of any instance falling into the defined regions (green region = class L, brown region = class N). kNN is a supervised learning algorithm that predicts the class of new instances based upon the classes of the most similar instances. Artificial neural networks are able to extract features during the classification process, and use these to predict the class of each input.
In this review, we primarily focus on popular supervised methods used for gene function prediction (summarized in Table 1). Most of the discussed methods use a specific approach for algorithm development (Fig. 1B), i.e., generation of a model using training data followed by cross‐validation. In addition, some of these approaches also test their models using completely distinct “test” or “challenge” data that the algorithm has not encountered during the training/validation process. Because there are many algorithms for feature‐based as well as deep learning (Fig. 2) (Li et al., 2019), it is not always possible to a priori determine the best algorithm for a given data set. Some studies (e.g., Lloyd et al., 2015) thus deploy a grid search or a random search approach to screen through multiple algorithms and their parameter combinations to identify the best‐performing algorithm using measures such as the area under the receiver operating characteristic (AUROC) curve, precision‐recall, and F‐score (Fig. 1B).
Table 1.
An overview of the tools and machine learning algorithms discussed in this review. The last column depicts some but not necessarily all features used by these tools for prediction.
| Task | Tools discussed | Typical algorithms used | Typical features used by feature‐based methods |
|---|---|---|---|
| Protein‐coding gene identification | MAKER (Augustus), BRAKER (GeneMark/Augustus), mGene | HMM a , SVM | HMM: Genomic sequences, mapped RNA‐seq transcripts, orthologous sequences; SVM: sequence signals (e.g., transcription start sites), sequence composition, sequence length |
| Cis‐regulatory element identification | Alipanahi et al., 2015; Umarov and Solovyev, 2017 | CNN | Sequence segments, experimentally determined binding scores from protein‐binding arrays, ChIP‐seq, etc. |
| Gene expression | Kakei et al., 2013; Wang et al., 2018; Washburn et al., 2019 | SVM, CNN, RF | Presence/absence of motifs and motif pairs for SVM/RF, direct genomic sequences for CNNs |
| Subcellular localization | TargetP, SignalP, Plant‐mPLoc | RNNs, ensemble clustering using kNN | Subcellular localization sequences, GO terms, domain composition |
| Protein–protein interactions | Rodgers‐Melnick et al., 2013; Liu et al., 2017; Ding and Kihara, 2019 | SVM, RF | Protein subcellular localization, expression patterns, domains, and features derived from protein structure, such as conserved interaction sites, hydrophobicity, etc. |
| Gene Ontology | NetGO, DeepGOPlus, GO‐At | CNN, decision trees, kNN, naïve Bayes | Gene expression, predicted secondary structure, homology, membership in enzyme families, interacting proteins, etc. |
| Metabolic pathways | Dale et al., 2010; Toubiana et al., 2019 | Naïve Bayes, decision trees, logistic regression, RF | Gene chromosomal location and neighbors, reaction evidence, pathway taxonomic range, integrated metabolomics‐transcriptomics correlation network properties |
| Phenotypes | Lloyd et al., 2015; Sprenger et al., 2018; Moore et al., 2019 | SVM, RF | Varied features derived from genomic, transcriptomic, and metabolomic data across species (e.g., tissue‐specific expression, correlation network connectivity) |
| Genomic prediction | Ornella et al., 2014; González‐Camacho et al., 2016 | SVM, probabilistic neural network | Presence/absence of genetic markers and pairs of genetic markers, direct genetic markers for PNN |
CNN = convolutional neural network; HMM = hidden Markov model; kNN = k‐nearest neighbors; RF = random forests; RNN = recurrent neural network; SVM = support vector machine.
For a newly sequenced species, gene predictions are made by first training HMMs to identify intron–exon structures using reference structures from a closely related species or from genome‐mapped RNA‐seq data, and then deploying these models on the species of interest.
This review covers recent attempts to define gene function using machine learning. We first review methods that identify structural regions in the genome, specifically focusing on protein‐coding genes and cis‐regulatory elements. We then cover methods that ascribe higher‐order properties to genes, e.g., gene expression patterns, subcellular localization, and PPIs. Next, we review integrative methods for predicting specific molecular or biological functions of genes, e.g., GO, involvement in metabolic networks, and genotype–phenotype associations. Finally, in each section and in the Discussion, we describe the current roadblocks in machine learning utilization in plant sciences, and suggest possible measures for addressing those roadblocks.
PREDICTION OF FUNCTIONAL GENOMIC REGIONS
The identification of the genomic sequences that constitute functional regions is the first step in genome annotation. Machine learning approaches have been developed to detect a number of features such as protein‐coding genes, microRNAs (miRNAs), long non‐coding RNAs (Sun et al., 2015), polyadenylation sites (Gao et al., 2018), DNAse I hypersensitive sites (Lyu et al., 2018), cis‐regulatory elements (CREs), and chromatin states (Ernst and Kellis, 2017). The detection of these genic elements may use machine learning in a classification setting: genic elements may be classified as “protein‐coding genes” or not, “miRNA” or not, or “CRE” or not. In these settings, machine learning’s ability to integrate large volumes of heterogeneous data may improve its accuracy over those of non–machine learning methods (Li et al., 2018). SVMs, RFs, and CNNs are, thus, used for genomic feature prediction (Fig. 2). SVMs have been shown to be efficient in binary classification problems (e.g., gene or not) and multi‐class problems (e.g., gene, miRNA, pseudogene, transposon) (Mathur and Foody, 2008) but, compared to RFs, have multiple parameters that need tuning. RFs may also have a better performance for most tasks (Caruana and Niculescu‐Mizil, 2006). Nonetheless, despite the popularity of these algorithms since the 1990s, the machine learning approach that works best for many structural feature prediction problems is hidden Markov models (HMM).
Protein‐coding gene prediction
Current pipelines for predicting protein‐coding genes in plant genomes (e.g., MAKER‐P, Campbell et al., 2014; MEGANTE, Numa and Itoh, 2014; BRAKER, Hoff et al., 2016) rely on the integration of a number of different features such as coding potential of nucleotide sequences, ab initio definition of intron–exon structures, transcript sequences, and orthologous genes from related species. Such integration can help address challenges arising from the genomic complexity such as multiple isoforms, alternative transcriptional start sites, duplicated genes, and the presence of pseudogenes and repeats (Schatz et al., 2012). The ab initio gene finders used in the above pipelines (e.g., Augustus, SNAP) primarily utilize HMMs, a probability‐based supervised learning algorithm described in the 1960s (Baum and Eagon, 1967) that predates the emergence of machine learning as a field. HMM algorithms can be trained iteratively to predict the functional class (e.g., splice site, exon, enhancer) of individual nucleotides based upon the class of their neighboring nucleotides. For example, if a nucleotide is predicted to be part of a splice site, its neighboring nucleotides must be an exon and an intron (Yip et al., 2013). Such nucleotide‐level predictions can be modeled using the inherent structure of HMMs, compared to slightly convoluted approaches that need to be implemented for SVMs or RFs.
