

In this review, Cherry et al. describe the current understanding of how splicing is regulated, discuss some classic and newer examples of how this regulation is altered in cancer, and then review recent insight into the breadth of splicing variability in cancer as well as the progress and obstacles in leveraging alternative splicing to increase our ability to classify and treat human metastatic disease.
Keywords: alternative splicing, cancer, transcriptomic analysis
Abstract
Over the past decade there has been increased awareness of the potential role of alternative splicing in the etiology of cancer. In particular, advances in RNA-Sequencing technology and analysis has led to a wave of discoveries in the last few years regarding the causes and functional relevance of alternative splicing in cancer. Here we discuss the current understanding of the connections between splicing and cancer, with a focus on the most recent findings. We also discuss remaining questions and challenges that must be addressed in order to use our knowledge of splicing to guide the diagnosis and treatment of cancer.
Alternative pre-mRNA splicing is a pervasive cellular process that expands the transcriptome relative to the genome through the differential use of exons or portions thereof (Nilsen and Graveley 2010; Blencowe 2017). As such, alternative splicing contributes to proteome complexity among higher eukaryotes and plays an important role in modulating the patterns of gene expression that govern many cell fate decisions. Despite the advantage that alternative splicing offers cells to expand and regulate function, aberrations or mistakes in splicing are typically deleterious to cells and result in cell death or dysregulated growth. Indeed, from the earliest discovery of pre-mRNA splicing, it was appreciated that mistakes in this process could alter gene expression in ways that lead to human disease (Atweh et al. 1987; Indik et al. 1989). However, only in recent years, with the advent of transcriptomic sequencing and the amassing of large databases of patient data, has it become apparent just how broadly splicing is altered in human cancers. This recognition has motivated much investigation of the causal contribution of altered splicing to cellular transformation, and how splicing maybe used as a biomarker for disease or target for therapy. Here we describe the current understanding of how splicing is regulated in general, discuss some classic and newer examples of how this regulation is altered in cancer, and then review recent insight into the breadth of splicing variability in cancer as well as the progress and obstacles in leveraging alternative splicing to increase our ability to classify and treat human metastatic disease.
Fundamental mechanisms of alternative splicing to regulate protein expression
Alternative splicing refers to any process by which all or part of an exon or intron from a nascent transcript is differentially included in the final mRNA during pre-mRNA splicing. Common patterns of alternative splicing include skipping or inclusion of a cassette exon, alternative 5′ or 3′ splice site choice, mutually exclusive exons, and intron retention, although many other complex patterns of alternative splicing can occur (Fig. 1). In all of these cases, the choice of splicing patterns can profoundly alter the expression and/or function of the encoded protein by changing the open reading frame of an mRNA or altering the presence of regulatory sequences in untranslated regions (UTRs) (Nilsen and Graveley 2010; Blencowe 2017). Importantly, the advent of high-throughput sequencing over the past decade has revealed that >95% of protein-coding genes in humans undergo some form of alternative splicing in a cell type- or condition-specific manner, with at least 80% of these changes altering the protein-coding potential of the transcript (Pan et al. 2008; Wang et al. 2008; Nilsen and Graveley 2010; Braunschweig et al. 2014; Ule and Blencowe 2019). As mentioned above, this ability to expand the coding potential of the genome is tremendously beneficial to cells. For example, transitions in alternative splicing are essential drivers of cell differentiation and fate decisions (Baralle and Giudice 2017; Fiszbein and Kornblihtt 2017) and contribute to the ability of cells to respond appropriately to signaling events such as metabolic changes, immune challenge, and neuronal stimulation (Lynch 2007; Fiszbein and Kornblihtt 2017; Ule and Blencowe 2019). However, mistakes in this process—leading to either aberrant splicing patterns or patterns expressed at the wrong time or place—can be profoundly deleterious to cells.
Figure 1.
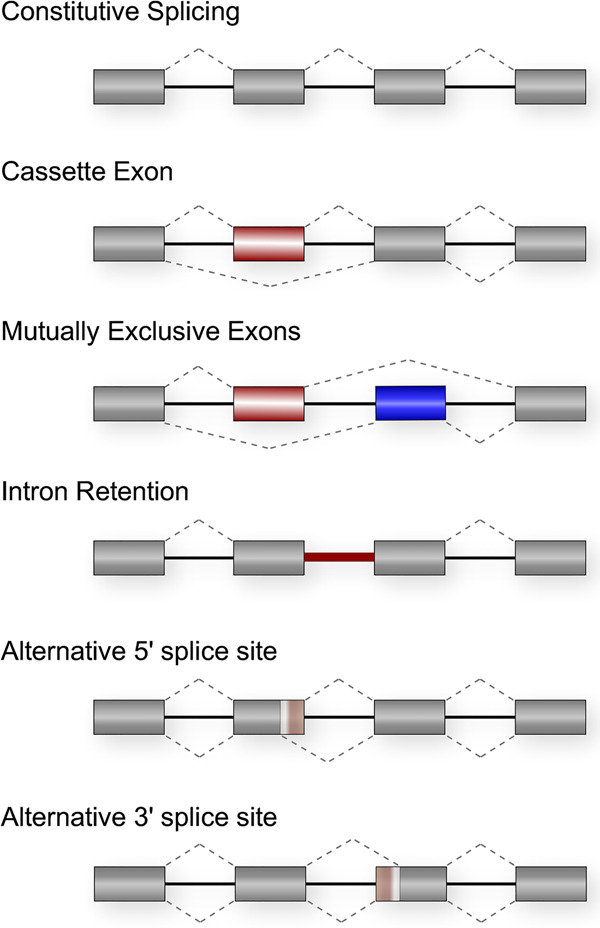
Common patterns of AS. Rectangles represent exons and lines represent introns. Dotted lines connect exons that are joined together to form the final mRNA. Red and blue regions are those that can be differentially included in the mature mRNA. Dotted lines above and below represent the two alternate possible splicing patterns.
The basic chemistry of splicing—that is, the removal of introns and joining of exons—is catalyzed by a multisubunit complex called the spliceosome, which assembles de novo on each intron through specific recognition of sequence elements at the exon–intron boundaries that mark the splice sites (Fig. 2A; for comprehensive reviews, see Wahl et al. 2009; Shi 2017). Importantly, however, the specificity of the interactions between the spliceosome and individual splice sites typically involves only a few Watson-Crick base pairs and thus is not sufficient to mediate the high level of fidelity required of splicing. Consistently, decades of study has revealed that efficient splice site recognition in human genes requires the additional action of RNA-binding proteins (RBPs) that bind adjacent to splice sites and promote recruitment of the spliceosome (for an extensive review, see Fu and Ares 2014; Ule and Blencowe 2019). Conversely, the inherently weak affinity of the individual spliceosomal components with the substrate means that these interactions are readily inhibited through competitive binding or sequestration of the splice sites by repressive RBPs. In sum, therefore, the ultimate decision of which splice sites are used and the resulting pattern of exon joining is determined by a complex interplay of RBPs that bind along exons and introns and direct the association of the splicing catalytic complex to appropriate sites of action (Fig. 2B).
Figure 2.

General mechanism of splicing and its regulation. (A) Spliceosome assembly. The 5′ end of introns are defined by the 5′ splice site (GU) and the 3′ end of introns are defined by the branch point sequence (BPS), polypyrimidine tract (PPT), and the 3′ splice site AG dinucleotides. The U1, U2, U4, U5, and U6 represent the snRNPs that assemble with substrate and each other as shown. The NTC is an additional snRNA-free spliceosomal subunit. The U2AF heterodimeric complex of a 65- and 35-kDa subunit, and the SF1 protein, recognize the 3′ endo of the intron prior to recruitment of the U2snRNP. SF3B1 is a component of the U2snRNP that makes direct contact with the substrate. (B) Regulation of alternative splicing. Enhancer auxiliary elements are denoted in green for exonic (ESE) or intronic (ISE) splicing enhancers. Silencer auxiliary elements are denoted in red for exonic (ESS) or intronic (ISS) splicing silencers. The activities of these auxiliary elements are often mediated through binding of SR and hnRNPs, two common families of RNA-binding proteins described in the text and Figure 3.
Splicing regulatory RBPs in humans can be divided into three major classes: SR proteins, canonical hnRNPs, and other hnRNP-like proteins (Fig. 3). SR proteins are defined by containing at least one RRM-type RNA-binding domain as well as a domain rich in serine–arginine (SR) dipeptides that makes homotypic interactions with other SR proteins and with core components of the spliceosome (Fu 1995; Ule and Blencowe 2019). SR proteins typically bind exonic sequences and promote spliceosome assembly, although this is not always the case. In contrast, canonical hnRNPs are defined not by structure, but rather by historic copurification as abundant nuclear factors (Pinol-Roma et al. 1988). These proteins are thus more structurally diverse than SR proteins, containing RRM-, KH-, and RGG-type RNA-binding domains and often one or more low-complexity regions (Fig. 3). HnRNP-like proteins share sequence similarity to the canonical hnRNPs but exhibit more tissue- or temporal-restricted expression and thus were not identified until more recently (Lunde et al. 2007; Hentze et al. 2018). For all three classes of RBPs, these protein bind to short sequence motifs with a range of promiscuity and redundancy (Ray et al. 2013; Dominguez et al. 2018; Ule and Blencowe 2019). This allows transcripts to simultaneous ensure the ability to bind a necessary subset of RBPs while still adhering to coding or other regulatory constraints.
