

Abstract
Modular, fully synthetic routes to structurally complex natural products provide useful avenues to access chemical diversity. Herein we report a concise route to virginiamycin M2, a member of the group A streptogramin class of natural products that inhibits bacterial protein synthesis. Our approach features a longest linear sequence of six steps from 7 simple building blocks, and is the shortest and highest yielding synthesis of any member of the streptogramin class reported to date. We believe this route will enable access to unexplored structural diversity and may serve as a useful tool to improve the therapeutic potential of the streptogramin class of antibiotics.
Keywords: Virginiamycin M2, Antibiotics, Bacterial resistance, Total synthesis, Ring-closing metathesis
Graphical Abstract
1. Introduction
Millions of lives have been saved since the advent of antibiotics in the last century.1 Due to the use and misuse of antibiotics in humans and animals, antibiotic resistance is becoming a critical worldwide health issue.2 There is an urgent need for the development of new antibiotics to overcome infections caused by multidrug-resistant, life-threatening pathogens.
The first pair of streptogramin antibiotics was isolated from Streptomyces pristinaperaelis by Charney in 1953.3 This class was divided into group A members featuring macrocyclic polyketide/nonribosomal peptide hybrids (e.g., 1-5, Fig. 1) and group B featuring 19-membered macrocyclic depsipeptides (e.g., virginiamycin S1 (6)).3,4 The activities and mechanisms of action of streptogramins have been well defined by biochemical and structural studies. Group A and group B streptogramins work in concert to inhibit protein synthesis by binding to adjacent sites in the bacterial ribosome.4 Although this synergy results in a potent antibacterial activity and has led to promising clinical candidates,5 only one streptogramin combination has been approved by the US Food and Drug Administration. Synercid is a 70:30 combination of dalfopristin (group A) and quinupristin (group B), which are derived semisynthetically from virginiamycin M1 (2) and virginiamycin S1 (6).6
Fig. 1.

Representive steptogramin antibiotics.
Due to their potent antibacterial activities and uncommon chemical architectures, streptogramin A antibiotics have received much attention from the chemistry community. In 1996, Schlessinger and colleagues published the first total synthesis of virginiamycin M2 (1) featuring vinylogous urethane chemistry.7a This report emerged concomitantly with the first total synthesis of madumycin II by the Meyers group,7b who also disclosed the first total synthesis of griseoviridin (5) in 2000.8 Recently, Panek reported an elegant route to virginiamycin M2 (1) from an allylsilane precursor in 6% overall yield in 10 linear steps by means of alkyne–alkyne reductive cross coupling.9 Other syntheses of these and related streptogramins have also emerged.10 In 2017, we reported a synthesis of four natural group A streptogramin antibiotics that proceeded in 7 linear steps (longest linear sequence) from 7 simple building blocks and featured an intramolecular Stille coupling reaction to construct the 23-membered macrocycle (Fig. 2A).11 Our strategy represented the shortest synthesis of virginiamycin M2 (1) and madumycin II (4), and also provided the first fully synthetic access to virginiamycin M1 (2) and madumycin I (3). Despite its brevity, our first generation route suffers from contextual limitations including acid sensitivity of intermediates, reliance on toxic organotin reagents (and generation of organotin byproducts), and a moderate-yielding Stille macrocyclization. Herein we report a shorter, higher-yielding, fully synthetic route to virginiamycin M2 (1) that overcomes these limitations and we anticipate it will serve to optimize this scaffold for improved therapeutic utility.
Fig. 2.

(A) The first generation route to virginiamycin M2. (B) Retrosynthetic analysis of the second generation route to virginiamycin M2.
2. Results and discussion
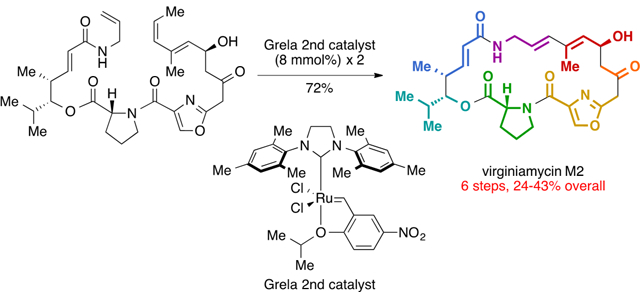
A retrosynthetic analysis for our revised route is described in Fig. 2B. We envisioned that the conjugated diene of 1 could be constructed by ring closing metathesis (RCM), which would not require organotin reagents and would shorten the overall strategy. RCM was first reported by Didier Villemin in 198012 and was expanded upon by R. H. Grubbs, R. R. Schrock, and Y. Chauvin to become a powerful method for the construction of alkene-containing ring systems.13,14 RCM precursor 8 could be assembled by peptide bond formation from amine 9 and acid 10 which we anticipated could be prepared using the framework of our first synthesis as a guideline.11
Our initial efforts toward a second generation route to viginiamycin M2 (1) are depicted in Scheme 1. The synthesis of the left half of the molecule (9) commenced with an oxazaborolidine-catalyzed, Mukaiyama-type vinylogous aldol reaction between isobutyraldehyde (11) and silyl dienol ether 12 to provide enoate 14 in 94% yield and 87% ee.11,15 Trimethylaluminum-mediated amidation of 14 with allyl amine (15) produced 16 in 90% yield. Coupling of 16 with Fmoc-D-Pro-OH (17) in presence of DCC and catalytic DMAP followed by removal of the Fmoc-group with diethylamine in one pot provided amine 9 in 96% yield. The synthesis of the left half (9) proceeds from building blocks 11 and 12 in three steps in 81% overall yield.
