

Abstract
The androgen receptor (AR) is the major driver of prostate cancer growth and survival. However, almost all patients relapse with castration resistant disease (CRPC) when treated with anti-androgen therapy. In CRPC, AR is often aberrantly activated independent of androgen. Targeting survival pathways downstream of AR could be a viable strategy to overcome CRPC. Surprisingly, little is known about how AR drives prostate cancer survival. Furthermore, CRPC tumors in which Pten is lost are also resistant to eradication by PI3K inhibitors. We sought to identify the mechanism by which AR drives tumor survival in CRPC to identify ways to overcome resistance to PI3K inhibition. We found that integrin α6β1 and Bnip3 are selectively elevated in CRPC downstream of AR. While integrin α6 promotes survival and is a direct transcriptional target of AR, the ability of AR to induce Bnip3 is dependent on adhesion to laminin and integrin α6β1-dependent nuclear translocation of HIF1α. Integrin α6β1 and Bnip3 were found to promote survival of CRPC cells selectively on laminin through the induction of autophagy and mitophagy. Furthermore, blocking Bnip3 or integrin α6β1 restored sensitivity to PI3K inhibitors in Pten-negative CRPC. We identified an AR driven pathway that cooperates with laminin and hypoxia to drive resistance to PI3K inhibitors. These findings can help explain in part why PI3K inhibitors have failed in clinical trials to overcome AR-dependent CRPC.
Keywords: prostate cancer, androgen receptor, integrin, PTEN, PI3K, drug resistance
INTRODUCTION
Death from prostate cancer is due to resistance to anti-androgen therapy (ADT), referred to as castration-resistant prostate cancer (CRPC). ADT blocks the transcriptional functions of the androgen receptor (AR) by depriving it of androgen. Despite the approval of two new ADT drugs, resistance to this therapy persists and almost all ADT-treated patients die from this disease. Many mechanisms of resistance have been proposed, but the most common theme is that AR remains functionally intact.13 DNA sequencing studies demonstrate that over 84% of CRPC tumors sustain genetic alterations in the AR gene or in members of the AR signaling pathway, such that the tumors remain dependent on AR but no longer require androgen. There has not been a single agent in clinical trials tested to date that can overcome this AR-dependent resistance, including immunotherapy.16, 51 Unfortunately, the signaling pathways downstream of AR that drive tumor cell survival during and after therapy resistance are essentially unknown.
Loss of Pten or constitutive activation of PI3K signaling occurs in ~60% of CRPC.30, 37 Thus, it was expected that pharmacological inhibitors of PI3K and/or downstream effectors, such as Akt or mTor, would be effective therapies for CRPC. While these drugs can repress cancer cell growth in vitro and in xenograft transplants, none have been effective as single agents in CRPC patients.11 The basis for this resistance is likely due to unknown AR-dependent survival pathways, since several in vitro studies have demonstrated that blocking PI3K increases AR-mediated survival.
In prostate cancer, integrin β4 is downregulated while integrin α6 is upregulated causing pairing of α6 with β1.10 We demonstrated that AR inhibits the expression of integrin β4 while directly inducing the expression of integrin α6, which further conferred a survival advantage to cells adherent to laminin.28 Because laminin is a major extracellular matrix component of the lymph node and bone microenvironment,18, 42 this pathway could be an important mechanism for escaping ADT treatment especially in the bone, the most common site of prostate cancer metastasis.32 ADT is known to induce a hypoxic environment within the prostate2, 6 and bone is known to be hypoxic.22 The hypoxic response is mediated by HIF1α/HIF2α and elevated levels of HIF1α and HIF2α have been reported in CRPC and metastatic prostate cancer.5 Furthermore, hypoxia directly stimulates the expression of integrin α6 and β1.6, 44, 46 Thus, hypoxia-induced integrin α6β1 expression may provide an additional mechanism for prostate cancer survival upon ADT treatment.
HIF1α/HIF2α mediate their survival effects under hypoxic conditions in part through direct transcriptional induction of target genes. One established target is Bnip3.19, 47 Bnip3 is reportedly elevated in castration-resistant tumor cell lines after passage in vivo in castrated mice,8 and elevated Bnip3 expression in human prostate cancers predicts for poor outcome.9, 54 Bnip3, a BH3 family member, is localized on mitochondria and reportedly induces apoptosis, autophagy, or mitophagy.55 The mechanism by which it induces apoptosis seems to primarily be a result of excessive overexpression.4 Bnip3 is proposed to control autophagy through its ability to interact with Bcl-xL, thus removing the negative constraint on the pro-autophagy protein, Beclin.3 Bnip3 also contains an LC3-interacting region (LIR), which binds LC3 on nascent autophagy membranes and thereby recruits mitochondria into autophagosomes.57 Genetic evidence demonstrates a role for Bnip3, and its homologue Nix/Bnip3L, in regulating mitochondrial clearance.17,56 In this study, we tested the hypothesis that Bnip3 links the AR/integrin α6β1 pathway and the hypoxia pathway to selectively promote the survival of CRPC.
RESULTS
Elevated Integrin α6β1 and Bnip3 expression positively correlate with CRPC.
Gene expression data from two human patient-derived xenograft (PDX) LuCaP tumor models, comparing androgen-sensitive lines to their castration-resistant variants, revealed ~2-fold increase in integrin α6 and β1 mRNA in CRPC (Fig. 1A). Elevated integrin α6 protein expression, based on IHC staining, was previously observed in CRPC LuCaP PDX tumors.46 Basal integrin α6 expression was higher in castration-resistant C4–2 and C4–2B cells adherent to laminin relative to their parental androgen-sensitive LNCaP cells (Fig. 1B).24, 50, 53 Stimulation with synthetic androgen, R1881, increased both integrin α6 and β1 expression in all cell lines, but to a higher degree in the C4–2 and C4–2B cells (Fig. 1B). Conversely, integrin β4 was decreased by androgen as previously reported.28
Figure 1: Integrin α6β1 and Bnip3 are elevated in CRPC.

A) Expression of integrin α6 (ITGα6), integrin β1 (ITGβ1), and BNIP3 mRNA in castration-resistant (CR) human-derived xenografts (PDX) of LuCaP 23.1 and LuCaP 35 normalized to expression in androgen-sensitive respective parental tumors. B) Levels of androgen receptor (AR), integrin α6 (ITGα6), integrin β1 (ITGβ1), integrin β4 (ITGβ4), Bnip3, PSA, and tubulin in laminin-adherent LNCaP, C4–2, and C4–2B cells treated with (+) or without (-) 10nM R1881 for 24 h as assessed by immunoblotting. C) Levels of Bnip3 mRNA in laminin-adherent LNCaP and C4–2 cells treated with (+) or without (-) 10nM R1881 for 24 h as measured by qRT-PCR. D) Levels of Bnip3 expression in androgen-sensitive vs castrate-resistant (CR) LuCaP PDX tumors as assessed by IHC. LuCaP 35/CR, LuCaP 77/CR, LuCaP 105/CR, and LuCaP 86.2/CR. E) C4–2, C4–2B, and LNCaP cells adherent to laminin treated with (+) or without (-) 10nM R1881 for 24 h and the levels of HIF1α, HIF2α, Bnip3, Bcl-XL, and tubulin assessed by immunoblotting. *p<0.05, **p<0.01, ***p< 0.005, n=3 biological replicates, error bars = SD
Gene expression data from the PDX LuCaP tumor models, revealed an increase in Bnip3 mRNA, specifically in the CRPC tumors (Fig. 1A). Microarray data comparing LNCaP and C4–2 cells previously identified a 2.4-fold increase in Bnip3 mRNA in C4–2 cells.8 We similarly observed increased Bnip3 expression in C4–2 and C4–2B cells adherent to laminin relative to LNCaP, at both the transcript and protein level following androgen stimulation (Fig. 1B,C). IHC staining of androgen-sensitive versus castrate-resistant LuCaP PDX tumors similarly revealed elevated Bnip3 expression in CRPC (Fig. 1D). Thus, elevated integrin α6β1 and Bnip3 expression is associated with castration resistance and both are stimulated by androgen.
