

Abstract
Sea cucumber species are abundant (>1400 species) and widely distributed globally. mtDNA sequencing is frequently used to identify the phylogenetic and evolutionary relationships among species. However, there are no reports on the mitochondrial genome of Phyllophorus liuwutiensis. Here, we performed mtDNA sequencing of P. liuwutiensis to examine its phylogenetic relationships with other echinoderms. Its mitochondrial genome (15 969 bp) contains 37 coding genes, including 13 protein‐coding genes, 22 tRNA genes and 2 rRNA genes. Except for one protein‐coding gene (nad6) and five tRNA genes encoded on the negative strand, all other genes were encoded on the positive strand. The mitochondrial bases of P. liuwutiensis were composed of 29.55% T, 22.16% C, 35.64% A and 12.64% G. The putative control region was 703 bp in length. Seven overlapping regions (1–10 bp) were found. The noncoding region between the genes ranged from 1 to 130 bp in length. One putative control region has been found in the P. liuwutiensis mitogenome. All of the tRNA genes were predicted to fold into a cloverleaf structure. In addition, we compared the gene arrangements of six echinoderms, revealing that the gene order of P. liuwutiensis was a new arrangement.
Keywords: mitochondrial genome, Phyllophorus liuwutiensis, sea cucumber, sequence analysis, structure characteristic
In this paper, we analyzed the mitochondrial genome of Phyllophorus liuwutiensis. We examined genome composition, base composition, codon usage of protein‐coding genes and gene arrangement. In addition, we compared the gene arrangements of six echinoderms, revealing that the gene order of P. liuwutiensis was a new arrangement.
Abbreviations
- CR
control region
- PCG
protein‐coding gene
- RSCU
relative synonymous codon usage
Sea cucumbers belong to the phylum Echinodermata, which are an important food source for human, particularly in some parts of Asia [1]. Sea cucumbers are nocturnal feeding species, hiding by day in coral reef rocks or sand [2]. The species of sea cucumbers are abundant and widely distributed throughout the world. There are more than 1400 species of sea cucumbers in the world, which are distributed in shallow sea areas, trenches and other marine environments [3]. Despite the variety and wide distribution of sea cucumbers, the phylogenetic and evolutionary relationships of sea cucumbers remain largely unknown.
In metazoan animals, the mitochondrial genomes are characterized by exposed circular double‐stranded DNA molecules [4]. It has been found that most mitochondrial genomes are self‐replicated and inherited in the maternal line. mtDNA sequencing is frequently used to identify the phylogenetic and evolutionary relationships among species [5, 6, 7]. Because of its rapid evolution and the lack of genetic recombination, mtDNA can provide important information about rearrangement laws and phylogenetic relationship [8]. The mtDNA contains 37 genes, including 13 protein‐coding genes (PCGs), 2 rRNA genes and 22 tRNA genes [9, 10]. At present, many studies have investigated the phylogeny and evolution of echinoderms. Mu et al. [3] have explored the adaptation of sea cucumber in a deep‐sea environment based on the research of the mtDNA. Fan et al. [11] have found a new sequence of genes in the mitochondrial genome of the Stichopus horrens. However, no study has been carried out on the mitochondrial genome of Phyllophorus liuwutiensis. Therefore, we, in this study, aimed to clarify the structure and composition of the mitochondrial genome of P. liuwutiensis, and its phylogeny and evolution were also discussed.
The sea cucumbers, P. liuwutiensis (Holothuroidea: Dendrochirotida: Phyllophoridae: Phyllophorus), are popularly distributed in the intertidal sandy bottom. In China, P. liuwutiensis is distributed in only the two provinces of Fujian and Guangdong. P. liuwutiensis has a thin, cylinder‐shaped body with a length of 90–200 mm and a diameter of 10–28 mm [12]. The whole body of P. liuwutiensis is covered by tube feet. It has a polian vesicle with a length of about 40 mm. The body wall bone is termed as a table, and the chassis has a round or irregular shape. Moreover, it has a hole in the center, and there are eight (or more than eight) other holes in the peripheral area. The rim of the chassis is an undulating shape, and the diameter of the disc ranges from 50 to 80 μm [12].
Materials and methods
Sample collection and identification
All animal handling procedures were reviewed and approved by the ethics committee of the ‘Regulations for the Administration of Affairs Concerning Experimental Animals’. The Institutional Animal Care and Use Committee of Fisheries Research Institute of Fujian, China, approved the experiments.
P. liuwutiensis were collected in Huandao Road, Xiamen City, Fujian Province, China. The back muscle of sea cucumber was clipped, fixed by anhydrous ethanol and stored at −20 °C [11]. The back wall bone fragment was used to identify the species using a scanning electron microscope [13]. The body wall of the sea cucumber was treated with a 10% NaClO solution for 1–2 min. Then the white precipitate was washed with distilled water four times. The samples were dried and then gold‐plated by an ion sputtering device. Finally, the samples were examined using a scanning electron microscope [11].
DNA extraction
DNA was extracted from 30 mg back muscle tissue using the TIANamp Marine Animals DNA Kit (Tiangen Biochemical Technology, Beijing, Co. Ltd., Beijing, China) according to the manufacturer's instructions. The obtained DNA was stored at −20 °C prior to further analysis.
PCR amplification and sequencing
The complete mitochondrial nucleotide sequence of P. liuwutiensis was obtained by general and long‐range PCR amplification using the specific primers (Table 1). The primers were designed to match the generally conserved regions of target mtDNA, and the short fragments of cox1, cox2, atp6, cox 3, nad 4, cytb, 12S, nad 1 and 16S were amplified. In brief, the amplifications were carried out with 40 cycles at a melting temperature of 94 °C for 30 s, an annealing temperature of 50 °C for 30 s and an extension temperature of 72°C for 1 min per 1 kb. The final MgCl2 concentration in the reaction was 2.0 mmol·L−1. PCR products were cloned into a pMD18‐T vector (Takara, Beijing, China) and then sequenced using the dideoxynucleotide procedure by ABI 3730 automatic sequencer. Sequences were assembled by dnastar software [14] and manually adjusted to generate the complete sequence of mtDNA.
Table 1.