Figure 3.
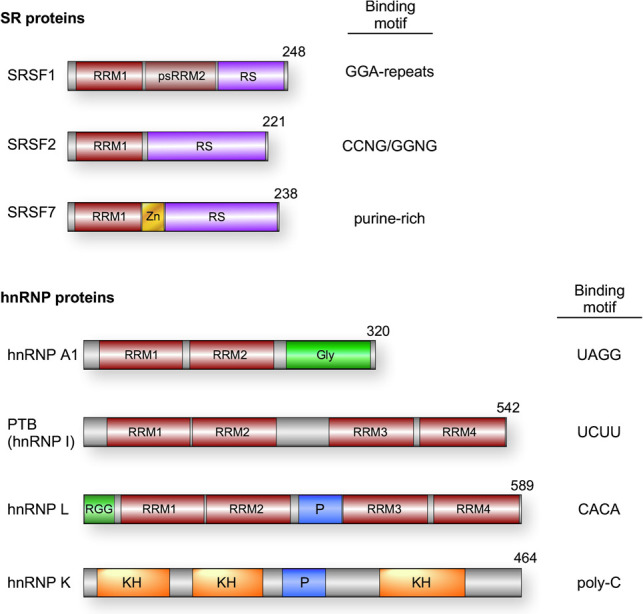
Common splicing regulatory RNA-binding proteins. Domain schematics for each factor are displayed. (RRM) RNA recognition motif; (psRRM) pseudo-RRM; (RS) arginine/serine-rich; (Zn) zinc finger; (Gly) glycine-rich region; (P) proline-rich region; (RGG) arginine/glycine/glycine repeat region; (RS) arginine/serine-rich; (KH) K homology domain. Binding preferences for each factor are specified for SRSF1 (Ray et al. 2013), SRSF2 (Kim et al. 2015; Zhang et al. 2015), SRSF7 (Ray et al. 2013), hnRNP A1 (Ray et al. 2013), PTB (Xue et al. 2009), hnRNP L (Hui et al. 2005), and hnRNP K (Klimek-Tomczak et al. 2004).
Importantly, the RBPs described above function in a highly combinatorial manner such that even modest changes in the abundance or activity of individual RBPs or core spliceosomal proteins can result in large changes in splicing pattern of particular transcripts (Fu and Ares 2014; Baralle and Giudice 2017; Ule and Blencowe 2019). Moreover, each gene is regulated by a discrete subset of RBPs, with each RBP typically regulating several hundred splicing events (Fu and Ares 2014; Baralle and Giudice 2017; Ule and Blencowe 2019). This results in a complex network of coregulation of splicing mediated by a web of RBPs in which any change in RBP expression or activity can potentially impact the splicing of some or many transcripts. Indeed, many developmental transitions in alternative splicing are due to changes in the expression of one or a few RBPs during cellular differentiation (Baralle and Giudice 2017; Fiszbein and Kornblihtt 2017). Similarly, signal responsive changes in splicing can be mediated by changes in RBP abundance or localization due to regulated transcription or mRNA stability of the message or changes in activity mediated by posttranslational modification (Heyd and Lynch 2011). In addition, the splicing of individual transcripts can also be altered by genetic mutations in the sequences recognized either by the spliceosome components themselves or by regulatory RBPs (Motta-Mena et al. 2011; Supek et al. 2014; Jayasinghe et al. 2018). While the impact of cis-mutations is often relatively easy to predict, and thus has been studied in cancer for many years, the role of mutation or dysregulation of RBPs in cancer has only more recently begun to be appreciated.
Functional implications of alternative splicing to cancer
Historically, much of the work on the molecular basis of cancer has focused on chromosomal abnormalities or genetic mutations. However, even many years ago it was clear that aberrations in splicing could contribute to disease phenotype in cancer through changing isoform expression of key enzymes in apoptosis, metabolism, and cell signaling (see Chen and Weiss 2015; Anczukow and Krainer 2016; Lee et al. 2016 for several recent comprehensive reviews). For example, alternate isoforms of pyruvate kinase M (PKM) are expressed in cancer versus normal cells to regulate metabolism as well as impact transcriptional programs (David et al. 2010; David and Manley 2010; Luo and Semenza 2012). PKM is the rate-limiting enzyme in the biosynthesis of pyruvate. The gene encoding PKM contains two mutually exclusive exons (see Fig. 1) 9 and 10, whose inclusion results in the encoding of PKM1 or PKM2, respectively. PKM1 and PKM2 differ by 23 amino acids that impact the dimerization state of the molecule and well as susceptibility to allosteric regulators and posttranslational modifications (Luo and Semenza 2012). Most adult tissues express PKM1, while embryos and cancer cells express predominantly PKM2. PKM2 expression is required for the aerobic glycolysis that is part of the Warburg effect, as replacement of PKM2 with PKM1 in cancer cells increases the alternate pathway of oxidative phosphorylation and reduces cell growth (Christofk et al. 2008). Importantly, the regulation of PKM splicing is well understood to be regulated by a delicate balance of the canonical hnRNPs PTB, hnRNP A1 and hnRNP A2. When highly expressed, as is typical in cancers, PTB, hnRNP A1, and hnRNP A2 bind to repressive sequences around exon 9 to inhibit its use (David et al. 2010; Chen et al. 2012), while lower levels of these proteins shift to repression of exon 10 and release of exon 9 repression (Chen et al. 2012). This concentration-dependent activity of hnRNPs is consistent with the combinatorial control of splicing discussed above.
Splicing can also phenocopy loss-of-function genetic mutations by decreasing the expression full-length proteins. Classic examples of this have been observed in tumor suppressor genes, as discussed below, such as TP53, the gene that encodes the classic tumor suppressor p53 protein. More recently, studies in AML has found that aberrant inclusion of cryptic exon 9b in the gene encoding EZH2 leads to a premature stop codon and reduced expression of the protein EZH2 (Kim et al. 2015; Rahman et al. 2020). Notably, while ∼5% of AML patients contain loss-of-function coding mutations in EZH2, the patients that include exon 9b do not appear to have additional EZH2 mutations, therefore the altered splicing provides an additional mechanism for disruption of this gene beyond genetic mutation. Recent work from our laboratories has expanded on this theme, uncovering evidence for altered splicing of transcripts from many AML-related genes, that occurs independently of changes in gene expression or mutation (Rivera et al. 2020). These studies and others demonstrate the importance of considering the splicing pattern of genes, in addition to common genetic and expression defects, in assessing the contribution of a given gene to a particular cancer type.
Beyond linking altered splicing of individual transcripts to the development and progression of cancer, recent advances in RNA-seq, including increased depth of sequencing and better tools for data analysis, has revealed that many cancers exhibit widespread alterations and variability in splicing patterns (for some recent examples, see Jung et al. 2015; Yoshimi et al. 2019; Calabrese et al. 2020; Phillips et al. 2020). Some of this large-scale disruption of splicing could be a consequence of dysregulated cell growth, rather than causal in the pathology of cancer. Nevertheless, the fact that genes involved in apoptosis, metabolism and gene expression, including known oncogenes and tumor suppressor genes, are often enriched amongst the genes that exhibit altered splicing (Supek et al. 2014; Chen and Weiss 2015; Jung et al. 2015; Phillips et al. 2020), suggests that at least some of the altered splicing contributes to metastatic growth. As described above, splicing patterns impact gene function. Thus, deeper analysis of consistent splicing changes in cancer may uncover additional, unrecognized, cancer-related genes for which gain or loss of function is dictated by splicing rather than mutation. In addition, it has been hypothesized that broad dysregulation of splicing across much of the transcriptome may, itself, cause toxicity as a generalized stress to cell physiology, perhaps through sequestration of RNA-binding proteins to aberrant intron-containing transcripts or inhibition of transcription, the generation of extensive R-loops in the genome and resulting replication stress (Lee et al. 2016; Chen et al. 2018; Nguyen et al. 2018).
Given the proven and potential functional links of alternative splicing to metastatic growth and disease pathology, an essential question then becomes what drives widespread or specific changes in splicing in cancer? Certainly, in the case of individual genes, mutations in splice sites or regulatory elements can lead to splicing alterations. Moreover, given our current understanding of the mechanisms that regulate splicing (see above), an obvious model to explain broader splicing dysregulation is through changes in the expression or function of specific RBPs. Indeed, examples of both cis and trans mutations driving alternative splicing have been described in the literature, as discussed below. However, additional mechanisms are also emerging, including changes in the core splicing machinery or changes in the activity of RBPs through altered expression or cell signaling.
