

Abstract
Background and Purpose:
In addition to the central nervous system-mediated action, leptin also directly induces fatty acid oxidation in skeletal muscle. Rapid induction of FAO by leptin is mediated by the AMP-activated protein kinase (AMPK) pathway, but the mechanism of prolonged FAO by leptin was previously unknown. In an earlier study, we showed that free fatty acids increase transcription of small ubiquitin-like modifier (SUMO) specific protease 2 (SENP2) in skeletal muscle, and that SENP2 stimulates expression of FAO-associated enzymes by deSUMOylating peroxisome proliferator-activated receptors, PPARδ and PPARγ. In this study, we examine whether SENP2 is involved in prolonged stimulation of FAO by leptin.
Methods:
The Effect of leptin on expression of SENP2 and on SENP2-mediated FAO was investigated by using western blotting and real time qPCR of C2C12 myotubes, and of C2C12 myotubes in which expression of specific genes was knocked down using siRNAs. Additionally, muscle-specific SENP2 knockout mice were generated to test the involvement of SENP2 in leptin-induced FAO in vivo.
Results:
We show that leptin treatment of C2C12 myotubes causes signal transducer and activator of transcription 3 (STAT3) to bind to the Senp2 promoter, inducing SENP2 expression. We also show that leptin increases the binding of PPARδ and PPARγ to PPRE sites in the promoters of two FAO-associated genes: long-chain acyl-CoA synthetase (Acsl1) or carnitine palmitoyl transferase 1b (Cpt1b). When SENP2 is knocked down in myotubes, leptin-induced expression of FAO-associated enzymes and prolonged increase of FAO are suppressed, but rapid increase of FAO is unaffected. In addition, leptin-induced expression of FAO-associated enzymes was not observed in muscle tissue of SENP2 knockout mice.
Conclusions:
We demonstrate that the peripheral actions of leptin on FAO are mediated by two different pathways: AMPK causes a rapid increase in FAO, and SENP2 of the STAT3 pathway causes a slow, prolonged increase in FAO.
Keywords: Leptin, Fatty acid oxidation, PPAR, SUMO-specific protease, SUMO
1. Introduction
Leptin is an adipokine secreted from adipocytes that plays an important role in the regulation of food intake and energy expenditure [1,2]. In the central nervous system, leptin regulates the synthesis of neuropeptides involved in appetite control and energy balance [3,4]. Through the hypothalamic-pituitary-adrenal axis, leptin plays an important role in glucose homeostasis, causing a shift from carbohydrate to fat metabolism during starvation [5]. Leptin also directly acts on peripheral tissues: leptin regulates hepatic gluconeogenesis and insulin sensitivity, and increases energy expenditure in skeletal muscle [6,7]. Binding of leptin to the leptin receptor, which is located in cell membranes, results in activation of various signaling pathways, including Janus kinase (JAK)/ signal transducer and activator of transcription 3 (STAT3) pathway in the central nervous system and peripheral tissues [8,9]. The JAK/STAT3 pathway is initiated when leptin binds the long isoform leptin receptor (LepRb). Binding of leptin to LepRb induces phosphorylation of the C-terminal domain of LepRb and JAK, which leads to phosphorylation of STAT3 [10–12]. Phosphorylated STAT3 is translocated to the nucleus, where it regulates the transcription of its target genes.
Leptin increases insulin sensitivity though the mechanisms have been unclear [13]. Leptin also increases fatty acid oxidation (FAO) in skeletal muscle. Leptin produces biphasic activation of AMP-activated protein kinase (AMPK) in red skeletal muscle such as soleus muscle. Rapid AMPK activation, which occurs within 15 minutes of leptin treatment, is believed to be directly mediated by leptin, while slow activation, which occurs several hours after leptin treatment, is believed to be mediated by the sympathetic nervous system [7,14]. AMPK phosphorylates acetyl-CoA carboxylase (ACC), thereby inactivating it. ACC inactivation results in the reduction of malonyl CoA, an inhibitor of carnitine palmitoyl transferase 1 (CPT1), causing an increase in FAO [15,16]. AMPK also stimulates translocation of CD36 to the plasma membrane, resulting in fatty acid uptake [17]. In C2C12 myotubes and primary myotubes, sustained leptin-induced FAO is associated with increased mRNA levels for FAO-associated enzymes such as CPT1b, medium chain acyl-CoA dehydrogenase (MCAD), and uncoupling protein 2 (UCP2) [18]. Prolonged leptin-induced FAO in myotubes appears to be regulated by the JAK2/STAT3 pathway, but the downstream part of this pathway has not been elucidated.
Small ubiquitin like modifier (SUMO) modification, or SUMOylaiont, is a type of post-translational modification that regulates the activity, cellular localization, and stability of proteins [19,20]. SUMOylation can be reversed by SUMO-specific proteases (SENPs), six of which have been identified in mammals (SENP1, 2, 3, 5, 6, and 7) [21,22]. We have shown that SENP2 increases the activities of peroxisome proliferator-activated receptor (PPAR) γ and PPARδ by deSUMOylation of these proteins [23,24]. In addition, SENP2 overexpression increases mRNA levels of FAO-associated proteins, long-chain acyl-CoA synthetase 1 (ACSL1), and CPT1b, through deSUMOylation of PPARγ and PPARδ, resulting in the increase of FAO and energy expenditure in skeletal muscle. Transgenic mice that overexpress SENP2 in muscle are protected from high fat diet-induced obesity and insulin resistance [24]. In myotubes, palmitate causes an increase in SENP2 expression through the toll-like receptor (TLF) 4 /MyD88/ NFκB signaling pathway. Therefore, we have suggested that SENP2 is a sensor of high levels of fat, and regulates lipid metabolism in skeletal muscle. In this study, we tested whether SENP2 is involved in leptin-induced FAO in skeletal muscle.
2. Materials and Methods
2.1. Plasmids and antibodies
Leptin was purchased from Sigma-Aldrich (St. Louis, MO, USA). The mouse Senp2 promoter-luciferase reporter vectors, mSenp2(−1980)-Luc, and mSenp2(−868)-Luc, were described previously [25]. To generate mutations at the STAT3 binding site in the Senp2 promoter, the nucleotide sequence from −1178 bp to −1170 bp, 5′-TTCCAGGAC-3′, was replaced with 5′-TTAAAGGAC-3′ by use of a site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA), and the vector harboring the mutation was named mSenp2(−1980)mt-Luc. The mouse Acsl1 promoter region from −1051 bp to −45 bp (from the start codon) was ligated to the pGL2-basic to generate mAcsl1(−1051)-Luc. APPAR response element (PPRE) between −217 bp and −205 bp in the mAcsl1 promoter (5′-TTACCTTTAGCCC-3′) was mutated to 5′-TTACCTTTAGGGC-3′ in the mAcsl1(−1051)mt-Luc. Expression vectors for PPARδ, PPARδK104R, PPARγ, PPARγK107R, SENP2, and PGC1α were previously described [23,24]. All small interfering (si) RNAs were purchased from Dharmacon (Lafayette, CO, USA). Antibodies against the leptin receptor (sc-8391), STAT3, pSTAT3, SENP2, PPARδ, and PPARγ were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). An antibody against γ-tubulin was purchased from Sigma-Aldrich. AMPK and pAMPK antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA).
2.2. Cell culture and leptin treatment
C2C12 myoblasts were maintained in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS) (Invitrogen, Carlsbad, CA, USA). Differentiation to myotubes was induced by culturing in DMEM supplemented with 2% horse serum (Invitrogen) for 4–5 days. For leptin treatment, myotubes were washed with phosphate-buffered saline and then serum-free DMEM containing the indicated amount of leptin was added.
