

Summary
B cell depletion potently reduces episodes of inflammatory demyelination in multiple sclerosis (MS), predominantly through loss of innate rather than adaptive immunity. However, molecular mechanisms controlling innate versus adaptive B cell function are poorly understood. N-glycan branching, via interactions with galectins, controls endocytosis and signaling of cell surface receptors to control cell function. Here we report that N-glycan branching in B cells dose dependently reduces pro-inflammatory innate responses by titrating decreases in Toll-like receptor-4 (TLR4) and TLR2 surface expression via endocytosis. In contrast, a minimal level of N-glycan branching maximizes surface retention of the B cell receptor (BCR) and the CD19 co-receptor to promote adaptive immunity. Branched N-glycans inhibit antigen presentation by B cells to reduce T helper cell-17 (TH17)/TH1 differentiation and inflammatory demyelination in mice. Thus, N-glycan branching negatively regulates B cell innate function while promoting/maintaining adaptive immunity via BCR, providing an attractive therapeutic target for MS.
Subject Areas: Immunology, Molecular Biology, Neuroscience
Graphical Abstract
Highlights
-
•
B cell TLR2/TLR4 inflammatory cell surface signaling is reduced by N-glycan branching
-
•
B cell antigen-presenting cell function is inhibited by N-glycan branching
-
•
Optimal B cell receptor and CD19 co-receptor signaling requires N-glycan branching
-
•
Inflammatory demyelination in mice is inhibited by N-glycan branching in B cells
Immunology; Molecular Biology; Neuroscience
Introduction
Multiple sclerosis (MS) is the most common autoimmune disease of the central nervous system, causing inflammatory demyelination and neurodegeneration (Noseworthy et al., 2000). Long-standing data from animal models of MS, particularly experimental autoimmune encephalomyelitis (EAE), implicate CD4+ T cells and their differentiation into pro-inflammatory TH1 and TH17 cells as critical to inflammatory demyelination (Ben-Nun et al., 2014; Hohlfeld and Steinman, 2017). Prominent TH1 and TH17 responses are also observed in patients with MS (Kaskow and Baecher-Allan, 2018; Steinman, 2014). Genetic analysis in MS has largely been consistent with this data, highlighting the relevance of the immune system, particularly T cells, in promoting the development of MS (Gregory et al., 2007; Hafler et al., 2007; Hussman et al., 2016; Lundmark et al., 2007; Sawcer et al., 2011). This is best exemplified by the strongest genetic risk factor for MS being variants of the molecules that present antigen to T cells, namely, the human leukocyte antigen complex (Canto and Oksenberg, 2018; Hafler et al., 2007).
We have previously shown that branched N-glycans produced by the sequential action of the N-acetylglucosaminyl transferase enzymes Mgat1, 2, 4, and 5 play a critical role in directly limiting pro-inflammatory T cell responses and autoimmunity (Araujo et al., 2017; Chen et al., 2007; Demetriou et al., 2001; Dennis et al., 2009; Grigorian and Demetriou, 2011; Grigorian et al., 2009; Lau et al., 2007; Mkhikian et al., 2011; Morgan et al., 2004; Zhou et al., 2014). Galectins bind the T cell receptor (TCR) and other glycoproteins at the cell surface in proportion to the branching, extension, and number of complex N-glycans, forming a molecular lattice that controls clustering, signaling, and endocytosis of surface receptors and transporters in a coordinated fashion. Molecularly, the galectin lattice regulates the segregation of surface glycoproteins to different membrane microdomains by acting as a brake on movement directed by intracellular actin microfilaments. For example, the galectin lattice excludes the TCR from GM3+ lipid rafts, while having the opposite effect on the phosphatase CD45, both by opposing the activity of actin microfilaments tethered to cytoplasmic domains of the respective proteins (Chen et al., 2007). Functionally, N-glycan branching negatively regulates TCR clustering/signaling (Chen et al., 2007; Demetriou et al., 2001; Grigorian et al., 2009; Zhou et al., 2014), promotes surface retention of anti-autoimmune cytotoxic T-lymphocyte antigen 4 (Lau et al., 2007), directly inhibits TH1 and TH17 while promoting anti-autoimmune-induced T regulatory cell differentiation (Araujo et al., 2017; Morgan et al., 2004), suppresses inflammatory demyelinating disease in mice (Demetriou et al., 2001; Grigorian et al., 2007, 2011; Lee et al., 2007), and is associated with risk of MS and other autoimmune diseases (Brynedal et al., 2010; Grigorian et al., 2012; Li et al., 2013; Mkhikian et al., 2011; Yu et al., 2012, 2014). The extent of N-glycan branching is regulated by multiple signals, including alterations in gene expression (e.g., Mgat1, Mgat5) induced by signals from multiple different cell surface receptors as well as supply of metabolic precursors for the Golgi branching enzymes (Araujo et al., 2017; Chen et al., 2009; Lau et al., 2007; Mkhikian et al., 2011).
Recently, the dominant role of T cells in MS has been challenged by the potent therapeutic activity of B cell-depleting therapies in MS (Sabatino et al., 2018), with the anti-CD20 monoclonal antibody ocrelizumab shown to be superior to high-dose interferon-β-1a (Rebif) at reducing relapse rates as well as displaying positive activity in progressive MS (Hauser et al., 2017a, 2017b). B cells are unique within the immune system, having both innate and adaptive immune activities, with the former exemplified by activation via pattern recognition receptors like Toll-like receptors (TLRs) and the ability to directly present antigen to CD4+ T cells. The therapeutic benefit of B cell depletion in MS appears to largely arise from altered innate activity and reduced antigen presentation to CD4+ T cells, rather than reduced B cell adaptive immune responses and autoantibody production (Hauser, 2015). For example, anti-CD20 therapy reduces pro-inflammatory TH1 and TH17 responses as well as B and T cell numbers but not IgG concentration, IgG index, IgG synthesis rate or oligoclonal band number in the cerebrospinal fluid of patients with MS who have been treated (Bar-Or et al., 2010; Cross et al., 2006). Data in EAE also support this conclusion, with B cells promoting TH1 and/or TH17 responses as well as inflammatory demyelination via their antigen-presenting cell (APC) function rather than through antibody production (Molnarfi et al., 2013; Monson et al., 2011; Pierson et al., 2014; Weber et al., 2010). B cells may also play an important role in MS via TLR4 and/or TLR2 signaling-mediated alterations in anti-inflammatory interleukin (IL)-10 production (Correale and Farez, 2009; Iwata et al., 2011; Okada et al., 2018), a cytokine utilized by B regulatory cells to inhibit autoimmunity (Fillatreau et al., 2002).
The regulatory mechanisms in B cells that drive innate activity and inflammatory demyelination in MS are poorly understood. TLR2 and TLR4 play critical roles in initiating innate inflammatory responses, and as the only TLR family members expressed at the cell surface of B cells (Kawai and Akira, 2010; Leifer and Medvedev, 2016), they may be regulated by N-glycan branching. Cell surface TLR4 and TLR2, which heterodimerizes with TLR1 or TLR6, are pro-inflammatory via MyD88/IRAK/TRAF6/AP-1/NF-κB, whereas signaling from endosomes is anti-inflammatory via TRIF/IRF3 (Siegemund and Sauer, 2012; Stack et al., 2014; Yamamoto et al., 2003). Thus, regulating TLR2/4 endocytosis is critical to pro- versus anti-inflammatory innate responses in B cells. CD14 controls lipopolysaccharide (LPS)-induced TLR4 endocytosis and associated anti-inflammatory signaling in myeloid cells (Zanoni et al., 2011); however, B cells do not express CD14.
