

HSV-1 is a common cause of ocular infections worldwide and a significant cause of preventable blindness. Corneal scarring and blindness are consequences of the immune response induced by repeated reactivation events. Therefore, HSV-1 therapeutic approaches should focus on preventing latency and reactivation. Our data suggest that the immune function of HVEM plays an important role in the HSV-1 latency and reactivation cycle that is independent of HVEM binding to gD.
KEYWORDS: cornea, virus replication, transgenic, exhaustion, eye disease, herpes simplex virus 1, HSV-1, herpesvirus entry mediator, HVEM, latency, ocular infection, reactivation
ABSTRACT
The immune modulatory protein herpes virus entry mediator (HVEM) is one of several cellular receptors used by herpes simplex virus 1 (HSV-1) for cell entry. HVEM binds to HSV-1 glycoprotein D (gD) but is not necessary for HSV-1 replication in vitro or in vivo. Previously, we showed that although HSV-1 replication was similar in wild-type (WT) control and HVEM−/− mice, HSV-1 does not establish latency or reactivate effectively in mice lacking HVEM, suggesting that HVEM is important for these functions. It is not known whether HVEM immunomodulatory functions contribute to latency and reactivation or whether its binding to gD is necessary. We used HVEM−/− mice to establish three transgenic mouse lines that express either human WT HVEM or human or mouse HVEM with a point mutation that ablates its ability to bind to gD. Here, we show that HVEM immune function, not its ability to bind gD, is required for WT levels of latency and reactivation. We further show that HVEM binding to gD does not affect expression of the HVEM ligands BTLA, CD160, or LIGHT. Interestingly, our results suggest that binding of HVEM to gD may contribute to efficient upregulation of CD8α but not PD1, TIM-3, CTLA4, or interleukin 2 (IL-2). Together, our results establish that HVEM immune function, not binding to gD, mediates establishment of latency and reactivation.
IMPORTANCE HSV-1 is a common cause of ocular infections worldwide and a significant cause of preventable blindness. Corneal scarring and blindness are consequences of the immune response induced by repeated reactivation events. Therefore, HSV-1 therapeutic approaches should focus on preventing latency and reactivation. Our data suggest that the immune function of HVEM plays an important role in the HSV-1 latency and reactivation cycle that is independent of HVEM binding to gD.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) can infect a multitude of cell and tissue types, causing mucocutaneous lesions, ocular infections, and, in rare cases, encephalitis (1–3). It is estimated that 60% to 90% of the population is seropositive for HSV-1, and HSV-1 is the leading cause of viral eye infections and infectious corneal blindness (4, 5). After initial infection and replication in the eye, HSV-1 travels to the trigeminal ganglia (TG), where it establishes lifelong latency and from which it can periodically reactivate (4). Recurrent ocular infections can cause corneal scarring (CS), leading to loss of visual acuity (1–3, 6, 7).
HSV-1 entry into cells requires the coordinated action of several of its surface glycoproteins (8–13). Viral attachment occurs via binding of viral glycoprotein B (gB) or gC to host heparan sulfate proteoglycans (14, 15). This interaction is followed by binding of gD to one of its several known cell surface receptors, such as nectin-1 (HveC) (16), nectin-2 (HveA) (17, 18), herpesvirus entry mediator (HVEM; TNFRSF14) (19–21), or a modified form of heparan sulfate (3-OST-3) (15). Binding of gD to its receptor activates a heterodimeric gH-gL complex, leading to fusion of the viral envelope with the host membrane (22). The ability of HSV-1 to use several receptors for entry likely explains its ability to infect a variety of tissues (9, 11, 15, 20, 21, 23, 24).
HVEM is a member of the tumor necrosis factor (TNF) receptor protein family (25, 26). It is expressed in the cornea, nervous tissue, and epithelial tissues of the lungs and gut (27–29). In addition, leukocytes such as T and B cells, natural killer (NK) cells, dendritic cells (DCs), and myeloid cells express HVEM (26). HVEM ligands include the immunoglobulin domain-containing cell surface receptor B- and T-lymphocyte attenuator (BTLA), CD160, and the TNF superfamily members lymphotoxin-related inducible ligand that competes for glycoprotein D binding to HVEM on T cells (LIGHT) and lymphotoxin alpha (Ltα) (30–32). HVEM binding to LIGHT or Ltα acts as a costimulatory signal for several cell types, including T cells and antigen-presenting cells, whereas binding of HVEM to BTLA or CD160 results in T cell inhibition (33–38). Evidence suggests that gD binding to HVEM inhibits T cell proliferation, activation, and effector functions and competes with BTLA for binding to HVEM (39).
Our studies suggest that HVEM plays an important role in the HSV-1 latency and reactivation cycle (29). We demonstrated that HVEM, but no other HSV-1 protein receptor, is upregulated during latency. Although initial HSV-1 replication was normal, HVEM−/− mice had less latency and slower reactivation than their parental wild-type (WT) C57BL/6 mice (29, 40). Interestingly, we have also demonstrated that BTLA, LIGHT, and CD160 play roles in efficient establishment of latency but not in reactivation (40). LTα, on the other hand, does not affect the establishment of latency (41). Currently, the mechanism of HVEM function in the latency and reactivation cycle is unclear. To determine whether HVEM immune function or gD binding plays a role in the latency and reactivation cycle, we established HVEM rescue mice by introducing either a human WT HVEM (HVEMhWT) gene, a human HVEM gene with a point mutation that disrupts binding to gD (HVEMhY23A), or a mouse HVEM gene with a point mutation that disrupts binding to gD (HVEMmY23A) into HVEM−/− mice. We demonstrated HVEM expression in the TG, corneas, and brain of these transgenic mice and were able to rescue HVEM immune functions. We also showed that latency and reactivation depend on HVEM immune function, not on binding to gD, because expression of a mutant HVEM can rescue both latency and reactivation to WT levels. We further showed that upregulation of CD8α and the exhaustion marker PD1, but not TIM-3, is a consequence of HVEM immune function, not binding to gD. However, upregulation of another exhaustion marker, CTLA4, may be affected by gD binding to human but not mouse HVEM. These results suggest that HVEM immune functions drive establishment of latency and reactivation and suggest a differential effect of gD binding to mouse versus human HVEM.
RESULTS
HVEM is expressed in the TG, brain, and corneas of HVEM transgenic mice.
HVEM transgenic mice were established by embryonic injection of a plasmid containing either human WT HVEM or human HVEM with a missense mutation that changes tyrosine to alanine at position 23 as described in Materials and Methods. The presence of the transgene was verified by PCR of tail cut DNA. PCR for human HVEM in HVEMhWT and HVEMhY23A transgenic mice produced a DNA fragment of 376 bp (Fig. 1A). Human and mouse HVEM share ∼45% homology, and therefore, the function of human HVEM may not be identical to that of mouse HVEM. To address this point, a transgenic mouse strain expressing mouse HVEM with a point mutation that abolishes its ability to bind gD (HVEMmY23A) was also established, and the presence of the transgene in pups was verified by PCR using primers specific for mouse HVEM. The presence of the 453-bp PCR product indicated successful integration of the transgene (Fig. 1B). The resulting pups were bred and screened for the presence of the transgene for at least four generations before being used in experiments. Mouse litters were born in mendelian frequencies and appeared normal in size and development.
FIG 1.

Validation of HVEMhWT, HVEMhY23A, and HVEMmY23A transgenic mice. (A) Tail cut DNAs from HVEMhWT and HVEMhY23A transgenic pups were amplified by PCR with primers that bind to human HVEM. A band of 376 bp indicates successful integration of the transgene. (B) Tail cut DNAs of HVEMmY23A pups were amplified by PCR using primers that bind to the mutated region of mouse HVEM. A 453-bp band indicated successful transgene integration. (C) Whole TG, cornea, and brain sections were harvested from HVEMhWT and HVEMhY23A mice. Total mRNA was isolated from individual mouse samples and amplified by qRT-PCR using primers against human HVEM. GAPDH expression was used to normalize HVEM expression, and fold change compared to mouse HVEM expression in WT C57BL/6 mice was determined and plotted. Results represent the mean for six TG, three corneas, and three brains ± SEM. (D) TG, cornea, and brain sections were harvested from a HVEMmY23A mouse, and total mRNA was isolated to determine mouse HVEMmY23A expression by qRT-PCR as described for panel C. The fold change compared to mouse HVEM expression in WT C57BL/6 mice is plotted. Results represent the mean for six TG, three corneas, and three brains ± SEM. (E to G) TG (E), cornea (F), and brain (G) sections were harvested from HVEM−/−, HVEMhWT, and HVEMhY23A mice. Homogenates were analyzed by Western blotting using an antibody against human HVEM. β-Actin was used as a loading control.
