

Abstract
We tested the activity of a p53 carboxy-terminal peptide containing the PARC-interacting region in cancer cells with wild type cytoplasmic p53. Peptide delivery was achieved by fusing it to the TAT transduction domain (TAT-p53-C-ter peptide). In a two-hybrid assay, the tetramerization domain (TD) of p53 was necessary and sufficient to bind PARC. The TAT-p53-C-ter peptide disrupted the PARC-p53 complex. Peptide treatment caused p53 nuclear relocation, p53-dependent changes in gene expression and enhancement of etoposide-induced apoptosis. These studies suggest that PARC-interacting peptides are promising candidates for the enhancement of p53-dependent apoptosis in tumours with wt cytoplasmic p53.
Keywords: p53, PARC, TAT, apoptosis, etoposide
Introduction
In approximately 50 % of all malignancies, the normal activity of p53 is compromised by inactivating mutations [1]. In specific tumours, p53 is present in the wild type form but its activity is prevented by inappropriate cellular localization [2].
Recently, PARC has been described as a p53-interacting protein involved in the p53 cytoplasmic sequestration that occurs in certain tumours [3]. PARC functions as ubiquitin ligase and forms a multiprotein complex of approximately 1 million kDa in the cytoplasm [3]. p53 is not a substrate for ubiquitination by PARC, although the C-terminus of p53 binds to a well defined N-terminal portion of PARC termed CPH [4]. Expression of PARC is ubiquitous, but high expression has been described in some tumour types [3].
Several approaches based on the use of small molecules [5] and peptides [6] have been used to reactivate p53 function in tumours. A peptide derived from the C-terminus of p53 (C1 peptide) previously shown to activate specifically p53 DNA binding in vitro by an unknown mechanism was used in pre-clinical models of peritoneal carcinomatosis [7]. Striking reduction of metastatic burden or complete eradication of tumours was achieved by this strategy, demonstrating the feasibility of p53 reactivation even in cases of advanced cancer.
The TAT protein transduction domain is a short stretch of 11 aminoacids that renders cell-permeable virtually any peptide [8]. The TAT sequence was identified in the HIV protein Tat and other short sequences with analogous capability were recognized in other organisms [8]. Rapid and efficient TAT-mediated intracellular delivery was demonstrated utilizing in vitro [9]and in vivo systems [8].
In this paper, we attempted p53 reactivation in tumour cells harbouring cytoplasmic wild type p53 and expressing PARC by using a peptide containing the C-terminus of p53 fused with the TAT transduction domain. We show that this peptide promotes the relocation of endogenous p53 into the nucleus by disrupting its binding to PARC. p53 relocation results, in turn, in the activation of p53 transcription targets and in the enhancement of apoptosis caused by genotoxic stress.
Materials and Methods
Cell culture:
Human neuroblastoma (NB) cell lines HTLA-230, KCNR, SK-N-BE2c, SK-N-BE, LAN-5, GI-CA-N, SH-EP, SK-N-AS, RN-GA and SY-5Y(N) were cultured as described [10]; 32D-BCR/ABL PG13 and 32D-BCR/ABL MG15 [11; 12] were grown in Iscove’s Modified D-MEM medium supplemented with 10% Fetal Calf Serum (FCS). Phoenix cells [13] were grown in D-MEM with 10% FCS.
Protein analysis.
Cellular proteins were extracted, and analyzed by western blotting as described [10]. Antibodies used were: anti-Bax (N-20) (Santa Cruz Biotechnology, CA, U.S.A.); anti-β actin (AC-15) (Sigma-Aldrich, St. Louis, MO, USA); anti-p21 SC-756 (Santa Cruz); anti-MDM2 SC-965 (Santa Cruz); anti-HSP-70 (SPA-820) (Stressgen Bioreagents Corp., Victoria, BC , Canada); anti-HA (Covance, Denver, PA, USA)
Peptide preparation and treatments:
The p53-C-ter sequence was cloned into the prokaryotic expression vector pTAT-HA [14] by RT-PCR using the primers:
p53-C-ter forward: (5’-CTCGAGCTCGAGCGCAAGAAAGGGGAGCCTCA-3’) and
p53-C-ter reverse (5’-GAATTCGAATTCTCAGTCTGAGTCAGGCCCTTC-3’) to amplify a cDNA sequence encoding amino acids 290–393 of the human p53 protein.
The amplification product was cloned in the TAT-HA plasmid. After purification, recombinant TAT-p53-C-ter was administered directly into culture media.
siRNA treatment:
U2OS cells were transfected with validated p53 siRNA or control siRNA (QIAGEN, Valencia, CA, U.S.A.) at 5 nM concentration utilizing HiPerFect Reagent (QIAGEN). After 48 hours, medium was changed and cells were left untreated or treated with the TAT-p53-C-ter peptide at the indicated concentrations for 2 hours.
Luciferase Assays:
Assays were carried out with Dual-Glo kit (Promega, Madison, WI, USA) using Wallac Victor 1420 plate reader (Perkin Elmer, Waltham, MA, USA)
Caspase activity assays:
U2OS cells were analyzed using CaspaseGlo 3/7 kit (Promega) according to the instructions. Readings were carried out using Wallac Victor 1420 plate reader (Perkin Elmer)
Flow cytometry:
Cells were harvested, fixed and stained with Propidium Iodide according to standard procedures. Samples were analyzed using a FACScalibur flow cytometer (BD Biosciences, San Jose, CA, USA). Cell cycle distribution was calculated by Cell Quest software (BD Biosciences).
