

SUMMARY
The BCL-2 family is composed of anti- and pro-apoptotic members that respectively protect or disrupt mitochondrial integrity. Anti-apoptotic overexpression can promote oncogenesis by trapping the BCL-2 homology 3 (BH3) “killer domains” of pro-apoptotic proteins in a surface groove, blocking apoptosis. Groove inhibitors, like the relatively large BCL-2 drug venetoclax (868 Da), have emerged as cancer therapies. BFL-1 remains an undrugged oncogenic protein and can cause venetoclax resistance. Having identified a unique C55 residue in the BFL-1 groove, we performed a disulfide tethering screen to determine if C55 reactivity could enable smaller molecules to block BFL-1’s BH3-binding functionality. We found that a disulfide-bearing N-acetyltryptophan analog (304 Da adduct) effectively targeted BFL-1 C55 and reversed BFL-1-mediated suppression of mitochondrial apoptosis. Structural analyses implicated the conserved leucine-binding pocket of BFL-1 as the interaction site, resulting in conformational remodeling. Thus, therapeutic targeting of BFL-1 may be achievable through the design of small, cysteine-reactive drugs.
Keywords: BFL-1/A1; BCL-2 family, BH3, anti-apoptotic; mitochondria; apoptosis, disulfide tethering; small molecule; inhibitor
eTOC
Harvey et al. perform a disulfide tethering screen of BFL-1, a BCL-2 protein implicated in cancer, in order to identify molecules capable of covalently targeting a unique C55-residue of the anti-apoptotic groove. 4E14 derivatization (304 Da moiety) effectively blocks BFL-1, informing the design of cysteine-reactive drugs to selectively target BFL-1.
Graphical Abstract
INTRODUCTION
The response to physiologic or pathologic stress is regulated by BCL-2 family proteins, which render a cellular life or death decision that is processed at the mitochondrial outer membrane (MOM) (Kalkavan and Green, 2018). Stress-induced activation of pro-apoptotic proteins, such as BAX or BAK, leads to transient exposure of their BCL-2 homology 3 (BH3) helices, which participate in converting BAX/BAK monomers into toxic oligomers that pierce the MOM and release apoptogenic factors into the cytosol (Walensky and Gavathiotis, 2011). Anti-apoptotic BCL-2 proteins contain a surface groove that can bind and trap the exposed BH3 helices of BAX and BAK, and thereby neutralize the killing mechanism (Sattler et al., 1997). An additional layer of regulation involves the BH3-only members, a heterogeneous subclass of pro-apoptotic BCL-2 proteins that only share homology in the BH3 region (O’Connor et al., 1998; Wang et al., 1996; Yang et al., 1995). BH3-only proteins can deploy their conserved helices to directly trigger pro-apoptotic BAX and BAK or serve as decoys to block the anti-apoptotic grooves (Cheng et al., 2001). The latter inhibit-the-inhibitor mechanism of BH3-only pro-apoptotic activity provided a blueprint for the development of small molecule drugs to reactivate apoptosis in cancer by targeting the anti-apoptotic groove (Oltersdorf et al., 2005).
Of the six anti-apoptotic BCL-2 family targets, small molecules have been developed to effectively block BCL-2, BCL-XL, and MCL-1. A selective BCL-2 inhibitor, venetoclax, is FDA-approved for the treatment of CLL, and other BCL-XL and MCL-1 inhibitors are currently in clinical trials (Kotschy et al., 2016; Souers et al., 2013; Tse et al., 2008). Such molecules are mimics of BH3 peptide helices, which engage a large, flat, and shallow groove on the surface of anti-apoptotic BCL-2 proteins. Correspondingly, the inhibitory molecules are relatively large (e.g. venetoclax, MW 868; navitoclax, MW 975; S63845, MW 829), which can pose “beyond-the-rule-of-5” (bRo5) pharmacologic challenges during development (DeGoey et al., 2018). BFL-1 (also known as BCL2A1 or A1) is another BCL-2 family anti-apoptotic protein that has been implicated in the pathogenesis of human cancers, including leukemia, lymphoma, melanoma, and gastric cancer (Choi et al., 1995; Fan et al., 2010; Haq et al., 2013; Morales et al., 2005). BH3 helices have been shown to engage individual, dual, or multiple anti-apoptotic targets (Certo et al., 2006; Chen et al., 2005; Stewart et al., 2010) and, therefore, a tunable spectrum of molecular targeting capability may be achievable. Nevertheless, the current trend favors selectivity to avoid the potential toxicity of broad targeting. For example, the dual BCL-2/BCL-XL inhibitor, nativoclax, was found to induce rapid thrombocytopenia as a result of on-mechanism toxicity associated with blockade of BCL-XL in platelets, motivating the development of selective BCL-2 inhibitors to avoid the risk of hemorrhage in cancer patients (Mason et al., 2007; Souers et al., 2013). However, resistance against precision anti-apoptotic inhibitors such as venetoclax has already emerged, including BCL-2 protein mutations and alternative anti-apoptotic expression, including MCL-1 and BFL-1, portending a “whack-a-mole” challenge for the field (Birkinshaw et al., 2019; Yecies et al., 2010). Thus, developing molecular inhibitors for the complete array of anti-apoptotic proteins, particularly those like BFL-1 that remain undrugged, is a high priority goal.
In surveying design opportunities for selective BFL-1 targeting, we noted a cysteine within its BH3-binding groove (Huhn et al., 2016). Strikingly, no other BCL-2 family protein has an analogous cysteine at the critical regulatory groove. Guided by a native cysteine pair in the complex between NOXA BH3 and BFL-1 (PDB: 3MQP), we generated a series of cysteine-reactive stabilized alpha-helices of BCL-2 domains (SAHBs), which were shown by biochemical, structural, and cellular means to target BFL-1 through combined covalent and non-covalent interaction, and thereby reactivate apoptosis in BFL-1-driven cancers (Guerra et al., 2018; Harvey et al., 2018; Huhn et al., 2016). To investigate if small molecules could do the same, we turned to disulfide tethering, which has emerged as a powerful screening method for identifying molecular fragments that engage a target binding site in the vicinity of a natural or installed cysteine residue (Erlanson et al., 2000; Ostrem et al., 2013). We were especially interested in determining if much smaller compounds, when endowed with a cysteine-reactive moiety, could achieve BH3-mimetic functionality. Here, we surprisingly identified a disulfide-bearing N-acetyltryptophan analog that forms an adduct of only 304 Da in size upon C55-targeting yet exerts structural and biochemical consequences analogous to BH3-mediated BFL-1 inhibition.
RESULTS
A Disulfide Tethering Screen Identifies Molecular Fragments that Conjugate to BFL-1 C55
We performed a disulfide tethering screen to identify molecules capable of engaging the BH3-binding groove of BFL-1 in the context of disulfide formation with BFL-1 C55 (Figure 1A). Specifically, we tested a 1600-member library (Burlingame et al., 2011; Hallenbeck et al., 2018) for interaction with BFL-1ΔC C4S/C19S, a construct lacking the C-terminal α9 helix and the two native cysteine residues located outside of the BH3-binding groove. Compounds were incubated with BFL-1 protein at room temperature for one hour in the presence of 500 μM β-mercaptoethanol (BME) and then percent tethering was determined by intact mass spectrometry. We identified 31 compounds with percent tethering values of two standard deviations above the mean, reflecting 51-73% BFL-1 derivatization under the reducing experimental conditions (Figure 1B). A common theme among the chemical structures of the hits is the presence of an aryl, benzyl, pyridyl, or indolyl group separated from the reactive cysteine by a linker of 4 to 6 bond lengths (Table S1). To verify the hits and identify the most potent molecular competitors of BH3/BFL-1 interaction, we performed a fluorescence polarization (FP) secondary screen based on the comparative binding of a fluoresceinated BID BH3 peptide to apo vs. fragment-conjugated BFL-1 protein. Whereas all of the identified compounds shifted the FITC-BID BH3 binding isotherm to the right upon BFL-1 conjugation, 4E14, an N-acetyltryptophan analog of 408 Da, emerged as the most effective competitor (~56-fold shift in EC50) (Figure 1C). Intact mass spectrometry documented complete conjugation of 4E14 to BFL-1ΔC (M+304) under the experimental conditions (Figure 1D). To evaluate the C55 dependence of 4E14 binding activity and compound selectivity, we repeated the competitive FP using BFL-1 protein that lacked C55 but retained the native C4 and C19 residues (BFL-1ΔC C55S), and alternative anti-apoptotic members, BCL-XLΔC and MCL-1ΔNΔC, which contain cysteine residues but not within their BH3-binding grooves. In each case, exposing the proteins to 4E14 under conjugating conditions had no effect on the capacity of FITC-BID BH3 to bind to the respective anti-apoptotic targets (Figure 1E–F). Thus, disulfide tethering yielded a lead molecular fragment capable of direct and selective C55 derivatization under reducing conditions, resulting in impaired BH3-binding interaction at the canonical BFL-1 groove.
Figure 1. A disulfide tethering screen identifies covalent BFL-1 inhibitor molecules that disrupt BH3-binding activity.
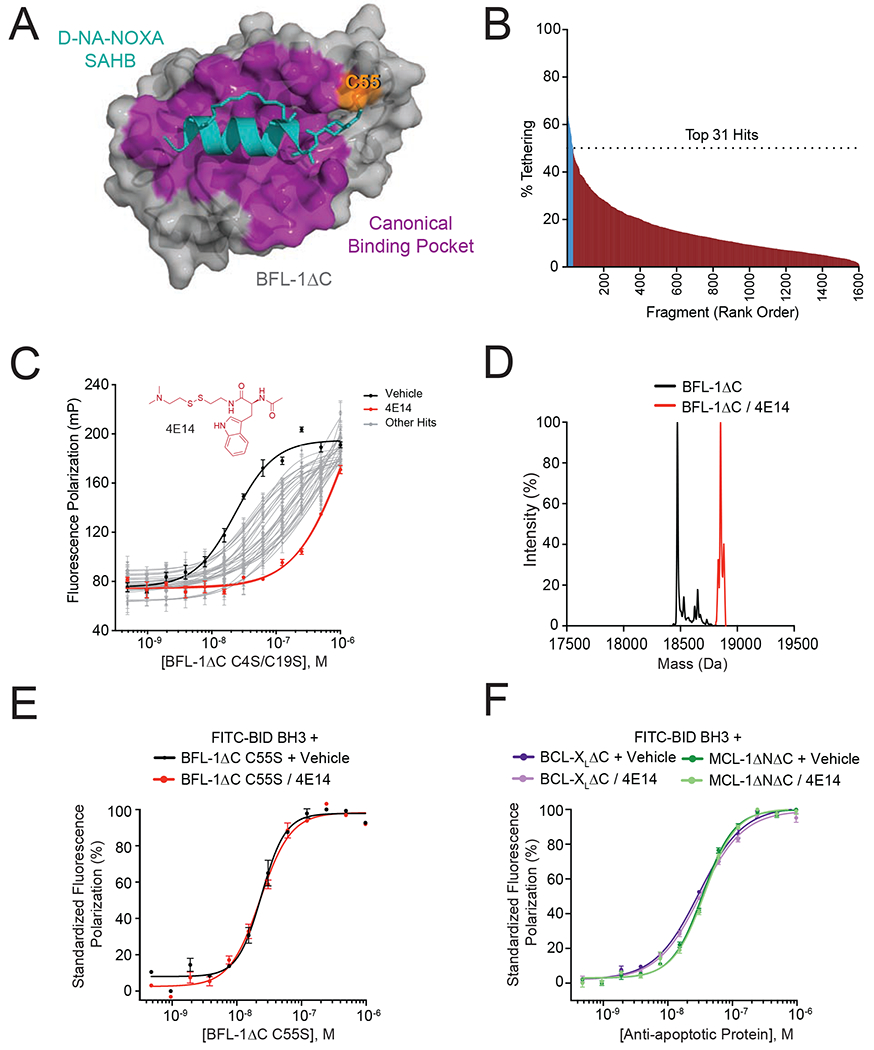
(A) Crystal structure of the complex between anti-apoptotic BFL-1ΔC (grey) and a cysteine-reactive stapled NOXA BH3 peptide (cyan) (PDB: 5WHH), highlighting the site of covalent attachment to C55 (orange) and interaction at the canonical BH3-binding groove (purple).