For example, the mGene software (Schweikert et al., 2009) implemented an SVM approach for the detection of genic elements using specially‐made kernel functions. These kernel functions allow for DNA sequences to be represented in a linear/non‐linear vector space, helping produce decision boundaries for classifying input sequences into their discrete categories (see Fig. 2B for additional SVM details) (Sonnenburg et al., 2007; Ben‐Hur et al., 2008). The authors found that SVMs could detect transcription start sites and splice‐sites with higher accuracy than HMMs, and therefore trained eight SVMs to each detect one genic feature (acceptor/donor splice sites, translation start/stop sites, transcription start/stop sites, polyadenylation sites, and trans‐splicing events), whose scores were then combined for predicting the overall gene feature. Although more convoluted than HMMs, the SVM approach allows for efficient incorporation of heterogeneous data such as RNA‐seq read counts. mGene.ngs, which incorporates gene expression information with genome sequence–based predictions, was used to annotate the genomes of 18 A. thaliana accessions (Gan et al., 2011). Another study trained an SVM classifier for a novel application in Triticum aestivum L. (bread wheat) (Brenchley et al., 2012). This plant has a notoriously large (17 Gbp) hexaploid genome, composed of three subgenomes that, for machine learning, can be defined as three different classes. The authors trained the SVM to recognize and differentiate between gene homologs belonging to one of these three subgenomes, and achieved a moderate (~59%) classification accuracy. The authors note that misclassified genes were those with high sequence similarity between homologs.
The mGene example above illustrates the power of machine learning approaches in incorporating a variety of heterogeneous data sets in prediction tasks. Such data can reduce the potential false positives in gene prediction, e.g., due to fragmented genes resulting from incomplete genome assemblies (Salzberg, 2019). With the advent of long read sequencing, genomes and transcriptomes of non‐model organisms are relatively easy to obtain (Schmutzer et al., 2017). It remains to be seen if SVMs trained in distant model species perform better than HMMs trained in distant species; nonetheless, homology information—already modeled in software such as OrthoFiller (Dunne and Kelly, 2017)—can play a greater role in machine learning–based gene prediction algorithms in the future.
Cis‐regulatory element prediction
Popular high‐throughput methods of CRE determination such as k‐mer enrichment, expectation maximization, and Gibbs sampling (D’haeseleer, 2006) identify over‐represented motifs in genomic sequences of interest, themselves generated from ChIP‐seq experiments or co‐expression analyses. However, these approaches have a high false positive rate for complex eukaryotic promoter sequences (Tompa et al., 2005; MacIsaac and Fraenkel, 2006). Among deep learning architectures, CNNs (Fig. 2D) are most popular for identifying CREs. Their use for CRE identification from DNA sequences is analogous to their common application in pattern recognition from two‐dimensional images. CNNs are also robust to variation in the location of the pattern of interest in the two‐dimensional image, which is not unlike variation in the position and percent identity of the CRE in the promoter region. In one such study, a CNN was trained to identify the binding motifs of transcription factors studied in ChIP/CLIP‐seq, protein‐binding microarrays, or systematic evolution of ligands by exponential enrichment (SELEX) experiments, using the sequences generated in these experiments as input data (Alipanahi et al., 2015). The authors used CNNs to find binding motifs in sequences without prior knowledge of their specific location, and found that their method outperformed all other methods submitted to the DREAM5 Transcription Factor‐DNA Motif Recognition Challenge (Weirauch et al., 2013).
In plants, CNNs were highly successful at detecting A. thaliana promoters—achieving sensitivity rates of 95% for promoters with a TATA‐box, and 94% for those without (Umarov and Solovyev, 2017). Within the identified promoters, authors then randomly substituted nucleotides in six‐nucleotide windows and estimated the drop in sensitivity of promoter detection by the CNN. Large drops in accuracy were considered indicative of potentially functional CREs with conserved positions in promoters, such as TATA boxes.
Although many studies have investigated CRE prediction in reference plant species, the ability to predict CREs from sequence inputs using CNNs potentially allows their deployment in non‐model species. Development of lineage‐specific CNNs may be required in this context and can be facilitated by targeted development of molecular resources in “anchor species” (Moghe and Kruse, 2018), as noted in the Discussion section. In addition, feature‐based approaches such as RF—incorporating elements such as physicochemical properties of binding sites, preferential presence in open‐chromatin regions/introns/untranslated regions (UTRs), and conservation between orthologous promoters—may serve as an orthogonal approach to CNNs for CRE identification (Khamis et al., 2018), if enough information about a transcription factor–binding site is available. Nonetheless, prediction of CREs is the first step toward modeling higher‐order genomic events such as transcription factor binding and gene expression, which themselves are steps toward prediction of organismal phenotypes. In the next two sections, we discuss such higher‐order interactions in greater detail.
PREDICTION OF HIGHER‐ORDER FUNCTIONAL CHARACTERISTICS
Once structural genomic elements are distinguished from putatively non‐functional sequences, prediction of the interactions between the gene‐encoded products (e.g., knowledge of a gene’s expression and a protein’s subcellular localization and interacting partners) can provide a greater degree of functional information. Here we discuss machine learning approaches devised to predict such higher‐order interactions.
Prediction of gene expression
Gene expression lends itself well to feature‐based learning because expression is dependent on multiple well‐understood features that can be experimentally determined, namely CREs, methylation and histone modification marks, and transcription factor expression. These features have been utilized to predict expression in yeast and animal species to varying degrees of success (Beer and Tavazoie, 2004; Karlić et al., 2010). Projects such as ENCODE (The ENCODE Project Consortium, 2012) and the Roadmap Epigenomics Project (Roadmap Epigenomics Consortium et al., 2015) have generated high‐resolution, cell‐type‐specific gene expression data in humans, and have facilitated development of efficient machine learning algorithms for feature‐based (Natarajan et al., 2012) and deep learning–based (Singh et al., 2016) prediction of gene expression. For example, one study (Wang et al., 2018) utilized ChIP‐seq data sets of two transcription factors available from the ENCODE project and determined the number of ENCODE motifs of other transcription factors co‐occurring with them. Using these motif counts as features and cell‐type specificity of ChIP‐seq peaks as class labels, the authors successfully trained an RF model to predict the cell‐type specificity of transcription factor binding based on co‐occurring motif combinations (Wang et al., 2018). In plants, one focus of machine learning studies has been on using sequence‐derived information from gene promoters. For example, promoter regions of genes up‐ or down‐regulated in response to ABA and glucose were analyzed using a relevance vector machine (Fig. 2B)—a Bayesian relative of SVM that exhibits certain properties conducive to identifying relevant motif combinations—in order to detect the most discriminatory motifs (Li et al., 2006). Combinations of these motifs were successfully used to predict gene up‐/down‐regulation. In a similar study in rice (Kakei et al., 2013), authors initially identified genes that were differentially regulated under iron deficiency, then detected enriched CREs in this set using motif similarity and frequency in co‐expressed genes. The authors next developed an SVM model to use the presence/absence of these motifs and motif combinations in a gene’s promoter for predicting whether the gene would be up‐regulated under iron deficiency. More recently, a unique strategy was developed that completely ignores motif discovery by simply feeding a CNN with promoter and terminator sequences to predict gene expression in maize (Washburn et al., 2019). With this method, the authors achieved an 86.6% accuracy when determining whether a particular gene was not expressed or highly expressed in a given condition, and found that the 3′ UTR of a gene was more informative in predicting mRNA abundance than the 5′ UTR. In the future, this approach could be used in a regression setting to predict specific expression level under a given condition and/or in a particular tissue type.