Scheme 1.

Initial efforts to synthesize virginiamycin M2 (1) by ring closing metathesis. DCC = dicyclohexylcarbodiimide, DCM = dichloromethane, DMAP = 4- dimethylaminopyridine, Fmoc = 9-fluorenylmethoxycarbonyl, HATU = 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium-3- oxidhexafluorophosphate, Im = imidazole, TBS = tert-butyldimethylsilyl, TfO = trifluoromethanesulfonate, TBAF = tetra-n-butylammonium fluoride, TMS = trimethylsilyl.
For the right half of the molecule (10), we initially targeted acid 10a, which contains a diene with minimal substitution (R = H). We found that the conditions used in our previous synthesis (TiCl4 and iPr2EtN)11,16 were unsuitable for the coupling of dienal 18a with acetyl thiazolidinethione 19, as they led to retro-aldol reaction after quenching with water or pH = 7 buffer. When Sn(OTf)2 and N-ethylpiperidine were used, 20a was produced in 74% yield as a single diastereomer.17 The minimal toxicity of tin(II) salts compared to organotin compounds18 made this an acceptable concession in the current route. Silylation of 20a with tert-butyldimethylsilyl trifluoromethanesulfonate in the presence of 2,6-lutidine delivered 21a in 93% yield. Cleavage of the thiazolidinethione auxiliary by dianion of oxazole 22 proceeded in 70% yield to provide the right half 10a. The route to the right half (10a) proceeds in 48% yield over three steps from 18a and 19. Then the left half (9) and the right half (10a) are coupled by means of HATU and iPr2EtN, providing RCM precursor 8a yield. Using the same general strategy, we also prepared precursor 8b, which contains an additional trans-methyl group on the diene function.
Efforts to cyclize the 23-membered ring of virginiamycin M2 are summarized in Table 1. Attempted cyclization of 8a with Grubbs I/II and Hoveyda-Grubbs I/II ([Ru]-1–4) catalysts at 23 °C resulted no conversion by 1H NMR analysis (entry 1). At higher temperature (70 °C), 8a was consumed but no product was detected (entry 2). Trans-methyl-substituted precursor 8b produced similar results with [Ru]-2 (entries 3 and 4). Our first promising result came when RCM precursor 8b was exposed to more active, phosphine-free Hoveyda-Grubbs II catalyst ([Ru]-4), resulting in 15% yield of macrocycle 23 at ambient temperature determined by 1H NMR with 1,4-dinitrobenzene as an internal standard.
Table 1.
Preliminary investigations into the ring closing metathesis of macrocycle precursors 8a and 8b.
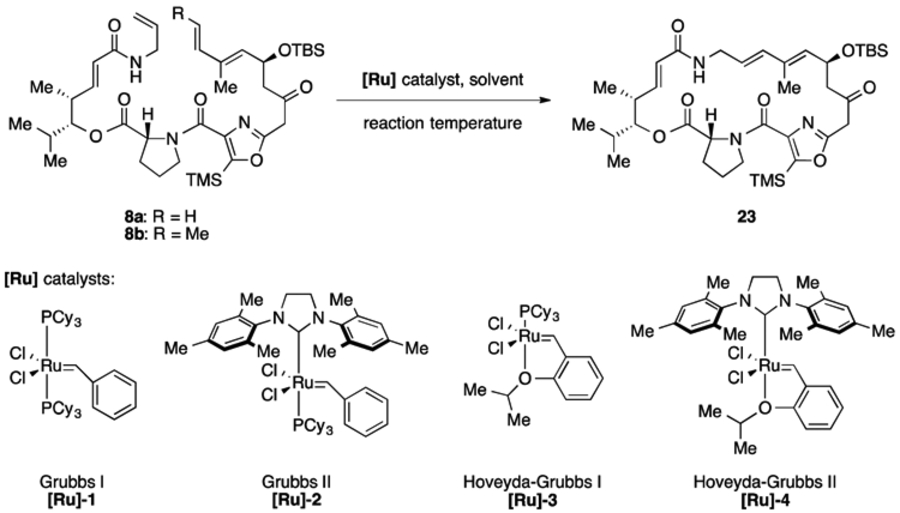 | |||||
|---|---|---|---|---|---|
| Entry | RCM precursor | Catalyst | Solvent | T(°C) | Yield |
| 1 | 8a | [Ru]-1, [Ru]-2, [Ru]-3 or [Ru]-4 (20 mmol%) | Tol | 23 | -a |
| 2 | 8a | [Ru]-1, [Ru]-2, [Ru]-3 or [Ru]-4 (20 mmol%) | Tol | 70 | -a |
| 3 | 8b | [Ru]-2 (20 mmol%) | Tol | 23 | -a |
| 4 | 8b | [Ru]-2 (20 mmol%) | Tol | 70 | -a |
| 5 | 8b | [Ru]-4 (20 mmol%) | Tol | 23 | 15%b |
No product detected.