(B) A disulfide tethering screen of 1600 disulfide fragments against BFL-1ΔC C4S/C19S yielded 31 fragments with percent tethering of at least two standard deviations above the mean, as measured by mass spectrometry.
(C) Fluorescence polarization (FP) assays comparing the relative capacity of the 31 identified fragments to compete with FITC-BID BH3 peptide for interaction at the BFL-1ΔC C4S/C19S groove. 4E14 emerged as the most potent competitive inhibitor of the FITC-BID BH3/BFL-1 interaction (EC50: FITC-BID BH3/BFL-1, 23 nM; FITC-BID BH3/BFL-1–4E14, 1.3 μM). Data are mean ± S.D. for experiments performed in technical triplicate and repeated two times with independent preparations of ligand and protein with similar results.
(D) Complete derivatization of His6-BFL-1ΔC by 4E14, as measured by intact mass spectrometry (molecule:protein, 20:1). Left peak (black): 18,473 Da (BFL-1 minus Met plus H2O); right peak (red): 18,777 Da (M+304).
(E-F) FP analysis evaluating the capacity of 4E14 to compete with FITC-BID BH3 for interaction with BFL-1ΔC C55S (E) and BCL-XLΔC and MCL-1ΔNΔC (F). Data are mean ± S.E.M. for experiments performed in technical triplicate and repeated two times with independent preparations of ligand and protein with similar results.
See also Table S1.
Conformational Consequences of the Covalent 4E14/BFL-1 Interaction
To assess the impact of disulfide bond formation between 4E14 and C55 on the structural dynamics of BFL-1ΔC C4S/C19S, we performed hydrogen deuterium exchange mass spectrometry (HXMS) analyses (Barclay et al., 2015; Engen, 2009; Lee et al., 2016). HXMS interrogates the conformation of proteins by measuring the relative deuterium uptake of backbone amides. Protein regions involved in ligand interaction can become protected or structured, resulting in decreased deuterium exchange of the corresponding backbone amide hydrogens. In contrast, deuterium exchange increases in areas of a protein that become deprotected, unstructured, or more dynamic upon ligand interaction. Here, we incubated BFL-1ΔC C4S/C19S with 4E14 or vehicle at room temperature for one hour and then performed HXMS analyses at 10 seconds and 10 minutes of deuteration. A reducing agent (tris(2-carboxyethyl)phosphine [TCEP]) was included in the quench buffer in order to remove C55-bound 4E14, thereby allowing for peptide comparisons between the vehicle and 4E14 conditions. In the presence of 4E14, the distal region of α2 and proximal portion of α3 were strongly protected from deuterium exchange at the 10 second labeling time point, indicative of molecular engagement of the very structures that comprise the upper portion of the canonical groove, including C55 (Figures 1A, 2A–B, Table S2, Data File S1). At 10 minutes of deuterium labeling, additional adjacent regions become protected from deuterium exchange, including a portion of the α1–α2 loop, proximal α2, and the middle of α4 (Figures 2A,C, Table S2, Data File S1). These data are consistent with 4E14 targeting the canonical groove and reducing the conformational dynamics of the associated components, spanning from the α1–α2 loop to α4 (Figures 2B–C, Table S2, Data File S1).
Figure 2. Conformational consequences of 4E14 derivatization of BFL-1.
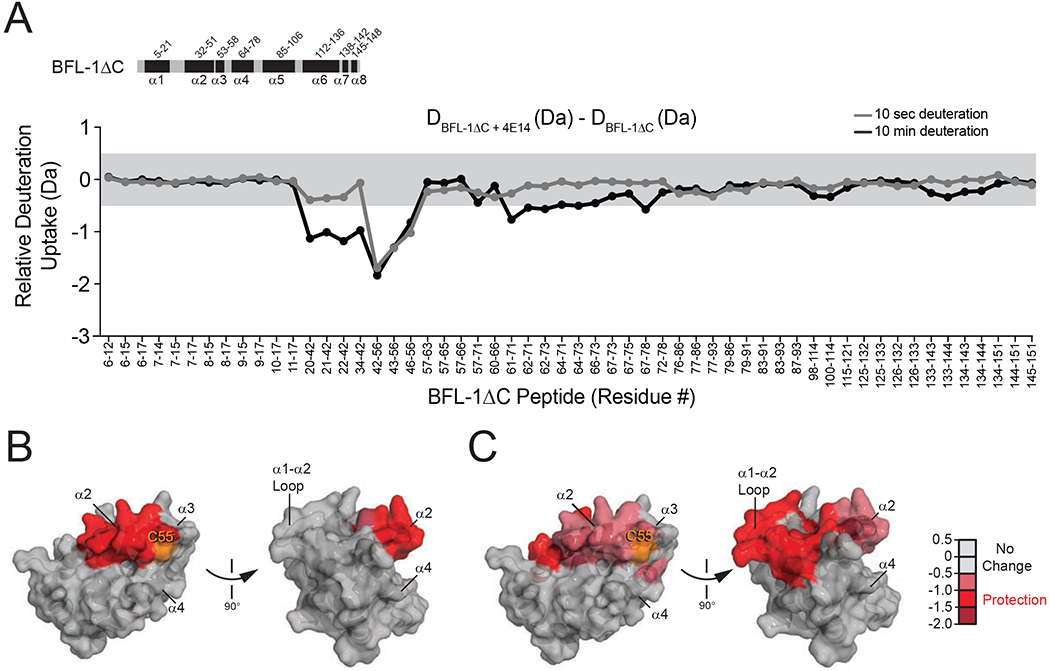
(A) A deuterium difference plot showing the relative deuterium incorporation of BFL-1ΔC C4S/C19S conjugated to 4E14 minus that of apo BFL-1ΔC, as measured at the indicated time points. Each identified peptide is listed in order on the x-axis with the relative protection displayed on the y-axis. The changes outside of the grey shaded region are above the significance threshold of 0.5 Da. Data are representative of three biological replicates. All HXMS data used to create this figure can be found in Data File S1.
(B-C) The regions of relative change in deuterium uptake for BFL-1ΔC/4E14 vs. apo BFL-1ΔC are mapped onto the structure of BFL-1ΔC for the indicated 10 second (B) and 10 minute (C) time points. The relative level of protection is indicated by the color scale.
See also Figure S1, Table S2, and Data File S1.
Next, to distinguish between the influence of 4E14 interaction with BFL-1ΔC in the presence and absence of disulfide bond formation, we repeated the HXMS analysis with BFL-1ΔC C55S. In contrast to the prominent protection of the C55 region of the canonical groove upon disulfide bond formation, 4E14 engagement of BFL-1ΔC in the absence of disulfide formation caused subtle deprotection of the proximal portion of α2 (Figure S1, Table S2, Data File S1). Since initial non-covalent interaction at the canonical groove by individual BH3 helices and small molecules is believed to open the groove to allow for ligand accommodation (Harvey et al., 2018; Lee et al., 2007), it is plausible that the subtle α2 deprotection observed by HXMS could reflect an analogous structural perturbation upon weak non-covalent binding of 4E14 in the absence of C55.
Structure of the 4E14/BFL-1 Complex
In an effort to determine the binding mode of the 4E14/BFL-1 interaction at the canonical groove, we solved a crystal structure of the complex at 1.74 Å resolution (Figure 3A, Table 1, PDB: 6VO4). Notably, BFL-1 α3 residues 55-57 and the molecular fragment itself were not visualized, likely due to flexibility of the disulfide linkage and/or 4E14 binding mode. Nevertheless, alignment with the structure of apo BFL-1 (PDB: 5WHI) and the complex between BFL-1 and the cysteine-reactive stapled peptide D-NA-NOXA SAHB (PDB: 5WHH) revealed that the positioning of α2 in the 4E14/BFL-1 complex is intermediate between unbound and BH3-bound BFL-1 (Figure 3B). This suggests that 4E14 is capable of inducing a conformational opening of the BFL-1 canonical pocket in a manner similar to that observed for a BH3 helix (Harvey et al., 2018). Integrating the crystallography and HXMS results, it appears that non-covalent 4E14 binding may remodel and transiently expose α2 to “open” the groove, followed by conformational stabilization of the region upon disulfide-bond formation between 4E14 and C55 and thus reinforcement of molecular interaction at the groove (Figure 2, Figure S1, Figure 3A–B).
Figure 3. Structural analyses of the 4E14/BFL-1ΔC interaction.
