

Abstract
The CDC7 kinase is essential for the activation of DNA replication origins and has been implicated in the replication stress response. Using a highly specific chemical inhibitor and a chemical genetic approach, we now show that CDC7 activity is required to coordinate multiple MRE11‐dependent processes occurring at replication forks, independently from its role in origin firing. CDC7 localizes at replication forks and, similarly to MRE11, mediates active slowing of fork progression upon mild topoisomerase inhibition. Both proteins are also retained on stalled forks, where they promote fork processing and restart. Moreover, MRE11 phosphorylation and localization at replication factories are progressively lost upon CDC7 inhibition. Finally, CDC7 activity at reversed forks is required for their pathological MRE11‐dependent degradation in BRCA2‐deficient cells. Thus, upon replication interference CDC7 is a key regulator of fork progression, processing and integrity. These results highlight a dual role for CDC7 in replication, modulating both initiation and elongation steps of DNA synthesis, and identify a key intervention point for anticancer therapies exploiting replication interference.
Keywords: DNA replication, fork protection, genome stability, kinase inhibitor
Subject Categories: DNA Replication, Repair & Recombination
CDC7 kinase acts at paused forks where it coordinates MRE11‐dependent fork processing, contributing to fork restart and modulating fork speed. Upon prolonged arrest and compromised fork protection, CDC7 promotes fork degradation contributing to replication‐dependent chromosome breakage.
Introduction
In eukaryotic cells, DNA replication initiates from multiple origins 1, 2. CDC7 kinase is required for the firing of replication origins, by phosphorylating multiple subunits of the MCM2–7 helicase complex 3, 4, 5, thus initiating bidirectional replication 6, 7. In addition, CDC7 kinase has important and yet poorly characterized roles in the replication stress response. For instance, in human cells CDC7‐dependent phosphorylation of the mediator protein CLASPIN is important for full activation of CHK1 by ATR and for maintaining cell viability in the presence of drugs that affect replication fork progression 8, 9, 10, 11. More recently, CDC7 has been shown to play a role in processing stalled replication forks by promoting limited resection that may be important for efficient replication fork restart and possibly by controlling EXO1 nuclease 12. Furthermore, the dephosphorylation of the MCM complex—which is mediated by protein phosphatase 1 and RIF1, counteracting CDC7—has been associated with loss of replisome stability in Xenopus and human cells 13. RIF1/PP1 also protects stalled forks from DNA2‐dependent degradation 13, 14, 15.
The idea that CDC7 may contribute to the regulation of nucleases is supported by work in budding yeast where CDC7 directly controls the activation of the Mus81 nuclease in mitosis by direct phosphorylation of the Mms4 subunit of the Mus81–Mms4 complex, thus promoting the disentanglement of DNA joint molecules 16. Furthermore, several laboratories have elucidated the role of yeast CDC7 kinase in controlling the formation of DNA double‐strand breaks (DSBs) during meiotic DNA replication to promote meiotic recombination 17, 18, 19, 20, 21, 22. However, whether CDC7 plays a direct role at forks and how it contributes to the control of replication fork processing are largely elusive.
Many CDC7 inhibitors have been developed as potential anticancer agents; among these, PHA‐767491 and XL413 have become tool compounds that have also been widely used to test CDC7 functions. Direct comparison of these two compounds in biochemical assays showed that XL413 is more specific than PHA‐767491 23. In order to better elucidate CDC7 functions, we have recently generated and characterized an engineered cell line, based on the principle of the analogue‐sensitive kinase 24, in which CDC7 activity can be partially but very specifically reduced using a bulky ATP competitor, 3MB‐PP1. Importantly, all the phenotypes observed by treating WT cells with 10 μM XL413 are recapitulated by treatment with 10 μM 3MB‐PP1 in this AS‐CDC7 cell line, including reduction of MCM2 phosphorylation at CDC7‐dependent sites and a mild delay in progressing through S‐phase 25.
Drugs that target DNA replication fork progression such as hydroxyurea (HU), DNA polymerases and DNA topoisomerase inhibitors, and DNA‐damaging compounds slow down forks causing their reversal into a four‐branched structure that can be detected by electron microscopy, also described as chicken‐foot structure 26, 27, 28, 29. Formation of these structures is promoted by several enzymatic activities, leading to BRCA1‐ and BRCA2‐dependent deposition of a RAD51 filament on the regressed arm of the fork (reviewed in 28, 29).
Fork reversal also occurs at endogenous obstacles in the absence of genotoxic drugs and can be triggered by oncogene activation 30, 31. It was thus proposed that the overall speed of each individual fork is determined by the combination of processive DNA synthesis, pausing into a chicken‐foot conformation and subsequent restart 28.
Once regressed arms are formed, they are stabilized by BRCA2‐dependent assembly of RAD51 filaments on their ssDNA portion, thus preventing nucleolytic attack and extensive fork degradation that is mediated by either CTIP, MRE11 or EXO1 (reviewed in 32, 33).
Lack of reversed fork protection in BRCA2‐deficient cells leads to a high level of genome instability and in particular to chromosomal breakage, as well as sensitivity to various chemotherapeutic drugs 34, 35. Thus, in genetic backgrounds with compromised fork stability, reversed forks represent crucial entry points for processing events that mediate chemosensitivity.
In this work, using the specific inhibitor XL413 and the AS‐CDC7 cell line, we describe the role of the CDC7 kinase in the dynamics of established replication forks. We find that CDC7 kinase promotes MRE11‐dependent processing of reversed replication forks and, by doing so, contributes to the modulation of replication fork speed and to chromosomal instability upon fork stalling, which is a hallmark of BRCA2‐deficient cells.
Results
CDC7 promotes replication fork processing and collapse in HU
CDC7 kinase is known to be involved in DNA replication initiation and checkpoint signalling 8, 9, 36, 37, 38, 39. To further characterize CDC7‐dependent events in the replication stress response, we treated U2OS osteosarcoma‐derived cancer cells and MCF10A breast‐derived immortalized cells with HU in the presence or absence of the CDC7 inhibitor XL413. With both cell lines, we observed that HU‐induced phosphorylation of RPA2—the middle subunit of the ssDNA‐binding protein RPA—was greatly reduced, as assessed by either altered electrophoretic mobility or specific anti‐phosphopeptide antibodies. In a time‐course experiment, the suppression of RPA phosphorylation in CDC7i‐treated cells was observed as early as 2 h post‐treatment and throughout the course of the experiment (Fig 1A). Intriguingly, the phosphorylation of H2AX at serine 139 (γH2AX) that occurs upon fork stalling and is further amplified upon fork collapse and formation of DNA double‐strand breaks (DSBs) 40 was also partially suppressed in XL413‐treated U2OS (Fig 1A). Similar results were obtained with breast‐derived immortalized MCF10A cells (Fig EV1A). In order to confirm that the suppression of RPA2 and H2AX phosphorylation in response to HU was specifically due to CDC7 kinase inhibition, we used an analogue‐sensitive CDC7 cell line in which the activity of the kinase can be partially downregulated with the bulky ATP competitive inhibitor 3MB‐PP1 25. Treatment of AS‐CDC7 cells with 3MB‐PP1 reduced both RPA2 and H2AX phosphorylation occurring in HU‐treated cells to a similar extent as XL413 in the isogenic MCF10A cells (Fig EV1A and B), suggesting that when CDC7 is inhibited, fewer DNA DSBs are generated and/or stalled forks are differently processed.
Figure 1. CDC7 inhibition suppresses histone H2AX and RPA2 phosphorylation and DNA double‐strand break formation upon fork stalling.
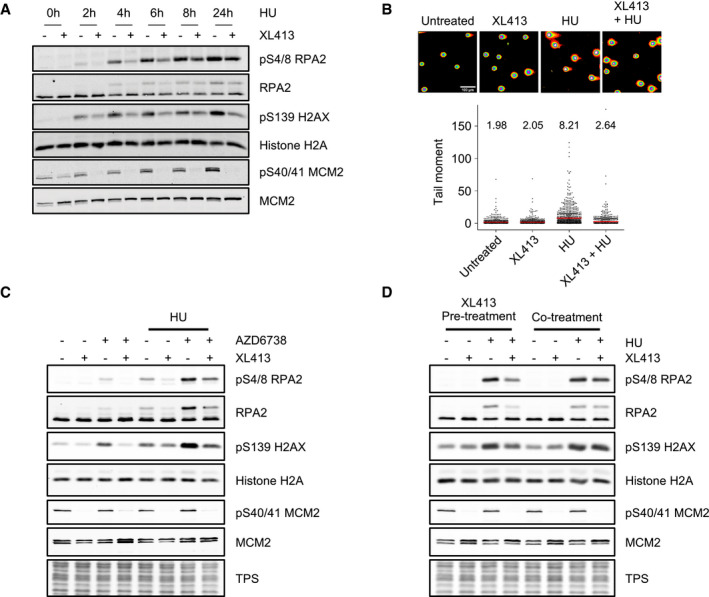
- U2OS cells were either mock‐treated or treated with 10 μM XL413 for 30 min, at which point 4 mM HU was added and cells further incubated for the indicated times. Whole‐cell extracts were then analysed by Western blotting with the indicated antibodies. Reduction of phosphorylation of Ser40/41 on MCM2 is indicative of CDC7 inhibition by XL413. Data are representative of two independent experiments.
- U2OS cells were either mock‐treated or treated with 10 μM XL413, 4 mM HU or both for 24 h before performing neutral comet assays. Representative images of comets are shown. Scale bar = 100 μm. In the dot plots, ˜800 comets per each condition were analysed, means are indicated with a red line, and their values are shown above the plot. Data are from two independent experiments.
- U2OS cells were either untreated or treated with 10 μM XL413 or with 5 μM AZD6738 alone or in combination and, where indicated, after 30 min, 4 mM HU was added for a further 2 h. Whole‐cell extracts were analysed by Western blotting with the indicated antibodies. Total protein stain (TPS) is as a loading control. Data are representative of two independent experiments.
- U2OS cells were either untreated or treated with 4 mM HU for 5 h in the presence or absence of 10 μM XL413, which had been added to cells either 30 min prior to (pre‐treatment) or during (cotreatment) the addition of HU. Whole‐cell extracts were analysed by Western blotting with the indicated antibodies, and total protein stain (TPS) is displayed as a loading control. Data are representative of two independent experiments.
Source data are available online for this figure.
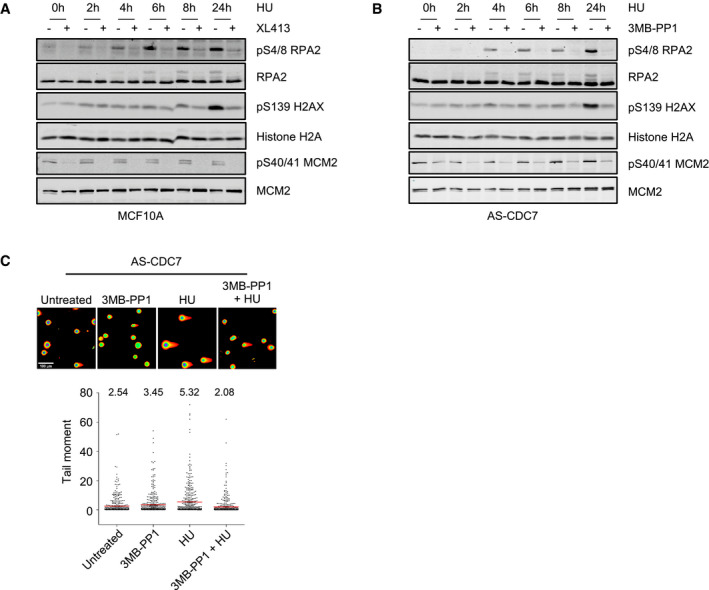
Figure EV1. (related to Fig 1): Inhibition of CDC7 with XL413 in MCF10A cells and with 3MB‐PP1 in AS‐CDC7 cells suppresses histone H2AX and RPA2 phosphorylation and DNA double‐strand break formation in HU.