Yield determined by 1H NMR analysis of the crude reaction mixture using 1,4-dinitrobenzene as an internal standard. Tol = toluene.
During preliminary experiments, batches of trans-methyl diene precursor 8b were contaminated with small amounts of the corresponding cis-methyl precursor (data not shown). 1H-NMR analysis of crude RCM reaction mixtures indicated this contaminant was completely consumed, even when the majority of 8b remained unreacted. Thus, we explored the possibility that the cis-trans diene might serve as a more effective precursor for macrocyclization than the trans-methyl diene in 8b. We were readily able to obtain significant quantities of cis-methyl precursor 8c using stereoisomerically pure dienal 18c as a starting material (Scheme 2). Following the same strategy as depicted in Scheme 1, we produced macrocycle precursor 8c in 41% from building block 18c or in 73% yield from building block 11 over 4 steps.
Scheme 2.

Optimized synthesis of virginiamycin M2 (1) by means of a macrocycle precursor containing a Z alkene. DCC = dicyclohexylcarbodiimide, DCM = dichloromethane, DMAP = 4- dimethylaminopyridine, Fmoc = 9-fluorenylmethoxycarbonyl, HATU = 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5- b]pyridinium-3- oxidhexafluorophosphate, Im = imidazole, TBS = tert-butyldimethylsilyl, TfO = trifluoromethanesulfonate, TBAF = tetra-n-butylammonium fluoride, TMS = trimethylsilyl.
To our delight, when 8c was treated with [Ru]-4 in tolene at 23 °C, the yield was greatly improved (38% 1H NMR yield, Table 2, entry 4). Unsurprisingly, we observed as a byproduct the trans-methyl macrocyclization precursor 8b, which results from isomerization of 8c. Such isomerizations are well-documented for olefin metathesis,19 and we next sought conditions that would favor the cyclization pathway over the unproductive isomerization pathway. Catalysts with pyridyl coordinating groups (e.g., [Ru] −520 and [Ru] −6,21 Table 2, entries 5 and 6) or with smaller carbene ligands (e.g., [Ru]-7,22 entry 7) did not improve the yield of macrocycle 23. We found that batchwise addition of catalyst [Ru]-4 (2 × 8 mol%) and a more polar solvent (benzenetrifluoride) led to small improvements (entries 8 and 9). Finally, a screen of metathesis catalysts that are known to increase initial reaction rate (entries 10–14)23 revealed that catalyst [Ru]-11, which was developed by Grela and coworkers,24 provided the highest yield of macrocycle 23 from precursor 8c (49% isolated yield). Interestingly, we found that removal of the silyl groups in prior to macrocizliation (8c → 8d) greatly increased the yield of the macrocyclization, providing virginiamycin M2 (1) in 72% isolated yield (Table 2, entry 14). While the cause of this improvement is unclear, it is possible that it arises from catalyst coordination to the exposed allylic alcohol25 or from favorable conformational bias in the desilylated macrocyclic precursor 8d relative to 8c. It is worth noting that this desilylation strategy did not improve the yield of the Stille macrocyclization in our 1st generation route.11
Table 2.
Optimization of ring closing metathesis on macrocycle precursors 8c and 8d, which contain Z alkenes.
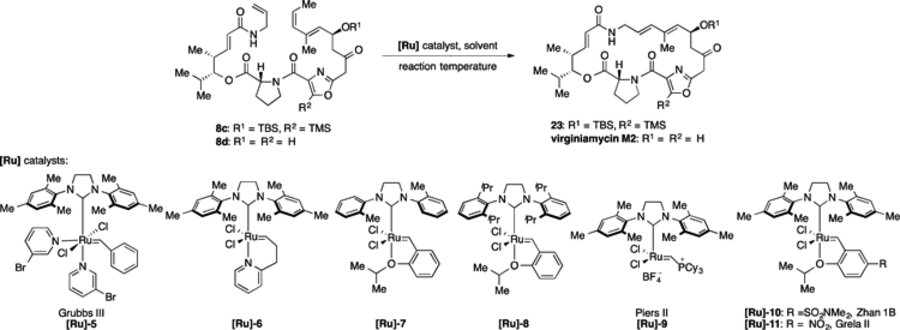 | |||||
|---|---|---|---|---|---|
| Entry | RCM precursor | Catalyst | Solvent | T(°C) | Yield |
| 1 | 8c | [Ru]-1 (20 mmol%) | Tol | 23 | -a |
| 2 | 8c | [Ru]-2 (20 mmol%) | Tol | 23 | 25%b |
| 3 | 8c | [Ru]-3 (20 mmol%) | Tol | 23 | -a |
| 4 | 8c | [Ru]-4 (20 mmol%) | Tol | 23 | 38%b |
| 5 | 8c | [Ru]-5 (20 mmol%) | Tol | 23 | -a |
| 6 | 8c | [Ru]-6 (20 mmol%) | Tol | 23 | -a |
| 7 | 8c | [Ru]-7 (20 mmol%) | Tol | 23 | 27%b |
| 8 | 8c | [Ru]-4 (8 mmol%) × 2 | Tol | 23 | 40%b |
| 9 | 8c | [Ru]-4 (8 mmol%) × 2 | BTF | 23 | 42%b |
| 10 | 8c | [Ru]-8 (8 mmol%) × 2 | BTF | 23 | 33%b |
| 11 | 8c | [Ru]-9 (8 mmol%) × 2 | BTF | 0 | -a |
| 12 | 8c | [Ru]-10 (8 mmol%) × 2 | BTF | 23 | 28%b |
| 13 | 8c | [Ru]-11 (8mmol%) × 2 | BTF | 23 | 49%c |
| 14 | 8d | [Ru]-11 (8 mmol%) × 2 | DCM | 23 | 72%c |
No product detected.
