

Abstract
Pulmonary disease after World Trade Center particulate matter (WTC-PM) exposure is associated with dyslipidemia and the receptor for advanced glycation end products (RAGE); however, the mechanisms are not well understood. We used a murine model and a multiomics assessment to understand the role of RAGE in the pulmonary long-term effects of a single high-intensity exposure to WTC-PM. After 1 month, WTC-PM–exposed wild-type (WT) mice had airway hyperreactivity, whereas RAGE-deficient (Ager−/−) mice were protected. PM-exposed WT mice also had histologic evidence of airspace disease, whereas Ager−/− mice remained unchanged. Inflammatory mediators such as G-CSF (granulocyte colony–stimulating factor), IP-10 (IFN-γ–induced protein 10), and KC (keratinocyte chemoattractant) were differentially expressed after WTC-PM exposure. WTC-PM induced α-SMA, DIAPH1 (protein diaphanous homolog 1), RAGE, and significant lung collagen deposition in WT compared with Ager−/− mice. Compared with WT mice with PM exposure, relative expression of phosphorylated to total CREB (cAMP response element–binding protein) and JNK (c-Jun N-terminal kinase) was significantly increased in the lung of PM-exposed Ager−/− mice, whereas Akt (protein kinase B) was decreased. Random forests of the refined lung metabolomic profile classified subjects with 92% accuracy; principal component analysis captured 86.7% of the variance in three components and demonstrated prominent subpathway involvement, including known mediators of lung disease such as vitamin B6 metabolites, sphingolipids, fatty acids, and phosphatidylcholines. Treatment with a partial RAGE antagonist, pioglitazone, yielded similar fold-change expression of metabolites (N6-carboxymethyllysine, 1-methylnicotinamide, N1+N8-acetylspermidine, and succinylcarnitine [C4-DC]) between WT and Ager−/− mice exposed to WTC-PM. RAGE can mediate WTC-PM–induced airway hyperreactivity and warrants further investigation.
Keywords: occupational exposure, particulate matter, airway hyperreactivity, murine models, lung injury
Clinical Relevance
This study adds to the body of literature investigating the role of receptor for advanced glycation end products in airway hyperreactivity and represents one of the few specifically investigating particulate matter–induced lung disease. The extensive multiomics design also further highlights novel related pathways through metabolomics.
The development of airway hyperreactivity (AHR) due to particulate matter (PM) exposure is a global health concern (1, 2). During the events of September 11, 2001 (9/11), Fire Department of New York (FDNY) firefighters were exposed to World Trade Center particulate matter (WTC-PM), a heterogeneous mixture of bioactive compounds (3, 4). In WTC-PM–exposed firefighters, metabolically active biomarkers are associated with the development of WTC lung injury (WTC-LI) (5–10). Increasing evidence supports the importance of the receptor for advanced glycation end products (RAGE), also known as the advanced glycation end product receptor (AGER), in obstructive airway disease (11). However, the mechanisms of WTC-PM–associated persistent AHR and the role of RAGE are not well characterized.
RAGE expression is highest in the lung and is localized to alveolar epithelial cells/macrophages, vascular endothelial cells, and airway smooth muscle cells (12–14). RAGE, a member of the immunoglobulin superfamily, binds diverse ligands, including products of metabolic stress, such as the advanced glycation end products (AGEs) and HMGB1 (high mobility group box 1 protein). The membrane-bound form is a key mediator of inflammation and metabolic syndrome (15–17). Soluble RAGE is being explored as a diagnostic biomarker of emphysema, chronic inflammatory diseases, and WTC-PM–associated FEV1 loss (18, 19). The role of RAGE was examined in occupational and idiopathic lung diseases such as chronic obstructive pulmonary disease (COPD) and pulmonary fibrosis (20, 21). In murine models of asthma, deletion and inhibition of RAGE-deficient (Ager−/−) mice was protective (22–24).
PPARγ (peroxisome proliferator-activated receptor-γ) agonists such as pioglitazone (Pio) have been investigated for antiinflammatory effects through the downregulation of RAGE and NF-κB (25–28). PPARγ is a nuclear hormone receptor present in lung tissue and alveolar macrophages that is involved in adipocyte differentiation and macrophage activation (29, 30). Polymorphisms of PPARγ have been associated with the development of COPD and asthma (31, 32). Pretreatment of aortic smooth muscle cells with PPARγ agonists downregulated RAGE expression and inhibited proliferation in response to RAGE agonists (33, 34). In addition to the attenuating effects that Pio has on RAGE, its effects on the metabolome have been studied in metabolically relevant conditions such as diabetes and dyslipidemia (35). These metabolically active conditions have been found to be predictive of a loss of lung function in WTC exposure (36–38). PPARγ agonists are being investigated as a possible therapy for COPD (31, 39, 40). Agonistic induction of PPARγ reverses cigarette smoke–induced emphysema and counteracts IL-8 release (41, 42).
WTC-PM exposure induced an inflammatory phenotype 24 hours (24-H) after exposure in both in vitro and in vivo models (11). Because WTC-PM exposure intensity is related to arrival time and loss of lung function in the FDNY cohort, we have again chosen to use a single high-dose exposure in our murine WTC-PM exposure model as we did in our prior work (11). Earlier investigations revealed that a single-time oropharyngeal aspiration of WTC-PM acutely causes AHR and FEV1 loss after 24-H, as observed in the human WTC-exposed cohort (3, 11, 43). Therefore, the long-term effects on repair and remodeling after WTC-PM exposure are critical endpoints to examine. Thus, we hypothesized that a single exposure to WTC-PM would yield a persistent change in pulmonary mechanics sustained over 1 month (1-M). Furthermore, we predicted that deletion of Ager in a murine model would protect mice from the development of WTC-PM–associated lung function changes. Finally, we predicted that there would be a distinct biomarker profile (phenome) expressed 1-M after WTC-PM exposure in wild-type (WT) mice compared with Ager−/− mice.
Methods
Ager refers to the murine gene, whereas AGER refers to the human gene and the human/murine protein.
WTC-PM Exposure
Female WT C57BL/6 mice (The Jackson Laboratory) more than 12 weeks old, matched for age and weight and cohoused with Ager−/− mice (A.M.S.), had free access to food and water and were maintained on a 12-h/12-h light/dark cycle (11, 43, 44). WTC-PM53 (200 μg in 100 μl of sterile PBS) or an equal volume of PBS (control animals) was oropharyngeally aspirated (intratracheally) with mice under isoflurane anesthesia (New York University Institutional Animal Care and Use Committee protocol s16-00447) (11, 43, 45, 46). The dose of WTC-PM <53 μm (aerodynamic diameter) reflects the measured concentration and particle size at the 9/11 debris pile (47).
Pio Treatment
We have previously determined that mice exposed to 100 μg of WTC-PM53 developed AHR after 24-H, a concentration that was considered equivalent to a rescue worker being exposed to 425 μg/m3 of WTC-PM over an 8-hour shift (11, 43). Mice were gavaged for 6 days with 60 mg/kg Pio (Santa Cruz Biotechnology) or an equal volume of 0.5% carboxymethylcellulose vehicle (CMC-Veh) (n = 3/exposure) and exposed on Day 6 to WTC-PM53 (100 μg) or an equal volume of PBS. This dose and route have been effective in ameliorating lung injury after exposure (48).
Lung Mechanics
Mice were anesthetized by intraperitoneal injection (ketamine/xylazine 100/10 mg/ml; 0.11 ml/10 g; Troy Laboratories), tracheostomized (18-gauge needle; BD Biosciences), and placed in a plethysmograph. Then they received 10 ml/kg tidal volume (Vt) at 150 breaths/min and positive end-expiratory pressure of 3 cm H2O (flexiVent; SCIREQ) after 24-H for Pio treatment (PioRx) and after 1-M for all other mice. Automated deep inflation, SnapShot-150, Quick Prime-3, pressure–volume, and negative pressure-driven forced expiration were measured; averages of at least three measures/subject were obtained (11, 49–52). Methacholine (Mch) (Santa Cruz Biotechnology) dilutions were nebulized (Aerogen), and Mch dose delivered (Mdel) was interpolated at which resistance (R) was 200% (PC200) of the mean response to PBS (45, 53, 54). N values in legends reflect exclusions made on the basis of quality controls.
Sample Acquisition/Microscopy
Lungs were lavaged (1 ml of cold PBS), and the cytospin preparation was stained with hematoxylin and eosin (H&E) (Hema 3; Fisher Scientific) (55). Plasma was collected (18-gauge cardiac puncture; heparinized 1-ml syringe) (11). Lungs were snap frozen, fixed in situ (4% paraformaldehyde; MilliporeSigma) at 25 cm H2O and embedded in paraffin (43, 56). Coronal 5-μm sections underwent H&E staining for primary assessment of architecture, Gömöri trichrome staining (Thermo Scientific) for collagen deposition, and periodic acid–Schiff staining (PAS; Abcam) for visualization of mucin (43, 57–60). Investigators were blinded to experimental conditions during selection and all measurements (11, 43, 56). Immunohistochemistry of 4-μm sections was performed with the primary antibodies antimammalian DIAPH1 (protein diaphanous homolog 1; Abcam), AGER, and anti–α-SMA, secondary antibodies, and DAPI (Santa Cruz Biotechnology). Fluorescence imaging (Nikon Instruments fluorescence upright microscope) and intensity scoring were performed by three blinded investigators. To examine morphology, a grid (520 × 520 μm) was laid over the entire section; at 20× magnification, every fifth field from left to right was chosen until 10 fields were selected; area fraction (AF) and mean linear intercept (MLI) were quantified (11, 57, 58, 60–64).
Collagen
Whole-mount glass slides (n = 3/group) were imaged (Carl Zeiss Microscopy) and digitized (NanoZoomer 2.0-HT; Hamamatsu Photonics) at 40× magnification, and collagen was assessed by support vector machine learning (Orbit Image Analysis; www.orbit.bio) (65). Digitized whole images (n = 12) were trained using manually annotated pixels for alveolar tissue/collagen/background classification. The proportion of classified collagen (green) to the entire classified area within the respective annotation was quantified (as a percentage) and compared (Welch t test for differing variances and Shapiro-Wilk test to confirm normality). The model classification was validated. Hydroxyproline was quantified per protocol (ab222941; Abcam).
Omics
Analytes
BAL and plasma were assayed for MCYTMAG-70K-PX25 (G-CSF [granulocyte colony-stimulating factor]; GM-CSF [granulocyte-macrophage colony-stimulating factor]; IFN-γ; IL-1α, -1β, -2, -4, -7, -9, -10, -12 [p40/p70], -13, -15, and -17; IP-10 [IFN-γ–induced protein 10], KC [keratinocyte chemoattractant], MCP-1 [monocyte chemoattractant protein 1]; MIP-1α [macrophage inflammatory protein-1α], -1β, and -2; RANTES [regulated upon activation, normal T cell expressed and secreted]; and TNF-α), MGAMMAG-300E (IgE) and -300K (IgA, IgG1, IgG2A, IgG2B, IgG3, and IgM) (Luminex 200IS; MilliporeSigma), and histamine (ELISA; Beckman Coulter).
Transcription factors
Phosphorylated/total CREB (cAMP response element–binding protein), NF-κB, Akt (protein kinase B), p70S6K, JNK (c-Jun N-terminal kinase), p38, ERK1/2 (extracellular signal-regulated kinase 1/2), STAT3/5 (signal transducer and activator of transcription 3/5) (48-680/1MAG), and total β-tubulin (46-713MAG) were quantified in lung homogenates (11). The metabolome of 100-mg snap-frozen lung sections was assessed (Metabolon Inc.) (5, 66–68).