-
A, BMCF10A or AS‐CDC7 cells were either mock‐treated or treated with 10 μM XL413 for 30 min, at which point 4 mM HU was added and cells further incubated for the indicated times. Whole‐cell extracts were then analysed by Western blotting with the indicated antibodies. Data are representative of at least two independent experiments.
-
CAS‐CDC7 cells were either mock‐treated or treated with 10 μM 3MB‐PP1, 4 mM HU or both for 24 h before performing neutral comet assays. Representative images of cells are shown. Scale bar = 100 μm. In the dot plots, ˜400 comets per each condition were analysed, means are indicated with red lines, and their values are shown above the plots. Data are from two independent experiments.
Source data are available online for this figure.
To directly assess if CDC7 promotes formation of DNA DSBs from stalled forks, we then performed neutral comet assays on U2OS cells. Following exposure of cells to HU for 24 h with or without XL413, we observed that CDC7 inhibition significantly reduced the tail moment, which is proportional to the number of DNA DSBs in the cell (Fig 1B). The formation of DNA DSBs in HU was also suppressed when CDC7 was inhibited with 3MB‐PP1 in the AS‐CDC7 cell line (Fig EV1C).
As DNA DSBs in HU can arise from replication fork collapse, which is actively prevented by ATR signalling 41, 42, we tested the effects of CDC7i in the presence of the ATR inhibitor AZD6738 finding that the addition of XL413 strongly decreased RPA2 and H2AX hyper‐phosphorylation (Fig 1C).
In all previous experiments, the CDC7 inhibitor was added 30 min before HU, as we had previously shown that CDC7 inhibition can only delay the onset of checkpoint signalling (i.e. CHK1 phosphorylation) before it is established 8. Consistent with these early findings, we found that unlike a short pre‐treatment with the CDC7i, simultaneous cotreatment of cells with CDC7i and HU was unable to prevent the phosphorylation of neither RPA2 nor H2AX (Fig 1D).
These observations suggest that CDC7 activity is required for replication fork processing or collapse upon sustained fork arrest, particularly in the absence of proper checkpoint signalling. However, as CDC7 is also required for origin firing, they may also reflect fewer active forks present at the moment of HU addition in cells cotreated with CDC7i, thus limiting fork‐associated phosphorylation events.
CDC7 drives H2AX phosphorylation in HU independently from origin firing and in a MRE11‐dependent manner
In order to possibly uncouple CDC7 function in initiation and at elongating forks, we first performed a titration from 0.123 to 10 μM of the CDC7 inhibitor XL413 in U2OS cells and accurately measured by flow cytometry the amount of DNA synthesis occurring at different doses as well as the amount of H2AX phosphorylation when CDC7i‐treated cells were also challenged with HU. This analysis indicated that very low levels of CDC7i had almost no effects on the rate of EdU incorporation which decreased then in a dose‐dependent manner at higher concentrations. Conversely, already at the lowest concentration of XL413, we detected a profound reduction on HU‐induced H2AX phosphorylation. These data suggest a different level of requirement for CDC7 activity in promoting efficient DNA synthesis and in driving HU‐dependent H2AX phosphorylation and suggest that key targets of CDC7 activity in initiation and at forks may be different or differently regulated (Fig EV2).
Figure EV2. Partial inhibition of CDC7 has a minor effect on DNA synthesis but markedly affects pS139 H2AX phosphorylation.
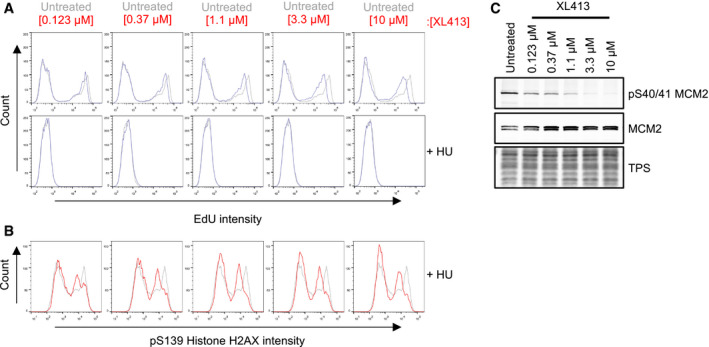
- Mono‐parametric analysis of cell count against EdU intensity. Histograms are overlaid to appreciate changes in EdU intensity upon treatment with XL413 (blue lines) relative to the untreated control (grey lines).
- Mono‐parametric analysis of cell count against pS139 histone H2AX intensity in HU‐treated cells. Histograms are overlaid to appreciate changes in pS139 histone H2AX intensity upon treatment (red lines) relative to untreated controls (grey lines).
- Whole‐cell extracts were analysed by Western blotting with the indicated antibodies, and total protein stain (TPS) is used as a loading control. Reduction of phosphorylation of Ser40/41 on MCM2 is indicative of CDC7 inhibition. Data are representative of two repeat experiments.
If CDC7 is indeed required to promote fork collapse independently from its role in initiation, we reasoned that allowing cells to enter S‐phase and be held in HU in the presence of CDC7i would allow the following events to occur: (i) the arrest of existing forks; (ii) checkpoint activation, which would prevent further origin firing and further loading of the initiator factor CDC45, as well as checkpoint‐dependent contribution in protecting existing forks from collapse; and (iii) further fork stabilization caused by CDC7 inhibition. Under these conditions, the subsequent removal of the CDC7i would possibly cause fork destabilization, while keeping strong checkpoint signalling and HU‐mediated prevention of further initiation events. Conversely, checkpoint inhibition would simultaneously cause both fork collapse and origin activation.
To test these hypotheses, U2OS cells were treated with XL413 and HU for 24 h; then, still in the presence of HU, XL413 was either kept or washed off and cells incubated for a further 2 h (Fig 2A). As a control for the checkpoint‐dependent suppression of both origin firing and replication fork stabilization, the ATRi AZD6738 was included. Proteins were then analysed by Western blotting (Fig 2B) and the levels of H2AX phosphorylation quantitatively assessed by flow cytometry (Fig 2C). In this assay, treatment with HU and XL413 for 24–26 h arrested cells in S‐phase with active checkpoint and low levels of γH2AX (Fig 2B, lanes 4 and 6; and Fig 2D). However, removal of CDC7i after 24 h clearly caused 2 h later a marked increase in γH2AX and CHK1 phosphorylation (Fig 2B, lanes 4 and 5; and Fig 2D). Importantly, this was not accompanied by an increase in chromatin‐bound CDC45, which is a surrogate marker for origin firing. Conversely, CDC45 was clearly and expectedly increased upon treatment with the ATRi AZD6738 and occurred in the absence of detectable EdU incorporation (Appendix Fig S1).
Figure 2. CDC7 inhibition suppresses histone H2AX phosphorylation independently from origin firing.
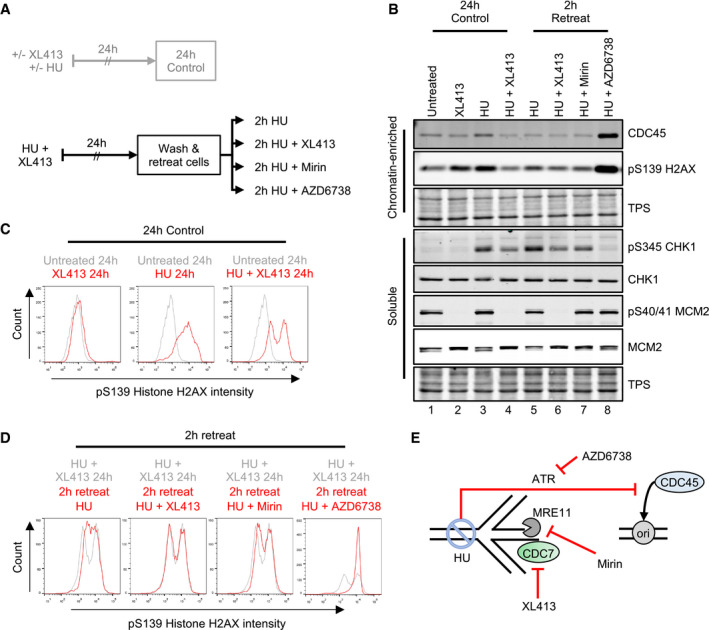
-
AOutline of experimental procedure. U2OS cells were either untreated or treated with 10 μM XL413 or with 4 mM HU in the presence or absence of XL413 for 24 h. Cells were EdU‐labelled for 30 min before harvesting. Cells that had been treated with both XL413 and HU for 24 h were washed with equilibrated media and retreated for a further 2 h. All further treatments included 4 mM HU, to prevent further DNA synthesis in either the absence or presence of CDC7 (XL413), MRE11 (Mirin) or ATR (AZD6738) inhibitors.
-
BCSK soluble and chromatin‐enriched protein fractions were analysed by Western blotting with the indicated antibodies. Data are representative of two independent experiments.
-
C, DFlow cytometry analysis to assess the levels of pS139 histone H2AX (c). Histograms show the mono‐parametric analysis of cell count against pS139 histone H2AX intensity. Histograms are overlaid to appreciate changes in pS139 histone H2AX intensity upon treatment (red lines) relative to appropriate experimental baseline controls (grey lines). Data are representative of two independent experiments.
-
EGraphical concept: cells treated with HU and XL413 for 24 h would have stalled forks, which exist in a stable state due to inhibition of CDC7 and with an active ATR‐dependent origin firing checkpoint. Upon washing and retreating of cells in the presence of HU, the late origin checkpoint is maintained, in an ATR‐dependent manner, and accumulation of DNA damage (pS139 H2AX) can be monitored independently from origin firing in the absence or presence of CDC7 (XL413) or MRE11 (Mirin) inhibitor. AZD6738 was used as a control to investigate molecular events occurring upon loss of checkpoint caused by both fork collapse and loss of the inhibition of origin firing. The increase in chromatin binding of CDC45 was used as a surrogate marker for origin (ori) activation, pS139 histone H2AX was used to monitor fork stability, pS345 CHK1 was used to monitor the ATR‐checkpoint signalling, and pS40/41 MCM2 was used to monitor CDC7 kinase activity.
Source data are available online for this figure.
Fork collapse and DSB formation are thought to be due to the loss of protection from the attack of several active nucleases, including MRE11 and EXO1. While EXO1 levels were previously reported to be downregulated by a promiscuous CDC7 inhibitor, PHA‐767491 12, these were not affected by XL413 (Appendix Fig S2). We therefore tested MRE11 involvement in H2AX phosphorylation after the removal of CDC7 inhibition in HU. Indeed, MRE11 inhibition by mirin 43 strongly limited histone H2AX and CHK1 phosphorylation to almost similar levels as maintaining the CDC7 inhibition (Fig 2B, lanes 5–7; and Fig 2D).