Immunocytochemistry:
Immunocytochemistry was performed according to standard procedures. Primary antibodies were : anti-HA (y-11) (Santa Cruz) and anti-p53 (DO-1) (Santa Cruz). Secondary antibodies were: anti-mouse (Alexa Fluor 488) and anti-rabbit (Alexa Fluor 568) (Molecular Probes, Eugene, Oregon, USA). Analysis was performed utilizing a confocal Nikon EZ-C1 microscope supported by EZ-C1 software (Nikon, Chiyoda-ku, Tokyo, Japan).
PARC-p53 binding assay.
PARC-p53 binding was assessed by the Checkmate system (Promega). The human PARC CPH domain was cloned into pFN10A vector (pFN10A-CPH-PARC). p53-C-ter and its truncated forms ΔC1, ΔTD and ΔNLS were cloned into pFN11A vector after PCR amplification with the Pfu enzyme (Promega), using these primers:
CPH-PARC forward: (5’-ATTGGCGATCGCCCCCAGAAGACAAGGGTGGGT-3’)
CPH-PARC reverse: (5’-TGGTGTTTAAACATCCTCAGTGGCTTCCTCAG-3’)
Common p53-C-ter forward:
(5’-TCTCGCGATCGCCCGCAAGAAAGGGGAGCCTCACCA-3’)
p53-C-ter reverse: (5’-GTCGGTTTAAACGTCTGAGTCAGGCCCTTCTGTCTT-3’)
Reverse ΔC1: (5’-GACGGTTTAAACCCCCCCTGGCTCCTTCCCAGC-3’)
Reverse ΔTD: (5’-GGCGGTTTAAACTCCATCCAGTGGTTTCTTCTTTGGCT-3)
Reverse ΔNLS: (5’-GGCGGTTTAAACGTTGTTGGGCAGTGCTCGCT-3’)
Clonings were carried out according to the manufacturer instructions. Each plasmid was checked by dideoxy-sequencing. Luminometric assays were carried out using Wallac Victor 1420 plate reader (Perkin Elmer).
Real-time PCR.
Total RNA was prepared by RNAwiz (Ambion, Austin, TX, USA). Retro-transcribed cDNA was used for real-time quantitative PCR to detect MDM-2 using the human MDM2 forward (5’-CTATTGGAAATGCACTTCATGCA-3’) and human MDM2 reverse (5’-CGAAGGGCCCAACATCTGT-3’) primers and Sybr-Green master mix (Applied Biosystems, Foster City, CA, USA)
Results and Discussion
Approximately 50% of all human tumours bear a mutation of p53 that prevents its normal function [1]. In most NB tumours and cell lines, p53 is not mutated but frequently misplaced in the cytoplasm especially in the undifferentiated forms [2]. In the U2OS osteosarcoma cells, p53 is also wt and resides in the cytoplasm [3]. Recently, PARC has been described as a cytoplasm-localized protein that binds p53 and, in some cellular contexts, prevents p53 from entering the nucleus [3]. We assessed PARC expression in 10 human NB cell lines, in Phoenix and in U2OS cells by western blotting (Fig. 1A). PARC protein was detectable in 7 of 10 NB cell lines, in U2OS and only barely in Phoenix cells. p53 is wt in all neuroblastoma lines except in SK-N-BE2c and SK-N-AS [15; 16]. We also tested the ability of PARC protein to bind p53 in LAN-5 cells by immunoprecipitation. As expected, p53 coimmunoprecipitated with PARC (Fig. 1B). We reasoned that in NB cells with wt p53 and PARC expression it may be possible to reactivate p53 by disrupting the p53-PARC interaction. Such interaction requires the NH2 portion of PARC and the C-terminus of p53 [17]. The C-terminus of human p53 encoding amino acids 292–393 was cloned in-frame with the 6-histidine tag, an HA tag and a TAT sequence corresponding to 11 amino acids of the TAT protein [8] in a prokaryotic expression vector. The TAT sequence confers cell permeability to proteins to which is fused. The TAT-containing p53 (TAT-p53-C-ter) peptide was tested for its ability to enter into NB cells. The TAT-p53-C-ter peptide was detected intracellularly in a dose-dependent manner when added to cells in a concentration range of 0.5 to 10 μg/ml (Fig. 2A). Peptide stability was evaluated by adding the TAT-p53-C-ter peptide to the cells (5 μg/ml) for 1 hour and removing it thereafter. Cells were harvested at different times and the TAT-p53-C-ter peptide detected by western blotting. Levels of TAT-p53-C-ter peptide were markedly decreased at 1 and 2 hours and undetectable at 4 hours (Fig. 2A), consistent with a short half life.
Fig. 1.