Cis-acting mutations linked to alternative splicing in cancer
Early studies to identify somatic mutations associated with the development of cancer focused on missense, nonsense, and frameshift mutations. However, as methodology has improved an increasing number of mutations have been found in cancer that alter splicing, including those that hinder splicing of individual introns and those that generate new splice sites (Supek et al. 2014; Jung et al. 2015; Jayasinghe et al. 2018). Indeed, one recent study suggests that many cancer-related mutations that have previously been annotated as missense or nonsense, actually alter splicing (Jayasinghe et al. 2018), while another study demonstrated that synonymous mutations in cancer can impact gene expression through altered splicing (Supek et al. 2014). In all cases, these mutations impact splicing through alteration of any of the splice-directing sequences discussed above (Fig. 2), including the splice sites themselves as well as the enhancer or silencer sequences that recruit RBPs (Supek et al. 2014; Jung et al. 2015; Jayasinghe et al. 2018).
Tumor suppressor genes, in particular, contain numerous identified splice-altering mutations (Jung et al. 2015; Jayasinghe et al. 2018). Most notably, almost 30 distinct splice-altering mutations have been identified in TP53 across a diverse array of cancer types (Supek et al. 2014; Jayasinghe et al. 2018). In one specific example, a recent study found that in colorectal cancer, 5% of loss-of-function mutations in TP53 do not directly alter the coding sequence but rather cause altered splicing patterns (Smeby et al. 2019). Common aberrant splicing patterns in TP53 include inefficient removal of introns (aka intron retention) or shifts in splice site usage that result in a frameshift. In either case, these splicing changes introduce novel stop codons that truncate the protein. Interestingly, many of splice-altering mutations fall in the region between exons 5 and 8 of TP53, which is also the primary location for coding mutations. Therefore, the protein products predicted to result from altered splicing closely mimic those generated by nonsense and frameshift mutations in this region (Smeby et al. 2019).
More broadly, intron retention is now recognized to be a frequent consequence of somatic mutations across a wide range of cancers and genes (Jung et al. 2015). This is perhaps not surprising given the highly combinatorial and cooperative nature of splicing described above, in which mutation of any number of intronic or exonic sequences can reduce the efficiency with which the spliceosome can assemble on an intron to promote its removal (see Fig. 2). Most instances of intron removal result in the introduction of stop codons, thus resulting in loss of protein function through protein truncation or nonsense-mediated decay of the mRNA (Ge and Porse 2014). It is worth noting, however, that intron retention can be particularly difficult to accurately quantify (see below) and thus the true extent to which intron retention impacts cancer pathology may still be over or underestimated.
Trans-acting mutations linked to alternative splicing in cancer
Splicing patterns are sensitive not only to mutations in cis-acting sequences, but also to mutations in the regulatory proteins that bind these sequences. Such trans-acting mutations conceivably have a larger impact on cellular splicing patterns than cis-mutations, as they can alter the splicing of a whole network of target transcripts rather than just dysregulate one splicing event. It is not surprising, therefore, that mutations in many RBPs have been causally linked to cancer (for recent comprehensive reviews, see Anczukow and Krainer 2016; Dvinge et al. 2016).
Perhaps the best-characterized example of a mutant splicing regulatory RBP in cancer is the mutation of proline 95 (P95) of the SR protein SRSF2 (previously known as SC35), which has a prevalence of almost 50% in chronic myelomonocytic leukemia (CMML) and is a common mutation in many other leukemias and hematologic dysplasias (Yoshida et al. 2011; Anczukow and Krainer 2016; Papaemmanuil et al. 2016). The P95 residues lay between the RRM and RS domains of SRSF2 (Fig. 3). Mutation of this residue, typically to histidine or arginine, impacts the structural conformation of the RRM (Kim et al. 2015) and alters the sequence specificity of RNA binding (Kim et al. 2015; Zhang et al. 2015). While the wild-type protein binds both CCNG and GGNG motifs, proteins with the P95H mutation bind preferentially to CCNG elements (Kim et al. 2015; Zhang et al. 2015). Like most SR proteins, SRSF2 commonly binds exonic sequences to enhance exonic inclusion, and regulates hundreds of genes (Fu and Ares 2014). The shift in the binding specificity for SRSF2 is thus predicted to change splicing patterns by reducing inclusion of GGNG-containing exons and increased inclusion of CCNG-containing exons. Indeed, several studies have observed this predicted impact of SRSF2 mutations on splicing, including changes in splicing of transcripts encoding the transcription regulatory proteins EZH2 and BCOR that have independently been implicated in cancer (Kim et al. 2015; Zhang et al. 2015; Rahman et al. 2020).
Mutations have also been widely observed in cancer in core components of the spliceosome (Anczukow and Krainer 2016; Inoue et al. 2016) including the small subunit of the U2AF complex (U2AF1) and a substrate-binding subunit of the U2 snRNP (SF3B1). Perhaps surprisingly, mutations in these proteins do not cause a complete transcriptome-wide inhibition of splicing but rather exhibit substrate-specific effects that range from intron retention to skipping of specific exons or use of alternative 3′ splice sites (see Fig. 1). These specific effects speak to the combinatorial nature of splicing regulation, in which even individual core components of the spliceosome can be dispensable if other features of the assembly are optimized (Park et al. 2004; Fu and Ares 2014).
The two most commonly mutated residues of U2AF1 are serine 34 (S34) and glutamate 157 (Q157), both of which fall in the Zn knuckles of the protein. As has been observed for P95 mutations in SRSF2, both of these mutations of U2AF1 alter its RNA sequence specificity, thus shifting the relative recognition of alternate 3′ splice sites (Brooks et al. 2014; Ilagan et al. 2015; Okeyo-Owuor et al. 2015; Fei et al. 2016). By comparison, the functional consequence of the SF3B1 mutations, which fall in the HEAT domain repeats, are less well understood; although, this protein is also involved in 3′ splice site recognition as part of the U2 snRNP (see Fig. 2) and thus, not surprisingly, mutations in SF3B1 also result in alternate usage of cryptic 3′ splice sites (Darman et al. 2015; Zhang et al. 2019). Recently, compelling data has implicated a previously unappreciated protein–protein interaction between SF3B1 and SUGP1 as critical to the functional effects of the HEAT repeat mutations of SF3B1 (Zhang et al. 2019). SUGP1 is an adaptor protein involved in 3′splice site binding by the U2 snRNP. It directly interacts with SF1 and the U2AF complex, both of which are involved in initial recognition of the 3′splice site, and activates an unknown helicase to promote release of SF1 and binding of the U2 snRNP to the substrate (Zhang et al. 2019). SF3B1, a component of the U2 snRNP, also interacts with SUGP1 and recruit it to the 3′ splice site region. Common cancer-causing mutations within the HEAT repeats of SF3B1, such as the K700E mutation, disrupt this interaction (Zhang et al. 2019). Remarkably, knockdown of SUGP1 or mutation of the helicase-activating domain phenocopy mutant SF3B1, while overexpression of SUGP1 partially rescues the splicing defect caused by K700E SF3B1 (Zhang et al. 2019). Moreover, mutations of SUGP1 that disrupt its function have also now been observed in cancer and phenocopy the splicing defect observed in SF3B1-mutant cells (Liu et al. 2020).
Although the data described above demonstrates that mutations in RBPs and splicing factors can lead to broad programs of altered splicing, it is still not fully clear exactly how such widespread changes in splicing lead to pathology. A simple model is that disrupted splicing of a few key transcripts leads to phenotypic changes that drive metastasis (see section on functional relevance below). However, there is little overlap in the genes that are dysregulated by mutations in the various RBPs, even in the case of proteins such as SRSF2, U2AF1, and SF3B1, all of which impact 3′ splice site selection and are commonly mutated in the same disease (MDS) (Pellagatti et al. 2018). It is possible that while these protein mutations impact distinct genes they impinge on similar pathways to give a similar phenotype (Pellagatti et al. 2018). Alternatively, as mentioned above, simply the burden of abundant misprocessed transcripts may be deleterious to normal cellular growth and trigger cell stress response pathways.
Altered expression of splicing regulatory proteins in cancer
Given that splicing is regulated through a balance of competing factors (Fig. 2), it is not surprising that in addition to mutations of RBPs, overexpression or underexpression of RBPs has also been shown to drive pathogenic changes in splicing. Indeed, the splicing of PKM, described above, is determined by the expression level of PTB, hnRNP A1, and hnRNP A2, all of which are misregulated in several cancers (Anczukow and Krainer 2016). One of the first RPBs shown to function as an oncogene was SRSF1 (originally known as ASF/SF2), which was found to be overexpressed in many solid tumors (Watermann et al. 2006; Karni et al. 2007; Das et al. 2012). Importantly, it has been shown in experimental systems that even modest overexpression of SRSF1 is sufficient to promote transformation of immortalized fibrobasts, human epithelial cells and organoid cultures (Karni et al. 2007; Anczukow et al. 2012; Anczukow et al. 2015). Similar functional consequences have now been observed for many other splicing factors, especially SR and hnRNP proteins (Anczukow and Krainer 2016). In some cancers, chromosomal rearrangements lead to an increased copy number of genes encoding RBPs (Karni et al. 2007), while in other instances increases in transcription factors such as Myc have been shown to drive increased transcription of specific RBP genes leading to increased protein expression (Das et al. 2012).