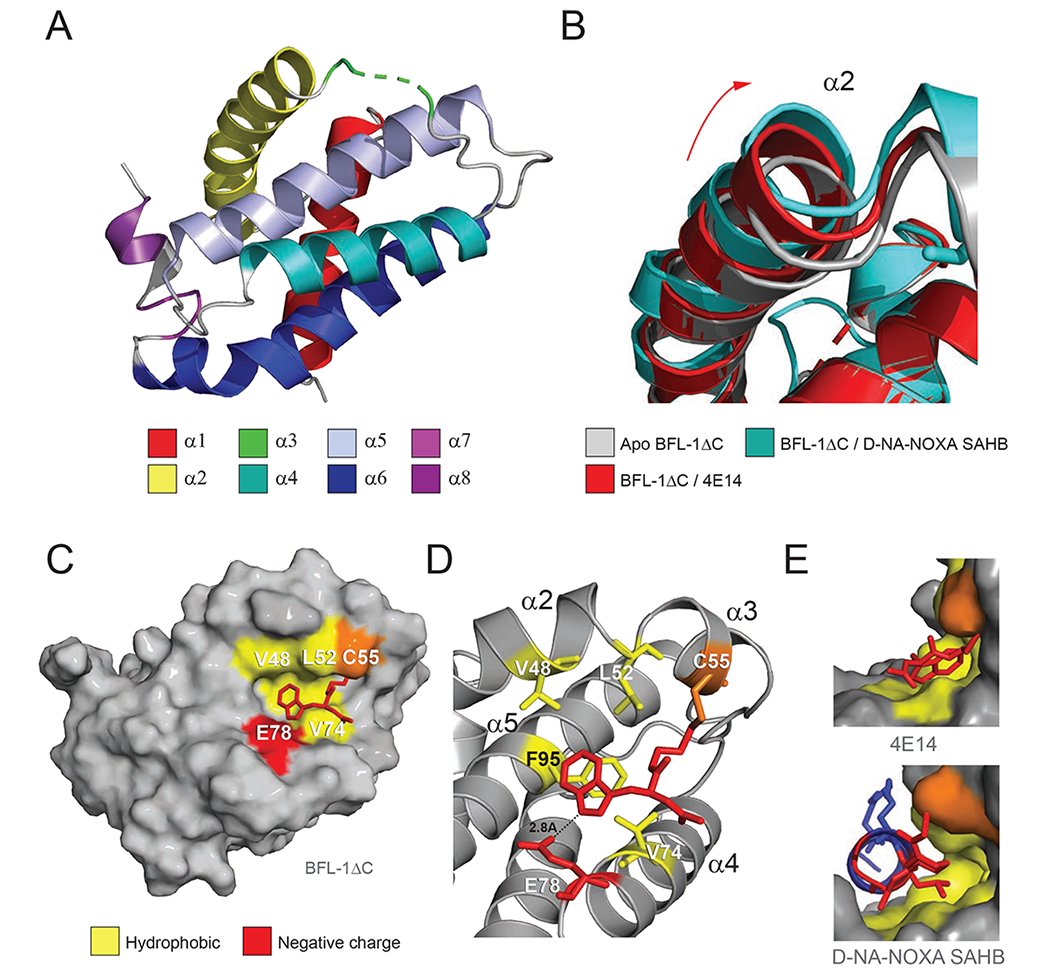
(A) Structure of His6-BFL-1ΔC C4S/C19S when conjugated to 4E14 via C55 (PDB ID: 6VO4). Amino acid residues 55-57 of α3, including the 4E14-C55 conjugate, are not resolved in the structure and instead represented by the green dotted line.
(B) Overlay of the α2 region in the crystal structures of apo BFL-1ΔC (PDB ID: 5WHI), and the complexes between BFL-1ΔC and D-NA-NOXA SAHB (PDB ID: 5WHH) and 4E14 (PDB ID: 6VO4), highlighting the dynamic nature of this region upon small molecule and peptide helix engagement.
(C) Calculated model structure of the 4E14/BFL-1ΔC interaction as determined by covalent docking and molecular dynamics simulations.
(D) Molecular interactions of 4E14 at the docked site on BFL-1ΔC, highlighting engagement of the indole moiety at the hydrophobic p2 region of BFL-1 and proximity of E78 to the indole NH, suggestive of a hydrogen-bonding interaction.
(E) Side views of the interaction between the BH3-binding groove of BFL-1 and 4E14 (top) and D-NA-NOXA SAHB (PDB ID: 5WHH) (bottom), demonstrating the covalent bond between C55 and the respective compounds, in addition to engagement of the disulfide linker (top) and D-nipecotic acid moiety (bottom) with the cryptic site that forms upon ligand-target interaction.
See also Figure S2.
Table 1.
Data Collection and Refinement Statistics
| Structure Name | BFL-1/4E14 |
|---|---|
| RCSB accession code | 6VO4 |
| Data collectiona | |
| Space group | P21 |
| Cell dimensions | |
| a, b, c (Å) | 39.55 43.24 43.49 |
| a, b, g (°) | 90.0 104.9 90.0 |
| Resolution (Å) | 32.78 - 1.74 (1.80 - 1.74)b |
| Rpim | 0.03546 (1.003) |
| I / σI | 11.20 (0.73) |
| Completeness (%) | 91.91 (87.60) |
| Redundancy | 3.2 (3.2) |
| Structure solution | |
| PDB entries used for molecular replacement | 3WHH |
| Refinement | |
| No. reflections, unique | 13520 (1286) |
| Rwork / Rfree | 0.2383/0.2830 |
| No. atoms | 1089 |
| Protein | 1069 |
| Water | 20 |
| B-factors | 60.06 |
| Protein | 60.09 |
| Water | 58.46 |
| R.m.s. deviations | |
| Bond lengths (Å) | 0.007 |
| Bond angles (°) | 0.86 |
| Ramachandran Plot | |
| Preferred | 96.2% |
| Allowed | 3.8% |
| Not Allowed | 0.0% |
A single crystal was used to collect data for the reported structure.
Values in parentheses are for highest-resolution shell.
To calculate a model structure of the 4E14 binding interaction, we performed a covalent docking analysis using the structure of BFL-1 bound to NOXA BH3 (PDB: 3MQP). The preferred pose, in addition to molecular dynamics simulations based on the docking analysis, positioned the indole moiety of 4E14 in the “p2” binding pocket of BFL-1, where the universally conserved leucine of BH3 domain helices engages anti-apoptotic targets (Acoca et al., 2011). 4E14 is predicted to interact with hydrophobic BFL-1 residues V48, L52, V74, and F95 of the p2 region, with the indole hydrogen forming a hydrogen bond with E78 (Figure 3C–E, Figure S2). With the exception of F95 of α5, peptides that contain these α2-α4 residues were all observed to have reduced deuterium exchange by HXMS analysis (Figure 2), supporting the calculated model structure. Taken together, our HXMS, crystallography, and computational studies indicate that upon disulfide bond formation with C55, 4E14 engages the adjacent p2 binding pocket of BFL-1, resulting in focal conformational stabilization of select canonical groove helices, with α2 maintaining an intermediate opening position between that of the apo and BH3-liganded conformations of BFL-1.
Functional Activity of the Covalent 4E14 Interaction
To generate a series of 4E14 analogs for functional testing, we developed a facile solid phase synthetic scheme involving four reaction phases, namely resin transformation, linker addition, monophore core coupling, and cleavage from the resin by disulfide exchange (Figure 4A). The first four synthetic steps transformed the resin’s amine functional group into a thiol that remained on the resin for the duration of the synthesis. The desired disulfide tethering linker was added in steps five and six by first activating the thiol by disulfide exchange with 2,2’-dipyridyldisulfide and then exchanging the activated disulfide with a thiol bearing a linker of desired length. The monophore core of interest was then attached to the linker by HATU coupling (step 7), yielding the disulfide fragment after cleavage from the resin by disulfide exchange (step 8) and LC-MS purification. Using this scheme, we generated 4E14 and a series of derivatives, including N-acetyl-D-tryptophan (D-4E14), N-acetyl-5-methyl-tryptophan (5-Me-4E14), and N-acetyl-D-2-naphthylalanine (D-Nal-4E14) (Figure 4B).
Figure 4. Covalent targeting by 4E14 blocks BFL-1 suppression of BAX-mediated mitochondrial apoptosis.
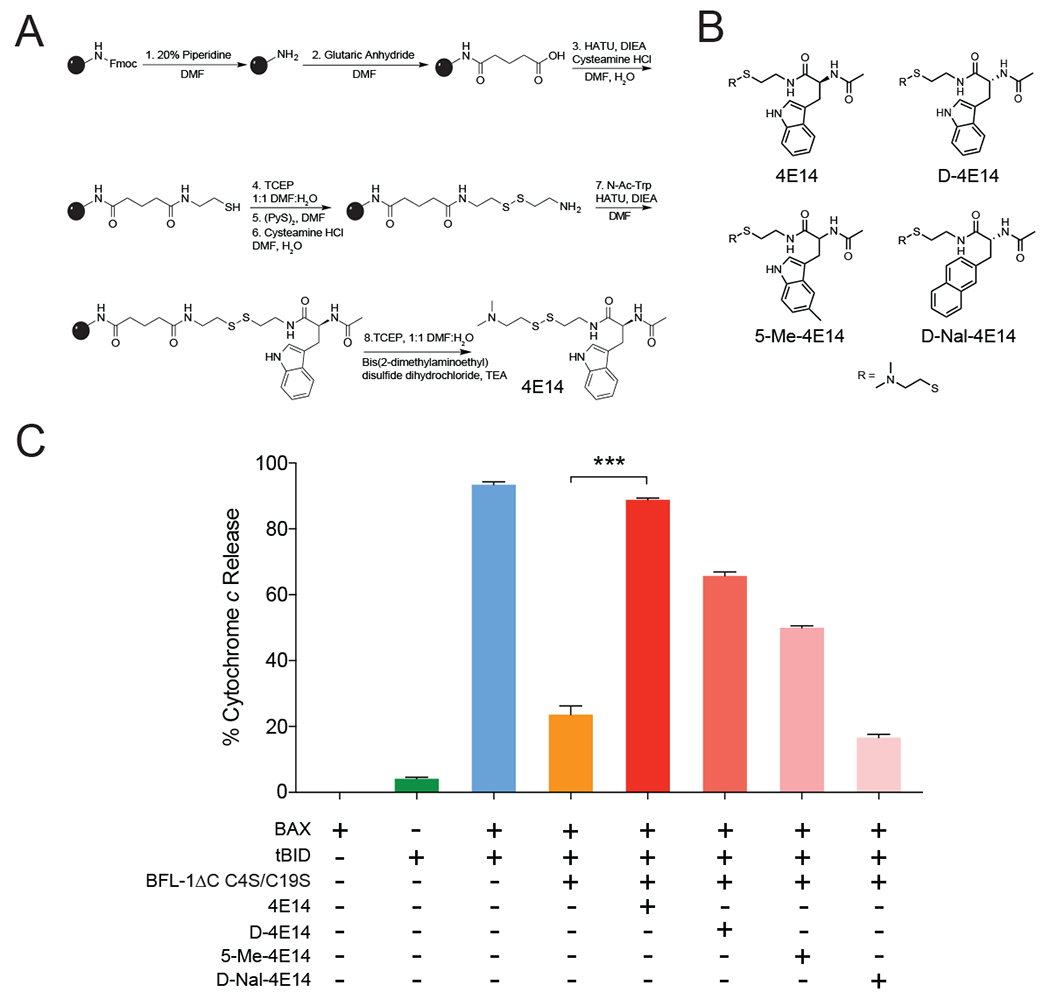
(A) Solid phase synthetic scheme for the production of diverse disulfide tethering molecular fragments on resin.