Database Management/Statistics
IBM SPSS Statistics version 23 (IBM Corp.), flexiWare version 7.5.4 (SCIREQ), MasterPlex QT (MiraiBio/Hitachi), and Prism version 7 (GraphPad Software) software was used. Statistical comparisons were made using the Mann-Whitney U test, and a two-tailed P < 0.05 was considered significant. Tissue resistance at each frequency was compared by multiple t tests with the Holm-Šídák correction. Qualified/curated metabolites were subjected to random forests (RF) analysis (randomForest package R 3.4.3; R Foundation for Statistical Computing) (69, 70). A refined metabolite profile (top 5% based on mean decrease accuracy) was developed (69). Principal component analysis (PCA) was used for feature projection/data visualization of mean-centered, normalized attributes. Variance explained by PCA was quantified as the summation of percentage of total variance. Unsupervised, agglomerative, two-way hierarchical clustering was performed on the refined profile (Spearman correlation, average linkage) (MATLAB R2018a; MathWorks). Pathway enrichment and volcano plots were assessed. Detailed methods are provided in the data supplement.
Results
WTC-PM Exposure–induced AGER Expression
Consistent with putative roles for RAGE in WTC-PM responses, 1-M after WTC-PM exposure, there was a significant induction of RAGE expression in WT mice compared with that in control animals (Figures 1A and 1B; Figure E1A in the data supplement). Of note, RAGE expression was not detected in Ager−/− mice (Figures 1C, 1D, and E1A). These findings indicate that even 1-M after exposure, expression of RAGE remains elevated, suggesting that RAGE-dependent pathways may exert long-term effects.
Figure 1.

Expression of advanced glycation end product receptor (AGER) in the lung is induced after World Trade Center particulate matter (WTC-PM) exposure, whereas expression is attenuated in receptor for advanced glycation end products (RAGE)-deficient (Ager−/−) mice. (A–D) AGER was stained (green); scale bars, 100 μm. All sections were costained with DAPI (blue) for nuclei visualization. n = 3. Ager−/− mice are protected from airway hyperreactivity, increased tissue resistance, and mean linear intercept (MLI) increase 1 month (1-M) after WTC-PM exposure. (E1) Methacholine (Mch) dose–response curve shows raw data (fine points), average resistance, and delivered Mch per nominal Mch dose, genotype, and exposure group (heavy points). *P < 0.05 between wild-type (WT)-PBS and WT-PM mice at doses of 50, 100, and 200 mg/ml nominal Mch and not significant between Ager−/−-PBS and Ager−/−-PM mice by multiple t test by row. (E2) Airway hyperreactivity (Mch dose delivered interpolated at which resistance was 200% [PC200]) was assessed in the following four groups: WT-PBS (n = 7), WT-PM (n = 10), Ager−/−-PBS (n = 5), and Ager−/−-PM (n = 5). *P < 0.05. (F) WTC-PM exposure affects small airways to a greater degree in WT mice than in Ager−/− mice. WT-PM mice had significantly elevated tissue resistance at 1 and 1.5 Hz (*P < 0.001) compared with WT-PBS mice. Ager−/−-PM mice had significantly decreased tissue resistance at 1 Hz (†P < 0.01) compared with the WT-PM mice. WT-PBS (n = 7), WT-PM (n = 15), Ager−/−-PBS (n = 8), and Ager−/−-PM (n = 7). Points and error bars represent mean Rtis and standard error. (G–J) Histology; scale bars, 1,428.57 μm. Light microscopic examination of representative hematoxylin and eosin (H&E)-stained sections of whole lung costained with Gömöri trichrome for collagen assessment (see Figure 2); scale bars, 200 μm. (G′–J′) Representative images magnified section; scale bars, 100 μm. (G and G′) WT-PBS. (H and H′) WT-PM. (I and I′) Ager−/−-PBS. (J and J′) Ager−/−-PM. H&E-stained images were used to assess (K) MLI and (L) area fraction. Columns and error bars represent mean and standard error. *P < 0.05. NS = not significant.
Ager−/− Mice Are Protected from AHR 1-M after WTC-PM Exposure
WT and Ager−/− mice exhibited no difference in multiple lung function parameters 1-M after WTC-PM exposure, including FEV, resistance, compliance, and tissue damping, contrasting with our earlier findings at 24-H (11). WT mice express an AHR phenotype 1-M after PM exposure (P < 0.05 for WT-PBS vs. WT-PM at doses of 50, 100, and 200 nominal Mch [mg/ml] and not significant between Ager−/−-PBS and Ager−/−-PM by multiple t test by row) (Figure 1E1). WT-PM mice had a significantly lower PC200, a marker of AHR (median [interquartile range (IQR)], 37.9 [22.3–44.9] mg/ml), than control animals (113.7 [55.4–160.1] mg/ml; P = 0.014). Ager−/− mice demonstrated no difference in PC200 between PM and control animals (median [IQR], (67.0 [40.4–196.0] mg/ml and 110.6 [39.1–219.0] mg/ml Mch, respectively; P = not significant) (Figure 1E2). Taken together, these data at 1-M indicate that there is persistence of AHR after WTC-PM exposure, whereas other WTC-PM–associated changes seen after 24-H resolved (11).
Because mucus production is a phenotypic finding in AHR, we next determined if WTC-PM exposure results in increased mucin production. There was no significant difference in the accumulation of PAS-stained material 1-M after WTC-PM exposure in either WT or Ager−/− mice (data not shown). In contrast, after 24-H, WTC-PM–exposed WT mice showed accumulation of PAS-stained material in the airway epithelial cells (Figures E2B, E2D, and E2F) when compared with PBS-WT mice (Figures E2A, E2C, and E2E), indicating that WTC-PM exposure resulted in mucin production by goblet cells. These findings indicate that WTC-PM induces acute mucus production in 24-H, whereas the presence of mucus has resolved by 1-M.
Changes in Respiratory Mechanics 1-M after WTC-PM Exposure Occur in the Peripheral Airways
To distinguish the effects of PM on central and peripheral airways, tissue resistance was derived at frequencies between 1 and 20.5 Hz from raw input impedance data collected during baseline mechanics. Lower frequencies are associated with smaller caliber peripheral airways and alveoli, whereas higher frequencies reflect the larger central airway resistance. PM-exposed WT mice had significantly increased mean tissue resistance at both 1 and 1.5 Hz compared with control animals, whereas PM-exposed Ager−/− mice had only significantly elevated 1-Hz resistance (Figure 1F).
Ager−/− Mice Are Protected from an Increase in MLI but Not from an Increase in AF 1-M after WTC-PM Exposure
We quantified MLI, the mean free distance of gas exchange surfaces within the acinar surface complex, and AF, the biovolume/airspace ratio using the H&E-stained images (representative images in Figures 1G–1J; low-power images from which Figures 1G–1J were extracted are shown in Figures 2A–2D) (11). WT mice exposed to WTC-PM had significantly increased MLI after 1-M compared with control animals (Figure 1K). Ager−/− mice exposed to WTC-PM did not have a significant difference in MLI 1-M after exposure when compared with control animals. AF, a measure of biovolume/airspace ratio, was significantly increased in both PM-exposed WT and Ager−/− mice compared with control animals (Figure 1L).
Figure 2.
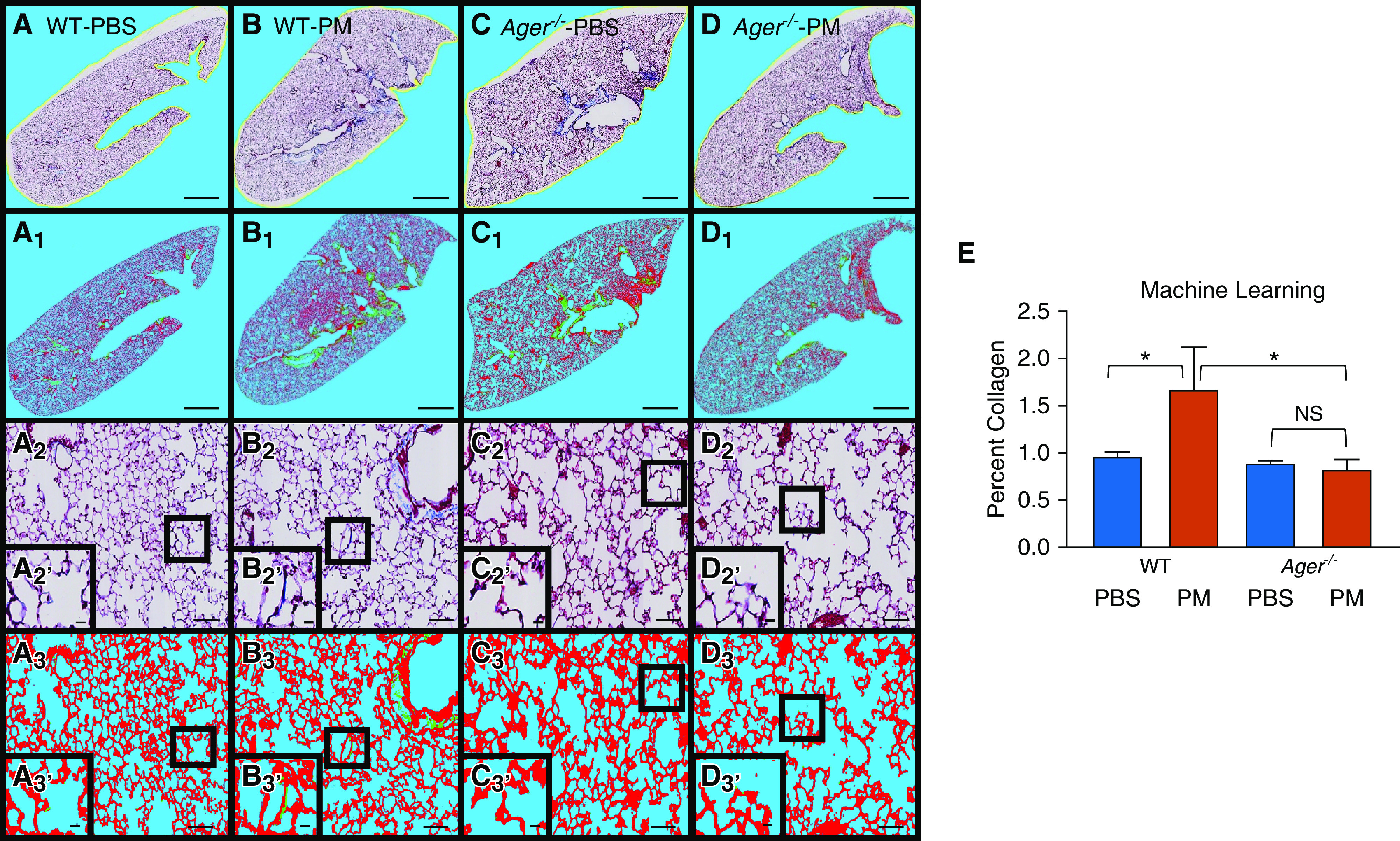
Ager−/− mice are protected from PM-induced collagen deposition whereas PM-exposed WT mice are not according to spectral analysis. Representative images using Gömöri trichrome staining to assess collagen. n = 3/group. (A–D) Unclassified whole lung of WT and Ager−/− specimens that were PM exposed and unexposed (A–D) are shown with their pixel-classified counterparts (A1–D1); scale bars, 1,428.57 μm. (A2–D2) Alveolar wall of WT and Ager−/− PM-exposed mice and PBS control animals; scale bars, 102.56 μm. (A2′–D2′) High-magnification insets; scale bars, 16.62 μm. (A3–D3) After pixel classification; scale bars, 102.56 μm. (A3′–D3′) High-magnification insets; scale bars, 16.62 μm. Faint vertical lines in the micrographs are artifacts of the automated image-scanning software used to create these montage images (Leica Microsystems). (E) Median percent collagen as assessed by machine learning. Error bars represent interquartile range. *P < 0.01. False color key: blue (red:77, green:203, blue:246), background (low and high magnification), red (red:237, green:15, blue:42), alveolar tissue, green (red:0, green:255, blue:1), collagen.