Yield determined by 1H NMR analysis of the crude reaction mixture using 1,4-dinitrobenzene as an internal standard.
isolated yield after column chromatography. BTF = benzotrifluoride, DCM = dichloromethane, Tol = toluene.
The interesting reactivity patterns of the four macrocycle precursors (8a–d) merit further discussion. Z alkenes have been shown to improve the yield and stereoselectivity of ring-closing metatheses to provide Z-alkene-containing macrocycles.26 In the present context, however, metathesis of the Z alkene precursors 8c and 8d selectively deliver the E macrocycles 23 and 1. To the best of our knowledge this is the first example of a Z alkene serving as a more effective RCM precursor than an E alkene or a monosubstituted alkene to provide an E-alkene-containing macrocycle. This pattern of reactivity may be due in part to the conformational rigidity imparted by the abundance of sp2 atoms in the macrocycle precursors herein, or from the differing reactivity of dienes compared to monoenes in metatheses reactions. We believe, however, that these results may prove to be more general, and that inclusion of Z alkene precursors should be considered during the optimization of challenging RCM reactions.
Scheme 3 provides a comparison of our two routes to virginiamycin M2 (1). By leveraging olefin metathesis to close the 23-membered macrocycle, we were able to reduce the longest linear sequence (LLS) from 7 to 6 steps. This resulted in improvement of the yields of each half and in a higher yield for macrocyclization (59% → 72%). Moreover, the new route is not reliant on organotin reagents and does not produce organotin byproducts, although it does require tin(II) salts. The absense of vinyltin intermediates in the current route also expands the scope of reagents that may be used during the synthesis of streptogramin analogs (e.g., acidic reagents that would protodestannylate vinyltin compounds). It is worth noting that both routes required extensive optimization of the macrocyclization step,11 which is not uncommon in the synthesis of macrocycles.
Scheme 3.

Comparison of the two routes to the group A streptogramin virginiamycin M2 (1).
3. Conclusion
In summary, we have develeped a short, efficient route to virginiamycin M2 (1) that proceeds in 6 steps from building blocks 11 and 18c in 24–43% overall yield. This route features a ring closing metathesis to form the 23-membered ring from an enediene system. Three precursors, terminal diene, trans-methyldiene and cis-methyldiene were designed to perform ring-closing metathesis and various Ru-carbene catalysts were screened to facilitate this transformation. The current route is one step shorter than our first generation route and proceeds in higher yield. We believe it will serve as a useful platform for the synthesis of group A streptogramin analogs with improved properties for therapeutic use.
Experimental section
3.1. General
All reactions were performed in flame- or oven-dried glassware fitted with rubber septa under a positive pressure of nitrogen or argon, unless otherwise noted. All reaction mixtures were stirred throughout the course of each procedure using Teflon-coated magnetic stir bars. Air- and moisture-sensitive liquids were transferred via syringe or stainless steel cannula. Solutions were concentrated by rotary evaporation below 35 °C. Analytical thin- layer chromatography (TLC) was performed using glass plates pre-coated with silica gel (0.25-mm, 60-Å pore size, 230–400 mesh, SILICYCLE INC) impregnated with a fluorescent indicator (254 nm). TLC plates were visualized by exposure to ultraviolet light (UV), and then were stained by submersion in a basic aqueous solution of potassium permanganate or with an acidic ethanolic solution of anisaldehyde, followed by brief heating.
DCM and tetrahydrofuran (THF) to be used in anhydrous reaction mixtures were dried by passage through activated alumina columns immediately prior to use. Anhydrous toluene and benzotrifluoride (BTF) used were purchased from Sigma-Aldrich. Hexanes used were ≥85% n-hexane. Other commercial solvents and reagents were used as received, unless otherwise noted.