To verify that HVEM was expressed in our transgenic mice, human HVEM mRNA expression levels were determined in the TG, cornea, and brain by reverse transcription-quantitative PCR (qRT-PCR). HVEM levels were normalized to mouse HVEM in WT C57BL/6 mice. HVEM expression was higher in the TG, corneas, and brain of HVEMhWT mice than in WT mice (Fig. 1C). Similarly, HVEM expression was higher in the TG, corneas, and brain of HVEMhY23A mice than in WT or HVEMhWT mice (Fig. 1C). HVEM expression was higher in the brain of HVEMhWT and HVEMhY23A mice than in either the TG or corneas.
Expression of HVEM in HVEMmY23A pups was also verified using qRT-PCR (Fig. 1C). Expression of HVEM in the transgenic mouse TG, corneas, and brain was higher than that in WT mice, with brain having the highest HVEM expression (Fig. 1D). These results show that all three transgenic mouse lines express HVEM at high levels in the tested tissues.
To verify human HVEM protein expression in the HVEM−/−, HVEMhWT, and HVEMhY23A mice, the TG, brain, and corneas of naive mice were homogenized and lysates were analyzed by Western blotting with an antibody recognizing human HVEM (Fig. 1E to G). Similar to the qRT-PCR results, HVEM expression was higher in HVEMhY23A mouse TG (Fig. 1E), corneas (Fig. 1F), and brain (Fig. 1G) than in those from HVEMhWT mice. As expected, no band corresponding to HVEM was observed in HVEM−/− mice.
Virus replication and mouse survival are independent of HVEM expression and binding to gD.
To determine if HVEM binding to gD is necessary for viral replication, HVEM−/−, HVEMhWT, and HVEMhY23A mice were ocularly infected with 2 × 105 PFU of HSV-1 strain McKrae, and the amount of virus in tear films from each eye was determined on days 1 to 5 postinfection (p.i.). There were no significant differences in virus titer between HVEM−/− and HVEMhWT mice on days 1 to 4 p.i. (Fig. 2A, P > 0.05). On day 5 p.i., HVEMhWT mice had a slightly lower virus titer than HVEM−/− mice (Fig. 2A, P = 0.02). No significant differences were observed between HVEMhY23A and HVEM−/− mice or between HVEMhY23A and HVEMhWT mice at any time point (Fig. 2A, P > 0.05). To establish whether HVEM binding to gD affects mouse survival, survival was recorded for 28 days p.i. All 21 HVEM−/− and HVEMhWT mice survived, and all but 1 of the 21 HVEMhY23A mice survived (Fig. 2B, P > 0.05). These differences were not statistically significant, suggesting that HVEM expression or its binding to gD does not affect mouse survival.
FIG 2.

Virus replication and mouse survival are independent of HVEM expression and binding to gD. (A) HVEM−/−, HVEMhWT, and HVEMhY23A mice were ocularly infected with 2 × 105 PFU/eye of HSV-1 strain McKrae. Tear films were collected from 24 eyes, and virus titers were determined by a standard plaque assay. Average virus titers from two experiments are shown, and error bars indicate SEM. The P value was calculated using Student's t test and is considered significant at <0.05. (B) Survival of HVEM−/−, HVEMhWT, and HVEMhY23A mice infected as described for panel A was monitored for 28 days p.i. and plotted. (C) WT C57BL/6 and HVEMmY23A mice were ocularly infected, and virus titers were determined from tear films as described for panel A. The experiment was repeated twice for a total of 24 HVEM−/−, HVEMhWT, and HVEMhY23A mouse samples at each time point. Virus titers (PFU/ml) are shown as mean ± SEM. The P value was calculated using Student's t test and is considered significant at <0.05. (D) The survival of WT C57BL/6 and HVEMmY23A mice from panel C was monitored for 28 days p.i.
In parallel experiments, virus replication and survival of WT and HVEMmY23A mice were determined. On days 1 and 3 p.i., HVEMmY23A mice had higher viral titers than did WT mice (Fig. 2C, P = 0.006 and P = 0.04, respectively). We observed no differences in virus replication in HVEMmY23A and WT mice on day 2, 4, or 5 (Fig. 2C, P > 0.05). None of the 8 WT mice died, but 2 of the 21 HVEMmY23A mice died before day 10 p.i. (Fig. 2D, P > 0.05). These results agree with our previous data showing that HSV-1 replication and mouse survival are independent of HVEM (29, 40).
Viral latency and reactivation are rescued in HVEM transgenic mice independently of binding to gD.
We have shown previously that levels of HSV-1 latency are reduced in HVEM−/− mice (29, 40). However, it is not clear if HVEM immune function or binding to gD mediates the establishment of latency. To differentiate between these two possibilities, infected mice from the experiments described above were sacrificed on day 28 p.i., and the HSV-1 latency-associated transcript (LAT) copy number was determined as a measure of latency. The LAT copy number in TG of latently infected HVEMhWT and HVEMhY23A mice was significantly higher than that in HVEM−/− mice (Fig. 3A, 7.4 × 107, 6.4 × 107, and 1.3 × 106 copies LAT/μg TG RNA, respectively), whereas LAT copy numbers did not differ significantly between HVEMhWT and HVEMhY23A mice (Fig. 3A, P > 0.05).
FIG 3.

Viral latency and reactivation are rescued in HVEM transgenic mice independently of binding to gD. (A) HVEM−/−, HVEMhWT, and HVEMhY23A mice were infected ocularly with 2 × 105 PFU/eye of HSV-1 strain McKrae. On day 28 p.i., mice were euthanized and the LAT copy number in individual TG was calculated by qRT-PCR from standard curves generated using pGEM-LAT5317. GAPDH expression was used to normalize relative LAT expression. Each bar represents the mean result for 10 TG, and the error bars indicate SEM. (B) HVEM−/−, HVEMhWT, and HVEMhY23A mice were infected as described for panel A, and individual TG were harvested 28 days p.i. and cultured in tissue culture medium as described in Materials and Methods. Each day, an aliquot of medium was transferred onto indicator cells (RS cells) and monitored for virus reactivation. Results are shown as the number of TG that were activated each day, and the numbers indicate the average time (in days) at which the individual TG showed cytopathic effect (CPE) ± SEM. The results represent 28 TG from HVEM−/− mice, 24 TG from HVEMhWT mice, and 22 TG from HVEMhY23A mice. (C) WT C57BL/6 mice were ocularly infected, and the amount of latency was determined as described for panel A. Each bar represents a mean for 10 TG, and error bars indicate SEM. (D) WT C57BL/6 and HVEMmY23A mice were infected as described for panel A, and the average time to reactivation was plotted as described for panel B. The results represent 37 TG from WT C57BL/6 mice and 14 TG from HVEMmY23A mice.
Because latency levels correlate with time to reactivation, time to reactivation was determined in latently infected mouse TG using an ex vivo explant assay. As we have reported previously (29, 40), the average time to reactivation in HVEM−/− mice is 6.6 ± 0.4 days (Fig. 3B). Although the frequency of reactivation events was similar for all three groups of mice, the time to reactivation in HVEMhWT and HVEMhY23A mice was significantly faster (3.8 ± 0.2 and 4.4 ± 0.2 days, respectively) (Fig. 3B, P < 0.0001) than that seen in HVEM−/− mice. These results correlate with increased levels of latency seen in HVEMhWT and HVEMhY23A mice compared to HVEM−/− mice. There was no difference in average time to reactivation between HVEMhWT and HVEMhY23A mice (Fig. 3B, P > 0.05).