Ager−/− Mice Are Protected from Collagen Formation after WTC-PM Exposure
Using our model’s code, we assessed a median of 7.8 × 107 pixels (IQR, 6.3 × 107–9.7 × 107/image) (data supplement file 2). Collagen was assessed via machine learning algorithm using whole-lung images (Figures 2A–2D). We demonstrated significantly greater collagen deposition in WT lungs exposed to WTC-PM relative to control animals after 1-M (mean [SE], 1.8% [0.21] vs. 0.78% [0.17]; P = 0.021) (Figure 2E). In contrast, Ager−/− mice exhibited no difference in collagen deposition 1-M after PM exposure when compared with control animals. Collagen deposition was also greater in PM-exposed WT lungs than in PM-exposed Ager−/− mice (mean [SE], 1.8% [0.21] vs. 0.80% [0.09]; P = 0.013) (Figure 2E). Our model had a median (IQR) accuracy of 90% (85–95%) when compared with blinded observers. Lung homogenate hydroxyproline content was quantified by colorimetric assay, and as part of the metabolome as trans-4-hydroxyproline, it was not significantly different in WT and Ager−/− mice exposed to PM compared with control animals (Figure E5).
Ager−/− PM-exposed Mice Display Attenuated Expression of α-SMA in Lung Tissue
α-SMA is highly expressed in collagen-producing cells. WT α-SMA immunofluorescence intensity was significantly elevated after WTC-PM exposure (Figures 3A, 3B, and E1B). Although PM-exposed Ager−/− mice displayed a significantly induced expression of α-SMA compared with control animals (Figures 3C, 3D, and E1B), the Ager−/− mice demonstrated significantly attenuated expression of α-SMA after exposure to WTC-PM compared with WT.
Figure 3.

Expression of α-SMA and DIAPH1 (protein diaphanous homolog 1) in the lung is induced after WTC-PM exposure, whereas their expression is attenuated in Ager−/− mice. (A–D) α-SMA and (E–H) DIAPH1 are all stained green. Scale bars, 100 μm. (A′–H′) Inset scale bars, 200 μm. All sections were costained with DAPI (blue) for nuclei visualization.
Ager−/− Mice Have Attenuated DIAPH1 Expression after WTC-PM Exposure
DIAPH1 is the binding partner of the RAGE cytoplasmic domain, which has been implicated in RAGE signaling (71). We thus tested its expression in WTC-PM–exposed mice. Although WT mice displayed significant increased expression of DIAPH1 1-M after exposure to WTC-PM compared with PBS control animals (Figures 3E, 3F, and E1C), Ager−/− mice demonstrated no difference in the expression of DIAPH1 1-M after WTC-PM exposure compared with PBS control animals (Figures 3G, 3H, and E1C). Overall, Ager−/− mice displayed significantly attenuated expression of α-SMA, RAGE, and DIAPH1 compared with WT animals 1-M after WTC-PM exposure (Figures 3F, 3H, and E1C).
WTC-PM Altered the BAL and Plasma Cytokine Profile 1-M after Exposure
There was no significant difference in mean BAL differential macrophage predominance 1-M after PM exposure in WT and Ager−/− mice (mean differentials, macrophages [M], neutrophils [N], lymphocytes [L], and eosinophils [E] for WT-PBS, M100N0L0E0; for WT-PM, M98N1L1E0; for Ager−/−-PBS, M98N1L1E0; for Ager−/−-PM, M98N1L1E0).
In BAL, IL-6 and IL-1α were significantly increased in both PM-exposed WT and Ager−/− mice compared with control animals at 1-M. In addition, PM-induced elaboration of IL-6 and IL-1α also significantly differed between genotypes. G-CSF elaboration was significantly different from that in control animals only in WT mice (Figure 4A). IL-10 fold change was significantly higher in PM-exposed Ager−/− mice, but it was not significantly different in WT animals at 1-M. IL-9 (data not shown), IP-10, RANTES, and KC were significantly elevated in WT and Ager−/− mice exposed to WTC-PM compared with control animals. RANTES and KC fold changes were significantly attenuated in PM-exposed Ager−/− mice compared with WT mice after 1-M, whereas IP-10 was not (Figure 4A).
Figure 4.

WTC-PM altered the chemokine/cytokine concentrations 1-M after exposure in (A) BAL and (B) plasma. All values are expressed as fold change of PM-exposed WT and Ager−/− mice over their respective PBS control animals. Bars denote means. *Significant in WT-PBS versus WT-PM, **significant between fold change of WT-PM versus Ager−/−-PM, and †significant in Ager−/−-PBS versus Ager−/−-PM. G-CSF = granulocyte colony–stimulating factor; IP-10 = IFN-γ–induced protein 10; KC = keratinocyte chemoattractant; RANTES = regulated upon activation, normal T cell expressed and secreted.
In plasma, similarly to the BAL G-CSF, IP-10 and KC fold changes were significantly increased in WT mice 1-M after PM exposure, whereas G-CSF, IP-10, and KC were no different in PM-exposed plasma of Ager−/− mice when compared with PBS control animals (Figure 4B). G-CSF and KC elaboration in WT mice after WTC-PM exposure was higher than that of PM-exposed Ager−/− mice. PM-exposed WT IP-10 expression was significantly decreased relative to baseline and was lower than that of the Ager−/− counterparts (Figure 4B).
The levels of expression of the following analytes were not significantly altered in either tissue compartment: GM-CSF; IFN-γ; IL-1β, -2, -4, -5, -7, -9, -12(p40), -12(p70), -13, -15, and -17; MCP-1; MIP-1α, -1β, and -2; and TNF-α. In addition, IgA, IgG1, IgG2A, IgG2B, IgG3, IgM, and IgE and histamine concentrations were no different between WTC-PM exposures and PBS control conditions in both WT and Ager−/− mice (data not shown).
Ager−/− Mice Express Differential Transcription Factor Profiles 1-M after WTC-PM Exposure in Both BAL and Lung Homogenates
Ratios of phosphorylated to total concentrations of signaling and transcription factors, expressed as median fluorescence intensity relative to tubulin, were compared between WT and Ager−/− mice. In BAL, phospho-p38 and JNK were significantly higher, whereas p70S6K proteins (phosphorylated) were attenuated, in Ager−/− mice compared with WT mice 1-M after exposure to WTC-PM in BAL pellets (Figure 5A). In lung homogenates, the ratios of phosphorylated/total JNK and CREB were higher, whereas phosphorylated/total Akt was lower, in Ager−/− mice than in WT mice 1-M after PM exposure (Figure 5B).
Figure 5.

Transcription factors. Ratio of phosphorylated to total concentrations of transcription factors, expressed as median fluorescence intensity (MFI) relative to tubulin in (A) BAL pellet and (B) lung homogenate, plotted and compared between WT and Ager−/− fold change (mean, SEM) of the ratio (phosphorylated to total proteins) 1-M after WTC-PM exposure compared with PBS control animals. *P < 0.05, significant by Mann-Whitney U test. AKT = protein kinase B; CREB = cAMP response element–binding protein; JNK = c-Jun N-terminal kinase.
WTC-PM–associated AHR Exhibited a Distinct Metabolome in WT Mice Compared with Ager−/− Mice 1-M after Exposure
Finally, we assessed the metabolome in lung homogenates to further our understanding of the metabolite milieu present 1-M after WTC-PM exposure. Of 737 metabolites detected, 580 qualified and were included in further analysis. These metabolites are summarized in Table E1. Initial assessments included calculation of the pathway enrichment values (Figure E3). This showed that AGEs, carnitine, and thiamine metabolism had the greatest enrichment values when we compared PM-exposed WT to Ager−/− mice (Figure E3). In addition, the differential expression of metabolites in the WT and Ager−/− mice allowed comparison of the fold change of metabolites compared with their respective control animals (Figures E4A and E4B). In WT mice, lipids, amino acids, and nucleotides are differentially expressed after WTC-PM exposure compared with PBS control animals (Figure E4A). In Ager−/− PM-exposed mice, similar metabolite pathways were differentially expressed but with the addition of galactose (carbohydrate) when compared with PBS control animals (Figure E4B).
Further assessment included qualified metabolites, which were subjected to RF analysis to reduce the dimensionality of the data by discovering those metabolites most relevant to our outcome. Mean decrease accuracy was measured for every metabolite to assess variable importance, and the top 5% were included in the refined metabolite profile (Figure 6A and Table E1). RF analysis of the refined metabolite profile showed a 6.7% out-of-bag estimated error rate (92% estimated predictive accuracy) (Figure 6A). PCA of the refined metabolite profile captured 86.7% of variance in the three components retained on the basis of the scree plot (Figure 6B) and demonstrated clear class separation.
Figure 6.

Metabolomics model optimization. (A) Random forests (RF) were generated to create a refined metabolic profile. RF variable importance in projection is measured by mean decrease accuracy; the top 5% of metabolites important to class separation are shown. The confusion matrix shows classification accuracy of the refined profile. (B) Principal component analysis (PCA) scores plot. PCA of refined metabolic profile reveals class separation (three components capture 86.7% of the total variance). (C) Hierarchical clustering. Refined profile subjected to hierarchical clustering using Spearman correlation as a distance measure and average linkage. GPC = glycerophosphocholine; GPE = glycerophosphoethanolamine; GPS = glycerophosphoserine; PUFA = polyunsaturated fatty acid; TCA = tricarboxylic acid.
PioRx Attenuates Metabolites
Of the 733 metabolites identified, 532 metabolites were qualified, as described in the Methods section. The RF of the 1-M was cross-referenced with the metabolites that met the qualification criteria (present in >80% of mice and relative SD >15%) in the PioRx mice. We identified 23 metabolites that were consistent. We also investigated the AGE-related pathway compound that was identified, N6-carboxymethyllysine. Of these 24 metabolites (which are listed in bold in Table E1), we identified 4 metabolites in which fold-change expression of metabolites was similar between PioRx and Ager−/− mice exposed to PM but different from 1-M WT-PM exposure: N6-carboxymethyllysine, 1-methylnicotinamide, N1+N8-acetylspermidine, and succinylcarnitine (C4-DC). These metabolites had decreased fold-change expression in WT mice 1-M after PM exposure but were relatively increased in Ager−/− and PioRx mice after PM exposure. Only N6-carboxymethyllysine had no difference between CMC-Veh–treated PBS mice and WT-1-M mice, making the changes in AGE-related pathways less likely to be solely due to the effect of time or the dose of WTC-PM (P = 0.25).