Altogether, these experiments strongly support the notion that CDC7 activity promotes MRE11‐dependent processing of stalled replication forks, independently from its role in regulating origin firing (Fig 2E), which can lead to checkpoint signalling amplification.
CDC7 and MRE11 associate with nascent DNA, and CDC7 is required for maintenance of MRE11 localization at replication factories
To further corroborate the hypothesis that CDC7 and MRE11 may act directly at replication forks, we performed a series of experiments to assess whether these proteins could be specifically captured on nascent DNA by DNA‐mediated chromatin pull‐down (Dm‐ChP), a technique which is very similar to iPOND 44, 45. Firstly, in a pulse chase experiment we observed that the two proteins could be efficiently cross‐linked to newly synthesized DNA, while their levels were substantially reduced on mature chromatin (Fig 3A), which is also consistent with previous studies coupled to mass spectrometry 45, 46. Both proteins could be still detected at replication forks when their progression was arrested by HU up to 24 h, but CDC7 inhibition notably reduced their retention at forks (Fig 3B and C).
Figure 3. CDC7 is a replisome‐associated protein, and its inhibition affects the localization of MRE11 at replication factories.
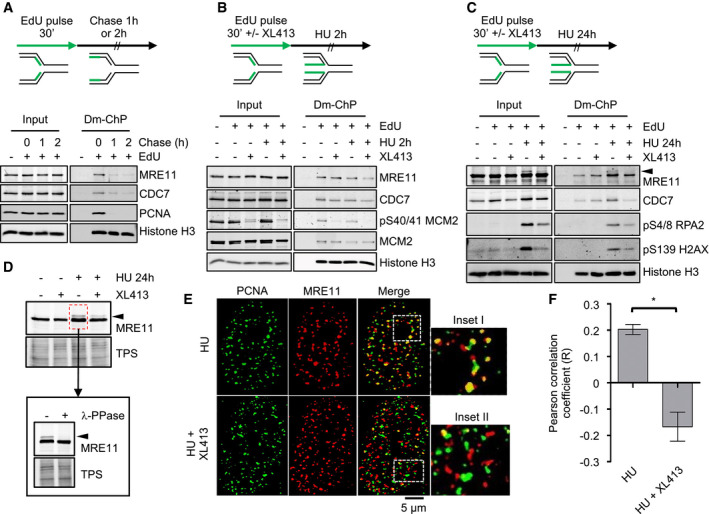
- U2OS cells were labelled with EdU for 30 min; then, EdU was washed, and cells were further incubated for either 1 or 2 h in the presence of thymidine. At the indicated time points, cells were fixed and proteins binding to EdU‐labelled DNA captured by the DNA‐mediated chromatin pull‐down technique (DmChP). Graphical experimental outline is shown above the analysis by Western blot of relevant proteins in both input and captured materials.
- U2OS cells were labelled with EdU for 30 min and then treated with 4 mM HU for 2 h in the presence or absence of 10 μM XL413. Proteins binding to EdU‐labelled DNA captured by DmChP were then analysed by Western blot as above.
- As in panel B, but incubation with HU was extended to 24 h. Black arrow indicates MRE11 electrophoretic mobility shift.
- U2OS cells were either mock‐treated or treated with 10 μM XL413 for 30 min, at which point 4 mM HU was added and cells incubated for a further 24 h. Extracts prepared from HU‐treated cells were then incubated in the presence or absence of λ‐phosphatase. Proteins were analysed by Western blotting with anti‐MRE11 antibodies. Total protein stain (TPS) is used as a loading control. Black arrow indicates MRE11 electrophoretic mobility shift.
- U2OS cells were treated with 4 mM HU in the presence or absence of 10 μM XL413 for 24 h. PCNA (green) and MRE11 (red) were detected by immunofluorescence. Insets I–II represent enlargements of selected region of the merged images.
- Quantification of PCNA and MRE11 colocalization was assessed with ImageJ in ˜70 randomly selected cells for each condition from four biological replicates and expressed as Pearson's correlation coefficient. Error bars represent SEM. Statistical significance was assessed by Student's t‐test (*P ˂ 0.05).
Source data are available online for this figure.
During the course of these experiments, we noticed that after a prolonged arrest in HU, a small fraction of MRE11 protein, which was also captured on stalled forks, was retarded in its electrophoretic mobility, and that its abundance was markedly decreased upon CDC7 inhibition (Fig 3C). Subsequently, we found that MRE11 altered electrophoretic mobility was sensitive to the method of protein extraction and that a mild treatment of cells with PFA before lysis greatly helped in its detection. We reasoned that this could be due to a post‐translational modification, most likely due to phosphorylation, which could be easily lost in native extracts. To test this hypothesis, new extracts were prepared from U2OS cells that had been either mock‐treated or treated with HU. Again, a slower migrating form of MRE11 was detected in HU‐treated cells, which was decreased by cotreatment with XL413. Importantly, MRE11 electrophoretic mobility shift was completely lost upon incubation of these extracts with purified lambda phosphatase (Fig 3D). Altogether, these experiments suggest that CDC7 may control MRE11 by regulating its phosphorylation status.
In order to refine our analysis, we assessed MRE11 localization by immunofluorescence microscopy. In these experiments, after drug treatments and fixation, cells were stained with anti‐MRE11 and anti‐PCNA antibodies. As previously described, MRE11 can be detected in a large number of nuclear foci and in HU‐arrested cells with a high degree of colocalization with PCNA, indicative of its association with replication factories 35, 47. Treatment with the CDC7 inhibitor markedly reduced the degree of colocalization of the two proteins (Fig 3E and F). Partial colocalization of MRE11 with RPA2 was also reduced by CDC7 inhibition, albeit to a lesser extent (Fig EV3).
Figure EV3. CDC7 inhibition decreases colocalization of MRE11 with RPA2.
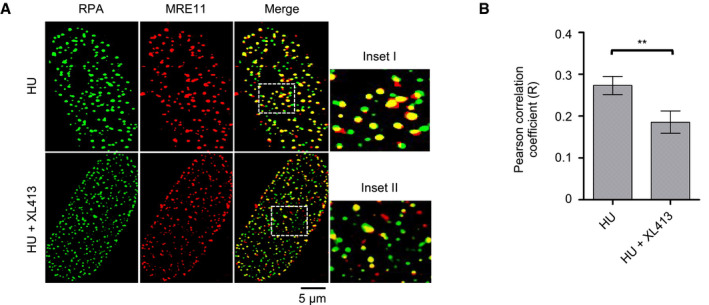
- U2OS cells were treated with 4 mM HU in the presence or absence of 10 μM XL413 for 24 h. RPA2 (green) and MRE11 (red) were detected by immunofluorescence. Insets I–II represent enlargements of selected region of the merged images.
- Quantification of RPA2 and MRE11 colocalization was assessed with ImageJ in ˜200 randomly selected cells for each condition from three biological replicates and expressed as Pearson's correlation coefficient. Error bars represent SEM. Statistical significance was assessed by Student's t‐test (**P ˂ 0.01).
Source data are available online for this figure.
We conclude that both CDC7 and MRE11 associate with nascent DNA and are retained at forks upon prolonged HU stalling. However, a discrete pool of MRE11 protein requires CDC7 activity to retain its localization at replication forks, possibly because of CDC7 control of MRE11 phosphorylation status.
CDC7 contributes to fork restart and determines fork speed
In order to evaluate the role of CDC7 at paused forks, before their collapse, we measured the efficiency of fork restart after a transient HU arrest. In a first set of experiments using MCF10A cells, ongoing forks were marked with IdU in the presence or absence of CDC7i, cells were then treated with HU for 2 h, after which DNA synthesis was allowed to resume by removing HU and new DNA synthesis was labelled with CldU. Notably, CDC7 inhibition markedly increased the fraction of forks that were unable to restart within the time frame of the experiment (Fig 4A). These results suggest that a CDC7‐dependent event is specifically required for efficient resumption of temporary arrested replication forks, and remind of a similar deficiency in fork restart described upon depletion of the MRE11 nuclease 48, which we confirmed in U2OS cells upon MRE11 inhibition with mirin (Fig 4B). The addition of CDC7i did not significantly exacerbate the fork restart defect induced by mirin (Fig 4B), suggesting that CDC7 and MRE11 are working at a common function in fork restart.
Figure 4. CDC7 is required for efficient fork restart and, together with MRE11, regulates fork speed.
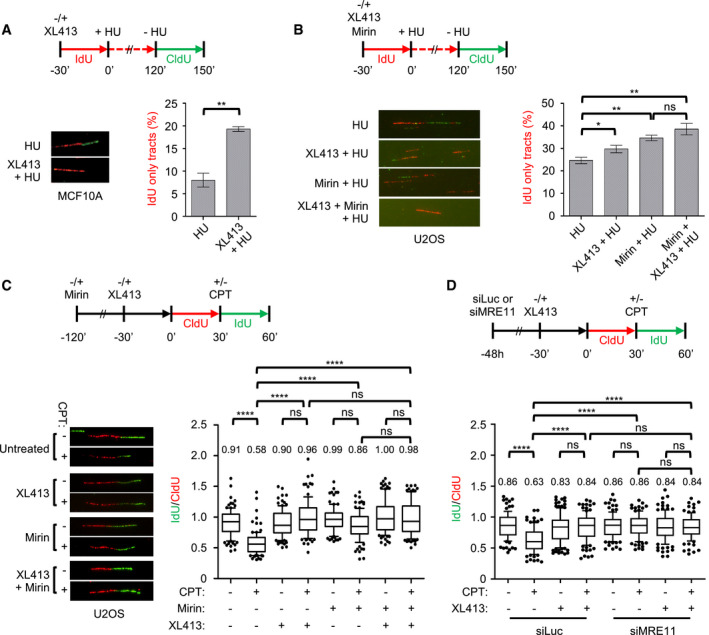
- MCF10A cells were either mock‐treated or treated with 10 μM XL413 and labelled with IdU (red) for 30 min, at which point 4 mM HU was added for 2 h. HU was then washed off and cells labelled with CldU (green) in the continued presence or absence of XL413. A set of representative DNA fibres from each condition is shown, and the percentage of IdU (red)‐only tracts is plotted. At least 200 replication forks were analysed for each condition. Error bars represent SEM from three biological repeats, and statistical significance was assessed by the Student t‐test (**P ˂ 0.01).
- U2OS cells were either mock‐treated or treated with 10 μM XL413, 50 μM Mirin or both and labelled with IdU (red) for 30 min, at which point 4 mM HU was added for 2 h. HU was then washed off and cells labelled with CldU (green) in the continued presence or absence of XL413 and/or Mirin. A set of representative DNA fibres from each condition is shown, and the percentage of IdU (red)‐only tracts is plotted. At least 200 replication forks were analysed for each condition. Error bars represent SEM from four biological repeats, and statistical significance was assessed by the Student t‐test (*P < 0.05, **P ˂ 0.01).
- U2OS cells were either mock‐treated or pre‐treated for the indicated times with 10 μM XL413, 50 μM Mirin or both and labelled with CldU (red). Then, 50 nM CPT was added and cells labelled with of IdU (green) for a further 30 min. A set of representative DNA fibres from each condition is shown and the IdU/CldU tract length ratio is plotted. The box extends from the 25th to the 75th percentile with the line in the box representing the median. Whiskers indicate the 10–90 percentiles with data outside this range for individual outliers being plotted as dots. At least 100 replication forks were analysed for each condition. P‐values were calculated using one‐way ANOVA, Kruskal–Wallis test and Dunn's multiple comparison post‐test (ns, not significant; ****P < 0.0001) and are related to the experiment shown. Similar results were obtained in a second independent experiment.