Proton nuclear magnetic resonance (1H NMR) spectra and carbon nuclear magnetic resonance (13C NMR) spectra were recorded on 400 MHz Bruker Avance III HD 2-channel instrument NMR spectrometers at 23 °C. Proton chemical shifts are expressed in parts per million (ppm, 03B4 scale) and are referenced to residual protium in the NMR solvent (CHC13: δ 7.26). Carbon chemical shifts are expressed in parts per million (ppm, δ scale) and are referenced to the carbon resonance of the NMR solvent (CDC13: δ 77.0). Data are represented as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, dd = doublet of doublets, dt = doublet of triplets, sxt = sextet, m = multiplet, br = broad, app = apparent), integration, and coupling constant (J) in hertz (Hz). Optical rotations were measured using a JASCO P-2000 polarimeter. High-resolution mass spectra were obtained on a Waters Acquity UPLC/Xevo G2-XS QTOF mass spectrometer with ESI ionization (special thanks to Ziyang Zhang in the Shokat Laboratory for assistance). Melting points were recorded on an Electrothermal IA6304 Melting Point Apparatus.
3.2. Experimental procedures and data for synthetic compounds
4.2.1. Mukaiyama aldol product 14
A 250-mL round-bottom flask was charged with phenylboronic acid (1.22 g, 10.0 mmol, 0.5 equiv) and (S)-diphenyl(pyrrolidin-2-yl)methanol (2.53 g, 10.0 mmol, 0.5 equiv). The vessel was equipped with a reflux condenser and a Dean-Stark apparatus, evacuated and flushed with nitrogen (the process of nitrogen exchange was repeated a total of 3 times). Toluene (50 mL) was added, and the resulting clear solution was brought to reflux by means of a 145 °C oil bath. After 12 h, the reaction mixture was allowed to cool to 23 °C and was concentrated. The resulting white solid was dried at ≤1 Torr for 1 h. The vessel was flushed with nitrogen and DCM (80 mL) was added. The resulting colorless solution was cooled to −78 °C and TfOH (0.80 mL, 8.99 mmol, 0.45 equiv) was added dropwise over 5 min by means of glass syringe (CAUTION: TfOH rapidly corrodes most plastic syringes!). Some of the TfOH freezes upon contact with the solution. After 1 h, the solids had dissolved, and a mixture of isobutyraldehyde (11, 1.82 mL, 20.0 mmol, 1 equiv), silyl dienol ether 12 (5.70 g, 25.0 mmol, 1.25 equiv), and isopropanol (1.68 mL, 22.0 mmol, 1.1 equiv) in DCM (20 mL) was added dropwise over 2 h by syringe pump. The mixture was stirred at −78 °C for another 1.5 h and saturated aqueous NaHCO3 solution (50 mL) was added in one portion. The vessel was removed from the cooling bath and the system was allowed to warm to 23 °C while it was rapidly stirred. The biphasic mixture was transferred to a separatory funnel and the layers were separated. The aqueous layer was extracted with DCM (2 × 30 mL). The organic layers were combined and the resulting solution was dried (Na2SO4). The dried solution was filtered and the filtrate was concentrated. The crude residue was purified by flash chromatography (silica gel, eluent: EtOAc:hexanes = 1:10 to 1:6) to afford Mukaiyama aldol product 14 (3.48 g, 94%) as a colorless oil. TLC (EtOAc:hexanes = 1:6): Rf = 0.25 (UV, KMnO4). [α]23D = + 23.5 (c = 1.0, CHCl3). 1H NMR (400 MHz, CDCl3) δ 6.92 (dd, J = 15.7, 8.1 Hz, 1H), 5.86 (dd, J = 15.7, 1.2 Hz, 1H), 3.72 (s, 3H), 3.26 (t, J = 5.8 Hz, 1H), 2.59 – 2.39 (m, 1H), 1.78–1.64 (m, 1H), 1.59 (br s, 1H), 1.09 (d, J = 6.7 Hz, 3H), 0.92 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 167.1, 152.2, 120.4, 80.0, 51.4, 39.9, 30.9, 19.6, 16.5, 13.9. HRMS-EI m/z calcd for C H +10 O +19 3 [M + H]+ 187.1329, found 187.1331.
Determination of enantiomeric excess:
To a solution of 14 (20 mg, 0.11 mmol, 1 equiv) in DCM (2 mL) at 23 °C was added successively Et3N (0.12 mL, 0.86 mmol, 8.0 equiv), DMAP (18 mg, 0.15 mmol, 1.4 equiv) and (S) or (R)- Mosher acid chloride (80 μL, 0.43 mmol, 4.0 equiv). After 2 h, the mixture was diluted with EtOAc (15 mL). The mixture was transferred to a separatory funnel and washed successively with 1 M aqueous KHSO4 solution (3 × 5 mL), 1 M aqueous NaOH solution (5 mL) and saturated aqueous NaHCO3 solution (3 × 5 mL). The organic phase was dried over MgSO4, the dried solution was filtered, and the filtrate concentrated. The crude residue was analyzed by 1H-NMR.
For (S)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoyl chloride: The enantiomeric excess was calculated from integration of the peaks at 5.82 ppm (major) and 5.84 ppm (minor). The ee was 87%.
For (R)-3,3,3-trifluoro-2-methoxy-2-phenylpropanoyl chloride: The enantiomeric excess was calculated from integration of the peaks at 5.84 ppm (major) and 5.82 ppm (minor). The ee was 87%.