2.3. Measurement of FAO
FAO rates in C2C12 myotubes or soleus muscle were measured using [1-14C] palmitate as described previously [24]. The radioactivity value was normalized by protein amount in each lysate.
2.4. RNA preparation and real-time polymerase chain reaction (PCR)
Total RNA was isolated using TRIzol® (Invitrogen), and real-time PCR was performed using the SYBR®-master mix (Takara, Mountain View, CA, USA) and the ABI 7500 real-time PCR system (Applied Biosystems, Foster City, CA, USA). Each cycle threshold (Ct) value was subtracted from the glyceraldehyde 3-phosphate dehydrogenase (Gapdh) Ct value of the same sample (dCt) and then subtracted from the dCt value of each control set (ddCt). Relative mRNA levels were expressed as 2−ddct. Nucleotide sequences of the primers are shown in Supplemental Table 1.
2.5. Transfection and reporter assays
For reporter assays, C2C12 myoblasts were transfected with 300 ng of mSenp2-Luc reporter vectors using Lipofectamine® with Plus Reagent (Invitrogen) and leptin was treated for 24 h. COS7 cells were transfected with mAcsl1(−1051)-Luc, and the expression vectors for PPARs, PGC1α, or SENP2. PGC1α expression vectors were transfected in all sets to increase basal levels of luciferase activity. All siRNAs were transfected into C2C12 myotubes using RNAiMAX™ reagent (Invitrogen).
2.6. Electrophoretic-mobility shift assays (EMSA)
The probe, representing the nucleotide sequences between −1190 bp and −1157 bp of the Senp2 promoter, was labeled with biotin (LightShift Chemiluminescent EMSA Kit, Thermo Fisher Scientific, Scotts Valley, CA, USA) and incubated with nuclear extracts (10 μg) of C2C12 myotubes that had been treated with leptin (100 ng/mL) for 24 h. For competition assays, unlabeled competitors were incubated in a 50-fold excess, and for super-shift analyses, antibody against STAT3 was added to the reaction. The nucleotide sequences of the probe and competitors are shown in Supplemental Table 1.
2.7. Chromatin immunoprecipitation (ChIP) coupled with quantitative PCR
C2C12 myotubes were treated with siRNAs against SENP2 (50 nmol/1) and the next day the cells were treated with leptin (100 ng/mL) for 24 h. After crosslinking and DNA fragmentation, immunoprecipitation was performed with antibodies against PPARδ and PPARγ. PCR was performed with primers flanking the PPRE (around −140 bp) in the Acsl1 or the PPRE (around −409 bp) in the Cpt1b promoters.
2.8. Animals
Male mice (C57BL/6J or db/db) (8 weeks old) were purchased from Samtako Bio Inc. (Osan, Korea). All aspects of animal care and experimentation were conducted in accordance with the Guide for Experimental Animal Research of the Laboratory for Experimental Animal Research, Clinical Research Institute, Seoul National University Bundang Hospital, Korea. (Permit Number: BA1403-149/013-01). Recombinant murine leptin (3 mg/kg body weight) (Eli Lilly, Indianapolis, IN, USA) dissolved in saline was injected intraperitoneally (i.p.) into 10 week-old mice. For the control group, saline was injected. Ten h or 24 h after the injections, the mice were killed, the soleus muscles were rapidly isolated, and FAO or mRNA levels were measured.
2.9. Ex vivo leptin treatment
Soleus muscles were isolated from male mice 16 h after food removal and transferred to a 12-well plate containing 2 ml of Krebs-Henseleit buffer, pH 7.4 (Sigma-Aldrich, St. Louis, MO) with 2 mmol/L pyruvate, 8 mmol/L mannitol, and 0.1% BSA, as previously described [26]. Muscles were incubated for 15 min in this buffer before replacing with the buffer containing leptin (300 ng/mL). Muscles were collected at the indicated time points and lysates were prepared. Briefly, muscles were homogenized in ice-cold RIPA buffer containing commercially available protease and phosphatase inhibitor cocktails (Roche) using glass teflon homogenizer. Protein content in lysates was measured by the bicinchoninic acid method (Pierce) and subject to immunostaining for detection of phospho- and total-STAT3.
2.10. Generation of SENP2 muscle specific-knockout mice
Animals bearing a LoxP-flanked Senp2 allele (Senp2flox/+ mice) were generated by inGenious Targeting Laboratory (Stony Brook, NY, USA). Briefly, a BAC clone (C57BL/6, RPC23:255D9 clone) containing an 8.36 kb fragment of Senp2 genomic DNA was used to generate a targeting vector (Supplemental Fig. 1). Six independent Senp2flox/+ ES clones were identified, which were injected into C57BL/6 blastocysts to generate chimeric mice. The chimeric mice were bred with wild type C57BL/6 mice for germline transmission. Heterozygous animals were then crossed with mice expressing flp-recombinase in the germline (Flipper mice, from Jackson lab, USA) to delete the FRT-flanked Neo cassette. Offspring of these mice were heterozygous for the desired Senp2flox/+ allele. SENP2 muscle specific-knockout mice (SENP2mKO, Senp2flox/flox, Mck-Cre) were generated by crossing with MCK-Cre (Jackson lab). Floxed (Senp2flox/flox) mice were used for the control group. 10-12 week-old mice were used for leptin injection experiments.
2.11. Statistical analyses
SPSS, version 22.0 (SPSS, Chicago, IL, USA) was used for statistical analysis. Differences between the two groups were assessed by Student’s t-test or two-way analysis of variance. Data were expressed as mean ± SEM. A P-value <0.05 denoted the presence of a statistically significant difference.
3. Results
3.1. Leptin increases expression of FAO-associated enzymes through the leptin receptor/STAT3 pathway in C2C12 myotubes.
We first tested whether the leptin receptor/STAT3 signaling pathway is activated by leptin in C2C12 myotubes, and mediates prolonged increase of FAO. Leptin treatment stimulated STAT3 phosphorylation in C2C12 myotubes, but this stimulation disappeared when leptin receptor was knocked down by using siRNAs of leptin receptor (siLepR) (Fig. 1A). A leptin-induced increase in FAO (24 h after the leptin treatment) that was observed in C2C12 myotubes was nearly abolished when leptin receptor or STAT3 was knocked down (Fig. 1B). Leptin-induced transcription of the FAO-associated enzymes ACSL1 and CPT1b was suppressed in the leptin receptor- or STAT3-knockdown myotubes (Fig. 1C and 1D). Knockdown of the leptin receptor or STAT3 by specific siRNAs was confirmed (Supplemental Fig. 2). These results suggest that the LepR/STAT3 signaling pathway induces expression of FAO enzymes when leptin binds the leptin receptor.
Figure 1. Leptin increases expression of FAO-associated enzymes through the leptin receptor/STAT3 pathway in C2C12 myotubes.