Androgen stimulates Bnip3 expression through Integrin α6 and HIF1α.
Bnip3 is a classic hypoxia target directly induced by HIF1α.19, 47 HIF1α and HIF2α are highly elevated in the two C4–2 castration-resistant lines relative to LNCaP (Fig. 1E). As seen previously, Bcl-xL is also induced by androgen28 and interestingly it is more elevated in the CRPC lines (Fig. 1E), consistent with increased AR signaling in these cells. We compared the kinetics of Bnip3 mRNA induction by R1881 to the known AR target, PSA. While PSA mRNA increased within 3–6 hours, Bnip3 transcript did not significantly increase until 18 hours (Fig. 2A). Furthermore, blocking protein synthesis with cycloheximide prevented R1881 from increasing Bnip3 transcription (Fig. 2B) indicating Bnip3 is not a direct target of AR. We monitored the localization and induction of HIF1α, integrin α6, and AR over time following androgen stimulation. In the first 6 hours, AR shifted into the nucleus and the direct targets of AR transcription, PSA and integrin α6, increased as expected (Fig. 2C). C4–2 cells constitutively express HIF1α, and while androgen does not significantly increase its expression (Fig. 1E), it did induce nuclear accumulation of HIF1α at 12 hours. Bnip3 protein did not increase until after 12 hours (Fig. 2C), indicating androgen induction of integrin α6 and HIF1α nuclear localization occurs prior to Bnip3 induction.
Figure 2: Androgen indirectly induces Bnip3 through integrin α6β1 and HIF1α.

A) Levels of PSA and Bnip3 mRNA in laminin-adherent C4–2 cells over a 24 h time course following treatment with 10nM R1881 measured by qRT-PCR and normalized to time 0. *p<0.05 relative to time 0, n=3 biological replicates, error bars = SD. B) Levels of Bnip3 mRNA in laminin-adherent VCaP, LNCaP, and C4–2 cells treated with (+) or without (-) 10nM R1881 in the absence (Veh) or presence of 10μg/mL cycloheximide (CHX) for 24 hours. Expression is relative to vehicle control. *p<0.05 relative to vehicle, n=3 biological replicates, error bars = SD. C) Levels of integrin α6 (ITGα6), Bnip3, PSA, and GAPDH in the cytosol (Cyto) and levels of androgen receptor (AR), HIF1α, and histone 3 (HH3) in the nucleus (Nuc) of laminin-adherent C4–2 cells treated with 10nM R1881 over a time course of 24 h as measured by immunoblotting. D) Laminin-adherent C4–2 cells stably expressing Tet-inducible shRNA targeting integrin α6 (Tet-shITGα6) were treated with (+) or without (-) doxycycline (Dox) for 48 h, and then stimulated with (+) or without (-) 10nM R1881 for 24 h. The levels of integrin α6 (ITGα6), Bnip3, PSA, and GAPDH in the cytosol (Cyto) and androgen receptor (AR), HIF1α, and histone 3 (HH3) in the nucleus (Nuc) were assessed by immunoblotting. E) Laminin-adherent C4–2 cells were transiently transfected with HIF1α or scrambled siRNA and 48 h later treated with 10nM R1881 for 24 h. The levels of Bnip3, PSA, and GAPDH in the cytosol (Cyto) and AR, HIF1α, and histone H3 (HH3) in the nucleus (Nuc) were assessed by immunoblotting.
Inhibition of integrin α6 expression by Tet-inducible shRNA prevented Bnip3 induction in response to androgen and blocked HIF1α nuclear localization (Fig. 2D). Inhibition of HIF1α by two different siRNAs prevented androgen-induced Bnip3 mRNA and protein expression (Fig. 2E, Supplemental Fig. S1A,B), but did not suppress integrin α6 expression (Supplemental Fig. S1B). These data indicate integrin α6 is required for androgen to stimulate HIF1α nuclear translocation to induce Bnip3 expression.
Integrin α6 and Bnip3 protect C4–2 cells from PI3K inhibition.
We found that C4–2 cells are ten times more resistant to the class I-specific PI3K inhibitor, PX-866, than the parental LNCaP cells; LNCaP LD50 is 40nM, while C4–2 is 400nM (Supplemental Fig. S2). Furthermore, treatment of C4–2 cells with androgen made them completely resistant to 500nM PX-866. One possibility is that androgen increases PI3K signaling or reduces the efficacy of PX-866 to block PI3K signaling. While, androgen did increase Akt activation, as previously reported,29, 36, 45 it did not prevent PX-866 from inhibiting Akt at the higher doses (Fig. 3A). To determine whether integrin α6 is required to protect C4–2 from PX-866, we blocked integrin α6 expression with shRNA or plated cells on fibronectin. Blocking either integrin α6 or adhesion to laminin blocked the ability of androgen to protect cells from PX-866 (Fig. 3B,C). Thus, integrin α6-mediated adhesion to laminin is required for androgen to protect C4–2 cells from PI3K inhibition.
Figure 3: AR confers resistance to PI3K inhibition via integrin α6β1.

A) Laminin-adherent C4–2 cells were treated with (+) or without (-) 10nM R1881 for 24 h prior to and in the presence of 0, 100, 200, and 400nM PX-866. Levels of activated Akt (P-Akt-308; P-Akt-473) and total Akt were measured by immunoblotting 48 h later. B) Laminin-adherent C4–2 cells stably expressing Tet-inducible shRNA targeting integrin α6 (Tet-shITGα6) were treated with (+) or without (-) 100ng/mL doxycycline (Dox) for 48 h, then treated with (+) or without (-) 10nM R1881 24 h prior to and in the presence (PX866) or absence (DMSO) of 500nM PX-866. Cell viability was assessed by trypan blue exclusion 48 h later. Levels of integrin α6 (ITGα6) and tubulin assessed by immunoblotting. C) C4–2 cells plated on laminin (LM +) or on fibronectin (LM -) were treated with (+) or without (-) 10nM R1881 for 24 h prior to and in the presence (PX866) or absence (DMSO) of 500nM PX-866. Cell viability was measured by trypan blue exclusion 48 h later. ****p<0.001 relative to PX866 without R1881, n=3 biological replicates, error bars = SD.
To determine whether Bnip3 is a downstream target of integrin α6-mediated survival, Bnip3 expression was inhibited with two different Tet-inducible shRNAs. Without Bnip3, androgen stimulation could not protect the cells from death induced by PX-866 (Fig. 4A,B). PX-866 effectively blocked Akt activation without inhibiting Bnip3 expression and the shRNAs effectively knocked down Bnip3 expression (Fig. 4C). Thus, Bnip3 is required to protect tumor cells from PI3K inhibition.
Figure 4: AR confers resistance to PI3K inhibition via Bnip3.

A, B) Laminin-adherent C4–2 cells stably expressing two different Tet-inducible Bnip3 shRNAs (Tet-shBnip3) were treated with (+) or without (-) 100ng/mL doxycycline (Dox) for 48, then with (+) or without (-) 10nM R1881 for 24 h prior to and in the presence (PX866) or absence (DMSO) of 500nM PX-866. Cell viability assessed by trypan blue exclusion 48 h later. C) Levels of activated Akt (P-Akt478), total Akt, Bnip3, and tubulin in (A) were assessed by immunoblotting. D-F) C4–2 cells stably expressing Tet-inducible Bnip3 shRNA (Tet-shBnip3) were injected orthotopically into prostates of male SCID mice. Mice were fed sucrose (Suc, Ctrl) or sucrose with doxycycline (D, Dox) for 2 weeks, and then half were further treated with PX-866 or vehicle for 4 more weeks. D) Tumors were harvested and weighed. *p<0.05; **p<0.01; ***p<0.005, n=7, error bars = SEM. E) Representative tumor tissues from mice were assessed for expression of Bnip3. F,G) Representative tumor tissues from mice were assessed for expression of F) cleaved caspase 3 or G) Ki67 by IHC and quantified. *p<0.05, **p<0.01, ***p<0.005, ****p< 0.0001, n=3 tumor samples, error bars = SD.