4.2.2. Amide 16
A 250-mL round-bottom flask was charged with allylamine (15, 3.2 mL, 43.0 mmol, 4.0 equiv) and dry DCM (72 mL) under nitrogen. The resulting colorless solution was cooled to 0 °C by means of an ice-water bath. A solution of AlMe3 in heptane (1 M, 43.0 mL, 43.0 mmol, 4.0 equiv) was added dropwise over 30 min (CAUTION: Gas evolution!). The mixture was allowed to warm to 23 °C. After stirring for 30 min, a solution of 14 (2.00 g, 10.7 mmol, 1 equiv) in DCM (20 mL) was added over 10 min (CAUTION: Gas evolution!). The vessel was equipped with a reflux condenser and the solution was brought to reflux by means of a 50 °C oil bath. After 3 h, the mixture was cooled to 0 °C by means of ice-water bath and MeOH (10 mL) was added (CAUTION: Gas evolution!). Once gas evolution ceased, saturated aqueous potassium sodium tartrate solution (100 mL) was added. After stirring for 1 hour, the biphasic mixture was transferred to a separatory funnel and the layers were separated. The aqueous layer was extracted with DCM (2 × 30 mL). The combined organic layers were washed with water (100 mL) and brine (100 mL) and the washed solution was dried (Na2SO4). The dried solution was filtered and the filtrate was concentrated. The crude residue was purified by flash chromatography (silica gel, eluent: EtOAc:hexanes = 1:1) to afford amide 16 (2.05 g, 90%) as a white solid. TLC (EtOAc:hexanes = 1:1): Rf = 0.20 (UV, KMnO4).1H NMR (400 MHz, CDCl3) δ 6.79 (dd, J = 15.4, 7.8 Hz, 1H), 5.91 (br t, J = 5.6 Hz, 1H), 5.89 – 5.77 (m, 2H), 5.18 (dq, J = 17.2, 1.6 Hz, 1H), 5.12 (dq, J = 10.2, 1.4 Hz, 1H), 3.92 (tt, J = 5.8, 1.6 Hz, 2H), 3.23 (q, J = 5.6 Hz, 1H), 2.47 (dddd, J = 8.1, 7.0, 5.7, 1.3 Hz, 1H), 1.98 (d, J = 5.3 Hz, 1H), 1.71 (dq, J = 13.1, 6.6 Hz, 1H), 1.06 (d, J = 6.7 Hz, 3H), 0.91 (d, J = 2.5 Hz, 3H), 0.89 (d, J = 2.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 165.9, 147.7, 134.1, 123.1, 116.5, 79.1, 41.9, 39.5, 30.8, 19.7, 16.9, 13.7. HRMS-ESI m/z calcd for C12H21NNaO2+ [M + Na]+ 234.1464, found 234.1473.
4.2.3. Amine 9
A oven-dried 250-mL round-bottom flask was charged with amide 16 (2.00 g, 9.46 mmol, 1.0 equiv), Fmoc-D-Pro-OH (17, 4.79 g, 14.2 mmol, 1.5 equiv) and DMAP (0.23 g, 1.89 mmol, 0.2 equiv). DCM (95 mL) was added, resulting in a colorless solution. DCC (3.12 g, 15.1 mmol, 1.6 equiv) was added in one portion by briefly removing the septum, resulting in a white suspension. After 5 h, the alcohol was entirely consumed as indicated by TLC analysis (eluent: EtOAc:hexanes = 1:3) and diethyl amine (48 mL) was added. After additional 3 h, the mixture was filtered through a pad of celite and the filter cake was washed with DCM (2 × 20 mL). The filtrate was concentrated and the crude residue was purified by flash chromatography (silica gel, eluent: NH4OH:MeOH:DCM = 0.2:1:100 to 0.2:1:50) to afford proline ester 9 (2.80 g, 96 %) as a light yellow, waxy solid. TLC (MeOH:DCM = 1:20): Rf = 0.30 (UV, KMnO4). 1H NMR (400 MHz, CDCl3) δ 6.68 (dd, J = 15.5, 7.7 Hz, 1H), 5.91 2013 5.67 (m, 3H), 5.18 (dd, J = 17.2, 1.6 Hz, 1H), 5.15 – 5.06 (m, 1H), 4.80 (t, J = 6.2 Hz, 1H), 3.98 – 3.88 (m, 2H), 3.78 (dd, J = 8.5, 5.7 Hz, 1H), 3.07 (ddd, J = 10.3, 7.4, 6.2 Hz, 1H), 2.91 (dt, J = 10.3, 6.7 Hz, 1H), 2.72 – 2.58 (m, 1H), 2.52 (br s, 1H), 2.20 – 2.05 (m, 1H), 1.95 – 1.80 (m, 2H), 1.80 – 1.65 (m, 2H), 1.02 (d, J = 6.8 Hz, 3H), 0.88 (d, J = 6.3 Hz, 3H), 0.86 (d, J = 6.4 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 175.0, 165.4, 145.3, 134.1, 123.7, 116.5, 80.5, 59.8, 46.8, 41.9, 38.1, 30.4, 29.8, 25.3, 19.5, 16.9, 14.5. HRMS-ESI m/z calcd for C17H29N2O3+ [M + H]+ 309.2173, found 309.2180.