(B) Chemical structures of 4E14 and analogs.
(C) Percent cytochrome c release from BAX/BAK-deficient mouse liver mitochondria treated with tBID, BAX, and/or BFL-1ΔC C4S/C19S in the presence or absence of 4E14 and its analogs. Data are mean ± S.E.M. for experiments performed in technical triplicate and repeated (biological duplicate) with independent preparations of compounds, protein, and mitochondria with similar results. BAX, 100 nM; BFL-1ΔC C4S/C19S, 1 μM; tBID, 40 nM; 4E14 and analogs conjugated to BFL-1 at molecule:protein ratio of 5:1. ***, p < 0.0001 by unpaired Student’s t test
See also Figures S3 and S4.
To determine if the covalent 4E14 interaction with BFL-1 had functional consequences beyond mitigating BH3-domain binding, as detected by competitive FP (Figure 1C), we evaluated the effect of 4E14 and its analogs on mitochondrial cytochrome c release. We purified BAX/BAK-deficient mitochondria from the livers of AlbCreposBaxfl/flBak−/− mice (Walensky et al., 2006) and treated them with monomeric full-length BAX, tBID, or the BAX/tBID combination in the presence or absence of BFL-1ΔC C4S/C19S. Whereas monomeric BAX or tBID alone had little to no effect on the mitochondria, the combination induced robust cytochrome c release, which was significantly suppressed by the addition of BFL-1 (Figure 4C). Conditions that support complete conversion of BFL-1 to the fully 4E14-conjugated form effectively blocked the anti-apoptotic functionality of BFL-1 (Figure 4C), allowing freed catalytic tBID to trigger BAX, which propagates the signal through autoactivation and resultant cytochrome c release. Importantly, the observed inhibitory activity of 4E14 was abrogated upon C55S mutagenesis of BFL-1ΔC C4S/C19S (Figure S3). Taken together, these data indicate that a small molecular fragment that engages the groove and is reinforced by covalent attachment to C55 can inhibit BFL-1 function.
Finally, we tested several analogs of 4E14 to evaluate potential differences in conjugation and selectivity of functional engagement. Intriguingly, the D-enantiomer of 4E14 fully conjugated to BFL-1 but was ~30% less effective at inhibiting BFL-1 in the cytochrome c release assay compared to the L-enantiomer (Figure 4C), perhaps due to a less effective binding mode for BH3 competition, as suggested by docking analysis (Figure S4A–D). A 5-methylated analog of 4E14 likewise conjugated fully to BFL-1 but was even less active (Figure 4C), indicating that the added methyl group may have altered the indole interaction in a manner that further impaired BH3 competition (Figure S4E–F). Of note, aside from 4E14, no fused ring structures or substituted analogs thereof were found among the 31 hits from the screen (Table S1), consistent with these data. To evaluate the effect of increased hydrophobic bulk and elimination of the indole hydrogen, we replaced the indole moiety of the 4E14 D-enantiomer with naphthalene, which resulted in complete abrogation of conjugation and BFL-1-inhibitory activity (Figure 4C, Figure S4G). These data support the relative specificity of 4E14 for functional BFL-1 interaction, with impairment of activity by only minor modifications of the chemical structure indicative of a discrete structure-activity relationship.
DISCUSSION
BFL-1 is an undrugged BCL-2 family protein implicated in both the pathogenesis and chemoresistance of human cancers (Fan et al., 2010; Haq et al., 2013; Mahadevan et al., 2005; Yecies et al., 2010). Thus, like BCL-2, BCL-XL, and MCL-1, BFL-1 is a high priority therapeutic target. Whereas relatively large small molecules have been developed to inhibit the canonical BH3-binding grooves of BCL-2, BCL-XL, and MCL-1 by non-covalent targeting, here we explored whether incorporating a covalent reaction with the unique C55 of the BFL-1 groove could enable smaller molecules to effectively block anti-apoptotic activity. Indeed, a disulfide tethering screen identified a series of small molecules that effectively competed with BID BH3 for binding to the BFL-1 groove. 4E14, a small N-acetyltryptophan analog of 408 Da that derivatized BFL-1 with a 304 Da moiety, surprisingly emerged as a lead fragment capable of effective BFL-1 blockade.
From a structural standpoint, 4E14 induced conformational changes in the BFL-1 groove that resembled those induced by a cysteine-reactive stapled peptide modeled after the NOXA BH3 domain (D-NA-NOXA SAHB), namely displacement of α2 to “open” and inhibit the groove. The amphipathic BH3 helices bind tightly to the canonical anti-apoptotic grooves by a series of hydrophobic contacts, which are reinforced by a perimeter of complementary charge-charge and hydrophilic interactions (Sattler et al., 1997; Stewart et al., 2010). One of the key hydrophobic interactions involves a leucine residue that is conserved across all BCL-2 family BH3 domains. Our calculated model structure of the 4E14/BFL-1 complex suggests that the indole moiety of 4E14 is positioned to both engage the conserved leucine-binding or p2 pocket and participate in a hydrogen-bonding interaction with E78 of BFL-1 α4. Interestingly, the installation of an indole moiety in venetoclax (based on the fortuitous interaction of W30 from an adjacent BCL-2 molecule with the p4 pocket seen in crystal packing) tailored its selectivity for BCL-2 by a hydrogen-bonding interaction between the indole moiety and D103 of BCL-2 α2 (Souers et al., 2013). In addition to the 4E14 indole interaction at the BFL-1 p2 pocket, the disulfide linker is observed to engage an adjacent cryptic site (involving L52, L56, V74, and F95) that forms below C55 upon covalent (Harvey et al., 2018) or non-covalent (PDB: 3MPQ) NOXA BH3 engagement of BFL-1. Importantly, this newfound hydrophobic pocket does not form upon NOXA BH3 interaction with MCL-1 (Harvey et al., 2018) and thus provides a further design opportunity to achieve selective targeting of BFL-1 by cysteine-reactive small molecules.
The incorporation of a cysteine-reactive moiety into a stapled NOXA BH3 peptide enabled truncation of the original template sequence from 22 amino acids (aa 22-43) to 15 amino acids (aa 26-40) (Guerra et al., 2018; Huhn et al., 2016). Whereas the shortened stapled sequence showed little to no non-covalent interaction with MCL-1 or BFL-1, incorporation of an acrylamide warhead conferred robust and BFL-1-selective binding activity (Guerra et al., 2018; Harvey et al., 2018). Here, we find that 4E14 reaction with BFL-1 C55 produces an M+304 Da adduct that is capable of blocking both BH3 interaction and the capacity of BFL-1 to suppress BAX-mediated cytochrome c release. Thus, our current dataset suggests that incorporation of a cysteine-reactive moiety into a matured non-covalent inhibitor of the anti-apoptotic groove could yield potent and selective molecular inhibitors of BFL-1 that are appreciably smaller than venetoclax, navitoclax, and S63845 molecules and potentially bypass the challenges associated with bRo5 compounds (DeGoey et al., 2018).
Disulfide tethering rapidly provided a starting point for proof-of-concept experiments to determine the feasibility of small molecule covalent targeting of BFL-1. To expand the diversity of analogs, we developed a synthetic scheme for producing monophores using the solid phase, which enables high throughput production of analogs, robust molecular diversification, and minimal purification requirements. For example, our use of solid-phase synthetic methods eliminated the need for aqueous workups and column chromatography, with residual solvents and salts removed upon HPLC purification. Further, the method was amenable to automation, with mixing and washing steps performed on an automated peptide synthesizer, which allowed for multiple syntheses to be readily performed in parallel. Thus, incorporating solid-phase methods to further diversify disulfide-tethering libraries may help expand the discovery potential of cysteine-reactive fragments that engage BFL-1 and the myriad of other targets that harbor cysteines in or around functionally-relevant sites of protein interaction.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact Loren Walensky (loren_walensky@dfci.harvard.edu).
Materials Availability
Plasmids and compounds generated in this study are available upon request to the lead contact.
Data and Code Availability
The data supporting the findings of this study are available within the article and its supplementary materials. X-ray crystallography data was deposited to the PDB under accession number 6VO4 (BFL-1ΔC C4S/C19S / 4E14 complex).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Microbe Strains
Recombinant proteins were expressed in either E. coli LOBSTR BL21(DE3) or E. coli BL21(DE3) bacteria, which were grown in Luria Broth (LB) and respectively maintained at 16°C and 37°C after induction, with shaking at 220 rpm.
METHOD DETAILS
Peptide Synthesis
Synthetic BID BH3 (DIIRNIARHLAQVGDSBDRSI) peptide derivatized at the N-terminus with FITC-β-Ala was synthesized, purified, and quantified as previously described in detail (Bird et al., 2008; Lee et al., 2016). Briefly, FITC-BID BH3 peptide was synthesized by sequential amino acid addition to Rink Amide AM resin (EMD Millipore) using Fmoc chemistry and N-terminal derivatization with FITC, followed by peptide deprotection, cleavage from the resin, purification by reverse phase high performance liquid chromatography-mass spectrometry (LC-MS), and quantitation by amino acid analysis.
Recombinant Protein Expression and Purification
Recombinant, N-terminal hexahistidine-tagged BFL-1ΔC (aa 1-151) and the indicated cysteine-mutant analogs generated by Q5 site-directed mutagenesis (New England Biolabs) were cloned into pET17b (Novagen), expressed in E. coli LOBSTR BL21(DE3) cells (Kerafast) overnight at 16°C, and purified by sequential Ni-affinity and size-exclusion chromatography (Harvey et al., 2018). Protein expression was induced using 0.5 mM isopropyl β-D-1-thiogalactopyranoside (IPTG, Gold Biotechnology). Bacterial pellets were resuspended in lysis buffer (20 mM Tris pH 7.5, 250 mM NaCl) containing two complete protease inhibitor tablets (Roche). Bacteria were lysed using a microfluidizer (M-110L, Microfluidics) and centrifuged at 20,000 x RPM for 45 minutes to remove insoluble debris. Clarified lysate was passed over a Ni-NTA (Qiagen) column equilibrated with lysis buffer. The column was sequentially washed with lysis buffer and then lysis buffer containing 10 mM, 20 mM, 35 mM, and 50 mM imidazole. His-BFL-1ΔC was eluted in buffer containing 150 mM imidazole. The BFL-1 containing fraction was then dialyzed overnight in lysis buffer, concentrated, and purified by size exclusion chromatography using a Superdex S-75 (GE Healthcare) gel filtration system. Protein purity and identity was confirmed by Coomassie staining and western blot analysis using a mouse monoclonal anti-His6 tag antibody (Abcam, RRID: Ab_444306) and sheep anti-mouse secondary antibody (Biorad, RRID: 321929).