Bnip3 promotes CRPC tumor growth and survival.
A total of 1 × 106 C4–2 cells harboring Tet-inducible Bnip3 shRNA were injected orthotopically into the ventral prostates of male SCID mice. Immediately after tumor cell injection, half of the mice were fed doxycycline in 5% sucrose in their drinking water, the other half only received sucrose. After 2 weeks, half the mice in each group were then treated with 2mg/kg PX-866 or vehicle for 4 weeks.25 The tumors from mice treated with doxycycline (i.e. loss of Bnip3) or PX-866 (inhibition of PI3K) were reduced in size 1.7-fold relative to control (Fig. 4D). Tumors treated with both Dox and PX-866 were reduced 3-fold relative to control. We verified decreased Bnip3 expression in the doxycycline-treated tumors (Fig. 4E). PX-866 treatment alone resulted in a significant increase in Bnip3 expression, some of which was suppressed upon Dox-induced shBnip3 (Fig. 4E). Loss of Bnip3 and inhibition of PI3K each increased cell death, as measured by quantification of cleaved caspase 3 staining (Fig. 4E). Only loss of Bnip3 was effective at reducing proliferation as quantified by Ki67 immunostaining (Fig. 4F). The ability of PX-866 and Dox-induced Bnip3 to further reduce tumor growth is likely a result of combined enhanced cell death and reduced proliferation. Thus, Bnip3 contributes to castration-resistant tumor growth and survival and its inhibition can overcome resistance to PI3K inhibition.
Adhesion to laminin via integrin α6 is required for androgen-induced autophagy in CRPC.
Bnip3 could be promoting CRPC survival through autophagy and/or mitophagy55, but either mechanism requires an overall increase in autophagy. We first measured autophagy by examining the accumulation of GFP-labelled LC3B puncta on autophagosomes in LNCaP versus C4–2 cells adherent to laminin following androgen stimulation with and without the lysosomal inhibitor bafilomycin A1.27 At steady state, both LNCaP and C4–2 have similar numbers of autophagosomes, and was not significantly changed by the addition of androgen (Fig. 5A,B). However, treatment with bafilomycin, to stop lysosomal turnover of autophagosomes and LC3B-II degradation, reveals that autophagosome accumulation occurs to a significantly higher extent in C4–2 cells, which is further enhanced by androgen, compared to LNCaP cells (Fig. 5A,B).
Figure 5: Androgen-induced autophagy in CRPC requires laminin.

A) LNCaP and C4–2 cells stably expressing GFP-LC3 and adherent to laminin were treated with (+) or without (-) 10nM R1881 for 24 h in the absence (-) or presence (+) of 100ng/mL Bafilomycin A1 (BafA1) during the last 2 h. Fixed cells were stained with Hoechst and imaged by epifluorescence microscopy. B) Quantification of puncta from (A). GFP puncta were counted as positive if the signal was 10 standard deviations greater than background fluorescence. Average is the number of puncta in at least 50 randomly selected individual cells under each condition. *p<0.05, **p<0.01 relative to untreated, n=50 cells repeated 3x, error bars = SD. C, D) LNCaP and C4–2 cells adherent to C) laminin or D) fibronectin were treated with (+) or without (-) 10nM R1881 for 24 h and in the absence (-) or presence (+) of 100ng/mL BafA1 during the last 2 h. Levels of Bnip3, LC3B-I/II, and tubulin as assessed by immunoblotting.
To evaluate the contribution of integrin α6, we first compared androgen-induced autophagy on laminin versus fibronectin by measuring the conversion of LC3B-I to LC3B-II by immunoblotting. Androgen alone did not induce LC3B-II, but androgen plus bafilomycin resulted in a significant increase in LC3B-II levels specifically in C4–2 cells on laminin (Fig. 5C, Supplementary Fig. S3A). Combined androgen and bafilomycin did not cause any significant change in the rate of autophagy in either cell line plated on fibronectin (Fig. 5D). Because Bnip3 binds to LC3B,20, 57 it is also degraded upon fusion of the autophagosome with the lysosome. Bnip3, like LC3B-II, accumulated significantly in C4–2 cells upon combined androgen and bafilomycin treatment (Fig. 5C, Supplementary Fig. S3B). This happened only on laminin and not on fibronectin (Fig. 5D), further supporting the laminin-specific induction of autophagy in response to androgen. To determine if the androgen-induced autophagic flux occurring on laminin is mediated by integrin α6, we generated C4–2 cells stably expressing scrambled or integrin α6 shRNA. Blocking integrin α6 expression prevented the accumulation of LC3B-II and Bnip3 in C4–2 cells in the presence of androgen and bafilomycin (Fig. 6A, Supplementary Fig. S3C).
Figure 6: Androgen-induced autophagy requires integrin α6 and Bnip3.
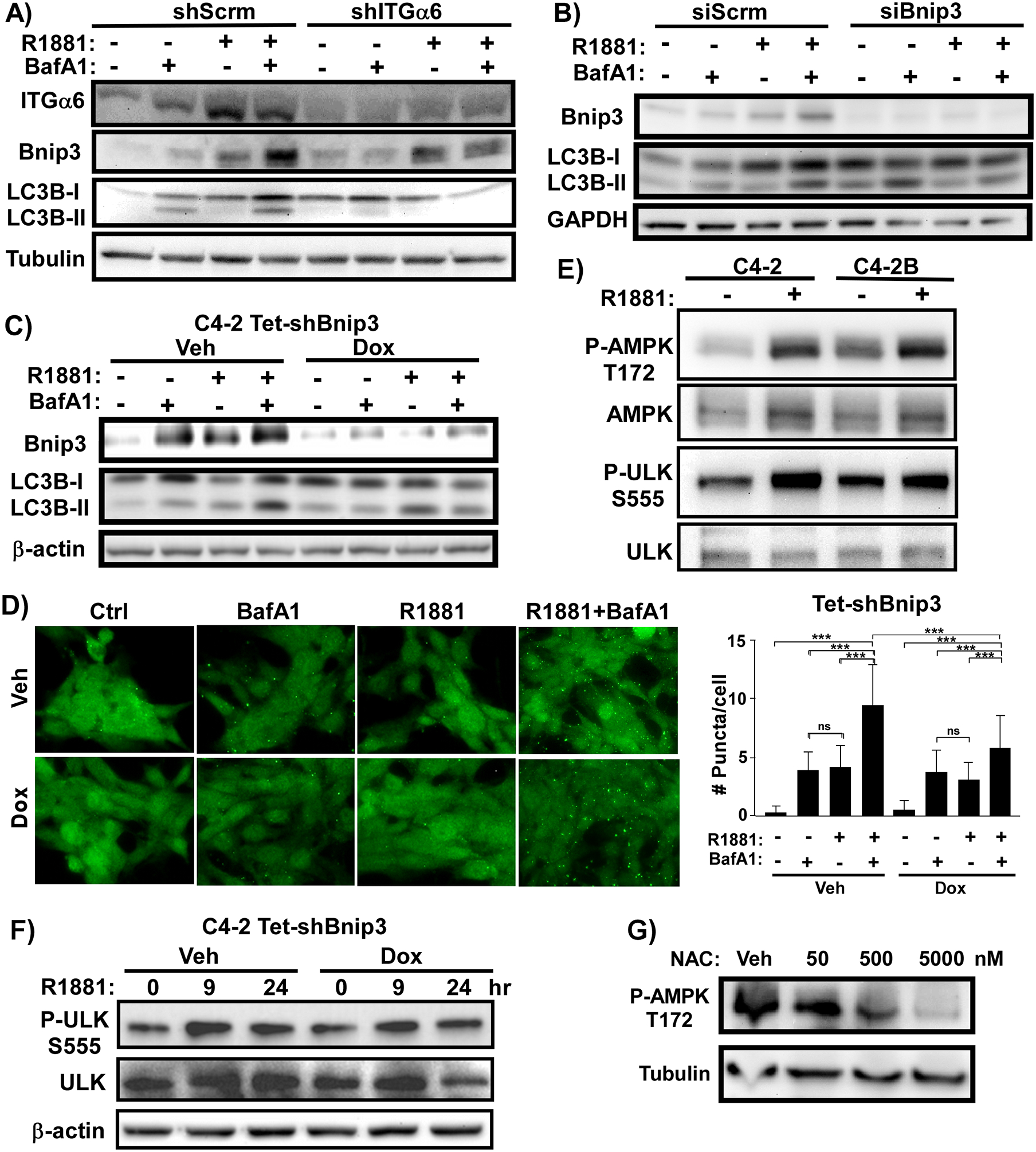