Latency and time to reactivation were also determined in HVEMmY23A mice and compared with those of the parental WT C57BL/6 mice. Similar to previous reports (29, 40), the LAT copy number in WT mice was approximately 9.4 × 107 copies per μg TG RNA, and the LAT copy number in TG of HVEMmY23A mice was nearly identical, at 9.1 × 107 copies per μg TG RNA (Fig. 3C, P > 0.05). Consistent with these findings, there was no difference in time to reactivation between WT and HVEMmY23A mice (Fig. 3D, 4.7 ± 0.03 and 4.0 ± 0.1 days, respectively, P > 0.05). Together, these results establish that restoring HVEM immune function in the absence of gD binding is sufficient to rescue both latency and reactivation in ocularly infected mice.
Binding of HVEM to gD is not required for expression of CD8α, immune exhaustion, and HVEM immune function.
We have shown previously that expression of the T cell marker CD8α and immune exhaustion markers PD1 and TIM-3 were reduced in HVEM−/− mice (40). In addition, HVEM activation has been shown to result in nuclear translocation of NF-κB (42, 43). To determine if these phenomena require HVEM immune function or binding to gD, the effect of HVEM on the relative levels of CD8α, markers of exhaustion (PD-1, TIM-3, CTLA4), NF-κB, and cytokines whose levels may be altered by exhaustion (interleukin 2 [IL-2], gamma interferon [IFN-γ], tumor necrosis factor alpha [TNF-α]) were determined by qRT-PCR of total TG extracts of WT, HVEM−/−, and transgenic mice infected as described above. The results are shown as the fold increase (or decrease) compared to baseline mRNA levels in TG from uninfected naive counterpart mice. As we have demonstrated previously (40), CD8α expression was significantly higher in WT mice than in HVEM−/− mice (Fig. 4A, P < 0.0001). CD8α expression was partially rescued in all three transgenic mouse lines but not in HVEM−/− mice (Fig. 4A, P < 0.0001). However, CD8α expression in the transgenic mice did not reach the levels seen in WT mice (Fig. 4A, P < 0.0001), and CD8α expression was significantly lower in HVEMhY23A than in HVEMhWT or HVEMmY23A mice (Fig. 4A, P < 0.0001).
FIG 4.
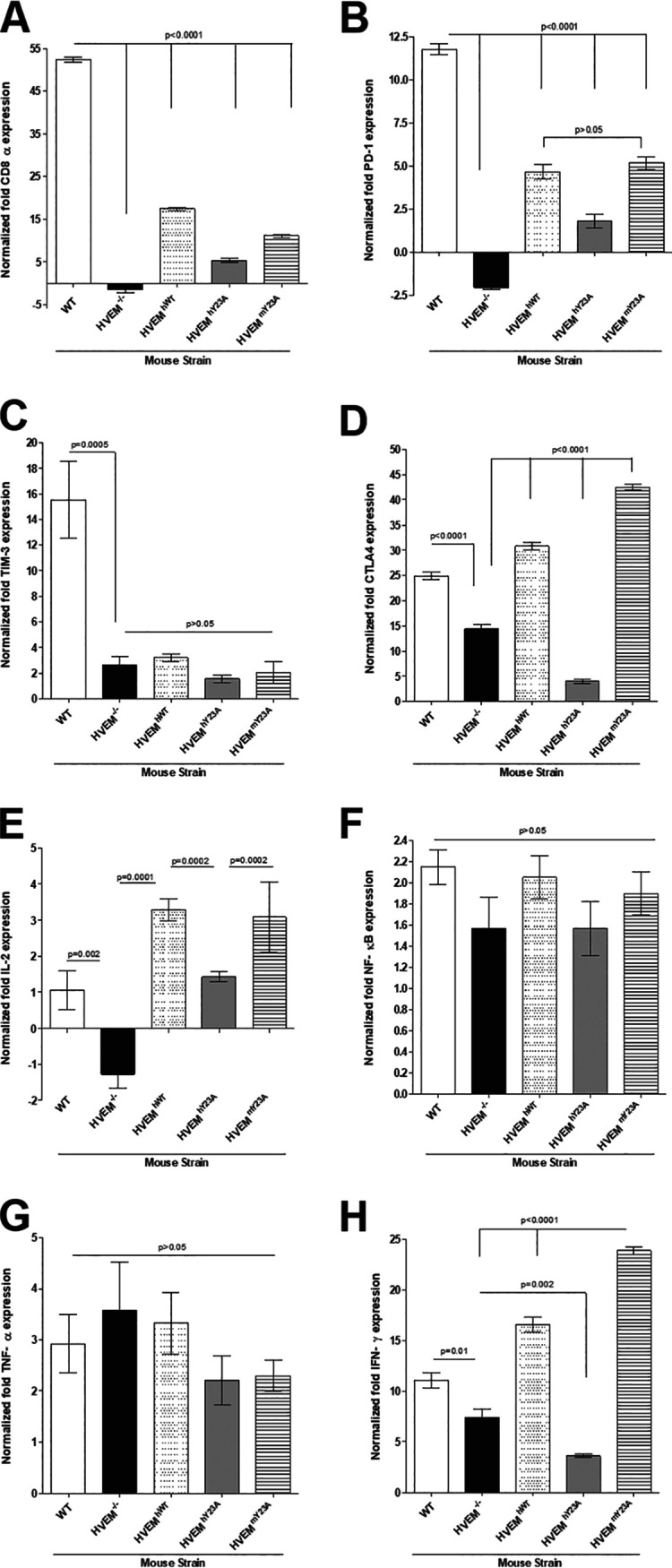
HVEM binding to gD is dispensable for expression of immune exhaustion markers and HVEM immunological activity. HVEM−/−, HVEMhWT, and HVEMhY23A mice were infected ocularly with 2 × 105 PFU/eye of HSV-1 strain McKrae. On day 28 p.i., mice were euthanized, individual TG were collected, and RNA was harvested. Levels of CD8 (A), PD1 (B), TIM-3 (C), CTLA4 (D), IL-2 (E), NF-κB (F), TNF-α (G), and IFN-γ (H) transcripts were measured by qRT-PCR, and fold differences were calculated by comparing transcript levels to those in their uninfected counterparts. GAPDH was used to normalize transcript expression in each sample. Results represent mean values for 8 TG from C57BL/6, 6 from HVEM−/−, 12 TG from HVEMhWT, 12 TG from HVEMhY23A, and 12 TG from HVEMmY23A mice (error bars indicate SEM).
Similar to CD8α expression, expression of PD1 was significantly lower in HVEM−/− mice than in WT mice (Fig. 4B, P < 0.0001), and its expression was partially rescued in all three transgenic mouse lines but not to the levels seen WT mice (Fig. 4B, P < 0.0001). Additionally, PD1 expression was lower in HVEMhY23A mice, but not HVEMmY23A mice, than in HVEMhWT mice (Fig. 4B, P < 0.0001 and P > 0.05, respectively). As we demonstrated previously (29, 40), TIM-3 expression was significantly lower in HVEM−/− mice than in WT mice (Fig. 4C, P = 0.0005). TIM-3 expression levels in HVEM−/− mice and in all three transgenic mouse lines were similar (Fig. 4C, P > 0.05), suggesting its expression is not rescued in any of the transgenic mice. CTLA4 expression was also lower in HVEM−/− mice than in WT mice (Fig. 4D, P < 0.0001), and it was rescued in HVEMhWT and HVEMmY23A mice (Fig. 4D, P < 0.0001). CTLA4 expression was lower in HVEMhY23A mice than in HVEM−/− mice (Fig. 4D, P < 0.0001).
Expression of IL-2, NF-κB, TNF-α, and IFN-γ were also measured by qRT-PCR in mice latently infected with HSV-1. IL-2 expression was significantly lower in HVEM−/− mice than in WT mice (Fig. 4E, P = 0.002) or the three transgenic mouse lines (Fig. 4E, P < 0.0001). In contrast, IL-2 expression was significantly higher in HVEMhWT and HVEMmY23A mice than in HVEMhY23A mice (Fig. 4E, P = 0.0002). No differences were seen in expression of NF-κB (Fig. 4F, P > 0.05) or TNF-α (Fig. 4G, P > 0.05) in WT, HVEM−/−, HVEMhWT, or HVEMmY23A mice. As we have shown before, IFN-γ expression was lower in HVEM−/− mice than in WT mice (Fig. 4H, P = 0.001), and IFN-γ expression was rescued in HVEMhWT and HVEMmY23A (Fig. 4H, P < 0.0001) mice but not in HVEMhY23A mice (Fig. 4H, P = 0.002).