Sphingolipids, Phospholipids, and Fatty Acids Were Differentially Expressed among Ager−/− and WT Mice
The heat map of the refined profile suggests metabolite associations through clustering patterns (Figure 6C). Overall, we identified three clusters of metabolites based on a linkage threshold of 0.5 (Figure 6C). Generally, metabolites in cluster A were elevated in Ager−/− compared with WT mice 1-M after PM. Cluster A was the most diverse cluster, containing several classes of lipids—fatty acids, phospholipids, and glycerolipids—as well as polyamine, glutamate, nicotinamide, nicotinate, histidine, and tricarboxylic acid cycle–related metabolites. Cluster B metabolites were elevated in PBS-exposed mice compared with those in mice exposed to WTC-PM. Phospholipids predominated in this cluster and were accompanied by a pyrimidine and fatty acid metabolite. Finally, metabolites in cluster C were decreased in Ager−/− mice compared with WT mice. Cluster C consisted primarily of fatty acids, as well as a monoacylglycerol and metabolites of vitamin B6, purine, and the urea cycle. In addition, sphinganine, a sphingolipid, appeared in the refined profile and was decreased in Ager−/− mice compared with WT mice.
Discussion
WTC-PM exposure is linked to lung injury and inflammation. Inflammatory mediators such as soluble RAGE, C-reactive protein, and lysophosphatidic acid (ligand of RAGE) have been associated with WTC-LI (11). The mechanisms by which WTC-PM yields chronic inflammation and alteration in lung function are poorly understood. RAGE, a biologically plausible mediator of these effects of WTC-PM–induced lung disease, was our focus.
The present study reveals novel findings. We demonstrated that a single exposure of WTC-PM yielded persistent inflammation-related lung injury in PM-exposed WT mice associated with the upregulation of RAGE protein and AHR injury; in contrast, Ager−/− mice were protected from AHR. Furthermore, WT animals had a significantly lower PC200 at 1-M than at 24-H (11). Finally, there is a distinct phenome that is expressed in the Ager−/− mice providing protection from WTC-PM exposure.
Lack of RAGE has been linked with protection from allergen, cigarette smoke, and environmental exposures (11, 24, 57, 72, 73). Our findings could be compared with the vast literature investigating the role of RAGE in allergic T-helper type 2 cell–driven inflammation and AHR/asthma. Ager−/− mice exposed to environmental allergens, specifically IL-33, house dust mite extract, or Alternaria alternata (a fungal allergen extract) did not develop AHR as assessed by Mch challenge (22, 74). Ullah and colleagues suggested that dendritic cells are key mediators in airway inflammation and showed that an adoptive transfer of house dust mite–exposed RAGE+/+ dendritic cells to RAGE−/− mice could recapitulate an allergic phenotype with increased BAL IL-13, eosinophils, neutrophils, and total IgE (75). Other studies investigating direct allergen exposure also found that administration of intranasal recombinant IL-5/IL-13 induced airway inflammation and mucus metaplasia in WT mice, whereas RAGE−/− mice were protected (72). Furthermore, a small-molecule antagonist of RAGE, FPS-ZM1, could pharmacologically block the effects by inhibiting STAT6 activation and led to reduction of the inflammatory profile in WT mice exposed to IL-5/IL-13 (72).
WTC-PM induces RAGE expression in the lungs of WT murine lungs after 1-M more than in control animals, suggesting that WTC-PM–induced lung injury is mediated at least partially through RAGE and that RAGE may be associated with the more chronic effects. RAGE not only is important to the acute effects of WTC-PM exposure but also is relevant to subacute WTC-PM effects in various tissue compartments. We found that the smaller airways are more involved even after 1-M in WT mice. This implies that PM-induced AHR is persistent and leads to permanent lung impairment. In parallel to the AHR and small airway changes, we saw that after 1-M, MLI and AF were increased in PM-exposed WT mice. Furthermore, although Ager−/− mice were protected from MLI increase after PM exposure, their AF remained significantly greater 1-M after a single PM exposure. These changes are unique and suggest a sustained morphometric pulmonary remodeling, implying that RAGE may be required for alveolar enlargement.
Mucus production, common in asthma, is often a marker of the severity of inflammation. Therefore, our finding that 1-M after PM exposure and in the context of AHR there is no evidence of mucus in the airway epithelium is phenotypically important. This finding is in contrast to those seen at 24-H, when PM-exposed WT mice had a phenotype of mucus accumulation on the epithelial cells and mucin production by goblet cells.
The development of AHR after a single PM exposure involves several cellular intermediates and intrinsic airway remodeling. WTC-PM exposure led to not only persistent AHR but also significant collagen deposition (as measured by assessment of digitized whole-lung images). In contrast, Ager−/− mice were protected from collagen deposition. To further explore this finding, we assessed the expression of α-SMA an actin isoform that predominates within vascular smooth muscle cells and plays an important role in fibrogenesis. The expression of α-SMA correlates with the activation of myofibroblasts, which are metabolically and morphologically distinctive fibroblasts expressing α-SMA and their activation plays a key role in development of the fibrotic response (76). WT mouse lung exposed to WTC-PM expresses higher concentrations of α-SMA than PBS-exposed mouse lung 1-M after exposure. Ager−/− mouse lungs were partially protected from α-SMA expression 1-M after WTC-PM exposure when compared with PBS-exposed Ager−/− mouse lungs, thereby suggesting that mice lacking Ager were protected from collagen formation after WTC-PM exposure through control of α-SMA, which is associated with fibroblast activation, therefore implying that α-SMA expression may be responsible for lung tissue fibrosis.
The role of RAGE and lung collagen deposition is also an area of active investigation. One study showed that RAGE promoted α-SMA expression and lung collagen deposition after exposure to bleomycin in an interstitial pulmonary fibrosis model, which initially may seem to contradict our findings (77, 78). However, it is important to note that the Ager−/− mice were bred on a CD-1 background that spontaneously developed lung fibrosis, which could indicate alternative pathways of fibrosis and not necessarily due to the exposure. Studies investigating similar bleomycin-induced pulmonary fibrosis in C57BL/6 mice found relatively decreased collagen deposition when knocking down HMGB1, a molecule that is well described as positively correlated with RAGE (79). Our findings are further bolstered by concordant data from other investigations that, similar to ours, used mice on a C57BL/6 background and found Ager−/− mice showed protection against pulmonary insults such as hyperoxia, allergens, elastase, and renal interstitial fibrosis (23, 80–82).
In this article, we have assayed hydroxyproline (a measure of collagen content) by colorimetric assay and in a global metabolomic panel (trans-4-hydroxyproline, which is equivalent to hydroxyproline) (83). We found that hydroxyproline was equivocal in all exposure groups. The hydroxyproline assay surveys collagen concentration across the whole lung. This includes bronchial tissue and other large airways, which comprise a majority of the lung’s collagen content. Our histological analysis explicitly excludes large airways to examine collagen deposition in alveolar tissue. Therefore, although the collagen deposition across the entire lung may not be significantly different according to hydroxyproline concentration measured in whole-lung homogenate, our histologic assessment using digital images captured nuanced differences in alveolar collagen deposition at this time point (65). There may be a possibility that elevated hydroxyproline content may be measurable at a later time point.
Although we did not see the difference in 4-hydroxyproline content, which is a major component of collagen, comprising around 13.5% of its amino acid composition (84), we noticed a significant difference in alveolar collagen deposition that was quantified by computational method. Furthermore, our data and the previous studies suggest that RAGE may be responsible for fibrosis either directly through collagen formation upon elevated DIAPH1 or indirectly upon activation of α-SMA in the lung. Besides explicitly binding AGER, RAGE also interacts with a number of ligands, including proinflammatory cytokine-like mediators of the S100/calgranulins and HMGB1 protein (85). By binding AGER and other ligands, RAGE contributes to a variety of diseases, including organ fibrosis (14). Furthermore, a calcium-binding protein, S100A8/A9, is a damage-associated molecular pattern that can activate RAGE. Activation of RAGE is involved in the progression of renal fibrosis (86).
To further our understanding of how the RAGE axis may be involved in the pulmonary and systemic effects seen after PM exposure, we assessed the expression DIAPH1, a signaling intermediate of RAGE. We found that PM exposure induced the expression of DIAPH1 but that there was attenuated DIAPH1 expression in pulmonary tissue of Ager−/− mice after PM exposure. RAGE inhibitors have focused on the cytoplasmic tail of the RAGE–intracellular effector DIAPH1 axis. Lack of RAGE eliminates the ligand with which DIAPH1 interacts, thus downregulating its production. Small-molecule inhibitors of RAGE–DIAPH1 interaction have been developed and are potentially bioactive therapeutic agents that may be suitable for future in vitro and in vivo investigations (15, 87).
Similarly to our prior finding at 24-H, several chemokines/cytokines were involved in the inflammatory process in both WT and Ager−/− mice 1-M after PM exposure. Specifically, in our prior work, human alveolar macrophages from healthy volunteers expressed higher concentrations of IL-1α and IL-10 at 24-H after exposure to WTC-PM (88). In our present investigation, in BAL, even 1-M after the exposure, there was differential expression of pro-(G-CSF, IL-6, IL-1α) and antiinflammatory (IL-10) cytokines and chemokines (RANTES, KC) in WT and Ager−/− mice. Similar differential expression of proinflammatory mediators in WT and Ager−/− mice was observed in serum of WT and Ager−/− mice. When compared with the respective PM-altered plasma and BAL chemokine/cytokine profiles at 24-H, there were upregulated early and late innate proinflammatory mediators (G-CSF, IL-6, IP-10, RANTES, and KC) and a downregulated antiinflammatory mediator (IL-10) with associated systemic inflammation either maintained or further differentiated between the two time points after a single dose of WTC-PM (11).
RAGE is expressed in alveolar-type epithelium and macrophages (12–14). To further explore the systemic inflammation seen 1-M after PM exposure, we investigated biologically plausible mediators of RAGE signaling. The mitogen-activated protein kinases p38 and JNK are activated upon stimulation of macrophages (89). Additional intermediates involved in RAGE signaling include Akt and JAK/STATs (90, 91). IL-1α and IL-10 induce STAT3 signaling; however, similar to what was seen at 24-H, WT mice did not show an induction in STAT3 at 1-M. In contrast, Ager−/− mice showed a significant increase in phosphorylated STAT3 after 24-H, but not after 1-M. Ager−/− lungs exposed to PM had higher expression of phosphorylated CREB and JNK than did WT-PM; this was similar to what was seen after 24-H.
In contrast to our earlier findings at 24-H, phosphorylated Akt/Akt was significantly attenuated in Ager−/− mice 1-M after PM. Previous studies demonstrated that inhibition of Akt can effectively inhibit fibrosis through the PI3K/Akt pathway, and p70S6K regulated lung fibrotic tissue formation through TGF-α (transforming growth factor-α) expression (92).Therefore, the increased concentrations of Akt and p70S6K upon PM exposure may contribute to the collagen acquisition and eventually to fibrotic scar formation (92).
In BAL, macrophages predominate, and JNK was similarly significantly elevated in lung homogenate, which is in line with our cytokine findings. Furthermore, in our BAL cellular profile, phosphorylated p38 and p70S6K expression was reduced in Ager−/−-PM mice in comparison to WT animals. Higher ratios of phosphorylated JNK, p38, and CREB were found in both the BAL pellet and lung of Ager−/− mice than in WT mice. The p38 and JNK pathways, despite both being stress activated, often have opposing effects; JNK activity is associated with proinflammatory states and increased cell proliferation, whereas many studies show that p38 has antiproliferative functions by arresting cell cycle progression (93). One possible explanation of the alternative relationship is that JNK signaling proteins have several isoforms with potentially opposing roles, namely JNK1 or JNK2 can increase or inhibit cell proliferation, respectively. Alternatively, CREB transcription factors have been linked to neuronal death and inhibition of NF-κB activation, limiting proinflammatory responses (94). In a murine hypoxic pulmonary hypertension model, RAGE-mediated TGF-β1 has proved to be a potent modulator of collagen synthesis, and the ablation of RAGE could inhibit TGF-β1–driven collagen deposition (95).