Prediction of gene expression from sequence is one of the holy grails of bioinformatics. As we better understand the various features important for expression regulation, such as transcription factor expression levels and binding affinities, methylation and other epigenetic marks, state of chromatin, and cell‐type specificity of transcription, the performance of machine learning algorithms in this arena will improve. Large‐scale studies of tissue‐specific gene expression have been carried out for several plant species to date, and these will also be instrumental in expanding the applicability of these algorithms beyond well‐studied model species.
Prediction of subcellular localization
Subcellular localization is an important determinant in the function of a protein, as it dictates factors such as the protein’s interaction partners, substrates, and optimum pH. Prediction of localization is essentially a classification problem and is therefore well‐suited for machine learning. Current popular prediction approaches such as SignalP and TargetP utilize signals within the proteins’ amino acid sequences—such as N‐terminal transit peptides for chloroplast and mitochondrial import, nuclear localization signals, secretory pathway signals, or peroxisomal target sequences—for prediction. Recent versions of these tools use ANNs instead of HMM (SignalP). For example, SignalP v5.0 was modified to utilize an RNN architecture (Fig. 2D) (Armenteros et al., 2019b). RNNs may fare better than CNNs in capturing long‐range interactions in ordered sequential data, thus enabling SignalP to capture long‐range motif interactions important for localization that may otherwise be missed. The algorithm was trained on high‐quality data sets of proteins with known subcellular localization obtained from the UniProt (UniProt Consortium, 2019) and PROSITE (https://prosite.expasy.org/) databases. To improve predictive power for organism groups with little data, the new version also implemented transfer learning, enabling models learned in one taxonomic group with substantial data to inform model development in the group with less data. The shift to RNNs from HMMs improved SignalP’s ability to recognize sequence motifs with varying length, and improved accuracy on archaeal data sets. Another tool, TargetP 2.0, which predicts chloroplast targeting signals, uses a very similar approach and achieved a high accuracy (85%) on plant proteins, and a 90% accuracy on non‐plant proteins (Armenteros et al., 2019a).
Plant‐mPLoc (Chou and Shen, 2010), a plant‐specific algorithm, integrated a number of functional descriptors such as GO, domain composition, and evolutionary information using a k‐nearest neighbors ensemble clustering approach (Fig. 2C), and can be used to identify proteins targeted to 12 different subcellular locations. The ensemble clustering approach enabled combining predictions made by different component classifiers designed for predicting specific descriptors into one single prediction. Although Plant‐mPLoc had better prediction accuracies than TargetP 2.0, the success rate of correct prediction was variable for the different subcellular locations, ranging from 10.3% to 89.5% for a benchmark set of proteins. Using specific sequence signals that are directly used by the organisms—in this case to transport a protein to its correct subcellular location—for prediction has an advantage over biological features that are only indirectly indicative of a certain function (e.g., gene expression or expression correlation). Nonetheless, the Plant‐mPLoc example highlights the importance of having enough experimentally validated training data to build accurate models. The generation of such data sets is still a laborious process, but availability of new mass spectrometry–based proteomics methods (Orre et al., 2019) can assist with generation of high‐resolution data sets and help improve prediction of subcellular localization.
Prediction of protein–protein interactions
Similar to subcellular localization, prediction of PPIs can also be conceived as a binary classification problem (two proteins interacting or not), and thus well‐suited for classification‐based machine learning algorithms. Protein interactomes can be constructed by first obtaining data regarding different features such as protein properties (e.g., solvent‐accessible surface area, domain information, surface hydrophobicity), evolutionary information (e.g., interacting homologs, co‐inheritance), expression profiles, or proxies for functional similarity (e.g., GO categories), and then integrating them into a unified prediction using supervised algorithms such as SVMs or RFs. Predicting interactomes based upon interolog information (i.e., determining if interacting proteins in an organism with a pre‐existing interactome have orthologs in the species of interest) is popular in plants, as interactomes for A. thaliana, rice, maize, Brassica rapa L., and tomato have been created in this manner. However, this approach fails to accurately detect interactions between non‐conserved and/or frequently duplicating and diversifying proteins. Interestingly, machine learning methods have shown promise in cross‐species deployment of models trained in A. thaliana (Rodgers‐Melnick et al., 2013; Zhu et al., 2016; Liu et al., 2017; Ding and Kihara, 2019).
Four PPI networks have been predicted for A. thaliana, due to better availability of higher‐confidence positive training examples. Two studies used A. thaliana to train models for PPI identification, and subsequently deployed them on the genomes of other plants. The first study incorporated features such as proteins’ pairwise domain information and subcellular localization for training an RF model to predict if a given pair of proteins interact (Rodgers‐Melnick et al., 2013). For A. thaliana, this model achieved an almost‐perfect AUROC of 0.96 (maximum possible being 1). This trained model was then used to predict PPIs within Populus trichocarpa Torr. & A. Gray. A second method trained an SVM and an RF model to predict PPIs from A. thaliana training data, and used this model to identify PPIs among proteins predicted to be co‐localized in soybean and maize (Ding and Kihara, 2019). The authors filtered results to consider only PPIs predicted by both machine learning models, and found that their method achieved a higher AUROC than STRING (Szklarczyk et al., 2019), a popular, rule‐based PPI prediction method. An RF‐based model generated in rice also achieved higher performance than STRING in predicting experimentally validated PPIs, despite having a small positive set of only 327 experimentally generated PPIs (Liu et al., 2017). However, this method applied imputation methods to generate additional positive instances. The authors further applied their method to predict a protein’s importance to an agronomically important trait (i.e., flowering time) by summing its interactions with known genes influencing the trait. Finally, in generating the maize interactome (Zhu et al., 2016), the authors made a distinction between physically interacting proteins (predicted via the interolog approach) and functionally interacting proteins, defined as any two proteins that are in the same metabolic pathway. An SVM was trained to distinguish between functionally interacting and non‐interacting proteins, using positive examples of proteins performing adjacent metabolic reactions from MaizeCyc, and negative examples of non‐adjacent proteins. After validating predictions by considering their GO term overlap and co‐occurrence in the same metabolic pathway, this model achieved an accuracy of 80% and an AUROC of 0.86, much better than random expectation (AUROC = 0.5).
As is evident in the discussion above, SVM and RF are the most popular methods for predicting PPIs. These are feature‐based methods, and hence a better understanding of the structural and physico‐chemical properties that affect these interactions will help further improve the models. An integrative model that, for example, can accurately predict a gene’s domain of expression, its product’s subcellular localization, and its interacting partners can fundamentally alter how we study the molecular basis of complex plant traits. These examples demonstrate that feature‐based machine learning approaches can not only produce predictive models but can also provide deeper biological insights into the underlying biological phenomenon.