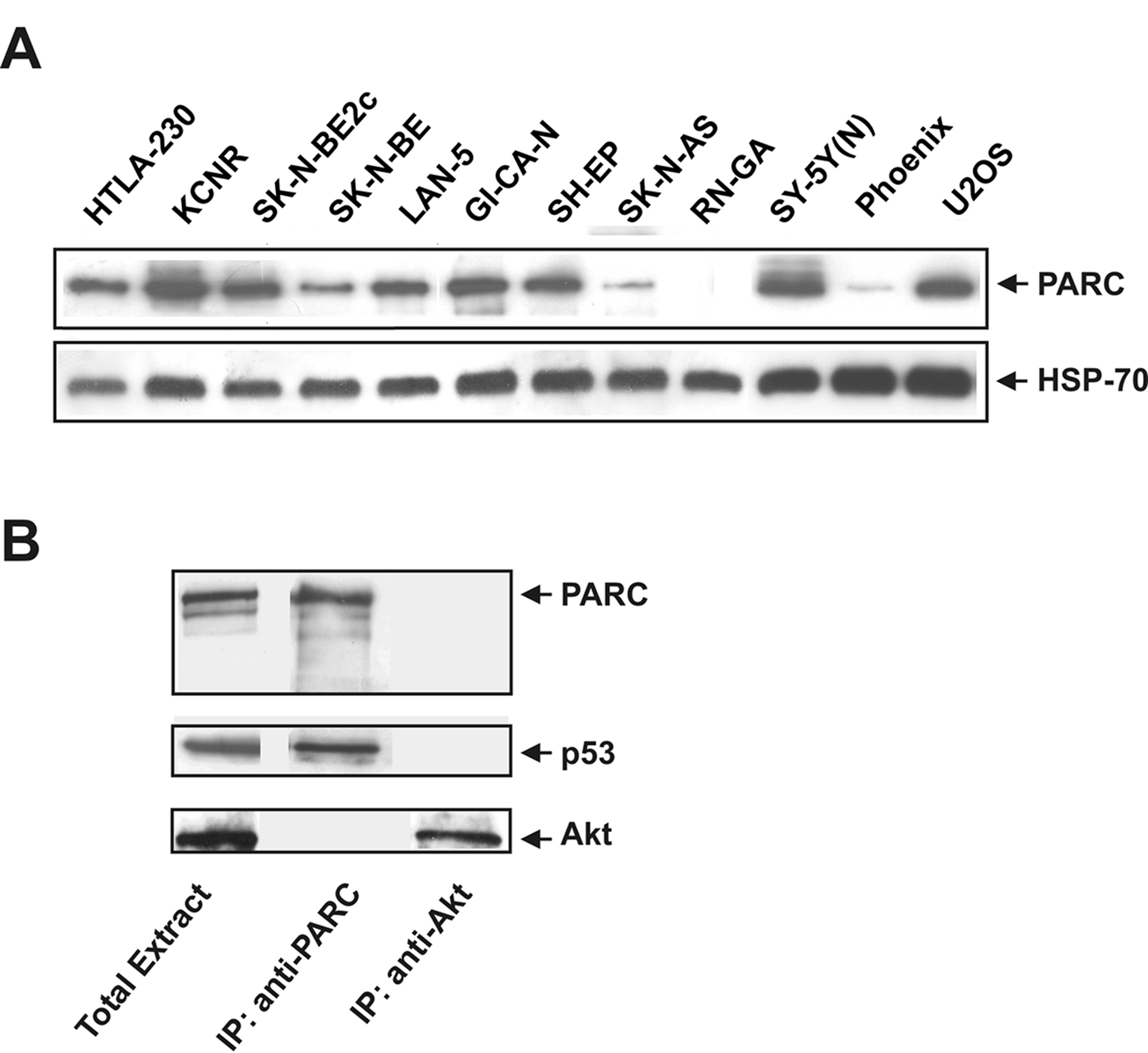
A: endogenous PARC expression in U2OS, Phoenix and 10 human NB cell lines. HSP-70 was used for normalization. B: p53 coimmunoprecipitates with PARC. PARC was immunoprecipitated from LAN-5 cell lysate using an anti-PARC antibody. Control immunoprecipitation was performed using an anti-Akt antibody. Western blots were performed on immunoprecipitates to detect PARC, p53 and Akt.
Fig. 2.
