

Abstract
Freshwater polyps of the genus Hydra do not age. However, temperature stress induces aging and a shift from reproduction by asexual budding to sexual gamete production in a cold-sensitive (CS) strain of H. oligactis. We sequenced the transcriptome of a male CS strain before and after this life history shift and compared changes in gene expression relative to those seen in a cold-resistant (CR) strain that does not undergo a life history shift in response to altered temperature. We found that the switch from non-aging asexual reproduction to aging and sexual reproduction involves upregulation of genes not only involved in gametogenesis but also genes involved in cellular senescence, apoptosis, and DNA repair accompanied by a downregulation of genes involved in stem cell maintenance. These results suggest that aging is a byproduct of sexual reproduction-associated cellular reprogramming and underscore the power of these H. oligactis strains to identify intrinsic mechanisms of aging.
Electronic supplementary material
The online version of this article (10.1007/s11357-020-00214-z) contains supplementary material, which is available to authorized users.
Keywords: Hydra oligactis, Transcriptome, Cold-sensitive, Gametogenesis, Aging, DNA repair
Introduction
Age-related degeneration and death are universal processes affecting virtually all metazoans. Yet, among the simplest of animals, most notably cnidarians, cases of immortality have been described. For example, individuals of the freshwater cnidarian, Hydra vulgaris, have been monitored for years and showed no signs of increased age-related mortality or any decrease in reproductive rate. Immortality among Hydra species has been ascribed to the continuous renewal of somatic cells from stem cells (Martinez 1998). However, one strain of Hydra oligactis undergoes aging under certain environmental conditions. Specifically, when transferred from the standard maintenance temperature of 18 to 10 °C this strain of H. oligactis showed multiple symptoms of aging, including Gompertzian increasing mortality, decreasing reproductive rate, and somatic degeneration and functional decline in prey capture and spontaneous movements (Yoshida et al. 2006). The correlation of aging with sexual reproduction has also been reported for other cnidarians and even for unicellular protozoa (Petralia et al. 2014). It is possible that an increased production of germ cells leads to a compensatory decline in the production of differentiated somatic cells from the same stem cell pool.
Sexual reproduction appears to often be related to environmental stress. Indeed, under benign environmental conditions, Hydra species mainly reproduce by asexual budding and do not undergo detectible gametogenesis (Martinez 1998; Schaible et al. 2015). Sexual reproduction, as defined by increased production of sperm and/or eggs from interstitial stem cells, can be induced in Hydra by environmental stressors such as food shortage, crowding, or low temperature, much like the conditions that accompany the beginning of winter (Littlefield et al. 1991). We were interested in the possible effect of this transition from a mainly vegetative state into a sexual state, on transcriptional regulation within cells of the organism. For this purpose, we examined a male laboratory strain of the species H. oligactis that is cold-sensitive (CS), that is responds to cold temperature by initiating aging. In addition to the signs of aging described above, this strain also exhibits disorganization of epithelial cell myofibers, deterioration of the apical nervous system, and due to loss of interstitial stem cells, generalized organ atrophy (Tomczyk et al. 2019; Tomczyk et al. 2015). We compared the transcriptome of the CS strain at both 18 and 10 °C with a closely related, cold-resistant (CR) H. oligactis strain that does not undergo these life history shifts when transferred to colder temperatures for the insight they might provide into mechanisms of potential immortality and prevention of aging. The results indicate a switch in transcriptional emphasis from regeneration to sexual reproduction and increased somatic maintenance.
Results
We performed directional deep total RNA sequencing (> 38 M reads per sample) on 3 individual hydras from CS and CR strains at both 18 and 10 °C (Fig. S1a). The genomes of three Hydra species (H. magnipapillata [now called H. vulgaris], H. oligactis, and H. viridissima) have been sequenced (Chapman et al. 2010; Vogg et al. 2019). However, sequence differences between H. oligactis and the other two species constrained the possibility of using their genomes as reference sequences. Moreover, the number of scaffolds in the H. oligactis genome was relatively high at 447,337, suggesting it would be a limiting factor in identifying full coding regions. Hence, to analyze our data, we first assembled a de novo H. oligactis transcriptome with quality-filtered reads from all sequence samples (Fig. S1b). The initial transcriptome assembly yielded 421,568 contigs with a median length of 289 bp and an N50 of 418 bp.
To reduce potential small non-coding RNAs, we removed contigs shorter than 400 bp and contigs without any similarity to the NCBI non-redundant database (Blastx, e-value < 1e−5), as done in previous studies (Petersen et al. 2015), thereby retaining only contigs with protein-coding potential. To further reduce transcriptome complexity, we extracted and retained only the longest isoform for each candidate gene in the contigs, resulting in a total of 31,585 contigs with a median length of 796 bp and N50 of 1575 bp. Notably, this number is in a similar range and has 93.4% homology compared with published contigs for other Hydra transcriptomes (Table S1) (Chapman et al. 2010; Juliano et al. 2014; Petersen et al. 2015) (Fig. 1a). These results indicate high genetic similarity among these Hydra species despite 60–100 MY divergence (Martinez et al. 2010; Schwentner and Bosch 2015), especially compared with 85% homology between human and mouse with a similar divergence time (Batzoglou et al. 2000; Mouse Genome Sequencing et al. 2002). However, it also indicated the differences between H. oligactis and other Hydra species if we consider the only 95% homology between human and chimpanzee under 7–10 MY divergence (Britten 2002; White et al. 2009). Phylogenetic analysis confirmed that H. oligactis is indeed the outgroup among the other three published transcriptomes (Fig. 1b) (Hemmrich et al. 2012). In addition, we found only one spliced leader (SL) RNA in the H. oligactis transcriptome compared to seven in H. vulgaris. As the SL RNAs are used to resolve polycistronic transcripts into individual units and to add 5′ cap structures for increasing mRNA stability and translation rate, the lesser diversity of SL RNAs in H. oligactis suggests a lesser capacity to cope with stress in this particular species (Michaeli 2011). In plotting the length distribution of the contigs, we found 99.8% of the assembled transcripts were shorter than 10 kb (Fig. 1c). To assess the quality of our assembly without effects of lowly expressed contigs, we calculated the N50 of 90% of the expressed sequences (Ex90N50), as 2255 bp, indicating that highly expressed sequences are well assembled (Fig. 1d). We annotated our contigs to human protein (GRCh38.p12, GCF_000001405.38) in order to assign putative gene function to the contigs in our transcriptome assembly. Finally, we performed BUSCO analysis for transcriptome completeness (Simao et al. 2015). We compared our assembly to the metazoan core orthologs (n = 978) and found 93.9% (n = 919) and 2.9% (n = 28) of orthologs were completed and fragmented, respectively, illustrating a high degree of completeness.