PREDICTION OF BIOLOGICAL FUNCTION
The availability of large‐scale phenotypic data is paving the way for machine learning approaches to predict phenotypes from genotype. The best example of this is precision medicine, which seeks to improve clinical decision‐making based on individual patient genotypes (Cruz and Wishart, 2007; Kourou et al., 2015). The quest for precision medicine, especially in cancer research, has seen substantial improvements in machine learning accuracy with advances in genomics and availability of well‐recorded genotype and phenotype data sets such as The Cancer Genome Atlas database (The Cancer Genome Atlas Research Network et al., 2013). There is a notable paucity of such phenotype databases with well‐recorded metadata in the plant sciences, impeding distributed innovation in large‐scale machine learning–based data analyses. Nonetheless, machine learning methods have been implemented to obtain novel biological insights and develop predictive models. Here we describe four applications of machine learning–based biological function prediction in plants: predicting GO categories, associating genes with metabolic pathways, prediction of phenotype from genotype, and genomic prediction.
Prediction of Gene Ontology category
GO categories—divided into cellular component, molecular function, and biological process—are the most popular, albeit incomplete and broad, form of functional annotation. Typically, in non‐model organisms, GO categories are assigned using sequence similarity (Götz et al., 2008). However, machine learning–based methods have demonstrated an improvement over this approach. For example, Schietgat et al. (2010) developed and tested an algorithm for automated gene annotation in Mus musculus, Saccharomyces cerevisiae, and A. thaliana. In A. thaliana, the authors used features generated from gene expression, predicted class in the Structural Classification of Proteins database (http://scop.mrc‐lmb.cam.ac.uk/), putative secondary structure, domains, presence in enzyme families, and homology, for building hierarchical, multi‐label decision trees (Fig. 2A). The authors used this approach to mimic the structure of GO, where each gene may have multiple GO categories (multiple labels) organized in a hierarchical fashion. The software performance was found to be comparable to other commonly used learning methods (e.g., SVM). Another program, GO‐At, which was designed to perform in silico gene function prediction in A. thaliana, uses features derived from five data types: gene expression, PPI, protein sequence, phylogenetic profile, and genomic data (Bradford et al., 2010). Taking advantage of high‐quality training data composed of genes with pre‐determined functions, the software uses a naïve Bayes classifier to learn probabilistic rules that are then applied to unknown genes. For example, if two proteins in the training data that share a common motif also share a common function, GO‐At learns to associate the function with the motif.
Recent methods such as DeepGOPlus (Kulmanov and Hoehndorf, 2020), DeepText2GO (You et al., 2018), multi‐task deep neural networks (Fa et al., 2018), and NetGO (You et al., 2019) have expanded the feature sets and methods used for GO prediction. NetGO and DeepGOPlus—the best‐performing algorithms in the Critical Assessment of protein Function Annotation challenge (CAFA, Zhou et al., 2019)—integrate sequence motif–based, similarity‐based, and/or protein interaction network–based features using k‐nearest neighbor (NetGO) or CNN (DeepGOPlus), respectively (Figs. 2C, D). This ensemble learning approach is made possible by the availability of a large volume of sequence data, as well as powerful computers running graphics processing units (GPUs) that are critical for most neural network–based learning. It is important to note, however, that the best algorithm for prediction of GO molecular function and GO biological process (NetGO) had the highest F‐score of 0.63 and 0.34, respectively, with an F‐score of 1 indicating perfect prediction. Thus, significant scope still exists for better prediction of GO categories.
Prediction of metabolic pathways
Plant metabolic pathways may be divided into two types, primary and specialized, with genes involved in the latter undergoing frequent lineage‐specific duplications and functional divergence. Thus, prediction of these genes’ function using sequence similarity—as is typically done for gene function annotation after genome sequencing—frequently leads to incorrect, incomplete, or no annotation of the gene. A combination of inputs from gene and protein structure analysis, expression patterns, metabolic profiles, and homology using machine learning can better associate a metabolic gene to its pathway and even to specific enzyme activity. In one study (Dale et al., 2010), the authors utilized 123 features, including reaction evidence, pathway holes (i.e., steps with unknown enzymes), pathway connectivity, gene chromosomal location and neighbors, pathway variants (i.e., alternative routes to an end product), and taxonomic range, to predict metabolic pathways in an organism from its genome‐wide gene complement. Four popular machine learning approaches (naïve Bayes, decision trees, logistic regression, and ensemble methods) were used on these data. The authors reported an improvement over the performance of their previous, non–machine learning algorithm called PathoLogic (Karp et al., 2010). They note the importance of machine learning being a data‐driven, scalable, and tunable approach vs. the hard‐coded rules embedded in the core PathoLogic algorithm that are not conducive to scaling with increasing quantity of training data. One of the major factors limiting the performance of machine learning–based pathway prediction methods is still the availability of data sets matching enzymes to the reaction they catalyze, a problem partially being addressed by the PlantCyc database (Schläpfer et al., 2017).
While the PathoLogic tool approaches pathway prediction using publicly available genomic data, a recent study (Toubiana et al., 2019) used a combination of transcriptomics and untargeted metabolomics data from tomato in an integrative RF framework to identify and validate novel metabolic enzymes. In particular, they predicted and experimentally validated the presence of a novel pathway for melibiose degradation in plants. This application of a machine learning–based prediction approach demonstrates the potential for deploying machine learning to gain new biological insights.
Prediction of phenotypes and biological processes
Compared to traditional statistical approaches that have difficulty scaling to multiple variables and benefit from dimensionality reduction, machine learning methods are well‐suited for phenotype prediction tasks, which are often characterized by a combination of various features and an often‐significant non‐independence between subsets of features. The ability of machine learning to combine disparate pieces of input features is particularly useful when the data come from completely different methods. For example, a recent study integrated gas chromatography–mass spectrometry (GC‐MS) profiling, reverse transcription–quantitative PCR (RT‐qPCR), and RNA‐seq for predicting drought resistance in potato via an RF approach (Sprenger et al., 2018). This method identified 20 metabolite and transcript markers that were indicative of drought resistance, independent of seasonal or regional agronomic conditions.
A combination of features was also used by Lloyd et al. (2015) for prediction of genes that may produce a lethal phenotype upon inactivation, by testing predictive accuracy across an ensemble of machine learning models. Features such as gene copy number, time since duplication, expression level and pattern, evolutionary rate, and connectivity in molecular networks were found to discriminate between essential and non‐essential genes in A. thaliana. Using these features, the authors developed an RF algorithm that was able to successfully identify essential genes not just in A. thaliana, but also in rice and yeast with a high success rate, illustrating the applicability of some models developed in one species to other species sometimes separated by hundreds of millions of years of evolution. A similar study utilized 50 different features including duplication pattern, sequence conservation, expression level, protein domain content, and gene network properties to identify—using RF and SVM—genes in A. thaliana that were part of general (primary) or specialized metabolism (Moore et al., 2019). The authors found that specialized metabolic genes are characterized by their origination from tandem duplications, tendency to be more specifically expressed in certain tissues, tendency to be co‐expressed with their paralogs, lower overall expression, and lower network connectivity in comparison to primary metabolism genes. This example also demonstrates the biological interpretability of feature‐based machine learning models. When the above features were used together in an integrative machine learning framework, the model could predict if a gene belongs to primary or specialized metabolism with a true positive rate of 87% and a true negative rate of 71%.