Other sources of alternative splicing in cancer
It is important to note that alternative splicing does not happen in a vacuum, but rather is mechanistically tied to other cellular processes that are often altered in cancer, such as cell signaling and transcription (Heyd and Lynch 2011; Braunschweig et al. 2013). Numerous studies have demonstrated that oncogenic signaling pathways such as JNK, MEK, and AKT alter the expression and/or activity of splicing regulatory RBPs (Weg-Remers et al. 2001; Matter et al. 2002; Blaustein et al. 2005; Pelisch et al. 2005; Lynch 2007; Zhou et al. 2012; Martinez et al. 2015). For example, phosphorylation of SR proteins, which regulates their splicing activity, has been shown to be enhanced in the presence of growth factors such as EGF through activation of AKT (Blaustein et al. 2005; Zhou et al. 2012). AKT can directly phosphorylate SR proteins such as the above-mentioned SRSF1, and also activates the SR-protein kinase SRPK1 to promote additional phosphorylation (Blaustein et al. 2005; Zhou et al. 2012). Consistent with the pathologic implications of SRSF1 overexpression in cancer described above, overexpression of SRPK1 is also widely observed in cancers, where it leads to hyperactivation of SR proteins such as SRSF1 and drives altered splicing programs that are functionally implicated in tumorgenesis (Hayes et al. 2007; Corkery et al. 2015).
Another post-translational modification that controls RBP function is arginine methylation, which typically occurs within RGG domains of these proteins. Methylation of RBPs have been observed to regulate subcellular localization, protein–protein, and protein–RNA interactions (Bedford and Richard 2005; Sinha et al. 2010; Radzisheuskaya et al. 2019). In particular, the RBP methyltransferase PRMT-5 has been shown be required for the proper splicing of many transcripts encoding proteins related to cell cycle progression and tumorgenesis (Bezzi et al. 2013; Braun et al. 2017), while inhibitors to PRMT-5 have utility as anticancer agents (Wang et al. 2018; Fong et al. 2019). A recent investigation specifically identified several targets of PRMT-5 methylation in AML, again including the aforementioned SRSF1 (Radzisheuskaya et al. 2019). The authors went on to demonstrate that methylation controls the splicing activity of SRSF1 through altering protein–protein and protei–RNA interactions. Inhibition of PRMT-5 or mutation of methylated residues in SRSF1 resulted in changes in alternative splicing and growth inhibition of AML cell lines (Radzisheuskaya et al. 2019), thus providing at least one definitive link between RBP methylation, splicing, and cancer progression.
Changes in transcription elongation rate and epigenetic marks have also been shown in many model systems to impact splicing patterns. In what is known as the “window of opportunity” model, elongation speed is thought to impact splice site choice by controlling the time between when two competing splice sites are transcribed and are available to bind the spliceosome (Fong et al. 2014). In the simplest case, the longer the lag between transcription of a suboptimal splice site (i.e., one not bound efficiently by splicing factors) and a competing optimal splice site, the more the suboptimal site is used. Similarly, epigenetic marks, including histone and DNA modifications, have been shown to determine splicing patterns by pausing polymerase or recruiting RBPs to the vicinity of the nascent transcript, both of which impact the association of RBPs nearby splice sites (Luco et al. 2010; Shukla et al. 2011; Braunschweig et al. 2013). Interestingly, a recent study found that AML-associated mutations in IDH2 result in widespread changes in splicing (Yoshimi et al. 2019). Mutations in IDH2 increase DNA methylation through generation of a competitive inhibitor of the TET enzymes (Dang et al. 2009). The resultant hypermethylation of DNA alters the kinetics of transcription elongation, giving rise to changes in alternative splicing as predicted from previous studies (Marina et al. 2016). Interestingly, mutations in IDH2 were found to be highly correlated with the above-described P95 mutations in SRSF2, and patients harboring the double mutation exhibited a profile of alternative splicing distinct from that observed with either individual mutation alone (Yoshimi et al. 2019). Aberrant splicing of one of these unique targets, INT3, was specifically shown to be due to the combined impact of increased methylation of the gene and the altered binding specificity of SRSF2 (Yoshimi et al. 2019). The functional impact of this combined effect was emphasized by the demonstration that overexpression of wild-type INT3 in patient cells partially rescued the phenotypic defect of the IDH2/SRSF2 double mutation (Yoshimi et al. 2019).
Current questions and challenges in alternative splicing and cancer
While evidence abounds that alternative splicing is altered in cancer and can contribute to pathology, many questions remain. Most notably, current work is largely focused on our ability to identify which among the many changes in splicing observed in cancer are most relevant for disease and in determining how to use our knowledge of splicing to develop and guide improved treatments for this disease. While much remains to be done in both of these areas, exciting progress has been made even in the past year or two.
Identifying functionally relevant splicing aberrations in cancer
With the increase in ease and depth of RNA-Sequencing it is becoming commonplace to analyze transcriptomic variation in any cancer type of interest. However, choice of analysis pipeline and thresholds of detection differ widely between studies and can dramatically impact conclusions. Pipelines that are broadly trusted in the splicing field include rMATS (Shen et al. 2014) and MAJIQ (Vaquero-Garcia et al. 2016), both of which have been extensively used and validated in cancer studies (Sotillo et al. 2015; Black et al. 2018; Park et al. 2018; Phillips et al. 2020), as well as JUM (Wang and Rio 2018), which is particularly useful for quantifying intron retention and Leafcutter (Li et al. 2018). Even with an optimal pipeline, experimental design and choice of analysis parameters are critical. The accuracy of splicing quantification is highly dependent on sequencing depth, with a minimum of 60 million reads serving as an accepted standard in the field (Shen et al. 2012; Vaquero-Garcia et al. 2016). A second broad standard in the field is reporting only those splicing events that exhibit a difference of 10%–20% between samples as a meaningful change, as this is seen as the limit of accurate detection and validation in orthogonal assays (Shen et al. 2014; Vaquero-Garcia et al. 2016), although this does not always guarantee functional significance.
It is worth noting, however, that even with these established standards at least three major challenges remain in the analysis of splicing across cancer samples. The first is that the accuracy with which one can assess intron retention remains controversial and challenging (Wang and Rio 2018; Broseus and Ritchie 2020) as mapping of reads across introns can be inaccurate due to the often repetitive nature of intronic sequences. Moreover, careful cellular fractionation is required to isolate nascent transcripts (i.e., those that have not completed splicing) from those that retain introns in the final processed transcript (Bhatt et al. 2012). Some of the difficulties with regard to intron retention will be helped by increased use of long-read sequencing, as has been demonstrated recently in a study of SF3B1 mutations (Tang et al. 2020); however, this technology is not yet easily scaled for large patient cohorts.
A second major challenge is that while batch correction is recognized as critical in gene expression analysis (Gilad and Mizrahi-Man 2015), and has been observed as a confounding factor in quantifying differential splicing (Van Nostrand et al. 2018), there currently exists no publicly available methods for batch correction in splicing analysis. This lack of batch correction is particularly an issue as the field seeks to leverage the extensive sequencing data that exists for large patient cohorts and to compare different cancer types using data from different studies. Finally, identifying cancer-relevant splicing events is also hindered by difficulties in defining and isolating a relevant control sample. Some cancers, such as AML, are considered to be driven through aberrant cellular development and thus do not have a corresponding “normal” cell type (Quek et al. 2016). Alternatively, cancers in complex organs such as brain are derived from a single cell type, while the surrounding “normal” tissue is more heterogeneous. Therefore, caution must be used in claiming which splicing changes are correlated with disease and which may simply reflect cell type variability.