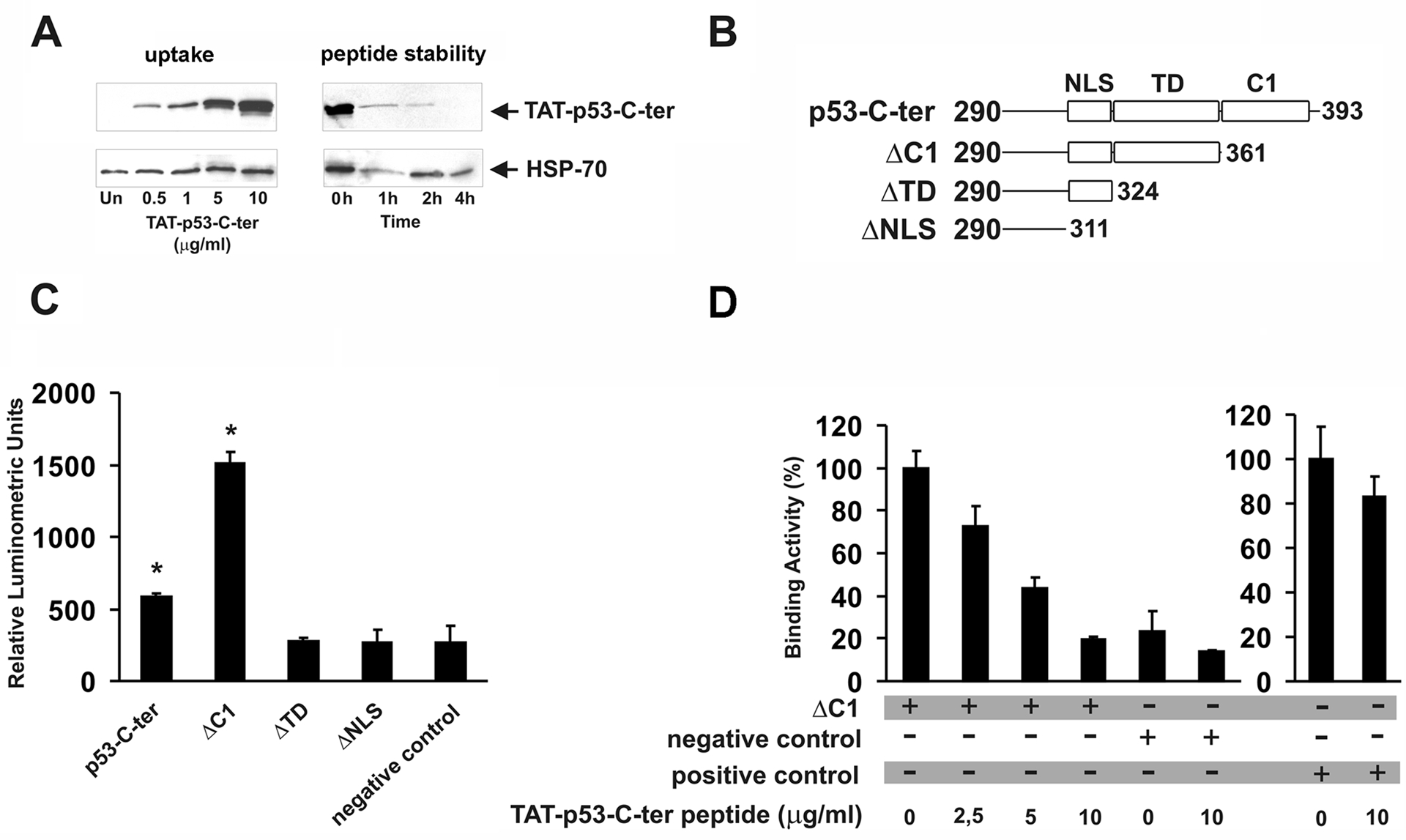
A: dose-dependent uptake of the TAT-p53-C-ter peptide. Cells were treated for 1 hour with the indicated peptide amounts; intracellular TAT-p53-C-ter peptide stability in cells treated with medium containing 10 μg/ml peptide for 1 hour (0h) and in cells cultured for the indicated times in peptide-free medium. In A, peptides were detected with an anti-HA antibody. HSP-70 content was used for normalization. Blots are representative of two different experiments with similar results. B: representation of p53 peptides cloned in the pFN11A vector. NLS= Nuclear Localization Signal; TD= Tetramerization Domain; C1= C1 peptide. Numbers refer to the aminoacids of human p53 protein cloned in each plasmid. C: PARC/p53 binding assay. Phoenix cells were transfected with the reporter vector pGL4.31, pFN10A-CPH-PARC and the pFN11A plasmids containing the p53 peptides described in B. Thirty-six hours after transfection, luciferase activity was determined. Negative control was carried out by transfecting Phoenix cells with pGL4.31 and pACT and pBIND non-interacting vectors. D: Phoenix cells were transfected with pGL4.31, pFN10A-CPH-PARC and pFN11A-ΔC1 plasmids and treated for 6 hours with the indicated amounts of TAT-p53-C-ter peptide (administration every 2 hours) before luminometric detection. Binding activity of untreated cells was taken as 100. Negative control experiments were designed by transfecting Phoenix cells with pGL4.31 and pACT and pBIND non-interacting vectors. As positive control (right histogram), Phoenix cells were transfected with pGL4.31 and pACT-MyoD and pBIND-Id interacting vectors. Binding activity of positive control left untreated was taken as 100. Samples in C and D were run in triplicate. Values ± SD are reported. Asterisks indicate statistically significant differences (p<0.05) compared to negative control. Experiments were repeated twice with similar results.
TAT-p53-C-ter peptide can displace the binding between the CPH domain of PARC and the tetramerization domain of p53.