The role of N-glycan branching in B cells is unknown. Here we report that N-glycan branching has a unique dual activity in B cells, suppressing innate activity by dose-dependently promoting TLR2 and TLR4 endocytosis while simultaneously fostering adaptive immunity by being required for optimal surface retention of B cell receptor (BCR) and its co-receptor CD19. N-glycan branching reduces B cell-triggered pro-inflammatory TH1/TH17 differentiation of T cells and inflammatory demyelination in mice.
Results
N-Glycan Branching Inhibits Pro-inflammatory TLR4 and TLR2 Signaling in B Cells
As N-glycan branching regulates the endocytosis of multiple glycoproteins, we hypothesized that branching may regulate TLR4 and TLR2 endocytosis in B cells to control pro- versus anti-inflammatory innate responses. To directly assess this hypothesis, we generated mice with B cell-specific deletion of Mgat1 or Mgat2 (i.e., Mgat1f/fCD19cre+/- and Mgat2f/fCD19cre+/-). Mgat1 deficiency is expected to produce a more severe phenotype than Mgat2 deficiency, as the former eliminates, whereas the latter only reduces branching (Figure S1A) (Mkhikian et al., 2016; Zhou et al., 2014). Indeed, whereas CD19-driven cre-deletion of Mgat2 had no impact on B cell development, deletion of Mgat1 completely blocked B cell development (Mortales et al., 2020). Therefore, to examine Mgat1 deficiency in mature peripheral B cells, we utilized Mgat1f/f/tetO-cre/ROSA-rtTA mice (Zhou et al., 2014), where doxycycline consistently induces Mgat1 deficiency in ∼35%–40% of mature ex vivo splenic B cells. Mgat1- and Mgat2-deleted B cells (97%–99% pure) were isolated from doxycycline-treated Mgat1f/f/tetO-cre/ROSA-rtTA or Mgat2f/fCD19cre+/- mice, respectively, by negative selection with Phaseolus vulgaris leukoagglutinin (L-PHA)+ cell depletion (Figure S1B). L-PHA binds β1,6-branched N-glycans, structures that are eliminated by Mgat1 or Mgat2 deficiency (Demetriou et al., 2001). For ease of nomenclature, we refer to these purified cells as Mgat1−/− and Mgat2−/− B cells in the following discussion. To address any potential complications from doxycycline treatment, we also phenocopied Mgat1 deletion by treating wild-type B cells with the mannosidase 1 inhibitor kifunensine (KIF). As the Mgat1 enzyme requires mannosidase 1 to remove mannose and initiate GlcNAc branching (Figure S1A), KIF blocks Mgat1 enzyme activity to prevent N-glycan branching.
Mgat1−/− and Mgat2−/− B cells hypo-proliferated in response to both LPS (TLR4 agonist) and the bacterial lipopeptides Pam2CSK4 (TLR2:6 heterodimer agonist) and Pam3CSK4 (TLR2:1 heterodimer agonist), as measured by Carboxyfluorescein succinimidyl ester (CFSE) dilution (Figure 1A). The magnitude of hypo-proliferation was less in Mgat2−/− than Mgat1−/− B cells, consistent with a less-severe loss of branching in the former. Remarkably, despite hypo-proliferation, up-regulation of the activation marker CD69 induced by TLR4 and TLR2 agonists was enhanced by both Mgat1 and Mgat2 deficiency (Figure 1B). Blocking N-glycan branching in wild-type B cells with KIF similarly enhanced CD69 induction by TLR4 and TLR2 agonists (Figure S1C). Consistent with this, pro-inflammatory NF-κB activation by cell surface TLR4 and TLR2 signaling was enhanced by Mgat1 and Mgat2 deficiency (Figure 1C). TLR4 also signals through SYK to activate ERK and AKT (Schweighoffer et al., 2017), and branching deficiency led to enhanced LPS-induced SYK, ERK, and AKT phosphorylation (Figures S1D and S1E). In contrast, anti-inflammatory endosomal TLR4 signaling via TBK1 and TRAF3 was unchanged by Mgat1 deficiency (Figure 1D). NF-κB activation drives secretion of pro-inflammatory tumor necrosis factor (TNF)-α over anti-inflammatory IL-10 (Fillatreau et al., 2002). Mgat1−/− B cells stimulated with TLR4 or TLR2 agonists displayed increased TNFα and reduced IL-10 secretion compared with control B cells (Figure 1E). Consistent with this, Mgat1 and Mgat2 deletion also greatly reduces the number of endogenous IL-10-producing B10 regulatory B cells in vivo (Figure S1F). B cells express the T cell co-stimulatory ligands CD80 and CD86, which promote pro-inflammatory TH1 and humoral/immunomodulatory TH2 responses, respectively, to influence inflammatory demyelination in EAE (Kuchroo et al., 1995; Zhang and Vignali, 2016). CD80 exists as a dimer, whereas CD86 is a monomer (Bhatia et al., 2006), allowing CD80 to bind CD28 with much greater affinity and thereby drive demyelinating disease (Kuchroo et al., 1995; Zhang and Vignali, 2016). LPS-stimulated Mgat1−/− and Mgat2−/− B cells displayed increased induction of CD80 and less CD86 on their cell surface compared with controls (Figure 1F). Induction of MHCII was also elevated in Mgat1−/− and Mgat2−/− B cells (Figure 1G). In contrast, CD80, CD86, and MHCII surface levels on resting B cells were unaltered by Mgat1 or Mgat2 deficiency (Figures S1G and S1H). Together, these data demonstrate that N-glycan branching inhibits TLR4 and TLR2 pro-inflammatory cell surface signaling in B cells.
Figure 1.

N-Glycan Branching Inhibits Pro-inflammatory TLR4 and TLR2 Activation in B Cells
(A, B, F, and G) TLR4- and TLR2-stimulated B cells were assessed by flow cytometry for proliferation by CFSE dilution after 2 days (A), CD69 expression after 1 day (B), surface expression of CD80/CD86 after 1–3 days (F), and MHCII after 3 days (G). Histograms in (A and B) represent highest agonist dose.
(C and D) Western blot analysis of phospho-NFκB (p65) (C) and total TRAF3 protein and phospho-TBK1 (D) in B cells stimulated for 2 days.
(E) TNFα and IL-10 secretion measure by ELISA from culture supernatants of stimulated B cells over 1–3 days.
Data shown are mean ± SEM of cells stimulated in triplicate (A, B, E, and F) and representative of n ≥ 3 experiments. Each symbol represents one mouse from three different experiments; horizontal line represents the mean; repeated-measures ANOVA with false discovery rate correction (Benjamini, Krieger, and Yekutieli) for multiple comparisons (G). NS, not significant; ∗p < 0.05. Divided (%), CFSE dilution by half or more. MFI, mean fluorescence intensity.
N-Glycan Branching Suppresses TLR4 and TLR2 Signaling by Enhancing Loss to Endocytosis
Next, we investigated the mechanism for enhanced TLR4 and TLR2 cell surface signaling. In resting B cells, cell surface expression of TLR4 is minimal (Figure S2A). Resting Mgat1−/− B cells displayed no significant difference in TLR4 or TLR2 surface expression, endocytosis rate, or mRNA levels (Figures S2A–S2C). Binding of LPS to cell surface TLR4 was also unaltered by Mgat1 deficiency, indicating that loss of branching did not impact the interaction of LPS with TLR4 (Figure S2D). However, 2–3 days after TLR4 or TLR2 stimulation, N-glycan branching-deficient B cells had greater TLR4 and TLR2 surface expression than control cells, with Mgat1 > Mgat2 deficiency (Figure 2A). This coincided with a TLR4- or TLR2-induced physiological increase in N-glycan branching in activated wild-type B cells (Figure 2B). Comparing cell surface versus total TLR4 and TLR2 by flow cytometry indicated that loss of branching did not alter total TLR protein levels but specifically enhanced cell surface TLR4 and TLR2 expression (Figure 2C). Western blotting confirmed that total TLR4 protein was not altered (Figure S2E). Consistent with increased surface levels, the rate of TLR4 and TLR2 endocytosis in activated B cells was significantly reduced by Mgat1 > Mgat2 deficiency (Figure 2D) as well as blocking branching in wild-type B cells with KIF (Figure S2F). Like mouse B cells, resting human B cells express little TLR4 at the cell surface; however, inflammatory conditions and activation via BCR, CD40, and TLR9 (CpG) induced TLR4 in human B cells (Ganley-Leal et al., 2010; Jagannathan et al., 2009; Mita et al., 2002; Noronha et al., 2008). Consistent with these data and our mouse data, blocking branching in human B cells with KIF enhanced TLR4 surface expression induced by co-stimulation with CpG + CD40L or CpG + anti-IgM/G/A (Figure S2G). Collectively, these data reveal that N-glycan branching is markedly up-regulated by TLR4 and TLR2 signaling and serves to inhibit pro-inflammatory TLR4 and TLR2 signaling in activated B cells by enhancing surface loss to endocytosis.