Thus, compared with HVEM−/− mice, all tested HVEM immune functions were restored in HVEM transgenic mice. However, compared to WT mice, some, but not all, of these functions were restored. Our results also suggest that functional differences may exist between human and mouse HVEM.
Abolishing the gD binding site does not affect expression of HVEM ligands BTLA, CD160, and LIGHT.
Previously, we showed that HVEM, but not its ligands BTLA, CD160, or LIGHT, was upregulated during latent HSV-1 ocular infection in a LAT-dependent manner (40). We also demonstrated that efficient levels of latency, but not reactivation, depended on BTLA, CD160, and LIGHT (40). On the other hand, expression of LTα did not affect latency levels (41). To determine whether rescue of latency and reactivation in HVEMhWT, HVEMhY23A, or HVEMmY23A transgenic mice is due to differential effects on known HVEM binding partners in HVEM−/− and transgenic mice, expression of BTLA, CD160, and LIGHT was measured in the TG of mice latently infected with HSV-1 by qRT-PCR as described above. BTLA expression was higher in all five mouse strains than in their uninfected counterparts (Fig. 5A). BTLA expression levels were similar in the different mouse strains (Fig. 5A, P > 0.05). Similar to BTLA, CD160 expression was upregulated in all mouse strains, and no differences were observed between strains of mice (Fig. 5B, P > 0.05). LIGHT expression was upregulated in all mouse strains, but its upregulation in HVEMhY23A and HVEMmY23A mice was slightly less than that in WT, HVEM−/−, and HVEMhWT mice, although these differences did not reach statistical significance (Fig. 5C, P > 0.05). These results suggest that blocking gD binding to HVEM did not affect expression of the HVEM binding partners BTLA, CD160, and LIGHT.
FIG 5.

Absence of the gD binding site does not affect expression of the HVEM ligands BTLA (A), CD160 (B), and LIGHT (C). WT, HVEM−/−, HVEMhWT, HVEMhY23A, and HVEMmY23A mice were infected ocularly with 2 × 105 PFU/eye of HSV-1 strain McKrae as described in the legend to Fig. 4. On day 28 p.i., mice were euthanized, individual TG were collected, and RNA was harvested. Levels of BTLA, CD160, and LIGHT were measured by qRT-PCR, and the fold difference was calculated based on their transcript levels in uninfected counterparts. Bars represent six TG from each group, and error bars indicate SEM.
Overexpression of mouse, but not human, HVEM may exacerbate corneal scarring in the absence of binding to gD.
We previously found similar levels of CS in HVEM−/− and WT mice (29, 40), suggesting that the absence of HVEM signaling does not protect against CS. To determine the effect of HVEM overexpression and HVEM binding to gD on CS, WT, HVEM−/−, HVEMhWT, HVEMhY23A, and HVEMmY23A mice were infected as described above and scored in a blinded fashion on day 28 p.i. There were no significant differences in CS scores between WT, HVEM−/−, HVEMhWT, and HVEMhY23A mice (Fig. 6A and B, P > 0.05). However, CS scores were significantly higher in HVEMmY23A than in WT mice (Fig. 6A, P = 0.008). In agreement with CS scores, angiogenesis scores were similar in WT, HVEM−/−, HVEMhWT, and HVEMhY23A mice (Fig. 6B, P > 0.05) but higher in HVEMmY23A mice (Fig. 6B, P = 0.01), suggesting that overexpression of mouse, but not human, HVEM may play a role in increased pathology independently of gD binding to HVEM. This may be a reflection of the relatively low homology (45%) between human and mouse HVEM.
FIG 6.

Overexpression of mouse, but not human, HVEM may exacerbate corneal scarring independently of binding to gD. WT, HVEM−/−, HVEMhWT, HVEMhY23A, and HVEMmY23A mice were infected ocularly with 2 × 105 PFU of HSV-1 strain McKrae. On day 28 p.i., eye disease severity was assessed by split lamp biomicroscopy and scored in a blinded fashion. (A) CS was scored on a scale of 0 to 4 (0 = no disease, 1 = mild hazing, 2 = moderate opacity, 3 = severe corneal opacity but iris visible, 4 = opaque and corneal ulceration). (B) Angiogenesis was scored by quantifying new vessel growth in each of four corneal quadrants on a scale of 0 to 4. The scores of each quadrant were summed, and the mean ± SEM is shown. Results represent mean ± SEM from 16 WT, 12 HVEM−/−, 22 HVEMhWT, 22 HVEMhY23A, and 22 HVEMmY23A mouse eyes.
DISCUSSION
Corneal scarring and the consequent loss of visual acuity associated with HSV-1 ocular infections are generally attributed to host immune responses to recurrent HSV-1 infections (44, 45). On the other hand, latency levels correlate with reactivation (46–48), and we have shown that HVEM plays a critical role in latency and reactivation (29, 40). We showed that HSV-1 LAT upregulates HVEM during latency, and in the absence of HVEM or LAT, latency and reactivation are reduced. However, the mechanism of HVEM’s role in latency and reactivation is not well understood. Although both nectin-1 and HVEM are expressed in epithelial cells and neurons, HSV-1 preferentially uses nectin-1 for entry into those cells. For example, antibodies against nectin-1, but not HVEM, inhibit entry of HSV-1 into cultured neurons (16, 21, 49). Additionally, mutations in gD that inhibit its binding to nectin-1 prevent infection of neurons, while mutations that eliminate crucial HVEM binding sites in gD do not (50). As HVEM is not the primary HSV-1 receptor in vivo, and is not necessary for HSV-1 infectivity, it is possible that the interaction of HVEM with gD is more important in modulating the immune response than in entry. Because HVEM binds to gD, and we have shown that gD is expressed during latency (40), we asked whether HVEM-gD binding or HVEM immune activity plays a role in latency and reactivation.
To determine whether HVEM-gD binding or HVEM immune activity is responsible for efficient latency and reactivation, we established three transgenic mouse lines expressing either WT human HVEM or human or mouse mutant HVEM that cannot bind gD in the absence of endogenous HVEM. We validated the presence of the transgenes in tail cut DNA of mice by PCR and detected HVEM mRNA expression in the TG, corneas, and brain of HVEMhWT, HVEMhY23A, and HVEMmY23A mice. We also showed the presence of HVEM protein in the TG, corneas, and brain of HVEMhWT and HVEMhY23A mice.
The immunomodulatory activities of HVEM are affected by its binding partners. Signaling via HVEM can either activate or inhibit T cells, depending on which ligand is engaged. For example, binding of BTLA or CD160 to the cysteine-rich domain 1 (CRD1) of HVEM represses T cell activation. On the other hand, binding of LIGHT and LTα to the CRD2 and CRD3 domains of HVEM serve as T cell activating signals. HSV-1 gD competes with binding of host HVEM ligands by binding to the CRD1 domain of HVEM, mimicking BTLA binding. Crystal structures of HVEM bound to its ligands have been solved, and subsequent single-amino-acid mutagenesis assays have identified three amino acids in the HVEM CRD1 domain that are critical for binding to gD: G22, Y23, and C37 (51). Previous work from the Cohen group established that the Y23A mutation inhibits gD binding to HVEM but does not interfere with HVEM localization to the cell surface in vitro (51). HVEM host ligand BTLA also binds to the CRD1 domain via amino acid Y23, and the HVEM Y23A mutant fails to bind BTLA in vitro (36). We have seen reduced latency but no delayed reactivation in BTLA, CD160, and LIGHT knockout (KO) mice and no effect on latency in Ltα KO mice (40, 41).