Primers used for amplification of complete mitochondrial genome of P. liuwutiensis.
| Fragment no. | Gene or region | Primer name | Sequence (5′–3′) | Length (bp) |
|---|---|---|---|---|
| F1 | cox1 | MIF1 | CGAACAGAACTAGCCCAACC | 1411 |
| MIR1 | CTTTGAATGTGTGGTGAGATGG | |||
| F2 | cox1‐cox2 | MIF2 | TCCATCTCCTCCATAGGATC | 708 |
| MIR2 | GCAATAGAATATTGATAAGAGG | |||
| F3 | cox2 | MIF3 | GAGCACAAATTGGATTACAAG | 595 |
| MIR3 | GCTCCACAGATTTCTGAGCA | |||
| F4 | cox2‐atp6 | MIF4 | GAGTTAAGATGGATGGAGTTC | 584 |
| MIR4 | GAAGCTTTGTGGCTTCTAGG | |||
| F5 | atp6 | MIF5 | CGACACAATAGGATTTCTCC | 607 |
| MIR5 | GTAAGCTTGGATACATGCAAC | |||
| F6 | atp6‐cox3 | MIF6 | GCCACCTGAGTCCTTAGATC | 322 |
| MIR6 | CATCATCTTATAGCAGTTAGG | |||
| F7 | cox3 | MIF7 | GTTGATCAAAGACCATGACC | 711 |
| MIR7 | GACGTCAACGAAGTGTCAGT | |||
| F8 | cox3‐nd4 | MIF8 | CAACCTTCCTAACAGTATGTC | 1404 |
| MIR8 | TAGGGAGCCTAGGGCACAGAAG | |||
| F9 | nad4 | MIF9 | CCAAAGGCCCACGTAGAAGC | 228 |
| MIR9 | GTGGCCTACAGAAGAATATGC | |||
| F10 | nad4‐cytb | MIF10 | CCTACTCCGACTCTTCCCTC | 3446 |
| MIR10 | CTTGATTTATGTAGGATCCA | |||
| F11 | cytb | MIF11 | GCACTACACCGCTGACATAAC | 880 |
| MIR11 | AGGTTCTTCTACTGGTTGGC | |||
| F12 | cytb‐12S | MIF12 | CCAATCATCCTCCTTTCGAC | 646 |
| MIR12 | GTATAGCGGGGTATCTAATCC | |||
| F13 | 12S | MIF13 | CACGTTAACCTTTAGCTAAAG | 570 |
| MIR13 | GGTACACCTACTTTGTTACG | |||
| F14 | 12S‐nad1 | MIF14 | GTACCTCCTTAAAGAAATAAG | 2206 |
| MIR14 | GATCATAAAGCAATTGCTAAAG | |||
| F15 | nad1 | MIF15 | GTAGTTGGGCCATACGGATTAC | 725 |
| MIR15 | CGTGGGTAGGAGGCTCGTAC | |||
| F16 | nad1‐16S | MIF16 | CTAGGCGGTAGAAGACCATTC | 1955 |
| MIR16 | CTTGGTTTTTGTTTATGTTTCC | |||
| F17 | 16S | MIF17 | CCTTTAGTAGACCTAAAAGC | 863 |
| MIR17 | GGTCCTTTCGTACTAAAGAAGG | |||
| F18 | 16S‐cox1 | MIF18 | GTAACCAAAGGGTGCAGCAG | 568 |
| MIR18 | CAATCATTAGTGGAATTAGTC |
Sequence analysis and gene annotation
After the quality proofing of the obtained fragments, the mtDNA sequences were manually assembled using dnastar v7.1 software [14]. First, the raw mtDNA sequences were imported into MITOS web servers to determine the approximate boundaries of genes. Exact positions of PCGs were found by searching open reading frames. All tRNA genes were identified using ARWEN, DOGMA and MITOS [15, 16, 17]. MEGAX was used to calculate the DNA base composition and codon preference of the mitochondrial genome of P. liuwutiensis [18]. Formula with GC‐skew = (G − C)/(G + C) and the AT‐skew = (A − T)/(A +T) were calculated for base's preferences [19].
Phylogenetic analysis
The P. liuwutiensis and another 25 echinoderm mitogenomes (obtained from GenBank; https://www.ncbi.nlm.nih.gov/) were used for phylogenetic analysis. Balanoglossus carnosus (Enteropneusta) was rooted as the outgroup. Echinoderms were divided into five classes as follows: Holothuroidea, Echinoidea, Asteroidea, Ophiuroidea and Crinoidea [20]. Therefore, the species of these five classes were selected to construct the evolutionary tree, among which more sea cucumber species were selected to study the phylogeny and evolution of the P. liuwutiensis. The megax [18] was used to perform the alignment of 13 PCGs. A Bayesian approach using mrbayes 3.1.2 version [21, 22] was employed to analyze the aligned datasets and trees. Analyses had two parallel runs with four chains of each (three hot chains and one cold chain), which were carried out for 1 000 000 generations (sampling every 100 generations). After the first 1000 ‘burn in’ trees were discarded, the remaining 9000 sampled trees were used to estimate the 50% majority rule consensus tree and the Bayesian posterior probabilities.
Results
Identification of P. liuwutiensis
We found four types of table body in the body wall of the sea cucumber (Fig. 1), which was in accordance with a previous study [12]. Based on the morphological characteristics, the sea cucumber was identified to be P. liuwutiensis.
Fig. 1.

The dorsal wall of the P. liuwutiensis. Tabular form (A), plates of sea cucumber (B), type rosettes (C) and type buttons (D). Scale bars: 10 μm (A, C, D); 50 μm (B).
Genome organization
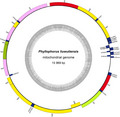
The complete mtDNA sequence of P. liuwutiensis was obtained by general and long‐range PCR. Similar to other deuterostomes, the mtDNA of P. liuwutiensis was a closed double‐stranded loop and composed of 15 969 bp (Fig. 2). The genome encoded 37 genes, including 13 PCGs, 2 rRNA genes and 22 tRNA genes. Among these genes, 31 genes were encoded on the positive strand, whereas others (such as nad6, tRNAser(tga), tRNAGln, tRNAAla, tRNAVal and tRNAAsp) were encoded on the negative strand. The base composition of the mtDNA of P. liuwutiensis is 29.55% T, 22.16% C, 35.64% A and 12.64% G.
Fig. 2.

Gene map of the complete mitochondrial genome of P. liuwutiensis. Genes encoded on the positive and negative strands are shown outside and inside the circular gene map, respectively.
Base composition and AT/GC‐skew of mtDNA
Table 2 shows the base composition of P. liuwutiensis and 25 species of echinoderms. We also found that the contents of the bases varied among species, with a T content of 31.63% ranging from 25.24% (Acanthaster planci) to 46.46% (Phanogenia gracilis), a C content of 21.00% ranging from 11.44% (P. gracilis) to 27.95% (A. planci), an A content of 32.25% ranging from 25.82% (P. gracilis) to 35.82% (Echinaster brasiliensis) and a G content of 15.12% ranging from 10.65% (Freyastera benthophila) to 18.22% (Strongylocentrotus droebachiensis; Table 2). The length of the P. liuwutiensis mtDNA was shorter compared with other echinoderms. The higher content of base A (than T) and the lower content of base G (than C) were found in most echinoderms. The data (Table 2) showed that all GC skewness of the P. liuwutiensis mtDNA was negative, and all AT skewness was positive, indicating that the mtDNA of P. liuwutiensis had a preference for A and C.