Figure 2.

N-Glycan Branching Reduces TLR4 and TLR2 Surface Levels by Promoting Endocytosis
(A–D) Flow cytometric analysis of B cells stimulated for 1–3 days and assessed for TLR4 and TLR2 surface expression (A), L-PHA binding (B), TLR4 and TLR2 surface-to-total ratio (C), and TLR4 and TLR2 endocytosis rates (D). Endocytosis rate over 1.5 h was calculated by dividing the MFI of acid-washed cells by the MFI of fluorescence-activated cell sorting (FACS) buffer-washed cells divided by 1.5 h (D). Data shown are mean ± SEM of cells stimulated in triplicate and representative of n ≥ 3 experiments. MFI, mean fluorescence intensity.
N-Glycan Branching Promotes BCR Signaling Independently of CD22 via Enhanced BCR and CD19 Surface Retention
As N-glycan branching regulates both basal and ligand-induced TCR signaling, we explored whether branching similarly regulates BCR signaling. Mgat1−/− B cells hypo-proliferated in response to anti-IgM F(ab’)2 compared with control B cells (Figure 3A), a result opposite to that in T cells but similar to TLR4- and TLR2-stimulated B cell proliferation. A potential mechanism for altered BCR signaling in Mgat1-deficient B cells is via CD22, a negative regulator of BCR signaling. CD22 is a Siglec (sialic-acid-binding immunoglobulin-like lectin) that binds α2,6-linked sialic acid, a terminal sugar attached to galactose in a wide variety of glycans including N-glycans, O-glycans, and glycolipids. CD22-deficient B cells hyper-proliferate in response to anti-IgM F(ab’)2 by preventing delivery of the phosphatase SHP-1 to BCR (Nitschke, 2005; O'Keefe et al., 1996). Deficiency of ST6Gal-I, the enzyme that generates the CD22 ligand (α2,6-linked sialic acid), promotes association of CD22 with BCR. This interaction promotes BCR endocytosis, inhibits BCR signaling, and reduces B cell proliferation (Collins et al., 2006; Grewal et al., 2006; Han et al., 2005; Hennet et al., 1998). As Mgat1 deficiency eliminates terminal sialic acids on N-glycans, but not other glycan types, it is possible that loss of branching may inhibit BCR signaling by promoting association of CD22 with BCR. To assess this hypothesis, we generated CD22−/−/Mgat1f/f/tetO-cre/ROSA-rtTA mice, where Mgat1−/−CD22−/− B cells are purified following doxycycline treatment. As previously reported, CD22−/− B cells were hyper-proliferative in response to anti-IgM F(ab’)2. However, eliminating CD22 on N-glycan branching-deficient B cells failed to rescue proliferation (Figure 3A). This is in contrast to ST6Gal-I deficiency, where CD22 loss reversed the negative effects of α2,6-linked sialic acid deficiency on BCR signaling to rescue proliferation (Collins et al., 2006).
Figure 3.

N-Glycan Branching Promotes Activation Signaling via CD19 and the B Cell Receptor Independent of CD22
(A, B, and D–F) Flow cytometric analysis of anti-IgM F(ab’)2-induced proliferation by CFSE dilution after 2 days (A) and Ca2+ mobilization over 4 min (B), and ex vivo B cells for IgM surface expression (D), CD19 surface expression (E), and CD19 endocytosis (F). Histograms in (A) represent highest stimulation concentration, arrow in (B) indicates addition of 2.5 μg/mL anti-IgM F(ab’)2, and immunofluorescent images in (E) were acquired on an Amnis ImageStream Imaging Flow Cytometer. Endocytosis rate over 1.5 h was calculated by dividing the MFI of acid-washed cells by the MFI of FACS buffer-washed cells divided by 1.5 h (F).
(C) Western blot analysis of phospho-CD19, phospho-Syk, and phospho-PLCγ in B cells stimulated with 10 μg/mL anti-IgM F(ab’)2. Data shown are mean ± SEM of cells stimulated in triplicate (A and F) and representative of n ≥ 3 experiments. Each symbol represents one mouse, and horizontal line represents the mean (D and E). Repeated-measures ANOVA with false discovery rate correction (Benjamini, Krieger, and Yekutieli) for multiple comparisons (D and E) and unpaired two-tailed t test (F). NS, not significant; ∗∗∗∗'p < 0.0001. MFI, mean fluorescence intensity.
Next, we explored whether N-glycan branching directly alters BCR signaling via reductions in surface expression of BCR and/or the CD19 co-receptor. CD19 facilitates the phosphorylation of immunoreceptor tyrosine-based activation motifs on Igα/Igβ at the BCR complex by recruiting the protein kinases Lyn and Syk to initiate signaling cascades including PLCγ/Ca2+-dependent proliferation (Dal Porto et al., 2004; Monroe, 2006). Mgat1−/− B cells displayed reduced anti-IgM F(ab’)2 induced Ca2+ flux (Figure 3B) and phosphorylation of CD19, Syk, and PLCγ2 (Figure 3C). Cell surface levels of IgM were similarly reduced in Mgat1−/−, CD22−/−, and Mgat1−/−CD22−/− B cells compared with control (Figure 3D), a phenotype comparable to ST6Gal-I and/or CD22 deficiency (Collins et al., 2006). There was also a reduction in CD22 surface levels in Mgat1−/− B cells (Figure S3A). However, Mgat1−/− and Mgat1−/−CD22−/− B cells had markedly reduced surface levels of CD19, whereas CD22−/− B cells had no significant difference compared with control (Figure 3E). Indeed, CD19 surface loss to endocytosis was enhanced by Mgat1 deficiency (Figure 3F) and KIF treatment (Figure S3B). There was no difference in CD19 mRNA levels (Figure S3C), whereas total CD19 protein was reduced in Mgat1−/− B cells (Figure S3D), consistent with protein loss via degradation of endocytosed protein. These results demonstrate that N-glycan branching promotes BCR signaling and proliferation by enhancing BCR and CD19 surface expression independent of regulation by CD22.
In contrast to Mgat1 deficiency and KIF treatment, which completely block all branching, limiting N-glycans to a single branch via Mgat2 deficiency in B cells did not alter anti-IgM F(ab’)2-induced proliferation, Ca2+ mobilization, or IgM and CD19 surface levels (Figures S3E–S3H). In T cells, the reduction in branching induced by Mgat2 deletion is partially compensated for by poly-N-acetyllactosamine (poly-LacNAc) extension of the remaining single GlcNAc branch, which leads to the same level of cell surface galectin-3 binding and TCR hyper-activity as Mgat5−/− T cells (Mkhikian et al., 2016). Mgat2−/− B cells similarly displayed up-regulated poly-LacNAc expression, as measured by flow cytometry with Lycopersicon esculentum lectin (LEA) (Figure S3I) (Mkhikian et al., 2016). Together these results demonstrate that a minimal threshold level of LacNAc (galectin ligand) within branched N-glycans is required to maintain CD19 at the cell surface and drive robust adaptive immunity BCR responses.