Based on these results, we predicted that the absence of one of the host HVEM ligands would reduce competition for gD binding to HVEM, thereby reducing latency. However, the absence of differences in latency or reactivation in mice expressing WT or Y23A mutant-containing HVEM suggests that HVEM immune function, not gD binding, is critical for these processes. Also, as we did not detect any effects on expression of the HVEM ligands BTLA, CD160, or LIGHT, it is possible that BTLA can bind the HVEM Y23A mutant in vivo. Alternatively, the absence of gD binding combined with a reduction in or absence of BTLA binding might change signaling dynamics, especially considering the complex interactions HVEM can support. For example, LIGHT, which binds to the CRD2 and CRD3 domains of HVEM and is therefore not likely to be affected by the Y23A mutation, competes with gD for binding to HVEM. Some evidence suggests that HVEM can bind BTLA and LIGHT simultaneously, although the effects on T cell activation are not clear (52). It is also possible that HVEM recognizes additional viral ligands. Previously, we have shown that overexpression of HSV-1 gK resulted in altered expression of 3-OS-HS, PILR-α, nectin-1, and nectin-2 in the TG of C57BL/6 mice during latency (53). Alternatively, we cannot rule out the possibility that in vivo, HSV-1 may have adapted to the loss of the Y23 residue, a possibility that will be addressed in ongoing work.
HVEM is known to play a role in apoptosis by activating prosurvival signaling via NF-κB activation upon binding to either BTLA, CD160, LIGHT, or gD (42, 43). Studies in cells treated with monoclonal antibodies that block the HVEM-gD interaction suggest that the HVEM-gD interaction is necessary for NF-κB-mediated apoptosis and reduced viral yield (42, 43). Our results contradict this finding, as we did not see reduced virus yield in the absence of HVEM-gD binding. We showed that NF-κB expression was elevated in the TG of WT, HVEM−/−, HVEMhWT, HVEMhY23A, and HVEMmY23A mice during latent ocular infection with HSV-1, and there were no differences between the different strains of mice (Fig. 4F), suggesting that HVEM-gD binding is not necessary for NF-κB activation during latent HSV-1 ocular infection in mice. Differences between our findings and those of Sciortino et al. (42) may be due to the previous studies being done in vitro and the use of an antibody that could have off-target effects such as blocking the binding of other HVEM ligands.
Our results suggest that expression of CD8α and the immune exhaustion marker PD1 are largely restored in HVEM transgenic mice. HVEM binding by LIGHT or LTα results in T cell activation and proliferation and secretion of immune cytokines such as IFN-γ (22, 52, 54). Latent HSV-1 infections lead to CD8+ T cell exhaustion, characterized by overexpression of the immune exhaustion markers PD1, TIM-3, and CTLA4 (46, 55). We and others have reported a correlation between increased latency and exhaustion (46, 56, 57). Our finding that CD8α and PD-1 expression was rescued in all transgenic mice indicate that the transgenes can replace HVEM function in these mice independently of HVEM binding to gD. Elevated exhaustion markers also correlated with restored latency and reactivation, consistent with the literature (46, 57) and our previous reports establishing a crucial role for CD8α+ DC cells in driving latency (58). Increased CD8α and PD1 levels were independent of viral load, because we did not observe any differences in LAT copy number between the different mouse groups. While PD1 levels were restored, at least partially, in all three transgenic mouse groups regardless of the ability to bind gD, TIM-3 expression was not rescued in any of the three transgenic mouse groups, possibly due to elevated HVEM expression in the transgenic mice. Conversely, CTLA4 expression was rescued in HVEMhWT and HVEMmY23A, but not in HVEMhY23A, mice. We also observed an increase in eye disease in HVEMmY23A mice in comparison to HVEMhY23A mice, suggesting possible differences in the use of mouse and human HVEM, which could be due to the relatively low homology (∼45%) between the two proteins.
HVEM activation is known to activate expression of inflammatory cytokines (22). For example, IFN-γ, TNF-α, and IL-2 are upregulated in response to betaherpesvirus cytomegalovirus (CMV) infection (31). We also recently showed that HVEM mediated the upregulation of IFN-γ, IL-2, and IL-21, but not TNF-α, during latent ocular infection with HSV-1 (40). In the current study, we observed rescue of IFN-γ expression in HVEMhWT and HVEMmY23A mice but not in HVEMhY23A mice, suggesting that the HVEM-gD interaction may be necessary to induce IFN-γ expression by human but not mouse HVEM. Our finding that IL-2 expression was rescued in our transgenic mice is consistent with restoring HVEM immune function and with findings from Edwards et al. (59), who used a virus incapable of binding HVEM to show that upregulation of several cytokines, including IL-2, is independent of the HVEM-gD interaction. IL-2 expression exceeded that in WT C57BL/6 mice, likely due to higher HVEM expression in transgenic mice. We did not see differences in TNF-α expression in the five mouse strains. These results are consistent with our previous report that showed no difference in TNF-α expression between WT and HVEM−/− mice during latency (40).
In summary, restoring HVEM immune function restores all effects of HVEM absence in the context of HSV-1 latency and reactivation. Interestingly, our findings also suggest that there may be differences in the function of mouse and human HVEM depending on the usage of the HVEM-gD binding site.
MATERIALS AND METHODS
Ethics statement.
All animal procedures were performed in strict accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and the NIH Guide for the Care and Use of Laboratory Animals (60). The animal research protocol was approved by the Institutional Animal Care and Use Committee of Cedars-Sinai Medical Center (protocol numbers 5030 and 8837).
Virus and cells.
Plaque-purified virulent HSV-1 strain McKrae was used to infect mice as described previously (48, 61). Rabbit skin (RS) cells were used to prepare virus stocks, culture mouse tear swabs, and determine growth kinetics. RS cells were grown in Eagle’s minimal essential medium (MEM) supplemented with 5% fetal bovine serum (FBS).
Construction of transgenic mice.
Wild-type human HVEM (HVEMhWT) (GenBank accession number [Gene ID] 8764), human HVEM with a Y-to-A mutation in amino acid (aa) 23 (HVEMY23A), and mouse HVEM with a Y-to-A mutation in aa 23 (HVEMY23A) were generous gifts from Gary Cohen (University of Pennsylvania) (51). Transgenic constructs were introduced into fertilized eggs harvested from HVEM knockout or wild-type C57BL/6J mice by pronuclear microinjection, followed by embryo transfer as described previously (62). Briefly, each construct was size-fractionated in low-gelling-temperature agarose (NuSieve GTG; Lonza, Basel Switzerland), purified by phenol-chloroform extraction, ethanol precipitated, resuspended in injection buffer (0.1 mM EDTA, 10 mM Tris-HCl, 100 mM NaCl) at a 2-ng/μl concentration, and used for injection. The transgenic status of the founders and subsequent progeny was confirmed by PCR genotyping on toe or ear tissue samples using a KAPA genotyping kit (KK7352; KAPA Biosystems, Wilmington, MA). The presence of HVEMhWT and HVEMhY23A transgenes was determined by PCR using the primer sequences Fwd, 5′-CCACCCCCAGAACCGACGTC-3′, and Rev, 5′-CGGCACGCGGCGCAGTGGTC-3′, producing a band migrating at 357 bp on a 1% agarose gel. The presence of the HVEMY23A transgene was determined using primer sequences Fwd, 5′-GCAACCCAGGTGCCCATGTG-3′, and Rev, 5′-CGACCTGGGAGGAGCAGGTGG-3′, producing a band migrating at 453 bp on a 1% agarose gel.
After initial characterization of each transgenic line, the three transgenic lines were established, expanded, and used for subsequent experiments after four generations of screening. The three transgenic mouse lines expressing WT human HVEM, mutated human HVEM, and mutated mouse HVEM were designated HVEMhWT, HVEMhY23A, and HVEMmY23A, respectively. Both male and female mice, 6 to 8 weeks old, were used in these studies. All mice are in a C57BL/6 background, and WT C57BL/6 and HVEM−/− mice were used as controls.
Ocular infection and virus titration in tear films.
Mice were infected with 2 × 105 PFU per eye of HSV-1 strain McKrae in 2.8 μl tissue culture medium (5% FBS–MEM) as an eye drop without corneal scarification. Tears were collected on days 1 to 5 p.i. using cotton swabs (63, 64). Individual swabs were placed in 1 ml tissue culture medium. Viral titers were determined using a standard plaque assay on RS cells.