4.2.2. General procedures for preparation of aldol products 20a–c
A round-bottom flask charged with Sn(OTf)2 (1.3 equiv) was exposed to vacuum (≤ 1 Torr). The vessel was heated with a heat gun for 1 minute, and was allowed to cool for 2 minutes. This process was repeated 5 times, after which the vessel was flushed with nitrogen and sealed with a rubber septum. Dry DCM was added (10 mL for each mmol substrate 18), resulting in a white suspension and the vessel was cooled to −40 °C by means of a dry ice-acetonitrile bath. N-ethylpiperidine (1.3 equiv) was added dropwise, resulting in a deep yellow solution. After 5 min, a solution of 19 (1.2 equiv) in dry DCM (1.0 M) was added dropwise, and the resulting yellow solution was stirred for 5 hous at −40 °C. The reaction mixture was cooled to −78 °C by means of a dry ice- acetone bath. A solution of aldehyde 18 (1.0 equiv) in DCM (10 mL per mmol 18) was added via syringe pump over 30 min. After 3 hours (monitored by TLC plate), pH = 7.0 phosphate buffer (100 mL) was added. The vessel was removed from the cooling bath and the system was allowed to warm to 23 °C while the mixture was rapidly stirred. The biphasic mixture was filtered, the filtrate was transferred to a separatory funnel and the layers were separated. The aqueous layer was extracted with DCM (2 × 30 mL). The combined organic layers were washed with water (2 × 100 mL) and brine (100 mL) and the washed solution was dried (Na2SO4). The dried solution was filtered and the filtrate was concentrated. The crude residue was purified by flash chromatography (silica gel, eluent: EtOAc:hexanes = 1:6 to 1:2.5) to afford aldol product 20.
4.2.2.1. Aldol product 20a
Aldol product 20a was prepared according to the general procedure from Sn(OTf)2 (0.54 g, 1.30 mmol, 1.3 equiv), N- ethylpiperidine (0.18 mL, 1.30 mmol, 1.3 equiv), 19 (0.24 g, 1.2 mmol, 1.2 equiv) and 18a (0.096 g, 1.0 mmol, 1 equiv). 20a (0.22 g, 74%) was obtained as a yellow oil. TLC (EtOAc:hexanes = 1:2.5): Rf = 0.40 (UV, KMnO4). 1H NMR (400 MHz, CDCl3) δ 6.36 (dd, J = 17.4, 10.7 Hz, 1H), 5.53 (d, J = 8.5 Hz, 1H), 5.23 (d, J = 17.4 Hz, 1H), 5.15 (ddd, J = 7.6, 6.2, 1.0 Hz, 1H), 5.07 (d, J = 10.8 Hz, 1H), 5.07 – 4.97 (m, 1H), 3.58 (ddd, J = 17.7, 3.1, 1.0 Hz, 1H), 3.53 (dd, J = 11.5, 8.0 Hz, 1H), 3.35 (dd, J = 17.7, 8.7 Hz, 1H), 3.04 (dd, J = 11.5, 1.1 Hz, 1H), 2.78 (dd, J = 4.1, 2.2 Hz, 1H), 2.44 – 2.29 (m, J = 6.8 Hz, 1H), 1.83 (d, J = 1.2 Hz, 3H), 1.07 (d, J = 6.8 Hz, 3H), 0.99 (d, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 202.9, 172.4, 140.6, 136.16, 132.0, 113.6, 71.4, 65.1, 45.3, 30.8, 30.62, 19.1, 17.8, 12.3. HRMS-ESI m/z calcd for C14H20NOS2+ [M – OH]+ 282.0981, found 282.0989.
4.2.2.2. Aldol product 20b
Aldol product 20b was prepared according to the general procedure from Sn(OTf)2 (1.60 g, 3.90 mmol, 1.3 equiv), N- ethylpiperidine (0.53 mL, 3.90 mmol, 1.3 equiv), 19 (0.73 g, 3.60 mmol, 1.2 equiv) and 18b (0.33 g, 3.0 mmol, 1 equiv). 20b (0.60 g, 64%) was obtained as a yellow oil. TLC (EtOAc:hexanes = 1:2.5): Rf = 0.40 (UV, KMnO4). H NMR (400 MHz, CDCl3) δ 6.05 (ddt, J = 15.5, 2.1, 1.4 Hz, 1H), 5.72 (dq, J = 15.5, 6.6 Hz, 1H), 5.39 (d, J = 8.6 Hz, 1H), 5.14 (ddd, J = 8.0, 6.1, 1.1 Hz, 1H), 5.00 (td, J = 8.7, 3.1 Hz, 1H), 3.57 – 3.48 (m, 2H), 3.34 (dd, J = 17.6, 8.8 Hz, 1H), 3.02 (dd, J = 11.5, 1.1 Hz, 1H), 2.74 (br s, 1H), 2.36 (dq, J = 13.6, 6.8 Hz, 1H), 1.80 (d, J = 1.3 Hz, 3H), 1.76 (dd, J = 6.6, 1.5 Hz, 3H), 1.06 (d, J = 6.8 Hz, 3H), 0.98 (d, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 202.9, 172.5, 136.0, 135.0, 129.1, 125.3, 71.4, 65.0, 45.5, 30.8, 30.6, 19.0, 18.2, 17.7, 13.0. HRMS-ESI m/z calcd for C15H22NOS +2 [M – OH]+ 296.1137, found 296.1133.