Recombinant anti-apoptotic BCL-XLΔC (aa 1-212) and MCL-1ΔNΔC (aa 172-329) were cloned into pGEX-4T-1 (GE Healthcare), expressed in E. coli BL21(DE3) cells (Invitrogen) for four hours at 37°C, and purified by sequential glutathione affinity and size exclusion chromatography (Harvey et al., 2018; Huhn et al., 2016; Pitter et al., 2008). After bacterial lysis and centrifugation, GST-tagged BCL-XLΔC and MCL-1ΔNΔC were purified using a glutathione chromatography column equilibrated with PBS containing 1% Triton X-100. The column was washed twice with PBS/1% Triton X-100 and then two more times with PBS alone. BCL-XLΔC and MCL-1ΔNΔC were cleaved from their GST tags using thrombin (12-15 units) dissolved in PBS. Eluted protein was concentrated and purified by size exclusion chromatography using a Superdex S-75 (GE Healthcare) gel filtration system. Protein purity and identity was confirmed by Coomassie staining and western blot analysis using a goat polyclonal anti-GST antibody (GE Healthcare, RRID: 771432) and donkey anti-goat secondary antibody (Santa Cruz Biotechnology, RRID: Ab_631728).
Disulfide Tethering Fragment Screening by Mass Spectrometry
A 1600-member disulfide fragment library (Burlingame et al., 2011; Hallenbeck et al., 2018) was screened by incubating BFL-1ΔC C4S/C19S protein (1 μM) with each compound (100 μM) for 1 hour at room temperature in the presence of 500 μM BME (Thermo Fisher Scientific). Each well was then screened by UPLC-MS for a change in mass that corresponded with the individual fragment. The compounds were then ranked based on percent tethering to C55 by comparing the relative abundance of conjugated vs. unconjugated BFL-1ΔC C4S/C19S protein. The criteria for pursuing a hit was set at a percent tethering value of at least two standard deviations above the mean for the 1600 fragments tested.
Solid Phase Synthesis of Disulfide Tethering Fragments
Disulfide tethering fragments were synthesized in accordance with the scheme presented in Figure 4A. Briefly, 0.5 mmol Rink Amide AM resin was rinsed once with dimethylformamide (DMF) and then dichloromethane (DCM), deprotected with 20% piperidine in DMF for 10 minutes, and then washed three times with DMF. Glutarylation was performed by adding 2.5 mmol (5 eq) glutaric anhydride dissolved in 5 mL DMF to the deprotected resin, stirring the mixture for 45 minutes, and then washing the resin three times with DMF. Cysteamine was then used to amidate the glutarylated resin as follows: N,N-diisopropylethylamine (DIEA; 1.5 mmol [3 eq]) in 5 mL DCM was added to the resin, stirred for 5 minutes, and removed by vacuum filtration; the resin was then mixed with HATU (1.5 mmol [3 eq]) in 5 mL DMF, the solution removed after 10 minutes, and then cysteamine HCl (2.5 mmol [5 eq]) added with 4 mL DMF, 0.33 mmol DIEA, and water (dropwise) until the solids dissolved. The mixture was stirred for 45 minutes and the resin washed three times with DMF. To reverse any oxidation of the nascent free thiol, TCEP reduction of the resin was performed for 30 minutes using TCEP HCl (0.5 mmol) dissolved in a degassed solution of 1:1 DMF:water. To remove residual TCEP and cysteamine, the resin was washed three times with the following sequence of solvents: DMF, methanol, water, methanol, DMF. Next, the thiol was converted to an activated sulfide by mixing the resin with 2,2’dithiodipyridine (2.5 mmol [5 eq]) dissolved in 5 mL DMF for 20 minutes to afford the 2-thiopyridyl disulfide adduct. To remove excess 2-thiopyridyl disulfide, the resin was washed 5 times with a sequence of DMF and DCM. Next, cysteamine HCl (1 mmol [2 eq]) dissolved in 5 mL of DMF, with minimal water (dropwise addition) for solubility, was used to exchange disulfides for 30 minutes. Following disulfide exchange, the resin was washed 5 times with a sequence of DMF and DCM. At this step of the synthesis, the resin was divided for monophore diversification. Monophore core conjugation to the tether was carried out by adding 3 equivalents of the appropriate carboxylic acid ((S)-2-acetamido-3-(1H-indol-3-yl)propanoic acid for 4E14; (R)-2-acetamido-3-(1H-indol-3-yl)propanoic acid for D-4E14; 2-acetamido-3-(5-methyl-1H-indol-3-yl)propanoic acid for 5-Me-4E14; and (R)-2-acetamido-3-(naphthalen-2-yl) for D-Nal-4E14) and 3.5 eq of HATU and 3.5 eq of DIEA in 5 mL DMF to the resin, followed by mixing overnight for 16 hours. Cleavage of disulfide tethering fragments from the resin was performed using Bis(2-dimethylaminoethyl) disulfide dihydrochloride (1.75 mmol [3.5 eq]) dissolved in a degassed solution of DMF:water (5 mL, 1:1), triethylamine (TEA) (3.5 mmol [7 eq]), and TCEP HCl (0.25 mmol [0.5 eq]). Compounds were purified on an Agilent 1260 Infinity HPLC-MS using a C18 column and a 5–95% acetonitrile gradient in water with 0.1% formic acid, and identities confirmed by high-resolution MS and proton (1H) NMR. 1H NMR was performed using a 500 MHz Bruker Avance III instrument outfitted with a BBFO room-temperature probe. 1H frequencies were referenced to tetramethylsilane (TMS). Proton spectra were acquired with a 45° pulse and a 4.3-s recycle delay at 0.3 Hz per point resolution.
(S)-2-acetamido-N-(2-((2-(dimethylamino)ethyl)disulfanyl)ethyl)-3-(1H-indol-3-yl)propanamide (4E14). 1H NMR (500MHz, MeCN-d3) δ = 9.17 (br. s., 1 H), 7.52 (d, J = 7.9 Hz, 1 H), 7.31 (d, J = 8.2 Hz, 1 H), 7.06 - 7.03 (m, 2 H), 6.99 - 6.95 (m, 1 H), 6.85 (br. s., 1 H), 6.66 (d, J = 7.3 Hz, 1 H), 4.48 - 4.43 (m, 1 H), 3.35 - 3.26 (m, 2 H), 3.11 (dd, J = 6.0, 14.5 Hz, 1 H), 3.00 (dd, J = 7.3, 14.6 Hz, 1 H), 2.84 (d, J = 9.8 Hz, 4 H), 2.68 - 2.57 (m, 2 H), 2.39 (s, 6 H), 1.77 (s, 3 H).
(R)-2-acetamido-N-(2-((2-(dimethylamino)ethyl)disulfanyl)ethyl)-3-(1H-indol-3-yl)propanamide (D-4E14). 1H NMR (500MHz, MeCN-d3) δ = 9.14 (br. s., 1 H), 7.51 (d, J = 7.9 Hz, 1 H), 7.30 (d, J = 7.9 Hz, 1 H), 7.06 - 7.01 (m, 2 H), 6.98 - 6.94 (m, 1 H), 6.78 (br. s., 1 H), 6.60 (d, J = 7.3 Hz, 1 H), 4.46 - 4.42 (m, 1 H), 3.35 - 3.23 (m, 2 H), 3.09 (dd, J = 6.0, 14.5 Hz, 1 H), 2.98 (dd, J = 7.5, 14.8 Hz, 1 H), 2.82 - 2.76 (m, 2 H), 2.67 - 2.62 (m, 2 H), 2.62 - 2.53 (m, 2 H), 2.26 (s, 6 H), 1.76 (s, 3 H).
2-acetamido-N-(2-((2-(dimethylamino)ethyl)disulfanyl)ethyl)-3-(5-methyl-1H-indol-3-yl)propanamide (5-Me-4E14). 1H NMR (500 MHz, DMSO-d6) δ = 10.65 (d, J = 2.4 Hz, 1H), 8.30 (s, 1H), 8.13 (t, J = 5.7 Hz, 1H), 8.02 (d, J = 8.2 Hz, 1H), 7.35 (dd, J = 1.7, 0.9 Hz, 1H), 7.20 (d, J = 8.1 Hz, 1H), 7.06 (d, J = 2.3 Hz, 1H), 6.88 (dd, J = 8.3, 1.6 Hz, 1H), 4.44 (td, J = 8.5, 5.4 Hz, 1H), 3.39 - 3.25 (m, 2H), 3.05 (dd, J = 14.5, 5.4 Hz, 1H), 2.87 (d, J = 8.7 Hz, 1H), 2.86 - 2.80 (m, 2H), 2.75 - 2.63 (m, 2H), 2.48 (s, 1H), 2.38 (s, 3H), 2.15 (s, 6H), 1.79 (s, 3H).
(R)-2-acetamido-N-(2-((2-(dimethylamino)ethyl)disulfanyl)ethyl)-3-(naphthalen-1-yl)propenamide (D-Nal-4E14). 1H NMR (500 MHz, DMSO-d6) δ = 8.37 (s, 3H), 8.28 - 8.14 (m, 1H), 7.90 - 7.77 (m, 3H), 7.75 - 7.68 (m, 1H), 7.53 - 7.38 (m, 3H), 6.28 (s, 1H), 4.55 (td, J = 8.9, 5.3 Hz, 1H), 3.18 (s, 2H), 3.12 (dd, J = 13.7, 5.3 Hz, 3H), 2.95 - 2.87 (m, 2H), 2.87 - 2.77 (m, 2H), 2.74 - 2.62 (m, 2H), 2.47 (d, J = 6.9 Hz, 2H), 2.39 - 2.31 (m, 1H), 2.18 - 2.09 (m, 6H), 1.80 - 1.73 (m, 3H).