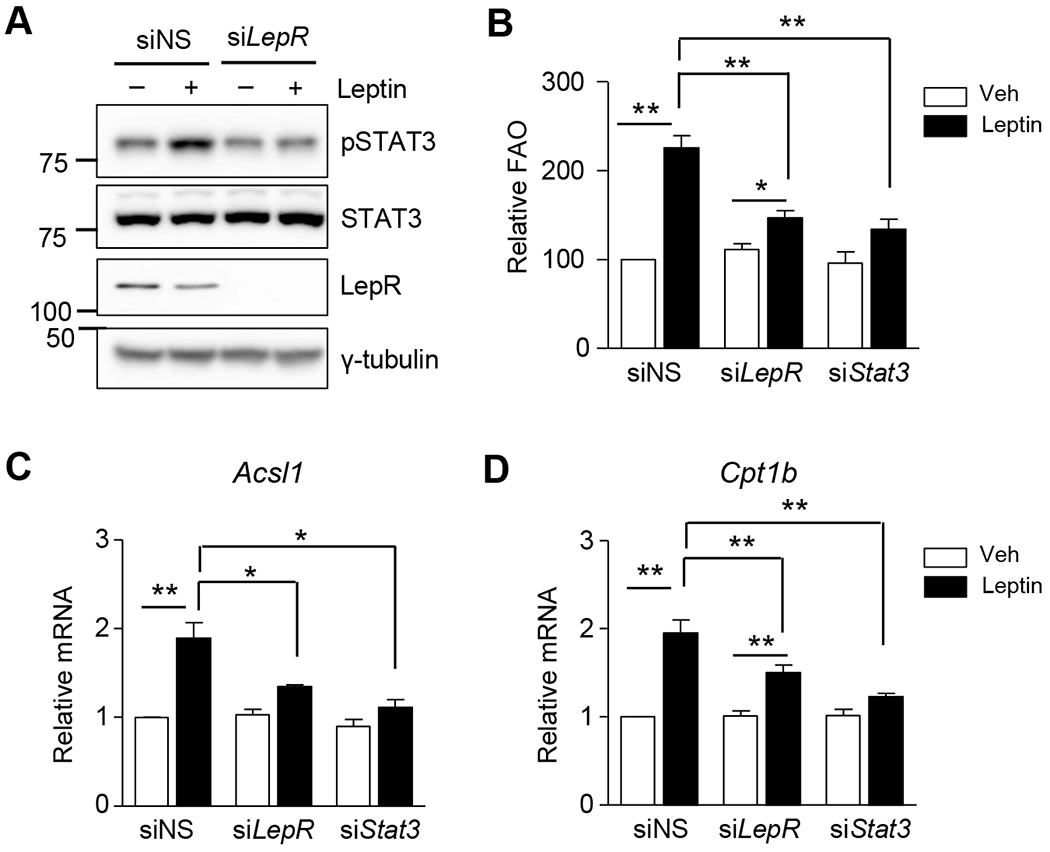
(A) C2C12 myotubes were treated with siRNAs (siNS or siLepR, 100 nM) for 24 h and treated with leptin for 3 h. The lysates were subjected to western blot analysis. Phosphorylated STAT3 (pSTAT3), total STAT3 (STAT3) and Leptin receptor (LepR) were detected. (B-D) C2C12 myotubes were treated with siRNAs (siNS, siLepR or siSTAT3) for 24 h and then treated with leptin (100 ng/mL) for another 24 h. Fatty acid oxidation (B) was measured and the cpm value/μg protein in the siNS treated cells without leptin (vehicle [Veh]) was expressed as 100, and others were expressed as its relative values (n = 5). Total mRNAs were prepared and real time quantitative PCR was performed using primers specific to Acsl1(C) and Cpt1b (D). The mRNA levels of leptin-untreated siNS cells were expressed as 1, and the others were expressed as its relative values (n = 4). *P < 0.05, **P < 0.01. Data are presented as mean ± SEM.
3.2. Leptin increases SENP2 expression through the leptin receptor/STAT3 pathway.
Leptin treatment of C2C12 myotubes caused a gradual, dose-dependent increase in SENP2 mRNA and protein levels over a 24 hour period (Fig. 2A and 2B). This leptin-induced increase in SENP2 expression was abolished when the leptin receptor or STAT3 was knocked down (Fig. 2C and 2D). A STAT3 binding site at the −1178 bp region of the Senp2 promoter was identified using transient transfection and reporter assays (Fig. 2E). The nucleotide sequence at this locus was similar to the consensus sequence for the STAT3 binding site (see the upper box in Fig. 2F). Leptin increased promoter activity in a construct containing the Senp2 promoter (−1980 bp/ +93 bp), but did not increase promoter activity in a construct lacking the STAT3 binding site (−868 bp/ +93 bp) or in a construct with a mutated STAT3 binding site (Fig. 2E). Leptin-induced binding of STAT3 to the −1178 bp region of the Senp2 promoter was confirmed by EMSA using biotin-labeled oligomers (self) and nuclear extracts (N.E.) of leptin-treated C2C12 myotubes (lane 3) (Fig. 2F). Oligomers containing the consensus sequence for STAT3 binding site (Cons) competed for the binding (lane 6). An antibody against STAT3 (α-STAT3) caused a super shift (lane 7). Together, these results demonstrate that STAT3 specifically binds to the region, and leptin increases Senp2 transcription by facilitating binding of STAT3 to the Senp2 promoter.
Figure 2. Leptin increases SENP2 expression through the leptin receptor/STAT3 pathway.
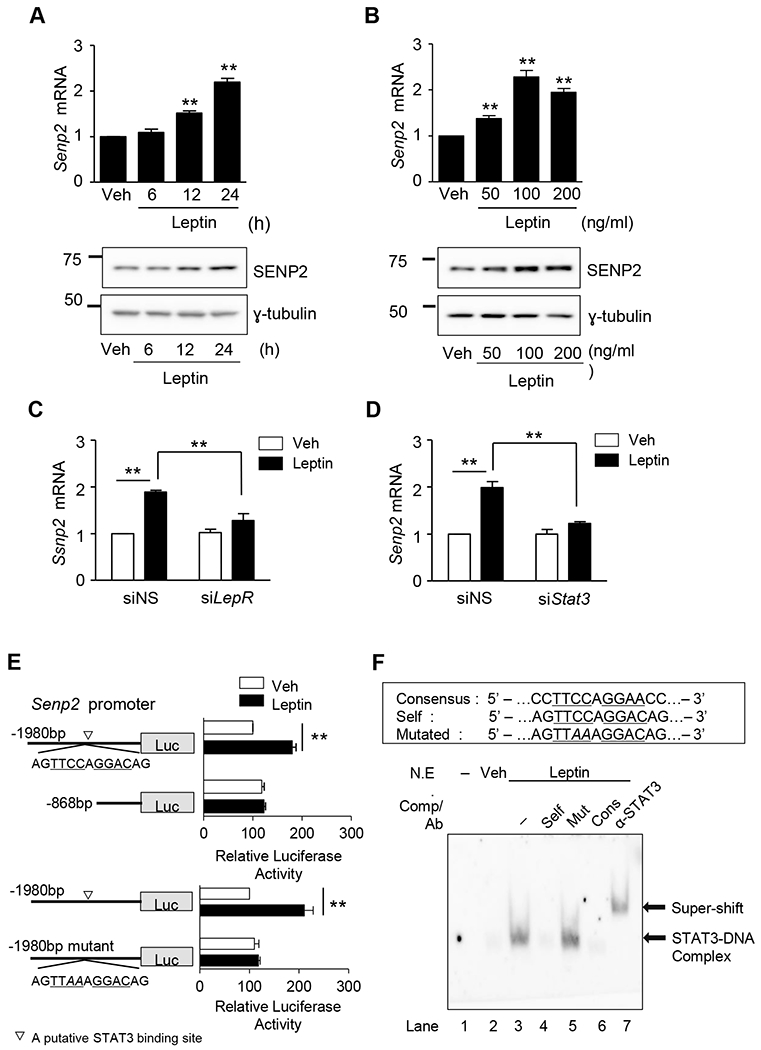
(A) C2C12 myotubes were treated with leptin (100 ng/mL) for different periods. The mRNA level of Senp2 was measured by real time PCR (upper panel). The mRNA level of leptin-untreated cells (Veh) was expressed as 1, and the others were expressed as its relative values (n=4). Western blotting was also performed using an anti-SENP2 antibody (lower panel). (B) C2C12 myotubes were treated with various concentration of leptin for 24 h, and real time PCR (upper panel) (n = 4) and western blotting (lower panel) were performed. (C, D) C2C12 myotubes were treated with siRNAs (100 nmol/1) against leptin receptor (siLepR) (C) or STAT3 (siStat3) (D) for 24 h and then Senp2 mRNA levels were measured (n = 4). (E) C2C12 myoblasts were transfected with one of the Senp2 promoter-Luc constructs and then treated with leptin (100 ng/ml) for 24 h. Normalized luciferase activity of the cells, transfected with mSenp2(−1980)-Luc without leptin, was expressed as 100 and the others were expressed as its relative values (n = 5). A putative STAT3 binding site was indicated (▽). *P < 0.05, **P < 0.01. Data are presented as mean ± SEM. (F) Oligonucleotides (Self) representing the STAT3 binding site identified in Fig. 2E were labeled with biotin and incubated with nuclear extracts of C2C12 myotubes treated with leptin. Unlabeled oligonucleotides (a 50-fold excess) (Self, mutated [Mut], consensus [Cons]) were used as competitors (Comp), and an anti-STAT3 antibody was used for super-shift. Lane 1, the probe alone.