Table 2.
List of species used for species identification based on the whole mitochondrial genome and the analysis of the base composition of each species.
| Species | T (%) | C (%) | A (%) | G (%) | A + T (%) | GC‐skew | AT‐skew |
|---|---|---|---|---|---|---|---|
| P. liuwutiensis | 29.55 | 22.16 | 35.64 | 12.64 | 65.19 | −0.273 | 0.093 |
| Amphiura digitula | 30.89 | 22.98 | 32.74 | 13.39 | 63.63 | −0.263 | 0.029 |
| Apostichopus japonicus | 30.14 | 20.21 | 31.75 | 17.89 | 61.89 | −0.061 | 0.026 |
| Parastichopus nigripunctatus | 30.14 | 20.15 | 31.68 | 18.03 | 61.82 | −0.055 | 0.025 |
| Parastichopus parvimensis | 29.91 | 20.38 | 31.78 | 17.93 | 61.69 | −0.064 | 0.030 |
| Parastichopus californicus | 29.87 | 20.57 | 31.53 | 18.04 | 61.40 | −0.066 | 0.027 |
| Stichopus sp. SF‐2010 | 29.29 | 23.76 | 30.95 | 16.01 | 60.24 | −0.195 | 0.028 |
| Stichopus horrens | 29.31 | 23.72 | 30.80 | 16.17 | 60.11 | −0.189 | 0.025 |
| Holothuria scabra | 27.07 | 24.34 | 32.67 | 15.91 | 59.74 | −0.209 | 0.094 |
| Holothuria forskali | 30.82 | 21.41 | 31.40 | 16.37 | 62.22 | −0.134 | 0.009 |
| Cucumaria miniata | 28.09 | 22.95 | 35.74 | 13.22 | 63.83 | −0.269 | 0.120 |
| Benthodytes marianensis | 36.91 | 17.83 | 32.33 | 12.93 | 69.24 | −0.160 | −0.066 |
| Freyastera benthophila | 33.53 | 21.13 | 34.70 | 10.65 | 68.23 | −0.330 | 0.017 |
| Astropecten polyacanthus | 31.51 | 22.44 | 32.50 | 13.55 | 64.01 | −0.247 | 0.015 |
| Acanthaster planci | 25.24 | 27.95 | 31.10 | 15.71 | 56.34 | −0.280 | 0.104 |
| Echinaster brasiliensis | 25.83 | 26.43 | 35.82 | 11.91 | 61.65 | −0.379 | 0.162 |
| Heliocidaris crassispina | 27.86 | 24.37 | 31.03 | 16.74 | 58.89 | −0.186 | 0.054 |
| Heterocentrotus mammillatus | 29.02 | 23.55 | 29.89 | 17.54 | 58.91 | −0.146 | 0.015 |
| Strongylocentrotus droebachiensis | 30.18 | 22.80 | 28.80 | 18.22 | 58.98 | −0.112 | −0.023 |
| Hemicentrotus pulcherrimus | 30.58 | 22.43 | 29.19 | 17.79 | 59.77 | −0.115 | −0.023 |
| Florometra serratissima | 46.37 | 11.57 | 26.45 | 15.61 | 72.82 | 0.149 | −0.274 |
| Phanogenia gracilis | 46.46 | 11.44 | 25.82 | 16.27 | 72.28 | 0.174 | −0.286 |
| Ophiura lutkeni | 33.11 | 19.23 | 32.77 | 14.90 | 65.88 | −0.127 | −0.005 |
Overlapping and noncoding regions
In this study, we identified seven overlapping regions of genes (trnP/trnQ, trnL1/trnA, trnY/trnG, atp8/atp6, cox3/trnS2, nad4/trnH and trnS1/nad5) in the complete genome of P. liuwutiensis (Table 3). The length of these overlaps varied from 1 to 10 bp, with the longest length between nad4 and trnH (Table 3). Among the 20 noncoding regions, the length was 1–703 bp (Table 3). The longest noncoding sequence (703 bp; AT% = 69.30%) was found between tRNAthr and tRNApro. Due to its location and AT richness, the noncoding sequence in P. liuwutiensis was identified as the putative control regions (CRs; Table 3), which were similar to that in the genomic studies of Stichopus sp. [23].
Table 3.
Annotation of the mitochondrial genome of P. liuwutiensis.
| Gene | Strand | Sequence location | Size (bp) | Start codon | Stop codon | Intergenic region |
|---|---|---|---|---|---|---|
| trna‐pro | + | 1–66 | 66 | −4 | ||
| trna‐gln | − | 63–132 | 70 | 6 | ||
| trna‐asn | + | 139–207 | 69 | 2 | ||
| trna‐leu(tag) | + | 210–282 | 73 | −1 | ||
| trna‐ala | − | 282–348 | 67 | 1 | ||
| trna‐trp | + | 350–419 | 70 | 0 | ||
| trna‐cys | + | 420–484 | 65 | 1 | ||
| trna‐val | − | 486–555 | 70 | 4 | ||
| trna‐asp | − | 560–624 | 65 | 57 | ||
| trna‐met | + | 682–750 | 69 | 7 | ||
| trna‐tyr | + | 758–826 | 69 | −1 | ||
| trna‐gly | + | 826–896 | 71 | 0 | ||
| trna‐leu(taa) | + | 897–967 | 71 | 0 | ||
| nad1 | + | 968–1939 | 972 | GTG | TAA | 5 |
| trna‐ile | + | 1945–2012 | 68 | 2 | ||
| nad2 | + | 2015–3055 | 1041 | ATG | TAA | 0 |
| 16S | + | 3056–4403 | 1348 | 130 | ||
| cox1 | + | 4534–6087 | 1554 | ATG | TAA | 1 |
| trna‐arg | + | 6089–6153 | 65 | 0 | ||
| nad4L | + | 6154–6450 | 297 | ATG | TAA | 0 |
| cox2 | + | 6451–7140 | 690 | ATG | TAA | 2 |
| trna‐lys | + | 7143–7206 | 64 | 0 | ||
| atp8 | + | 7207–7371 | 165 | ATG | TAA | −7 |
| atp6 | + | 7365–8048 | 684 | ATG | TAA | 2 |
| cox3 | + | 8051–8833 | 783 | ATG | TAA | −1 |
| trna‐ser(tga) | − | 8833–8903 | 71 | 39 | ||
| nad3 | + | 8943–9287 | 345 | ATG | TAA | 3 |
| nad4 | + | 9291–10 652 | 1362 | ATG | TAG | −10 |
| trna‐his | + | 10 643–10 710 | 68 | 1 | ||
| trna‐ser(gct) | + | 10 712–10 778 | 67 | −3 | ||
| nad5 | + | 10 776–12 605 | 1830 | TTG | TAA | 13 |
| nad6 | − | 12 619–13 107 | 489 | CTA | CAT | 8 |
| cytb | + | 13 116–14 255 | 1140 | ATG | TAA | 1 |
| trna‐phe | + | 14 257–14 327 | 71 | 0 | ||
| 12S | + | 14 328–15 128 | 801 | 0 | ||
| trna‐glu | + | 15 129–15 196 | 68 | 0 | ||
| trna‐thr | + | 15 197–15 266 | 70 | 0 | ||
| Putative CR | + | 15 267–15 969 | 703 | 0 |
PCGs and codon usage
The length of PCGs in the mtDNA of P. liuwutiensis was 11 352 bp. The longest (1830 bp) and shortest (165 bp) lengths were nad5 and atp8, respectively (Table 3). The bases C and A were found to be predominant in most genes, while the base T was predominant in some genes (Table 4). Except for nad6, which was encoded on the negative strand, all PCGs were encoded on the positive strand (Table 3 and Fig. 2), which was similar to the results found in other sea cucumber species [3]. ATG was found to be the start codon in most PCGs, except for nad1 (GTG as the start codon), nad5 (TTG) and nad6 (CTA), while TAA was the termination codon of most PCGs, excepting nad4 (TAG as the start codon) and nad6 (CAT; Table 3).