Finally, we assessed the metabolome of he lung 1-M after WTC-PM. In the metabolome, we found that polyunsaturated fatty acids, phospholipids, and sphingolipids were differentially expressed among the different mouse genotypes. The regulation of these metabolites indicates downstream phenotypical variations among the mice. Eicosapentaenoate (EPA) is a precursor to various eicosanoids, oxidized fatty acids that act as attenuators of the inflammatory response and collagen deposition (96–98). The opposing patterns of relative EPA expression in our 1-M murine metabolomes, together with our pulmonary function and histological assays, suggest that PM-exposed Ager−/− mice are protected from the inflammatory effects of WTC-PM observed in the exposed WT mice and do not require the antiinflammatory properties of EPA, owing to preexisting protection from the PM-induced inflammatory phenotype observed in the WT group. In addition, we found several phospholipids, including phosphatidylcholines, phosphatidylserines, and phosphatidylethanolamines, which direct induced macrophage-mediated apoptosis/phagocytosis and signal collagen deposition (99–102). With respect to WTC-PM exposure, phosphatidylcholines were correlated with acylcarnitine species in clusters A and B, which may be active in the hexose monophosphate shunt (HMS) (103). The presence of these acylcarnitine species together with the aforementioned phosphatidylcholines, nucleic acids (cytosine and ADP), and 1-methylnicotinamide indicates a correlation between WTC-PM exposure, the presence of RAGE, and metabolic activity, including the citric acid cycle, the HMS, and β-oxidation (103–106).
The interactions of these metabolic pathways in our given model suggest that WTC-PM–related oxidative stress and subsequent DNA damage initiate glutathione production via the HMS and membrane-associated lipid metabolism via β-oxidation/citric acid cycle. The metabolic profile of Ager−/− shifts toward a phenotype protected from inflammation and collagen accumulation: Antioxidants are produced, and markers of collagen deposition decrease, presumptively due to the elimination of RAGE itself. Finally, the decrease of sphinganine in WT-PM mice compared with WT-PBS, Ager−/−PBS, and Ager−/−-PM mice support the idea that cell membrane components in the lung play a key role in the lung’s response to PM. To further understand how inhibition of RAGE may affect the metabolome, we used Pio, a RAGE antagonist, and found a similar fold-change expression of metabolites (N6-carboxymethyllysine, N1+N8-acetylspermidine, and C4-DC) between PioRx and Ager−/− mice exposed to PM.
The refined profile of metabolites is similar to that observed in human studies, especially the profile observed in resistance to WTC-LI. Specifically, EPA, together with other fatty acids (docosapentaenoate, oleoylcholine, and 2-hydroxystearate), and sphinganine were seen to be important to WTC-LI in humans and have been linked to dyslipidemia and oxidative stress (5, 107, 108). Interestingly, the present metabolomics study was performed on lung homogenates, whereas previous human studies used the serum metabolome; therefore, the present study supports the link between the lung and metabolites previously observed in human serum.
Our study has several limitations. Although several optimal methods of PM exposure are discussed in the literature, we chose an oropharyngeal aspirational technique to deliver WTC-PM. This exposure model was successfully tested in both our prior work and several other investigations (11, 109, 110). The initial exposure that occurred at the WTC site was not only of PM suspended in the inhaled air; there was also a significant amount of PM that was in the upper airway that was then further inhaled. Therefore, the exposure at the WTC site on 9/11 was an acute oropharyngeal aspiration. In previous studies, oropharyngeal aspiration has proved to be equivalent to intratracheal instillation in PM deposition efficiency (45, 46, 111, 112). Those who arrived at the WTC site on the morning of 9/11 presented for pulmonary evaluation more often than those who arrived at later time points (113). Therefore, we aimed to replicate the acute oropharyngeal aspiration of WTC-PM by rescue workers during this time. To this end, a murine oropharyngeal aspiration of 100–200 μg of WTC-PM is estimated to cause adverse pulmonary effects similar to those of a human exposure to 425–850 μg/m3 over an 8-hour work period (43). This dose represents exposure on the lower end of intensity because the PM concentration in lower Manhattan and the WTC site is estimated to have reached the thousands of μg/m3 range for 4–8 hours after the WTC collapse (47, 114). The PioRx experiments were focused on a 24-H time point and used a 100-μg dose of WTC-PM that was previously found to cause AHR (11). However, our results regarding the AGE metabolite N6-carboxymethyllysine are bolstered by the fact that the control (CMC-Veh) PM, when compared with 1-M PM, showed no difference in this metabolite. This suggests that AGE metabolites were not affected by the lower dose or the earlier time point.
Although there is no gold standard of assessing AHR, Mch challenge testing is a widely accepted direct method and diagnostic aid in assessment of one of the cardinal features of asthma (115, 116). In humans, a Mch dose at which bronchoconstriction induces FEV1 to drop by at least 20% and 200 ml is known as PC20. In mice, the FEV is approximated by a passive negative pressure forced expiration maneuver because mice cannot exhale on command (117). However, this maneuver is imperfect because it rapidly deflates the murine lung and in itself affects respiratory mechanics, causing a significant increase in all parameters except resistance (118). Thus, a PC200 from the Mch challenge is an accepted and suggested measure of murine AHR (11, 45, 51, 52, 119, 120).
The results of at least several studies contrast with our results in regard to Ager−/−. Taniguchi and colleagues reported that naive Ager−/− mice have elevated resistance after Mch challenge at baseline and after inhaled ovalbumin compared with WT animals (23). Interestingly, Ager−/− mice did not have increased AHR after sensitization with intraperitoneal ovalbumin before inhalational challenge (23). One reason for this discrepancy may be that Taniguchi and colleagues elected to compare the WT and Ager−/− mice directly, whereas prior studies, including our own, compared Ager−/− mice with their own PBS control animals (22, 23, 74). Specifically, we chose to compare pulmonary measures only between Ager−/− mice exposed to PM and their PBS control animals, because Ager−/− mice have relatively larger alveolar spaces than WT mice do (11, 57). In a model of murine viral pneumonia infection, RAGE-deficient mice (similarly provided by A.M.S. and similarly on a C57BL/6 background) in a murine model of respiratory syncytial virus exposure in early life had subsequent airway smooth muscle remodeling (121). Subsequent reinfection of Ager−/− mice with the virus led to an increase in airway resistance compared with their control animals as measured by Mch challenge (121). This study strengthens the understanding that Ager−/− mice may be capable of developing AHR as assessed by an Mch challenge but, after other exposures, have relative protection from environmental challenge. Ager−/− mice may have other pathways that are attenuated because of the loss of RAGE protein from developmental stages. There may be other anatomic and physiologic differences that confer the relative respiratory protection from exogenous stimuli as seen in much of the literature. Therefore, translatability of our murine findings to our WTC-exposed human cohort may be limited and may be affected by confounders such as diet.
In summary, our results demonstrate that the acute effect in the pulmonary environment caused by a single WTC-PM exposure is persistent and may lead to pulmonary inflammation, cellular injury, and remodeling. Our study suggests that Ager−/− mice display partial protection against WTC-PM exposure–induced lung disease and hyperactivity by decreasing lung injury, inflammation, and collagen deposition. In addition, our data emphasize important roles for RAGE in the structural and functional deterioration in PM-induced lung injury. RAGE may be key therapeutic target after high-intensity particulate exposures. Although our study was focused on the inhibition of RAGE as a possible therapeutic intervention for the negative effects of PM exposure, we agree that further investigations are needed not only to further define mechanisms but also to fully develop targeted therapies. Specifically, competitive RAGE inhibitors tailored to the interaction of the cytoplasmic tail of RAGE and intracellular DIAPH1 may be suitable agents for future investigations (15, 87, 122). Finally, because PM-associated lung disease may not be best treated by a single agent, and diet-affected metabolites are associated with the development of WTC-LI, we are currently conducting a randomized clinical trial, the FIREHOUSE trial (Food Intake Restriction for Health Outcome Support and Education; clinicaltrials.gov/ct2/show/NCT0358106). The primary goal is to determine if lung function, metabolic syndrome, and metabolites can be positively affected in WTC-PM–exposed firefighters using a technology-assisted low-calorie Mediterranean diet.
Acknowledgments
Acknowledgment
The authors thank the FDNY first responders for their selflessness and sacrifice in the line of duty and dedicate our work to them.
Footnotes
Supported by U.S. National Heart, Lung, and Blood Institute grant R01HL119326 (A.N.); CDC/National Institute for Occupational Safety and Health grant U01-OH11300 (A.N.); Clinical Center of Excellence grant 200-2017-93426; and Data Center grant 200-2017-93326.
Author Contributions: S.H.H., A.V., A.M.S., and A.N. participated in study conception and design. A.N. was the primary investigator. S.H.H., A.V., E.J.C., D.O., M.M., R.L., M.S., and A.N. were responsible for data collection. S.K., D.J.P., and A.N. were responsible for data validation. S.H.H., A.V., G.C., S.K., and A.N. participated in data analysis. G.C., Y.W., S.K., M.L., and A.N. undertook the statistical analysis. All authors participated in data interpretation, writing and revision of the report, and approval of the final version.
This article has a data supplement, which is accessible from this issue’s table of contents at www.atsjournals.org.
Originally Published in Press as DOI: 10.1165/rcmb.2019-0064OC on April 21, 2020
Author disclosures are available with the text of this article at www.atsjournals.org.