3.3. SENP2 mediates leptin-induced stimulation of FAO.
Transfection of C2C12 cells with siSenp2 resulted in a decrease in SENP2 mRNA and protein levels, confirming the SENP2 was knocked down (Fig. 3A and 3B). After 24 hours of leptin treatment, FAO was much lower in SENP2-knockdown myotubes (Fig. 3C). Furthermore, the leptin-induced increase of mRNA levels for FAO-associated enzymes, including ACSL1 and CPT1b, and E1CP3 was not observed in Senp2 knockdown myotubes (Fig. 3D). These results indicate that SENP2 plays an important role in the leptin-mediated increase in FAO in C2C12 myotubes.
Figure 3. SENP2 plays an important role in leptin-induced FAO in C2C12 myotubes.
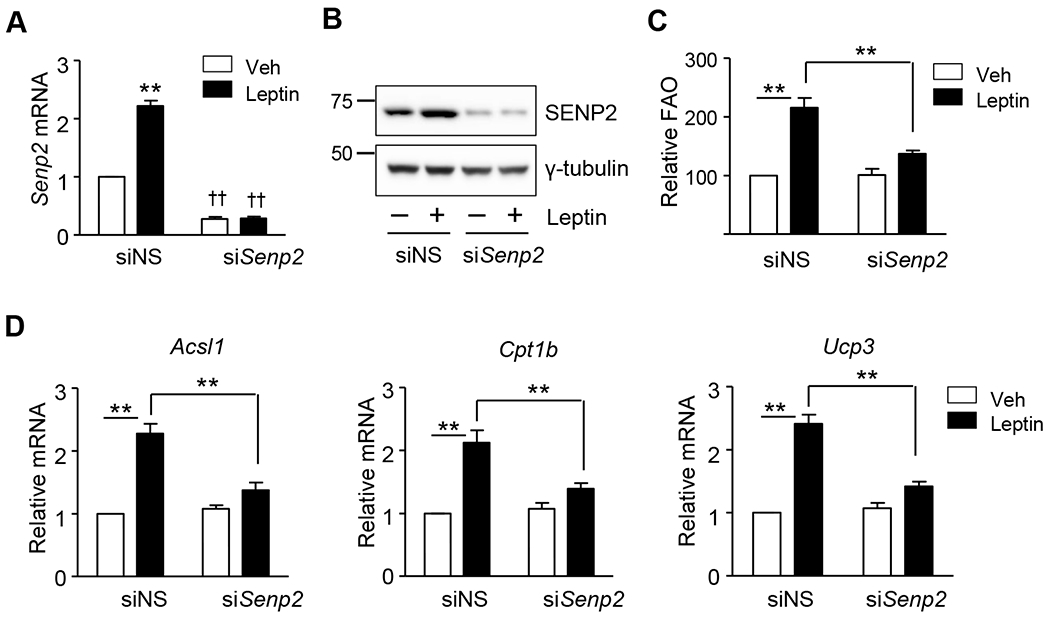
C2C12 myotubes were treated with siRNAs against Senp2 (siSenp2, 50 nmol/1), and the next day leptin was treated for another 24 h. (A) SENP2 knockdown was confirmed by real time PCR (n = 5). **P < 0.01 vs. Veh; ††P < 0.01 vs. siNS. (B) SENP2 knockdown was confirmed by western blot analysis. (C) FAO was measured (n =5). (D) The mRNA levels of Acsl1, Cpt1b and Ucp3 were measured by real time PCR. The mRNA levels of leptin-untreated siNS cells were expressed as 1, and the others were expressed as its relative values (n = 5). *P < 0.05, **P < 0.01. Data are presented as mean ± SEM.
3.4. Leptin induces transcription of FAO-associated enzymes through SENP2-mediated activation of PPARδ and PPARγ.
To confirm that PPARδ and PPARγ are involved in the leptin-induced transcriptional increases of FAO-associated enzymes, Acsl1 promoter activity was measured in the presence or absence of leptin treatment in C2C12 myotubes. A leptin-induced increase in promoter activity was observed for the construct mAcsl1(−1051)-Luc, which contains residues −1051 bp to −45 bp of the mouse Acsl1 gene, but was not observed when a potential PPRE site at −210 bp of this construct was eliminated by mutagenesis (Fig. 4A). Co-transfection with Pparδ and Pparγ expression vectors resulted in an increase in mAcsl1(−105l)-Luc promoter activity. Promoter activity was further enhanced when a Senp2 expression vector was included along with the Pparδ and Pparγ expression vectors (Fig. 4B). Promoter activity was also enhanced by co-transfection with PPARδ and PPARγ expression vectors containing mutations at the SUMO conjugation sites (K104R in PPARδ and K107R in PPARγ). Together, these results suggest that SENP2 mediates deSUMOylation of PPARδ and PPARγ, resulting in increase of the Acsl1 promoter activity.
Figure 4. SENP2 mediates leptin effects through the activation of PPARδ and PPARγ.