Table 4.
The base composition and preference of mitochondrial gene in P. liuwutiensis.
| Gene | T (%) | C (%) | A (%) | G (%) | G + C (%) | Total (bp) | GC‐skew | AT‐skew |
|---|---|---|---|---|---|---|---|---|
| PCGs | 30.20 | 23.60 | 34.10 | 12.10 | 35.70 | 11 352 | −0.322 | 0.061 |
| nad6 | 18.40 | 22.90 | 50.51 | 8.18 | 31.08 | 489 | −0.474 | 0.466 |
| nad5 | 28.96 | 22.79 | 38.25 | 10.00 | 32.79 | 1830 | −0.390 | 0.138 |
| nad4L | 35.35 | 23.91 | 32.32 | 8.42 | 32.32 | 297 | −0.479 | −0.045 |
| nad4 | 29.59 | 25.11 | 35.17 | 10.13 | 35.24 | 1362 | −0.425 | 0.086 |
| nad3 | 34.20 | 23.77 | 30.14 | 11.88 | 35.65 | 345 | −0.333 | −0.063 |
| nad2 | 34.49 | 21.13 | 33.14 | 11.24 | 32.37 | 1041 | −0.306 | −0.020 |
| nad1 | 34.67 | 20.88 | 30.76 | 13.68 | 34.57 | 972 | −0.208 | −0.060 |
| cytb | 28.33 | 25.70 | 33.51 | 12.46 | 38.16 | 1140 | −0.347 | 0.084 |
| cox3 | 29.76 | 25.93 | 29.50 | 14.81 | 40.74 | 783 | −0.273 | −0.004 |
| cox2 | 30.72 | 24.20 | 31.74 | 13.33 | 37.54 | 690 | −0.290 | 0.016 |
| cox1 | 29.99 | 23.42 | 30.24 | 16.34 | 39.77 | 1554 | −0.178 | 0.004 |
| atp8 | 23.64 | 16.36 | 49.09 | 10.91 | 27.27 | 165 | −0.200 | 0.350 |
| atp6 | 30.70 | 26.02 | 32.31 | 10.96 | 36.99 | 684 | −0.407 | 0.026 |
| trna‐val | 32.86 | 21.43 | 30.00 | 15.71 | 37.14 | 70 | −0.154 | −0.045 |
| trna‐tyr | 27.54 | 15.94 | 37.68 | 18.84 | 34.78 | 69 | 0.083 | 0.156 |
| trna‐trp | 34.29 | 12.86 | 44.29 | 8.57 | 21.43 | 70 | −0.200 | 0.127 |
| trna‐thr | 35.71 | 11.43 | 41.43 | 11.43 | 22.86 | 70 | 0.000 | 0.074 |
| trna‐ser | 29.58 | 26.76 | 29.58 | 14.08 | 40.85 | 71 | −0.310 | 0 |
| trna‐ser(gct) | 31.34 | 17.91 | 28.36 | 22.39 | 40.30 | 67 | 0.111 | −0.050 |
| trna‐pro | 36.36 | 9.09 | 36.36 | 18.18 | 27.27 | 66 | 0.333 | 0.000 |
| trna‐phe | 25.35 | 16.90 | 40.85 | 16.90 | 33.80 | 71 | 0 | 0.234 |
| trna‐met | 33.33 | 18.84 | 33.33 | 14.49 | 33.33 | 69 | −0.130 | 0 |
| trna‐lys | 31.25 | 17.19 | 39.06 | 12.50 | 29.69 | 64 | −0.158 | 0.111 |
| trna‐leu | 34.25 | 16.44 | 32.88 | 16.44 | 32.88 | 73 | 0.000 | −0.020 |
| trna‐leu(taa) | 29.58 | 16.90 | 33.80 | 19.72 | 36.62 | 71 | 0.077 | 0.067 |
| trna‐ile | 32.35 | 19.12 | 32.35 | 16.18 | 35.29 | 68 | −0.083 | 0 |
| trna‐his | 32.35 | 16.18 | 38.24 | 13.24 | 29.41 | 68 | −0.100 | 0.083 |
| trna‐gly | 30.99 | 15.49 | 40.85 | 12.68 | 28.17 | 71 | −0.100 | 0.137 |
| trna‐glu | 26.47 | 19.12 | 36.76 | 17.65 | 36.76 | 68 | −0.040 | 0.163 |
| trna‐gln | 31.43 | 20.00 | 35.71 | 12.86 | 32.86 | 70 | −0.217 | 0.064 |
| trna‐cys | 41.54 | 9.23 | 33.85 | 15.38 | 24.62 | 65 | 0.250 | −0.102 |
| trna‐asp | 36.92 | 21.54 | 26.15 | 15.38 | 36.92 | 65 | −0.167 | −0.171 |
| trna‐asn | 24.64 | 17.39 | 37.68 | 20.29 | 37.68 | 69 | 0.077 | 0.209 |
| trna‐arg | 32.31 | 15.38 | 38.46 | 13.85 | 29.23 | 65 | −0.053 | 0.087 |
| trna‐ala | 35.82 | 13.43 | 38.81 | 11.94 | 25.37 | 67 | −0.059 | 0.040 |
| 16S | 27.74 | 17.36 | 39.09 | 15.80 | 33.16 | 1348 | −0.047 | 0.170 |
| 12S | 21.22 | 21.72 | 41.57 | 15.48 | 37.20 | 801 | −0.168 | 0.324 |
| CRs | 26.60 | 21.30 | 42.70 | 9.40 | 30.70 | 703 | −0.388 | 0.232 |
Table 5 and Fig. 3 summarize the relative synonymous codon usage (RSCU) values for the 13 PCGs. The mtDNA of P. liuwutiensis contained 5323 codons. Among the 13 PCGs, the most frequently used amino acid was Ser (12.91%), followed by Leu (10.82%), Lys (8.34%), Pro (7.06%) and Thr (6.01%). A common feature in most metazoan mtDNA is a bias toward a higher representation of nucleotides A and T, leading to a subsequent bias in the corresponding encoded amino acids [8, 24]. The content of A + T in the 13 PCGs was 64.3% and the AT‐skew was positive, indicating a higher occurrence of A than T (Table 4).