To determine whether the TAT-p53-C-ter peptide disrupts the physical interaction of p53 and PARC, we used a mammalian two-hybrid system. The CPH domain of human PARC that is necessary for binding p53 [17] was cloned in -frame with the VP16 transactivation domain in the expression vector vector pFN10A. The p53-C-ter peptide and portions of the same lacking the C1 peptide region (ΔC1), the tetramerization domain (ΔTD) and the nuclear localization signal (ΔNLS) were cloned in- frame with the GAL4 DNA binding domain in the vector pFN11A (see Fig. 2B). We co-transfected Phoenix cells with the PARC-CPH- containing vector and each of the p53 plasmids together with a luciferase reporter vector (pGL4.31) in which the promoter region has multiple GAL4 binding sites. Only the p53 peptides containing the tetramerization domain were able to interact with the CPH domain of PARC and promote transcription from the reporter (Fig. 2C). Of interest, ΔC1 peptide in which the C-terminal region downstream of the TD domain was deleted interacted more efficiently with CPH. The deletion could affect binding efficiency by destabilizing the peptide and/or by causing a steric hindrance which renders more difficult the physical contact between the TD and CPH domains. Furthermore, we tested the ability of the TAT-p53-C-ter peptide to inhibit the physical interaction between PARC-CPH and the tetramerization domain of p53. To this end, we performed luciferase assays in cells transfected with ΔC1, PARC-CPH and the reporter vector pGL4.31, and exposed to pulsed administrations (1 every 2 hours) of TAT-p53-C-ter peptide at increasing concentrations (2.5, 5.0 and 10.0 μg/ml). Luciferase activity was reduced by the TAT-p53-C-ter peptide in a dose-dependent manner, indicating that it was able to interfere with the binding between PARC-CPH and p53 (Fig. 2D). This interference was specific for PARC/p53 interaction since binding between MyoD and Id was not decreased by treatment with the TAT-p53-C-ter peptide (Fig. 2D). To our knowledge, this is the first demonstration that a p53-C-ter peptide can displace the binding of p53 to PARC. Of interest, our assay could be extremely useful to screen other peptides or small molecules for their ability to interfere with PARC/p53 interaction.
Treatment with the TAT-p53-C-ter peptide stimulates transcription from p53-dependent promoters.
Myeloid precursor 32D-BCR/ABL cells [11] stably transfected with a plasmid in which the luciferase gene is under the control of wt or mutated p53 binding sites (32D-BCR/ABL PG13 and 32D-BCR/ABL MG15, respectively) were treated with 5 μg/ml of the TAT-p53-C-ter peptide every 2 hours for 6 hours. Transcription was increased by approximately 3-fold in 32D- BCR/ABL PG13 (wt p53 binding sites) but not in 32D-BCR/ABL MG15 (mutated p53 binding sites) cells (Fig. 3A). As positive control, cells were treated with 0.2 μg/ml doxorubicin (Fig. 3A). Similarly, co-transfection of NB LAN-5 cells with a mammalian expression vector encoding the p53-C-ter peptide and with the reporter vector PG13 containing a promoter with multiple p53-binding sites revealed that the p53-C-ter peptide was able to stimulate p53-dependent transcription (Fig. 3B).
Fig. 3.

A: 32D-BCR/ABL myeloid precursor cells stably transfected with reporter vector PG13 or MG15 (luciferase gene driven by a promoter with wt or mutated p53 binding sites respectively) were treated every 2 hours for 6 hours with 5 μg/ml of TAT-p53-C-ter peptide. As positive control, cells were treated for 16 hours with 0.2 μg/ml doxorubicin. B: human NB LAN-5 cells were co-transfected with reporter vectors PG13 or MG15 and expression vector pcDNA3-p53-C-ter-HA. Luciferase activity was determined 36 hours after transfection. As positive control, cells transfected with PG13 and empty vector pcDNA3 were treated for 16 hours with 2 μg/ml doxorubicin. To evaluate additive effects of the p53-C-ter peptide and doxorubicin, cells transfected with PG13 and pcDNA3-p53-C-ter-HA were treated for 16 hours with doxorubicin at the same concentration indicated above. Samples were run in triplicate. Values ± SD are reported. C: Cellular localization of endogenous p53 and TAT-p53-C-ter peptide. U2OS cells, untreated (a, b, c, d), or treated for 1 hour with 10 μg/ml TAT-p53-C-ter peptide (e, f, g and h), or for 1 hour with the peptide followed by 1 hour in peptide-free medium (i, j, k, l), were fixed and processed for immunodetection of endogenous p53 (b, f and j) or the TAT-p53-C-ter peptide (c, g and k). Nuclei were stained with DAPI (a, e and i). Merged fluorescences are shown in d, h and l. Inset in d represents a negative control carried out by treating the cells only with secondary antibodies.
To visualize the subcellular localization of p53 after treatment of U2OS cells with the TAT-p53-C-ter peptide, immunofluorescence experiments were performed using an anti-p53 antibody that recognizes the p53 NH2 terminus and an anti-HA antibody that detects the TAT-p53-C-ter peptide. p53 was partially localized in the cytoplasm of untreated U2OS cells (Fig. 3C, b and d), but it was largely relocated into the nucleus upon treatment with the TAT-p53-C-ter peptide (Fig. 3C, f and h). The TAT-p53-C-ter peptide was detected in the cytoplasm and the nucleus of treated cells (Fig. 3C, g and h). Cells were also treated with TAT-p53-C-ter peptide for 1 hour, washed to remove the peptide and, 1 hour later, subjected to immunofluorescence. p53 was still localized in the nucleus (Fig. 3C, j and l) but the TAT-p53-C-ter peptide was no longer detectable (Fig. 3C, k). It has been reported that p53-TD-containing peptides mask the Nuclear Export Signal embedded in the TD region, thus preventing nuclear export which is active in NB cells [18]. In our experiments, the peptide was given in pulsed administrations; since the TAT-p53-C-ter peptide is labile, it is conceivable that the formation of inactive heterodimers is only transient, allowing functional p53 tetramerization after degradation of the peptide. In agreement with this hypothesis, immunofluorescence experiments detected the TAT-p53-C-ter peptide in the nucleus 1 hour after administration, but not after two hours (Fig. 3C, compare g with k), a time point at which the endogenous p53 was still predominantly nuclear (Fig 3C, j). Therefore, it is unlikely that pulsed TAT-p53-C-ter peptide delivery could promote the stable sequestration of p53 molecules in a non-functional complex that has been described by stable expression of p53-C-ter peptides [19]
p53 transcription targets are up-regulated after treatment with the TAT-p53-C-ter peptide.