Fig. 1.

De novo transcriptome assembly of H. oligactis. a The number of contigs in H. oligactis and H. vulgaris transcriptomes. About 93.4% of assembled H. oligactis contigs shows homology to H. vulgaris. Only 2% of the mRNAs published in NCBI by Chapman et al. was missing, which reflect putative species-specific genes. b Phylogenetic tree of Hydra transcriptomes. The H. vulgaris from Chapman et al. originated from a different subgroup of H. vulgaris. c Length distribution of final assembled contigs. 60.3% of contigs were shorter than 1 kb, while only 0.2% were longer than 10 kb. d The distribution of N50 of the contigs with an expression value that represented percentage of the total expression data (ExN50). About 90% of the expression data is represented by contigs of an N50 of 2255 bp
To search for global changes in gene expression during the temperature shift in CS and CR strains, we performed differential expression analysis against our 31,585 contigs. As expected, the temperature shift had a greater expression effect in the CS, relative to the CR, strain (Fig. 2a). Specifically, we found ~ 1500 differentially expressed contigs (DECs) in the CS strain due to the temperature shift (approximately one-third more of these upregulated than downregulated). By contrast, we found only 103 DECs in response to the temperature shift in the CR strain. Indeed, comparison of the two strains at the colder 10 °C temperature revealed ~ 2600 DECs, most with higher expression in the CS strain. At the higher temperature, we found ~ 1100 DECs, most of which were higher in the CR strain. In sum, these results indicate substantial differences in gene expression between the two strains, even at the standard laboratory temperature of 18 °C. These results were confirmed by unsupervised hierarchical clustering analysis using the DECs, which showed that the gene expression profile in the aging CS Hydra at 10 °C was distinct from the other three groups of budding, non-aging animals (Fig. 2b).
Fig. 2.

Differentially expressed contigs (DECs) in H. oligactis. a The number of DECs in each comparison group. We computed DECs between the strains to obtain the contigs related to specific processes. The comparisons between different temperatures (10 °C and 18 °C) reflect temperature shift-specific differences, while DECs between different strains (CR and CS) represent strain-specific differences. Also, we expected a higher number of DECs between CS at 10 °C and other asexual strains as these DECs will also represent the change of spermatogenesis and aging. Because of only 103 DECs between the CR strain at different temperatures, there is no need for comparison between CR at 18 °C and CS at 10 °C. b Heat map of correlation between hydra samples. Expression values for all samples were compared, and Pearson correlation values were plotted. Asexual samples (CS and CR both at 18 °C) cluster according to Pearson correlation values, indicating high similarity. CS at 18 °C is an outlier among asexual samples due to strain differences. c Venn diagram of DECs in group comparisons. We overlapped the DECs from (a) to identify key DECs related to the temperature shift in CS. d GO term enrichment results using 1131 DECs. Size of each dot represents Gene Ratio enrichment for each category while color indicates Padj value.
We extracted the overlap of DECs between a comparison of CS animals at both temperatures and a comparison of CS and CR animals both at 10 °C; these two comparisons, therefore, were between non-aging and aging individuals. This yielded a total of 1131 DECs, which we reasoned would be informative with respect to the temperature-induced life history switch (Fig. 2c). Gene ontology (GO) analysis of the 884 upregulated and 247 downregulated DECs showed a significant enrichment for biological processes related to the reproductive switch from asexual budding to meiotic gamete production (Fig. 2d; Table S2). For instance, the enrichment in “reproduction” and “cell cycle” points towards a shift to gametogenesis (Table S3). In addition, in “reproduction” we observed upregulation of meiosis-related genes contributing to recombination in spermatogenesis (Table S3). The upregulation of these genes accompanying meiotic gamete production confirms a long-standing hypothesis that genetic diversity such as that generated by sexual reproduction is most advantageous under changing environmental conditions such as the onset of fall and winter (Williams 1975).
We also observed gene expression changes related to the shift from a non-aging to an aging phenotype. Specifically, in the CS strain when undergoing aging at 10 °C, we saw upregulation of CCNA1 (Cyclin A1), CDC14B, and FGF2, all pointing to the emergence of cellular senescence (Table S4) (Davalos et al. 2010; Huang et al. 2016; Su 2006; Takahashi et al. 2012). FGF2 has been shown to be up-regulated in senescent cells and are involved in the senescence-associated secretory phenotype (SASP) (Coppe et al. 2010). Interestingly enough, the process of cellular senescence has never previously been suggested in any Hydra species.