A) Laminin-adherent C4–2 cells stably expressing an shRNA targeting integrin α6 (shITGα6) or a control scrambled shRNA (shScrm) treated with (+) or without (-) 10nM R1881 for 24 h in the absence (-) and presence (+) of 100ng/mL BafA1 during the last 2 h. Levels of Bnip3, LC3B-I/II, and tubulin as assessed by immunoblotting. B) Laminin-adherent C4–2 cells transfected with siRNA targeting Bnip3 (siBnip3) or a control scrambled siRNA (siScrm) treated with (+) or without (-) 10nM R1881 for 24 h in the absence (-) or presence (+) of 100ng/mL BafA1 during the last 2 h. Levels of Bnip3, LC3B-I/II, and GAPDH as assessed by immunoblotting. C) Laminin-adherent C4–2 cells stably expressing Tet-inducible shRNA targeting Bnip3 (Tet-shBnip3) were treated with (Dox) or without (Veh) 100ng/ml doxycycline for 48 h and then treated with (+) or without (-) 10nM R1881 for 24 h in the absence (-) or presence (+) of 100ng/mL BafA1 during the last 6 h. Levels of Bnip3, LC3B-I/II, and β-actin as assessed by immunoblotting. D) Laminin-adherent C4–2 cells stably expressing Tet-inducible shRNA targeting Bnip3 (Tet-shBnip3) were treated with (Dox) or without (Veh) 100ng/ml doxycycline for 48 hours, and then treated with (+) or without (-) 10nM R1881 for 24 h and in the absence (-) or presence (+) of 100ng/mL BafA1 during the last 6 h. Endogenous LC3-positive puncta were visualized by immunostaining and quantified. ***p<0.005, n=100 cells, error bars = SD. E) Laminin-adherent C4–2 or C4–2B cells treated with (+) or without (-) 10nM R1881 for 24 h. Levels of activated (P-AMPK T172), total AMPK, activated (P-ULK S555), and total ULK as assessed by immunoblotting. F) Laminin-adherent C4–2 cells stably expressing Tet-inducible shRNA targeting Bnip3 (Tet-shBnip3) treated with (Dox) or without (Veh) 100ng/ml doxycycline for 48 hours and then stimulated with (+) or without (-) 10nM R1881 for 24 h in the presence (+) or absence (-) of 100ng/mL BafA1 during the last 6 h. Levels of activated ULK (P-ULK-S555), total ULK, and β-actin as assessed by immunoblotting. G) C4–2 cells adherent to laminin treated with 10nM R1881 were further treated with increasing concentrations of NAC. Levels of activated AMPK (P-AMPK-T172) and tubulin as assessed by immunoblotting.
Bnip3 facilitates autophagosome accumulation by androgen in CRPC.
To determine if Bnip3 is required for androgen-induced autophagy on laminin, we blocked Bnip3 expression with siRNA or Tet-inducible shRNA and measured LC3B-II accumulation in the presence of androgen and bafilomycin. Bnip3 loss only partially reduced androgen-induced accumulation of LC3B-II in the presence of bafilomycin (Fig. 6B,C, Supplementary Fig. S3D).
To determine if Bnip3 is required for the induction of autophagosomes, we measured endogenous LC3B accumulation in puncta by immunostaining. Combined treatment with Bafilomycin and androgen significantly increased puncta formation (Fig. 6D). Inhibition of Bnip3 by Tet-inducible shRNA only partially reduced the extent of puncta formation.
AMPK phosphorylation at Thr172 and is induced in response to low levels of ATP, which is a trigger for autophagy. AMPK in turn phosphorylates ULK to initiate autophagy.1 Stimulation of CRPC cells with androgen induced AMPK and ULK phosphorylation (Fig. 6E) around 6 hours and continued to increase over 24 hours (Supplementary Fig. S4A). However, suppression of Bnip3 did not inhibit the ability of androgen to induce ULK phosphorylation (Fig. 6F). Androgen signaling in castration-resistant cells leads to elevated ROS,49 which can stimulate AMPK activation.41 Indeed, treatment of C4–2 cells with the ROS scavenger, N-acetyl cysteine (NAC), decreased androgen-induced AMPK phosphorylation in C4–2 cells (Fig. 6G).
All together these data indicate that androgen-induced autophagic flux upon integrin α6-dependent adhesion to laminin is controlled in part by Bnip3 through its contribution to the autophagosome assembly, downstream of ROS, AMPK, and ULK, as previously suggested based on its interaction with Beclin.31
Bnip3-mediated mitophagy protects CRPC cells from PI3K inhibition.
The N-terminus of Bnip3 has an LC3-interacting region (LIR) responsible for bringing mitochondria into the autophagosome for degradation. Mutating LIR abrogates Bnip3-induced mitophagy.20, 57 To test whether Bnip3 promotes cell survival through mitophagy, we generated a C4–2 cell line stably expressing a constitutive shRNA targeting the 3’ UTR of Bnip3. We then used a Tet-inducible vector to re-express WT or an LIR Bnip3 mutant (ΔLIR) in the Bnip3-knocked down cells. Because endogenous Bnip3 is constitutively suppressed in these cells, androgen could not rescue them from death induced by PX-866 treatment (Fig. 7). Inducing WT Bnip3 expression with doxycycline protected the cells from PX-866-induced death (Fig. 7A,C). However, the ΔLIR mutant prevented this rescue (Fig. 7B,C). Thus, the pro-mitophagy function of Bnip3 is required to promote R1881-dependent survival and protects CRPC cells from PI3K inhibition.
Figure 7: Bnip3 LC3-interaction domain is required to promote resistance to PI3K inhibition.