Even when it is clear that robust differences in splicing exist in cancer samples, a major hurdle remains in determining the functional relevance of these splicing changes to disease pathology (Lynch 2015). Historically, the study of the functional impact of splicing on cancer has involved the analysis of individual genes on a case-by-case basis, such as those studies described above. While impactful, such studies are both slow and biased to investigating genes already implicated in cancer biology. Excitingly, several groups are starting to leverage the power of CRISPR-based screening to carry out more unbiased analysis of splicing and its impact on disease (Gonatopoulos-Pournatzis et al. 2018; Thomas et al. 2020). Specifically with respect to cancer, Bradley and colleagues (Thomas et al. 2020) recently described use of a CRISPR-based method they term pgFARM (paired guide RNAs for alternative exon removal) to specifically force the removal of >500 highly conserved poison exons (those that introduce premature stop codons). They applied this screen to lung adenocarcinoma xenographs to show that ∼10% of the poison exons targeted appear to promote tumor growth, while another 10% inhibit growth (Thomas et al. 2020). In a more directed study, CRISPR-mediated knock out of 235 genes for which SF3B1 mutations result in poison-exon inclusion revealed the functional relevance of the chromatin-modifying protein BRD9 (Inoue et al. 2019). Notably, direct knockout of BRD9 is sufficient to transformation in a number of cancer models, while prevention of the use of the BRD9 poison exon in SF3B1 mutant cells suppressed tumorgenesis (Inoue et al. 2019). Expanding such multiplexed CRISPR methodology to other groups of splicing events and other cancers holds tremendous promise for expanding our understanding of the true extent to which splicing dysregulation is an active contributor to the development and progression of metastatic disease.
Leveraging alternative splicing to improve cancer therapy
Finally, beyond simply understanding how splicing may drive or shape cancer, identifying and characterizing splicing dysregulation in cancer holds significant value in the diagnosis and treatment of disease. In terms of therapy, as we observe more and more dysregulated splicing in cancer, it has not escaped notice that cancer-specific splicing events may result in tumor-specific “neoepitopes” that can be targeted by immunotherapy (Jayasinghe et al. 2018; Kahles et al. 2018; Smith et al. 2019). On the other hand, understanding potential alternative splicing patterns is critical to predict or understand mechanisms by which cancer cells may evade immunotherapy (Sotillo et al. 2015). Moreover, in those cases in which specific splicing events have been shown to promote tumorgenesis, such as the examples of PKM, EZH2, INT3, and BRD9 mentioned above, it is increasingly feasible to correct aberrant splicing patterns through the use of antisense oligonucleotides (Mogilevsky et al. 2018; Scharner et al. 2019). Several detailed reviews of such approaches for the therapeutic targeting of cancer have been published recently (Lee and Abdel-Wahab 2016; Urbanski et al. 2018; Smith et al. 2019).
In addition to expanding our therapeutic approaches to cancer, analysis of splicing potentially adds an important further level of information to the characterization of patients that may inform prognosis or drug sensitivity. Several recent studies have correlated splicing patterns with disease outcome to demonstrate that particular splicing events have prognostic value with regard to survival and disease progression (Tian et al. 2019; Anande et al. 2020; Xie et al. 2020). Even more excitingly, two studies have demonstrated a correlation between expression of RBPs and/or specific splicing patterns in leukemia, with sensitivity to specific chemotherapeutics (Sciarrillo et al. 2020; Sinnakannu et al. 2020). If validated in broader studies, the potential for alternative splicing analysis, either alone or coupled with gene expression, to differentiate responders from nonresponders of various therapies, would be of tremendous value in guiding effective personalized medicine approaches in cancer treatment.
In sum, several decades of work have culminated in the clear recognition in the past few years that alternative splicing is highly dysregulated in cancer and contributes to at least some of the disease pathology. Many questions yet remain, however, such as how much of the alternative splicing in cancer is “real” versus detection “noise” and which splicing changes are most relevant for cancer diagnosis and disease. In addition, while recent work has suggested the utility of splicing in guiding therapeutics, we still have a long way to go to prove we can leverage splicing differences between cancer and normal tissues to treat patients or even to meaningfully predict what patients will respond best to which therapy. Addressing such questions will involve significant collaboration between basic scientists and clinicians. The good news is that the recent developments in methodology and insight provide every confidence that we are making good progress toward answering these questions and developing such collaborations and thus are one step closer to the goal of defeating this devastating disease.
Acknowledgments
K.W.L. acknowledges funding from R35 GM118048.
Footnotes
Article is online at http://www.genesdev.org/cgi/doi/10.1101/gad.338962.120.
References
- Anande G, Deshpande NP, Mareschal S, Batcha AMN, Hampton HR, Herold T, Lehmann S, Wilkins MR, Wong JWH, Unnikrishnan A, et al. 2020. RNA splicing alterations induce a cellular stress response associated with poor prognosis in acute myeloid leukemia. Clin Cancer Res 10.1158/1078-0432.CCR-20-0184 [DOI] [PubMed] [Google Scholar]
- Anczuków O, Krainer AR. 2016. Splicing-factor alterations in cancers. RNA 22: 1285–1301. 10.1261/rna.057919.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anczuków O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, Muthuswamy SK, Krainer AR. 2012. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol 19: 220–228. 10.1038/nsmb.2207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anczuków O, Akerman M, Cléry A, Wu J, Shen C, Shirole NH, Raimer A, Sun S, Jensen MA, Hua Y, et al. 2015. SRSF1-regulated alternative splicing in breast cancer. Mol Cell 60: 105–117. 10.1016/j.molcel.2015.09.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atweh GF, Wong C, Reed R, Antonarakis SE, Zhu D, Ghosh PK, Maniatis T, Forget BG, Kazazian HH Jr. 1987. A new mutation in IVS-1 of the human β globin gene causing β thalassemia due to abnormal splicing. Blood 70: 147–151. 10.1182/blood.V70.1.147.147 [DOI] [PubMed] [Google Scholar]
- Baralle FE, Giudice J. 2017. Alternative splicing as a regulator of development and tissue identity. Nature reviews 18: 437–451. 10.1038/nrm.2017.27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford MT, Richard S. 2005. Arginine methylation an emerging regulator of protein function. Mol Cell 18: 263–272. 10.1016/j.molcel.2005.04.003 [DOI] [PubMed] [Google Scholar]
- Bezzi M, Teo SX, Muller J, Mok WC, Sahu SK, Vardy LA, Bonday ZQ, Guccione E. 2013. Regulation of constitutive and alternative splicing by PRMT5 reveals a role for Mdm4 pre-mRNA in sensing defects in the spliceosomal machinery. Genes Dev 27: 1903–1916. 10.1101/gad.219899.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatt DM, Pandya-Jones A, Tong AJ, Barozzi I, Lissner MM, Natoli G, Black DL, Smale ST. 2012. Transcript dynamics of proinflammatory genes revealed by sequence analysis of subcellular RNA fractions. Cell 150: 279–290. 10.1016/j.cell.2012.05.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black KL, Naqvi AS, Asnani M, Hayer KE, Yang SY, Gillespie E, Bagashev A, Pillai V, Tasian SK, Gazzara MR, et al. 2018. Aberrant splicing in B-cell acute lymphoblastic leukemia. Nucleic Acids Res 46: 11357–11369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein M, Pelisch F, Tanos T, Muñoz MJ, Wengier D, Quadrana L, Sanford JR, Muschietti JP, Kornblihtt AR, Cáceres JF, et al. 2005. Concerted regulation of nuclear and cytoplasmic activities of SR proteins by AKT. Nat Struct Mol Biol 12: 1037–1044. 10.1038/nsmb1020 [DOI] [PubMed] [Google Scholar]
- Blencowe BJ. 2017. The relationship between alternative splicing and proteomic complexity. Trends Biochem Sci 42: 407–408. 10.1016/j.tibs.2017.04.001 [DOI] [PubMed] [Google Scholar]