N-Glycan Branching Deficiency in B Cells Promotes Pro-inflammatory APC Function
As Mgat2 deficiency impacted innate TLR2 and TLR4, but not adaptive BCR/CD19, responses, we utilized this model to explore the impact of branching on APC function of B cells. LPS-induced up-regulation of MHCII and CD80 in Mgat2−/− B cells should increase B cell-triggered T cell activation. To examine this, LPS pre-stimulated and non-LPS pre-stimulated Mgat2−/− and control B cells from C57BL/6 mice were co-cultured with wild-type allogeneic splenic CD4+ T cells from PL/J mice under neutral, TH1-, TH17-, or TREG-inducing conditions. Mgat2 branching deficiency in LPS > non-LPS pre-stimulated B cells markedly enhanced the differentiation of wild-type CD4+ T cells to TH1 and TH17 cells under their respective conditions (Figure 4A, 4B, S4A, and S4B) while also inhibiting TREG differentiation under TREG-inducing conditions (Figure 4C and S4C). Under neutral conditions, addition of LPS also directly enhanced TH1 differentiation by Mgat2−/− B cells (Figure S4D). To confirm these results in an antigen-specific in vitro model, we utilized congenic 2D2 TCRMOG transgenic mice that harbor CD4+ T cells specific for the human MOG35-55 peptide (hMOG35-55). Relative to control B cells, Mgat2−/− B cells significantly increased the differentiation of 2D2 CD4+ T cells to TH1 and TH17 cells in response to hMOG35-55 (Figures 4D, 4E, S4E, and S4F), but had little effect on TREG differentiation (Figure 4F and S4G). LPS pre-treatment was required for enhanced TH1 differentiation of 2D2 CD4+ T cells by Mgat2−/− B cells, but was less critical to enhanced TH17 differentiation (Figures S4E and S4F). 2D2 CD4+ T cell differentiation in response to hMOG35-55 was dose dependent (Figure S4H), whereas OVA323-339 peptide did not induce robust CD4+ T cell responses under any conditions (Figure S4I). Thus, N-glycan branching deficiency directly enhances the ability of B cells to induce pro-autoimmune TH1 and TH17 responses via co-stimulatory APC function.
Figure 4.

N-Glycan Branching Deficiency in B Cells Promotes Pro-inflammatory APC Function
(A–F) LPS-stimulated B cells were co-cultured with allogeneic CD4+ T cells (A–C) or congenic 2D2 TCR transgenic CD4+ T cells +2.5 μg/mL hMOG35-55 (D–F) under TH1/TH17/TREG-inducing conditions. CD4+ T cells were assessed by flow cytometry for intracellular staining of IFNγ after 3 days (A and D) and IL-17A (B and E) and FOXP3 (C and F) after 4 days. Data shown are mean ± SEM of cells stimulated in triplicate and representative of n = 3 experiments. Unpaired one-tailed t tests. NS, not significant; ∗p < 0.05; ∗∗p < 0.01.
N-Glycan Branching Deficiency in B Cells Promotes Inflammatory Demyelination
Next, we explored whether the enhanced innate activity induced by Mgat2 deficiency in B cells promotes inflammatory demyelination in the MS model EAE. As C57BL/6 mice lacking B cells are resistant to EAE induced with whole recombinant human myelin oligodendrocyte glycoprotein protein (rhMOG) but remain sensitive to EAE induced by the MOG35-55 peptide and whole recombinant mouse MOG (Fillatreau et al., 2002; Molnarfi et al., 2013), we utilized rhMOG to induce B cell-dependent EAE in Mgat2f/fCD19cre+/- and control CD19cre+/- C57BL/6 mice. Mgat2 deficiency in B cells significantly enhanced the incidence and severity of clinical EAE (Figures 5A–5C) as well as infiltration of the spinal cord by CD4+ T cells and B220+ B cells (Figure 5D). To evaluate whether Mgat2 deletion enhanced B cell APC function to promote induction of TH1 and TH17 cells in vivo, we examined splenocytes isolated from EAE mice. Consistent with our in vitro co-culture data, Mgat2 deficiency in B cells of EAE mice enhanced rhMOG induction of TH1 and TH17 cells, but not TREG cells, in vivo (Figure 5E).
Figure 5.

N-Glycan Branching Deficiency in B Cells Promotes Inflammatory Demyelination
(A–C) EAE was induced in 8- to 12-week-old congenic C57BL/6 mice with day 0 indicating the time of immunization and clinical score (A), disease incidence (B), and highest clinical score (C) monitored over 35 days (n = 12 (F7,M5) CD19cre+/-, n = 11 (F6,M5) Mgat2f/fCD19cre+/-).
(D and E) On day 35, mice induced with EAE were used for flow cytometric analysis of infiltrating CD4+ T cell and B220+ B cell in spinal cords (D) and in vivo cytokine production of splenic CD4+ T cells (E). Each symbol represents one mouse; horizontal line represents the mean.
Two-way ANOVA (A and B) and unpaired one-tailed t tests (C–E). NS, not significant; ∗p < 0.05; ∗∗p < 0.01.
Discussion
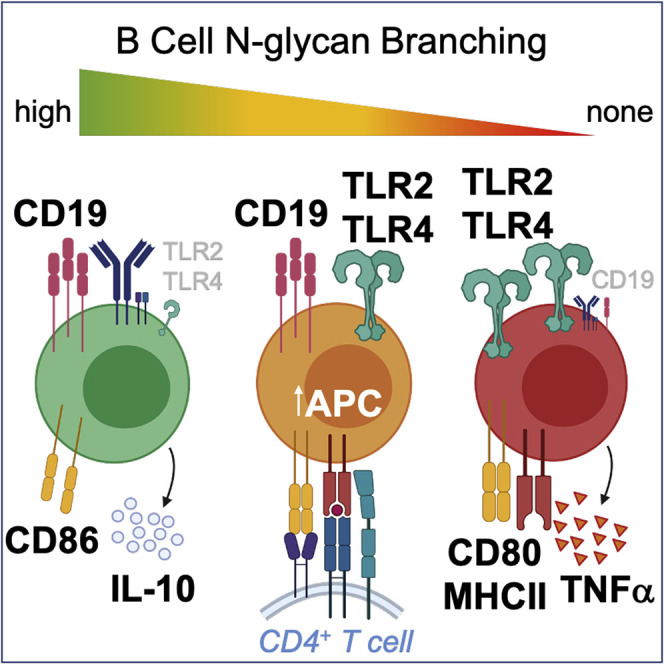
B cells play an important role in MS pathogenesis (Hauser et al., 2017a, 2017b). Here we identify N-glycan branching as a regulatory mechanism in B cells that decouples innate pro-inflammatory cell surface TLR function from adaptive activity via BCR/CD19, the former negatively regulating B cell APC-induced pro-inflammatory TH1/TH17 T cell responses and demyelinating disease (Figure 6). N-glycan branching is markedly increased by TLR4 and TLR2 signaling, which titrates reductions in surface expression of TLR4 and TLR2 via endocytosis over a broad continuum. In contrast, a single GlcNAc branch maximizes BCR and CD19 retention at the cell surface to drive adaptive immune responses, with higher levels of branching having no additional impact. The APC function of B cells is inhibited by N-glycan branching in part via suppression of TLR4 and TLR2 signaling-induced up-regulation of MHCII, CD80, and pro-inflammatory cytokines. Co-suppression of MHCII and CD80 by N-glycan branching reduces both primary TCR signaling as well as co-stimulatory signals via CD28, thereby inhibiting B cell triggered pro-inflammatory TH1/TH17 responses and inflammatory demyelination.