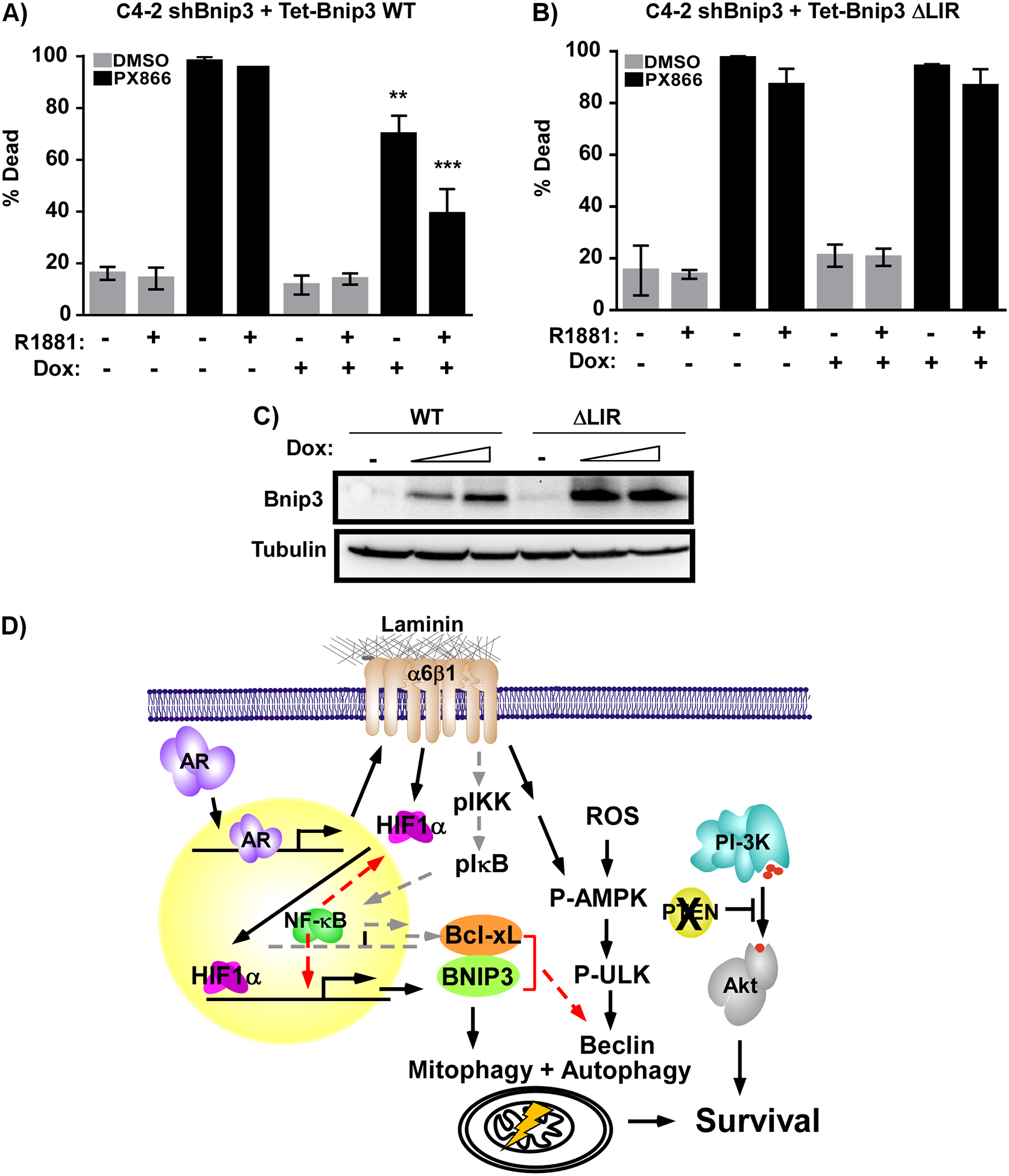
C4–2 cells stably expressing constitutive shRNA targeting the 3’-UTR of Bnip3 (C4–2-shBnip3) and A) Tet-inducible WT (Tet-Bnip3 WT) or B) ΔLIR mutant Bnip3 (Tet-Bnip3 ΔLIR) adherent to laminin were treated with (+) or without (-) 10nM R1881 in the presence (+) or absence (-) of 100ng/mL doxycycline (Dox) for 24 h prior to and in the absence (DMSO) and presence (PX866) of 500nM PX-866. Cell viability assessed by trypan blue exclusion 48 hours later. n=3 biological replicates; **p<0.01, ***p<0.005 relative to PX866-treated cells in the absence of Dox, error bars = SD. C) As in A and B, except cells were stimulated with 100ng/ml or 1000ng/ml doxycycline (Dox) for 48 hours and levels of Bnip3 and tubulin measured by immunoblotting. D) Proposed model for how AR/integrin α6 signaling to Bnip3 promotes resistance to PI3K inhibition (black arrows) along with previously identified underlying NF-κB signaling pathway (grey arrows). Potential crosstalk between these pathways is indicated by red arrows.
DISCUSSION
There is a marked selection for increased integrin α6β1 expression and decreased integrin β4 expression in prostate cancer.10, 11, 21 Elevated integrin α6β1 is associated with increased invasiveness, lymph node metastasis, and bone metastasis.21, 34 We previously demonstrated that AR is responsible for directly inducing integrin α6 transcription and expression in tumor cells.28 In this study, we demonstrate that integrin α6β1 expression is further elevated in CRPC. We previously demonstrated that adhesion to laminin via integrin α6β1, confers resistance to PI3K inhibition in Pten-null prostate cancer cells by inducing NF-κB signaling and up-regulating the anti-apoptotic protein Bcl-XL (Fig. 7D, grey arrows).28 In this study, we identified another integrin α6β1-mediated survival pathway that is selectively operating in CRPC (Fig. 7D, black arrows). Castration-resistant cells harboring active AR stimulate integrin α6β1 expression. Engagement of laminin by integrin α6β1 promotes nuclear translocation of elevated HIF1α already present in the CRPC cells leading to the induction of Bnip3. Simultaneously, elevated ROS and integrin α6 triggers AMPK/ULK1 activity to initiate autophagy. Bnip3 assists in the assembly of autophagosomes and recruits mitochondria for targeted degradation. Presumably, this reduces the level of damaged mitochondria to prevent apoptosis and promote survival. Independently and in parallel, constitutive activation of PI3K by Pten loss also promotes survival. Blocking both pathways is required to effectively induce tumor cell death. Previously published data suggest there may be additional interactions within this pathway (Fig. 7D, red arrows). NF-κB can increase expression of HIF1α and Beclin, but also binds the promoter of Bnip3 to limit its transcription.33, 40, 48 Finally, increased Bcl-XL/Bnip3 complexes can promote autophagy by releasing Beclin from Bcl-XL.3
The association of castration-resistance with elevated Bnip3 expression is intriguing and our finding that AR and integrin α6β1 control Bnip3 transcription suggests an interesting mechanism for both acquiring and maintaining a castration-resistant state. Androgen deprivation therapy, particularly in the bone, leads to increased hypoxia39 and thus elevated HIF1α expression. Recent studies indicate that not only is Bnip3 a direct transcriptional target of HIF1α, so is integrin α6β1.6, 44 Thus, under conditions of hypoxia and no androgen, both integrin α6β1 and Bnip3 are elevated, which promotes survival of tumor cells by limiting ROS production through selective removal of mitochondria. Prostate tumors that amplify AR or gain androgen-independent AR function (through mutation or splice variants), can further sustain and amplify this Bnip3-dependent survival pathway. These castration-resistant tumors may even become adapted to tolerating elevated ROS. In fact, AR increases ROS and ROS induces HIF1α and AMPK signaling.26, 49 Thus, ROS production caused by AR signaling, particularly in cells with elevated AR expression, may be responsible for causing the increasing both HIF1α signaling to Bnip3 while simultaneously stimulating autophagy and mitophagy.
Our studies emphasize the importance of the tumor microenvironment when attempting to understand mechanisms of castration-resistance. Laminin is a major ECM component in lymph nodes and bone, the major sites for prostate cancer metastasis,18, 42 and integrin α6β1 is the major integrin expressed on prostate tumors in these tissues.21, 34 Our previous study demonstrated that activation of the NF-κB/Bcl-XL survival pathway is only observed on laminin, not fibronectin or collagen.28 Here we demonstrate that adhesion to laminin, but not fibronectin, induces Bnip3 and autophagy uniquely in castration-resistant cells. Moreover, we previously showed that only adhesion to laminin, but not fibronectin, generates the resistance to PI3K inhibition in the presence of androgen.28 Interestingly, adhesion to collagen creates resistance to PI3K inhibition independently of AR activation.28 We are currently exploring this collagen-dependent pathway and find that neither Bcl-XL or Bnip3 is induced on collagen.
It is well-established that autophagy promotes therapeutic resistance in cancer, and targeting autophagy has provided some promising results in other cancers,52 but has not necessarily translated well to patients. This is complicated by constitutive PI3K signaling in prostate cancer due to loss of Pten or constitutively activated PI3K mutants,30 which suppresses autophagy. Given the high level of PI3K activation in prostate cancer it is surprising that PI3K/mTor inhibitors have not been effective as single agents, or even in combination with anti-androgen therapies.11 Our data indicate that single agent PI3K inhibitors won’t work because AR/α6β1 integrin signaling is still active, which suppresses apoptosis via Bcl-XL28 and promotes Bnip3-mediated autophagy and mitophagy. Furthermore, PI3K inhibition will further stimulate autophagy to augment AR/α6β1 signaling. In combination with anti-androgen therapies, this pathway will still be active due to acquisition of androgen-independent AR signaling and/or gain in hypoxia/ROS signaling. Thus, a multi-prong approach that includes targeting PI3K, hypoxia, and other AR-downstream targets, like integrin α6β1, will be necessary to effectively suppress bone metastatic CRPC.