Ex vivo explant reactivation assay.
Individual TG were removed from mice on day 28 p.i., placed in tissue culture medium, and cultured in a humidified 37°C incubator supplemented with 5% CO2 (65, 66). Aliquots of culture medium from the explants were transferred onto indicator RS cells daily for 15 days, and the cells were monitored for virus reactivation. Because the medium from explanted TG cultures was plated daily, the time at which reactivated virus first appeared in the explanted TG cultures could be determined.
Western blotting.
Tissues from the corneas, TG, and brain were harvested from naive mice and snap-frozen on dry ice. Tissue samples were homogenized in radioimmunoprecipitation (RIPA) buffer (ThermoFisher, Waltham, MA) supplemented with protease inhibitors (Roche Life Sciences, Penzberg, Germany) using TissueLyser LT (Qiagen, Hilden, Germany). Lysates were clarified by centrifugation, and proteins were separated by SDS-PAGE, followed by transfer to a nitrocellulose membrane. The membrane was blocked using 5% nonfat dry milk in phosphate-buffered saline with Tween 20 (PBST) and incubated with primary antibodies (rabbit anti-TNFRSF14; Ab47677; Abcam, Cambridge, United Kingdom), rabbit anti-β-actin (4967S; Cell Signaling, Danvers, MA), followed by horseradish peroxidase (HRP)-conjugated secondary antibodies (Invitrogen, Carlsbad, CA). Images were acquired using FluorChem E (Protein Simple, San Jose, CA).
RNA extraction, cDNA synthesis, and TaqMan RT-PCR.
Infected and mock-infected mice were euthanized 28 days p.i. Individual TG were harvested, suspended in TRIzol (Qiagen, Hilden, Germany), and stored at –80°C until processed. Individual TG were homogenized using a TissueLyzer LT (Qiagen, Hilden, Germany), and RNA extraction was done by chloroform (Ambion, Austin, TX) extraction, followed by purification using RNeasy columns (Qiagen, Hilden, Germany) as we have described previously (67, 68). RNA (500 ng/sample) was reverse transcribed using random hexamer primers and murine leukemia virus (MuLV) reverse transcriptase from a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA), according to the manufacturer’s recommendations. Expression levels of various RNAs were determined using TaqMan gene expression assays (Applied Biosystems, Foster City, CA), as follows: (i) CD8α Mm01182108_m1; amplicon size, 67 bp; (ii) PD-1 Mm01285676_m1; amplicon size, 64 bp; (iii) TIM-3 Mm00454540_m1; amplicon size, 98 bp; (iv) CTLA4 Mm00486849_m1; amplicon size, 71 bp; (v) BTLA Mm00616981_m1; amplicon size, 71 bp; (vi) CD160 Mm00444461_m1; amplicon size, 61 bp; (vii) LIGHT Mm00444567_m1; amplicon size, 68 bp; (viii) GAPDH Mm99999915_g1; amplicon size, 107 bp; (ix) hHVEM Hs00187058_m; amplicon size, 76 bp; (x) mHVEM Mm00619239_m1; amplicon size, 65 bp; (xi) TNF-α Mm00443258_m1; amplicon size, 81 bp; (xii) IFN-γ Mm00801778_m1; amplicon size, 101 bp; (xiii) IL-2 Mm00434256_m1; amplicon size, 82 bp. Primer-probe sets consisted of two unlabeled PCR primers and the 6-carboxyfluorescein (FAM) dye-labeled TaqMan MGB probe in a single mixture. All amplicons included an intron-exon junction to eliminate signal from genomic DNA contamination. Latency levels were determined by measuring HSV-1 LAT expression using custom-made TaqMan gene expression assays (Applied Biosystems, Foster City, CA) with the following components in a single mixture: forward primer, 5′-GGGTGGGCTCGTGTTACAG-3′; reverse primer, 5′-GGACGGGTAAGTAACAGAGTCTCTA-3′; and probe, 5′-FAM-ACACCAGCCCGTTCTTT-3′. The LAT amplicon length was 81 bp and corresponds to LAT nucleotides 119553 to 119634. Relative LAT copy numbers were calculated using standard curves generated from the plasmid pGem-LAT5317.
Monitoring corneal scarring and angiogenesis.
Infected mice were assessed for the presence of eye disease in a blinded fashion. CS was scored on a scale of 0 to 4 (0 = no disease, 1 = mild hazing, 2 = moderate opacity, 3 = severe corneal opacity but iris visible, 4 = opaque and corneal ulceration). The angiogenesis score was determined by assessing for the presence of new vessel growth in each of the four corneal quadrants on a scale from 0 to 4. The scores of each quadrant were summed, and the mean ± standard error of the mean (SEM) are shown (69).
Statistical analysis.
Student's t test and chi-square tests were performed using the computer program Instat (GraphPad, San Diego, CA). Results were considered statistically significant when the P value was <0.05.
ACKNOWLEDGMENTS
This study was supported by Public Health Service NIH grant numbers RO1 EY029160 and RO1 EY024649.
REFERENCES
- 1.Liesegang TJ. 1999. Classification of herpes simplex virus keratitis and anterior uveitis. Cornea 18:127–143. doi: 10.1097/00003226-199903000-00001. [DOI] [PubMed] [Google Scholar]
- 2.Liesegang TJ. 2001. Herpes simplex virus epidemiology and ocular importance. Cornea 20:1–13. doi: 10.1097/00003226-200101000-00001. [DOI] [PubMed] [Google Scholar]
- 3.Liesegang TJ, Melton LJ III, Daly PJ, Ilstrup DM. 1989. Epidemiology of ocular herpes simplex. Incidence in Rochester, Minn, 1950 through 1982. Arch Ophthalmol 107:1155–1159. doi: 10.1001/archopht.1989.01070020221029. [DOI] [PubMed] [Google Scholar]
- 4.Perng GC, Jones C. 2010. Towards an understanding of the herpes simplex virus type 1 latency-reactivation cycle. Interdiscip Perspect Infect Dis 2010:262415. doi: 10.1155/2010/262415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Koujah L, Suryawanshi RK, Shukla D. 2019. Pathological processes activated by herpes simplex virus-1 (HSV-1) infection in the cornea. Cell Mol Life Sci 76:405–419. doi: 10.1007/s00018-018-2938-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Binder PS. 1984. A review of the treatment of ocular herpes simplex infections in the neonate and immunocompromised host. Cornea 3:178–182. [PubMed] [Google Scholar]
- 7.Young RC, Hodge DO, Liesegang TJ, Baratz KH. 2010. Incidence, recurrence, and outcomes of herpes simplex virus eye disease in Olmsted County, Minnesota, 1976-2007: the effect of oral antiviral prophylaxis. Arch Ophthalmol 128:1178–1183. doi: 10.1001/archophthalmol.2010.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hadigal S, Shukla D. 2013. Exploiting herpes simplex virus entry for novel therapeutics. Viruses 5:1447–1465. doi: 10.3390/v5061447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Satoh T, Arii J, Suenaga T, Wang J, Kogure A, Uehori J, Arase N, Shiratori I, Tanaka S, Kawaguchi Y, Spear PG, Lanier LL, Arase H. 2008. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 132:935–944. doi: 10.1016/j.cell.2008.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Spear PG, Eisenberg RJ, Cohen GH. 2000. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 275:1–8. doi: 10.1006/viro.2000.0529. [DOI] [PubMed] [Google Scholar]
- 11.Taylor JM, Lin E, Susmarski N, Yoon M, Zago A, Ware CF, Pfeffer K, Miyoshi J, Takai Y, Spear PG. 2007. Alternative entry receptors for herpes simplex virus and their roles in disease. Cell Host Microbe 2:19–28. doi: 10.1016/j.chom.2007.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862. doi: 10.1038/nature09420. [DOI] [PubMed] [Google Scholar]