Table 5.
The codon number and RSCU in P. liuwutiensis mitochondrial protein‐coding genes.
| Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU | Codon | Count | RSCU |
|---|---|---|---|---|---|---|---|---|---|---|---|
| UUU(F) | 171 | 1.14 | UCU(S) | 162 | 1.83 | UAU(Y) | 212 | 1.39 | UGU(C) | 30 | 1.07 |
| UUC(F) | 130 | 0.86 | UCC(S) | 121 | 1.36 | UAC(Y) | 92 | 0.61 | UGC(C) | 26 | 0.93 |
| UUA(L) | 169 | 1.76 | UCA(S) | 97 | 1.09 | UAA(*) | 244 | 1.71 | UGA(W) | 74 | 0.52 |
| UUG(L) | 61 | 0.64 | UCG(S) | 18 | 0.20 | UAG(*) | 111 | 0.78 | UGG(W) | 32 | 1.00 |
| CUU(L) | 134 | 1.40 | CCU(P) | 141 | 1.50 | CAU(H) | 96 | 1.19 | CGU(R) | 19 | 0.50 |
| CUC(L) | 73 | 0.76 | CCC(P) | 99 | 1.05 | CAC(H) | 65 | 0.81 | CGC(R) | 16 | 0.42 |
| CUA(L) | 112 | 1.17 | CCA(P) | 107 | 1.14 | CAA(Q) | 151 | 1.43 | CGA(R) | 27 | 0.71 |
| CUG(L) | 27 | 0.28 | CCG(P) | 29 | 0.31 | CAG(Q) | 60 | 0.57 | CGG(R) | 12 | 0.31 |
| AUU(I) | 124 | 1.15 | ACU(T) | 81 | 1.01 | AAU(N) | 153 | 1.13 | AGU(S) | 57 | 0.64 |
| AUC(I) | 74 | 0.69 | ACC(T) | 101 | 1.26 | AAC(N) | 118 | 0.87 | AGC(S) | 77 | 0.87 |
| AUA(M) | 126 | 1.17 | ACA(T) | 104 | 1.30 | AAA(K) | 340 | 1.53 | AGA(S) | 94 | 2.46 |
| AUG(M) | 71 | 1.00 | ACG(T) | 34 | 0.43 | AAG(K) | 104 | 0.47 | AGG(S) | 61 | 1.60 |
| GUU(V) | 43 | 1.29 | GCU(A) | 40 | 1.23 | GAU(D) | 76 | 1.11 | GGU(G) | 25 | 0.83 |
| GUC(V) | 27 | 0.81 | GCC(A) | 55 | 1.69 | GAC(D) | 61 | 0.89 | GGC(G) | 12 | 0.40 |
| GUA(V) | 55 | 1.65 | GCA(A) | 31 | 0.95 | GAA(E) | 127 | 1.53 | GGA(G) | 66 | 2.20 |
| GUG(V) | 8 | 0.24 | GCG(A) | 4 | 0.12 | GAG(E) | 39 | 0.47 | GGG(G) | 17 | 0.57 |
Fig. 3.

RSCU in P. liuwutiensis mitogenome.
rRNA and tRNA genes
The results revealed that the two rRNA of P. liuwutiensis were encoded on the positive strand, and the 16S and 12S genes were 1348 bp (G + C% = 33.16%) and 801 bp (G + C% = 37.2%) in length, respectively (Tables 3 and 4). The 16S gene was between nad2 and cox1, and 12S was between trnF and trnE. The results showed that 5/22 tRNA genes, including trna‐gln, trna‐ala, trna‐ser (tga), trna‐asp and trna‐val, were encoded on the negative strand, while the remaining genes were encoded on the positive strand. The length of the tRNA genes ranged from 64 to 73 bp, and the shortest (64 bp) and longest (73 bp) lengths were found from trna‐lys and trna‐leu(tag), respectively (Table 3). In addition, Table 4 shows the base composition of tRNA A + T bias. All 22 tRNA genes were predicted to be capable of folding into a cloverleaf secondary structure using the MITOS web server (Fig. 4).
Fig. 4.

Secondary structures of the 22 tRNA genes of P. liuwutiensis.
Phylogenetic analysis
Phylogenetic analysis revealed that the complete mtDNA of P. liuwutiensis and the other 25 echinoderm species were separated into five major clades as follows: Holothuroidea, Echinoidea, Asteroidea, Crinoidea and Ophiuroidea (Fig. 5). Almost all of the clades were strongly supported. The two species, P. liuwutiensis and Cucumaria miniata, formed a cluster, which were distinguished from other species, indicating that these two species together belonged to the same order (Dendrochirotida).
Fig. 5.
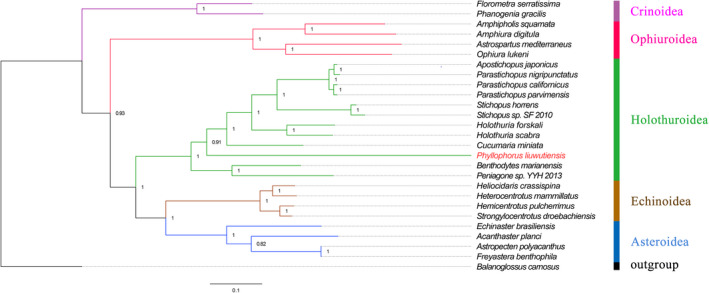
Phylogenetic trees based on the concatenated amino acid of 13 PCGs. The Balanoglossus carnosus (NC001887.1) is used as outgroup. The red name highlights the species sequenced in this study. P. liuwutiensis (MN198190), Amphiura digitula (MH791160.1), Apostichopus japonicus (FJ986223.1), P. nigripunctatus (AB525762.1), Parastichopus parvimensis (KU168761.1), Parastichopus californicus (KP398509.1), Stichopus sp. SF‐2010 (HM853683.2), Stichopus horrens (HQ000092.1), Holothuria scabra (KP257577.1), Holothuria forskali (FN562582.1), Cucumaria miniata (AY182376.1), Benthodytes marianensis (MH208310.1), Freyastera benthophila (MG563681.1), Astropecten polyacanthus (AB183560.1), Acanthaster planci (AB231475.1), Echinaster brasiliensis (MG636999.1), Heliocidaris crassispina (KC479025.1), Heterocentrotus mammillatus (KJ680292.1), Strongylocentrotus droebachiensis (EU054306.1), Hemicentrotus pulcherrimus (KC490911.1), Florometra serratissima (NC001878.1), Phanogenia gracilis (DQ068952.1), Ophiura lutkeni (AY184223.1), Peniagone sp. YYH‐2013 (KF915304.1), Amphipholis squamata (FN562578.1) and Astrospartus mediterraneus (NC013878.1).