A major driver of cellular senescence is DNA damage due to reactive oxygen species (ROS) production, an inescapable byproduct of oxidative metabolism (Sohal and Weindruch 1996). Expression changes in the CS strain, e.g. TRAP1 and H2AX (H2AFX), during the shift to 10 °C point towards increased ROS (Chen et al. 2015a; Lisanti et al. 2014) (Table S5). We also noticed that in ROS-related genes, SOD genes and CAT are upregulated, whereas most GPX genes are downregulated (Fig. S2). SOD genes convert superoxide anion to hydrogen peroxide and molecular oxygen, while CAT and GPX genes breakdown the hydrogen peroxide to the water and molecular oxygen in peroxisomes breakdowns the mitochondria, respectively (Ighodaro and Akinloye 2018). The changes of these genes indicated high levels of ROS, especially in mitochondria, suggesting that senescence and the SASP are activated as part of the switch from non-aging to aging. This was confirmed by the observed upregulation of genes involved in genome maintenance (Table S6), a protective mechanism in the sexual reproduction phase to delay senescence.
Conversely, we also looked for possible differences that could explain the resistance of the CR strain to cold-induced aging and a shift to sexual reproduction. We observed that one contig, assigned as cold-inducible RNA binding protein (CIRBP), displayed reduced expression in both CR and CS strains (Fig. S3). However, the reduction was significantly greater in the CS than in the CR strain (2.3-fold vs 1.2-fold, respectively). Hence, it is plausible that relatively high expression of the CIRBP orthologous contig contributes to senescence bypass in this cold-resistant strain (Lleonart 2010).
To understand the global response of gene expression changes during the temperature shift, we performed weighted gene co-expression network analysis (WGCNA) on the annotated contigs (see Experimental Procedures) (Langfelder and Horvath 2008). We find four modules highly correlated with sexual reproduction and aging—black, red, blue, and green—while one module—brown—was highly correlated with asexual budding and the absence of aging (Fig. 3a, b). The sexual reproduction/aging modules were highly enriched for genes related to sperm production and motility (black and red modules) as well as cell cycle transition, DNA replication, and recombination (green and blue modules). The brown module associated with budding/non-aging was highly enriched for proteins regulating extracellular matrix, such as WNT and MMP genes (Table S7). Thus, there are clear gene sets that drive the observed sexual reproduction/aging phenotype during the temperature shift.
Fig. 3.

Weighted gene co-expression network analysis a The relationships between gene modules and sample traits. The values in the top are the Pearson correlation values, while the values in the bracket are the p-values. b The correlations between gene modules and every sample are plotted. Positive values indicate positive correlation between module and sample. c The upregulation of spermatogenesis-related gene sets in CS at 10 °C to promote sexual reproduction
Next, we used gene set enrichment analysis (GSEA) to test for possible enrichment of pathways involved in aging, using the Molecular Signatures Database (MSigDB) hallmark, GO, KEGG, and REACTOME gene sets (Liberzon et al. 2015). We first compared the CS strain when aging at 10 °C with the three other strain/conditions in which aging was lacking and obtained 346 upregulated and 405 downregulated gene sets (Table S8). Reassuringly, given the development of male gonads accompanying aging in this experimental system, our results indicated upregulation of spermatogenesis-related gene sets in the sexual reproduction/aging hydra, including “GO sperm part,” “GO spermatid differentiation,” “GO sperm flagellum,” and “GO sperm motility” (Fig. 3c), which is consistent with the results of the DEC analysis and WGCNA described above. With respect to relevance to aging, we also observed upregulation of DNA repair gene sets, including “GO DNA repair,” “GO double-strand break repair,” “GO nucleotide excision repair DNA damage recognition,” GO DNA synthesis involved in DNA repair,” and “GO recombinational repair” (Fig. 4a). Notably, we observed that “REACTOME regulation of apoptosis” and “REACTOME apoptosis” gene sets were upregulated in aging CS Hydra (Fig. 4b) as was “Hallmark E2F targets.” The latter are critical regulators of autophagy and apoptosis (Chen et al. 2015b). The enrichment map of analyzed GSEA gene sets showed that a cluster of spermatogenesis gene sets was linked to DNA repair gene sets through sets like “GO synapsis” and “GO homologous chromosome regulation” (Fig. 5). We also observed links between DNA repair-related gene sets and apoptosis-related sets, for example, gene sets of “GO DNA replication” and “REACTOME synthesis of DNA.” We conclude that upregulation of spermatogenesis is inherently associated with increased DNA repair.
Fig. 4.

Gene set enrichment analysis. a The upregulation of five DNA repair gene sets in CS at 10 °C (FDR < 0.25). b The upregulation of three apoptosis gene sets in CS at 10 °C (FDR < 0.25)
Fig. 5.

Enrichment map of GSEA gene sets. We could observe association between spermatogenesis (green circle) and repair gene sets (purple circle), and repair and apoptosis gene sets (orange circle). The gene sets with 50% overlap were linked in the figure. The red dots and blue dots are gene sets upregulated and downregulated, respectively by CS at 10 °C
A hallmark gene set found downregulated in CS at 10 °C is “Hallmark IL6/JAK/STAT3 signaling” (Fig. S4a). JAK/STAT3 signaling regulated by IL6 is considered one of the key pathways in regulating multiple types of stem cell self-renewal (Bharti et al. 2016; Chen et al. 2015b; Hirano et al. 2000). Also, the results showed that in CS at 10 °C the “GO stem cell differentiation,” “GO stem cell proliferation,” and “GO regulation of notch signaling pathway” gene sets are all downregulated, which likely weakens somatic cell renewal capacity and possibly promoting senescence. This conclusion was further confirmed by the observed downregulation in CS at 10 °C of FOXO3 (Fig. S4b). FOXO3 appeared to be a critical regulator of stem cell maintenance in immortal Hydra (Boehm et al. 2012). Other key markers of stem cell maintenance, such as POU5F1 (OCT3/4) and WNT1, were also significantly downregulated (Fig. S4b) (Masui et al. 2007; Williams et al. 2010).