We tested whether expression of some p53 transcriptional targets was modified after treatment with the TAT-p53-C-ter peptide. NB cells were treated with pulsed administrations (1 administration/hour) of peptide (10 μg/ml) for 2 hours. Expression of the cell cycle inhibitor p21 and of the pro-apoptotic protein Bax were increased after TAT-p53-C-ter peptide treatment of LAN-5 and HTLA-230 cells that carry a wt p53 but not of the p53-mutated SK-N-AS cell line (Fig. 4A). We assessed the expression of MDM2, another target of p53 [20], by quantitative PCR. After peptide treatment, an increase of MDM2 mRNA levels was detected only in LAN-5 and HTLA-230 cells (Fig. 4B). Together, these data show that the TAT-p53-C-ter peptide activates the expression of p53 targets in tumour cells with wt cytoplasmic p53. To demonstrate that TAT-p53-C-ter peptide elicits its activity in a p53-dependent manner, we down-regulated endogenous p53 in U2OS cells by RNA interference. After siRNA treatment, cells were exposed to TAT-p53-C-ter peptide (10 μg/ml) for 2 hours or left untreated. Immunoblotting analysis shows that after p53 inhibition and TAT-p53-C-ter peptide treatment, the levels of p53 targets were unchanged (Bax and MDM2) or were strongly reduced (p21) (Fig. 4C). On the contrary, in cells treated with control siRNA, the TAT-p53-C-ter peptide induced the up-regulation of p21, Bax and MDM2 levels (Fig. 4C). Together, these results indicate that the peptide elicits its activity through wt p53.
Fig. 4.
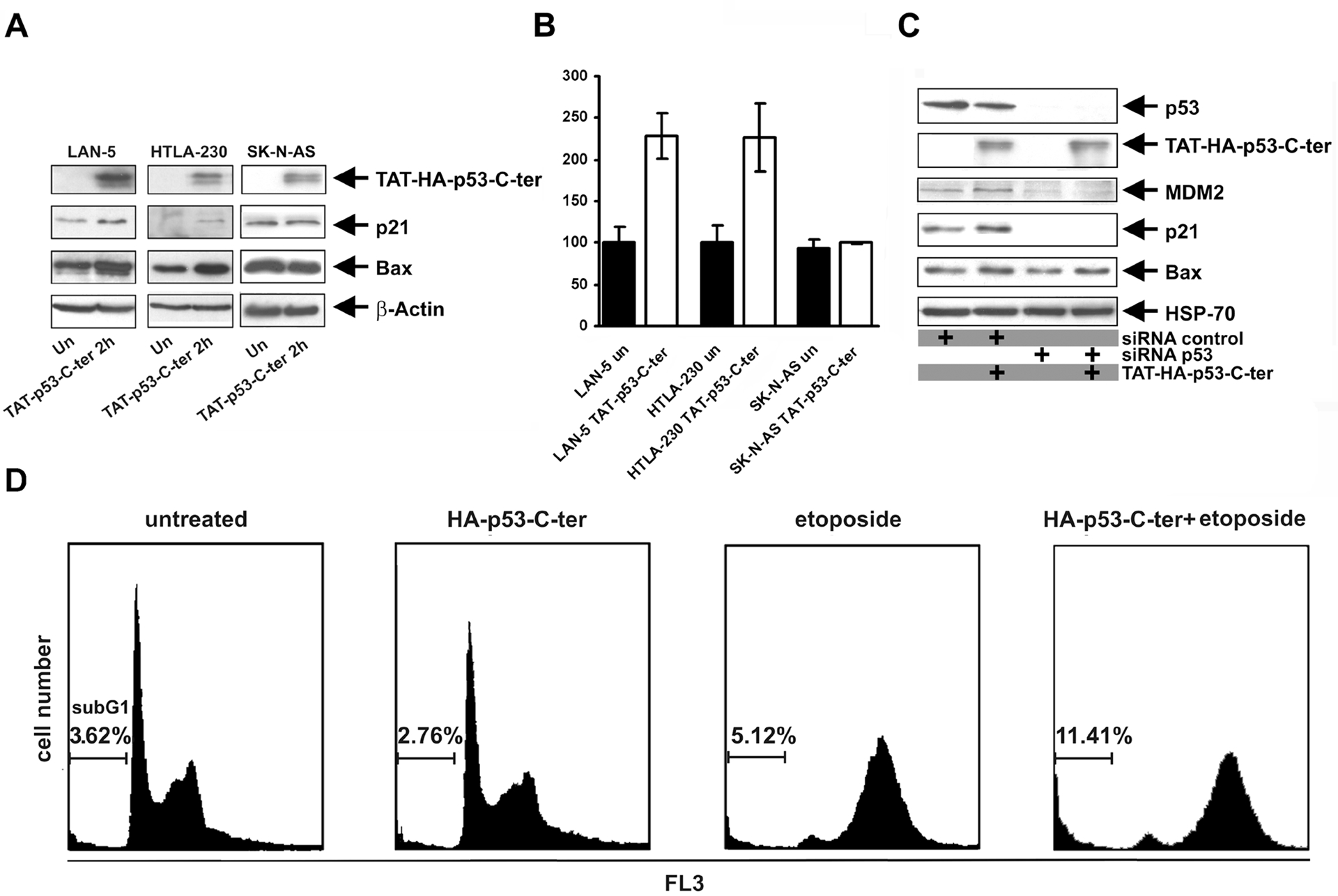
p53 transcription targets are up-regulated by the TAT-p53-C-ter peptide. A: indicated cell lines were treated for 2 hours with 10 μg/ml TAT-p53-C-ter peptide added every hour to the medium. Immunodetection was carried out with the anti-HA (to detect TAT-p53-C-ter peptide), anti-p21, anti-Bax and anti-β-actin antibodies. B: total RNA was extracted from cells treated for 2 hours with 10 μg/ml TAT-p53-C-ter peptide. Quantitative real-time PCR was performed to detect MDM2 expression. Values are normalized for β-actin transcripts of each sample. Reactions were run in triplicate, values ± SD are reported. C: Imunodetection of p53 targets in U2OS cells transfected with p53-siRNA or control-siRNA, untreated or treated with TAT-p53-C-ter peptide (see text for details). D: Flow cytometric analysis of U2OS cells transfected with pcDNA3-p53-C-ter-HA, or control pcDNA3 and pCMVEGFP (4:1) vectors. 24 hours after transfection, cells were treated for 36 hours with 1 μM etoposide or left untreated before fixation and processing for cytometric evaluation. DNA content analyses were performed by gating 20 × 104 green fluorescent cells. Experiments were repeated twice with similar results.
TAT-p53-C-ter peptide treatment enhances genotoxic stress-induced apoptosis.