Figure 6.

N-glycan Branching Decouples Endocytosis of Surface TLR and CD19/BCR to Suppress Innate and Promote Adaptive Immunity
Branching in B cells promotes endocytic loss of surface TLR (TLR4 and TLR2) to suppress ligand-induced surface TLR signaling and downstream APC activity in B cells via reductions in MHCII, CD80 over CD86 and pro-inflammatory TNFa, which in turn inhibits B cell-dependent CD4+ TH1/TH17 differentiation and inflammatory demyelination. Concurrently, branching promotes CD19/BCR surface-retention to drive Ca2+ flux and proliferation in response to BCR ligands, thereby promoting adaptive B cell function. While intermediate reductions in branching enhance surface TLR signaling, CD19/BCR surface expression/signaling is not impacted. Rather, complete absence of branched N-glycans is required to drive CD19 endocytosis and inhibit BCR responses. Thus, the impact of branching on TLR/APC activity occurs over a large continuum, whereas a minimal level of branching is sufficient to promote BCR responses. Figure created with Biorender.com.
B cell depletion with anti-CD20 antibodies inhibits relapses and progression in MS, a blunt tool that non-specifically reduces both innate and adaptive immunity. In contrast, enhancing N-glycan branching in B cells may provide a safer therapeutic strategy by suppressing APC function and innate immunity while maintaining adaptive immunity via BCR/CD19. In this regard, we have previously demonstrated that metabolically supplementing T cells with the simple sugar N-acetylglucosamine (GlcNAc) increases N-glycan branching via enhanced substrate supply to the Golgi (Mgat) branching enzymes (Araujo et al., 2017; Grigorian et al., 2007; Lau et al., 2007; Lee et al., 2019; Mkhikian et al., 2011). GlcNAc is orally active, and when fed to mice, it suppresses pro-inflammatory T cell responses in vivo and inhibits/treats EAE and autoimmune diabetes (Grigorian et al., 2007, 2011). Based on this activity, we are currently conducting a dose-finding Phase 1 clinical trial of GlcNAc in MS. Our data on B cells raise the possibility that in addition to direct effects on T cells, oral GlcNAc may also function to suppress inflammatory demyelination by raising branching in B cells in vivo. Further investigations are warranted to characterize branching/poly-LacNAc expression of B cells in patients with MS, and determine the effects of GlcNAc on B cell activity.
In myeloid cells, CD14 controls TLR4 endocytosis to regulate pro-inflammatory (cell surface) versus anti-inflammatory (endosomal) signaling (Zanoni et al., 2011). As B cells lack CD14, how TLR4 cell surface versus endosomal signaling is regulated in B cells was poorly understood. Our data identify N-glycan branching in activated B cells as a critical determinant of pro- versus anti-inflammatory TLR4 signaling. Conditions that drive low branching would be expected to trigger TLR4 surface expression and innate pro-inflammatory B cell activity. This has implications for a wide diversity of inflammatory conditions where B cell innate activity is critical. Whether branching also regulates TLR4/2 surface expression in other APCs requires further investigation.
In contrast to promoting TLR4/2 endocytosis, N-glycan branching inhibits CD19 endocytosis to support BCR signaling and downstream adaptive immunity. N-glycan branching–galectin interactions regulate the segregation of transmembrane glycoproteins to different membrane microdomains, such as endocytic pits, by opposing the movement driven by intra-cellular actin-microfilament networks (Chen et al., 2007). This molecular mechanism likely explains how N-glycan branching simultaneously enhances TLR4/TLR2, but inhibits CD19 endocytosis. If actin tethering to cytoplasmic domains drives a glycoprotein to endocytic pits, N-glycan branching (galectin lattice) opposes this to inhibit endocytosis. However, if actin has the opposite effect on a glycoprotein and drives it away from endocytic pits, then N-glycan branching would oppose this and promote endocytosis. In unpublished data using a cell surface whole proteome mass-spectrometric analysis of T cells, we observed that of 208 transmembrane proteins identified, 71 were reduced at the cell surface without N-glycan branching, whereas 27 were increased. These results were confirmed by flow cytometry, with 11 of 13 replicated (6 down, 5 up). Thus, the opposing effects of N-glycan branching on TLR and CD19 are not unique; however, additional experiments are required to further delineate molecular details.
Although N-glycan branching deficiency in B cells augments TLR2/TLR4 surface expression and pro-inflammatory signaling, LPS-induced proliferation was reduced. CD19 plays an important role in LPS-induced proliferation, with CD19 knockout inhibiting and CD19 over-expression enhancing LPS-induced B cell proliferation (Engel et al., 1995). Consistent with this, the marked reduction in CD19 induced by Mgat1 deficiency is associated with reduced LPS-triggered proliferation. In contrast, Mgat2 deficiency has little impact on CD19 surface levels and LPS-induced proliferation is only marginally impacted in Mgat2−/− B cells. Poly-LacNAc extension of N-glycans is markedly increased in Mgat2−/− B cells, which serves to enhance galectin avidity and thereby likely contributes to maintaining surface CD19 and signaling similar to control. Capping branched N-glycans with α2,6-sialic acid also inhibits galectin binding (Zhuo and Bellis, 2011), and this may be another important contributor to the regulation of TLR2/4 versus CD19 surface retention.
Human naive, germinal center (GC), and memory B cells express bi-, tri-, and tetra-antennary complex N-glycans containing poly-LacNAc, thereby promoting binding of galectin-9 to BCR and inhibiting BCR signaling (Cao et al., 2018; Giovannone et al., 2018). Mgat2 deficiency induces loss of bi-, tri-, and tetra-antennary branched N-glycans, but concurrent up-regulation of poly-LacNAc expression serves to maintain galectin ligand availability (Mkhikian et al., 2016). Thus, Mgat2 deficiency should have little impact on galectin-9-mediated suppression of BCR signaling, which is consistent with the lack of altered BCR signaling observed in Mgat2−/− B cells. In contrast, Mgat1 deficiency eliminates both branching and poly-LacNAc and is expected to prevent binding of galectin-9 and enhance BCR signaling. However, the dramatic reduction in BCR and CD19 surface expression induced by Mgat1 (but not Mgat2) deficiency is dominant, thereby reducing BCR signaling despite a potential decrease in galectin-9 binding to BCR. Consistent with this, we recently observed that deleting Mgat1 in pro/pre-bone-marrow B cells completely blocks development of mature B cells by reducing CD19/BCR signaling (Mortales et al., 2020).
The effects of branching on TLR4 surface retention were greater than TLR2. This may be explained by differences in the number of attached N-glycosylation sites, which also determines interaction with galectins (Lau et al., 2007). TLR4 has 14 and TLR2 has 3. The TLR2 binding partners TLR1 and TLR6 have 9 and 10, respectively. When respective agonists bind, these TLRs dimerize to form TLR4:4 with 28, TLR2:1 with 12, and TLR2:6 with 13 combined N-glycosylation sites, respectively. Thus, TLR4:4 homodimers have more than double the number of N-glycans, making the consequences for complete loss of N-glycan branching more severe than that of TLR2:1 or TLR2:6 heterodimers.
Others have reported that loss of BCR inhibits LPS-induced Syk activation in short-term signaling assays (i.e., 15 minutes) (Schweighoffer et al., 2017). In contrast, we observed that Mgat1 deficiency enhanced Syk activation following 2 days of LPS stimulation despite the concurrent reduction in BCR and CD19 surface levels. The reason for this difference requires further investigation, but our data suggest that the gain of TLR4 surface levels in blasting B cells is dominant over negative regulation of Syk activation from reduced BCR/CD19.