References
- 1.Dockery DW, Pope CA, III, Xu X, Spengler JD, Ware JH, Fay ME, et al. An association between air pollution and mortality in six U.S. cities. N Engl J Med. 1993;329:1753–1759. doi: 10.1056/NEJM199312093292401. [DOI] [PubMed] [Google Scholar]
- 2.Schwartz J. Short term fluctuations in air pollution and hospital admissions of the elderly for respiratory disease. Thorax. 1995;50:531–538. doi: 10.1136/thx.50.5.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aldrich TK, Gustave J, Hall CB, Cohen HW, Webber MP, Zeig-Owens R, et al. Lung function in rescue workers at the World Trade Center after 7 years. N Engl J Med. 2010;362:1263–1272. doi: 10.1056/NEJMoa0910087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Prezant DJ, Weiden M, Banauch GI, McGuinness G, Rom WN, Aldrich TK, et al. Cough and bronchial responsiveness in firefighters at the World Trade Center site. N Engl J Med. 2002;347:806–815. doi: 10.1056/NEJMoa021300. [DOI] [PubMed] [Google Scholar]
- 5.Crowley G, Kwon S, Haider SH, Caraher EJ, Lam R, St-Jules DE, et al. Metabolomics of World Trade Center-lung injury: a machine learning approach. BMJ Open Respir Res. 2018;5:e000274. doi: 10.1136/bmjresp-2017-000274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Naveed B, Weiden MD, Kwon S, Gracely EJ, Comfort AL, Ferrier N, et al. Metabolic syndrome biomarkers predict lung function impairment: a nested case-control study. Am J Respir Crit Care Med. 2012;185:392–399. doi: 10.1164/rccm.201109-1672OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsukiji J, Cho SJ, Echevarria GC, Kwon S, Joseph P, Schenck EJ, et al. Lysophosphatidic acid and apolipoprotein A1 predict increased risk of developing World Trade Center-lung injury: a nested case-control study. Biomarkers. 2014;19:159–165. doi: 10.3109/1354750X.2014.891047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holguin F. The metabolic syndrome as a risk factor for lung function decline. Am J Respir Crit Care Med. 2012;185:352–353. doi: 10.1164/rccm.201112-2172ED. [DOI] [PubMed] [Google Scholar]
- 9.Balmes JR. Can we predict who will develop chronic sequelae of acute inhalational injury? Chest. 2012;142:278–279. doi: 10.1378/chest.12-0126. [DOI] [PubMed] [Google Scholar]
- 10.Antao VC. The World Trade Center disaster: a tragic source of medical advancement. Eur Respir J. 2013;41:999–1001. doi: 10.1183/09031936.00181112. [DOI] [PubMed] [Google Scholar]
- 11.Caraher EJ, Kwon S, Haider SH, Crowley G, Lee A, Ebrahim M, et al. Receptor for advanced glycation end-products and World Trade Center particulate induced lung function loss: a case-cohort study and murine model of acute particulate exposure. PLoS One. 2017;12:e0184331. doi: 10.1371/journal.pone.0184331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Demling N, Ehrhardt C, Kasper M, Laue M, Knels L, Rieber EP. Promotion of cell adherence and spreading: a novel function of RAGE, the highly selective differentiation marker of human alveolar epithelial type I cells. Cell Tissue Res. 2006;323:475–488. doi: 10.1007/s00441-005-0069-0. [DOI] [PubMed] [Google Scholar]
- 13.Katsuoka F, Kawakami Y, Arai T, Imuta H, Fujiwara M, Kanma H, et al. Type II alveolar epithelial cells in lung express receptor for advanced glycation end products (RAGE) gene. Biochem Biophys Res Commun. 1997;238:512–516. doi: 10.1006/bbrc.1997.7263. [DOI] [PubMed] [Google Scholar]
- 14.Buckley ST, Ehrhardt C. The receptor for advanced glycation end products (RAGE) and the lung. J Biomed Biotechnol. 2010;2010:917108. doi: 10.1155/2010/917108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yan SF, Ramasamy R, Schmidt AM. The RAGE axis: a fundamental mechanism signaling danger to the vulnerable vasculature. Circ Res. 2010;106:842–853. doi: 10.1161/CIRCRESAHA.109.212217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ge X, Xu XY, Feng CH, Wang Y, Li YL, Feng B. Relationships among serum C-reactive protein, receptor for advanced glycation products, metabolic dysfunction, and cognitive impairments. BMC Neurol. 2013;13:110. doi: 10.1186/1471-2377-13-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kierdorf K, Fritz G. RAGE regulation and signaling in inflammation and beyond. J Leukoc Biol. 2013;94:55–68. doi: 10.1189/jlb.1012519. [DOI] [PubMed] [Google Scholar]
- 18.Yonchuk JG, Silverman EK, Bowler RP, Agustí A, Lomas DA, Miller BE, et al. Circulating soluble receptor for advanced glycation end products (sRAGE) as a biomarker of emphysema and the RAGE axis in the lung. Am J Respir Crit Care Med. 2015;192:785–792. doi: 10.1164/rccm.201501-0137PP. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt AM. Soluble RAGEs: prospects for treating & tracking metabolic and inflammatory disease. Vascul Pharmacol. 2015;72:1–8. doi: 10.1016/j.vph.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Robinson AB, Johnson KD, Bennion BG, Reynolds PR. RAGE signaling by alveolar macrophages influences tobacco smoke-induced inflammation. Am J Physiol Lung Cell Mol Physiol. 2012;302:L1192–L1199. doi: 10.1152/ajplung.00099.2012. [DOI] [PubMed] [Google Scholar]
- 21.Englert JM, Hanford LE, Kaminski N, Tobolewski JM, Tan RJ, Fattman CL, et al. A role for the receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Am J Pathol. 2008;172:583–591. doi: 10.2353/ajpath.2008.070569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milutinovic PS, Alcorn JF, Englert JM, Crum LT, Oury TD. The receptor for advanced glycation end products is a central mediator of asthma pathogenesis. Am J Pathol. 2012;181:1215–1225. doi: 10.1016/j.ajpath.2012.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taniguchi A, Miyahara N, Waseda K, Kurimoto E, Fujii U, Tanimoto Y, et al. Contrasting roles for the receptor for advanced glycation end-products on structural cells in allergic airway inflammation vs. airway hyperresponsiveness. Am J Physiol Lung Cell Mol Physiol. 2015;309:L789–L800. doi: 10.1152/ajplung.00087.2015. [DOI] [PubMed] [Google Scholar]
- 24.Akirav EM, Henegariu O, Preston-Hurlburt P, Schmidt AM, Clynes R, Herold KC. The receptor for advanced glycation end products (RAGE) affects T cell differentiation in OVA induced asthma. PLoS One. 2014;9:e95678. doi: 10.1371/journal.pone.0095678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asada K, Sasaki S, Suda T, Chida K, Nakamura H. Antiinflammatory roles of peroxisome proliferator-activated receptor gamma in human alveolar macrophages. Am J Respir Crit Care Med. 2004;169:195–200. doi: 10.1164/rccm.200207-740OC. [DOI] [PubMed] [Google Scholar]
- 26.Spears M, McSharry C, Thomson NC. Peroxisome proliferator-activated receptor-gamma agonists as potential anti-inflammatory agents in asthma and chronic obstructive pulmonary disease. Clin Exp Allergy. 2006;36:1494–1504. doi: 10.1111/j.1365-2222.2006.02604.x. [DOI] [PubMed] [Google Scholar]
- 27.Ma C, Zhang Y, Li YQ, Chen C, Cai W, Zeng YL. The role of PPARγ in advanced glycation end products-induced inflammatory response in human chondrocytes. PLoS One. 2015;10:e0125776. doi: 10.1371/journal.pone.0125776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mangoni M, Sottili M, Gerini C, Bonomo P, Bottoncetti A, Castiglione F, et al. A PPAR-gamma agonist attenuates pulmonary injury induced by irradiation in a murine model. Lung Cancer. 2015;90:405–409. doi: 10.1016/j.lungcan.2015.11.005. [DOI] [PubMed] [Google Scholar]
- 29.Szatmari I, Nagy L. Nuclear receptor signalling in dendritic cells connects lipids, the genome and immune function. EMBO J. 2008;27:2353–2362. doi: 10.1038/emboj.2008.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reddy RC. Immunomodulatory role of PPAR-gamma in alveolar macrophages. J Investig Med. 2008;56:522–527. doi: 10.2310/JIM.0b013e3181659972. [DOI] [PubMed] [Google Scholar]
- 31.Standiford TJ, Keshamouni VG, Reddy RC. Peroxisome proliferator-activated receptor-gamma as a regulator of lung inflammation and repair. Proc Am Thorac Soc. 2005;2:226–231. doi: 10.1513/pats.200501-010AC. [DOI] [PubMed] [Google Scholar]
- 32.Penyige A, Poliska S, Csanky E, Scholtz B, Dezso B, Schmelczer I, et al. Analyses of association between PPAR gamma and EPHX1 polymorphisms and susceptibility to COPD in a Hungarian cohort, a case-control study. BMC Med Genet. 2010;11:152. doi: 10.1186/1471-2350-11-152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang K, Zhou Z, Zhang M, Fan L, Forudi F, Zhou X, et al. Peroxisome proliferator-activated receptor gamma down-regulates receptor for advanced glycation end products and inhibits smooth muscle cell proliferation in a diabetic and nondiabetic rat carotid artery injury model. J Pharmacol Exp Ther. 2006;317:37–43. doi: 10.1124/jpet.105.095125. [DOI] [PubMed] [Google Scholar]
- 34.Wang G, Wei J, Guan Y, Jin N, Mao J, Wang X. Peroxisome proliferator-activated receptor-gamma agonist rosiglitazone reduces clinical inflammatory responses in type 2 diabetes with coronary artery disease after coronary angioplasty. Metabolism. 2005;54:590–597. doi: 10.1016/j.metabol.2004.11.017. [DOI] [PubMed] [Google Scholar]
- 35.Ament Z, Masoodi M, Griffin JL. Applications of metabolomics for understanding the action of peroxisome proliferator-activated receptors (PPARs) in diabetes, obesity and cancer. Genome Med. 2012;4:32. doi: 10.1186/gm331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Webber MP, Yip J, Zeig-Owens R, Moir W, Ungprasert P, Crowson CS, et al. Post-9/11 sarcoidosis in WTC-exposed firefighters and emergency medical service workers. Respir Med. 2017;132:232–237. doi: 10.1016/j.rmed.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kwon S, Crowley G, Caraher EJ, Haider SH, Lam R, Veerappan A, et al. Validation of predictive metabolic syndrome biomarkers of World Trade Center lung injury: a 16-year longitudinal study. Chest. 2019;156:486–496. doi: 10.1016/j.chest.2019.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kwon S, Crowley G, Mikhail M, Lam R, Clementi E, Zeig-Owens R, et al. Metabolic syndrome biomarkers of World Trade Center airway hyperreactivity: a 16-year prospective cohort study. Int J Environ Res Public Health. 2019;16:1486. doi: 10.3390/ijerph16091486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Caito S, Yang SR, Kode A, Edirisinghe I, Rajendrasozhan S, Phipps RP, et al. Rosiglitazone and 15-deoxy-Δ12,14-prostaglandin J2, PPARγ agonists, differentially regulate cigarette smoke-mediated pro-inflammatory cytokine release in monocytes/macrophages. Antioxid Redox Signal. 2008;10:253–260. doi: 10.1089/ars.2007.1889. [DOI] [PubMed] [Google Scholar]
- 40.Patel HJ, Belvisi MG, Bishop-Bailey D, Yacoub MH, Mitchell JA. Activation of peroxisome proliferator-activated receptors in human airway smooth muscle cells has a superior anti-inflammatory profile to corticosteroids: relevance for chronic obstructive pulmonary disease therapy. J Immunol. 2003;170:2663–2669. doi: 10.4049/jimmunol.170.5.2663. [DOI] [PubMed] [Google Scholar]