- U2OS cells were transfected with control (siLuc) or MRE11‐targeting siRNA (siMRE11). After 48 h, cells were either mock‐treated or pre‐treated with 10 μM XL413 and labelled with CldU (red) for 30 min. Where indicated, 50 nM CPT was added, and then, cells were labelled with IdU (green). A set of representative DNA fibres from each condition is shown, and the IdU/CldU tract length ratio is plotted. The box extends from the 25th to the 75th percentile with the line in the box representing the median. Whiskers indicate the 10–90 percentiles with data outside this range for individual outliers being plotted as dots. At least 100 replication forks were analysed for each condition. P‐values were calculated using one‐way ANOVA, Kruskal–Wallis test and Dunn's multiple comparison post‐test (ns, not significant; ****P < 0.0001) and are related to the experiment shown. Similar results were obtained in two other independent experiments.
Source data are available online for this figure.
We then asked if CDC7 activity was also required upon treatments that slow down but do not terminally arrest forks. Low levels of camptothecin (CPT), a topoisomerase I inhibitor, have been shown to induce a large number of reversed forks and substantially reduce replication fork speed in the absence of detectable chromosomal breakage 27. As fork slowdown under these conditions was shown to genetically depend on fork reversal activities 26, 49, it was suggested that CPT induces fork slowing by increasing topological stress and transiently pausing DNA synthesis at forks converted in a chicken‐foot conformation. Using DNA fibre assay, we therefore assessed the effect of CDC7 and MRE11 inhibition in combination with low levels of CPT. In these experiments, replication intermediates were first labelled with CldU, and then, 50 nM CPT was added together with IdU; the change in the velocity of the forks during drug treatment was measured as the ratio between CldU‐ and IdU‐containing tracks in the same DNA fibre (Fig 4C). As previously shown, CPT treatment drastically reduced the length of the DNA molecules synthesized during the second labelling period, consistent with reduced fork speed. Remarkably, when XL413 was added prior to CPT, IdU‐labelled tracks were of similar length to those observed in mock‐treated cells. Strikingly, the addition of mirin (Fig 4C) or MRE11 depletion (Fig 4D) was also sufficient to allow unrestrained fork progression in the presence of CPT. The combination of XL413 with mirin or MRE11 depletion did not result in further enhancement of fork speed compared to XL413 alone (Fig 4C and D).
Altogether, these results indicate that CDC7 and MRE11 act epistatically in the processing of replication forks facing a transient arrest. As neither CDC7 nor MRE11 inhibition was shown to affect damage‐induced formation of reversed forks 50, 51, these data also strongly suggest that, in addition to fork remodelling, active slowing of replication forks requires CDC7‐ and MRE11‐dependent processing of newly synthesized DNA.
CDC7 promotes the degradation of reversed forks
Previous studies have indicated that HU‐arrested replication forks undergo reversal 28. Regressed arms at these remodelled forks are coated by RAD51 filaments in a BRCA2‐dependent manner and, upon BRCA2 deficiency, are readily subjected to aberrant nucleolytic attack and MRE11‐dependent degradation 35, 50, 52, 53, 54, 55.
In order to monitor the effects of CDC7 inhibition on arrested replication forks, we implemented a fork degradation assay based on DNA fibre spreading. For this purpose, U2OS cells were labelled with two consecutive 20‐min pulses of nucleoside analogues (CldU and IdU) and then treated with HU for 5 h in the presence or absence of XL413 (Fig 5A). The degradation of arrested forks was then assessed by calculating the ratio between the length of IdU‐ over CldU‐containing tracks in individual fibres. These experiments were performed in cells transfected with control or BRCA2‐targeting siRNAs, with the goal to assess the impact of CDC7 inhibition on fork protection mechanisms (Fig 5B). We observed that CDC7 inhibition had no obvious effect in U2OS cells transfected with control siRNA and that, as expected, forks were degraded in BRCA2‐depleted cells. Notably, we found that the addition of the CDC7 inhibitor together with HU fully protected replication forks from degradation (Fig 5C and D).
Figure 5. CDC7 promotes the nucleolytic degradation of reversed forks in BRCA2‐deficient cells.
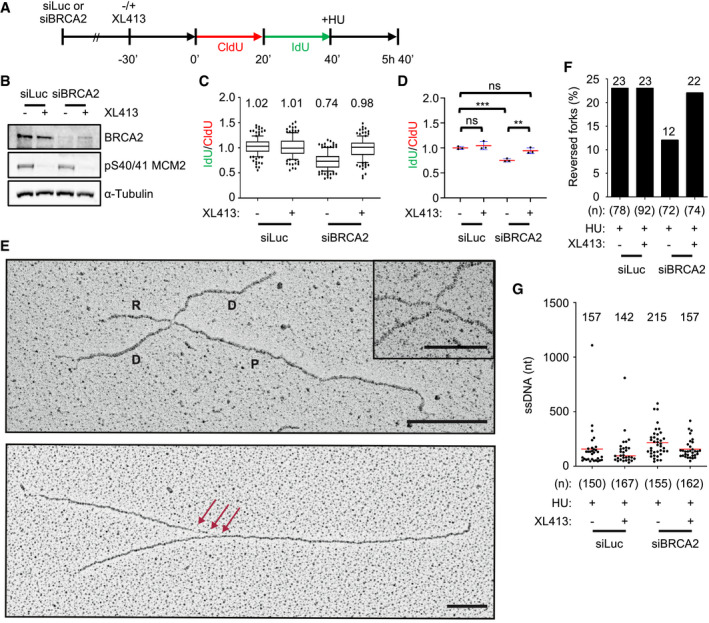
- U2OS cells were transfected with control (siLuc) or BRCA2‐targeting siRNA (siBRCA2). After 48 h, cells were either mock‐treated or treated with 10 μM XL413 for 30 min and labelled consecutively with CldU (red) and IdU (green), followed by treatment with 4 mM HU for a further 5 h.
- Representative Western blot of indicated proteins obtained from whole‐cell extracts. pS40/41 on MCM2 indicates effective CDC7 inhibition by XL413.
- The ratios of IdU/CldU tract length are plotted. The box extends from the 25th to the 75th percentile with the line in the box representing the median. Whiskers indicate the 10–90 percentiles with data outside this range for individual outliers being plotted as dots. Mean values of IdU/CldU ratios are indicated above the plot. At least 100 replication forks were analysed for each condition.
- The mean values of IdU/CldU tract length ratios from three independent experiments are plotted, with the standard deviation (blue lines) and mean (red line). Statistical analysis: Student's t‐test; ns, not significant; **P < 0.01; ***P < 0.001
- Electron micrographs of representative replication forks from U2OS cells: A reversed fork is labelled: P = parental strand; D = daughter strand; and R = regressed arm; the four‐way junction at the reversed fork is magnified in the inset. The red arrows indicate single‐stranded DNA at the fork. Scale bar is 200 nm (= 460 bp) and 100 nm (= 230 bp) in the inset.
- Frequency of reversed replication forks isolated from mock‐depleted (siLuc) and BRCA2‐depleted (siBRCA2) U2OS cells upon 5‐h treatment with 4 mM HU in the presence or absence of 10 μM XL413. The number of replication intermediates analysed is indicated in parentheses. Similar results were obtained in two independent experiments (Appendix Table S1).
- Amount of ssDNA length at the junction (red arrows in Fig 4D) in siLuc and siBRCA2 U2OS cells treated with 4 mM of HU for 5 h in the presence or absence of XL413. N indicates the number of forks observed, and only molecules with detectable ssDNA stretches are included in the analysis. The lines show the mean length of ssDNA regions at the fork, and the value is displayed above the plot.
Source data are available online for this figure.
In principle, the rescue from degradation by CDC7i in BRCA2‐deficient cells could be consistent with defective formation of reversed forks—i.e. the entry point for fork degradation—or with induced protection of reversed forks from nuclease digestion despite the BRCA2 defect. To discriminate among these possibilities, we used electron microscopy (EM) to directly analyse the replication intermediates that accumulate in HU‐treated cells (Fig 5E). When specifically assessing the percentage of detectable reversed forks in the different samples, we found that in wild‐type cells, the inclusion of the CDC7 inhibitor during the treatment with HU did not significantly alter their frequency, in keeping with previous evidence upon DNA‐damaging agents 51. As previously shown 35, 50, 52, 53, 54, 55, BRCA2 depletion causes the loss of reversed fork structures in the sample. However, we strikingly found that CDC7 inhibition completely rescues the decrease in reversed fork frequency observed in BRCA2‐depleted cells (Fig 5F and Appendix Table S1). In agreement with the results obtained with the DNA fibre assay, EM analysis also indicated that CDC7 inhibition abolishes the accumulation of extended ssDNA regions, which are observed at stalled forks upon BRCA2 depletion (Fig 5G). Furthermore, immunofluorescence microscopy analysis of HU‐treated BRCA2‐depleted cells showed that CDC7 inhibition did not rescue the focal recruitment of RAD51, indicating that in these conditions, arrested forks are not protected by enforcing RAD51 filament formation (Appendix Fig S3), and further suggesting a role for CDC7 in fork resection. We therefore conclude that CDC7 kinase does not affect the formation of reversed forks but promotes their degradation when fork protection is lost.
CDC7 contributes to replication stress‐induced chromosomal breakages
As fork stalling is a main source of chromosomal damage and genome instability (reviewed in 56), we assessed the effects of CDC7 inhibition on the integrity of mitotic chromosomes in WT and BRCA2‐depleted cells optionally treated with HU.
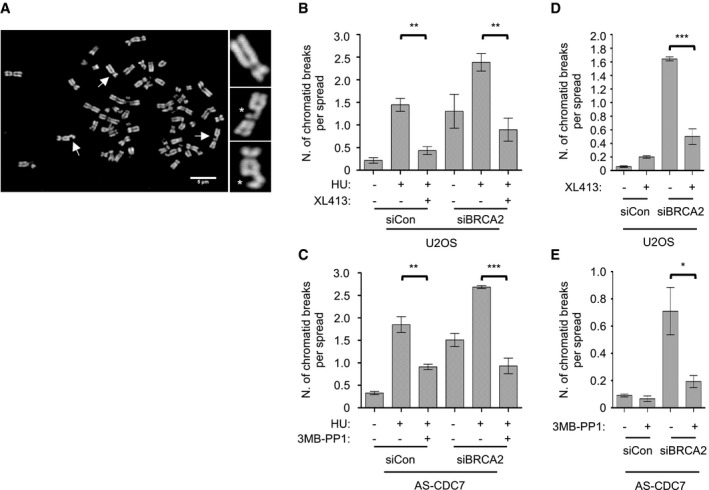
In a first set of experiments, U2OS cells and AS‐CDC7 MCF10A cells were incubated with HU for 5 h with DMSO as control or with XL413 or 3MB‐PP1, respectively, and then released into nocodazole for 16 h, in order to complete DNA synthesis before arresting in mitosis. Metaphase spreads were prepared, and chromosome integrity was assessed by fluorescence microscopy (Fig 6A). HU treatment greatly increased the average number of chromatid breaks in both cell lines. However, when a CDC7 inhibitor was included in the medium, the levels of chromosome breaks were dramatically reduced, regardless of their BRCA2 status (Fig 6B and C).