U2OS cells were treated with increasing amounts of TAT-p53-C-ter peptide (2.5, 5 and 10 μg/ml) for 24 hours and caspase 3/7 activation was measured by a chemo-luminescent assay. Exposure of U2OS cells to TAT-p53-C-ter peptide (2.5, 5,0 and 10.0 μg/ml/6 hours) caused a modest increase of caspase 3/7 activity (29 ± 6, 29 ± 2 and 42% ± 2 respectively compared to untreated cells), consistent with the up-regulation of pro-apoptotic Bax protein in NB cells (Fig. 4A). The enhanced expression of p21 and Bax, together with the Caspase 3/7 activation observed after treatment of wt p53 tumour cells with the TAT-p53-C-ter peptide could be indicative of variations in apoptotic rate. To test these possibilities over a long time span that is difficult to achieve with the labile TAT-p53-C-ter peptide, the HA-tagged p53-C-ter peptide was cloned in a mammalian expression vector and transfected in the osteosarcoma U2OS cell line that has wt cytoplasmic p53 and detectable expression of PARC (Fig. 1 and [3]) at a 4:1 ratio with the EGFP-Spectrin vector (pCMVEGFP) [21]. The ability of the mammalian vector to express HA-tagged p53-C-ter peptide was tested by immunoblotting of the transfected cells (data not shown). 24 hours after transfection, cells were treated with 1μM etoposide or left untreated. After additional 36 hours, cells were processed for flow cytometric analysis. Cells transfected with the empty vector or the vector encoding the p53-C-ter peptide did not show differences in the apoptotic rate (sub G1 peak) (Fig. 4D). However, etoposide treatment induced apoptosis with greater efficiency in cells transfected with the vector encoding the p53-C-ter peptide than in controls (sub G1 peaks 11.41 and 5.12% respectively, Fig. 4D). Together, these experiments indicate that treatment with the p53-C-ter peptide renders tumour cells more susceptible to apoptosis induced by genotoxic stress.
In conclusion, p53 reactivation can be obtained in tumour cells that have wt cytoplasmic p53 by using a PARC-interacting peptide. The use of the TAT transduction sequence renders this strategy particularly flexible and potentially applicable in many cellular contexts. The screening of other PARC/p53-interacting molecules with appropriate stability and pharmacokinetic features, by using the mammalian two-hybrid assay presented here, would allow the exploitation of these compounds as potentially useful therapeutic tools.
Acknowledgments
This work was partially supported by grants from “Fondazione Italiana per la Lotta al Neuroblastoma” (G.R.), Italian Ministry of Health (84/2004) (C.D), “Io…domani - Associazione per la Lotta contro i Tumori Infantili” (ALTI) (C.D.) and NCI RO1 CA95111 (B.C). B.T. was a fellow of Bambino Gesù Children’s Hospital, Rome. R.V. is a fellow of “Fondazione Italiana per la Lotta al Neuroblastoma”. We thank Dr. Marco Ranalli (University of “Tor Vergata”, Rome, Italy) for his help with confocal microscopy.