- Braun CJ, Stanciu M, Boutz PL, Patterson JC, Calligaris D, Higuchi F, Neupane R, Fenoglio S, Cahill DP, Wakimoto H, et al. 2017. Coordinated splicing of regulatory detained introns within oncogenic transcripts creates an exploitable vulnerability in malignant glioma. Cancer Cell 32: 411–426.e11. 10.1016/j.ccell.2017.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig U, Gueroussov S, Plocik AM, Graveley BR, Blencowe BJ. 2013. Dynamic integration of splicing within gene regulatory pathways. Cell 152: 1252–1269. 10.1016/j.cell.2013.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos-Pournatzis T, Frey B, Irimia M, Blencowe BJ. 2014. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res 24: 1774–1786. 10.1101/gr.177790.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AN, Choi PS, de Waal L, Sharifnia T, Imielinski M, Saksena G, Pedamallu CS, Sivachenko A, Rosenberg M, Chmielecki J, et al. 2014. A pan-cancer analysis of transcriptome changes associated with somatic mutations in U2AF1 reveals commonly altered splicing events. PLoS One 9: e87361 10.1371/journal.pone.0087361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broseus L, Ritchie W. 2020. Challenges in detecting and quantifying intron retention from next generation sequencing data. Comput Struct Biotechnol J 18: 501–508. 10.1016/j.csbj.2020.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese C, Davidson NR, Demircioğlu D, Fonseca NA, He Y, Kahles A, Lehmann K-V, Liu F, Shiraishi Y, Soulette CM, et al. 2020. Genomic basis for RNA alterations in cancer. Nature 578: 129–136. 10.1038/s41586-020-1970-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Weiss WA. 2015. Alternative splicing in cancer: implications for biology and therapy. Oncogene 34: 1–14. 10.1038/onc.2013.570 [DOI] [PubMed] [Google Scholar]
- Chen M, David CJ, Manley JL. 2012. Concentration-dependent control of pyruvate kinase M mutually exclusive splicing by hnRNP proteins. Nat Struct Mol Biol 19: 346–354. 10.1038/nsmb.2219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chen JY, Huang YJ, Gu Y, Qiu J, Qian H, Shao C, Zhang X, Hu J, Li H, et al. 2018. The augmented R-Loop is a unifying mechanism for myelodysplastic syndromes induced by high-risk splicing factor mutations. Mol Cell 69: 412–425.e6. 10.1016/j.molcel.2017.12.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christofk HR, Vander Heiden MG, Harris MH, Ramanathan A, Gerszten RE, Wei R, Fleming MD, Schreiber SL, Cantley LC. 2008. The M2 splice isoform of pyruvate kinase is important for cancer metabolism and tumour growth. Nature 452: 230–233. 10.1038/nature06734 [DOI] [PubMed] [Google Scholar]
- Corkery DP, Holly AC, Lahsaee S, Dellaire G. 2015. Connecting the speckles: splicing kinases and their role in tumorigenesis and treatment response. Nucleus 6: 279–288. 10.1080/19491034.2015.1062194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang L, White DW, Gross S, Bennett BD, Bittinger MA, Driggers EM, Fantin VR, Jang HG, Jin S, Keenan MC, et al. 2009. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature 462: 739–744. 10.1038/nature08617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darman RB, Seiler M, Agrawal AA, Lim KH, Peng S, Aird D, Bailey SL, Bhavsar EB, Chan B, Colla S, et al. 2015. Cancer-associated SF3B1 hotspot mutations induce cryptic 3′ splice site selection through use of a different branch point. Cell Rep 13: 1033–1045. 10.1016/j.celrep.2015.09.053 [DOI] [PubMed] [Google Scholar]
- Das S, Anczuków O, Akerman M, Krainer AR. 2012. Oncogenic splicing factor SRSF1 is a critical transcriptional target of MYC. Cell Rep 1: 110–117. 10.1016/j.celrep.2011.12.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Manley JL. 2010. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 24: 2343–2364. 10.1101/gad.1973010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- David CJ, Chen M, Assanah M, Canoll P, Manley JL. 2010. HnRNP proteins controlled by c-Myc deregulate pyruvate kinase mRNA splicing in cancer. Nature 463: 364–368. 10.1038/nature08697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez D, Freese P, Alexis MS, Su A, Hochman M, Palden T, Bazile C, Lambert NJ, Van Nostrand EL, Pratt GA, et al. 2018. Sequence, structure, and context preferences of human RNA binding proteins. Mol Cell 70: 854–867.e9. 10.1016/j.molcel.2018.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvinge H, Kim E, Abdel-Wahab O, Bradley RK. 2016. RNA splicing factors as oncoproteins and tumour suppressors. Nat Rev Cancer 16: 413–430. 10.1038/nrc.2016.51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei DL, Motowski H, Chatrikhi R, Prasad S, Yu J, Gao S, Kielkopf CL, Bradley RK, Varmus H. 2016. Wild-type U2AF1 antagonizes the splicing program characteristic of U2AF1-mutant tumors and is required for cell survival. PLoS Genet 12: e1006384 10.1371/journal.pgen.1006384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiszbein A, Kornblihtt AR. 2017. Alternative splicing switches: important players in cell differentiation. Bioessays 39: 1600157 10.1002/bies.201600157 [DOI] [PubMed] [Google Scholar]
- Fong N, Kim H, Zhou Y, Ji X, Qiu J, Saldi T, Diener K, Jones K, Fu XD, Bentley DL. 2014. Pre-mRNA splicing is facilitated by an optimal RNA polymerase II elongation rate. Genes Dev 28: 2663–2676. 10.1101/gad.252106.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fong JY, Pignata L, Goy PA, Kawabata KC, Lee SC, Koh CM, Musiani D, Massignani E, Kotini AG, Penson A, et al. 2019. Therapeutic targeting of RNA splicing catalysis through inhibition of protein arginine methylation. Cancer Cell 36: 194–209.e9. 10.1016/j.ccell.2019.07.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X-D. 1995. The superfamily of arginine/serine-rich splicing factors. RNA 1: 663–680. [PMC free article] [PubMed] [Google Scholar]
- Fu XD, Ares M Jr. 2014. Context-dependent control of alternative splicing by RNA-binding proteins. Nat Rev Genet 15: 689–701. 10.1038/nrg3778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge Y, Porse BT. 2014. The functional consequences of intron retention: alternative splicing coupled to NMD as a regulator of gene expression. Bioessays 36: 236–243. 10.1002/bies.201300156 [DOI] [PubMed] [Google Scholar]
- Gilad Y, Mizrahi-Man O. 2015. A reanalysis of mouse ENCODE comparative gene expression data [version 1; peer review: 3 approved, 1 approved with reservations]. F1000Res 4: 121 10.12688/f1000research.6536.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonatopoulos-Pournatzis T, Wu M, Braunschweig U, Roth J, Han H, Best AJ, Raj B, Aregger M, O'Hanlon D, Ellis JD, et al. 2018. Genome-wide CRISPR-Cas9 interrogation of splicing networks reveals a mechanism for recognition of autism-misregulated neuronal microexons. Mol Cell 72: 510–524.e12. 10.1016/j.molcel.2018.10.008 [DOI] [PubMed] [Google Scholar]
- Hayes GM, Carrigan PE, Miller LJ. 2007. Serine-arginine protein kinase 1 overexpression is associated with tumorigenic imbalance in mitogen-activated protein kinase pathways in breast, colonic, and pancreatic carcinomas. Cancer Res 67: 2072–2080. 10.1158/0008-5472.CAN-06-2969 [DOI] [PubMed] [Google Scholar]
- Hentze MW, Castello A, Schwarzl T, Preiss T. 2018. A brave new world of RNA-binding proteins. Nature reviews 19: 327–341. 10.1038/nrm.2017.130 [DOI] [PubMed] [Google Scholar]
- Heyd F, Lynch KW. 2011. Degrade, move, regroup: signaling control of splicing proteins. Trends Biochem Sci 36: 397–404. 10.1016/j.tibs.2011.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui J, Hung LH, Heiner M, Schreiner S, Neumüller N, Reither G, Haas SA, Bindereif A. 2005. Intronic CA-repeat and CA-rich elements: a new class of regulators of mammalian alternative splicing. Embo J 24: 1988–1998. 10.1038/sj.emboj.7600677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilagan JO, Ramakrishnan A, Hayes B, Murphy ME, Zebari AS, Bradley P, Bradley RK. 2015. U2AF1 mutations alter splice site recognition in hematological malignancies. Genome Res 25: 14–26. 10.1101/gr.181016.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Indik Z, Yeh H, Ornstein-Goldstein N, Kucich U, Abrams W, Rosenbloom JC, Rosenbloom J. 1989. Structure of the elastin gene and alternative splicing of elastin mRNA: implications for human disease. Am J Med Genet 34: 81–90. 10.1002/ajmg.1320340115 [DOI] [PubMed] [Google Scholar]
- Inoue D, Bradley RK, Abdel-Wahab O. 2016. Spliceosomal gene mutations in myelodysplasia: molecular links to clonal abnormalities of hematopoiesis. Genes Dev 30: 989–1001. 10.1101/gad.278424.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue D, Chew GL, Liu B, Michel BC, Pangallo J, D'Avino AR, Hitchman T, North K, Lee SC, Bitner L, et al. 2019. Spliceosomal disruption of the non-canonical BAF complex in cancer. Nature 574: 432–436. 10.1038/s41586-019-1646-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayasinghe RG, Cao S, Gao Q, Wendl MC, Vo NS, Reynolds SM, Zhao Y, Climente-González H, Chai S, Wang F, et al. 2018. Systematic analysis of splice-site-creating mutations in cancer. Cell Rep 23: 270–281.e3. 10.1016/j.celrep.2018.03.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung H, Lee D, Lee J, Park D, Kim YJ, Park W-Y, Hong D, Park PJ, Lee E. 2015. Intron retention is a widespread mechanism of tumor-suppressor inactivation. Nat Genet 47: 1242–1248. 10.1038/ng.3414 [DOI] [PubMed] [Google Scholar]