- 41.Shan M, You R, Yuan X, Frazier MV, Porter P, Seryshev A, et al. Agonistic induction of PPARγ reverses cigarette smoke-induced emphysema. J Clin Invest. 2014;124:1371–1381. doi: 10.1172/JCI70587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yin Y, Hou G, Li ER, Wang QY, Kang J. PPARγ agonists regulate tobacco smoke-induced Toll like receptor 4 expression in alveolar macrophages. Respir Res. 2014;15:28. doi: 10.1186/1465-9921-15-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gavett SH, Haykal-Coates N, Highfill JW, Ledbetter AD, Chen LC, Cohen MD, et al. World Trade Center fine particulate matter causes respiratory tract hyperresponsiveness in mice. Environ Health Perspect. 2003;111:981–991. doi: 10.1289/ehp.5931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song F, Hurtado del Pozo C, Rosario R, Zou YS, Ananthakrishnan R, Xu X, et al. RAGE regulates the metabolic and inflammatory response to high-fat feeding in mice. Diabetes. 2014;63:1948–1965. doi: 10.2337/db13-1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Card JW, Carey MA, Bradbury JA, DeGraff LM, Morgan DL, Moorman MP, et al. Gender differences in murine airway responsiveness and lipopolysaccharide-induced inflammation. J Immunol. 2006;177:621–630. doi: 10.4049/jimmunol.177.1.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McGee JK, Chen LC, Cohen MD, Chee GR, Prophete CM, Haykal-Coates N, et al. Chemical analysis of World Trade Center fine particulate matter for use in toxicologic assessment. Environ Health Perspect. 2003;111:972–980. doi: 10.1289/ehp.5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geyh AS, Chillrud S, Williams DL, Herbstman J, Symons JM, Rees K, et al. Assessing truck driver exposure at the World Trade Center disaster site: personal and area monitoring for particulate matter and volatile organic compounds during October 2001 and April 2002. J Occup Environ Hyg. 2005;2:179–193. doi: 10.1080/15459620590923154. [DOI] [PubMed] [Google Scholar]
- 48.Bauer CM, Zavitz CC, Botelho FM, Lambert KN, Brown EG, Mossman KL, et al. Treating viral exacerbations of chronic obstructive pulmonary disease: insights from a mouse model of cigarette smoke and H1N1 influenza infection. PLoS One. 2010;5:e13251. doi: 10.1371/journal.pone.0013251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rivera-Sanchez YM, Johnston RA, Schwartzman IN, Valone J, Silverman ES, Fredberg JJ, et al. Differential effects of ozone on airway and tissue mechanics in obese mice. J Appl Physiol (1985) 2004;96:2200–2206. doi: 10.1152/japplphysiol.00960.2003. [DOI] [PubMed] [Google Scholar]
- 50.Hartney JM, Robichaud A. Assessment of airway hyperresponsiveness in mouse models of allergic lung disease using detailed measurements of respiratory mechanics. Methods Mol Biol. 2013;1032:205–217. doi: 10.1007/978-1-62703-496-8_16. [DOI] [PubMed] [Google Scholar]
- 51.Shalaby KH, Gold LG, Schuessler TF, Martin JG, Robichaud A. Combined forced oscillation and forced expiration measurements in mice for the assessment of airway hyperresponsiveness. Respir Res. 2010;11:82. doi: 10.1186/1465-9921-11-82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vanoirbeek JA, Rinaldi M, De Vooght V, Haenen S, Bobic S, Gayan-Ramirez G, et al. Noninvasive and invasive pulmonary function in mouse models of obstructive and restrictive respiratory diseases. Am J Respir Cell Mol Biol. 2010;42:96–104. doi: 10.1165/rcmb.2008-0487OC. [DOI] [PubMed] [Google Scholar]
- 53.Ryu MH, Jha A, Ojo OO, Mahood TH, Basu S, Detillieux KA, et al. Chronic exposure to perfluorinated compounds: impact on airway hyperresponsiveness and inflammation. Am J Physiol Lung Cell Mol Physiol. 2014;307:L765–L774. doi: 10.1152/ajplung.00100.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Robichaud A, Fereydoonzad L, Schuessler TF. Delivered dose estimate to standardize airway hyperresponsiveness assessment in mice. Am J Physiol Lung Cell Mol Physiol. 2015;308:L837–L846. doi: 10.1152/ajplung.00343.2014. [DOI] [PubMed] [Google Scholar]
- 55.Nolan A, Weiden MD, Thurston G, Gold JA. Vascular endothelial growth factor blockade reduces plasma cytokines in a murine model of polymicrobial sepsis. Inflammation. 2004;28:271–278. doi: 10.1007/s10753-004-6050-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shvedova AA, Yanamala N, Kisin ER, Tkach AV, Murray AR, Hubbs A, et al. Long-term effects of carbon containing engineered nanomaterials and asbestos in the lung: one year postexposure comparisons. Am J Physiol Lung Cell Mol Physiol. 2014;306:L170–L182. doi: 10.1152/ajplung.00167.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sambamurthy N, Leme AS, Oury TD, Shapiro SD. The receptor for advanced glycation end products (RAGE) contributes to the progression of emphysema in mice. PLoS One. 2015;10:e0118979. doi: 10.1371/journal.pone.0118979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Soutiere SE, Tankersley CG, Mitzner W. Differences in alveolar size in inbred mouse strains. Respir Physiol Neurobiol. 2004;140:283–291. doi: 10.1016/j.resp.2004.02.003. [DOI] [PubMed] [Google Scholar]
- 59.Kurimoto E, Miyahara N, Kanehiro A, Waseda K, Taniguchi A, Ikeda G, et al. IL-17A is essential to the development of elastase-induced pulmonary inflammation and emphysema in mice. Respir Res. 2013;14:5. doi: 10.1186/1465-9921-14-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hsia CC, Hyde DM, Ochs M, Weibel ER ATS/ERS Joint Task Force on Quantitative Assessment of Lung Structure. An official research policy statement of the American Thoracic Society/European Respiratory Society: standards for quantitative assessment of lung structure. Am J Respir Crit Care Med. 2010;181:394–418. doi: 10.1164/rccm.200809-1522ST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oldmixon EH, Butler JP, Hoppin FG. Semi-automated measurement of true chord length distributions and moments by video microscopy and image analysis. J Microsc. 1994;175:60–69. doi: 10.1111/j.1365-2818.1994.tb04788.x. [DOI] [PubMed] [Google Scholar]
- 62.Hamakawa H, Bartolák-Suki E, Parameswaran H, Majumdar A, Lutchen KR, Suki B. Structure-function relations in an elastase-induced mouse model of emphysema. Am J Respir Cell Mol Biol. 2011;45:517–524. doi: 10.1165/rcmb.2010-0473OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dunnill MS. Quantitative methods in the study of pulmonary pathology. Thorax. 1962;17:320–328. doi: 10.1136/thx.17.4.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muñoz-Barrutia A, Ceresa M, Artaechevarria X, Montuenga LM, Ortiz-de-Solorzano C. Quantification of lung damage in an elastase-induced mouse model of emphysema. Int J Biomed Imaging. 2012;2012:734734. doi: 10.1155/2012/734734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Seger S, Stritt M, Vezzali E, Nayler O, Hess P, Groenen PMA, et al. A fully automated image analysis method to quantify lung fibrosis in the bleomycin-induced rat model. PLoS One. 2018;13:e0193057. doi: 10.1371/journal.pone.0193057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Boudonck KJ, Mitchell MW, Német L, Keresztes L, Nyska A, Shinar D, et al. Discovery of metabolomics biomarkers for early detection of nephrotoxicity. Toxicol Pathol. 2009;37:280–292. doi: 10.1177/0192623309332992. [DOI] [PubMed] [Google Scholar]
- 67.Nielsen KA, Tattersall DB, Jones PR, Møller BL. Metabolon formation in dhurrin biosynthesis. Phytochemistry. 2008;69:88–98. doi: 10.1016/j.phytochem.2007.06.033. [DOI] [PubMed] [Google Scholar]
- 68.Ryals J, Lawton K, Stevens D, Milburn M. Metabolon, Inc. Pharmacogenomics. 2007;8:863–866. doi: 10.2217/14622416.8.7.863. [DOI] [PubMed] [Google Scholar]
- 69.Breiman L. Random forests. Mach Learn. 2001;45:5–32. [Google Scholar]
- 70.Hackstadt AJ, Hess AM. Filtering for increased power for microarray data analysis. BMC Bioinformatics. 2009;10:11. doi: 10.1186/1471-2105-10-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hudson BI, Kalea AZ, Del Mar Arriero M, Harja E, Boulanger E, D’Agati V, et al. Interaction of the RAGE cytoplasmic domain with diaphanous-1 is required for ligand-stimulated cellular migration through activation of Rac1 and Cdc42. J Biol Chem. 2008;283:34457–34468. doi: 10.1074/jbc.M801465200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Perkins TN, Oczypok EA, Dutz RE, Donnell ML, Myerburg MM, Oury TD. The receptor for advanced glycation end products is a critical mediator of type 2 cytokine signaling in the lungs. J Allergy Clin Immunol. 2019;144:796–808, e12. doi: 10.1016/j.jaci.2019.03.019. [DOI] [PubMed] [Google Scholar]
- 73.Wolf L, Herr C, Niederstraßer J, Beisswenger C, Bals R. Receptor for advanced glycation endproducts (RAGE) maintains pulmonary structure and regulates the response to cigarette smoke. PLoS One. 2017;12:e0180092. doi: 10.1371/journal.pone.0180092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Oczypok EA, Milutinovic PS, Alcorn JF, Khare A, Crum LT, Manni ML, et al. Pulmonary receptor for advanced glycation end-products promotes asthma pathogenesis through IL-33 and accumulation of group 2 innate lymphoid cells. J Allergy Clin Immunol. 2015;136:747–756, e4. doi: 10.1016/j.jaci.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ullah MA, Loh Z, Gan WJ, Zhang V, Yang H, Li JH, et al. Receptor for advanced glycation end products and its ligand high-mobility group box-1 mediate allergic airway sensitization and airway inflammation. J Allergy Clin Immunol. 2014;134:440–450. doi: 10.1016/j.jaci.2013.12.1035. [DOI] [PubMed] [Google Scholar]
- 76.Kawasaki Y, Imaizumi T, Matsuura H, Ohara S, Takano K, Suyama K, et al. Renal expression of alpha-smooth muscle actin and c-Met in children with Henoch-Schönlein purpura nephritis. Pediatr Nephrol. 2008;23:913–919. doi: 10.1007/s00467-008-0749-6. [DOI] [PubMed] [Google Scholar]
- 77.Ding H, Ji X, Chen R, Ma T, Tang Z, Fen Y, et al. Antifibrotic properties of receptor for advanced glycation end products in idiopathic pulmonary fibrosis. Pulm Pharmacol Ther. 2015;35:34–41. doi: 10.1016/j.pupt.2015.10.010. [DOI] [PubMed] [Google Scholar]
- 78.Myint KM, Yamamoto Y, Doi T, Kato I, Harashima A, Yonekura H, et al. RAGE control of diabetic nephropathy in a mouse model: effects of RAGE gene disruption and administration of low-molecular weight heparin. Diabetes. 2006;55:2510–2522. doi: 10.2337/db06-0221. [DOI] [PubMed] [Google Scholar]
- 79.Hamada N, Maeyama T, Kawaguchi T, Yoshimi M, Fukumoto J, Yamada M, et al. The role of high mobility group box1 in pulmonary fibrosis. Am J Respir Cell Mol Biol. 2008;39:440–447. doi: 10.1165/rcmb.2007-0330OC. [DOI] [PubMed] [Google Scholar]
- 80.Gasparitsch M, Arndt AK, Pawlitschek F, Oberle S, Keller U, Kasper M, et al. RAGE-mediated interstitial fibrosis in neonatal obstructive nephropathy is independent of NF-κB activation. Kidney Int. 2013;84:911–919. doi: 10.1038/ki.2013.171. [DOI] [PubMed] [Google Scholar]