These examples illustrate that machine learning–based approaches can easily be adapted to use a large variety of different data types (e.g., gene expression, sequence‐based features, metabolomic profiles, evolutionary information), and that the models developed in one species can be used—depending on the predictive task—in distant non‐model species. The ability to use and integrate different data types is also a crucial factor for applying such strategies to non‐model systems, as standardized, large‐scale data sets are often not available in such species.
Genomic prediction
Genomic prediction is the process of predicting the value of a complex (quantitative) trait of an organism on the basis of its combination of genetic markers, such as single nucleotide polymorphisms (SNPs), and is typically performed in crops, livestock, and humans. In plants, the phenotypic traits studied are typically of agronomic value (e.g., yield, interval between male/female flowering time, disease resistance). Genomic prediction models are trained on a population that is both genotyped and phenotyped, in order to identify the effects of SNP combinations on the phenotype under study. These models are then used to predict the phenotypes of a test population in which every organism has been genotyped, and its predicted values are then measured against the actual values. Genomic prediction can save breeders both time and money, as only a fraction of the individuals in a population need to be phenotyped and it shortens the breeding cycle through accelerated identification of preferable genotypes. However, genetic complexities such as low trait heritability, large numbers of loci underlying traits, influence of genotype × environment interactions, and the high dimensionality of data sets can lower the accuracy of predictive models (Crossa et al., 2017).
In particular, high dimensionality (caused by having more markers than individuals under study) makes traditional linear regression inappropriate for genomic prediction. Although algorithms for dimensionality reduction through variable (here, marker) selection, such as Bayesian LASSO or ridge regression, may account for this, they still fail to capture any non‐linearity present in the phenotypic response. Some machine learning algorithms are better able to capture these varied responses, and hence have been employed for both classification and regression tasks. For classification, algorithms are trained to distinguish between markers yielding the top N% (e.g., 10%) of the population and those yielding lower values. A comprehensive study (Ornella et al., 2014) comparing the performance of different machine learning classification and regression models in maize and wheat revealed that predicting a binary phenotypic outcome can be more accurate than predicting an individual’s specific trait value, with SVM models performing the best. A similar study (González‐Camacho et al., 2016) assessed the performance of ANNs in the classification of a very similar set of maize and wheat data sets. The authors found that a probabilistic neural network (Fig. 2D) had the best performance for classifying individuals into three phenotypic classes (e.g., low, middle, or high trait value). ANNs have recently been applied to genomic prediction problems with high frequency (Crossa et al., 2017), as advances in computing power have allowed for additional neuron layers (Fig. 2D), which provide increased power to capture inter‐marker correlations and interactions (Gianola et al., 2011). With increasing ease of obtaining SNP data, genomic prediction models could potentially be used in applications beyond plant breeding, such as to identify loci influencing differences in morphology/biochemistry in natural populations of plant species.
DISCUSSION
In this review, we detail multiple machine learning algorithms that define different aspects of gene function. Despite these innovations, the power of machine learning has not been adequately tapped in the plant sciences. One of the biggest roadblocks is the relative lack of large data sets and, in particular, a lack of positive training instances, which results in significantly imbalanced data sets. Recently, the advent of a new deep learning method—generative adversarial networks, infamous for the so‐called “deepfake” images and videos—has also spurred studies seeking to use the method to artificially generate biologically realistic data (e.g., Liu et al., 2019) and minimize data imbalance for machine learning. However, for the near future at least, a strong foundation of data generated through wet‐lab experiments is absolutely needed. For machine learning, and especially for ANNs, this requirement scales up to need big data. In human models, projects such as ENCODE (The ENCODE Project Consortium, 2012), the Roadmap Epigenomics Mapping Project (Bernstein et al., 2010), and the Cancer Genome Atlas (TCGA; Tomczak et al., 2015) have aimed to catalog not only the depth but also the breadth of various features relevant to human biology. These experiments generate “gold standard” validated data sets containing positive instances, on the scale applicable for machine learning. For example, TCGA includes genomic, transcriptomic, epigenomic, proteomic, histopathological, and clinical information for ~33 different tumors collected from ~12,000 samples, and has spawned a series of machine learning–based studies that have produced valuable insights in cancer biology (e.g., Malta et al., 2018). In order to reach the scales required for breakthrough insights in plant sciences, the breadth and depth of available data must increase. For example, most of the current big data sets in plants come primarily from A. thaliana, maize, and rice. Efforts to make systematic, easily accessible data compendia in these species (e.g., AtGenExpress [Kilian et al., 2007] in A. thaliana or EBI Expression Atlas [Kapushesky et al., 2012]) can foster new machine learning studies. It may also be useful to focus on some “anchor species” (e.g., tomato, soybean) for generation of more in‐depth genomic and post‐genomic data sets (Moghe and Kruse, 2018). These species could serve as phylogenetic anchors for understanding and utilizing evolutionary variance as a feature set for machine learning, potentially increasing the breadth of applicability of machine learning models developed in one species to other species in their phylogenetic neighborhood. The prediction of metabolic pathways in the PathoLogic software and its machine learning variant demonstrates the value of using evolutionary relatedness for filling “gaps” or unknown reactions in metabolic pathways (Dale et al., 2010; Karp et al., 2010).
The TCGA example also shows the value of central localization of disparate data types in improving the data’s accessibility and usability. Although such an endeavor would likely be an enormous undertaking for plant‐related data, linking disparate resources together through application programming interfaces (APIs) or inter‐database links may be a feasible and useful option. Several tools and databases are currently used in plant genomics for annotating different aspects of genic features (reviewed in Bolger et al., 2018). Integrating heterogeneous genomic and post‐genomic data spread out across different databases can help develop more complex models for predicting genotype–phenotype relationships. Such data need not necessarily be molecular—experimental metadata such as treatment types, organs/tissues, or genotypes could also serve as additional, potentially useful features for making genotype‐to‐phenotype machine learning models. As highlighted by some studies in this collection (Mirnezami et al., 2020; Théroux‐Rancourt et al., 2020), machine learning algorithms can also be used to generate useful phenotypic trait data. If associated properly with metadata as well as genomic and other omics data, this recent revolution in high‐throughput phenomics could help machine learning algorithm development and provide novel insights about complex traits (Großkinsky et al., 2015). Indeed, the addition of metadata in a consistent format across different databases is instrumental for the integration of different genomic features in machine learning model training.
In addition to data and databases, the availability of good cyberinfrastructure and good training opportunities in programming, statistics, and machine learning is also critical for utilizing the power of machine learning in the plant sciences. The National Science Foundation–funded CyVerse (https://cyverse.org/) and the Extreme Science and Engineering Discovery Environment (XSEDE; https://portal.xsede.org/) are two excellent resources that allow scientists and educators in the United States who do not have access to their own high‐performance computing servers to test out and implement various machine learning applications, including deep learning algorithms. It is also important that students be well‐trained in programming languages, especially Python and R, which currently serve as the starting point for many machine learning–oriented packages (e.g., scikit‐learn [Pedregosa et al., 2011], Keras [https://github.com/fchollet/keras], caret [Kuhn, 2008]). Such training may take the shape of semester‐wide courses, focused workshops, or machine learning–related classes offered by many online educational platforms. There are currently more than 2000 genomics‐related models stored in the open access Kipoi model repository (Avsec et al., 2019) that could be used for such training activities. However, for botanists and life scientists who do not have such platforms in place already, organizations and competitions that facilitate collaborations with data scientists—such as the Herbarium2019 competition noted in this collection (Little et al., 2020)—are a valuable resource.