- Kahles A, Lehmann KV, Toussaint NC, Hüser M, Stark SG, Sachsenberg T, Stegle O, Kohlbacher O, Sander C, Cancer Genome Atlas Research Network et al. 2018. Comprehensive analysis of alternative splicing across tumors from 8,705 patients. Cancer Cell 34: 211–224.e6. 10.1016/j.ccell.2018.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. 2007. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol 14: 185–193. 10.1038/nsmb1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Ilagan JO, Liang Y, Daubner GM, Lee SC, Ramakrishnan A, Li Y, Chung YR, Micol JB, Murphy ME, et al. 2015. SRSF2 mutations contribute to myelodysplasia by mutant-specific effects on exon recognition. Cancer Cell 27: 617–630. 10.1016/j.ccell.2015.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klimek-Tomczak K, Wyrwicz LS, Jain S, Bomsztyk K, Ostrowski J. 2004. Characterization of hnRNP K protein–RNA interactions. J Mol Biol 342: 1131–1141. 10.1016/j.jmb.2004.07.099 [DOI] [PubMed] [Google Scholar]
- Lee SC, Abdel-Wahab O. 2016. Therapeutic targeting of splicing in cancer. Nat Med 22: 976–986. 10.1038/nm.4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Dvinge H, Kim E, Cho H, Micol JB, Chung YR, Durham BH, Yoshimi A, Kim YJ, Thomas M, et al. 2016. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat Med 22: 672–678. 10.1038/nm.4097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YI, Knowles DA, Humphrey J, Barbeira AN, Dickinson SP, Im HK, Pritchard JK. 2018. Annotation-free quantification of RNA splicing using LeafCutter. Nat Genet 50: 151–158. 10.1038/s41588-017-0004-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Zhang J, Sun Y, Perea-Chamblee TE, Manley JL, Rabadan R. 2020. Pan-cancer analysis identifies mutations in SUGP1 that recapitulate mutant SF3B1 splicing dysregulation. Proc Natl Acad Sci 117: 10305–10312. 10.1073/pnas.1922622117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luco RF, Pan Q, Tominaga K, Blencowe BJ, Pereira-Smith OM, Misteli T. 2010. Regulation of alternative splicing by histone modifications. Science 327: 996–1000. 10.1126/science.1184208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lunde BM, Moore C, Varani G. 2007. RNA-binding proteins: modular design for efficient function. Nature reviews 8: 479–490. 10.1038/nrm2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo W, Semenza GL. 2012. Emerging roles of PKM2 in cell metabolism and cancer progression. Trends Endocrinol Metab 23: 560–566. 10.1016/j.tem.2012.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch KW. 2007. Regulation of alternative splicing by signal transduction pathways. Adv Exp Med Biol 623: 161–174. 10.1007/978-0-387-77374-2_10 [DOI] [PubMed] [Google Scholar]
- Lynch KW. 2015. Thoughts on NGS, alternative splicing and what we still need to know. RNA 21: 683–684. 10.1261/rna.050419.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marina RJ, Sturgill D, Bailly MA, Thenoz M, Varma G, Prigge MF, Nanan KK, Shukla S, Haque N, Oberdoerffer S. 2016. TET-catalyzed oxidation of intragenic 5-methylcytosine regulates CTCF-dependent alternative splicing. Embo j 35: 335–355. 10.15252/embj.201593235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez NM, Agosto L, Qiu J, Mallory MJ, Gazzara MR, Barash Y, Fu XD, Lynch KW. 2015. Widespread JNK-dependent alternative splicing induces a positive feedback loop through CELF2-mediated regulation of MKK7 during T-cell activation. Genes Dev 29: 2054–2066. 10.1101/gad.267245.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matter N, Herrlich P, König H. 2002. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 420: 691–695. 10.1038/nature01153 [DOI] [PubMed] [Google Scholar]
- Mogilevsky M, Shimshon O, Kumar S, Mogilevsky A, Keshet E, Yavin E, Heyd F, Karni R. 2018. Modulation of MKNK2 alternative splicing by splice-switching oligonucleotides as a novel approach for glioblastoma treatment. Nucleic Acids Res 46: 11396–11404. 10.1093/nar/gky921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motta-Mena LB, Smith SA, Mallory MJ, Jackson J, Wang J, Lynch KW. 2011. A disease-associated polymorphism alters splicing of the human CD45 phosphatase gene by disrupting combinatorial repression by heterogeneous nuclear ribonucleoproteins (hnRNPs). J Biol Chem 286: 20043–20053. 10.1074/jbc.M111.218727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen HD, Leong WY, Li W, Reddy PNG, Sullivan JD, Walter MJ, Zou L, Graubert TA. 2018. Spliceosome iutations induce R loop-associated sensitivity to ATR inhibition in myelodysplastic syndromes. Cancer Res 78: 5363–5374. 10.1158/0008-5472.CAN-17-3970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. 2010. Expansion of the eukaryotic proteome by alternative splicing. Nature 463: 457–463. 10.1038/nature08909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okeyo-Owuor T, White BS, Chatrikhi R, Mohan DR, Kim S, Griffith M, Ding L, Ketkar-Kulkarni S, Hundal J, Laird KM, et al. 2015. U2AF1 mutations alter sequence specificity of pre-mRNA binding and splicing. Leukemia 29: 909–917. 10.1038/leu.2014.303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Q, Shai O, Lee LJ, Frey BJ, Blencowe BJ. 2008. Deep surveying of alternative splicing complexity in the human transcriptome by high-throughput sequencing. Nat Genet 40: 1413–1415. 10.1038/ng.259 [DOI] [PubMed] [Google Scholar]
- Papaemmanuil E, Dohner H, Campbell PJ. 2016. Genomic classification in acute myeloid leukemia. N Engl J Med 375: 900–901. 10.1056/NEJMc1608739 [DOI] [PubMed] [Google Scholar]
- Park JW, Parisky K, Celotto AM, Reenan RA, Graveley BR. 2004. Identification of alternative splicing regulators by RNA interference in Drosophila. Proc Natl Acad Sci USA 101: 15974–15979. 10.1073/pnas.0407004101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park E, Pan Z, Zhang Z, Lin L, Xing Y. 2018. The expanding landscape of alternative splicing variation in human populations. Am J Hum Genet 102: 11–26. 10.1016/j.ajhg.2017.11.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelisch F, Blaustein M, Kornblihtt AR, Srebrow A. 2005. Cross-talk between signaling pathways regulates alternative splicing: a novel role for JNK. J Biol Chem 280: 25461–25469. 10.1074/jbc.M412007200 [DOI] [PubMed] [Google Scholar]
- Pellagatti A, Armstrong RN, Steeples V, Sharma E, Repapi E, Singh S, Sanchi A, Radujkovic A, Horn P, Dolatshad H, et al. 2018. Impact of spliceosome mutations on RNA splicing in myelodysplasia: dysregulated genes/pathways and clinical associations. Blood 132: 1225–1240. 10.1182/blood-2018-04-843771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JW, Pan Y, Tsai BL, Xie Z, Demirdjian L, Xiao W, Yang HT, Zhang Y, Lin CH, Cheng D, et al. 2020. Pathway-guided analysis identifies Myc-dependent alternative pre-mRNA splicing in aggressive prostate cancers. Proc Natl Acad Sci 117: 5269–5279. 10.1073/pnas.1915975117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinol-Roma S, Choi YD, Matunis MJ, Dreyfuss G. 1988. Immunopurification of heterogeneous nuclear ribonucleoprotein particles reveals an assortment of RNA-binding proteins. Genes Dev 2: 215–227. 10.1101/gad.2.2.215 [DOI] [PubMed] [Google Scholar]