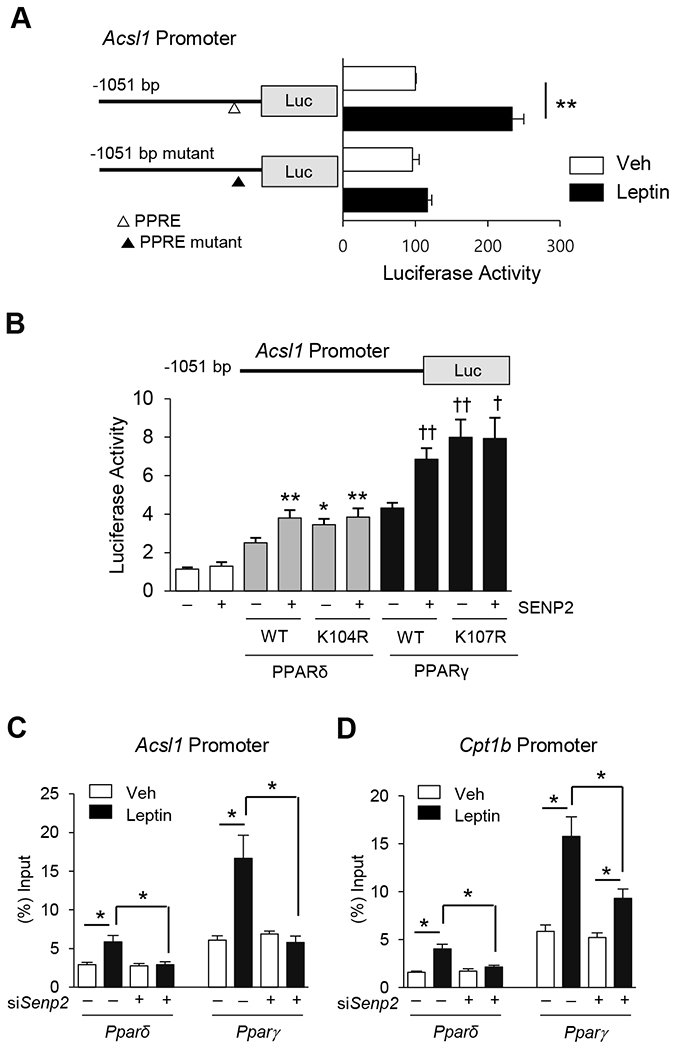
(A) C2C12 myoblasts were transfected with mAcsl1(−1051)-Luc or mAcsl1(−1051)mt-Luc containing mutations at a potential PPRE (−210 bp), and then treated with leptin for 24 h. Luciferase activity of the cells, transfected with mAcsl1(−1051)-Luc without leptin, was expressed as 100, and the others were expressed as its relative values (n = 5). **P < 0.01. △, a PPRE site; ▲, a mutation at the PPRE site. (B) COS7 cells were transfected with one of PPAR expression vectors (100 ng), the SENP2 expression vector (100 ng), and mAcsl1(−1051)-Luc (300 ng) (n = 4). *P < 0.05, **P < 0.01 vs. PPARδ WT without SENP2, †P < 0.05, ††P < 0.01 vs. PAPRγ WT without SENP2. (C, D) C2C12 myotubes were treated with siNS or siSenp2 for 24 h, followed by treatment of leptin for another 24 h, and then ChIP was performed using PPARδ and PPARγ antibodies. Real time PCR was performed with the primers representing the flanking regions of the PPREs in the Acsl1 promoter (C) or Cpt1b promoter (D) (n = 4). *P < 0.05. Data are presented as mean ± SEM.
ChIP coupled with qPCR, in which qPCR was used on the PPRE sites of Acsl1 and Cpt1b promoters that had been immunoprecipitated using PPARδ and PPARγ antibodies, show that leptin increased the affinity of PPARδ and PPARγ to the Acsl1 and Cpt1b promoters. However, SENP2 knockdown abolished or significantly suppressed the leptin-induced binding of PPARδ and PPARγ to the promoters (Fig. 4C and 4D). Together, these results show that leptin increases the binding affinity of PPARδ and PPARγ for the PPRE sites in the Acsl1 or Cpt1b promoters and that these increases mainly resulted from deSUMOylation of PPARδ and PPARγ by SENP2.
3.5. SENP2 mediates a Leptin-induced increase in FAO-associated enzyme expression in soleus muscle in vivo.
Real-time qPCR with leptin receptor long form-specific primers was used to determine LepR expression levels in several types of muscles and in the hypothalamus. Higher levels of LepR mRNAs were observed in soleus muscles compared to the gastrocnemius (GM) or quadriceps muscles (QM). High levels of LepR mRNAs were also observed in C2C12 myotubes originating in the soleus (Fig. 5A). Similar results were obtained using western blot analysis (Supplemental Fig. 3). To test whether leptin treatment activates the leptin receptor/STAT3 signaling pathway in soleus muscle, soleus muscles were isolated and treated with leptin ex vivo. STAT3 phosphorylation was detected between 0.5 and 2 hours after leptin treatment, indicating that leptin triggers STAT3 activation in soleus muscle (Fig. 5B). Next, we measured the expression level of SENP2 in soleus muscles of mice after treatment with leptin (3 mg/kg). An increase in Senp2 mRNA levels was observed 10 hours and sustained until 24 hours after the injection of leptin (Fig. 5C). Additionally, mRNA levels of FAO-associated enzymes ACSL1 and CPT1b increased significantly 24 hours after the leptin treatment (Fig. 5D). Consistently, a 1.8-fold increase in FAO activity was also observed 24 hours after the leptin administration (Fig. 5E). To confirm that the leptin-induced increase in transcription of Senp2 and FAO-associated enzymes is mediated by LepRb, db/db mice, which are LepRb deficient, were treated with leptin. No leptin-induced increase in the mRNA levels of Senp2, Acsl1 and Cpt1b in soleus muscle occurred in these mice, and no increase in FAO was observed (Fig. 5F and 5G). Muscle-specific SENP2 knockout mice (SENP2mKO) were generated by crossing Senp2 floxed mice and MCK-Cre mice. The knockout was confirmed by measuring in the SENP2 protein levels in skeletal muscle (Fig. 5H). A leptin-induced increase in Acsl1 and Cpt1b mRNA levels was not observed in the soleus muscles of SENP2mKO mice, but was observed in the floxed control mice (Fig. 5I). These results suggest that SENP2 plays an important role in leptin-induced FAO in skeletal muscle.
Figure 5. Effects of leptin on the transcription levels of SENP2 and FAO in vivo.
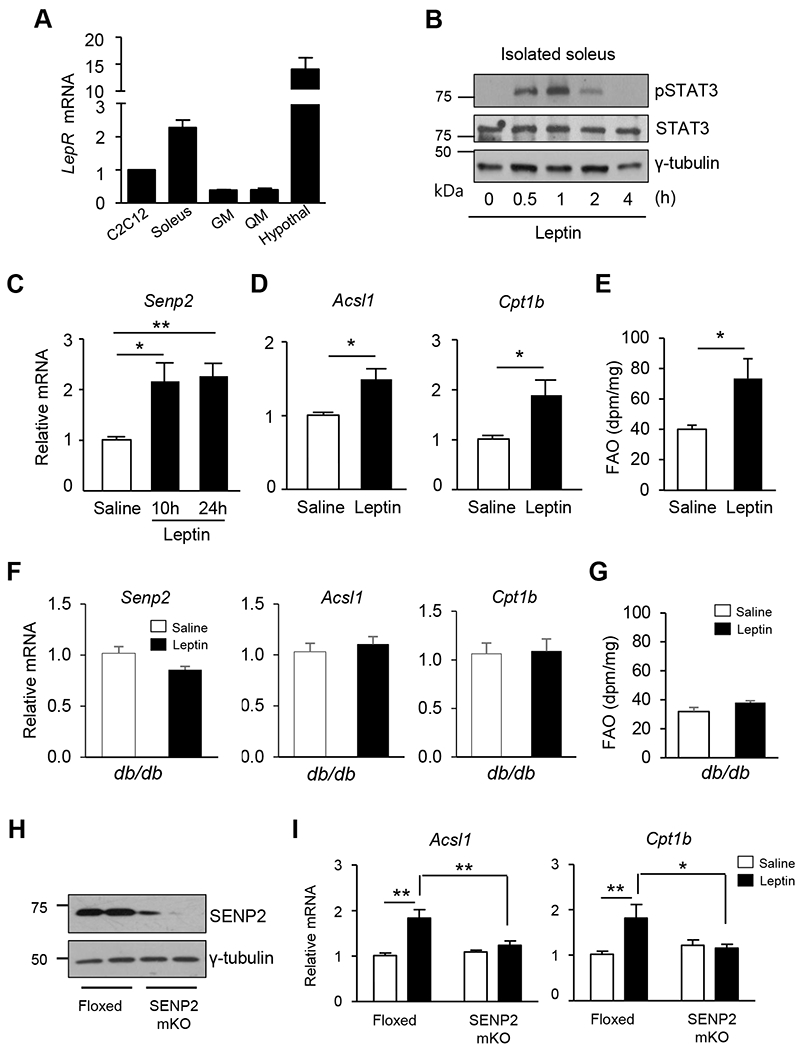
(A) Total RNAs were isolated from C2C12 myotubes or several tissues, and then real time PCR was performed using the primers specific to the leptin receptor long form (primers in the Supplemental table 1). The mRNA levels of the leptin receptor (normalized by Gapdh mRNA) in the C2C12 myotubes were expressed as 1, and the others were expressed as its relative values (n = 3). (B) Soleus muscles were isolated from mice, and then treated with leptin ex vivo. At the different time points after the treatment, muscles were homogenized, and proteins were subjected to immunoblot analysis. (C-E) C57BL/6J mice were treated with leptin (3 mg/kg, i.p.), and then the soleus muscles were isolated 10 h or 24 h after the administration. (C) SENP2 mRNA levels were determined (n = 7). (D) The mRNA levels of Acsl1 and Cpt1b, were determined in the soleus muscles prepared 24 h after the leptin treatment. The mRNA levels of saline-treated mice were set to 1 and the others were expressed as its relative values (n = 7). (E) FAO was measured 24 h after the leptin injection (n = 5). *P < 0.05, **P < 0.01 vs. saline. (F-G) Male db/db mice were treated with leptin for 24 h, and then mRNAs were prepared from soleus muscles. (F) The mRNA levels of Senp2, Acsl1 and Cpt1b were determined using real-time qPCR (n = 8). (G) FAO was measured (n = 5). (H) Tissue lysates from the skeletal muscles of control (Floxed) and SENP2 muscle specific knockout (SENP2mKO) mice were subjected to western blot analysis. (I) 10-12 week-old mice were used for the experiments. Leptin was administered to Floxed or SENP2mKO mice, and total RNAs of soleus muscles were isolated 24 h after the administration. The mRNA levels of saline-treated control mice were expressed as 1 and the others were expressed as its relative values (n =6-9). *P < 0.05, **P < 0.01. Data are presented as mean ± SEM.