MATERIALS AND METHODS
Cell Culture:
Tissue culture plates were coated with 10μg/mL laminin (Gibco: 23017–015) in Ca+/Mg+-free PBS overnight at 4°C and then blocked with 1% BSA at 37°C for 1 hour prior to plating cells. For the few experiments where cells were not plated on laminin, they were plated in serum, which contains fibronectin. LNCaP cells, purchased from ATCC, and C4–2 cells, obtained directly from Dr. Robert Sikes,8 were validated by STR analysis in our Genomics core. Cells were maintained on laminin in RPMI supplemented with 10% FBS, 1mM sodium pyruvate, 2mM glutamine, 0.3% glucose, 10mM HEPES, and 30U/mL Pen/Strep. HEK293FT cells (Clontech), used for lentivirus generation, were maintained in DMEM supplemented with 10% HIFBS and 30U/mL Pen/Strep. All cells were tested every 3 months for mycoplasma.
Drug treatments:
Prior to drug treatment, cells were placed in starvation media (Phenol red-free RPMI supplemented with 0.1% charcoal-stripped serum). When needed for shRNA induction, 100ng/mL doxycycline was added and after 24 hours, cells were stimulated with 10nM R1881 (Sigma) or ethanol and re-spiked with doxycycline. Twenty-four hours later, cells were then treated with PX-86623 or DMSO for 48 hours.
Cell death assay:
Attached and floating cells were collected, pooled, and stained with Trypan blue. For each individual experiment 3 wells per condition and at least 3 grids per well were counted on a hemocytometer.
qRT PCR:
RNA was isolated using Trizol or RNEasy kit from Qiagen. RNA was reverse transcribed using MuLV reverse transcriptase (New England Biolabs) with a mix of random hexamers and polyT primers. cDNA was amplified using FastStar Universal SYBR Green Master (Rox, Roche) in the Applied Biosystems 7500 RT PCR System. Target mRNAs were normalized to 18S ribosomal RNA. List of primers is in Supplementary Table S1. When used, cycloheximide in ethanol was added to a final concentration of 10μg/mL coincidental with R1881 treatment.
Immunoblotting:
30–60μg of protein were separated on precast tris-glycine gels (Invitrogen) and transferred to PVDF membrane. Membranes were blocked with 5% BSA/TBST. Primary antibodies, after incubation in 5% BSA/TBST, were detected using HRP conjugated secondary antibodies in chemiluminescent solution using the Quantity One imaging software on a Bio-Rad Gel Docking system. Primary antibodies: Rabbit mAb Bnip3 (EPR4034) from Abcam, rabbit anti-P-AktSer473, rabbit mAb P-Akt308 (C31E5E), rabbit mAb Akt (pan) (C67E7), mouse mAb Histone H3 (96C10), rabbit mAb P-Ser555 ULK (D1H4, #5869), rabbit mAb ULK (D8H5, #8054), rabbit mAb P-AMPKα (Thr172) (40H9, #2535), and rabbit anti-AMPKα (#2532) from Cell Signaling Technology, rabbit anti-LC3B (NB100–2200) from Novus Biologicals, mouse mAb GAPDH from Millipore, rat anti-integrin α6 (GoH3) and mouse anti-HIF1α from BD Pharmingen, mouse mAb AR (441) from Santa Cruz, mouse anti-α tubulin and β-actin-HRP mouse mAb from Sigma-Aldrich), and rabbit anti-integrin α6 (AA6A, A6NT).12, 35
Virus generation and infection:
For lentiviral constructs, 5 × 106 HEK293FT cells, and for retroviral constructs, 5 × 106 Phoenix-AMPO cells, were plated in DMEM with 10% HIFBS in a T75 flask that was coated with 2μg/mL Poly-D-lysine in PBS and left overnight at 37°C. Cells were transfected in Opti-MEM using Lipofectamine2000 with 5μg pLP1, 5μg pLP2, 5μg pVSV-G, and 5μg of target construct. After 24 hours, the media was changed to RPMI with 10% HIFBS and no antibiotics and returned to 37°C for 48 hours. Floating cells were spun out and the supernatant was passed through a 0.45μm filter. Polybrene, at 5μg/mL, was added to the filtered virus, this was added to target cells, and incubated at 32°C. Six hours later, cells were washed, returned to normal media, and subjected to antibiotic selection.
RNAi:
Twenty-four hours after plating on laminin, cells were transfected with 20nM siRNA from Dharmacon in antibiotic-free starvation media using siLentFect (BioRad). Twenty-four hours later, fresh starvation medium was added. siRNAs included two different HIF1α siRNAs (J-004018–07 and J-004018–08), one integrin α6 siRNA (5’-CGAGAAGGAAATCAAGACAAA-3’), one Bnip3 siRNA (J-004636–08-0005), and a non-targeting siRNA (D-001206–14). Doxycycline-inducible shRNA plasmids targeting integrin α6 and Bnip3 were generated by sub-cloning shRNA sequences into a Tet-inducible lentiviral vector, EZ-Tet-pLKO-Puro,14 available through Addgene (#85966). After infection of C4–2 cells, cells were selected in 2μg/mL puromycin and single cells were isolated to generate clonal lines. Targeting sequences are in Supplementary Table S2. The same integrin α6 targeting shRNA sequence was also cloned into pLKO.1 Puro (gift from Bob Weinberg (Addgene plasmid # 8453))43 and used to generate a stable constitutive shRNA cell line. A scrambled non-targeting shRNA was used as vector control.
LC3 quantification:
LNCaP C4–2 cells were infected with retrovirus containing pBABE-Puro GFP-LC3 (gift from Jayanta Debnath (Addgene plasmid # 22405))15 and selected with 2μg/mL puromycin. Cells on laminin-coated glass coverslips were serum starved for 24 h and then treated for 24 h with 10nM R1881 or vehicle (ethanol). During the last 2 hours, cells were treated with or without Bafilomycin A1 at 100ng/ml. Tet-shBnip3 C4–2 cells on laminin-coated glass coverslips in starvation medium were treated with or without 100ng/ml doxycycline for 48 h. Cells were then treated for 24 h with or without 10nM R1881 and during the last 6 hours treated with or without Bafilomycin A1 (100ng/ml). Cells were fixed in 4% paraformaldehyde in PBS at 4% for 10 min and then neutralized with 100mM glycine. Cells were permeabilized with 0.2% Triton-X for 3 min and blocked with 1% normal goat serum for 1–2 hours at RT. Cells were incubated with primary antibody to LC3B (NB600–1384 Novus) at 1:1000 in PBS/1%BSA overnight at 4°C. Secondary FITC-conjugated antibody at 1:500 in 1% BSA/PBS was added for 1 hr at RT. Puncta in 25 fields per condition per experiment were counted. Puncta were considered positive if they were 10 standard deviations brighter than background fluorescence and within the size range of an HBSS-treated positive control.
LC3-II immunoblot quantification:
Blot density was measured using ImageJ software. The density of LC3-II was normalized to tubulin density in the same lane. The ratio of the first lane was set to one, and subsequent lanes are relative to the control lane. Data was collected from 3 separate experiments.