Discussion
These present results provide the first comprehensive assembly and analysis of the H. oligactis transcriptome, comparing the non-aging, asexually reproducing condition with the aging, sexually reproducing condition. Collectively, our data demonstrate that the switch from a non-aging to an aging phenotype is accompanied by activation of a series of molecular pathways known to be characteristic for aging metazoans, such as cellular senescence, ROS-induced damage, and lack of stem cell maintenance. Our data also highlight pathways of interest for understanding the exceptional mechanisms by which Hydra achieve potential immortality. We find that upregulation of sexual reproduction pathways induces global changes in gene expression, particularly related to gamete production and DNA recombination, which may drive these “canonical” aging phenotypes.
One of the major findings of this study showed that a large number of DECs and pathways are associated with sexual reproduction and aging phenotypes between the non-aging and aging-inducible strain, while there are fewer DECs between non-aging and aging strains under pressure of temperature shift. Based on these findings, we propose that under benign conditions, Hydra display exceptional maintenance of their three stem cell types, viz., ectodermal and endodermal epithelial stem cells and interstitial stem cells, continually replacing all differentiated somatic cells (Fig. 6) (Tomczyk et al. 2015). However, the CS H. oligactis strain under sufficient environmental duress, enough to threaten somatic survival, undergoes a complete shift of stem cell activity away from maintenance to exclusive support of sexual reproduction, and by doing so maximizes gamete production at the cost of somatic aging and eventual death—a classical display of the disposable soma phenomenon (Kirkwood and Austad 2000). Sexual reproduction in the CS strains elicits persistent DNA damage induced by ROS, and the subsequent increase in DNA repair may not provide sufficient protection to prevent somatic mutation accumulation, loss of genome stability, cellular senescence, and cell death of the soma (Milholland et al. 2017). The CR strain, as well as other Hydra species, does not show such adaptive shifts, for unknown but possibly ecological and evolutionary reasons (Hemmrich et al. 2012).
Fig. 6.
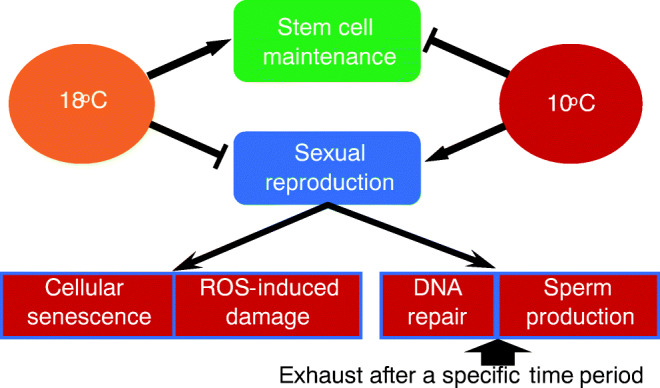
Schematic diagram of “trade-off” model. The trade-off between genetic evolutionary advantage and cellular maintenance in hydra is impacted by environmental duress, such as cold stress. Under such conditions, maintenance processes such as DNA repair and sperm production will be exhausted after a specific time period since the temperature shift. Hydra will die due to processes such as cellular senescence and ROS-induced damage.
Although our studies showed multiple pathway alterations induced by the temperature shift and how these pathways might contribute to aging phenotypes, it should be noted that the present study only included one specific environmental change, i.e., the temperature shift, within each of the CS and CR strains. Moreover, we noted the caveat that RNA was isolated from whole-bodies of hydra rather than individual tissues. We note that as we sampled these transcriptomes at a single time point, we have generated a snapshot rather than a full description of the shift from nonaging to aging phenotypes. As a result, we can only speculate that DNA repair mainly takes place in the germline given the upregulation of RAD51 family and MutS homologs (MSH4 and MSH5), which are essential for homologous recombination and DNA mismatch repair during spermatogenesis (Marcon and Moens 2005).
This first comprehensive H. oligactis transcriptome is a promising start to understanding how genome regulation can favor aging versus nonaging. Recent advances in single cell sequencing, combined with serial sampling of these strains over the nonaging to aging transition, will allow greater understanding of the importance of differential transcriptional regulation among different cell types (Covington et al. 2008; de Soysa et al. 2019). It would also be of interest to investigate the transcriptomics of similar life history transitions in response other environmental stressors such as food supply and crowding. Understanding the complex mechanisms behind each age-inducible stimuli would shed light on the dramatic changes occurring during the transition process towards canonical aging.
Our study confirms the strong relationships between aging phenotypes and sexual reproduction, which is also reported in various animals like nematodes, fruit flies, rodents, and primates (Harvanek et al. 2017). Further research could focus on the exact nature of transitions from activating sexual reproduction and enhancing DNA repair capability to reducing stem cell renewal and inducing cell senescence under certain environmental stressors, like the temperature shift.
Experimental procedures
Sample growth and collection
CR and CS Hydra oligactis were obtained from Brigitte Galliot’s laboratory in Geneva, Switzerland, and maintained in standard hydra medium at 18 °C ± 0.5 °C while being fed freshly hatched brine shrimp (Artemia sp.) larvae 3–5 times per week. To induce gametogenesis, animals of both strains were transferred to 10 °C ± 0.3 °C medium at which time feeding was reduced to 2 times per week. At 10 °C, the vast majority of CR strain animals ceased budding and began forming testes; the vast majority of CR strain animals continued budding although at a reduced rate.