In this review, we have highlighted some of the principal machine learning approaches used for functional prediction in plants. These examples are by no means exhaustive. One of machine learning’s most impressive characteristics is that it continues to be used in innovative ways and applied to different biological processes, a trend that will only increase in the future (for an excellent review, see Li et al., 2019). In addition, as evidenced in this review, well‐thought‐out experimental designs can also help us gain useful biological understanding from machine learning models, mitigating the “black box” characterization of machine learning methods. We believe that machine learning algorithmic development can be accelerated by improved generation and cataloging of phylogenetically broad experimental data—especially data on molecular interactions and phenotypes—as well as by reducing the infrastructural and skill barriers for machine learning implementation and training.
ACKNOWLEDGMENTS
This work was funded by Cornell University startup funds to G.D.M. and a Deutsche Forschungsgemeinschaft (DFG) award (#411255989) to L.H.K. The authors thank Dr. Suzy Strickler and two anonymous reviewers for valuable feedback on the manuscript.
Mahood, E. H. , Kruse L. H., and Moghe G. D.. 2020. Machine learning: A powerful tool for gene function prediction in plants. Applications in Plant Sciences 8(7): e11376.
LITERATURE CITED
- Alipanahi, B. , Delong A., Weirauch M. T., and Frey B. J.. 2015. Predicting the sequence specificities of DNA‐ and RNA‐binding proteins by deep learning. Nature Biotechnology 33: 831–838. [DOI] [PubMed] [Google Scholar]
- Armenteros, J. J. A. , Salvatore M., Emanuelsson O., Winther O., von Heijne G., Elofsson A., and Nielsen H.. 2019a. Detecting sequence signals in targeting peptides using deep learning. Life Science Alliance 2(5): e201900429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armenteros, J. J. A. , Tsirigos K. D., Sønderby C. K., Petersen T. N., Winther O., Brunak S., von Heijne G., and Nielsen H.. 2019b. SignalP 5.0 improves signal peptide predictions using deep neural networks. Nature Biotechnology 37: 420–423. [DOI] [PubMed] [Google Scholar]
- Avsec, Ž. , Kreuzhuber R., Israeli J., Xu N., Cheng J., Shrikumar A., Banerjee A., et al. 2019. The Kipoi repository accelerates community exchange and reuse of predictive models for genomics. Nature Biotechnology 37: 592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baum, L. E. , and Eagon J. A.. 1967. An inequality with applications to statistical estimation for probabilistic functions of Markov processes and to a model for ecology. Bulletin of the American Mathematical Society 73: 360–363. [Google Scholar]
- Beer, M. A. , and Tavazoie S.. 2004. Predicting gene expression from sequence. Cell 117: 185–198. [DOI] [PubMed] [Google Scholar]
- Ben‐Hur, A. , Ong C. S., Sonnenburg S., Schölkopf B., and Rätsch G.. 2008. Support vector machines and kernels for computational biology. PLoS Computational Biology 4: e1000173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein, B. E. , Stamatoyannopoulos J. A., Costello J. F., Ren B., Milosavljevic A., Meissner A., Kellis M., et al. 2010. The NIH Roadmap Epigenomics Mapping Consortium. Nature Biotechnology 28: 1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, M. E. , Arsova B., and Usadel B.. 2018. Plant genome and transcriptome annotations: From misconceptions to simple solutions. Briefings in Bioinformatics 19: 437–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford, J. R. , Needham C. J., Tedder P., Care M. A., Bulpitt A. J., and Westhead D. R.. 2010. GO‐At : In silico prediction of gene function in Arabidopsis thaliana by combining heterogeneous data. The Plant Journal 61: 713–721. [DOI] [PubMed] [Google Scholar]
- Brenchley, R. , Spannagl M., Pfeifer M., Barker G. L. A., D’Amore R., Allen A. M., McKenzie N., et al. 2012. Analysis of the bread wheat genome using whole‐genome shotgun sequencing. Nature 491: 705–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bzdok, D. , Altman N., and Krzywinski M.. 2018. Points of significance: Statistics versus machine learning. Nature Methods 15: 233–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, M. S. , Law M., Holt C., Stein J. C., Moghe G. D., Hufnagel D. E., Lei J., et al. 2014. MAKER‐P: A tool kit for the rapid creation, management, and quality control of plant genome annotations. Plant Physiology 164: 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caruana, R. , and Niculescu‐Mizil A.. 2006. An empirical comparison of supervised learning algorithms In Proceedings of the 23rd International Conference on Machine Learning (ICML ’06), 161–168. ACM, New York, New York, USA. [Google Scholar]
- Chou, K.‐C. , and Shen H.‐B.. 2010. Plant‐mPLoc: A top‐down strategy to augment the power for predicting plant protein subcellular localization. PLoS ONE 5: e11335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crossa, J. , Pérez‐Rodríguez P., Cuevas J., Montesinos‐López O., Jarquín D., de los Campos G., Burgueño J., et al. 2017. Genomic selection in plant breeding: Methods, models, and perspectives. Trends in Plant Science 22: 961–975. [DOI] [PubMed] [Google Scholar]
- Cruz, J. A. , and Wishart D. S.. 2007. Applications of machine learning in cancer prediction and prognosis. Cancer Informatics 2: 59–77. [PMC free article] [PubMed] [Google Scholar]
- Dale, J. M. , Popescu L., and Karp P. D.. 2010. Machine learning methods for metabolic pathway prediction. BMC Bioinformatics 11: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’haeseleer, P. 2006. How does DNA sequence motif discovery work? Nature Biotechnology 24: 959–961. [DOI] [PubMed] [Google Scholar]
- Ding, Z. , and Kihara D.. 2019. Computational identification of protein‐protein interactions in model plant proteomes. Scientific Reports 9: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunne, M. P. , and Kelly S.. 2017. OrthoFiller: Utilising data from multiple species to improve the completeness of genome annotations. BMC Genomics 18: 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst, J. , and Kellis M.. 2017. Chromatin‐state discovery and genome annotation with ChromHMM. Nature Protocols 12: 2478–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fa, R. , Cozzetto D., Wan C., and Jones D. T.. 2018. Predicting human protein function with multi‐task deep neural networks. PLoS ONE 13: e0198216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan, X. , Stegle O., Behr J., Steffen J. G., Drewe P., Hildebrand K. L., Lyngsoe R., et al. 2011. Multiple reference genomes and transcriptomes for Arabidopsis thaliana . Nature 477: 419–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, X. , Zhang J., Wei Z., and Hakonarson H.. 2018. DeepPolyA: A convolutional neural network approach for polyadenylation site prediction. IEEE Access 6: 24340–24349. [Google Scholar]
- Gerstein, M. B. , Bruce C., Rozowsky J. S., Zheng D., Du J., Korbel J. O., Emanuelsson O., et al. 2007. What is a gene, post‐ENCODE? History and updated definition. Genome Research 17: 669–681. [DOI] [PubMed] [Google Scholar]