- 81.Ota C, Ishizawa K, Yamada M, Tando Y, He M, Takahashi T, et al. Receptor for advanced glycation end products expressed on alveolar epithelial cells is the main target for hyperoxia-induced lung injury. Respir Investig. 2016;54:98–108. doi: 10.1016/j.resinv.2015.08.009. [DOI] [PubMed] [Google Scholar]
- 82.Waseda K, Miyahara N, Taniguchi A, Kurimoto E, Ikeda G, Koga H, et al. Emphysema requires the receptor for advanced glycation end-products triggering on structural cells. Am J Respir Cell Mol Biol. 2015;52:482–491. doi: 10.1165/rcmb.2014-0027OC. [DOI] [PubMed] [Google Scholar]
- 83.MacNee W, Rennard SI, Hunt JF, Edwards LD, Miller BE, Locantore NW, et al. Evaluation of exhaled breath condensate pH as a biomarker for COPD. Respir Med. 2011;105:1037–1045. doi: 10.1016/j.rmed.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 84.Neuman RE, Logan MA. The determination of hydroxyproline. J Biol Chem. 1950;184:299–306. [PubMed] [Google Scholar]
- 85.Schmidt AM, Yan SD, Yan SF, Stern DM. The multiligand receptor RAGE as a progression factor amplifying immune and inflammatory responses. J Clin Invest. 2001;108:949–955. doi: 10.1172/JCI14002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tammaro A, Florquin S, Brok M, Claessen N, Butter LM, Teske GJD, et al. S100A8/A9 promotes parenchymal damage and renal fibrosis in obstructive nephropathy. Clin Exp Immunol. 2018;193:361–375. doi: 10.1111/cei.13154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Manigrasso MB, Pan J, Rai V, Zhang J, Reverdatto S, Quadri N, et al. Small molecule inhibition of ligand-stimulated RAGE-DIAPH1 signal transduction. Sci Rep. 2016;6:22450. doi: 10.1038/srep22450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Weiden MD, Naveed B, Kwon S, Segal LN, Cho SJ, Tsukiji J, et al. Comparison of WTC dust size on macrophage inflammatory cytokine release in vivo and in vitro. PLoS One. 2012;7:e40016. doi: 10.1371/journal.pone.0040016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kokkola R, Andersson A, Mullins G, Ostberg T, Treutiger CJ, Arnold B, et al. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61:1–9. doi: 10.1111/j.0300-9475.2005.01534.x. [DOI] [PubMed] [Google Scholar]
- 90.Sakaguchi T, Yan SF, Yan SD, Belov D, Rong LL, Sousa M, et al. Central role of RAGE-dependent neointimal expansion in arterial restenosis. J Clin Invest. 2003;111:959–972. doi: 10.1172/JCI17115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Reddy MA, Li SL, Sahar S, Kim YS, Xu ZG, Lanting L, et al. Key role of Src kinase in S100B-induced activation of the receptor for advanced glycation end products in vascular smooth muscle cells. J Biol Chem. 2006;281:13685–13693. doi: 10.1074/jbc.M511425200. [DOI] [PubMed] [Google Scholar]
- 92.Hsu HS, Liu CC, Lin JH, Hsu TW, Hsu JW, Su K, et al. Involvement of ER stress, PI3K/AKT activation, and lung fibroblast proliferation in bleomycin-induced pulmonary fibrosis. Sci Rep. 2017;7:14272. doi: 10.1038/s41598-017-14612-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Aaron SD, Fergusson D, Dent R, Chen Y, Vandemheen KL, Dales RE. Effect of weight reduction on respiratory function and airway reactivity in obese women. Chest. 2004;125:2046–2052. doi: 10.1378/chest.125.6.2046. [DOI] [PubMed] [Google Scholar]
- 94.Wen AY, Sakamoto KM, Miller LS. The role of the transcription factor CREB in immune function. J Immunol. 2010;185:6413–6419. doi: 10.4049/jimmunol.1001829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jia D, He Y, Zhu Q, Liu H, Zuo C, Chen G, et al. RAGE-mediated extracellular matrix proteins accumulation exacerbates HySu-induced pulmonary hypertension. Cardiovasc Res. 2017;113:586–597. doi: 10.1093/cvr/cvx051. [DOI] [PubMed] [Google Scholar]
- 96.Yang P, Jiang Y, Fischer SM. Prostaglandin E3 metabolism and cancer. Cancer Lett. 2014;348:1–11. doi: 10.1016/j.canlet.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Serhan CN, Petasis NA. Resolvins and protectins in inflammation resolution. Chem Rev. 2011;111:5922–5943. doi: 10.1021/cr100396c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Innes JK, Calder PC. The differential effects of eicosapentaenoic acid and docosahexaenoic acid on cardiometabolic risk factors: a systematic review. Int J Mol Sci. 2018;19:532. doi: 10.3390/ijms19020532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yeon SH, Yang G, Lee HE, Lee JY. Oxidized phosphatidylcholine induces the activation of NLRP3 inflammasome in macrophages. J Leukoc Biol. 2017;101:205–215. doi: 10.1189/jlb.3VMA1215-579RR. [DOI] [PubMed] [Google Scholar]
- 100.Zargarian S, Shlomovitz I, Erlich Z, Hourizadeh A, Ofir-Birin Y, Croker BA, et al. Phosphatidylserine externalization, “necroptotic bodies” release, and phagocytosis during necroptosis. PLoS Biol. 2017;15:e2002711. doi: 10.1371/journal.pbio.2002711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Elvas F, Stroobants S, Wyffels L. Phosphatidylethanolamine targeting for cell death imaging in early treatment response evaluation and disease diagnosis. Apoptosis. 2017;22:971–987. doi: 10.1007/s10495-017-1384-0. [DOI] [PubMed] [Google Scholar]
- 102.Lentz BR. Exposure of platelet membrane phosphatidylserine regulates blood coagulation. Prog Lipid Res. 2003;42:423–438. doi: 10.1016/s0163-7827(03)00025-0. [DOI] [PubMed] [Google Scholar]
- 103.Kwon S, Haider SH, Caraher E, Lam R, Crowley G, Prezant DJ, et al. Validation of metabolic syndrome biomarkers of particulate induced lung injury in World Trade Center exposed firefighters: a 15 year longitudinal cohort study [abstract] Am J Respir Crit Care Med. 2020;201:A2584. [Google Scholar]
- 104.Marquis BJ, Hurren NM, Carvalho E, Kim IY, Schutzler S, Azhar G, et al. Skeletal muscle acute and chronic metabolic response to essential amino acid supplementation in hypertriglyceridemic older adults. Curr Dev Nutr. 2017;1:e002071. doi: 10.3945/cdn.117.002071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bhuiyan AK, Jackson S, Turnbull DM, Aynsley-Green A, Leonard JV, Bartlett K. The measurement of carnitine and acyl-carnitines: application to the investigation of patients with suspected inherited disorders of mitochondrial fatty acid oxidation. Clin Chim Acta. 1992;207:185–204. doi: 10.1016/0009-8981(92)90118-a. [DOI] [PubMed] [Google Scholar]
- 106.Mejía SA, Gutman LAB, Camarillo CO, Navarro RM, Becerra MCS, Santana LD, et al. Nicotinamide prevents sweet beverage-induced hepatic steatosis in rats by regulating the G6PD, NADPH/NADP+ and GSH/GSSG ratios and reducing oxidative and inflammatory stress. Eur J Pharmacol. 2018;818:499–507. doi: 10.1016/j.ejphar.2017.10.048. [DOI] [PubMed] [Google Scholar]
- 107.Hama H. Fatty acid 2-hydroxylation in mammalian sphingolipid biology. Biochim Biophys Acta. 2010;1801:405–414. doi: 10.1016/j.bbalip.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kumar SA, Sudhahar V, Varalakshmi P. Protective role of eicosapentaenoate-lipoate (EPA-LA) derivative in combating oxidative hepatocellular injury in hypercholesterolemic atherogenesis. Atherosclerosis. 2006;189:115–122. doi: 10.1016/j.atherosclerosis.2005.11.037. [DOI] [PubMed] [Google Scholar]
- 109.De Vooght V, Vanoirbeek JA, Haenen S, Verbeken E, Nemery B, Hoet PH. Oropharyngeal aspiration: an alternative route for challenging in a mouse model of chemical-induced asthma. Toxicology. 2009;259:84–89. doi: 10.1016/j.tox.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 110.Van Winkle LS, Bein K, Anderson D, Pinkerton KE, Tablin F, Wilson D, et al. Biological dose response to PM2.5: effect of particle extraction method on platelet and lung responses. Toxicol Sci. 2015;143:349–359. doi: 10.1093/toxsci/kfu230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Gavett SH, Madison SL, Stevens MA, Costa DL. Residual oil fly ash amplifies allergic cytokines, airway responsiveness, and inflammation in mice. Am J Respir Crit Care Med. 1999;160:1897–1904. doi: 10.1164/ajrccm.160.6.9901053. [DOI] [PubMed] [Google Scholar]
- 112.Kodavanti UP, Hauser R, Christiani DC, Meng ZH, McGee J, Ledbetter A, et al. Pulmonary responses to oil fly ash particles in the rat differ by virtue of their specific soluble metals. Toxicol Sci. 1998;43:204–212. doi: 10.1006/toxs.1998.2460. [DOI] [PubMed] [Google Scholar]
- 113.Weiden MD, Ferrier N, Nolan A, Rom WN, Comfort A, Gustave J, et al. Obstructive airways disease with air trapping among firefighters exposed to World Trade Center dust. Chest. 2010;137:566–574. doi: 10.1378/chest.09-1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lippmann M, Cohen MD, Chen LC. Health effects of World Trade Center (WTC) dust: an unprecedented disaster’s inadequate risk management. Crit Rev Toxicol. 2015;45:492–530. doi: 10.3109/10408444.2015.1044601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Crapo RO, Casaburi R, Coates AL, Enright PL, Hankinson JL, Irvin CG, et al. Guidelines for methacholine and exercise challenge testing—1999. Am J Respir Crit Care Med. 2000;161:309–329. doi: 10.1164/ajrccm.161.1.ats11-99. [DOI] [PubMed] [Google Scholar]
- 116.Coates AL, Wanger J, Cockcroft DW, Culver BH, Kai-Hakon C, Diamant Z, et al. Bronchoprovocation Testing Task Force. ERS technical standard on bronchial challenge testing: general considerations and performance of methacholine challenge tests. Eur Respir J. 2017;49:1601526. doi: 10.1183/13993003.01526-2016. [DOI] [PubMed] [Google Scholar]
- 117.Devos FC, Maaske A, Robichaud A, Pollaris L, Seys S, Lopez CA, et al. Forced expiration measurements in mouse models of obstructive and restrictive lung diseases. Respir Res. 2017;18:123. doi: 10.1186/s12931-017-0610-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Brannan JD, Lougheed MD. Airway hyperresponsiveness in asthma: mechanisms, clinical significance, and treatment. Front Physiol. 2012;3:460. doi: 10.3389/fphys.2012.00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Carey MA, Card JW, Bradbury JA, Moorman MP, Haykal-Coates N, Gavett SH, et al. Spontaneous airway hyperresponsiveness in estrogen receptor-alpha-deficient mice. Am J Respir Crit Care Med. 2007;175:126–135. doi: 10.1164/rccm.200509-1493OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.McGovern TK, Robichaud A, Fereydoonzad L, Schuessler TF, Martin JG. Evaluation of respiratory system mechanics in mice using the forced oscillation technique. J Vis Exp. 2013;(75):e50172. doi: 10.3791/50172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Arikkatt J, Ullah MA, Short KR, Zhang V, Gan WJ, Loh Z, et al. RAGE deficiency predisposes mice to virus-induced paucigranulocytic asthma. Elife. 2017;6:e21199. doi: 10.7554/eLife.21199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Yan SF, Ramasamy R, Schmidt AM. Soluble RAGE: therapy and biomarker in unraveling the RAGE axis in chronic disease and aging. Biochem Pharmacol. 2010;79:1379–1386. doi: 10.1016/j.bcp.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]