Gene order
Figure 6 illustrates the gene arrangement of P. liuwutiensis and seven other echinoderms. Similar gene arrangements exist in the species used to construct evolutionary trees or that had a few gene rearrangements. Holothuria scabra, Holothuria forskali, Apostichopus japonicus, Parastichopus nigripunctatus, Parastichopus parvimensis and Parastichopus californicus had the same gene arrangements [3]. Florometra serratissima and P. gracilis also had the same gene arrangement. The gene arrangement of Astropecten polyacanthus and A. planci is the same [9]. The gene order of A. japonicus (class Holothuroidea), S. droebachiensis (class Echinoidea) and P. liuwutiensis had similar changes with the inversion occurring between the genes trnD and trnM (Fig. 6), including the two conserved genetic blocks (cox1‐R‐nad4L‐cox2‐K‐atp8‐atp6‐cox3‐S2‐nad3‐nad4‐H‐S1‐nad5‐nad6‐cob‐F‐rrnS‐E‐T‐P‐Q‐N and Y‐G‐L2‐nad1‐I‐nad2‐rrnL‐L1‐A‐W‐C‐V), which were found in the same order. The gene order of F. serratissima (class Crinoidea) was significantly different from that of P. liuwutiensis, with more translocations and inversions, and two genes were rearranged (rrnL and V). Ophiura lutkeni (class Ophiuroidea) produced more translocations. Therefore, these 10 genes (G, rrnL, M, P, E, Y, D, cob, T and W) were involved in the gene rearrangement, and five conserved gene blocks (cox1‐R‐nad4L‐cox2‐K‐atp8‐atp6‐cox3‐S2‐nad3‐nad4‐H‐S1‐nad5‐nad6, C‐V, L1‐A, Q‐N and L2‐nad1‐I‐nad2) were found in the same order. In contrast, A. planci (class Asteroidea) had two parts of a wide range of gene location inversions (trnY‐rrnL and trnP‐trnV), and the translocations of these two parts were observed. The only two conserved genetic blocks (cox1‐R‐nad4L‐cox2‐K‐atp8‐atp6‐cox3‐S2‐nad3‐nad4‐H‐S1‐nad5‐nad6‐cob‐F‐rrnS‐E‐T and D‐M) were found in the same order. Four conserved gene blocks were found (cox1‐R, N‐L1, nad4L‐cox2‐K‐atp8‐atp6‐cox3‐S2‐nad3‐nad4‐H‐S1‐nad5‐nad6‐cob‐F‐rrnS, Y‐G‐L2‐nad1‐I‐nad2‐rrnL) in the gene arrangement of C. miniata (class Holothuroidea), and the inversion occurred between the genes trnD and trnM. In the sea cucumber Benthodytes marianensis (class Holothuroidea), five conserved gene blocks (atp6‐cox3‐S2‐nad3‐nad4‐H‐S1‐nad5‐nad6‐cob‐F‐rrnS‐E, A‐W, P‐Q‐N‐L1, C‐V‐D, L2‐nad1‐I‐nad2‐T‐rrnL) were identified. Meanwhile, compared with the gene arrangement of P. liuwutiensis, the translocation of three tRNAs (M, G and Y) can be found.
Fig. 6.

Linear representation of gene rearrangements of P. liuwutiensis, Apostichopus japonicus, Florometra serratissima, Strongylocentrotus droebachiensis, Ophiura lutkeni, Acanthaster planci, Cucumaria miniata and Benthodytes marianensis. Gene segments are not drawn to scale. All genes are transcribed from left to right except those indicated by underlining, which are transcribed from right to left. The circling arrows indicate inversions. tRNA genes are represented by the corresponding single‐letter amino acid code, especially S1 (AGN), S2 (UCN), L1 (CUN) and L2 (UUR). rrnL and rrnS are the large and small rRNA subunits, respectively.
Discussion
Phylogenetics studies the evolutionary relationships of organisms, and the phylogenetic tree is the topological structure that describes the evolutionary order of various groups of organisms [25, 26]. However, with the development and application of molecular biology, our understanding of genes and proteins has been constantly increasing, and a theoretical method has been gradually developed to study the evolutionary relationship of species based on the genetic information of biological macromolecules, such as DNA or protein sequence. Because most of the mitochondrial genes are matrilineal and the sequence variation is rapid, they are widely used to study the phylogenetic evolution [27, 28]. The most basic problem solved by phylogenetic research is to determine the taxonomic status of species [29, 30]. However, determining the relationship of species can further infer the biological characteristics of unknown species, which, of course, requires a lot of phylogenetic studies to prove [31]. Each newly sequenced species enriches the database, providing more information for studying the phylogeny between species.
In this study, we conducted a preliminary study on the mtDNA of P. liuwutiensis. Mitochondrial genomes are maternal mtDNA and do not recombine DNA. Therefore, individuals with the same mtDNA sequence are descended from the same female ancestor [32]. Consequently, mtDNA sequences can be used to determine the relationships between species. In this study, we aim to characterize the mtDNA of P. liuwutiensis, as well as to determine the taxonomic relationship between P. liuwutiensis and other echinoderm species.
The results showed that the contents of base A and base T were higher, the content of A + T was higher compared with G + C, and the lowest content of A + T was 56.34% (A. planci), indicating the characteristics of mtDNA sequence in invertebrates [33]. The CR was found to be the main noncoding region of the mtDNA, which is necessary for the initiation of mtDNA transcription and replication of metazoa [34, 35]. Its size and nucleotide sequence greatly varied. Most metazoa mtDNA contain only one CR, whereas in some sea cucumbers there are two duplicate CRs or two independent CRs with the same or highly similar nucleotide sequences [33]. The mtDNA of P. liuwutiensis was found to contain a CR between tRNAthr and tRNApro. Three mechanisms may contribute to mtDNA with duplicate CRs: tandem duplication, dimerization and illegitimate recombination. Many studies support the idea that mitogenome with repetitive CRs may replicate more efficiently than mitogenome with a single CR [36, 37]. However, because only two species of sea cucumber (B. marianensis and Cucumaria miniate) have been shown to have duplicate CRs, additional mitotic genomes are needed to elucidate the mechanism that causes this phenomenon [3].