- Gianola, D. , Okut H., Weigel K. A., and Rosa G. J.. 2011. Predicting complex quantitative traits with Bayesian neural networks: A case study with Jersey cows and wheat. BMC Genetics 12: 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González‐Camacho, J. M. , Crossa J., Pérez‐Rodríguez P., Ornella L., and Gianola D.. 2016. Genome‐enabled prediction using probabilistic neural network classifiers. BMC Genomics 17: 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Götz, S. , García‐Gómez J. M., Terol J., Williams T. D., Nagaraj S. H., Nueda M. J., Robles M., et al. 2008. High‐throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Research 36: 3420–3435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graur, D. , Zheng Y., Price N., Azevedo R. B. R., Zufall R. A., and Elhaik E.. 2013. On the immortality of television sets: “Function” in the human genome according to the evolution‐free gospel of ENCODE. Genome Biology and Evolution 5: 578–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Großkinsky, D. K. , Svensgaard J., Christensen S., and Roitsch T.. 2015. Plant phenomics and the need for physiological phenotyping across scales to narrow the genotype‐to‐phenotype knowledge gap. Journal of Experimental Botany 66: 5429–5440. [DOI] [PubMed] [Google Scholar]
- Hoff, K. J. , Lange S., Lomsadze A., Borodovsky M., and Stanke M.. 2016. BRAKER1: Unsupervised RNA‐Seq‐Based genome annotation with GeneMark‐ET and AUGUSTUS. Bioinformatics (Oxford, England) 32: 767–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kakei, Y. , Ogo Y., Itai R. N., Kobayashi T., Yamakawa T., Nakanishi H., and Nishizawa N. K.. 2013. Development of a novel prediction method of cis‐elements to hypothesize collaborative functions of cis‐element pairs in iron‐deficient rice. Rice 6: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapushesky, M. , Adamusiak T., Burdett T., Culhane A., Farne A., Filippov A., Holloway E., et al. 2012. Gene Expression Atlas update—A value‐added database of microarray and sequencing‐based functional genomics experiments. Nucleic Acids Research 40: D1077–D1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlić, R. , Chung H.‐R., Lasserre J., Vlahoviček K., and Vingron M.. 2010. Histone modification levels are predictive for gene expression. Proceedings of the National Academy of Sciences USA 107: 2926–2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karp, P. D. , Paley S. M., Krummenacker M., Latendresse M., Dale J. M., Lee T. J., Kaipa P., et al. 2010. Pathway Tools version 13.0: Integrated software for pathway/genome informatics and systems biology. Briefings in Bioinformatics 11: 40–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khamis, A. M. , Motwalli O., Oliva R., Jankovic B. R., Medvedeva Y. A., Ashoor H., Essack M., et al. 2018. A novel method for improved accuracy of transcription factor binding site prediction. Nucleic Acids Research 46: e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian, J. , Whitehead D., Horak J., Wanke D., Weinl S., Batistic O., D’Angelo C., et al. 2007. The AtGenExpress global stress expression data set: Protocols, evaluation and model data analysis of UV‐B light, drought and cold stress responses. The Plant Journal 50: 347–363. [DOI] [PubMed] [Google Scholar]
- Kourou, K. , Exarchos T. P., Exarchos K. P., Karamouzis M. V., and Fotiadis D. I.. 2015. Machine learning applications in cancer prognosis and prediction. Computational and Structural Biotechnology Journal 13: 8–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuhn, M. 2008. Building predictive models in R using the caret package. Journal of Statistical Software 28: 10.18637/jss.v028.i05. [DOI] [Google Scholar]
- Kulmanov, M. , and Hoehndorf R.. 2020. DeepGOPlus: Improved protein function prediction from sequence. Bioinformatics 36: 422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Lee K. K., Walsh S., Smith C., Hadingham S., Sorefan K., Cawley G., and Bevan M. W.. 2006. Establishing glucose‐ and ABA‐regulated transcription networks in Arabidopsis by microarray analysis and promoter classification using a Relevance Vector Machine. Genome Research 16: 414–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Shi W., and Wasserman W. W.. 2018. Genome‐wide prediction of cis‐regulatory regions using supervised deep learning methods. BMC Bioinformatics 19: 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Huang C., Ding L., Li Z., Pan Y., and Gao X.. 2019. Deep learning in bioinformatics: Introduction, application, and perspective in the big data era. Methods 166: 4–21. [DOI] [PubMed] [Google Scholar]
- Little, D. P. , Tulig M., Tan K. C., Liu Y., Belongie S., Kaeser‐Chen C., Michelangeli F. A., et al. 2020. An algorithm competition for automatic species identification from herbarium specimens. Applications in Plant Sciences 8(6): e11365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Q. , Lv H., and Jiang R.. 2019. hicGAN infers super resolution Hi‐C data with generative adversarial networks. Bioinformatics 35: i99–i107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S. , Liu Y., Zhao J., Cai S., Qian H., Zuo K., Zhao L., and Zhang L.. 2017. A computational interactome for prioritizing genes associated with complex agronomic traits in rice (Oryza sativa). The Plant Journal 90: 177–188. [DOI] [PubMed] [Google Scholar]
- Lloyd, J. P. , Seddon A. E., Moghe G. D., Simenc M. C., and Shiu S.‐H.. 2015. Characteristics of plant essential genes allow for within‐ and between‐species prediction of lethal mutant phenotypes. The Plant Cell 27: 2133–2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu, C. , Wang L., and Zhang J.. 2018. Deep learning for DNase I hypersensitive sites identification. BMC Genomics 19: 905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacIsaac, K. D. , and Fraenkel E.. 2006. Practical strategies for discovering regulatory DNA sequence motifs. PLoS Computational Biology 2: e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malta, T. M. , Sokolov A., Gentles A. J., Burzykowski T., Poisson L., Weinstein J. N., Kamińska B., et al. 2018. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 173: 338–354.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathur, A. , and Foody G. M.. 2008. Multiclass and binary SVM classification: Implications for training and classification users. IEEE Geoscience and Remote Sensing Letters 5: 241–245. [Google Scholar]
- Mirnezami, V. , Young T., Assefa T., Prichard S., Nagasubramanian K., Sandhu K., Sarkar S., et al. 2020. Automated trichome counting in soybean using advanced image‐processing techniques. Applications in Plant Sciences 8(7): e11375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghe, G. D. , and Kruse L. H.. 2018. The study of plant specialized metabolism: Challenges and prospects in the genomics era. American Journal of Botany 105: 959–962. [DOI] [PubMed] [Google Scholar]
- Moore, B. M. , Wang P., Fan P., Leong B., Schenck C. A., Lloyd J. P., Lehti‐Shiu M. D., et al. 2019. Robust predictions of specialized metabolism genes through machine learning. Proceedings of the National Academy of Sciences USA 116: 2344–2353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natarajan, A. , Yardımcı G. G., Sheffield N. C., Crawford G. E., and Ohler U.. 2012. Predicting cell‐type–specific gene expression from regions of open chromatin. Genome Research 22: 1711–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numa, H. , and Itoh T.. 2014. MEGANTE: A web‐based system for integrated plant genome annotation. Plant and Cell Physiology 55: e2 10.1093/pcp/pct157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ornella, L. , Pérez P., Tapia E., González‐Camacho J. M., Burgueño J., Zhang X., Singh S., et al. 2014. Genomic‐enabled prediction with classification algorithms. Heredity 112: 616–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orre, L. M. , Vesterlund M., Pan Y., Arslan T., Zhu Y., Fernandez Woodbridge A., Frings O., et al. 2019. SubCellBarCode: Proteome‐wide mapping of protein localization and relocalization. Molecular Cell 73: 166–182.e7. [DOI] [PubMed] [Google Scholar]