- Quek L, Otto GW, Garnett C, Lhermitte L, Karamitros D, Stoilova B, Lau IJ, Doondeea J, Usukhbayar B, Kennedy A, et al. 2016. Genetically distinct leukemic stem cells in human CD34− acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. J Exp Med 213: 1513–1535. 10.1084/jem.20151775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radzisheuskaya A, Shliaha PV, Grinev V, Lorenzini E, Kovalchuk S, Shlyueva D, Gorshkov V, Hendrickson RC, Jensen ON, Helin K. 2019. PRMT5 methylome profiling uncovers a direct link to splicing regulation in acute myeloid leukemia. Nat Struct Mol Biol 26: 999–1012. 10.1038/s41594-019-0313-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MA, Lin KT, Bradley RK, Abdel-Wahab O, Krainer AR. 2020. Recurrent SRSF2 mutations in MDS affect both splicing and NMD. Genes Dev 34: 413–427. 10.1101/gad.332270.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al. 2013. A compendium of RNA-binding motifs for decoding gene regulation. Nature 499: 172–177. 10.1038/nature12311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera OD, Mallory MJ, Quesnel-Vallieres M, Schultz DC, Carroll M, Barash Y, Cherry S, Lynch KW. 2020. Alternative splicing redefines landscape of commonly mutated genes in acute myeloid leukemia. bioRxiv 10.1101/2020.05.21.107557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharner J, Qi S, Rigo F, Bennett CF, Krainer AR. 2019. Delivery of GalNAc-conjugated splice-switching ASOs to non-hepatic cells through ectopic expression of asialoglycoprotein receptor. Mol Ther Nucleic Acids 16: 313–325. 10.1016/j.omtn.2019.02.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sciarrillo R, Wojtuszkiewicz A, Kooi IE, Leon LG, Sonneveld E, Kuiper RP, Jansen G, Giovannetti E, Kaspers GJL, Cloos J. 2020. Glucocorticoid resistant pediatric acute lymphoblastic leukemia samples display altered splicing profile and vulnerability to spliceosome modulation. Cancers (Basel) 12: 723 10.3390/cancers12030723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Huang J, Dittmar KA, Lu ZX, Zhou Q, Carstens RP, Xing Y. 2012. MATS: a Bayesian framework for flexible detection of differential alternative splicing from RNA-Seq data. Nucleic Acids Res 40: e61 10.1093/nar/gkr1291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen S, Park JW, Lu Z-x, Lin L, Henry MD, Wu YN, Zhou Q, Xing Y. 2014. rMATS: robust and flexible detection of differential alternative splicing from replicate RNA-Seq data. Proc Natl Acad Sci 111: E5593–E5601. 10.1073/pnas.1419161111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. 2017. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nature reviews 18: 655–670. 10.1038/nrm.2017.86 [DOI] [PubMed] [Google Scholar]
- Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M, Oberdoerffer P, Sandberg R, Oberdoerffer S. 2011. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature 479: 74–79. 10.1038/nature10442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha R, Allemand E, Zhang Z, Karni R, Myers MP, Krainer AR. 2010. Arginine methylation controls the subcellular localization and functions of the oncoprotein splicing factor SF2/ASF. Mol Cell Biol 30: 2762–2774. 10.1128/MCB.01270-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinnakannu JR, Lee KL, Cheng S, Li J, Yu M, Tan SP, Ong CCH, Li H, Than H, Anczukow-Camarda O, et al. 2020. SRSF1 mediates cytokine-induced impaired imatinib sensitivity in chronic myeloid leukemia. Leukemia 10.1038/s41375-020-0732-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smeby J, Sveen A, Eilertsen IA, Danielsen SA, Hoff AM, Eide PW, Johannessen B, Hektoen M, Skotheim RI, Guren MG, et al. 2019. Transcriptional and functional consequences of TP53 splice mutations in colorectal cancer. Oncogenesis 8: 35 10.1038/s41389-019-0141-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CC, Selitsky SR, Chai S, Armistead PM, Vincent BG, Serody JS. 2019. Alternative tumour-specific antigens. Nat Rev Cancer 19: 465–478. 10.1038/s41568-019-0162-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sotillo E, Barrett DM, Black KL, Bagashev A, Oldridge D, Wu G, Sussman R, Lanauze C, Ruella M, Gazzara MR, et al. 2015. Convergence of acquired mutations and alternative splicing of CD19 enables resistance to CART-19 immunotherapy. Cancer Discov 5: 1282–1295. 10.1158/2159-8290.CD-15-1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F, Miñana B, Valcárcel J, Gabaldón T, Lehner B. 2014. Synonymous mutations frequently act as driver mutations in human cancers. Cell 156: 1324–1335. 10.1016/j.cell.2014.01.051 [DOI] [PubMed] [Google Scholar]
- Tang AD, Soulette CM, van Baren MJ, Hart K, Hrabeta-Robinson E, Wu CJ, Brooks AN. 2020. Full-length transcript characterization of SF3B1 mutation in chronic lymphocytic leukemia reveals downregulation of retained introns. Nat Commun 11: 1438 10.1038/s41467-020-15171-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Polaski JT, Feng Q, De Neef EJ, Hoppe ER, McSharry MV, Pangallo J, Gabel AM, Belleville AE, Watson J, et al. 2020. RNA isoform screens uncover the essentiality and tumor-suppressor activity of ultraconserved poison exons. Nat Genet 52: 84–94. 10.1038/s41588-019-0555-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian J, Wang Z, Mei S, Yang N, Yang Y, Ke J, Zhu Y, Gong Y, Zou D, Peng X, et al. 2019. CancerSplicingQTL: a database for genome-wide identification of splicing QTLs in human cancer. Nucleic Acids Res 47: D909–D916. 10.1093/nar/gky954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ule J, Blencowe BJ. 2019. Alternative splicing regulatory networks: functions, mechanisms, and evolution. Mol Cell 76: 329–345. 10.1016/j.molcel.2019.09.017 [DOI] [PubMed] [Google Scholar]
- Urbanski LM, Leclair N, Anczuków O. 2018. Alternative-splicing defects in cancer: splicing regulators and their downstream targets, guiding the way to novel cancer therapeutics. Wiley Interdiscip Rev RNA 9: e1476 10.1002/wrna.1476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand EL, Freese P, Pratt GA, Wang X, Wei X, Xiao R, Blue SM, Chen J-Y, Cody NAL, Dominguez D, et al. 2018. A large-scale binding and functional map of human RNA binding boteins. bioRxiv 10.1101/179648 [DOI] [Google Scholar]
- Vaquero-Garcia J, Barrera A, Gazzara MR, González-Vallinas J, Lahens NF, Hogenesch JB, Lynch KW, Barash Y. 2016. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife 5: e11752 10.7554/eLife.11752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl MC, Will CL, Lührmann R. 2009. The spliceosome: design principles of a dynamic RNP machine. Cell 136: 701–718. 10.1016/j.cell.2009.02.009 [DOI] [PubMed] [Google Scholar]
- Wang Q, Rio DC. 2018. JUM is a computational method for comprehensive annotation-free analysis of alternative pre-mRNA splicing patterns. Proc Natl Acad Sci USA 115: E8181–E8190. 10.1073/pnas.1806018115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, Kingsmore SF, Schroth GP, Burge CB. 2008. Alternative isoform regulation in human tissue transcriptomes. Nature 456: 470–476. 10.1038/nature07509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Hu W, Yuan Y. 2018. Protein arginine methyltransferase 5 (PRMT5) as an anticancer target and its inhibitor discovery. J Med Chem 61: 9429–9441. 10.1021/acs.jmedchem.8b00598 [DOI] [PubMed] [Google Scholar]
- Watermann DO, Tang Y, Zur Hausen A, Jäger M, Stamm S, Stickeler E. 2006. Splicing factor Tra2-β1 is specifically induced in breast cancer and regulates alternative splicing of the CD44 gene. Cancer Res 66: 4774–4780. 10.1158/0008-5472.CAN-04-3294 [DOI] [PubMed] [Google Scholar]
- Weg-Remers S, Ponta H, Herrlich P, König H. 2001. Regulation of alternative pre-mRNA splicing by the ERK MAP-kinase pathway. Embo J 20: 4194–4203. 10.1093/emboj/20.15.4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie ZC, Gao L, Chen G, Ma J, Yang LH, He RQ, Li MW, Cai KT, Li TT, Peng ZG. 2020. Prognostic alternative splicing regulatory network of splicing events in acute myeloid leukemia patients based on SpliceSeq data from 136 cases. Neoplasma 67: 623–635. 10.4149/neo_2020_190917N922 [DOI] [PubMed] [Google Scholar]
- Xue Y, Zhou Y, Wu T, Zhu T, Ji X, Kwon YS, Zhang C, Yeo G, Black DL, Sun H, et al. 2009. Genome-wide analysis of PTB–RNA interactions reveals a strategy used by the general splicing repressor to modulate exon inclusion or skipping. Mol Cell 36: 996–1006. 10.1016/j.molcel.2009.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K, Sanada M, Shiraishi Y, Nowak D, Nagata Y, Yamamoto R, Sato Y, Sato-Otsubo A, Kon A, Nagasaki M, et al. 2011. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature 478: 64–69. 10.1038/nature10496 [DOI] [PubMed] [Google Scholar]
- Yoshimi A, Lin KT, Wiseman DH, Rahman MA, Pastore A, Wang B, Lee SC, Micol JB, Zhang XJ, de Botton S, et al. 2019. Coordinated alterations in RNA splicing and epigenetic regulation drive leukaemogenesis. Nature 574: 273–277. 10.1038/s41586-019-1618-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Lieu YK, Ali AM, Penson A, Reggio KS, Rabadan R, Raza A, Mukherjee S, Manley JL. 2015. Disease-associated mutation in SRSF2 misregulates splicing by altering RNA-binding affinities. Proc Natl Acad Sci USA 112: E4726–E4734. 10.1073/pnas.1514105112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Ali AM, Lieu YK, Liu Z, Gao J, Rabadan R, Raza A, Mukherjee S, Manley JL. 2019. Disease-causing mutations in SF3B1 alter splicing by disrupting interaction with SUGP1. Mol Cell 76: 82–95.e7. 10.1016/j.molcel.2019.07.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Qiu J, Liu W, Zhou Y, Plocinik RM, Li H, Hu Q, Ghosh G, Adams JA, Rosenfeld MG, et al. 2012. The Akt-SRPK-SR axis constitutes a major pathway in transducing EGF signaling to regulate alternative splicing in the nucleus. Mol Cell 47: 422–433. 10.1016/j.molcel.2012.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]