Synthesis of 4E14-2-thiopyridine
4E14-2-thiopyridine was generated to maximize 4E14 conjugation to BFL-1ΔC C4S/C19S for X-ray crystallography by replacing the N,N-dimethylcysteamine leaving group with the more efficient 2-mercaptopyridyl leaving group (i.e. molecule:protein ratio reduced to 4:1, allowing for decreased DMSO content in droplets). The synthesis of 2-(pyridin-2-yldisulfanyl)ethanamine was performed as previously reported (Lelle et al., 2017). Briefly, 2.5 g of 2,2’-dithiodipyridine (11.5 mmol, 5.75 eq) in 8 mL of methanol was degassed followed by dropwise addition under nitrogen of a cysteine solution, which was generated by adding 225 mg of cysteamine hydrochloride (2 mmol) to 8 mL of degassed methanol. The mixture was stirred at room temperature overnight. The solution was concentrated in vacuo, resuspended in 5 mL methanol, and then precipitated three times from 100 mL ice cold ether. The product, an off-white solid, was collected via filtration (260 mg, 62% yield). The NMR spectrum matched the published data (Lelle et al., 2017). To generate (S)-2-acetamido-3-(1H-indol-3-yl)-N-(2-(pyridin-2-yldisulfanyl)ethyl) propanamide (4E14-2-thiopyridine), 100 mg of 2-(pyridin-2-yldisulfanyl)ethanamine (537 μmol, 1 eq) was dissolved in 5 mL DMF, followed by 100 mg N-acetyltryptophan (400 μmol, 0.75 eq), 220 mg HATU (580 μmol, 0.9 eq), and 0.25 mL of N,N-diisopropylethylamine (1.5 mmol, 3 eq). The mixture was stirred overnight at room temperature. The DMF solution was poured into 50 mL of ethyl acetate and washed 3 times with 50 mL HCl (0.2 M). The organic layer was dried over sodium sulfate, concentrated, and HPLC purified as described above to afford 46 mg of yellow oil (25% yield). 1H NMR (500 MHz ,DMSO-d6) δ = 10.81 (br. s., 1 H), 8.47 (d, J = 4.3 Hz, 1 H), 8.19 (t, J = 5.5 Hz, 1 H), 8.07 (d, J = 7.9 Hz, 1 H), 7.81 (dd, J = 1.7, 7.5 Hz, 1 H), 7.78 - 7.72 (m, 1 H), 7.58 (d, J = 7.6 Hz, 1 H), 7.32 (d, J = 8.2 Hz, 1 H), 7.25 (dd, J = 4.9, 6.7 Hz, 1 H), 7.14 (d, J = 2.1 Hz, 1 H), 7.09 - 6.92 (m, 2 H), 4.47 (d, J = 5.5 Hz, 1 H), 3.38 - 3.25 (m, 2 H), 3.10 (dd, J = 5.3, 14.5 Hz, 1 H), 2.96 - 2.87 (m, 1 H), 2.84 - 2.72 (m, 2 H), 1.80 (s, 3 H). 13C NMR (126 MHz, DMSO-d6) δ = 172.3, 169.6, 159.6, 150.1, 138.3, 136.5, 127.8, 124.0, 121.7, 121.3, 119.7, 118.9, 118.6, 111.8, 110.7, 53.8, 38.4, 37.6, 28.4, 23.1
Fluorescence Polarization Assays
The indicated BFL-1ΔC constructs, MCL-1ΔNΔC, or BCL-XLΔC (1 μM) were incubated with 25× 4E14 for one hour at room temperature. The proteins were then serially diluted into fluorescence polarization assay buffer (50 mM Tris pH 8, 100 mM NaCl) and incubated with FITC-peptide (60 nM). Fluorescence polarization was measured at equilibrium using a SpectraMax M5 microplate reader, and nonlinear regression analysis of dose-response curves was performed using Prism software 7 (GraphPad).
Mitochondrial Cytochrome c Release Assays
Liver mitochondria from AlbCreBaxf/fBak−/− mice were isolated by dounce homogenization and release assays performed as described (Walensky et al., 2006). Briefly, the indicated BFL-1ΔC construct was incubated with 4E14 or its analogs (molecule: protein, 5:1) or vehicle for 1 hour and then desalted into BFL-1 FPLC buffer using Micro Bio-Spin 6 columns (Biorad) to remove excess 4E14. Mitochondria (1 mg/mL) were incubated with recombinant BAX (100 nM) in the presence or absence of tBID (40 nM) and BFL-1 protein (1 μM) for 45 minutes at room temperature in experimental buffer (200 mM mannitol, 68 mM sucrose, 10 mM HEPES-KOH pH 7.4, 110 mM KCl, 1 mM EDTA, protease inhibitor). The pellet and supernatant fractions were isolated by centrifugation, and cytochrome c was quantitated using a colorimetric Rat/Mouse Cytochrome c Quantikine ELISA assay (R&D Systems), per the manufacturer’s protocol. Briefly, cytochrome c conjugate (75 μL) and sample (50 μL) were added to the ELISA plate and incubated at room temperature for 2 hours. Each well was then aspirated and washed four times using the manufacturer-supplied wash buffer. Substrate solution (100 μL) was added to the plate, which was incubated in the dark for 30 minutes. Stop solution (100 μL) was then added and absorbance read at 450 nm with wavelength correction at 540 nm using a SpectraMax M5 microplate reader. Percent cytochrome c released into the supernatant (%cyto c release) was calculated according to the following equation: %cyto c release = [cyto csup]/ [cyto cmax]*100, where cyto csup and cyto cmax represent the amount of cytochrome c detected in the supernatant upon treatment with the indicated conditions or 1% (v/v) Triton X-100, respectively.
X-ray Crystallography
Apo His6-BFL-1ΔC C4S/C19S was expressed and purified as described (Harvey et al., 2018) and buffer exchanged into 20 mM HEPES pH 7.5, 300 mM NaCl, 10% glycerol, and 50 mM arginine. His6-BFL-1ΔC C4S/C19 in complex with 4E14 was generated by incubating 4E14-2-thiopyridine with His6-BFL-1ΔC C4S/C19 (250 μM) at room temperature for 1 hour. Intact mass spectrometry was used to confirm complete BFL-1 labeling by 4E14. An equal volume (100 nL) of 4.65 mg/mL (250 μM) His6-BFL-1ΔC C4S/C19S/4E14 complex was mixed with reservoir buffer (2.8 M sodium acetate trihydrate, pH 7.0), and crystals were prepared in hanging drops at 20°C. The crystals were transferred into crystallization buffer containing 25% glycerol prior to flash-freezing in liquid nitrogen. Diffraction data from crystals of His6-BFL-1ΔC C4S/C19S conjugated to fragment 4E14 were collected at beamline 24ID-C of the NE-CAT at the Advanced Photon Source (Argonne National Laboratory), and data sets were integrated and scaled with the XIA2 package, which uses the programs Pointless and XDS (Evans, 2006; Kabsch, 2010; Winter et al., 2010). The structure was solved by molecular replacement using the program Phaser (McCoy et al., 2007) and the search model PDB: 5WHH. Iterative manual model building and refinement using Phenix (Adams et al., 2010) and Coot (Emsley and Cowtan, 2004) led to a model with excellent statistics, including maximum diffraction of 1.74 Å (Table 1, PDB: 6VO4).
Molecular Dynamics Simulation and Docking
Computational analyses were performed using the Schrodinger software suite (Version 2016-2). The 4E14 molecule was built and conformations were generated using MacroModel and the OPLS3 forcefield (Harder et al., 2016). BFL-1ΔC (PDB: 3MQP) was prepared using default parameters in the PrepWiz wizard in Maestro (Sastry et al., 2013). Briefly, the bound peptide and all water molecules were removed, the H-bond network of the protein was optimized, and the protein was subjected to an impref minimization using the OPLS3 forcefield. Protonation states were those predicted to occur at pH 7.0 using the Epik module (Shelley et al., 2007). The receptor grid (radius 1 nm) was defined at the center of C55. Docking was performed using the pose prediction mode of CovDock in Schrodinger, with the protein structure minimized in a 0.3 nm radius surrounding the docked molecule (Zhu et al., 2014).
Molecular dynamics calculations were performed using the 4E14/BFL-1 complex generated by molecular docking. The protein was prepared using the default parameters of the Protein Preparation Workflow in Maestro software (Schrödinger version 2016-2). Protonation states were those predicted to occur at pH 7.0 using the Epik module. Each protein was pre-soaked in a cubic box of TIP3P water molecules using the System Builder workflow in Desmond. The box was sized to leave all peptide atoms at least 1 nm from the boundaries. All overlapping solvent molecules were removed, the system was charge neutralized with appropriate counterions and 150 mM NaCl was added to simulate buffer conditions. All MD simulations were performed using the Desmond package (Jorgensen, 1983), with the OPLS3 forcefield used to model all interactions. Periodic boundary conditions were maintained throughout. Long-range electrostatic interactions were calculated using the particle-mesh Ewald method (Essmann, 1995), and van der Waals and short-range electrostatic interactions were smoothly truncated at 0.9 nm. Constant system temperature of 300 K was maintained using Nose-Hoover thermostats (Hoover, 1985), and system pressure was maintained at 1 atm using the Martyna-Tobias-Klein method (Martyna et al., 1994). The equations of motion were integrated using the RESPA integrator, with a 2.0 fs timestep for bonded and short-range interactions and a 6.0 fs timestep for non-bonded interactions beyond the 0.9 nm cutoff. The default parameters in Desmond were used to relax the system prior to simulation (Guo et al., 2010) and a 100 ns production simulation was then run. All simulations were judged to have converged on the basis of radius of gyration calculations and RMSD.
Hydrogen Deuterium Exchange Mass Spectrometry
His-BFL-1ΔC C4S/C19S or His-BFL-1ΔC C55S (25 μM) were conjugated with 4E14 (20x) or vehicle for 1 hour at room temperature. Samples (25 pmol, 1 μL) were deuterated for the indicated time points using an 18-fold excess of labeling buffer (50 mM Tris, 150 mM NaCl, pD 7.6), and labeling was stopped with an equal volume of quench buffer (4M guanidinium chloride, 200 mM potassium phosphate, 0.72 M TCEP, H2O, pH 2.34) (Table S2 and Data File S1). Proteolysis of BFL-1 protein was conducted using an online pepsin digestion column. Digested protein was injected into a Waters UPLC HDX system (Wales et al., 2008) maintained at 0°C to minimize back-exchange. Peptic peptides underwent a 3 min trap and desalting step using a VanGuard pre-column trap (2.1 × 5 mm, ACQUITY UPLC BEH C18, 1.7 μm) flowing at 100 μL/min. Peptides were then separated using an ACQUITY UPLC HSS T3, 1.8 μm, 1.0 × 50 mm analytical column (Waters Corporation) with a 5-35% gradient of increasing acetonitrile (0.1 % formic acid) over 6 min at a flow rate of 100 μL/min. Mass spectra were acquired using a Synapt G2-Si (Waters) in MSE (data independent acquisition) mode with 0.4 scans/sec over a 50-2000 m/z range with ion mobility engaged. BFL-1 peptides were identified using Protein Lynx Global Server (PLGS 3.0.1, Waters Corporation, RRID: SCR_016664) with undeuterated BFL-1 protein as a reference. Data analysis was performed using DynamX 3.0 (Waters Corporation), which included determination of centroid masses for each isotopic distribution, generation of deuterium uptake plots, and comparison between experimental states. Relative deuterium uptake for each peptide was determined by subtracting the average mass of each undeuterated peptide from the average mass of each deuterated peptide. Deuterium exchange was not corrected for back-exchange and thus expressed as relative deuterium uptake (Wales and Engen, 2006).