References
- [1].Soussi T, Kato S, Levy PP, Ishioka C, Reassessment of the TP53 mutation database in human disease by data mining with a library of TP53 missense mutations Hum. Mutat 25 (2005) 6–17. [DOI] [PubMed] [Google Scholar]
- [2].Moll UM, LaQuaglia M, Benard J, Riou G, Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors Proc. Natl. Acad. Sci. U.S.A 92 (1995) 4407–4411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Nikolaev AY, Li M, Puskas N, Qin J, Gu W, Parc: a cytoplasmic anchor for p53 Cell 112 (2003) 29–40. [DOI] [PubMed] [Google Scholar]
- [4].Kasper JS, Arai T, DeCaprio JA, A novel p53-binding domain in CUL7 Biochem. Biophys. Res. Commun 348 (2006) 132–138. [DOI] [PubMed] [Google Scholar]
- [5].Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, Kong N, Kammlott U, Lukacs C, Klein C, Fotouhi N, Liu EA, In vivo activation of the p53 pathway by small-molecule antagonists of MDM2 Science 303 (2004) 844–848. [DOI] [PubMed] [Google Scholar]
- [6].Hupp TR, Sparks A, Lane DP, Small peptides activate the latent sequence-specific DNA binding function of p53 Cell 83 (1995) 237–245. [DOI] [PubMed] [Google Scholar]
- [7].Snyder EL, Meade BR, Saenz CC, Dowdy SF, Treatment of terminal peritoneal carcinomatosis by a transducible p53-activating peptide PLoS. Biol 2 (2004) E36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ho A, Schwarze SR, Mermelstein SJ, Waksman G, Dowdy SF, Synthetic protein transduction domains: enhanced transduction potential in vitro and in vivo Cancer Res. 61 (2001) 474–477. [PubMed] [Google Scholar]
- [9].Caron NJ, Torrente Y, Camirand G, Bujold M, Chapdelaine P, Leriche K, Bresolin N, Tremblay JP, Intracellular delivery of a Tat-eGFP fusion protein into muscle cells Mol. Ther. 3 (2001) 310–318. [DOI] [PubMed] [Google Scholar]
- [10].Tanno B, Mancini C, Vitali R, Mancuso M, McDowell HP, Dominici C, Raschella G, Down-regulation of insulin-like growth factor I receptor activity by NVP-AEW541 has an antitumor effect on neuroblastoma cells in vitro and in vivo Clin. Cancer Res 12 (2006) 6772–6780. [DOI] [PubMed] [Google Scholar]
- [11].Skorski T, Bellacosa A, Nieborowska-Skorska M, Majewski M, Martinez R, Choi JK, Trotta R, Wlodarski P, Perrotti D, Chan TO, Wasik MA, Tsichlis PN, Calabretta B, Transformation of hematopoietic cells by BCR/ABL requires activation of a PI-3k/Akt-dependent pathway EMBO J. 16 (1997) 6151–6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].el-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B, WAF1, a potential mediator of p53 tumor suppression Cell 75 (1993) 817–825. [DOI] [PubMed] [Google Scholar]
- [13].Grignani F, Kinsella T, Mencarelli A, Valtieri M, Riganelli D, Grignani F, Lanfrancone L, Peschle C, Nolan GP, Pelicci PG, High-efficiency gene transfer and selection of human hematopoietic progenitor cells with a hybrid EBV/retroviral vector expressing the green fluorescence protein Cancer Res. 58 (1998) 14–19. [PubMed] [Google Scholar]
- [14].Nagahara H, Vocero-Akbani AM, Snyder EL, Ho A, Latham DG, Lissy NA, Becker-Hapak M, Ezhevsky SA, Dowdy SF, Transduction of full-length TAT fusion proteins into mammalian cells: TAT-p27Kip1 induces cell migration Nat. Med 4 (1998) 1449–1452. [DOI] [PubMed] [Google Scholar]
- [15].Karlsson J, Edsjo A, Pahlman S, Pettersson HM, Multidrug-resistant neuroblastoma cells are responsive to arsenic trioxide at both normoxia and hypoxia Mol. Cancer Ther. 4 (2005) 1128–1135. [DOI] [PubMed] [Google Scholar]
- [16].Nakamura Y, Ozaki T, Niizuma H, Ohira M, Kamijo T, Nakagawara A, Functional characterization of a new p53 mutant generated by homozygous deletion in a neuroblastoma cell line Biochem. Biophys. Res. Commun 354 (2007) 892–898. [DOI] [PubMed] [Google Scholar]
- [17].Kaustov L, Lukin J, Lemak A, Duan S, Ho M, Doherty R, Penn LZ, Arrowsmith CH, The conserved CPH domains of Cul7 and PARC are protein-protein interaction modules that bind the tetramerization domain of p53 J. Biol. Chem 282 (2007) 11300–11307. [DOI] [PubMed] [Google Scholar]
- [18].Stommel JM, Marchenko ND, Jimenez GS, Moll UM, Hope TJ, Wahl GM, A leucine-rich nuclear export signal in the p53 tetramerization domain: regulation of subcellular localization and p53 activity by NES masking EMBO J. 18 (1999) 1660–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ostermeyer AG, Runko E, Winkfield B, Ahn B, Moll UM, Cytoplasmically sequestered wild-type p53 protein in neuroblastoma is relocated to the nucleus by a C-terminal peptide Proc. Natl. Acad. Sci. U.S.A 93 (1996) 15190–15194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Iwakuma T, Lozano G, MDM2, an introduction Mol. Cancer Res 1 (2003) 993–1000. [PubMed] [Google Scholar]
- [21].Kalejta RF, Shenk T, Beavis AJ, Use of a membrane-localized green fluorescent protein allows simultaneous identification of transfected cells and cell cycle analysis by flow cytometry Cytometry 29 (1997) 286–291. [DOI] [PubMed] [Google Scholar]