Figure 6. CDC7 contributes to replication‐dependent chromosomal breakages.
-
ARepresentative metaphase spread from BRCA2‐depleted U2OS cells treated with HU. Scale bar = 5 μm. Arrows indicate the chromosomes enlarged in the insets, two of which show chromatid breaks (*).
-
B, CThe graphs show the average number of chromatid breaks per spread. In each experiment, 30 chromosome spreads for each condition were analysed and three independent experiments were performed. Error bars represent SEM. Statistical significance was assessed by Student's t‐test (**P ˂ 0.01, ***P < 0.001).
-
D, EU2OS and AS‐CDC7 cells were transfected with control (siCon) or BRCA2 (siBRCA2)‐targeting siRNAs. Forty‐eight hours post‐transfection, cells were either mock‐treated or treated with 10 μM XL413 or 10 μM 3MB‐PP1, respectively, for 24 h with nocodazole added for the last 16 h of the experiment. The graph shows the average number of chromatid breaks per spread. In each experiment, 30 chromosome spreads for each condition were analysed and three independent experiments were performed. Error bars represent SEM. Statistical significance was assessed by Student's t‐test (*P < 0.05, ***P < 0.001).
Since in BRCA2‐deficient cells chromatid breaks arise at higher frequency even without HU—possibly as a consequence of the loss of protection of forks stalling at endogenous obstacles and MRE11‐dependent degradation 34—we tested whether CDC7 also contributes to spontaneous chromosome aberrations. U2OS or AS‐CDC7 cells were transfected with a control or BRCA2‐targeting siRNAs and then either mock‐treated or treated for 24 h with XL413 or 3MB‐PP1, respectively, before metaphase spread analysis. Notably, partial inhibition of CDC7 strongly reduced chromosome damage also in untreated BRCA2‐depleted cells (Fig 6D and E).
Although it is possible that the effect of CDC7 inhibition on decreasing chromosome breaks might be in part mediated by a reduction in the number of active forks, these results are also consistent with the idea that, in the absence of fork protection, CDC7 kinase promotes MRE11 nuclease attack of reversed forks leading to chromosomal instability.
Discussion
The duplication of human genomic DNA poses numerous threats to genome integrity, as a large quantity of DNA needs to be rapidly synthesized, containing endogenous obstacles to replication fork progression. This task is achieved through the robust and temporally regulated activation of numerous origins of replication and by modulation of the speed of replication forks which temporarily pause and restart. Furthermore, several safeguard mechanisms evolved to protect fork integrity, repair DNA lesions arising at forks and modulate further origin activation.
CDC7 kinase and CDKs are the two master S‐phase‐promoting factors that control origin activation. It is well established that CDKs regulate origin firing with CDK‐dependent phosphorylation events regulating origin licensing and usage (reviewed in 57, 58). At the same time, CDKs control the activity of enzymes required for dealing with replication stress, including the CtIP and EXO1 nucleases and the MRN complex 59, 60.
In this work, we demonstrate that CDC7 kinase behaves similarly to CDKs, with the dual function of promoting origin firing and contributing to the processing of paused forks, leading to efficient restart and effective DNA synthesis. Our phenotypical analysis strongly points to the possibility that the primary target of CDC7 regulation in fork processing is the MRE11 nuclease. CDC7 inhibition mirrors many phenotypes that have been ascribed to MRE11 inhibition, strongly indicating that these two proteins act together, at both unchallenged and stalled forks.
MRE11 processing is important for fork restart, but uncontrolled nuclease activity can be extremely detrimental and multiple mechanisms exist to restrict MRE11 activity (reviewed in 61). At paused replication forks, this is achieved through RAD51‐ and BRCA2‐dependent protection, preventing pathological degradation of nascent DNA starting from reversed forks 35, 50, 52, 53, 54, 55, 62. As CDC7 inhibition does not affect the formation of reversed forks 51, nor focal recruitment of RAD51, the rescue of fork stability by CDC7i in BRCA2‐deficient cells is most likely due to absence of pathological resection, consistent with CDC7 allowing MRE11 action at unprotected reversed forks.
Unlike a previous report 12, in our experiments we did not observe fork degradation occurring upon HU treatment in WT cells, where fork protection is efficient, and therefore, we were unable to measure the effects of CDC7i in this context. However, the fact that CDC7 inhibition, also in WT cells, prevents the accumulation of DNA DSBs in response to HU and limits chromatid breaks that originated from replication stress is a strong indication that fork protection can occasionally fail also in BRCA2‐proficient cells, allowing access of nucleases and leading to fork collapse. Fork collapse in HU can be highly stimulated by deficiency in the ATR/CHK1 checkpoint signalling 41, 63; remarkably, we and others find that such excessive fork collapse in the presence of HU and checkpoint inhibitors can be limited by both promiscuous and specific CDC7 inhibitors (Fig 1C) 12, 63. Although this effect of CDC7 inhibition was previously attributed to reduced levels of origin firing and thereby of active forks prone to collapse, we now propose that CDC7 actively contributes to fork instability and DNA damage by promoting the aberrant processing of unprotected forks by MRE11.
Of particular relevance is the new finding that both MRE11 and CDC7 are required to slow down replication forks in response to topological stress, indicating that whenever a fork pauses in a chicken‐foot conformation, DNA processing occurs and represents a major determinant of the temporal delay in fork progression. Intriguingly, long treatments with CDC7 inhibitors have been reported to increase fork speed in the presence of sub‐limiting doses of HU 64 or in the absence of other drugs 65, 66. In both cases, this was suggested to be caused by an ill‐defined compensatory mechanism that allows forks to accelerate when origin firing is limited, as it was shown that forks emanating from distantly spaced origins tend to move faster than those associated with short replicons 67. We now propose that besides its coordination with the number of active forks, fork speed is directly modulated by the engagement of nucleolytic processing during transient fork pausing and reversal. Besides fork reversal activities, this mechanism requires both CDC7 and MRE11 and actively keeps the fork in an idle status, transiently incompetent for DNA synthesis.
We further show that both the collapse and the pathological degradation of unprotected reversed forks require active MRE11 and CDC7. These deleterious effects on genome stability are likely to be the price to pay in order to promote an efficient S‐phase progression, which requires regulated processing to efficiently restart forks that encounter obstacles in their progression, such as tightly bound chromatin proteins, difficult‐to‐replicate DNA structures or collisions with the transcriptional machinery 29, 68, 69, 70. In that respect, it is not surprising that the S‐phase‐promoting kinase CDC7 may play a complex role in initiation, elongation and efficient restart of DNA synthesis.
CDC7 has already been reported to influence MRE11 activity through phosphorylation of the HSP90 chaperone, thus stabilizing a ATR–HSP90–HCLK2–MRE11 complex 71. Based on our data, it is tempting to speculate that CDC7 may regulate MRE11‐dependent processes through direct phosphorylation, promoting MRE11 nuclease activity and, in the long range, its retention at replication factories. Such direct kinase substrate model will need further investigation, but is already supported by the observation that fork‐bound MRE11 in HU displays a CDC7‐dependent phosphorylation mobility shift. Furthermore, several of the reported MRE11 phosphorylation sites resemble CDC7 preferred sites, which include S/T just before an acidic residue, as well as S/TpSP sites. As CDC7 is largely cell cycle regulated and, as we now show, is also a component of the replisome, such direct mechanism provides an obvious way to regulate in time and space MRE11 function at forks while restraining its activity outside S‐phase. However, our data do not exclude that other proteins may also be key CDC7 targets at stalled forks. Among plausible candidates is the MCM complex, which, in the absence of a robust CDC7 activity, readily becomes dephosphorylated in a RIF1/PP1‐dependent manner, correlating with replisome instability and impaired capability of resuming DNA synthesis after fork stalling 13. Further studies will be required to fully understand how CDC7 acts at forks, uncovering the effects of specific CDC7‐dependent phosphorylation events on given substrates and at specific phosphorylation sites.
Finally, these results have strong implications on the use of CDC7 inhibitors in a clinical setting. This is particularly important when considering CDC7 inhibitors in combination therapies with S‐phase genotoxic agents, which cause fork stalling and whose efficacy is dependent on the amount of DNA damage that is produced upon fork processing, in both BRCA2‐deficient and BRCA2‐proficient patients.
Materials and Methods
Cell culture
All cells were maintained at 37°C with 5% CO2 in a humidified atmosphere. U2OS (HTB‐96; ATCC) cells were cultured in DMEM supplemented with 10% (v/v) foetal bovine serum, 50 U/ml penicillin and 50 μg/ml streptomycin. MCF10A (CRL‐10317; ATCC) and derivative cell lines were cultured in DMEM supplemented with 5% (v/v) horse serum, 100 ng/ml cholera toxin, 10 μg/ml insulin, 20 ng/ml epidermal growth factor (Peprotech), 500 ng/ml hydrocortisone, 50 U/ml penicillin and 50 μg/ml streptomycin. MCF10A cells expressing analogue‐sensitive CDC7 (MCF10A AS‐CDC7) were generated as previously described 25.
Chemicals
Cell culture reagents and chemicals were obtained from Sigma‐Aldrich unless otherwise stated. Hydroxyurea (HU, H8627) and camptothecin (CPT, C9911) were used at concentrations of 4 mM and 50 nM, respectively. PHA‐767491 (3140; TOCRIS Bioscience), XL413 (synthesized in‐house) and 3MB‐PP1 (Cayman Chemical Co., 17860) were all used at a concentration of 10 μM, while AZD6738 and mirin (HY‐19323 and HY‐19959; MedChemExpress) were used at 5 and 50 μM, respectively. DMSO was used as a vehicle control.
siRNA transfections
Transfections were performed according to the manufacturer's instructions. Cells were seeded in 6‐well plates and, after 24 h, were transfected with siRNA targeting either BRCA2 (UUGACUGAGGCUUGCUCAGUUdTdT) 50, EXO1 (SI02665145; Qiagen) or siControl (GCAUAUCGUCGUAUACUAUdTdT) 25 at a final concentration of 75 nM using JetPrime reagent (related to Fig 6 and Appendix Fig S2). Downregulation of MRE11 (Silencer Select, 4392420, IDs8961; Ambion) or BRCA2 (related to Figs 4 and 5, respectively) was performed in 100‐mm dishes over a 48 h period using 40 nM final concentration of siRNA and RNAiMAX transfection reagent with siLuciferase as a control (CGUACGCGGAAUACUUCGAUUdTdT) 50.