3.6. Leptin increases FAO through the leptin receptor by two different pathways.
It has been reported that leptin directly acts on skeletal muscle and increases FAO by AMPK activation [7]. In C2C12 myotubes, leptin induced a reasonably rapid increase in AMPK phosphorylation, occurring within 3 hours of the start of leptin treatment. The increase in AMPK phosphorylation was markedly suppressed with a knockdown of leptin receptor (Fig. 6A). In C2C12 myotubes, leptin-induced AMPK phosphorylation was sustained until 6 hours after the treatment, after which it decreased to the basal level (Fig. 6B). Next, C2C12 myotubes were transfected with siRNAs against Senp2, Ampkα, or both, and then treated with leptin (Supplemental Fig. 4). FAO was measured at various time points after leptin treatment. FAO increased 1.7-fold 6 hours after the start of leptin treatment, and continued to increase until 48 (Fig. 6C). Knockdown of SENP2 did not affect the increase in FAO that was observed at 6 hours, but suppressed the FAO increase that was observed at 24 and 48 hours. In contrast, when AMPKα was knocked down, little stimulation of FAO was observed at 6 hours, but stimulation of FAO was observed at 24 and 48 hours even though the fold induction was lower than that in the siNS-treated group. The results also showed that there were some overlapped effects by the AMPK- and SENP2-mediated pathway on FAO at 24 or 48 hours. Knockdown of both SENP2 and AMPKα eliminated leptin-induced stimulation of FAO (Fig. 6C). These results indicate that in myotubes, leptin increases FAO through the leptin receptor by two different pathways: a rapid, AMPK-mediated pathway and a delayed, SENP2-mediated pathway.
Figure 6. Leptin activates two different pathways through the leptin receptor, resulting in early and delayed effects on FAO.
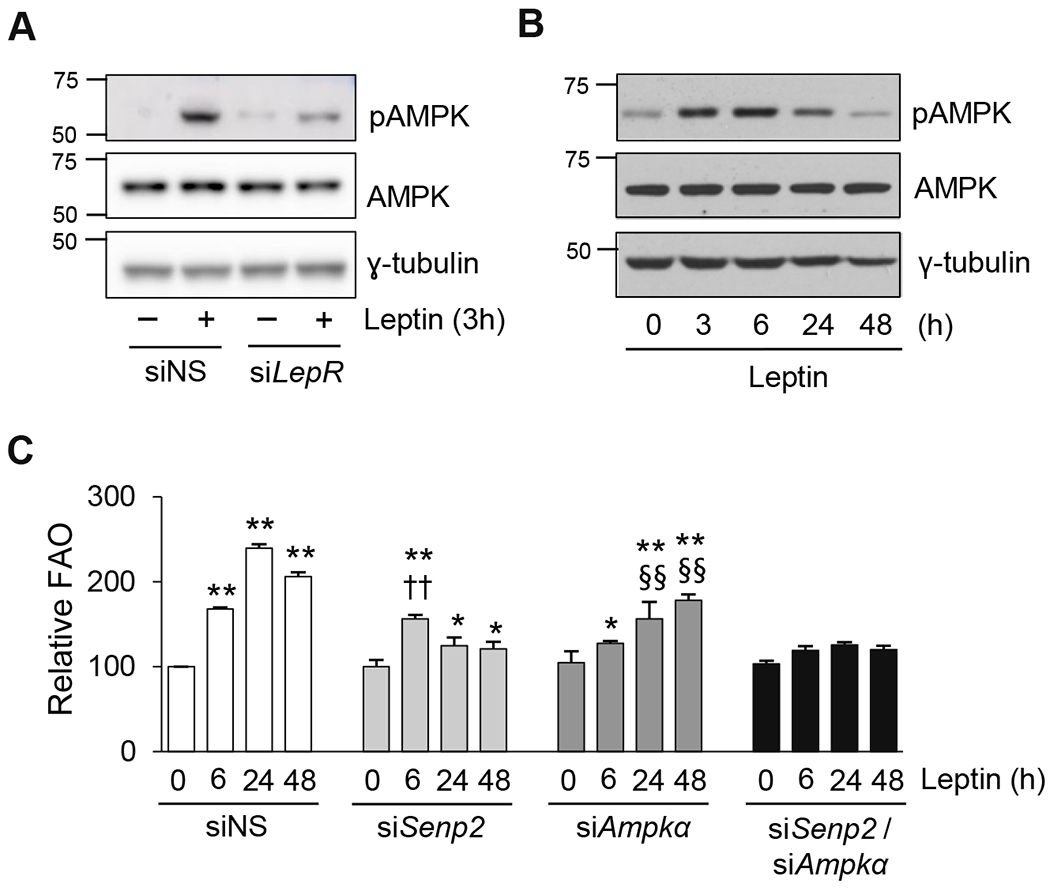
(A) C2C12 myotubes were treated with siNS or siLepR for 24 h, and then treated with leptin for 3 h. Cell lysates were subjected to western blotting using antibodies against phosphorylated AMPK (pAMPK) and total AMPK (AMPK). (B) C2C12 myotubes were treated with leptin, and cell lysates were collected at different time points for western blot analysis. (C) C2C12 myotubes were treated with siNS, siSenp2, siAmpkα, or both siSenp2 and siAmpkα for 24 h, followed by leptin treatment for the indicated periods, and then FAO was measured (n = 4). *P < 0.05, **P < 0.01 vs. siNS without leptin; ††P < 0.01 vs. siSenp2 without leptin; §§P < 0.01 vs. siAmpkα without leptin. Data are presented as mean ± SEM.