Library preparation and sequencing
As soon as testes were fully formed, we flash-froze single hydra, homogenized them in Qiazol, and purified the RNA using the miRNeasy Micro Kit (Qiagen). Total RNA quality was assessed using an Agilent 2100 Bioanalyzer; only samples with a RIN greater than 8.0 were used for subsequent analysis. Total RNA was treated with DNaseI, and again column purified using the miRNeasy Micro Kit (Qiagen). Ribosomal RNA was depleted using the Ribo-Zero Magnetic Gold Kit (Epicenter), followed by ethanol precipitation. Depleted RNA was converted to cDNA using the SuperScript IV First-Strand Synthesis Kit (Invitrogen) with 80 ng random hexamers and 50 μM oligo dT, and subsequently ethanol precipitated. Single-stranded cDNA was converted to dsDNA by DNA polymerase I using dU/VTPs (10 mM). Samples were then fragmented in 1X TE pH 8.0 to 200–300 bp using Covaris. Samples were then purified using the MinElute PCR purification kit (Qiagen). Fragmented samples underwent standard end-repair, dA-tailing, and adapter ligation using Illumina TruSeq adaptors for multiplexing. Adaptor-ligated cDNA was treated with uracil-DNA glycosylase followed by enrichment PCR using NEBNext HiFi polymerase (New England Biolabs) for 18 cycles. Libraries were size selected for 150–600 bp on a 2% low-melt ultra-low range agarose gel stained with SYBR Gold (Invitrogen). Purified libraries were then clustered (6 samples per flow cell lane) and sequenced on an Illumina HiSeq2000 for 100 bp paired-end reads.
Transcriptome de novo assembly
Raw reads were first trimmed by Trim Galore (version 0.4.1) to clip adapter and low-quality bases at 3′ ends. To avoid content biases in 5′ ends, we removed the first 7 bp from each read. The quality of reads before and after trimming was assessed using FastQC (version 0.11.4). To generate a de novo Hydra oligactis transcriptome, trimmed reads from all samples were combined and assembled utilizing Trinity (version 2.2.0) (Haas et al. 2013), including a total of 289 M paired-end reads. The cut-off for transcript length was set to 400 bp to avoid short potential reading frames. The transcripts were then compared to the NCBI non-redundant (Nr) database with Blastx (version ncbi-blast-2.6.0+: options: -max_target_seqs 20, -evalue 1e–5, -outfmt 5) to identify probable protein-coding transcripts. Homology search was performed using Tblastx (-evalue 1e–5). To construct the phylogenetic tree, we first extracted the long open read frames (ORFs) using TransDecoder (version 5.3.0) and identified homology of the ORFs to known proteins via Blastp (-max_target_seqs 1, -evalue 1e-5, -outfmt 6) to UniProt (Release 2018_04) (UniProt Consortium T 2018) and HMMscan (version 3.1b2) (Finn et al. 2011) to Pfam (version 31.0) (El-Gebali et al. 2019). We next predicted likely coding regions by integrating the Blastp and Pfam results with TransDecoder (--single_best_only) and identified single copy orthologs between the transcriptomes using Proteinortho (version 5.16b) (Lechner et al. 2011). The orthologous contigs were aligned using MAFFT (version 7.407) (Nakamura et al. 2018) and trimmed using Gblocks (version 0.91b) (Castresana 2000). The concatenated sequences were then used for phylogenetic tree construction with RAxML (version 8.2.4) (Stamatakis 2014) and visualized using the ggtree package (Yu et al. 2018). We examined published seven SL sequences in our 31,585 contigs using Blat (version 35) (Kent 2002). Transcript quantification was performed by mapping trimmed reads to the assembled de novo transcriptome using bowtie2 (version 2.2.3) (Langmead and Salzberg 2012) and gene expression calculated using RSEM (version 1.3.0) as Trinity pipeline. ExN50 values were then obtained and plotted using scripts from Trinity. For assessment of transcriptome assembly completeness, we used BUSCO v3 (Simao et al. 2015) with the metazoan orthologs (metazoa_odb9).
Functional annotation
Clean reads were mapped to the final transcriptome by utilizing STAR (version 2.6.0c; options: --outFilterScoreMinOverLread 0.25, --outFilterMatchNminOverLread 0.25) (Dobin et al. 2013). The counts of reads for each contig were extracted by HTSeq (version 0.6.1) (Anders et al. 2015). Contig expression levels were normalized to FPKM under a TMM method, and differential contig expression analysis was performed using edgeR (Robinson et al. 2010). To obtain the GO annotation, we annotated predicted coding regions using InterProScan (version 5.29–68.0; options: -dp, -iprlookup, -goterms) (Jones et al. 2014) and mapped GO annotations to assembled contigs based on Blastx results to Nr. All GO annotations were then merged by Blast2GO (version 5.2.0; cut-off for e-value hit: 1e−6) (Conesa et al. 2005). The differentially expressed contigs were then enriched using clusterProfiler (Yu et al. 2012).