Protein manipulations
Whole‐cell extracts for BRCA2 (related to Fig 5) were prepared by lysing cells in SDS buffer and resolved as previously described 50. Alternatively, cells were lysed in 1x Laemmli buffer for total cell lysates or fractionated using CSK buffer supplemented with protease and phosphatase inhibitors to obtain soluble and chromatin‐enriched cell fractionations as previously described 72. For analysis of proteins that were associated with nascent DNA, cells were plated at 9 × 106 cells/150 mm plate. Following treatments, cells were processed and analysed using the DNA‐mediated chromatin pull‐down (Dm‐ChP) technique as previously described 44. The MRE11 phosphorylation shift was detected using a protocol adapted from Dm‐ChP sample preparation. Briefly, after the appropriate treatments in culture, cells were rapidly washed twice with PBS and then incubated in media containing 1% PFA for 10 min at room temperature with rocking (40 rpm) prior to quenching with 0.125 M glycine for a further 10 min. Cells were then washed three times with ice‐cold PBS, harvested by scrapping and pelleted by centrifugation in microfuge tubes at 1,200 rpm for 5 min at 4°C. Cell pellets were lysed in CL buffer (50 mM HEPES [pH 7.8], 150 mM NaCl, 1.5 mM MgCl2, 0.5% NP‐40, 0.25% Triton X‐100, 10% glycerol) for 10 min at 4°C with rotation (12 rpm) before being centrifuged at 1,200 rpm for 5 min at 4°C. The supernatant was removed and the insoluble pellet was washed with wash buffer (10 mM Tris–HCl [pH 8.0], 140 mM NaCl, 0.5 mM DTT) for 10 min at 4°C with rotation (12 rpm) before being centrifuged at 1,200 rpm for 5 min at 4°C. The supernatant was removed and the insoluble pellet was resuspended in RIPA buffer (10 mM Tris–HCl [pH 8.0], 140 mM NaCl, 0.1% sodium deoxycholate, 0.1% SDS, 1% Triton X‐100) for 10 min at 4°C with rotation (12 rpm). Protein concentration was then determined using the BCA assay (ThermoFisher), and 20 μg of protein was analysed by SDS–PAGE. The lambda phosphatase assay was performed on RIPA lysates containing 20 μg of protein and involved incubating the samples for 30 min at 4°C with ~400 U of lambda phosphatase (P0753L, NEB) in 1x PMP (protein metallophosphatase) buffer containing 1 mM MnCl2. Samples were heat denatured at 95°C for 3 min in 1× Laemmli buffer and resolved on SDS–PAGE before Western blot analysis. Nitrocellulose membranes were stained with Fast Green (0.0001% w/v in 0.1% acetic acid) as a total protein stain and imaged at 700 nm on the Odyssey infrared imaging system (LI‐COR Biosciences). Membranes were destained (0.1 M NaOH, 30% methanol) for 10 min, washed with dH2O and then blocked (TBST, 5% milk) for 1 h, followed by sequential incubation with primary and secondary antibodies. Signals were acquired using the Odyssey infrared imaging system and analysed using Image Studio software, except in Fig 5B, where signals were detected using chemiluminescence and the Fusion Solo imaging system. Primary antibodies diluted in blocking buffer (TBST, 5% milk) included RPA2 (1:1,000, NA19L; Calbiochem), Histone H2A (1:1,000, 07‐146; Upstate), MRE11 (1:1,000, NB100‐142; Novus), EXO1 (1:1,000, A302‐640A; Bethyl Laboratories), BRCA2 (1:2,000, A303‐434A; Bethyl Laboratories; 1:500, OP 95; EMD Millipore Ab‐1), Histone H3 (1:1,000, A300‐823A; Bethyl Laboratories), CDC7 (1:1,000, K0070‐3; MBL), CHK1 (1:1,000, SC‐8408; Santa Cruz Biotech) and CDC45 (1:5, a gift from Broderick et al). Primary antibodies diluted in blocking buffer (TBST, 1% BSA) included pS40/41 MCM2 (in‐house, 1:3,000), pS4/8 RPA32 (1:1,000, A300‐254A; Bethyl Laboratories), pS139 H2AX (1:1,000; Cell Signaling Technology) and pS345 CHK1 (1:1,000, 2348; Cell Signaling Technology). Secondary antibodies (LI‐COR Biosciences) were diluted in blocking buffer (1:10,000, TBST, 5% milk) and included IRDye 800CW secondary anti‐rat (926–32,219), IRDye 800CW secondary anti‐rabbit (926–32,211) and IRDye 800CW secondary anti‐mouse (926–32,210).
Flow cytometry
For flow cytometry analysis of cell cycle phases in combination with pS139 histone H2AX, cells were treated as required followed by a 30 min pulse with EdU (10 μM) prior to harvest. Approximately 1 × 106 cells were processed by extraction of soluble proteins for 10 min on ice (PBS, 0.2% Triton X‐100). Following centrifugation (450 g, 5 min), cells were washed with PBS and fixed (PBS, 1% PFA) at room temperature for 10 min and then incubated with blocking buffer (PBS, 1% BSA). Cells were then sequentially incubated in blocking buffer containing 0.05% saponin at room temperature with a primary antibody against pSer139 histone H2AX (1:500, 9718; Cell Signaling Technology) for 2 h, followed by washing, and then with a secondary antibody (1:250, donkey α‐rabbit Alexa Fluor 647, A32795; ThermoFisher) for 30 min in the dark. Incorporated EdU was labelled with CLICK chemistry (10 μM 6‐carboxyfluorescein‐TEG‐azide, 10 mM sodium L‐ascorbate, 2 mM copper(II) sulphate) for 30 min in the dark. Cells were washed (PBS, 1% BSA, 5% Tween‐20) and DNA stained with DAPI (1 μg/ml, PBS, 1% BSA). Data were acquired on a BD FACSCanto II and analysed using FlowJo software.
Neutral comet assay
Cells were seeded at 3.5 × 105 cells/well in 60‐mm plates and incubated for 24 h. Following treatments, cells were harvested, counted and diluted to 1 × 105 cells/ml with low melting agarose (LMA; Trevigen) and spread across poly‐lysine‐coated slides. Slides with LMA‐embedded cells were incubated with lysis solution (Trevigen) overnight, followed by electrophoresis at 4°C for 1 h with an applied voltage of 1V/cm. DNA was stained with SYBR Green (1:10,000 in 10 mM Tris, pH 7.5, 1 mM EDTA) for 10 min at room temperature in the dark. Fluorescence images were captured with a DeltaVision Core wide‐field microscope (GE Healthcare, Image solution) using a 10× UPlanFL N, N/A 0.30. Excitation and emission for FITC were 475 and 523 nm, respectively. Images were processed in ImageJ/Fiji and analysed using CometScore software (version 2.0.0.38) to measure comet tail moments. Statistical analysis was performed using Prism 5 (GraphPad Software).
Chromosome spreads
Cells were seeded in 6‐well plates at 1 × 105 cells/well, treated as required and then washed three times with PBS, followed by addition of fresh media containing 200 ng/ml nocodazole for 16 h. Cells were harvested and swollen with 75 mM KCl for 20 min at 37 °C before being fixed with methanol:acetic acid (3:1). The fixing step was repeated two times. Cells were then dropped onto pre‐hydrated glass slides and air‐dried overnight before coverslips were mounted onto the slides with Vectashield containing DAPI. Images of metaphase spreads were acquired with a 60× objective using an Olympus IX51 microscope equipped with a Hamamatsu camera (C4742‐95). Images were processed using ImageJ/Fiji software 73, and visible chromatid breaks were scored. Statistical analysis was performed using Prism 5 (GraphPad Software).
Immunofluorescence
For PCNA/MRE11 colocalization studies, U2OS cells were seeded at 15,000 cells/well of Ibidi 8‐well cell culture chamber μ‐slide and allowed to recover for 24 h. Cells were treated as required, followed by three washes with PBS, fixation with ice‐cold methanol for 20 min, permeabilization with ice‐cold acetone for 30 s and three washes with PBS prior to blocking. For RAD51 or RPA/MRE11 colocalization studies, U2OS cells were seeded onto coverslips in 6‐well plates at 1 × 105 cells/well and treated as required. To detect RAD51, cells were fixed, permeabilized and blocked as previously reported 74. For RPA/MRE11 colocalization, soluble proteins were pre‐extracted by incubation on ice for 10 min (CSK buffer: 10 mM PIPES, pH 6.8, 10 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, 0.5% Triton X‐100) before being washed with PBS. Cells were then fixed (PBS, 4% PFA) for 20 min on ice and washed with PBS prior to blocking. For all immunofluorescence staining, cells were blocked (PBS, 1% BSA) for 30 min and then incubated with primary antibodies diluted in blocking buffer for 1 h at room temperature. Cells were washed three times with PBS, followed by incubation with secondary antibodies diluted in blocking buffer for 1 h at room temperature in combination with DAPI (0.5 μg/ml) to stain nuclei. Cells were washed three times with PBS, and fluorescence images were captured with a DeltaVision wide‐field microscope (GE Healthcare, Image solution) using an oil 100X UPlanFL N, N/A 1.30 objective. Excitations/emissions for DAPI, FITC and TRICT were 390/435 nm, 475/523 nm and 542/594 nm, respectively. Z‐stacks composed of twelve slices with a 500‐nm step were acquired for each image. Following acquisition, image restoration was applied and Huygens Professional (Scientific Volume Imaging) was used to deconvolve images, using the Classic Maximum Likelihood Estimation (CMLE) algorithm. Images were processed using ImageJ/Fiji software, and the degree of colocalization between proteins was analysed by measuring the Pearson correlation coefficient using ImageJ/Fiji 73. Statistical analysis was performed using Prism 5 (GraphPad Software). Primary antibodies included RPA2 (1:500, NA19L; Calbiochem), Mre11 (1:500, NB100‐142; Novus), PCNA (1:1,000, SC‐56; Santa Cruz) and RAD51 (1:1,000, PC130; Calbiochem). Secondary antibodies included goat anti‐rabbit Alexa Fluor 488 (A11008; Molecular Probes; 1:500) and goat anti‐mouse Alexa Fluor 546 (A11003; Molecular Probes; 1:500).
DNA fibre analysis
Following depletion and/or inhibition of proteins of interest, cells were sequentially pulse‐labelled with halogenated nucleotides (CldU and IdU) according to the treatment schemes displayed (Figs 4 and 5) and previously described protocols for preparation and analysis of DNA fibre spreads 25, 50. Briefly, cells were collected and resuspended in PBS at 2.5 × 105 cells per ml. The labelled cells were diluted 1:5 (v/v) with unlabelled cells, and 2.5 μl of cells was mixed with 7.5 μl of lysis buffer (200 mM Tris–HCl, pH 7.5, 50 mM EDTA, and 0.5% (w/v) SDS) on a glass slide. After 9 min, the slides were tilted at 15–45°, and the resulting DNA spreads were air‐dried and fixed in 3:1 methanol/acetic acid overnight at 4°C. The fibres were denatured with 2.5 M HCl for 1 h, washed with PBS and blocked with 0.2% Tween‐20 in 1% BSA/PBS for 40 min. The newly replicated CldU and IdU tracks were labelled (for 2.5 h in the dark, at RT) with anti‐BrdU antibodies recognizing CldU (1:500, ab6326; Abcam) and IdU (1:100, B44, 347580; BD), followed by 1‐h incubation with secondary antibodies at RT in the dark: anti‐mouse Alexa Fluor 488 (1:300, A11001; Invitrogen) and anti‐rat Cy3 (1:150, 712‐166‐153; Jackson ImmunoResearch Laboratories, Inc.). Fibres were visualized (IX81; Olympus; objective lenses: LC Plan Fluor 60 × 1.42 NA oil Olympus BX60 microscope) and analysed using ImageJ software. Statistical analysis was performed using Prism 5 (GraphPad Software). Alternatively, DNA was combed and analysed as previously described by Eykelenboom et al (2013) (Fig 4C) 75. Primary antibodies included BrdU (BU1/75 (ICR1)) rat monoclonal antibody (1:100, MA1‐82088; ThermoFisher), anti‐BrdU (B44) IgG1 mouse monoclonal antibody (1:100, 347580; BD Biosciences) and anti‐ssDNA (poly dT) IgG2aκ mouse monoclonal antibody (1:100, mab3034; Sigma‐Aldrich). Secondary antibodies included chicken anti‐rat Alexa Fluor 488 (1:300, A21470; ThermoFisher), goat anti‐mouse IgG1 Alexa Fluor 546 (1:300, A21123; ThermoFisher) and goat anti‐mouse IgG2a Alexa Fluor 647 (1:300, A21241; ThermoFisher).