Bnip3 re-expression:
pENTR223-Bnip3 (HsCD00366502) containing the Bnip3 cDNA, was obtained from the Harvard PlasmID repository.38 Site-directed mutagenesis was used to generate a stop codon in pENTR223-Bnip3. pENTR223-Bnip3 underwent two more rounds of site directed mutagenesis to eliminate the LC3-interacting region (LIR) to generate pENTR223-Bnip3 ΔLIR in which amino acids W18 and L21 were converted to alanine.20, 57 Mutagenesis primers are listed in Supplementary Table S3. Bnip3 WT and ΔLIR were each recombined into a Tet-inducible lentiviral vector, pLenti CMVTight Neo DEST (gift from Eric Campeau (Addgene plasmid # 26432)),7 using LR recombinase to generate the pLenti CMV-Tight Neo Bnip3 WT and ΔLIR. C4–2 cells selected in 2μg/mL puromycin and constitutively expressing Bnip3 shRNA targeting the 3’-UTR (SHCLNG-NM_004052, 5’-CCACGTCACTTGTGTTTATT-3’; Sigma-Aldrich) were infected with lentivirus expressing doxycycline-regulated rtTA in pLentiCMV rtTA3 Blast (Addgene #26429; Eric Campau)7 and further selected in 5μg/mL blasticidin. An isolated pool expressing rtTA was then infected with pLenti CMVTight Neo Bnip3 WT or ΔLIR lentivirus and further selected in 100ng/mL G418 (neomycin) to generate a triple antibiotic-resistant pool.
Mouse studies:
Mouse studies were conducted according to an IACUC approved protocol. 1 × 106 C4–2 Tet-shBnip3 cells in 10μL of DMEM were injected orthotopically into the prostates of 40 8-week-old castrated male SCID mice. Mice were randomly divided into 2 cohorts of 14 each. Half the mice received water containing 5% sucrose and the other half 5% sucrose with 2mg/ml doxycycline (Dox), replaced weekly. Two weeks after Dox-treatment, half the mice in each group were blindly randomized and given 2mg/kg PX866 three times a week by oral gavage for 4 weeks and the other half received drug diluent.25 Mice were sacrificed after the 4-week drug treatment. Prostate tumors were excised, weighed, and assessed by IHC. Ki67 and Caspase 3 IHC staining was quantified by counting number of positive cells in five random fields per sample. Counter was blinded to the treatment groups.
Immunohistochemistry:
Deparaffinized and rehydrated formalin fixed samples tumor samples were subjected to antigen retrieval using Dako S1699 Retrieval Buffer at pH 6.1 at 90oC for 30 mins. After a 5 min peroxidase blockade, slides were blocked for 10 min at RT with PBS containing 0.5% BSA, 5% goat serum, and 1X mouse/human FcR blocker (Miltenyi Biotec 130–092-575). Slides were incubated with primary anti-Bnip3 (EPR4034, Abcam) at 1:100 at RT for 30 min, rabbit mAb to cleaved Caspase-3 (Asp175) (5A1E) (Cell Signaling Technology) or anti-Ki67 (SP6) (Thermo-Scientific) at 1:100 overnight at 4oC. IHC was visualized by DAB staining kit (TL-015-HD by Invitrogen), counterstained with hematoxylin, and mounted in non-aqueous solution (Richard-Allen Scientific 4112).
Supplementary Material
ACKNOWLEDGEMENTS
We would like to thank Drs. Sander Frank and Don Tindall for feedback and constructive suggestions and Penny Berger for technical expertise. Special thanks to Scott Peterson for supplying the PX-866. These studies were supported by funding from NIH/NCI R01CA154835, P30CA023074 (CKM, SSG, EAN, VSS) and the Van Andel Research Institute. Additional support was provided by NIH/NCI CA159406 (AEC).
Footnotes
CONFLICT OF INTEREST
Authors declare no conflict of interest.
REFERENCES
- 1.Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol 2012; 32: 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anastasiadis AG, Stisser BC, Ghafar MA, Burchardt M, Buttyan R. Tumor hypoxia and the progression of prostate cancer. Curr Urol Rep 2002; 3: 222–228. [DOI] [PubMed] [Google Scholar]
- 3.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 2009; 29: 2570–2581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Berger PL, Frank SB, Schulz VV, Nollet EA, Edick MJ, Holly B et al. Transient induction of ING4 by Myc drives prostate epithelial cell differentiation and its disruption drives prostate tumorigenesis. Cancer Res 2014; 74: 3357–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boddy JL, Fox SB, Han C, Campo L, Turley H, Kanga S et al. The androgen receptor is significantly associated with vascular endothelial growth factor and hypoxia sensing via hypoxia-inducible factors HIF-1a, HIF-2a, and the prolyl hydroxylases in human prostate cancer. Clin Cancer Res 2005; 11: 7658–7663. [DOI] [PubMed] [Google Scholar]
- 6.Brooks DL, Schwab LP, Krutilina R, Parke DN, Sethuraman A, Hoogewijs D et al. ITGα6 is directly regulated by hypoxia-inducible factors and enriches for cancer stem cell activity and invasion in metastatic breast cancer models. Molecular cancer 2016; 15: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campeau E, Ruhl VE, Rodier F, Smith CL, Rahmberg BL, Fuss JO et al. A versatile viral system for expression and depletion of proteins in mammalian cells. PloS one 2009; 4: e6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Q, Watson JT, Marengo SR, Decker KS, Coleman I, Nelson PS et al. Gene expression in the LNCaP human prostate cancer progression model: progression associated expression in vitro corresponds to expression changes associated with prostate cancer progression in vivo. Cancer letters 2006; 244: 274–288. [DOI] [PubMed] [Google Scholar]
- 9.Chen X, Gong J, Zeng H, Chen N, Huang R, Huang Y et al. MicroRNA145 targets BNIP3 and suppresses prostate cancer progression. Cancer Res 2010; 70: 2728–2738. [DOI] [PubMed] [Google Scholar]
- 10.Cress AE, Rabinovitz I, Zhu W, Nagle RB. The α6β1 and α6β4 integrins in human prostate cancer progression. Cancer Metastasis Rev 1995; 14: 219–228. [DOI] [PubMed] [Google Scholar]
- 11.Crumbaker M, Khoja L, Joshua AM. AR signaling and the PI3K pathway in prostate cancer. Cancers 2017; 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demetriou MC, Pennington ME, Nagle RB, Cress AE. Extracellular α6 integrin cleavage by urokinase-type plasminogen activator in human prostate cancer. Experimental cell research 2004; 294: 550–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feng Q, He B. Androgen receptor signaling in the development of castration-resistant prostate cancer. Frontiers in oncology 2019; 9: 858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Frank SB, Schulz VV, Miranti CK. A streamlined method for the design and cloning of shRNAs into an optimized Dox-inducible lentiviral vector. BMC Biotechnol 2017; 17: 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fung C, Lock R, Gao S, Salas E, Debnath J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Molecular biology of the cell 2008; 19: 797–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galletti G, Leach BI, Lam L, Tagawa ST. Mechanisms of resistance to systemic therapy in metastatic castration-resistant prostate cancer. Cancer treatment reviews 2017; 57: 16–27. [DOI] [PubMed] [Google Scholar]
- 17.Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC et al. BNIP3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol 2012; 32: 2570–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gu YC, Kortesmaa J, Tryggvason K, Persson J, Ekblom P, Jacobsen SE et al. Laminin isoform-specific promotion of adhesion and migration of human bone marrow progenitor cells. Blood 2003; 101: 877–885. [DOI] [PubMed] [Google Scholar]
- 19.Guo K, Searfoss G, Krolikowski D, Pagnoni M, Franks C, Clark K et al. Hypoxia induces the expression of the pro-apoptotic gene BNIP3. Cell Death Differ 2001; 8: 367–376. [DOI] [PubMed] [Google Scholar]
- 20.Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 2012; 287: 19094–19104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Harryman WL, Hinton JP, Rubenstein CP, Singh P, Nagle RB, Parker SJ et al. The cohesive metastasis phenotype in human prostate cancer. Biochimica et biophysica acta 2016; 1866: 221–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hiraga T Hypoxic microenvironment and metastatic bone disease. Int J Mol Sci 2018; 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong DS, Bowles DW, Falchook GS, Messersmith WA, George GC, O’Bryant CL et al. A multicenter phase I trial of PX-866, an oral irreversible phosphatidylinositol 3-kinase inhibitor, in patients with advanced solid tumors. Clin Cancer Res 2012; 18: 4173–4182. [DOI] [PubMed] [Google Scholar]