4. Discussion
The hypothalamic-sympathetic nervous system-mediated effect of leptin on lipid metabolism in skeletal muscle occurs through AMPK activation after several hours after the leptin injection [7,27]. In addition, peripheral effects of leptin in skeletal muscle have been reported. Early activation of AMPK through direct action of leptin on skeletal muscle that occurs within 15 min after leptin treatment, which results in an increase in FAO (Fig. 7, left panel) [7]. In this study, we show that in myotubes and skeletal muscle, prolonged FAO is mediated by SENP2 in the LepR/STAT3 pathway. The JAK/STAT3 pathway is initiated by leptin binding to the leptin receptor in muscle, which induces phosphorylation of STAT3, thus causing STAT3 to translocate to the nucleus where it binds to the Senp2 promoter and increases Senp2 expression. The SENP2 protein deSUMOylates PPARδ and PPARγ, and these two proteins, in turn, stimulate transcription of FAO-associated genes such as Ascl1 and Cpt1b (Fig. 7, right panel). Based on our results, we propose that in skeletal muscle, leptin causes a rapid increase in FAO that is mediated by an AMPK phosphorylation pathway, and a slower, sustained increase in FAO that is mediated by the STAT3/SENP2 pathway (Fig. 7). As mentioned earlier, leptin also increases FAO in skeletal muscle indirectly through the hypothalamic-nervous system in vivo, which occurs after the early direct activation of AMPK. Because leptin activates several pathways at different time intervals, leptin-stimulated FAO may continue for a long period of time in skeletal muscle. Further study will be required to determine the overall contribution of each of the pathways to FAO in skeletal muscle.
Figure 7. A diagram of supposed leptin-induced peripheral signaling pathways in skeletal muscle.
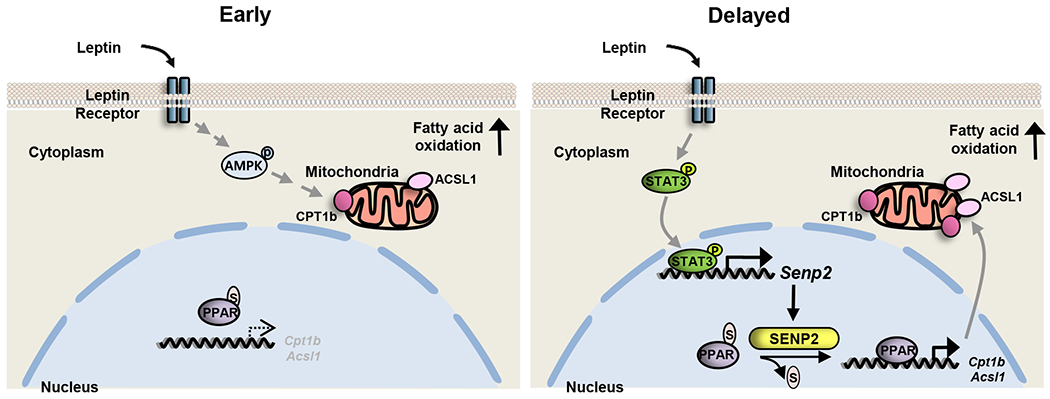
Leptin directly activates two different pathways through the leptin receptor, the AMPK and the STAT3/SENP2 pathways, resulting in fatty acid oxidation increase in skeletal muscle. P, phosphate; S, SUMO.
Previous studies have demonstrated that AMPK increases PGC1α expression and regulates mitochondria biogenesis [28–30]. Here we show that knockdown of AMPKα causes a decrease in the mRNA levels of Pgc1α1 and Pgc1α4 (about 20 % and 40 % decrease, respectively) (Supplemental Fig. 5A), but has little effect on expression of genes related to oxidative phosphorylation (supplemental Fig. 5B and 5C). Furthermore, knockdown of AMPKα or SENP2 did not alter the basal FAO levels in the absence of leptin (Fig. 6C), indicating that knockdown of these genes did not change mitochondrial capacity for FAO.
The SENP2-mediated increase in FAO that we observed here may not occur in all muscle types. It has been reported that among the leptin receptor isoforms, only LepRb has a STAT3 binding site and induces STAT3 activation [31,32]. LepRb was more highly expressed in soleus than in GM or QM (Fig. 5A and Supplemental Fig. 3), indicating that SENP2-mediated FAO increase may be smaller in these other kinds of muscle. The stimulation of FAO in soleus muscle may not cause a great change in whole body lipid metabolism. However, it is possible that leptin activates the SENP2-mediated signaling pathway in other tissues, such as liver and adipose tissue, containing the leptin receptor.
Although previous studies have suggested that Acsl1 mRNA levels are regulated by PPARα and PPARγ [33,34], a PPRE in the mouse Acsl1 promoter was not identified. In this study, we show that a PPRE sequence exists at around −210 bp in the mouse Acsl1 promoter (Fig. 4A, 4B and 4C). Previous studies have shown that ACSL1 is the primary ACSL isoform responsible for stimulating FAO [35,36], suggesting that ACSL1 is an important FAO regulator in addition to the more well-known FAO regulator, CPT1b. DeSUMOylation of PPARδ or PPARγ increases the affinity of these proteins to the PPRE site in the promoters of FAO-associated genes, and may also result in recruitment of a coactivator(s) such as p300 that increase transcription of the target genes [23,24]. Among SUMO-specific proteases, SENP2 has the highest PPAR deSUMOylation activity. However, PPAR SUMOylation can be regulated by other factors such as a SUMO E3 ligase. Further study is necessary to determine the role of PPARδ and PPARγ SUMOylation in whole body lipid metabolism.
We have previously shown that SENP2 transcription is regulated by free fatty acids through the TLR4/Myd88/NFkB signaling pathway in skeletal muscle [24]. The present study shows that leptin also regulates SENP2 expression and leads to FAO in skeletal muscle. In conclusion, we suggest that SENP2 is a sensor for elevated levels of circulating fatty acids and leptin, and SENP2-induced PPAR desSUMOylation is an important step in the regulation of lipid metabolism in skeletal muscle. Therefore, regulation of SENP2 activity in skeletal muscle can be a therapeutic target for alleviation of obesity or obesity-related metabolic syndromes.
Supplementary Material
Acknowledgements
This research was supported by grants from the BK21Plus Program of the National Research Foundation of Korea (NRF) funded by the Ministry of Education (10Z20130000017), NRF-2016R1A2B3010373, the National Institutes of Health (1R01DK083567 to Y.B.K.), and AstraZeneca. All authors of the manuscript have declared that no conflict of interest exists.