Electron microscopy analysis
The procedure was performed as recently described 50, with minor modifications described below. Following the depletion of the protein of interest, asynchronous sub‐confluent cells were treated with 4 mM HU for 5 h. Where indicated, cells were pre‐treated with XL413 for 30 min. Cells were collected, resuspended in PBS and cross‐linked with 4,5′,8‐trimethylpsoralen (10 μg ml‐1 final concentration), followed by irradiation pulses with UV 365‐nm monochromatic light (UV Stratalinker 1800; Agilent Technologies). For DNA extraction, cells were lysed (1.28 M sucrose, 40 mM Tris–HCl (pH 7.5), 20 mM MgCl2 and 4% Triton X‐100; Qiagen) and digested (800 mM guanidine‐HCl, 30 mM Tris–HCl (pH 8.0), 30 mM EDTA (pH 8.0), 5% Tween‐20 and 0.5% Triton X‐100) at 50°C for 2 h in the presence of 1 mg/ml proteinase K. The DNA was purified using chloroform/isoamyl alcohol (24:1) and precipitated in 0.7 volume of isopropanol. Finally, the DNA was washed with 70% EtOH and resuspended in 200 μl TE (Tris–EDTA) buffer. Restriction enzyme of 100 U (PvuII high fidelity; New England Biolabs) was used to digest 12 μg of mammalian genomic DNA for 4–5 h. Replication intermediate enrichment was performed by QIAGEN Plasmid Mini Kit columns. The QIAGEN‐tip 20 surface tension was reduced by applying 1 ml QBT buffer. The columns were washed and equilibrated with 10 mM Tris–HCl (pH 8.0) and 1M NaCl, followed by 10 mM Tris–HCl (pH 8.0) and 300 mM NaCl, respectively. DNA was then loaded onto the columns. The columns were then washed with high‐NaCl solution (10 mM Tris–HCl (pH 8.0) and 900 mM NaCl) and eluted in caffeine solution (10 mM Tris–HCl (pH 8.0), 1M NaCl and 1.8% (w/v) caffeine). To purify and concentrate the DNA, an Amicon size‐exclusion column was used. DNA was then resuspended in TE buffer. The benzyldimethylalkylammonium chloride method was used to spread the DNA on the water surface and then load it on carbon‐coated 400‐mesh copper grids. Subsequently, DNA was coated with platinum using a high‐vacuum evaporator (MED 020; BalTec). Microscopy was performed with a transmission electron microscope (Tecnai G2 Spirit; FEI; LaB6 filament; high tension ≤ 120 kV) and picture acquisition with a side‐mount charge‐coupled device camera (2,600 × 4,000 pixels; Orius 1000; Gatan, Inc.). For each experimental condition, at least 70 replication fork molecules were analysed. DigitalMicrograph version 1.83.842 (Gatan, Inc.) and ImageJ (National Institutes of Health) were used to process and analyse the images. Statistical analysis was performed using Prism 5 (GraphPad Software).
Author contributions
Conceptualization: MDR, ML and CS; Investigation: MDR, AQ, CC, SM, OM, JK and AG; Writing—original draft: CS; Writing—review & editing: MDR, ML and CS; Supervision: ML and CS; Funding acquisition: ML and CS.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Expanded View Figures PDF
Source Data for Appendix and Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6
Acknowledgements
The authors thank Kevin Sullivan and all the members of the Santocanale laboratory for discussion and support, Heinz Peter Nasheuer for gift of anti‐CDC45 antibodies and Matteo Berti for technical support. This work was supported by SFI grant 16/IA/4476. SM and the work in the Lopes laboratory were supported by the ERC Consolidator Grant 617102 and the Swiss Cancer League grant KFS‐3967‐08‐2016.
EMBO Reports (2020) 21: e48920
[The copyright line of this article was changed on 6 October 2020 after original online publication.]
References
- 1. Huberman JA, Riggs AD (1968) On the mechanism of DNA replication in mammalian chromosomes. J Mol Biol 32: 327–341 [DOI] [PubMed] [Google Scholar]
- 2. Cairns J (1966) Autoradiography of HeLa cell DNA. J Mol Biol 15: 372‐IN32 [DOI] [PubMed] [Google Scholar]
- 3. Masai H, Taniyama C, Ogino K, Matsui E, Kakusho N, Matsumoto S, Kim J‐M, Ishii A, Tanaka T, Kobayashi T et al (2006) Phosphorylation of MCM4 by Cdc7 Kinase Facilitates Its Interaction With Cdc45 on the chromatin. J Biol Chem 281: 39249–39261 [DOI] [PubMed] [Google Scholar]
- 4. Montagnoli A, Valsasina B, Brotherton D, Troiani S, Rainoldi S, Tenca P, Molinari A, Santocanale C (2006) Identification of Mcm2 phosphorylation sites by S‐phase‐regulating kinases. J Biol Chem 281: 10281–10290 [DOI] [PubMed] [Google Scholar]
- 5. Sheu Y‐J, Stillman B (2006) Cdc7‐Dbf4 phosphorylates MCM proteins via a docking site‐mediated mechanism to promote s phase progression. Mol Cell 24: 101–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bell SP, Dutta A (2002) DNA replication in eukaryotic cells. Annu Rev Biochem 71: 333–374 [DOI] [PubMed] [Google Scholar]
- 7. Sclafani RA, Holzen TM (2007) Cell cycle regulation of DNA replication. Annu Rev Genet 41: 237–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Rainey M, Harhen B, Wang G‐N, Murphy P, Santocanale C (2013) Cdc7‐dependent and ‐independent phosphorylation of Claspin in the induction of the DNA replication checkpoint. Cell Cycle 12: 1560–1568 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kim JM, Kakusho N, Yamada M, Kanoh Y, Takemoto N, Masai H (2008) Cdc7 kinase mediates claspin phosphorylation in DNA replication checkpoint. Oncogene 27: 3475–3482 [DOI] [PubMed] [Google Scholar]
- 10. Tenca P, Brotherton D, Montagnoli A, Rainoldi S, Albanese C, Santocanale C (2007) Cdc7 is an active kinase in human cancer cells undergoing replication stress. J Biol Chem 282: 208–215 [DOI] [PubMed] [Google Scholar]
- 11. Yang C‐C, Kato H, Shindo M, Masai H (2019) Cdc7 activates replication checkpoint by phosphorylating the Chk1‐binding domain of Claspin in human cells. Elife 8: e50796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sasi NK, Coquel F, Lin Y‐L, MacKeigan JP, Pasero P, Weinreich M (2018) DDK has a primary role in processing stalled replication forks to initiate downstream checkpoint signaling. Neoplasia 20: 985–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Alver RC, Chadha GS, Gillespie PJ, Blow JJ (2017) Reversal of DDK‐mediated MCM phosphorylation by Rif1‐PP1 regulates replication initiation and replisome stability independently of ATR/Chk1. Cell Rep 18: 2508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garzón J, Ursich S, Lopes M, Hiraga S‐I, Donaldson AD (2019) Human RIF1‐protein phosphatase 1 prevents degradation and breakage of nascent DNA on replication stalling. Cell Rep 27: 2558–2566.e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mukherjee C, Tripathi V, Manolika EM, Heijink AM, Ricci G, Merzouk S, de Boer HR, Demmers J, van Vugt MATM, Ray Chaudhuri A (2019) RIF1 promotes replication fork protection and efficient restart to maintain genome stability. Nat Commun 10: 3287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Princz LN, Wild P, Bittmann J, Aguado FJ, Blanco MG, Matos J, Pfander B (2017) Dbf4‐dependent kinase and the Rtt107 scaffold promote Mus81‐Mms4 resolvase activation during mitosis. EMBO J 36: 664–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lo H‐C, Kunz RC, Chen X, Marullo A, Gygi SP, Hollingsworth NM (2012) Cdc7‐Dbf4 is a gene‐specific regulator of meiotic transcription in yeast. Mol Cell Biol 32: 541–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Matos J, Lipp JJ, Bogdanova A, Guillot S, Okaz E, Junqueira M, Shevchenko A, Zachariae W (2008) Dbf4‐dependent Cdc7 kinase links DNA replication to the segregation of homologous chromosomes in meiosis I. Cell 135: 662–678 [DOI] [PubMed] [Google Scholar]
- 19. Murakami H, Keeney S (2014) Temporospatial coordination of meiotic DNA replication and recombination via DDK recruitment to replisomes. Cell 158: 861–873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sasanuma H, Hirota K, Fukuda T, Kakusho N, Kugou K, Kawasaki Y, Shibata T, Masai H, Ohta K (2008) Cdc7‐dependent phosphorylation of Mer2 facilitates initiation of yeast meiotic recombination. Genes Dev 22: 398–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Valentin G, Schwob E, Della SF (2006) Dual role of the Cdc7‐regulatory protein Dbf4 during yeast meiosis. J Biol Chem 281: 2828–2834 [DOI] [PubMed] [Google Scholar]
- 22. Wan L, Niu H, Futcher B, Zhang C, Shokat KM, Boulton SJ, Hollingsworth NM (2008) Cdc28‐Clb5 (CDK‐S) and Cdc7‐Dbf4 (DDK) collaborate to initiate meiotic recombination in yeast. Genes Dev 22: 386–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hughes S, Elustondo F, Di Fonzo A, Leroux FG, Wong AC, Snijders AP, Matthews SJ, Cherepanov P (2012) Crystal structure of human CDC7 kinase in complex with its activator DBF4. Nat Struct Mol Biol 19: 1101–1107 [DOI] [PubMed] [Google Scholar]
- 24. Bishop AC, Ubersax JA, Petsch DT, Matheos DP, Gray NS, Blethrow J, Shimizu E, Tsien JZ, Schultz PG, Rose MD et al (2000) A chemical switch for inhibitor‐sensitive alleles of any protein kinase. Nature 407: 395–401 [DOI] [PubMed] [Google Scholar]
- 25. Rainey MD, Quachthithu H, Gaboriau D, Santocanale C (2017) DNA replication dynamics and cellular responses to ATP competitive CDC7 kinase inhibitors. ACS Chem Biol 12: 1893–1902 [DOI] [PubMed] [Google Scholar]
- 26. Zellweger R, Dalcher D, Mutreja K, Berti M, Schmid JA, Herrador R, Vindigni A, Lopes M (2015) Rad51‐mediated replication fork reversal is a global response to genotoxic treatments in human cells. J Cell Biol 208: 563–579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ray Chaudhuri A, Hashimoto Y, Herrador R, Neelsen KJ, Fachinetti D, Bermejo R, Cocito A, Costanzo V, Lopes M (2012) Topoisomerase I poisoning results in PARP‐mediated replication fork reversal. Nat Struct Mol Biol 19: 417–423 [DOI] [PubMed] [Google Scholar]
- 28. Neelsen KJ, Lopes M (2015) Replication fork reversal in eukaryotes: from dead end to dynamic response. Nat Rev Mol Cell Biol 16: 207–220 [DOI] [PubMed] [Google Scholar]
- 29. Cortez D (2019) Replication‐coupled DNA repair. Mol Cell 74: 866–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Neelsen KJ, Zanini IMY, Herrador R, Lopes M (2013) Oncogenes induce genotoxic stress by mitotic processing of unusual replication intermediates. J Cell Biol 200: 699–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Follonier C, Oehler J, Herrador R, Lopes M (2013) Friedreich's ataxia–associated GAA repeats induce replication‐fork reversal and unusual molecular junctions. Nat Struct Mol Biol 20: 486–494 [DOI] [PubMed] [Google Scholar]