To interpret the function of assembled contigs, we annotated hydra contigs with human proteins (GRCh38.p12, GCF_000001405.38). The orthologs between predicted coding regions of hydra contigs and human proteins were identified in Proteinortho, resulting in ~ 5000 orthologs. We also aligned hydra contigs with human proteins using Blastx (-e-value 1e-5, -max_target_seqs 500; top 20 genes) to show the probable orthologous genes. To perform the gene set functional enrichment, the hydra contigs unexpressed (FPKM < 1) in any samples were removed, whereas duplicate annotations were filtered based first on blast e-values and second on the sum of expression values in all samples. Finally, we obtained 7730 human gene annotations and performed gene set enrichment analysis with GSEA (version 6.2) (Subramanian et al. 2005). The enriched gene set was identified as significant if FDR q-value was smaller than 0.25. Enrichment plots were performed using Rtoolbox (https://github.com/PeeperLab/Rtoolbox). The enrichment map of GSEA results was obtained in Cytoscape (version 3.6.1; nominal p < 0.05, FDR q-value < 0.25, overlap cut-off: 0.5) (Shannon et al. 2003).
Weighted gene correlation network analysis
All contigs in GSEA (with human gene annotation) were used to identify groups of co-regulated genes in an unsupervised way using WGCNA (Langfelder and Horvath 2008). R2 cut-off of 0.9 was used in the minimum power tested. A signed gene co-expression network was constructed with a soft threshold power of 30 and using biweight mid-correlation. The cutoff for merging modules was set as 0.25. Minimum genes per module was 30. The Pearson correlations between module eigengenes and traits were checked, and student asymptotic p-values were calculated. For chosen modules, GO enrichment analysis was performed in clusterProfiler.
Electronic supplementary material
{kind=link}
(a) Schematic overview of the experimental design to perform transcriptome analysis. The CS and CR strains at 18oC and 10oC were collected for RNA-seq. 313 million paired reads were obtained and used for transcriptome assembly. Functional analysis was performed based on assembled contigs. (b) Analysis pipeline to obtain a de novo transcriptome and perform functional enrichment. (PNG 268 kb)
{kind=link}
The expression level of antioxidants in ROS-related genes. Three homologs of SOD2 identified by Proteinortho are plotted. Most GPX genes breaking down hydrogen peroxide in the mitochondria are downregulated, indicating the CS at 10oC may accumulate the ROS after present of sexual reproduction. (PNG 147 kb)
{kind=link}
The expression level of cold-inducible RNA binding protein (CIRBP). The high level of CIRBP could bypass senescence and reduce oxidative damage. The differences between CR strains at the two temperatures are much smaller than in the CS strains, suggesting a possible way CR strains escape mortality. (PNG 23 kb)
{kind=link}
(a) Four gene sets related to stem cell maintenance were down-regulated in CS at 10oC. (b) Three important genes in relation to the stem cell self-renewal gene sets were down-regulated in CS at 10oC. (PNG 203 kb)
(XLSX 280 kb)
Acknowledgments
We are grateful to Brigitte Galliot for providing us with the Hydra strains. SNA is particularly indebted to Dr. Galliot for introducing him to the H. oligactis experimental system and to the details of Hydra husbandry.
Author contributions
SNA and JV conceived and supervised the study. KEF and RRW performed the experiments, SS and ZZ analyzed the data and SS, RRW, ZZ, JV and ZZ interpreted the data and wrote the manuscript.
Funding information
This work was supported by grants from the National Institutes of Health to JV (AG017242, CA180126, AG047200, AG038072 and the Glenn Foundation for Medical Research), and SNA (AG037962, AG0508860) and the Glenn Foundation for Medical Research.
Data availability
Raw RNAseq data from this manuscript were deposited to the Sequence Read Archive under BioProject PRJNA559195. This Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GHUC00000000. The version described in this paper is the first version, GHUC01000000.
Compliance with ethical standards
Conflict of interest
J.V. is the founder SingulOmics, Corp. The other authors declare no competing interests.
Code availability
Pipelines and code are available upon request.
Footnotes
Shixiang Sun and Ryan R. White are co-first authors.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Steven N. Austad, Email: austad@uab.edu
Jan Vijg, Email: jan.vijg@einsteinmed.org.
References
- Anders S, Pyl PT, Huber W. HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31:166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzoglou S, Pachter L, Mesirov JP, Berger B, Lander ES. Human and mouse gene structure: comparative analysis and application to exon prediction. Genome Res. 2000;10:950–958. doi: 10.1101/gr.10.7.950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharti R, Dey G, Mandal M. Cancer development, chemoresistance, epithelial to mesenchymal transition and stem cells: a snapshot of IL-6 mediated involvement. Cancer Lett. 2016;375:51–61. doi: 10.1016/j.canlet.2016.02.048. [DOI] [PubMed] [Google Scholar]
- Boehm AM, et al. FoxO is a critical regulator of stem cell maintenance in immortal. Hydra Proc Natl Acad Sci U S A. 2012;109:19697–19702. doi: 10.1073/pnas.1209714109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britten RJ. Divergence between samples of chimpanzee and human DNA sequences is 5%, counting indels. Proc Natl Acad Sci U S A. 2002;99:13633–13635. doi: 10.1073/pnas.172510699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castresana J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol. 2000;17:540–552. doi: 10.1093/oxfordjournals.molbev.a026334. [DOI] [PubMed] [Google Scholar]