QUANTIFICATION AND STATISTICAL ANALYSIS
The number of technical and biological replicates for each experiment are indicated in the corresponding figure legend. Mean ± S.D. or S.E.M. values were calculated using Prism software (Graphpad).
Supplementary Material
Data File S1, Related to Figures 2 and S1. HXMS data for analyses of BFL-1ΔC C4S/C19S and its conjugate with 4E14.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-His6 | Abcam | Cat# Ab18184; RRID:AB_444306 |
| Sheep anti-mouse IgG:HRP | Biorad | Cat# AAC10P; RRID:AB_321929 |
| Goat polyclonal anti-GST | GE Healthcare | Cat# 27-4577-01; RRID:AB_771432 |
| Donkey anti-goat IgG:HRP | Santa Cruz Biotech | Cat# Sc-2020; RRID:AB_631728 |
| Bacterial and Virus Strains | ||
| LOBSTR BL21(DE3) Competent Cells | Kerafast | Cat# EC001 |
| One Shot BL21(DE3) Competent Cells | Invitrogen | Cat# C600003 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| His6 BFL-1ΔC WT, C55S, C4S/C19S, C4S/C19S/C55S | Walensky Lab | N/A |
| GST MCL-1ΔNΔC | Walensky Lab | N/A |
| GST BCL-XLΔC | Walensky Lab | N/A |
| Complete Protease Inhibitor Tablet Cocktail | Sigma-Aldrich | Cat# 16829800 |
| IPTG | Gold Biotechnology | Cat# I2481C |
| β-mercaptoethanol (BME) | Thermo Fisher Scientific | Cat# PI35602 |
| 4E14 | Walensky Lab | N/A |
| 4E14-2-thiopyridine | Walensky Lab | N/A |
| D-4E14 | Walensky Lab | N/A |
| 5-Me-4E14 | Walensky Lab | N/A |
| D-Nal-4E14 | Walensky Lab | N/A |
| Disulfide Tethering Fragment Library | Wells Lab | N/A |
| Rink Amide AM Resin 200-400 mesh | Novabiochem | Cat# 855004 |
| Glutaric Anhydride | Sigma Aldrich | Cat# G3806 |
| N,N-Dimethylformamide | Pharmco | Cat# zh324UVHPLC |
| N,N-Diisopropylethylamine | Sigma-Aldrich | Cat# 387649 |
| Dichloromethane | Sigma-Aldrich | Cat# D65100 |
| HATU | ApexBio Tech LLC | Cat# A7022 |
| Cysteamine Hydrochloride | Sigma Aldrich | Cat# M6500 |
| Tris(2-carboxyethyl)phosphine hydrochloride | Sigma Aldrich | Cat# C4706 |
| Aldrithiol-2 | Sigma Aldrich | Cat# 143049 |
| Bis(2-dimethylaminoethyl) Disulfide Dihydrochloride | TCI | Cat# B2272 |
| Critical Commercial Assays | ||
| Q5 Site Directed Mutagenesis Kit | New England Biolabs | Cat# E0554S |
| Rat/Mouse Cytochrome c Quantikine ELISA Kit | R&D Systems | Cat# MCTC0 |
| Deposited Data | ||
| BFL-1/4E14 Crystal Structure | This Paper | PDB: 6VO4 |
| BFL-1/D-NA-NOXA SAHB Crystal Structure | Harvey et al., 2018 | PDB: 5WHH |
| Apo BFL-1 Crystal Structure | Harvey et al., 2018 | PDB: 5WHI |
| BFL-1/NOXA BH3 Crystal Structure | Protein Data Bank | PDB: 3MQP |
| Recombinant DNA | ||
| Plasmid PET17b | Novagen | Cat# 69663 |
| Plasmid PET17b_BFL1ΔC_WT | Walensky Lab | N/A |
| Plasmid PET17b_BFL1ΔC_C4S/C19S | Walensky Lab | N/A |
| Plasmid PET17b_BFL1ΔC_C4S/C19S/C55S | Walensky Lab | N/A |
| Plasmid PET17b_BFL1ΔC_C55S | Walensky Lab | N/A |
| Plasmid PGEX-4T-1 | GE Healthcare | Cat# 28-9545-49 |
| Plasmid PGEX-4T-1_MCL-1ΔNΔC | Walensky Lab | N/A |
| Plasmid PGEX-4T-1_BCL-XLΔC | Walensky Lab | N/A |
| Software and Algorithms | ||
| GraphPad Prism | Graphpad Software Inc. | https://www.graphpad.com/scientific-software/prism/ |
| XIA2 | Winter, 2010 | https://xia2.github.io |
| POINTLESS | Evans, 2006 | http://www.ccp4.ac.uk |
| XDS | Kabsch, 2010 | http://xds.mpimf-heidelberg.mpg.de/ |
| Phaser | McCoy et al., 2007 | https://www.phenix-online.org/documentation/reference/phaser.html |
| Phenix | Adams et al., 2010 | https://www.phenix-online.org/ |
| Coot | Emsley and Cowtan, 2004 | https://www2.mrc-lmb.cam.ac.uk/personal/pemsley/coot/ |
| Schrodinger Maestro | Schrodinger, Version 2016-2 | Schrodinger, LLC |
| Pymol | The PyMol Molecular Graphics System, Version 1.7.4.0 | Schrodinger, LLC |
| ProteinLynx Global Server (PLGS) 3.0.1 | Waters Corporation | http://www.waters.com/waters/en_US/ProteinLynx-Global-SERVER-(PLGS)/nav.htm?cid=513821&locale=en_US |
| DynamX 3.0 | Waters Corporation | http://www.waters.com/waters/library.htm?cid=511436&lid=134832928&locale=en_US |
| Other | ||
| Superdex 75 10/300 GL size exclusion column | GE Healthcare Life Sciences | Cat# 29148721 |
SIGNIFICANCE.
BCL-2 family proteins maintain the critical balance between cellular life and death and, when deregulated, can contribute to the development, maintenance, and chemoresistance of human cancer. Anti-apoptotic BCL-2 proteins suppress cell death by capturing the essential BH3 helix of pro-apoptotic proteins in a surface groove. Relatively large small molecules (800-1000 Da) effectively target the groove but necessitated overcoming beyond-the-rule-of-5 challenges during clinical development. Harnessing a unique cysteine in the canonical groove of anti-apoptotic BFL-1, we performed a disulfide tethering screen that identified a lead small molecule fragment capable of derivatizing BFL-1 with a 304 Da moiety, effectively blocking its BH3-binding and anti-apoptotic functionality. Thus, we demonstrate that disulfide tethering and a solid phase synthetic scheme to expand disulfide tethering libraries can provide a starting point for the development of selective, covalent inhibitors of BFL-1.
HIGHLIGHTS.
A disulfide tethering screen identifies small molecules that target BFL-1 C55
Structural analyses reveal the conformational consequences of disulfide formation
Lead molecule 4E14 effectively competes with BH3-binding at the BFL-1 groove
Covalent 4E14 targeting of C55 inhibits BFL-1 suppression of mitochondrial apoptosis
ACKNOWLEDGMENTS
We thank M. Godes for technical assistance, K. Korshavn for scientific discussions, and M. Tye for assistance with compound characterization by NMR. This research was supported by National Institutes of Health (NIH) grant R35CA197583 and a Leukemia and Lymphoma Society (LLS) Translational Research Program award to L.D.W.; a research collaboration between J.R.E. and the Waters Corporation; NIH grant 5F31CA210592 to E.P.H, with additional support from 5T32GM007306 and 5T32GM095450; NIH grant 5F31CA210590 to Z.J.H.; a National Science Foundation pre-doctoral research fellowship to R.M.G; NIH grant R50CA211399 to G.H.B.; NIH grant R01CA191018 and the Harry and Dianna Professorship in Pharmaceutical Sciences to J.A.W; NIH grant F31CA180378 and a Krevan predoctoral fellowship to T.J.R; and support to C.E.N. and J.L. from NIH grant T32GM007753. Structural analyses were based on X-ray data acquired at the Advanced Photon Source on the Northeastern Collaborative Access Team beamlines (NIGMS P41 GM103403). We are also indebted to the Wolpoff Family Foundation, Jim and Lisa LaTorre, the family of Ivo Coll, and the Todd J. Schwartz Memorial Fund for their steadfast financial contributions to our cancer chemical biology research.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors have no competing interests related to this work.