Limitations of the Study
There are several limitations of this study. First, although we confirmed that reducing N-glycan branching in human B cells similarly up-regulated TLR4 surface expression, we did not examine the functional consequences in human B cells. Second, we did not directly examine whether patients with MS have alterations in N-glycan branching that result in altered innate B cell function. Such studies require highly characterized cohorts of patients who are not on treatment, as the latter would impact our ability to assess immune function. Third, the precise mechanism by which N-glycan branching promotes TLR2/4 endocytosis while inhibiting CD19 endocytosis is not well defined. Given our results in T cells on the mechanism by which N-glycan branching differentially regulates TCR versus CD45 segregation to membrane microdomains, it is likely that different cytoplasmic microfilament tethers to TLR2/4 versus CD19 direct these proteins away from or toward endocytic pits, respectively, with N-glycan branching/galectin lattice opposing this force. Additional experiments will be required to investigate this hypothesis.
Resource Availability
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Michael Demetriou (mdemetri@uci.edu).
Materials Availability
All unique reagents/mice generated in this study will be made available on request, but may require a completed Materials Transfer Agreement.
Data and Code Availability
This study did not generate/analyze [datasets/code].
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
Research was supported by the National Institute of Allergy and Infectious Diseases (USA) via a research grant (R01AI144403) to M.D. and an immunology training award (T32AI060573) to C.-L.M.
Author Contributions
C.-L.M. performed the in vitro and in vivo animal experiments with assistance from S.-U.L. A.M. and K.H. co-wrote the manuscript with M.D. S.-U.L assisted C.-L.M. with experiments. A.M. assisted C.-L.M. with experiments. K.H. assisted C.-L.M. with experiments. M.D. conceived the study, coordinated the analysis, and wrote the manuscript together with C.-L.M.
Declaration of Interests
M.D. is an inventor on a patent that describes GlcNAc as a biomarker and potential therapeutic for MS and is a co-founder of Glixis Therapeutics, a company developing analogs of GlcNAc for MS and other autoimmune diseases.
Published: August 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101380.
Supplemental Information
References
- Araujo L., Khim P., Mkhikian H., Mortales C.L., Demetriou M. Glycolysis and glutaminolysis cooperatively control T cell function by limiting metabolite supply to N-glycosylation. eLife. 2017;6:1–16. doi: 10.7554/eLife.21330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Or A., Fawaz L., Fan B., Darlington P.J., Rieger A., Ghorayeb C., Calabresi P.A., Waubant E., Hauser S.L., Zhang J. Abnormal B-cell cytokine responses a trigger of T-cell-mediated disease in MS? Ann. Neurol. 2010;67:452–461. doi: 10.1002/ana.21939. [DOI] [PubMed] [Google Scholar]
- Ben-Nun A., Kaushansky N., Kawakami N., Krishnamoorthy G., Berer K., Liblau R., Hohlfeld R., Wekerle H. From classic to spontaneous and humanized models of multiple sclerosis: impact on understanding pathogenesis and drug development. J. Autoimmun. 2014;54:33–50. doi: 10.1016/j.jaut.2014.06.004. [DOI] [PubMed] [Google Scholar]
- Bhatia S., Edidin M., Almo S.C., Nathenson S.G. B7-1 and B7-2: similar costimulatory ligands with different biochemical, oligomeric and signaling properties. Immunol. Lett. 2006;104:70–75. doi: 10.1016/j.imlet.2005.11.019. [DOI] [PubMed] [Google Scholar]
- Brynedal B., Wojcik J., Esposito F., Debailleul V., Yaouanq J., Martinelli-Boneschi F., Edan G., Comi G., Hillert J., Abderrahim H. MGAT5 alters the severity of multiple sclerosis. J. Neuroimmunol. 2010;220:120–124. doi: 10.1016/j.jneuroim.2010.01.003. [DOI] [PubMed] [Google Scholar]
- Canto E., Oksenberg J.R. Multiple sclerosis genetics. Mult. Scler. 2018;24:75–79. doi: 10.1177/1352458517737371. [DOI] [PubMed] [Google Scholar]
- Cao A., Alluqmani N., Buhari F.H.M., Wasim L., Smith L.K., Quaile A.T., Shannon M., Hakim Z., Furmli H., Owen D.M. Galectin-9 binds IgM-BCR to regulate B cell signaling. Nat. Commun. 2018;9:3288. doi: 10.1038/s41467-018-05771-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.L., Li C.F., Grigorian A., Tian W., Demetriou M. T cell receptor signaling co-regulates multiple Golgi genes to enhance N-glycan branching. J. Biol. Chem. 2009;284:32454–32461. doi: 10.1074/jbc.M109.023630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I.J., Chen H.L., Demetriou M. Lateral compartmentalization of T cell receptor versus CD45 by galectin-N-glycan binding and microfilaments coordinate basal and activation signaling. J. Biol. Chem. 2007;282:35361–35372. doi: 10.1074/jbc.M706923200. [DOI] [PubMed] [Google Scholar]
- Collins B.E., Smith B.A., Bengtson P., Paulson J.C. Ablation of CD22 in ligand-deficient mice restores B cell receptor signaling. Nat. Immunol. 2006;7:199–206. doi: 10.1038/ni1283. [DOI] [PubMed] [Google Scholar]
- Correale J., Farez M. Helminth antigens modulate immune responses in cells from multiple sclerosis patients through TLR2-dependent mechanisms. J. Immunol. 2009;183:5999–6012. doi: 10.4049/jimmunol.0900897. [DOI] [PubMed] [Google Scholar]
- Cross A.H., Stark J.L., Lauber J., Ramsbottom M.J., Lyons J.A. Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J. Neuroimmunol. 2006;180:63–70. doi: 10.1016/j.jneuroim.2006.06.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dal Porto J.M., Gauld S.B., Merrell K.T., Mills D., Pugh-Bernard A.E., Cambier J. B cell antigen receptor signaling 101. Mol. Immunol. 2004;41:599–613. doi: 10.1016/j.molimm.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Demetriou M., Granovsky M., Quaggin S., Dennis J.W. Negative regulation of T-cell activation and autoimmunity by Mgat5 N-glycosylation. Nature. 2001;409:733–739. doi: 10.1038/35055582. [DOI] [PubMed] [Google Scholar]
- Dennis J.W., Nabi I.R., Demetriou M. Metabolism, cell surface organization, and disease. Cell. 2009;139:1229–1241. doi: 10.1016/j.cell.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel P., Zhou L.J., Ord D.C., Sato S., Koller B., Tedder T.F. Abnormal B lymphocyte development, activation, and differentiation in mice that lack or overexpress the CD19 signal transduction molecule. Immunity. 1995;3:39–50. doi: 10.1016/1074-7613(95)90157-4. [DOI] [PubMed] [Google Scholar]
- Fillatreau S., Sweenie C.H., McGeachy M.J., Gray D., Anderton S.M. B cells regulate autoimmunity by provision of IL-10. Nat. Immunol. 2002;3:944–950. doi: 10.1038/ni833. [DOI] [PubMed] [Google Scholar]