- 32. Quinet A, Lemaçon D, Vindigni A (2017) Replication fork reversal: players and guardians. Mol Cell 68: 830–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhat KP, Cortez D (2018) RPA and RAD51: fork reversal, fork protection, and genome stability. Nat Struct Mol Biol 25: 446–453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schlacher K, Christ N, Siaud N, Egashira A, Wu H, Jasin M (2011) Double‐strand break repair independent role for BRCA2 in blocking stalled replication fork degradation by MRE11. Cell 145: 529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee J‐E, Wong N, Lafarga V, Calvo JA, Panzarino NJ et al (2016) Replication fork stability confers chemoresistance in BRCA‐deficient cells. Nature 535: 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lee AY‐L, Chiba T, Truong LN, Cheng AN, Do J, Cho MJ, Chen L, Wu X (2012) Dbf4 is direct downstream target of ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3‐related (ATR) protein to regulate intra‐s‐phase checkpoint. J Biol Chem 287: 2531–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamada M, Masai H, Bartek J (2014) Regulation and roles of Cdc7 kinase under replication stress. Cell Cycle 13: 1859–1866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Swords R, Mahalingam D, O'Dwyer M, Santocanale C, Kelly K, Carew J, Giles F (2010) Cdc7 kinase – A new target for drug development. Eur J Cancer 46: 33–40 [DOI] [PubMed] [Google Scholar]
- 39. Labib K (2010) How do Cdc7 and cyclin‐dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev 24: 1208–1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Petermann E, Orta ML, Issaeva N, Schultz N, Helleday T (2010) Hydroxyurea‐stalled replication forks become progressively inactivated and require two different RAD51‐mediated pathways for restart and repair. Mol Cell 37: 492–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Dungrawala H, Rose KL, Bhat KP, Mohni KN, Glick GG, Couch FB, Cortez D (2015) The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol Cell 59: 998–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Couch FB, Bansbach CE, Driscoll R, Luzwick JW, Glick GG, Bétous R, Carroll CM, Jung SY, Qin J, Cimprich KA et al (2013) ATR phosphorylates SMARCAL1 to prevent replication fork collapse. Genes Dev 27: 1610–1623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Dupré A, Boyer‐Chatenet L, Sattler RM, Modi AP, Lee J‐H, Nicolette ML, Kopelovich L, Jasin M, Baer R, Paull TT et al (2008) A forward chemical genetic screen reveals an inhibitor of the Mre11–Rad50–Nbs1 complex. Nat Chem Biol 4: 119–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kliszczak AE, Rainey MD, Harhen B, Boisvert FM, Santocanale C (2011) DNA mediated chromatin pull‐down for the study of chromatin replication. Sci Rep 1: 95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sirbu BM, Couch FB, Feigerle JT, Bhaskara S, Hiebert SW, Cortez D (2011) Analysis of protein dynamics at active, stalled, and collapsed replication forks. Genes Dev 25: 1320–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Alabert C, Bukowski‐Wills J‐C, Lee S‐B, Kustatscher G, Nakamura K, de Lima AF, Menard P, Mejlvang J, Rappsilber J, Groth A (2014) Nascent chromatin capture proteomics determines chromatin dynamics during DNA replication and identifies unknown fork components. Nat Cell Biol 16: 281–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mirzoeva OK, Petrini JHJ (2003) DNA replication‐dependent nuclear dynamics of the Mre11 complex. Mol Cancer Res 1: 207–218 [PubMed] [Google Scholar]
- 48. Bryant HE, Petermann E, Schultz N, Jemth A‐S, Loseva O, Issaeva N, Johansson F, Fernandez S, McGlynn P, Helleday T (2009) PARP is activated at stalled forks to mediate Mre11‐dependent replication restart and recombination. EMBO J 28: 2601–2615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vujanovic M, Krietsch J, Raso MC, Terraneo N, Zellweger R, Schmid JA, Taglialatela A, Huang J‐W, Holland CL, Zwicky K et al (2017) Replication fork slowing and reversal upon DNA damage require PCNA polyubiquitination and ZRANB3 DNA translocase activity. Mol Cell 67: 882–890.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P et al (2017) Replication fork reversal triggers fork degradation in BRCA2‐defective cells. Nat Commun 8: 859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mutreja K, Krietsch J, Hess J, Ursich S, Berti M, Roessler FK, Zellweger R, Patra M, Gasser G, Lopes M (2018) ATR‐mediated global fork slowing and reversal assist fork traverse and prevent chromosomal breakage at DNA interstrand cross‐links. Cell Rep 24: 2629–2642.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Taglialatela A, Alvarez S, Leuzzi G, Sannino V, Ranjha L, Huang J‐W, Madubata C, Anand R, Levy B, Rabadan R et al (2017) Restoration of replication fork stability in BRCA1‐ and BRCA2‐deficient cells by inactivation of SNF2‐family fork remodelers. Mol Cell 68: 414–430.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Schlacher K, Wu H, Jasin M (2012) A distinct replication fork protection pathway connects fanconi anemia tumor suppressors to RAD51‐BRCA1/2. Cancer Cell 22: 106–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lemaçon D, Jackson J, Quinet A, Brickner JR, Li S, Yazinski S, You Z, Ira G, Zou L, Mosammaparast N et al (2017) MRE11 and EXO1 nucleases degrade reversed forks and elicit MUS81‐dependent fork rescue in BRCA2‐deficient cells. Nat Commun 8: 860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kolinjivadi AM, Sannino V, De Antoni A, Zadorozhny K, Kilkenny M, Técher H, Baldi G, Shen R, Ciccia A, Pellegrini L et al (2017) Smarcal1‐mediated fork reversal triggers Mre11‐dependent degradation of nascent DNA in the absence of Brca2 and stable Rad51 nucleofilaments. Mol Cell 67: 867–881.e7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Aguilera A, García‐Muse T (2013) Causes of genome instability. Annu Rev Genet 47: 1–32 [DOI] [PubMed] [Google Scholar]
- 57. Diffley JFX (2004) Regulation of early events in chromosome replication. Curr Biol 14: R778–R786 [DOI] [PubMed] [Google Scholar]
- 58. Moiseeva TN, Bakkenist CJ (2018) Regulation of the initiation of DNA replication in human cells. DNA Repair 72: 99–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ferretti LP, Lafranchi L, Sartori AA (2013) Controlling DNA‐end resection: a new task for CDKs. Front Genet 4: 99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Tomimatsu N, Mukherjee B, Catherine Hardebeck M, Ilcheva M, Vanessa Camacho C, Louise Harris J, Porteus M, Llorente B, Khanna KK, Burma S (2014) Phosphorylation of EXO1 by CDKs 1 and 2 regulates DNA end resection and repair pathway choice. Nat Commun 5: 3561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Syed A, Tainer JA (2018) The MRE11‐RAD50‐NBS1 complex conducts the orchestration of damage signaling and outcomes to stress in DNA replication and repair. Annu Rev Biochem 87: 263–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ying S, Hamdy FC, Helleday T (2012) Mre11‐dependent degradation of stalled DNA replication forks is prevented by BRCA2 and PARP1. Cancer Res 72: 2814–2821 [DOI] [PubMed] [Google Scholar]
- 63. Toledo LI, Altmeyer M, Rask M‐B, Lukas C, Larsen DH, Povlsen LK, Bekker‐Jensen S, Mailand N, Bartek J, Lukas J (2013) ATR prohibits replication catastrophe by preventing global exhaustion of RPA. Cell 155: 1088–1103 [DOI] [PubMed] [Google Scholar]
- 64. Rodriguez‐Acebes S, Mourón S, Méndez J (2018) Uncoupling fork speed and origin activity to identify the primary cause of replicative stress phenotypes. J Biol Chem 293: 12855–12861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Montagnoli A, Valsasina B, Croci V, Menichincheri M, Rainoldi S, Marchesi V, Tibolla M, Tenca P, Brotherton D, Albanese C et al (2008) A Cdc7 kinase inhibitor restricts initiation of DNA replication and has antitumor activity. Nat Chem Biol 4: 357–365 [DOI] [PubMed] [Google Scholar]
- 66. Iwai K, Nambu T, Dairiki R, Ohori M, Yu J, Burke K, Gotou M, Yamamoto Y, Ebara S, Shibata S et al (2019) Molecular mechanism and potential target indication of TAK‐931, a novel CDC7‐selective inhibitor. Sci Adv 5: eaav3660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Conti C, Saccà B, Herrick J, Lalou C, Pommier Y, Bensimon A (2007) Replication fork velocities at adjacent replication origins are coordinately modified during DNA replication in human cells. Mol Biol Cell 18: 3059–3067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gaillard H, García‐Muse T, Aguilera A (2015) Replication stress and cancer. Nat Rev Cancer 15: 276–289 [DOI] [PubMed] [Google Scholar]
- 69. Técher H, Koundrioukoff S, Nicolas A (2017) Debatisse M (2017) The impact of replication stress on replication dynamics and DNA damage in vertebrate cells. Nat Rev Genet 1818: 535–550 [DOI] [PubMed] [Google Scholar]
- 70. Zeman MK, Cimprich KA (2014) Causes and consequences of replication stress. Nat Cell Biol 16: 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Cheng AN, Fan C‐C, Lo Y‐K, Kuo C‐L, Wang H‐C, Lien I‐H, Lin S‐Y, Chen C‐H, Jiang SS, Chang I‐S et al (2017) Cdc7‐Dbf4‐mediated phosphorylation of HSP90‐S164 stabilizes HSP90‐HCLK2‐MRN complex to enhance ATR/ATM signaling that overcomes replication stress in cancer. Sci Rep 7: 17024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mailand N, Diffley JFX (2005) CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C‐dependent proteolysis. Cell 122: 915–926 [DOI] [PubMed] [Google Scholar]
- 73. Schindelin J, Arganda‐Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B et al (2012) Fiji: an open‐source platform for biological‐image analysis. Nat Methods 9: 676–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. O'Dea R, Santocanale C (2020) Non‐canonical regulation of homologous recombination DNA repair by the USP9X deubiquitylase. J Cell Sci 133: jcs233437 [DOI] [PubMed] [Google Scholar]
- 75. Eykelenboom JK, Harte EC, Canavan L, Pastor‐Peidro A, Calvo‐Asensio I, Llorens‐Agost M, Lowndes NF (2013) ATR Activates the S‐M checkpoint during unperturbed growth to ensure sufficient replication prior to mitotic onset. Cell Rep 5: 1095–1107 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Expanded View Figures PDF
Source Data for Appendix and Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 5
Source Data for Figure 6