- Pedregosa, F. , Varoquaux G., Gramfort A., Michel V., Thirion B., Grisel O., Blondel M., et al. 2011. Scikit‐learn: Machine learning in Python. Journal of Machine Learning Research 12: 2825–2830. [Google Scholar]
- Roadmap Epigenomics Consortium , Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi‐Moussavi A., et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518: 317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodgers‐Melnick, E. , Culp M., and DiFazio S. P.. 2013. Predicting whole genome protein interaction networks from primary sequence data in model and non‐model organisms using ENTS. BMC Genomics 14: 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg, S. L. 2019. Next‐generation genome annotation: We still struggle to get it right. Genome Biology 20: 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz, M. C. , Witkowski J., and McCombie W. R.. 2012. Current challenges in de novo plant genome sequencing and assembly. Genome Biology 13: 243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schietgat, L. , Vens C., Struyf J., Blockeel H., Kocev D., and Džeroski S.. 2010. Predicting gene function using hierarchical multi‐label decision tree ensembles. BMC Bioinformatics 11: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schläpfer, P. , Zhang P., Wang C., Kim T., Banf M., Chae L., Dreher K., et al. 2017. Genome‐wide prediction of metabolic enzymes, pathways, and gene clusters in plants. Plant Physiology 173: 2041–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutzer, T. , Bolger M. E., Rudd S., Chen J., Gundlach H., Arend D., Oppermann M., et al. 2017. Bioinformatics in the plant genomic and phenomic domain: The German contribution to resources, services and perspectives. Journal of Biotechnology 261: 37–45. [DOI] [PubMed] [Google Scholar]
- Schweikert, G. , Zien A., Zeller G., Behr J., Dieterich C., Ong C. S., Philips P., et al. 2009. mGene: Accurate SVM‐based gene finding with an application to nematode genomes. Genome Research 19: 2133–2143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh, R. , Lanchantin J., Robins G., and Qi Y.. 2016. DeepChrome: Deep‐learning for predicting gene expression from histone modifications. Bioinformatics 32: i639–i648. [DOI] [PubMed] [Google Scholar]
- Sonnenburg, S. , Schweikert G., Philips P., Behr J., and Rätsch G.. 2007. Accurate splice site prediction using support vector machines. BMC Bioinformatics 8: S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprenger, H. , Erban A., Seddig S., Rudack K., Thalhammer A., Le M. Q., Walther D., et al. 2018. Metabolite and transcript markers for the prediction of potato drought tolerance. Plant Biotechnology Journal 16: 939–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, L. , Liu H., Zhang L., and Meng J.. 2015. lncRScan‐SVM: A tool for predicting long non‐coding RNAs using support vector machine. PLoS ONE 10: e0139654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk, D. , Gable A. L., Lyon D., Junge A., Wyder S., Huerta‐Cepas J., Simonovic M., et al. 2019. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome‐wide experimental datasets. Nucleic Acids Research 47: D607–D613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAIR . 2019. The Arabidopsis Information Resource. Website https://www.arabidopsis.org/portals/genAnnotation/genome_snapshot.jsp [accessed 30 September 2019].
- The Cancer Genome Atlas Research Network , Weinstein J. N., Collisson E. A., Mills G. B., Shaw K. R. M., Ozenberger B. A., Ellrott K., et al. 2013. The Cancer Genome Atlas Pan‐Cancer analysis project. Nature Genetics 45: 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The ENCODE Project Consortium . 2012. An integrated encyclopedia of DNA elements in the human genome. Nature 489: 57–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Théroux‐Rancourt, G. , Jenkins M. R., Broderson C. R., McElrone A., Forrestel E. J., and Earles J. M.. 2020. Digitally deconstructing leaves in 3D using X‐ray microcomputed tomography and machine learning. Applications in Plant Sciences 8(7): e11380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomczak, K. , Czerwińska P., and Wiznerowicz M.. 2015. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemporary Oncology (Poznan, Poland) 19: A68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tompa, M. , Li N., Bailey T. L., Church G. M., De Moor B., Eskin E., Favorov A. V., et al. 2005. Assessing computational tools for the discovery of transcription factor binding sites. Nature Biotechnology 23: 137–144. [DOI] [PubMed] [Google Scholar]
- Toubiana, D. , Puzis R., Wen L., Sikron N., Kurmanbayeva A., Soltabayeva A., del M. R. Wilhelmi M., et al. 2019. Combined network analysis and machine learning allows the prediction of metabolic pathways from tomato metabolomics data. Communications Biology 2: 214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umarov, R. K. , and Solovyev V. V.. 2017. Recognition of prokaryotic and eukaryotic promoters using convolutional deep learning neural networks. PLoS ONE 12: e0171410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium . 2019. UniProt: A worldwide hub of protein knowledge. Nucleic Acids Research 47: D506–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, X. , Lin P., and Ho J. W. K.. 2018. Discovery of cell‐type specific DNA motif grammar in cis‐regulatory elements using random forest. BMC Genomics 19: 929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn, J. D. , Mejia‐Guerra M. K., Ramstein G., Kremling K. A., Valluru R., Buckler E. S., and Wang H.. 2019. Evolutionarily informed deep learning methods for predicting relative transcript abundance from DNA sequence. Proceedings of the National Academy of Sciences USA 116: 5542–5549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch, M. T. , Cote A., Norel R., Annala M., Zhao Y., Riley T. R., Saez‐Rodriguez J., et al. 2013. Evaluation of methods for modeling transcription factor sequence specificity. Nature Biotechnology 31: 126–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yip, K. Y. , Cheng C., and Gerstein M.. 2013. Machine learning and genome annotation: A match meant to be? Genome Biology 14: 205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You, R. , Huang X., and Zhu S.. 2018. DeepText2GO: Improving large‐scale protein function prediction with deep semantic text representation. Methods 145: 82–90. [DOI] [PubMed] [Google Scholar]
- You, R. , Yao S., Xiong Y., Huang X., Sun F., Mamitsuka H., and Zhu S.. 2019. NetGO: Improving large‐scale protein function prediction with massive network information. Nucleic Acids Research 47: W379–W387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, N. , Jiang Y., Bergquist T. R., Lee A. J., Kacsoh B. Z., Crocker A. W., Lewis K. A., et al. 2019. The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biology 20: 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, G. , Wu A., Xu X.‐J., Xiao P.‐P., Lu L., Liu J., Cao Y., et al. 2016. PPIM: A protein‐protein interaction database for maize. Plant Physiology 170: 618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]