- Ganley-Leal L.M., Liang Y., Jagannathan-Bogdan M., Farraye F.A., Nikolajczyk B.S. Differential regulation of TLR4 expression in human B cells and monocytes. Mol. Immunol. 2010;48:82–88. doi: 10.1016/j.molimm.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannone N., Liang J., Antonopoulos A., Geddes Sweeney J., King S.L., Pochebit S.M., Bhattacharyya N., Lee G.S., Dell A., Widlund H.R. Galectin-9 suppresses B cell receptor signaling and is regulated by I-branching of N-glycans. Nat. Commun. 2018;9:3287. doi: 10.1038/s41467-018-05770-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory S.G., Schmidt S., Seth P., Oksenberg J.R., Hart J., Prokop A., Caillier S.J., Ban M., Goris A., Barcellos L.F. Interleukin 7 receptor alpha chain (IL7R) shows allelic and functional association with multiple sclerosis. Nat. Genet. 2007;39:1083–1091. doi: 10.1038/ng2103. [DOI] [PubMed] [Google Scholar]
- Grewal P.K., Boton M., Ramirez K., Collins B.E., Saito A., Green R.S., Ohtsubo K., Chui D., Marth J.D. ST6Gal-I restrains CD22-dependent antigen receptor endocytosis and Shp-1 recruitment in normal and pathogenic immune signaling. Mol. Cell Biol. 2006;26:4970–4981. doi: 10.1128/MCB.00308-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorian A., Araujo L., Naidu N.N., Place D., Choudhury B., Demetriou M. N-acetylglucosamine inhibits T-helper 1 (Th1)/T-helper 17 (Th17) responses and treats experimental autoimmune encephalomyelitis. J. Biol. Chem. 2011;286:40133–40141. doi: 10.1074/jbc.M111.277814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorian A., Demetriou M. Mgat5 deficiency in T cells and experimental autoimmune encephalomyelitis. ISRN Neurol. 2011;2011:374314. doi: 10.5402/2011/374314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorian A., Lee S.U., Tian W., Chen I.J., Gao G., Mendelsohn R., Dennis J.W., Demetriou M. Control of T Cell-mediated autoimmunity by metabolite flux to N-glycan biosynthesis. J. Biol. Chem. 2007;282:20027–20035. doi: 10.1074/jbc.M701890200. [DOI] [PubMed] [Google Scholar]
- Grigorian A., Mkhikian H., Li C.F., Newton B.L., Zhou R.W., Demetriou M. Pathogenesis of multiple sclerosis via environmental and genetic dysregulation of N-glycosylation. Semin. Immunopathol. 2012;34:415–424. doi: 10.1007/s00281-012-0307-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorian A., Torossian S., Demetriou M. T-cell growth, cell surface organization, and the galectin-glycoprotein lattice. Immunol. Rev. 2009;230:232–246. doi: 10.1111/j.1600-065X.2009.00796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafler D.A., Compston A., Sawcer S., Lander E.S., Daly M.J., De Jager P.L., de Bakker P.I., Gabriel S.B., Mirel D.B., Ivinson A.J. Risk alleles for multiple sclerosis identified by a genomewide study. N. Engl. J. Med. 2007;357:851–862. doi: 10.1056/NEJMoa073493. [DOI] [PubMed] [Google Scholar]
- Han S., Collins B.E., Bengtson P., Paulson J.C. Homomultimeric complexes of CD22 in B cells revealed by protein-glycan cross-linking. Nat. Chem. Biol. 2005;1:93–97. doi: 10.1038/nchembio713. [DOI] [PubMed] [Google Scholar]
- Hauser S.L. The Charcot Lecture | beating MS: a story of B cells, with twists and turns. Mult. Scler. 2015;21:8–21. doi: 10.1177/1352458514561911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauser S.L., Bar-Or A., Comi G., Giovannoni G., Hartung H.P., Hemmer B., Lublin F., Montalban X., Rammohan K.W., Selmaj K. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N. Engl. J. Med. 2017;376:221–234. doi: 10.1056/NEJMoa1601277. [DOI] [PubMed] [Google Scholar]
- Hauser S.L., Belachew S., Kappos L. Ocrelizumab in primary progressive and relapsing multiple sclerosis. N. Engl. J. Med. 2017;376:1694. doi: 10.1056/NEJMc1702076. [DOI] [PubMed] [Google Scholar]
- Hennet T., Chui D., Paulson J.C., Marth J.D. Immune regulation by the ST6Gal sialyltransferase. Proc. Natl. Acad. Sci. U S A. 1998;95:4504–4509. doi: 10.1073/pnas.95.8.4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohlfeld R., Steinman L. T cell-transfer experimental autoimmune encephalomyelitis: pillar of multiple sclerosis and autoimmunity. J. Immunol. 2017;198:3381–3383. doi: 10.4049/jimmunol.1700346. [DOI] [PubMed] [Google Scholar]
- Hussman J.P., Beecham A.H., Schmidt M., Martin E.R., McCauley J.L., Vance J.M., Haines J.L., Pericak-Vance M.A. GWAS analysis implicates NF-kappaB-mediated induction of inflammatory T cells in multiple sclerosis. Genes Immun. 2016;17:305–312. doi: 10.1038/gene.2016.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwata Y., Matsushita T., Horikawa M., Dilillo D.J., Yanaba K., Venturi G.M., Szabolcs P.M., Bernstein S.H., Magro C.M., Williams A.D. Characterization of a rare IL-10-competent B-cell subset in humans that parallels mouse regulatory B10 cells. Blood. 2011;117:530–541. doi: 10.1182/blood-2010-07-294249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan M., Hasturk H., Liang Y., Shin H., Hetzel J.T., Kantarci A., Rubin D., McDonnell M.E., Van Dyke T.E., Ganley-Leal L.M. TLR cross-talk specifically regulates cytokine production by B cells from chronic inflammatory disease patients. J. Immunol. 2009;183:7461–7470. doi: 10.4049/jimmunol.0901517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaskow B.J., Baecher-Allan C. Effector T cells in multiple sclerosis. Cold Spring Harb. Perspect. Med. 2018;8:1–14. doi: 10.1101/cshperspect.a029025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawai T., Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- Kuchroo V.K., Das M.P., Brown J.A., Ranger A.M., Zamvil S.S., Sobel R.A., Weiner H.L., Nabavi N., Glimcher L.H. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 1995;80:707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- Lau K.S., Partridge E.A., Grigorian A., Silvescu C.I., Reinhold V.N., Demetriou M., Dennis J.W. Complex N-glycan number and degree of branching cooperate to regulate cell proliferation and differentiation. Cell. 2007;129:123–134. doi: 10.1016/j.cell.2007.01.049. [DOI] [PubMed] [Google Scholar]
- Lee S.U., Grigorian A., Pawling J., Chen I.J., Gao G., Mozaffar T., McKerlie C., Demetriou M. N-glycan processing deficiency promotes spontaneous inflammatory demyelination and neurodegeneration. J. Biol. Chem. 2007;282:33725–33734. doi: 10.1074/jbc.M704839200. [DOI] [PubMed] [Google Scholar]
- Lee S.U., Li C.F., Mortales C.L., Pawling J., Dennis J.W., Grigorian A., Demetriou M. Increasing cell permeability of N-acetylglucosamine via 6-acetylation enhances capacity to suppress T-helper 1 (TH1)/TH17 responses and autoimmunity. PLoS One. 2019;14:e0214253. doi: 10.1371/journal.pone.0214253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leifer C.A., Medvedev A.E. Molecular mechanisms of regulation of Toll-like receptor signaling. J. Leukoc. Biol. 2016;100:927–941. doi: 10.1189/jlb.2MR0316-117RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C.F., Zhou R.W., Mkhikian H., Newton B.L., Yu Z., Demetriou M. Hypomorphic MGAT5 polymorphisms promote multiple sclerosis cooperatively with MGAT1 and interleukin-2 and 7 receptor variants. J. Neuroimmunol. 2013;256:71–76. doi: 10.1016/j.jneuroim.2012.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundmark F., Duvefelt K., Iacobaeus E., Kockum I., Wallstrom E., Khademi M., Oturai A., Ryder L.P., Saarela J., Harbo H.F. Variation in interleukin 7 receptor alpha chain (IL7R) influences risk of multiple sclerosis. Nat. Genet. 2007;39:1108–1113. doi: 10.1038/ng2106. [DOI] [PubMed] [Google Scholar]