REFERENCES
- Acoca S, Cui Q, Shore GC, and Purisima EO (2011). Molecular dynamics study of small molecule inhibitors of the Bcl-2 family. Proteins 79, 2624–2636. [DOI] [PubMed] [Google Scholar]
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barclay LA, Wales TE, Garner TP, Wachter F, Lee S, Guerra RM, Stewart ML, Braun CR, Bird GH, Gavathiotis E, et al. (2015). Inhibition of pro-apoptotic BAX by a noncanonical interaction mechanism. Mol Cell 57, 873–886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird GH, Bernal F, Pitter K, and Walensky LD (2008). Synthesis and biophysical characterization of stabilized alpha-helices of BCL-2 domains. Methods Enzymol 446, 369–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birkinshaw RW, Gong JN, Luo CS, Lio D, White CA, Anderson MA, Blombery P, Lessene G, Majewski IJ, Thijssen R, et al. (2019). Structures of BCL-2 in complex with venetoclax reveal the molecular basis of resistance mutations. Nat Commun 10, 2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlingame MA, Tom CT, and Renslo AR (2011). Simple one-pot synthesis of disulfide fragments for use in disulfide-exchange screening. ACS Comb Sci 13, 205–208. [DOI] [PubMed] [Google Scholar]
- Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, and Letai A (2006). Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell 9, 351–365. [DOI] [PubMed] [Google Scholar]
- Chen L, Willis SN, Wei A, Smith BJ, Fletcher JI, Hinds MG, Colman PM, Day CL, Adams JM, and Huang DC (2005). Differential targeting of prosurvival Bcl-2 proteins by their BH3-only ligands allows complementary apoptotic function. Mol Cell 17, 393–403. [DOI] [PubMed] [Google Scholar]
- Cheng EH, Wei MC, Weiler S, Flavell RA, Mak TW, Lindsten T, and Korsmeyer SJ (2001). BCL-2, BCL-X(L) sequester BH3 domain-only molecules preventing BAX- and BAK-mediated mitochondrial apoptosis. Mol Cell 8, 705–711. [DOI] [PubMed] [Google Scholar]
- Choi SS, Park IC, Yun JW, Sung YC, Hong SI, and Shin HS (1995). A novel Bcl-2 related gene, Bfl-1, is overexpressed in stomach cancer and preferentially expressed in bone marrow. Oncogene 11, 1693–1698. [PubMed] [Google Scholar]
- DeGoey DA, Chen HJ, Cox PB, and Wendt MD (2018). Beyond the Rule of 5: Lessons Learned from AbbVie’s Drugs and Compound Collection. J Med Chem 61, 2636–2651. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Engen JR (2009). Analysis of protein conformation and dynamics by hydrogen/deuterium exchange MS. Anal Chem 81, 7870–7875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erlanson DA, Braisted AC, Raphael DR, Randal M, Stroud RM, Gordon EM, and Wells JA (2000). Site-directed ligand discovery. Proc Natl Acad Sci U S A 97, 9367–9372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essmann U, Perera L, & Berkowitz ML (1995). A mooth particle mesh Ewald method. J Chem Phys 103, 8577. [Google Scholar]
- Evans P (2006). Scaling and assessment of data quality. Acta Crystallogr D Biol Crystallogr 62, 72–82. [DOI] [PubMed] [Google Scholar]
- Fan G, Simmons MJ, Ge S, Dutta-Simmons J, Kucharczak J, Ron Y, Weissmann D, Chen CC, Mukherjee C, White E, et al. (2010). Defective ubiquitin-mediated degradation of antiapoptotic Bfl-1 predisposes to lymphoma. Blood 115, 3559–3569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra RM, Bird GH, Harvey EP, Dharia NV, Korshavn KJ, Prew MS, Stegmaier K, and Walensky LD (2018). Precision Targeting of BFL-1/A1 and an ATM Co-dependency in Human Cancer. Cell Rep 24, 3393–3403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Z, Mohanty U, Noehre J, Sawyer TK, Sherman W, and Krilov G (2010). Probing the alpha-helical structural stability of stapled p53 peptides: molecular dynamics simulations and analysis. Chem Biol Drug Des 75, 348–359. [DOI] [PubMed] [Google Scholar]
- Hallenbeck KK, Davies JL, Merron C, Ogden P, Sijbesma E, Ottmann C, Renslo AR, Wilson C, and Arkin MR (2018). A Liquid Chromatography/Mass Spectrometry Method for Screening Disulfide Tethering Fragments. SLAS Discov 23, 183–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haq R, Yokoyama S, Hawryluk EB, Jonsson GB, Frederick DT, McHenry K, Porter D, Tran TN, Love KT, Langer R, et al. (2013). BCL2A1 is a lineage-specific antiapoptotic melanoma oncogene that confers resistance to BRAF inhibition. Proc Natl Acad Sci U S A 110, 4321–4326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder E, Damm W, Maple J, Wu C, Reboul M, Xiang JY, Wang L, Lupyan D, Dahlgren MK, Knight JL, et al. (2016). OPLS3: A Force Field Providing Broad Coverage of Drug-like Small Molecules and Proteins. J Chem Theory Comput 12, 281–296. [DOI] [PubMed] [Google Scholar]
- Harvey EP, Seo HS, Guerra RM, Bird GH, Dhe-Paganon S, and Walensky LD (2018). Crystal Structures of Anti-apoptotic BFL-1 and Its Complex with a Covalent Stapled Peptide Inhibitor. Structure 26, 153–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoover WG (1985). Canonical dynamics: Equilibrium phase-space distributions. Phys Rev A Gen Phys 31, 1695–1697. [DOI] [PubMed] [Google Scholar]
- Huhn AJ, Guerra RM, Harvey EP, Bird GH, and Walensky LD (2016). Selective Covalent Targeting of Anti-Apoptotic BFL-1 by Cysteine-Reactive Stapled Peptide Inhibitors. Cell Chem Biol 23, 1123–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen WL, Chandrasekhar J, Madura JD, Impey RW, & Klein ML (1983). Comparison of simple potential functions for simulating liquid water. J Chem Phys 79, 926–935. [Google Scholar]
- Kabsch W (2010). Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr D Biol Crystallogr 66, 133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalkavan H, and Green DR (2018). MOMP, cell suicide as a BCL-2 family business. Cell Death Differ 25, 46–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotschy A, Szlavik Z, Murray J, Davidson J, Maragno AL, Le Toumelin-Braizat G, Chanrion M, Kelly GL, Gong JN, Moujalled DM, et al. (2016). The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature 538, 477–482. [DOI] [PubMed] [Google Scholar]
- Lee EF, Czabotar PE, Smith BJ, Deshayes K, Zobel K, Colman PM, and Fairlie WD (2007). Crystal structure of ABT-737 complexed with Bcl-xL: implications for selectivity of antagonists of the Bcl-2 family. Cell Death Differ 14, 1711–1713. [DOI] [PubMed] [Google Scholar]
- Lee S, Wales TE, Escudero S, Cohen DT, Luccarelli J, Gallagher CG, Cohen NA, Huhn AJ, Bird GH, Engen JR, et al. (2016). Allosteric inhibition of antiapoptotic MCL-1. Nat Struct Mol Biol 23, 600–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lelle M, Freidel C, Kaloyanova S, Tabujew I, Schramm A, Musheev M, Niehrs C, Mullen K, and Peneva K (2017). Overcoming drug resistance by cell-penetrating peptide-mediated delivery of a doxorubicin dimer with high DNA-binding affinity. Eur J Med Chem 130, 336–345. [DOI] [PubMed] [Google Scholar]
- Mahadevan D, Spier C, Della Croce K, Miller S, George B, Riley C, Warner S, Grogan TM, and Miller TP (2005). Transcript profiling in peripheral T-cell lymphoma, not otherwise specified, and diffuse large B-cell lymphoma identifies distinct tumor profile signatures. Mol Cancer Ther 4, 1867–1879. [DOI] [PubMed] [Google Scholar]
- Martyna GJ, Tobias DJ, and Klein ML (1994). Constant pressure molecular dynamics algorithms. J Chem Phys 101, 4177–4189. [Google Scholar]
- Mason KD, Carpinelli MR, Fletcher JI, Collinge JE, Hilton AA, Ellis S, Kelly PN, Ekert PG, Metcalf D, Roberts AW, et al. (2007). Programmed anuclear cell death delimits platelet life span. Cell 128, 1173–1186. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morales AA, Olsson A, Celsing F, Osterborg A, Jondal M, and Osorio LM (2005). High expression of bfl-1 contributes to the apoptosis resistant phenotype in B-cell chronic lymphocytic leukemia. Int J Cancer 113, 730–737. [DOI] [PubMed] [Google Scholar]
- O’Connor L, Strasser A, O’Reilly LA, Hausmann G, Adams JM, Cory S, and Huang DC (1998). Bim: a novel member of the Bcl-2 family that promotes apoptosis. EMBO J 17, 384–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, et al. (2005). An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435, 677–681. [DOI] [PubMed] [Google Scholar]
- Ostrem JM, Peters U, Sos ML, Wells JA, and Shokat KM (2013). K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 503, 548–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitter K, Bernal F, Labelle J, and Walensky LD (2008). Dissection of the BCL-2 family signaling network with stabilized alpha-helices of BCL-2 domains. Methods Enzymol 446, 387–408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sastry GM, Adzhigirey M, Day T, Annabhimoju R, and Sherman W (2013). Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des 27, 221–234. [DOI] [PubMed] [Google Scholar]
- Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, et al. (1997). Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science 275, 983–986. [DOI] [PubMed] [Google Scholar]
- Shelley JC, Cholleti A, Frye LL, Greenwood JR, Timlin MR, and Uchimaya M (2007). Epik: a software program for pK( a ) prediction and protonation state generation for drug-like molecules. J Comput Aided Mol Des 21, 681–691. [DOI] [PubMed] [Google Scholar]
- Souers AJ, Leverson JD, Boghaert ER, Ackler SL, Catron ND, Chen J, Dayton BD, Ding H, Enschede SH, Fairbrother WJ, et al. (2013). ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat Med 19, 202–208. [DOI] [PubMed] [Google Scholar]
- Stewart ML, Fire E, Keating AE, and Walensky LD (2010). The MCL-1 BH3 helix is an exclusive MCL-1 inhibitor and apoptosis sensitizer. Nat Chem Biol 6, 595–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse C, Shoemaker AR, Adickes J, Anderson MG, Chen J, Jin S, Johnson EF, Marsh KC, Mitten MJ, Nimmer P, et al. (2008). ABT-263: a potent and orally bioavailable Bcl-2 family inhibitor. Cancer Res 68, 3421–3428. [DOI] [PubMed] [Google Scholar]
- Walensky LD, and Gavathiotis E (2011). BAX unleashed: the biochemical transformation of an inactive cytosolic monomer into a toxic mitochondrial pore. Trends Biochem Sci 36, 642–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walensky LD, Pitter K, Morash J, Oh KJ, Barbuto S, Fisher J, Smith E, Verdine GL, and Korsmeyer SJ (2006). A stapled BID BH3 helix directly binds and activates BAX. Mol Cell 24, 199–210. [DOI] [PubMed] [Google Scholar]
- Wales TE, and Engen JR (2006). Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev 25, 158–170. [DOI] [PubMed] [Google Scholar]
- Wales TE, Fadgen KE, Gerhardt GC, and Engen JR (2008). High-speed and high-resolution UPLC separation at zero degrees Celsius. Anal Chem 80, 6815–6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Yin XM, Chao DT, Milliman CL, and Korsmeyer SJ (1996). BID: a novel BH3 domain-only death agonist. Genes Dev 10, 2859–2869. [DOI] [PubMed] [Google Scholar]
- Winter G, Dokel S, Jones AK, Scheerer P, Krauss N, Hohne W, and Friedrich B (2010). Crystallization and preliminary X-ray crystallographic analysis of the [NiFe]-hydrogenase maturation factor HypF1 from Ralstonia eutropha H16. Acta Crystallogr Sect F Struct Biol Cryst Commun 66, 452–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang E, Zha J, Jockel J, Boise LH, Thompson CB, and Korsmeyer SJ (1995). Bad, a heterodimeric partner for Bcl-XL and Bcl-2, displaces Bax and promotes cell death. Cell 80, 285–291. [DOI] [PubMed] [Google Scholar]
- Yecies D, Carlson NE, Deng J, and Letai A (2010). Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 115, 3304–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu K, Borrelli KW, Greenwood JR, Day T, Abel R, Farid RS, and Harder E (2014). Docking covalent inhibitors: a parameter free approach to pose prediction and scoring. J Chem Inf Model 54, 1932–1940. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data File S1, Related to Figures 2 and S1. HXMS data for analyses of BFL-1ΔC C4S/C19S and its conjugate with 4E14.
Data Availability Statement
The data supporting the findings of this study are available within the article and its supplementary materials. X-ray crystallography data was deposited to the PDB under accession number 6VO4 (BFL-1ΔC C4S/C19S / 4E14 complex).