Abbreviations:
- ACC
acetyl-CoA carboxylase
- ACSL
long-chain acyl-CoA synthetase
- AMPK
AMP-activated protein kinase
- ChIP
chromatin immunoprecipitation
- CPT
carnitine palmitoyl transferase
- FAO
fatty acid oxidation
- GM
gastrocnemius muscle
- MCAD
medium chain acyl-CoA dehydrogenase
- PPAR
peroxisome proliferator-activated receptor
- PPRE
PPAR response element
- QM
quadriceps muscle
- SENP
SUMO specific protease
- SUMO
small ubiquitin like modifier
- STAT
signal transducer and activator of transcription
- UCP
uncoupling protein
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Pelleymounter MA, Cullen MJ, Baker MB, Hecht R, Winters D, et al. (1995) Effects of the obese gene product on body weight regulation in ob/ob mice. Science 269: 540–543. [DOI] [PubMed] [Google Scholar]
- 2.Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, et al. (1994) Positional cloning of the mouse obese gene and its human homologue. Nature 372: 425–432. [DOI] [PubMed] [Google Scholar]
- 3.Meister B (2000) Control of food intake via leptin receptors in the hypothalamus. Vitam Horm 59: 265–304. [DOI] [PubMed] [Google Scholar]
- 4.Shiuchi T, Toda C, Okamoto S, Coutinho EA, Saito K, et al. (2017) Induction of glucose uptake in skeletal muscle by central leptin is mediated by muscle beta2-adrenergic receptor but not by AMPK. Sci Rep 7: 15141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perry RJ, Wang Y, Cline GW, Rabin-Court A, Song JD, et al. (2018) Leptin Mediates a Glucose-Fatty Acid Cycle to Maintain Glucose Homeostasis in Starvation. Cell 172: 234–248 e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marti A, Berraondo B, Martinez JA (1999) Leptin: physiological actions. J Physiol Biochem 55: 43–49. [PubMed] [Google Scholar]
- 7.Minokoshi Y, Kim YB, Peroni OD, Fryer LG, Muller C, et al. (2002) Leptin stimulates fatty-acid oxidation by activating AMP-activated protein kinase. Nature 415: 339–343. [DOI] [PubMed] [Google Scholar]
- 8.Park HK, Ahima RS (2014) Leptin signaling. F1000Prime Rep 6: 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Morris DL, Rui L (2009) Recent advances in understanding leptin signaling and leptin resistance. Am J Physiol Endocrinol Metab 297: E1247–1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bahrenberg G, Behrmann I, Barthel A, Hekerman R Heinrich PC, et al. (2002) Identification of the critical sequence elements in the cytoplasmic domain of leptin receptor isoforms required for Janus kinase/signal transducer and activator of transcription activation by receptor heterodimers. Mol Endocrinol 16: 859–872. [DOI] [PubMed] [Google Scholar]
- 11.Bjorbaek C, Uotani S, da Silva B, Flier JS (1997) Divergent signaling capacities of the long and short isoforms of the leptin receptor. J Biol Chem 272: 32686–32695. [DOI] [PubMed] [Google Scholar]
- 12.Leshan RL, Bjornholm M, Munzberg H, Myers MG Jr. (2006) Leptin receptor signaling and action in the central nervous system. Obesity (Silver Spring) 14 Suppl 5: 208S–212S. [DOI] [PubMed] [Google Scholar]
- 13.Sivitz WI, Walsh SA, Morgan DA, Thomas MJ, Haynes WG (1997) Effects of leptin on insulin sensitivity in normal rats. Endocrinology 138: 3395–3401. [DOI] [PubMed] [Google Scholar]
- 14.Muoio DM, Dohm GL, Fiedorek FT Jr., Tapscott EB, Coleman RA (1997) Leptin directly alters lipid partitioning in skeletal muscle. Diabetes 46: 1360–1363. [DOI] [PubMed] [Google Scholar]
- 15.Winder WW, Hardie DG (1999) AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol 277: E1–10. [DOI] [PubMed] [Google Scholar]
- 16.Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, Wakil SJ (2001) Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 291: 2613–2616. [DOI] [PubMed] [Google Scholar]
- 17.Momken I, Chabowski A, Dirkx E, Nabben M, Jain SS, et al. (2017) A new leptin-mediated mechanism for stimulating fatty acid oxidation: a pivotal role for sarcolemmal FAT/CD36. Biochem J 474: 149–162. [DOI] [PubMed] [Google Scholar]
- 18.Akasaka Y, Tsunoda M, Ide T, Murakami K (2009) Chronic leptin treatment stimulates lipid oxidation in immortalized and primary mouse skeletal muscle cells. Biochim Biophys Acta 1791: 103–109. [DOI] [PubMed] [Google Scholar]
- 19.Johnson ES (2004) Protein modification by SUMO. Annu Rev Biochem 73: 355–382. [DOI] [PubMed] [Google Scholar]
- 20.Hay RT (2005) SUMO: a history of modification. Mol Cell 18: 1–12. [DOI] [PubMed] [Google Scholar]
- 21.Yeh ET (2009) SUMOylation and De-SUMOylation: wrestling with life’s processes. J Biol Chem 284: 8223–8227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hickey CM, Wilson NR, Hochstrasser M (2012) Function and regulation of SUMO proteases. Nat Rev Mol Cell Biol 13: 755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chung SS, Ahn BY, Kim M, Kho JH, Jung HS, et al. (2011) SUMO modification selectively regulates transcriptional activity of peroxisome-proliferator-activated receptor gamma in C2C12 myotubes. Biochem J 433: 155–161. [DOI] [PubMed] [Google Scholar]
- 24.Koo YD, Choi JW, Kim M, Chae S, Ahn BY, et al. (2015) SUMO-Specific Protease 2 (SENP2) Is an Important Regulator of Fatty Acid Metabolism in Skeletal Muscle. Diabetes 64: 2420–2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chung SS, Ahn BY, Kim M, Choi HH, Park HS, et al. (2010) Control of adipogenesis by the SUMO-specific protease SENP2. Mol Cell Biol 30: 2135–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jorgensen SB, Viollet B, Andreelli F, Frosig C, Birk JB, et al. (2004) Knockout of the alpha2 but not alpha1 5′-AMP-activated protein kinase isoform abolishes 5-aminoimidazole-4-carboxamide-1-beta-4-ribofuranosidebut not contraction-induced glucose uptake in skeletal muscle. J Biol Chem 279: 1070–1079. [DOI] [PubMed] [Google Scholar]
- 27.Minokoshi Y, Kahn BB (2003) Role of AMP-activated protein kinase in leptin-induced fatty acid oxidation in muscle. Biochem Soc Trans 31: 196–201. [DOI] [PubMed] [Google Scholar]
- 28.Terada S, Goto M, Kato M, Kawanaka K, Shimokawa T, et al. (2002) Effects of low-intensity prolonged exercise on PGC-1 mRNA expression in rat epitrochlearis muscle. Biochem Biophys Res Commun 296: 350–354. [DOI] [PubMed] [Google Scholar]
- 29.Jager S, Handschin C, St-Pierre J, Spiegelman BM (2007) AMP-activated protein kinase (AMPK) action in skeletal muscle via direct phosphorylation of PGC-1alpha. Proc Natl Acad Sci U S A 104: 12017–12022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okamoto S, Asgar NF, Yokota S, Saito K, Minokoshi Y (2019) Role of the alpha2 subunit of AMP-activated protein kinase and its nuclear localization in mitochondria and energy metabolism-related gene expressions in C2C12 cells. Metabolism 90: 52–68. [DOI] [PubMed] [Google Scholar]
- 31.Chen H, Charlat O, Tartaglia LA, Woolf EA, Weng X, et al. (1996) Evidence that the diabetes gene encodes the leptin receptor: identification of a mutation in the leptin receptor gene in db/db mice. Cell 84: 491–495. [DOI] [PubMed] [Google Scholar]
- 32.Banks AS, Davis SM, Bates SH, Myers MG Jr. (2000) Activation of downstream signals by the long form of the leptin receptor. J Biol Chem 275: 14563–14572. [DOI] [PubMed] [Google Scholar]
- 33.Dunning KR, Anastasi MR, Zhang VJ, Russell DL, Robker RL (2014) Regulation of fatty acid oxidation in mouse cumulus-oocyte complexes during maturation and modulation by PPAR agonists. PLoS One 9: e87327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clemenz M, Frost N, Schupp M, Caron S, Foryst-Ludwig A, et al. (2008) Liver-specific peroxisome proliferator-activated receptor alpha target gene regulation by the angiotensin type 1 receptor blocker telmisartan. Diabetes 57: 1405–1413. [DOI] [PubMed] [Google Scholar]
- 35.Li LO, Grevengoed TJ, Paul DS, Ilkayeva O, Koves TR, et al. (2015) Compartmentalized acyl-CoA metabolism in skeletal muscle regulates systemic glucose homeostasis. Diabetes 64: 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ellis JM, Li LO, Wu PC, Koves TR, Ilkayeva O, et al. (2010) Adipose acyl-CoA synthetase-1 directs fatty acids toward beta-oxidation and is required for cold thermogenesis. Cell Metab 12: 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.