Based on the basic assumption that shared genetic arrangements imply a common ancestor, it is highly unlikely that the same sequence of genes will emerge independently in different lineages [9]. Therefore, comparative gene alignment may be a useful tool for phylogenetic studies, especially when some ancestral relationships are concerned. Over the past two decades, many studies have been reported on the mitochondrial gene sequence in echinoderms [38, 39]. There are four possible mechanisms for genome rearrangement: inversion (reversals), transposition, reverse transposition and tandem duplication random losses [3]. It is of great significance to explore the evolutionary history of mitochondrial gene rearrangement in echinoderms by comparing the gene arrangement in the mtDNA of echinoderms and studying the common sequence among different individuals [40, 41]. In this study, the gene arrangement of P. liuwutiensis was very similar to that of the other five echinoderm species, which all contained conserved gene blocks of 15 genes. In particular, A. japonicus and S. droebachiensis had an inversion of only two genes compared with P. liuwutiensis. Shen et al. [9] have hypothesized that the inclusion of a consensus nonavian vertebrate gene order does support the echinoid mtDNA gene order as the most likely representative of the echinoderm ground pattern. Genes and the environment act together on biological traits, with genes playing a dominant role. Therefore, the study of genes can infer the adaptability of species to the environment. Mu et al. [3] have studied the adaptation of sea cucumber to the deep‐sea environment through the mtDNA of sea cucumber, and predicted that nad2 and nad4 might be important candidate genes for the further study on the adaptation of B. marianensis to the deep‐sea environment. The different gene order may be related to certain ecological or morphological features of the species. Genes may produce different effects in different positions, and the interactions between genes have different results. These assumptions need to be tested by further research.
Conclusions
In this study, we characterized the structure of the mtDNA of P. liuwutiensis, and the results showed that the mtDNA (15 969 bp) encoded 37 genes, including 13 PCGs, 22 tRNA genes and 2 rRNA genes. One putative CR was found in the mitogenome of P. liuwutiensis. The mtDNA of P. liuwutiensis was clustered together with C. miniate. Moreover, the gene arrangement of P. liuwutiensis was also described in detail.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
QY and QL designed and supervised the research. FY performed most of the experiments and wrote the paper with assistance from C. Zhou, NTT, ZS, JW, HG, ZL, C. Zhong and ZZ. All authors made contributions to the final version of this manuscript. All authors read and approved the final manuscript.
Acknowledgements
We thank professor Cai Lizhe of Xiamen University for help in the identification of P. liuwutiensis. This work was supported by the specialized research foundation of Fujian Provincial Department of Science & Technology, China (Grant No. 2018R1003‐1 and 2019R1013‐5) and the project of marine fisheries of Fujian Provincial Department of Ocean and Fisheries, China (Grant No. 2020HYJG02 and FJHJF‐L‐2020‐4).
Contributor Information
Qiuhua Yang, Email: qhyang1314@163.com.
Qi Lin, Email: xmqlin@sina.com.
Data accessibility
All original sequence data in this study were submitted to the NCBI database under accession number MN198190.
References
- 1. Bordbar S, Anwar F and Saari N (2011) High‐value components and bioactives from sea cucumbers for functional foods‐A review. Marine Drugs 9, 1761–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fan S, Hu C, Zhang L, Sun H, Wen J and Luo P (2012) Complete mitochondrial genome of the sea cucumber Stichopus sp. and its application in the identification of this species. Aquac Res 43, 1306–1316. [Google Scholar]
- 3. Mu W, Liu J and Zhang H (2018) Complete mitochondrial genome of Benthodytes marianensis (Holothuroidea: Elasipodida: Psychropotidae): Insight into deep sea adaptation in the sea cucumber. PLoS One 13, e0208051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Boore JL (1999) Animal mitochondrial genomes. Nucleic Acids Res 27, 1767–1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Scouras A, Beckenbach K, Arndt A and Smith MJ (2004) Complete mitochondrial genome DNA sequence for two ophiuroids and a holothuroid: the utility of protein gene sequence and gene maps in the analyses of deep deuterostome phylogeny. Mol Phylogenet Evol 31, 50–65. [DOI] [PubMed] [Google Scholar]
- 6. Zhang D, Li WX, Zou H, Wu SG, Li M, Jakovlic I, Zhang J, Chen R and Wang GT (2018) Mitochondrial genomes of two diplectanids (Platyhelminthes: Monogenea) expose paraphyly of the order Dactylogyridea and extensive tRNA gene rearrangements. Parasit Vectors 11, 601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Červená B, Modrý D, Fecková B, Hrazdilová K, Foronda P, Alonso AM, Lee R, Walker J, Niebuhr CN, Malik R et al (2019) Low diversity of Angiostrongylus cantonensis complete mitochondrial DNA sequences from Australia, Hawaii, French Polynesia and the Canary Islands revealed using whole genome next‐generation sequencing. Parasites Vectors 12, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang Z, Wang Z, Shi X, Wu Q, Tao Y, Guo H, Ji C and Bai Y (2018) Complete mitochondrial genome of Parasesarma affine (Brachyura: Sesarmidae): Gene rearrangements in Sesarmidae and phylogenetic analysis of the Brachyura. Int J Biol Macromol 118, 31–40. [DOI] [PubMed] [Google Scholar]
- 9. Shen X, Tian M, Liu Z, Cheng H, Tan J, Meng X and Ren J (2009) Complete mitochondrial genome of the sea cucumber Apostichopus japonicus (Echinodermata: Holothuroidea): the first representative from the subclass Aspidochirotacea with the echinoderm ground pattern. Gene 439, 78–86. [DOI] [PubMed] [Google Scholar]
- 10. Fan S, Hu C, Wen J and Zhang L (2011) Characterization of mitochondrial genome of sea cucumber Stichopus horrens: a novel gene arrangement in Holothuroidea. Sci China Life Sci 54, 434–441. [DOI] [PubMed] [Google Scholar]
- 11. Lei R, Shore GD, Brenneman RA, Engberg SE, Sitzmann BD, Bailey CA and Louis EE (2010) Complete sequence and gene organization of the mitochondrial genome for Hubbard's sportive lemur (Lepilemur hubbardorum). Gene 464, 44–49. [DOI] [PubMed] [Google Scholar]
- 12. Liao Y (1997) Fauna Sinica: Phylum Echinodermata, Class Holothuroidea, pp. 215–216. Science Press, Beijing. [Google Scholar]