- Mita Y., Dobashi K., Endou K., Kawata T., Shimizu Y., Nakazawa T., Mori M. Toll-like receptor 4 surface expression on human monocytes and B cells is modulated by IL-2 and IL-4. Immunol. Lett. 2002;81:71–75. doi: 10.1016/s0165-2478(01)00328-5. [DOI] [PubMed] [Google Scholar]
- Mkhikian H., Grigorian A., Li C.F., Chen H.L., Newton B., Zhou R.W., Beeton C., Torossian S., Tatarian G.G., Lee S.U. Genetics and the environment converge to dysregulate N-glycosylation in multiple sclerosis. Nat. Commun. 2011;2:334. doi: 10.1038/ncomms1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mkhikian H., Mortales C.L., Zhou R.W., Khachikyan K., Wu G., Haslam S.M., Kavarian P., Dell A., Demetriou M. Golgi self-correction generates bioequivalent glycans to preserve cellular homeostasis. eLife. 2016;5:1–27. doi: 10.7554/eLife.14814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molnarfi N., Schulze-Topphoff U., Weber M.S., Patarroyo J.C., Prod'homme T., Varrin-Doyer M., Shetty A., Linington C., Slavin A.J., Hidalgo J. MHC class II-dependent B cell APC function is required for induction of CNS autoimmunity independent of myelin-specific antibodies. J. Exp. Med. 2013;210:2921–2937. doi: 10.1084/jem.20130699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monroe J.G. ITAM-mediated tonic signalling through pre-BCR and BCR complexes. Nat. Rev. Immunol. 2006;6:283–294. doi: 10.1038/nri1808. [DOI] [PubMed] [Google Scholar]
- Monson N.L., Cravens P., Hussain R., Harp C.T., Cummings M., de Pilar Martin M., Ben L.H., Do J., Lyons J.A., Lovette-Racke A. Rituximab therapy reduces organ-specific T cell responses and ameliorates experimental autoimmune encephalomyelitis. PLoS One. 2011;6:e17103. doi: 10.1371/journal.pone.0017103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan R., Gao G., Pawling J., Dennis J.W., Demetriou M., Li B. N-acetylglucosaminyltransferase V (Mgat5)-mediated N-glycosylation negatively regulates Th1 cytokine production by T cells. J. Immunol. 2004;173:7200–7208. doi: 10.4049/jimmunol.173.12.7200. [DOI] [PubMed] [Google Scholar]
- Mortales C.L., Lee S.U., Demetriou M. N-glycan branching is required for development of mature B cells. J. Immunol. 2020;205:630–636. doi: 10.4049/jimmunol.2000101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nitschke L. The role of CD22 and other inhibitory co-receptors in B-cell activation. Curr. Opin. Immunol. 2005;17:290–297. doi: 10.1016/j.coi.2005.03.005. [DOI] [PubMed] [Google Scholar]
- Noronha A., Liang Y., Harnett W., Harnett M., Stucchi A., Becker J., Farraye F., Ganley-Leal L. TILR2 and TLR4 regulate B cell responses in human inflammatory bowel disease. Inflamm. Bowel Dis. 2008;14:S8. [Google Scholar]
- Noseworthy J.H., Lucchinetti C., Rodriguez M., Weinshenker B.G. Multiple sclerosis. N. Engl. J. Med. 2000;343:938–952. doi: 10.1056/NEJM200009283431307. [DOI] [PubMed] [Google Scholar]
- O'Keefe T.L., Williams G.T., Davies S.L., Neuberger M.S. Hyperresponsive B cells in CD22-deficient mice. Science. 1996;274:798–801. doi: 10.1126/science.274.5288.798. [DOI] [PubMed] [Google Scholar]
- Okada Y., Ochi H., Fujii C., Hashi Y., Hamatani M., Ashida S., Kawamura K., Kusaka H., Matsumoto S., Nakagawa M. Signaling via toll-like receptor 4 and CD40 in B cells plays a regulatory role in the pathogenesis of multiple sclerosis through interleukin-10 production. J. Autoimmun. 2018;88:103–113. doi: 10.1016/j.jaut.2017.10.011. [DOI] [PubMed] [Google Scholar]
- Pierson E.R., Stromnes I.M., Goverman J.M. B cells promote induction of experimental autoimmune encephalomyelitis by facilitating reactivation of T cells in the central nervous system. J. Immunol. 2014;192:929–939. doi: 10.4049/jimmunol.1302171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatino J.J., Jr., Zamvil S.S., Hauser S.L. B-cell therapies in multiple sclerosis. Cold Spring Harb. Perspect. Med. 2018;9:a032037. doi: 10.1101/cshperspect.a032037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawcer S., Hellenthal G., Pirinen M., Spencer C.C., Patsopoulos N.A., Moutsianas L., Dilthey A., Su Z., Freeman C., Hunt S.E. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schweighoffer E., Nys J., Vanes L., Smithers N., Tybulewicz V.L.J. TLR4 signals in B lymphocytes are transduced via the B cell antigen receptor and SYK. J. Exp. Med. 2017;214:1269–1280. doi: 10.1084/jem.20161117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegemund S., Sauer K. Balancing pro- and anti-inflammatory TLR4 signaling. Nat. Immunol. 2012;13:1031–1033. doi: 10.1038/ni.2452. [DOI] [PubMed] [Google Scholar]
- Stack J., Doyle S.L., Connolly D.J., Reinert L.S., O'Keeffe K.M., McLoughlin R.M., Paludan S.R., Bowie A.G. TRAM is required for TLR2 endosomal signaling to type I IFN induction. J. Immunol. 2014;193:6090–6102. doi: 10.4049/jimmunol.1401605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinman L. Immunology of relapse and remission in multiple sclerosis. Annu. Rev. Immunol. 2014;32:257–281. doi: 10.1146/annurev-immunol-032713-120227. [DOI] [PubMed] [Google Scholar]
- Weber M.S., Prod'homme T., Patarroyo J.C., Molnarfi N., Karnezis T., Lehmann-Horn K., Danilenko D.M., Eastham-Anderson J., Slavin A.J., Linington C. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann. Neurol. 2010;68:369–383. doi: 10.1002/ana.22081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto M., Sato S., Hemmi H., Hoshino K., Kaisho T., Sanjo H., Takeuchi O., Sugiyama M., Okabe M., Takeda K. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–643. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- Yu Z., Gillen D., Li C.F., Demetriou M. Incorporating parental information into family-based association tests. Biostatistics. 2012;14:556–572. doi: 10.1093/biostatistics/kxs048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Z., Li C.F., Mkhikian H., Zhou R.W., Newton B.L., Demetriou M. Family studies of type 1 diabetes reveal additive and epistatic effects between MGAT1 and three other polymorphisms. Genes Immun. 2014;15:218–223. doi: 10.1038/gene.2014.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanoni I., Ostuni R., Marek L.R., Barresi S., Barbalat R., Barton G.M., Granucci F., Kagan J.C. CD14 controls the LPS-induced endocytosis of Toll-like receptor 4. Cell. 2011;147:868–880. doi: 10.1016/j.cell.2011.09.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q., Vignali D.A. Co-stimulatory and Co-inhibitory pathways in autoimmunity. Immunity. 2016;44:1034–1051. doi: 10.1016/j.immuni.2016.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R.W., Mkhikian H., Grigorian A., Hong A., Chen D., Arakelyan A., Demetriou M. N-glycosylation bidirectionally extends the boundaries of thymocyte positive selection by decoupling Lck from Ca(2)(+) signaling. Nat. Immunol. 2014;15:1038–1045. doi: 10.1038/ni.3007. [DOI] [PubMed] [Google Scholar]
- Zhuo Y., Bellis S.L. Emerging role of alpha2,6-sialic acid as a negative regulator of galectin binding and function. J. Biol. Chem. 2011;286:5935–5941. doi: 10.1074/jbc.R110.191429. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate/analyze [datasets/code].