- 24.Horoszewicz JS, Leong SS, Chu TM, Wajsman ZL, Friedman M, Papsidero L et al. The LNCaP cell line - a new model for studies on human prostatic carcinoma. Prog Clin Biol Res 1980; 37: 115–132. [PubMed] [Google Scholar]
- 25.Ihle NT, Paine-Murrieta G, Berggren MI, Baker A, Tate WR, Wipf P et al. The phosphatidylinositol-3-kinase inhibitor PX-866 overcomes resistance to the epidermal growth factor receptor inhibitor gefitinib in A-549 human non-small cell lung cancer xenografts. Mol Cancer Ther 2005; 4: 1349–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jung SN, Yang WK, Kim J, Kim HS, Kim EJ, Yun H et al. Reactive oxygen species stabilize hypoxia-inducible factor-1α protein and stimulate transcriptional activity via AMP-activated protein kinase in DU145 human prostate cancer cells. Carcinogenesis 2008; 29: 713–721. [DOI] [PubMed] [Google Scholar]
- 27.Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8: 445–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lamb LE, Zarif JC, Miranti CK. The androgen receptor induces integrin α6β1 to promote prostate tumor cell survival via NF-κB and Bcl-xL independently of PI3K signaling. Cancer Res 2011; 71: 2739–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee SH, Johnson D, Luong R, Sun Z. Crosstalking between androgen and PI3K/AKT signaling pathways in prostate cancer cells. J Biol Chem 2015; 290: 2759–2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Majumder PK, Sellers WR. Akt-regulated pathways in prostate cancer. Oncogene 2005; 24: 7465–7474. [DOI] [PubMed] [Google Scholar]
- 31.Mazure NM, Pouyssegur J. Atypical BH3-domains of BNIP3 and BNIP3L lead to autophagy in hypoxia. Autophagy 2009; 5: 868–869. [DOI] [PubMed] [Google Scholar]
- 32.Neri P, Bahlis NJ. Targeting of adhesion molecules as a therapeutic strategy in multiple myeloma. Current cancer drug targets 2012; 12: 776–796. [DOI] [PubMed] [Google Scholar]
- 33.Pietrocola F, Izzo V, Niso-Santano M, Vacchelli E, Galluzzi L, Maiuri MC et al. Regulation of autophagy by stress-responsive transcription factors. Seminars in cancer biology 2013; 23: 310–322. [DOI] [PubMed] [Google Scholar]
- 34.Pontes-Junior J, Reis ST, Dall’Oglio M, Neves de Oliveira LC, Cury J, Carvalho PA et al. Evaluation of the expression of integrins and cell adhesion molecules through tissue microarray in lymph node metastases of prostate cancer. Journal of carcinogenesis 2009; 8: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ports MO, Nagle RB, Pond GD, Cress AE. Extracellular engagement of α6 integrin inhibited urokinase-type plasminogen activator-mediated cleavage and delayed human prostate bone metastasis. Cancer Res 2009; 69: 5007–5014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Qi W, Morales C, Cooke LS, Johnson B, Somer B, Mahadevan D. Reciprocal feedback inhibition of the androgen receptor and PI3K as a novel therapy for castrate-sensitive and -resistant prostate cancer. Oncotarget 2015; 6: 41976–41987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Robinson D, Van Allen EM, Wu YM, Schultz N, Lonigro RJ, Mosquera JM et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015; 161: 1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rual JF, Hirozane-Kishikawa T, Hao T, Bertin N, Li S, Dricot A et al. Human ORFeome version 1.1: a platform for reverse proteomics. Genome Res 2004; 14: 2128–2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shabsigh A, Ghafar MA, de la Taille A, Burchardt M, Kaplan SA, Anastasiadis AG et al. Biomarker analysis demonstrates a hypoxic environment in the castrated rat ventral prostate gland. Journal of cellular biochemistry 2001; 81: 437–444. [PubMed] [Google Scholar]
- 40.Shaw J, Yurkova N, Zhang T, Gang H, Aguilar F, Weidman D et al. Antagonism of E2F-1 regulated Bnip3 transcription by NF-κB is essential for basal cell survival. Proceedings of the National Academy of Sciences of the United States of America 2008; 105: 20734–20739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sinha RA, Singh BK, Zhou J, Wu Y, Farah BL, Ohba K et al. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy 2015; 11: 1341–1357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sobocinski GP, Toy K, Bobrowski WF, Shaw S, Anderson AO, Kaldjian EP. Ultrastructural localization of extracellular matrix proteins of the lymph node cortex: evidence supporting the reticular network as a pathway for lymphocyte migration. BMC immunology 2010; 11: 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003; 9: 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang D, Yan T, Zhang J, Jiang X, Zhang D, Huang Y. Notch1 signaling contributes to hypoxia-induced high expression of integrin β1 in keratinocyte migration. Sci Rep 2017; 7: 43926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thomas C, Lamoureux F, Crafter C, Davies BR, Beraldi E, Fazli L et al. Synergistic targeting of PI3K/AKT pathway and androgen receptor axis significantly delays castration-resistant prostate cancer progression in vivo. Mol Cancer Ther 2013; 12: 2342–2355. [DOI] [PubMed] [Google Scholar]
- 46.Toth RK, Tran JD, Muldong MT, Nollet EA, Schulz VV, Jensen CC et al. Hypoxia-induced PIM kinase and laminin-activated integrin alpha6 mediate resistance to PI3K inhibitors in bone-metastatic CRPC. Am J Clin Exp Urol 2019; 7: 297–312. [PMC free article] [PubMed] [Google Scholar]
- 47.Tracy K, Dibling BC, Spike BT, Knabb JR, Schumacker P, Macleod KF. BNIP3 is an RB/E2F target gene required for hypoxia-induced autophagy. Mol Cell Biol 2007; 27: 6229–6242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.van Uden P, Kenneth NS, Rocha S. Regulation of hypoxia-inducible factor-1α by NF-κB. The Biochemical journal 2008; 412: 477–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Veeramani S, Yuan TC, Lin FF, Lin MF. Mitochondrial redox signaling by p66Shc is involved in regulating androgenic growth stimulation of human prostate cancer cells. Oncogene 2008; 27: 5057–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Veldscholte J, Berrevoets CA, Brinkmann AO, Grootegoed JA, Mulder E. Anti-androgens and the mutated androgen receptor of LNCaP cells: differential effects on binding affinity, heat-shock protein interaction, and transcription activation. Biochemistry 1992; 31: 2393–2399. [DOI] [PubMed] [Google Scholar]
- 51.Wang D, Tindall DJ. Androgen action during prostate carcinogenesis. Methods Mol Biol 2011; 776: 25–44. [DOI] [PubMed] [Google Scholar]
- 52.White E The role for autophagy in cancer. The Journal of clinical investigation 2015; 125: 42–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wu HC, Hsieh JT, Gleave ME, Brown NM, Pathak S, Chung LW. Derivation of androgen-independent human LNCaP prostatic cancer cell sublines: role of bone stromal cells. International journal of cancer 1994; 57: 406–412. [DOI] [PubMed] [Google Scholar]
- 54.Yoo NJ, Kim MS, Park SW, Seo SI, Song SY, Lee JY et al. Expression analysis of caspase-6, caspase-9 and BNIP3 in prostate cancer. Tumori 2010; 96: 138–142. [DOI] [PubMed] [Google Scholar]
- 55.Zhang J, Ney PA. Role of BNIP3 and NIX in cell death, autophagy, and mitophagy. Cell Death Differ 2009; 16: 939–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang J, Loyd MR, Randall MS, Waddell MB, Kriwacki RW, Ney PA. A short linear motif in BNIP3L (NIX) mediates mitochondrial clearance in reticulocytes. Autophagy 2012; 8: 1325–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Zhu Y, Massen S, Terenzio M, Lang V, Chen-Lindner S, Eils R et al. Modulation of serines 17 and 24 in the LC3-interacting region of Bnip3 determines pro-survival mitophagy versus apoptosis. J Biol Chem 2013; 288: 1099–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.