- 13. Utzeri VJ, Ribani A, Bovo S, Taurisano V, Calassanzio M, Baldo D and Fontanesi L (2019) Microscopic ossicle analyses and the complete mitochondrial genome sequence of Holothuria (Roweothuria) polii (Echinodermata; Holothuroidea) provide new information to support the phylogenetic positioning of this sea cucumber species. Mar Genomics 51, 100735. [DOI] [PubMed] [Google Scholar]
- 14. Burland TG (1999) DNASTAR's Lasergene sequence analysis software. Bioinformatics Methods and Protocols 132, 71–91. [DOI] [PubMed] [Google Scholar]
- 15. Bernt M, Donath A, Juhling F, Externbrink F, Florentz C, Fritzsch G, Putz J, Middendorf M and Stadler PF (2013) MITOS: improved de novo metazoan mitochondrial genome annotation. Mol Phylogenet Evol 69, 313–319. [DOI] [PubMed] [Google Scholar]
- 16. Laslett D and Canback B (2007) ARWEN: a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24, 172–175. [DOI] [PubMed] [Google Scholar]
- 17. Wyman SK, Jansen RK and Boore JL (2004) Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. [DOI] [PubMed] [Google Scholar]
- 18. Sudhir K, Koichiro T and Masatoshi N (2004) MEGA3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief Bioinform 2, 2. [DOI] [PubMed] [Google Scholar]
- 19. Perna NT and Kocher TD (1995) Patterns of nueleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J Mol Evol 41, 353–358. [DOI] [PubMed] [Google Scholar]
- 20. Arndt A, Marquez C, Lambert P and Smith MJ (1996) Molecular phylogeny of eastern pacific sea cucumbers (Echinodermata: Holothuroidea) based on mitochondrial DNA sequence. Mol Phylogenet Evol 6, 425–437. [DOI] [PubMed] [Google Scholar]
- 21. Sun XJ, Li Q and Kong LF (2010) Comparative mitochondrial genomics within sea cucumber (Apostichopus japonicus): provide new insights into relationships among color variants. Aquaculture 309, 280–285. [Google Scholar]
- 22. Ronquist F and Huelsenbeck JP (2003) MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. [DOI] [PubMed] [Google Scholar]
- 23. Wang Z, Shi X, Tao Y, Wu Q, Bai Y, Guo H and Tang D (2019) The complete mitochondrial genome of Parasesarma pictum (Brachyura: Grapsoidea: Sesarmidae) and comparison with other Brachyuran crabs. Genomics 111, 799–807. [DOI] [PubMed] [Google Scholar]
- 24. Perseke M, Bernhard D, Fritzsch G, Brummer F, Stadler PF and Schlegel M (2010) Mitochondrial genome evolution in Ophiuroidea, Echinoidea, and Holothuroidea: insights in phylogenetic relationships of Echinodermata. Mol Phylogenet Evol 56, 201–211. [DOI] [PubMed] [Google Scholar]
- 25. Tang BP, Xin ZZ, Liu Y, Zhang DZ, Zhang HB, Chai XY, Zhou CL and Liu QN (2017) The complete mitochondrial genome of Sesarmops sinensis reveals gene rearrangements and phylogenetic relationships in Brachyura. PLoS One 12, e0179800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li X, Xiao N, Zeng X and Yao W (2019) Molecular phylogeny and ossicle evolution analysis for 13 species of sea cucumber in China. J Fish Sci China 26, 416–426. [Google Scholar]
- 27. Takezaki N and Gojobori T (1999) Correct and incorrect vertebrate phylogenies obtained by the entire mitochondrial DNA sequences. Mol Biol Evol 5, 5. [DOI] [PubMed] [Google Scholar]
- 28. Yan D, Tang Y, Xue X, Wang M, Liu F and Fan J (2012) The complete mitochondrial genome sequence of the western flower thrips Frankliniella occidentalis (Thysanoptera: Thripidae) contains triplicate putative control regions. Gene 506, 117–124. [DOI] [PubMed] [Google Scholar]
- 29. Arndt A and Smith MJ (1998) Mitochondrial gene rearrangement in the sea cucumber genus Cucumaria . Mol Biol Evol 8, 8. [DOI] [PubMed] [Google Scholar]
- 30. Kumazawa Y, Ota H, Nishida M and Ozawa T (1996) Gene rearrangements in snake mitochondrial genomes: highly concerted evolution of control‐region‐like sequences duplicated and inserted into a tRNA gene cluster. Mol Biol Evol 9, 9. [DOI] [PubMed] [Google Scholar]
- 31. Shen H, Braband A and Scholtz G (2013) Mitogenomic analysis of decapod crustacean phylogeny corroborates traditional views on their relationships. Mol Phylogenet Evol 66, 776–789. [DOI] [PubMed] [Google Scholar]
- 32. Bronstein O and Kroh A (2019) The first mitochondrial genome of the model echinoid Lytechinus variegatus and insights into Odontophoran phylogenetics. Genomics 111, 710–718. [DOI] [PubMed] [Google Scholar]
- 33. Steel M (2005) Phylogenetic diversity and the greedy algorithm. Syst Biol 54, 527–529. [DOI] [PubMed] [Google Scholar]
- 34. Kilpert F and Podsiadlowski L (2006) The complete mitochondrial genome of the common sea slater, Ligia oceanica (Crustacea, Isopoda) bears a novel gene order and unusual control region features. BMC Genomics 7, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Andrea S and Smith MJ (2001) A novel mitochondrial gene order in the crinoid echinoderm Florometra serratissima . Mol Biol Evol 1, 1. [DOI] [PubMed] [Google Scholar]
- 36. Zou H, Jakovlic I, Chen R, Zhang D, Zhang J, Li WX and Wang GT (2017) The complete mitochondrial genome of parasitic nematode Camallanus cotti: extreme discontinuity in the rate of mitogenomic architecture evolution within the Chromadorea class. BMC Genom 18, 840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Uthicke S, Byrne M and Conand C (2010) Genetic barcoding of commercial Beche‐de‐mer species (Echinodermata: Holothuroidea). Mol Ecol Resour 10, 634–646. [DOI] [PubMed] [Google Scholar]
- 38. Zhang D, Zou H, Wu SG, Li M, Jakovlic I, Zhang J, Chen R, Li WX and Wang GT (2018) Three new Diplozoidae mitogenomes expose unusual compositional biases within the Monogenea class: implications for phylogenetic studies. BMC Evol Biol 18, 133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhang D, Zou H, Hua CJ, Li WX, Mahboob S, Al‐Ghanim KA, Al‐Misned F, Jakovlic I and Wang GT (2019) Mitochondrial architecture rearrangements produce asymmetrical nonadaptive mutational pressures that subvert the phylogenetic reconstruction in Isopoda. Genome Biol Evol 11, 1797–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ivey JL and Santos SR (2007) The complete mitochondrial genome of the Hawaiian anchialine shrimp Halocaridina rubra Holthuis, 1963 (Crustacea: Decapoda: Atyidae). Gene 394, 35–44. [DOI] [PubMed] [Google Scholar]
- 41. Li WX, Zhang D, Boyce K, Xi BW, Zou H, Wu SG, Li M and Wang GT (2017) The complete mitochondrial DNA of three monozoic tapeworms in the Caryophyllidea: a mitogenomic perspective on the phylogeny of eucestodes. Parasit Vectors 10, 314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All original sequence data in this study were submitted to the NCBI database under accession number MN198190.