- 13.Suenaga T, Satoh T, Somboonthum P, Kawaguchi Y, Mori Y, Arase H. 2010. Myelin-associated glycoprotein mediates membrane fusion and entry of neurotropic herpesviruses. Proc Natl Acad Sci U S A 107:866–871. doi: 10.1073/pnas.0913351107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Laquerre S, Argnani R, Anderson DB, Zucchini S, Manservigi R, Glorioso JC. 1998. Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins B and C, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J Virol 72:6119–6130. doi: 10.1128/JVI.72.7.6119-6130.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. 1999. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell 99:13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- 16.Cocchi F, Lopez M, Menotti L, Aoubala M, Dubreuil P, Campadelli-Fiume G. 1998. The V domain of herpesvirus Ig-like receptor (HIgR) contains a major functional region in herpes simplex virus-1 entry into cells and interacts physically with the viral glycoprotein D. Proc Natl Acad Sci U S A 95:15700–15705. doi: 10.1073/pnas.95.26.15700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- 18.Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. 1998. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology 246:179–189. doi: 10.1006/viro.1998.9218. [DOI] [PubMed] [Google Scholar]
- 19.Connolly SA, Landsburg DJ, Carfi A, Wiley DC, Cohen GH, Eisenberg RJ. 2003. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD-HveA/HVEM binding interface. J Virol 77:8127–8140. doi: 10.1128/jvi.77.14.8127-8140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoon M, Zago A, Shukla D, Spear PG. 2003. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J Virol 77:9221–9231. doi: 10.1128/jvi.77.17.9221-9231.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436. doi: 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- 22.Edwards RG, Longnecker R. 2017. Herpesvirus entry mediator and ocular herpesvirus infection: more than meets the eye. J Virol 91:e00115-17. doi: 10.1128/JVI.00115-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cheung TC, Humphreys IR, Potter KG, Norris PS, Shumway HM, Tran BR, Patterson G, Jean-Jacques R, Yoon M, Spear PG, Murphy KM, Lurain NS, Benedict CA, Ware CF. 2005. Evolutionarily divergent herpesviruses modulate T cell activation by targeting the herpesvirus entry mediator cosignaling pathway. Proc Natl Acad Sci U S A 102:13218–13223. doi: 10.1073/pnas.0506172102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.O'Donnell CD, Kovacs M, Akhtar J, Valyi-Nagy T, Shukla D. 2010. Expanding the role of 3-O sulfated heparan sulfate in herpes simplex virus type-1 entry. Virology 397:389–398. doi: 10.1016/j.virol.2009.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ware CF. 2011. The TNF receptor super family in immune regulation. Immunol Rev 244:5–8. doi: 10.1111/j.1600-065X.2011.01065.x. [DOI] [PubMed] [Google Scholar]
- 26.Murphy KM, Nelson CA, Sedy JR. 2006. Balancing co-stimulation and inhibition with BTLA and HVEM. Nat Rev Immunol 6:671–681. doi: 10.1038/nri1917. [DOI] [PubMed] [Google Scholar]
- 27.Shui JW, Kronenberg M. 2013. HVEM: an unusual TNF receptor family member important for mucosal innate immune responses to microbes. Gut Microbes 4:146–151. doi: 10.4161/gmic.23443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kopp SJ, Karaba AH, Cohen LK, Banisadr G, Miller RJ, Muller WJ. 2013. Pathogenesis of neonatal herpes simplex 2 disease in a mouse model is dependent on entry receptor expression and route of inoculation. J Virol 87:474–481. doi: 10.1128/JVI.01849-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allen SJ, Rhode-Kurnow A, Mott KR, Jiang X, Carpenter D, Rodriguez-Barbosa JI, Jones C, Wechsler SL, Ware CF, Ghiasi H. 2014. Regulatory interactions between herpesvirus entry mediator (TNFRSF14) and latency-associated transcript (LAT) during HSV-1 latency. J Virol 88:1961–1971. doi: 10.1128/JVI.02467-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mauri DN, Ebner R, Montgomery RI, Kochel KD, Cheung TC, Yu GL, Ruben S, Murphy M, Eisenberg RJ, Cohen GH, Spear PG, Ware CF. 1998. LIGHT, a new member of the TNF superfamily, and lymphotoxin alpha are ligands for herpesvirus entry mediator. Immunity 8:21–30. doi: 10.1016/s1074-7613(00)80455-0. [DOI] [PubMed] [Google Scholar]
- 31.Sedy JR, Gavrieli M, Potter KG, Hurchla MA, Lindsley RC, Hildner K, Scheu S, Pfeffer K, Ware CF, Murphy TL, Murphy KM. 2005. B and T lymphocyte attenuator regulates T cell activation through interaction with herpesvirus entry mediator. Nat Immunol 6:90–98. doi: 10.1038/ni1144. [DOI] [PubMed] [Google Scholar]
- 32.Cai G, Anumanthan A, Brown JA, Greenfield EA, Zhu B, Freeman GJ. 2008. CD160 inhibits activation of human CD4+ T cells through interaction with herpesvirus entry mediator. Nat Immunol 9:176–185. doi: 10.1038/ni1554. [DOI] [PubMed] [Google Scholar]
- 33.Wan X, Zhang J, Luo H, Shi G, Kapnik E, Kim S, Kanakaraj P, Wu J. 2002. A TNF family member LIGHT transduces costimulatory signals into human T cells. J Immunol 169:6813–6821. doi: 10.4049/jimmunol.169.12.6813. [DOI] [PubMed] [Google Scholar]
- 34.Tamada K, Chen L. 2000. T lymphocyte costimulatory molecules in host defense and immunologic diseases. Ann Allergy Asthma Immunol 85:164–175. doi: 10.1016/S1081-1206(10)62462-3. [DOI] [PubMed] [Google Scholar]
- 35.del Rio ML, Lucas CL, Buhler L, Rayat G, Rodriguez-Barbosa JI. 2010. HVEM/LIGHT/BTLA/CD160 cosignaling pathways as targets for immune regulation. J Leukoc Biol 87:223–235. doi: 10.1189/jlb.0809590. [DOI] [PubMed] [Google Scholar]
- 36.Compaan DM, Gonzalez LC, Tom I, Loyet KM, Eaton D, Hymowitz SG. 2005. Attenuating lymphocyte activity: the crystal structure of the BTLA-HVEM complex. J Biol Chem 280:39553–39561. doi: 10.1074/jbc.M507629200. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe N, Gavrieli M, Sedy JR, Yang J, Fallarino F, Loftin SK, Hurchla MA, Zimmerman N, Sim J, Zang X, Murphy TL, Russell JH, Allison JP, Murphy KM. 2003. BTLA is a lymphocyte inhibitory receptor with similarities to CTLA-4 and PD-1. Nat Immunol 4:670–679. doi: 10.1038/ni944. [DOI] [PubMed] [Google Scholar]
- 38.De Trez C, Schneider K, Potter K, Droin N, Fulton J, Norris PS, Ha SW, Fu YX, Murphy T, Murphy KM, Pfeffer K, Benedict CA, Ware CF. 2008. The inhibitory HVEM-BTLA pathway counter regulates lymphotoxin receptor signaling to achieve homeostasis of dendritic cells. J Immunol 180:238–248. doi: 10.4049/jimmunol.180.1.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stiles KM, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ, Krummenacher C. 2010. Herpes simplex virus glycoprotein D interferes with binding of herpesvirus entry mediator to its ligands through downregulation and direct competition. J Virol 84:11646–11660. doi: 10.1128/JVI.01550-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang S, Ljubimov AV, Jin L, Pfeffer K, Kronenberg M, Ghiasi H. 2018. Herpes simplex virus 1 latency and the kinetics of reactivation are regulated by a complex network of interactions between the herpesvirus entry mediator, its ligands (gD, BTLA, LIGHT, and CD160), and the latency-associated transcript. J Virol 92:e01451-18. doi: 10.1128/JVI.01451-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang S, Hirose S, Ghiasi H. 2019. The absence of lymphotoxin-alpha, an HVEM ligand, affects HSV-1 infection in vivo differently than the absence of other HVEM cellular ligands. J Virol 93:e00707-19. doi: 10.1128/JVI.00707-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sciortino MT, Medici MA, Marino-Merlo F, Zaccaria D, Giuffre-Cuculletto M, Venuti A, Grelli S, Bramanti P, Mastino A. 2008. Involvement of gD/HVEM interaction in NF-kB-dependent inhibition of apoptosis by HSV-1 gD. Biochem Pharmacol 76:1522–1532. doi: 10.1016/j.bcp.2008.07.030. [DOI] [PubMed] [Google Scholar]