- Chapman JA, et al. The dynamic genome of Hydra. Nature. 2010;464:592–596. doi: 10.1038/nature08830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Ruiz PD, McKimpson WM, Novikov L, Kitsis RN, Gamble MJ. MacroH2A1 and ATM play opposing roles in paracrine senescence and the senescence-associated secretory phenotype. Mol Cell. 2015;59:719–731. doi: 10.1016/j.molcel.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Ye S, Ying QL. Stem cell maintenance by manipulating signaling pathways: past, current and future. BMB Rep. 2015;48:668–676. doi: 10.5483/BMBRep.2015.48.12.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–3676. doi: 10.1093/bioinformatics/bti610. [DOI] [PubMed] [Google Scholar]
- Coppe JP, Desprez PY, Krtolica A, Campisi J. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu Rev Pathol. 2010;5:99–118. doi: 10.1146/annurev-pathol-121808-102144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covington MF, Maloof JN, Straume M, Kay SA, Harmer SL. Global transcriptome analysis reveals circadian regulation of key pathways in plant growth and development. Genome Biol. 2008;9:R130. doi: 10.1186/gb-2008-9-8-r130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davalos AR, Coppe J-P, Campisi J, Desprez P-Y. Senescent cells as a source of inflammatory factors for tumor progression. Cancer Metastasis Rev. 2010;29:273–283. doi: 10.1007/s10555-010-9220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Soysa TY, et al. Single-cell analysis of cardiogenesis reveals basis for organ-level developmental defects. Nature. 2019;572:120–124. doi: 10.1038/s41586-019-1414-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gebali S, et al. The Pfam protein families database in 2019. Nucleic Acids Res. 2019;47:D427–D432. doi: 10.1093/nar/gky995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Clements J, Eddy SR (2011) HMMER web server: interactive sequence similarity searching Nucleic Acids Res 39:W29–W37 doi:10.1093/nar/gkr367. [DOI] [PMC free article] [PubMed]
- Haas BJ et al. (2013) De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis Nat Protoc 8:1494-1512 doi:10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed]
- Harvanek ZM, Lyu Y, Gendron CM, Johnson JC, Kondo S, Promislow DEL, Pletcher SD. Perceptive costs of reproduction drive ageing and physiology in male Drosophila. Nat Ecol Evol. 2017;1:152. doi: 10.1038/s41559-017-0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemmrich G, et al. Molecular signatures of the three stem cell lineages in hydra and the emergence of stem cell function at the base of multicellularity. Mol Biol Evol. 2012;29:3267–3280. doi: 10.1093/molbev/mss134. [DOI] [PubMed] [Google Scholar]
- Hirano T, Ishihara K, Hibi M. Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene. 2000;19:2548–2556. doi: 10.1038/sj.onc.1203551. [DOI] [PubMed] [Google Scholar]
- Huang KC, et al. Cyclin A1 expression and paclitaxel resistance in human ovarian cancer cells. Eur J Cancer. 2016;67:152–163. doi: 10.1016/j.ejca.2016.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ighodaro O, Akinloye O. First line defence antioxidants-superoxide dismutase (SOD), catalase (CAT) and glutathione peroxidase (GPX): their fundamental role in the entire antioxidant defence grid Alexandria. J Med. 2018;54:287–293. [Google Scholar]
- Jones P, et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014;30:1236–1240. doi: 10.1093/bioinformatics/btu031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juliano CE, et al. PIWI proteins and PIWI-interacting RNAs function in Hydra somatic stem cells. Proc Natl Acad Sci U S A. 2014;111:337–342. doi: 10.1073/pnas.1320965111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ. BLAT--the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood TB, Austad SN. Why do we age? Nature. 2000;408:233–238. doi: 10.1038/35041682. [DOI] [PubMed] [Google Scholar]
- Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics. 2008;9:559. doi: 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechner M, Findeiss S, Steiner L, Marz M, Stadler PF, Prohaska SJ. Proteinortho: detection of (co-)orthologs in large-scale analysis. BMC Bioinformatics. 2011;12:124. doi: 10.1186/1471-2105-12-124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisanti S, et al. Deletion of the mitochondrial chaperone TRAP-1 uncovers global reprogramming of metabolic networks. Cell Rep. 2014;8:671–677. doi: 10.1016/j.celrep.2014.06.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Littlefield CL, Finkemeier C, Bode HR. Spermatogenesis in Hydra oligactis. II. How temperature controls the reciprocity of sexual and asexual reproduction. Dev Biol. 1991;146:292–300. doi: 10.1016/0012-1606(91)90231-q. [DOI] [PubMed] [Google Scholar]
- Lleonart ME. A new generation of proto-oncogenes: cold-inducible RNA binding proteins. Biochim Biophys Acta. 2010;1805:43–52. doi: 10.1016/j.bbcan.2009.11.001. [DOI] [PubMed] [Google Scholar]
- Marcon E, Moens PB. The evolution of meiosis: recruitment and modification of somatic DNA-repair proteins. Bioessays. 2005;27:795–808. doi: 10.1002/bies.20264. [DOI] [PubMed] [Google Scholar]
- Martinez DE. Mortality patterns suggest lack of senescence in hydra. Exp Gerontol. 1998;33:217–225. doi: 10.1016/S0531-5565(97)00113-7. [DOI] [PubMed] [Google Scholar]
- Martinez DE, Iniguez AR, Percell KM, Willner JB, Signorovitch J, Campbell RD. Phylogeny and biogeography of Hydra (Cnidaria: Hydridae) using mitochondrial and nuclear DNA sequences. Mol Phylogenet Evol. 2010;57:403–410. doi: 10.1016/j.ympev.2010.06.016. [DOI] [PubMed] [Google Scholar]
- Masui S, et al. Pluripotency governed by Sox2 via regulation of Oct3/4 expression in mouse embryonic stem cells. Nat Cell Biol. 2007;9:625–635. doi: 10.1038/ncb1589. [DOI] [PubMed] [Google Scholar]
- Michaeli S. Trans-splicing in trypanosomes: machinery and its impact on the parasite transcriptome. Future Microbiol. 2011;6:459–474. doi: 10.2217/fmb.11.20. [DOI] [PubMed] [Google Scholar]