- 43.Patel A, Hanson J, McLean TI, Olgiate J, Hilton M, Miller WE, Bachenheimer SL. 1998. Herpes simplex type 1 induction of persistent NF-kappa B nuclear translocation increases the efficiency of virus replication. Virology 247:212–222. doi: 10.1006/viro.1998.9243. [DOI] [PubMed] [Google Scholar]
- 44.Jaggi U, Wang S, Tormanen K, Matundan H, Ljubimov AV, Ghiasi H. 2018. Role of herpes simplex virus type 1 (HSV-1) glycoprotein K (gK) pathogenic CD8(+) T cells in exacerbation of eye disease. Front Immunol 9:2895. doi: 10.3389/fimmu.2018.02895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rajasagi NK, Rouse BT. 2019. The role of T cells in herpes stromal keratitis. Front Immunol 10:512. doi: 10.3389/fimmu.2019.00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Allen SJ, Hamrah P, Gate DM, Mott KR, Mantopoulos D, Zheng L, Town T, Jones C, von Andrian UH, Freeman GJ, Sharpe AH, Benmohamed L, Ahmed R, Wechsler SL, Ghiasi H. 2011. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus type 1. J Virol 85:4184–4197. doi: 10.1128/JVI.02290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matundan HH, Mott KR, Allen SJ, Wang S, Bresee CJ, Ghiasi YN, Town T, Wechsler SL, Ghiasi H. 2016. Interrelationship of primary virus replication, level of latency, and time to reactivation in the trigeminal ganglia of latently infected mice. J Virol 90:9533–9542. doi: 10.1128/JVI.01373-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol 68:8045–8055. doi: 10.1128/JVI.68.12.8045-8055.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simpson SA, Manchak MD, Hager EJ, Krummenacher C, Whitbeck JC, Levin MJ, Freed CR, Wilcox CL, Cohen GH, Eisenberg RJ, Pizer LI. 2005. Nectin-1/HveC mediates herpes simplex virus type 1 entry into primary human sensory neurons and fibroblasts. J Neurovirol 11:208–218. doi: 10.1080/13550280590924214. [DOI] [PubMed] [Google Scholar]
- 50.Manoj S, Jogger CR, Myscofski D, Yoon M, Spear PG. 2004. Mutations in herpes simplex virus glycoprotein D that prevent cell entry via nectins and alter cell tropism. Proc Natl Acad Sci U S A 101:12414–12421. doi: 10.1073/pnas.0404211101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Connolly SA, Landsburg DJ, Carfi A, Wiley DC, Eisenberg RJ, Cohen GH. 2002. Structure-based analysis of the herpes simplex virus glycoprotein D binding site present on herpesvirus entry mediator HveA (HVEM). J Virol 76:10894–10904. doi: 10.1128/jvi.76.21.10894-10904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cai G, Freeman GJ. 2009. The CD160, BTLA, LIGHT/HVEM pathway: a bidirectional switch regulating T-cell activation. Immunol Rev 229:244–258. doi: 10.1111/j.1600-065X.2009.00783.x. [DOI] [PubMed] [Google Scholar]
- 53.Allen SJ, Mott KR, Ghiasi H. 2014. Overexpression of herpes simplex virus glycoprotein K (gK) alters expression of HSV receptors in ocularly-infected mice. Invest Ophthalmol Vis Sci 55:2442–2451. doi: 10.1167/iovs.14-14013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Cohavy O, Zhou J, Granger SW, Ware CF, Targan SR. 2004. LIGHT expression by mucosal T cells may regulate IFN-gamma expression in the intestine. J Immunol 173:251–258. doi: 10.4049/jimmunol.173.1.251. [DOI] [PubMed] [Google Scholar]
- 55.Wherry EJ, Kurachi M. 2015. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol 15:486–499. doi: 10.1038/nri3862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mott KR, Bresee CJ, Allen SJ, BenMohamed L, Wechsler SL, Ghiasi H. 2009. Level of herpes simplex virus type 1 latency correlates with severity of corneal scarring and exhaustion of CD8+ T cells in trigeminal ganglia of latently infected mice. J Virol 83:2246–2254. doi: 10.1128/JVI.02234-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chentoufi AA, Kritzer E, Tran MV, Dasgupta G, Lim CH, Yu DC, Afifi RE, Jiang X, Carpenter D, Osorio N, Hsiang C, Nesburn AB, Wechsler SL, BenMohamed L. 2011. The herpes simplex virus 1 latency-associated transcript promotes functional exhaustion of virus-specific CD8+ T cells in latently infected trigeminal ganglia: a novel immune evasion mechanism. J Virol 85:9127–9138. doi: 10.1128/JVI.00587-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mott KR, Allen SJ, Zandian M, Konda B, Sharifi BG, Jones C, Wechsler SL, Town T, Ghiasi H. 2014. CD8a dendritic cells drive establishment of HSV-1 latency. PLoS One 9:e93444. doi: 10.1371/journal.pone.0093444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edwards RG, Kopp SJ, Karaba AH, Wilcox DR, Longnecker R. 2015. Herpesvirus entry mediator on radiation-resistant cell lineages promotes ocular herpes simplex virus 1 pathogenesis in an entry-independent manner. mBio 6:e01532-15. doi: 10.1128/mBio.01532-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.National Research Council. 1996. Guide for the care and use of laboratory animals. National Academies Press, Washington, DC. [Google Scholar]
- 61.Osorio Y, Ghiasi H. 2003. Comparison of adjuvant efficacy of herpes simplex virus type 1 recombinant viruses expressing TH1 and TH2 cytokine genes. J Virol 77:5774–5783. doi: 10.1128/jvi.77.10.5774-5783.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reilly MP, McCune SL, Ryan TM, Townes TM, Katsumata M, Asakura T. 1994. Preparation of recombinant hemoglobin in transgenic mice. Methods Enzymol 231:403–434. doi: 10.1016/0076-6879(94)31028-9. [DOI] [PubMed] [Google Scholar]
- 63.Ghiasi H, Bahri S, Nesburn AB, Wechsler SL. 1995. Protection against herpes simplex virus-induced eye disease after vaccination with seven individually expressed herpes simplex virus 1 glycoproteins. Invest Ophthalmol Vis Sci 36:1352–1360. [PubMed] [Google Scholar]
- 64.Ghiasi H, Wechsler SL, Kaiwar R, Nesburn AB, Hofman FM. 1995. Local expression of tumor necrosis factor alpha and interleukin-2 correlates with protection against corneal scarring after ocular challenge of vaccinated mice with herpes simplex virus type 1. J Virol 69:334–340. doi: 10.1128/JVI.69.1.334-340.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mott KR, Ghiasi H. 2008. Role of dendritic cells in enhancement of herpes simplex virus type 1 latency and reactivation in vaccinated mice. Clin Vaccine Immunol 15:1859–1867. doi: 10.1128/CVI.00318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mott KR, UnderHill D, Wechsler SL, Ghiasi H. 2008. Lymphoid-related CD11c+ CD8a+ dendritic cells are involved in enhancing herpes simplex virus type 1 latency. J Virol 82:9870–9879. doi: 10.1128/JVI.00566-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mott KR, Perng GC, Osorio Y, Kousoulas KG, Ghiasi H. 2007. A recombinant herpes simplex virus type 1 expressing two additional copies of gK is more pathogenic than wild-type virus in two different strains of mice. J Virol 81:12962–12972. doi: 10.1128/JVI.01442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Tormanen K, Allen S, Mott KR, Ghiasi H. 2019. The latency-associated transcript inhibits apoptosis via downregulation of components of the type I interferon pathway during latent herpes simplex virus 1 ocular infection. J Virol 93:e00103-19. doi: 10.1128/JVI.00103-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jaggi U, Varanasi SK, Bhela S, Rouse BT. 2018. On the role of retinoic acid in virus induced inflammatory response in cornea. Microbes Infect 20:337–345. doi: 10.1016/j.micinf.2018.04.007. [DOI] [PubMed] [Google Scholar]