- Milholland B, Suh Y, Vijg J. Mutation and catastrophe in the aging genome. Exp Gerontol. 2017;94:34–40. doi: 10.1016/j.exger.2017.02.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouse Genome Sequencing C et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Yamada KD, Tomii K, Katoh K. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics. 2018;34:2490–2492. doi: 10.1093/bioinformatics/bty121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen HO, Höger SK, Looso M, Lengfeld T, Kuhn A, Warnken U, Nishimiya-Fujisawa C, Schnölzer M, Krüger M, Özbek S, Simakov O, Holstein TW. A comprehensive transcriptomic and proteomic analysis of Hydra head regeneration. Mol Biol Evol. 2015;32:1928–1947. doi: 10.1093/molbev/msv079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petralia RS, Mattson MP, Yao PJ. Aging and longevity in the simplest animals and the quest for immortality. Ageing Res Rev. 2014;16:66–82. doi: 10.1016/j.arr.2014.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaible R, Scheuerlein A, Danko MJ, Gampe J, Martinez DE, Vaupel JW. Constant mortality and fertility over age in Hydra. Proc Natl Acad Sci U S A. 2015;112:15701–15706. doi: 10.1073/pnas.1521002112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwentner M, Bosch TC. Revisiting the age, evolutionary history and species level diversity of the genus Hydra (Cnidaria: Hydrozoa) Mol Phylogenet Evol. 2015;91:41–55. doi: 10.1016/j.ympev.2015.05.013. [DOI] [PubMed] [Google Scholar]
- Shannon P, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498–2504. doi: 10.1101/gr.1239303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simao FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–3212. doi: 10.1093/bioinformatics/btv351. [DOI] [PubMed] [Google Scholar]
- Sohal RS, Weindruch R. Oxidative stress, caloric restriction, and aging. Science. 1996;273:59–63. doi: 10.1126/science.273.5271.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30:1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su TT. Cellular responses to DNA damage: one signal, multiple choices. Annu Rev Genet. 2006;40:187–208. doi: 10.1146/annurev.genet.40.110405.090428. [DOI] [PubMed] [Google Scholar]
- Subramanian A, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi A, et al. DNA damage signaling triggers degradation of histone methyltransferases through APC/C(Cdh1) in senescent cells. Mol Cell. 2012;45:123–131. doi: 10.1016/j.molcel.2011.10.018. [DOI] [PubMed] [Google Scholar]
- Tomczyk S, Buzgariu W, Perruchoud C, Fisher K, Austad S, Galliot B. Loss of Neurogenesis in Aging Hydra. Dev Neurobiol. 2019;79:479–496. doi: 10.1002/dneu.22676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomczyk S, Fischer K, Austad S, Galliot B. Hydra, a powerful model for aging studies. Invertebr Reprod Dev. 2015;59:11–16. doi: 10.1080/07924259.2014.927805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UniProt Consortium T UniProt: the universal protein knowledgebase. Nucleic Acids Res. 2018;46:2699. doi: 10.1093/nar/gky092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogg MC, et al. An evolutionarily-conserved Wnt3/beta-catenin/Sp5 feedback loop restricts head organizer activity in Hydra. Nat Commun. 2019;10:312. doi: 10.1038/s41467-018-08242-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White TD, Asfaw B, Beyene Y, Haile-Selassie Y, Lovejoy CO, Suwa G, WoldeGabriel G. Ardipithecus ramidus and the paleobiology of early hominids. Science. 2009;326:75–86. [PubMed] [Google Scholar]
- Williams GC (1975) Sex and evolution. Monogr Popul Biol:3–200. [PubMed]
- Williams JM, Oh SH, Jorgensen M, Steiger N, Darwiche H, Shupe T, Petersen BE. The role of the Wnt family of secreted proteins in rat oval "stem" cell-based liver regeneration: Wnt1 drives differentiation. Am J Pathol. 2010;176:2732–2742. doi: 10.2353/ajpath.2010.080486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Fujisawa T, Hwang JS, Ikeo K, Gojobori T. Degeneration after sexual differentiation in hydra and its relevance to the evolution of aging. Gene. 2006;385:64–70. doi: 10.1016/j.gene.2006.06.031. [DOI] [PubMed] [Google Scholar]
- Yu G, Lam TT, Zhu H, Guan Y. Two Methods for Mapping and Visualizing Associated Data on Phylogeny Using Ggtree. Mol Biol Evol. 2018;35:3041–3043. doi: 10.1093/molbev/msy194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(a) Schematic overview of the experimental design to perform transcriptome analysis. The CS and CR strains at 18oC and 10oC were collected for RNA-seq. 313 million paired reads were obtained and used for transcriptome assembly. Functional analysis was performed based on assembled contigs. (b) Analysis pipeline to obtain a de novo transcriptome and perform functional enrichment. (PNG 268 kb)
The expression level of antioxidants in ROS-related genes. Three homologs of SOD2 identified by Proteinortho are plotted. Most GPX genes breaking down hydrogen peroxide in the mitochondria are downregulated, indicating the CS at 10oC may accumulate the ROS after present of sexual reproduction. (PNG 147 kb)
The expression level of cold-inducible RNA binding protein (CIRBP). The high level of CIRBP could bypass senescence and reduce oxidative damage. The differences between CR strains at the two temperatures are much smaller than in the CS strains, suggesting a possible way CR strains escape mortality. (PNG 23 kb)
(a) Four gene sets related to stem cell maintenance were down-regulated in CS at 10oC. (b) Three important genes in relation to the stem cell self-renewal gene sets were down-regulated in CS at 10oC. (PNG 203 kb)
(XLSX 280 kb)
Data Availability Statement
Raw RNAseq data from this manuscript were deposited to the Sequence Read Archive under BioProject PRJNA559195. This Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GHUC00000000. The version described in this paper is the first version, GHUC01000000.