4.2.2.3. Aldol product 20c
Aldol product 20c was prepared according to the general procedure from Sn(OTf)2 (5.9 g, 14.20 mmol, 1.3 equiv), N- ethylpiperidine (1.95 mL, 14.20 mmol, 1.3 equiv), 19 (2.66 g, 13.1 mmol, 1.2 equiv) and 18c (1.20 g, 10.9 mmol, 1 equiv). 20c (2.40 g, 70%) was obtained as a yellow oil. TLC (EtOAc:hexanes = 1:2.5): Rf = 0.40 (UV, KMnO4). 1H NMR (400 MHz, CDCl3) δ 5.81 (d, J = 11.7 Hz, 1H), 5.50 (dq, J = 11.5, 7.2 Hz, 1H), 5.40 (d, J = 8.5 Hz, 1H), 5.18 – 5.10 (m, 1H), 4.97 (td, J = 8.7, 2.9 Hz, 1H), 3.58 (dd, J = 17.6, 2.7 Hz, 1H), 3.52 (dd, J = 11.5, 8.0 Hz, 1H), 3.37 (dd, J = 17.6, 8.7 Hz, 1H), 3.03 (d, J = 11.5 Hz, 1H), 2.74 (br s, 1H), 2.45 – 2.30 (m, J = 6.8 Hz, 1H), 1.85 (s, 3H), 1.79 (dd, J = 7.2, 1.8 Hz, 3H), 1.06 (d, J = 6.8 Hz, 3H), 0.99 (d, J = 6.9 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 202.9, 172.5, 135.4, 132.7, 129.9, 125.6, 71.4, 65.1, 45.4, 30.8, 30.6, 19.1, 17.8, 17.4, 14.7. HRMS-ESI m/z calcd for C15H22NOS +2 [M – OH]+ 296.1137, found 296.1133.
4.2.3. General procedures for preparation of TBS ether 21a–c
A round-bottom flask charged with 20 (1 equiv) was evacuated and flushed with nitrogen (this process was repeated a total of 3 times) and was sealed with a rubber septum. DCM (10 mL per mmol of 20) was added followed by 2,6-lutidine (2.0 equiv), resulting a yellow solution. The vessel was cooled to 0 °C by means of ice-water bath. TBSOTf (1.25 equiv) was added dropwise over 10 min. After stirring for 30 min, DCM (10 mL per mmol of 20) was added and the mixture was transferred to a separatory funnel and washed with water (2 × 100 mL) and brine (100 mL). The washed solution was dried (Na2SO4). The dried solution was filtered and the filtrate was concentrated. The resulting crude residue was purified by flash chromatography (silica gel, eluent: EtOAc:hexanes = 1:40) to afford TBS ether 20.
4.2.3.1. TBS ether 21a
TBS ether 21a was prepared according to the general procedure from 20a (0.86 g, 2.90 mmol, 1 equiv), 2,6-lutidine (0.67 mL, 5.70 mmol, 2.0 equiv) and TBSOTf (0.82 mL, 3.6 mmol, 1.25 equiv). 21a (1.10 g, 93%) was obtained as a yellow oil. TLC (EtOAc:hexanes = 1:10): Rf = 0.30 (UV, KMnO4). 1H NMR (400 MHz, CDCl3) δ 6.33 (dd, J = 17.4, 10.7 Hz, 1H), 5.45 (d, J = 8.9 Hz, 1H), 5.18 (d, J = 17.4 Hz, 1H), 5.12 (td, J = 8.8, 3.8 Hz, 1H), 5.04 (d, J = 10.1 Hz, 1H), 5.03 – 4.98 (m, 1H), 3.74 (dd, J = 16.2, 8.8 Hz, 1H), 3.46 (dd, J = 11.4, 7.8 Hz, 1H), 3.07 – 3.02 (m, 1H), 3.01 (s, 1H), 2.44 – 2.30 (m, J = 6.9 Hz, 1H), 1.80 (d, J = 1.2 Hz, 3H), 1.05 (d, J = 6.8 Hz, 3H), 0.97 (d, J = 7.0 Hz, 3H), 0.83 (s, 9H), 0.03 (s, 3H), 0.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 202.8, 171.1, 140.9, 134.5, 133.5, 112.9, 71.7, 66.8, 45.8, 30.9, 30.8, 25.7, 19.1, 18.0, 17.8, 12.2, −4.4, −5.0. HRMS-ESI m/z calcd for C20H35NNaO2S2Si+ [M + Na]+ 436.1771, found 436.1783.
4.2..2. TBS ether 21b
TBS ether 21b was prepared according to the general procedure from 20b (0.60 g, 1.90 mmol, 1 equiv), 2,6-lutidine (0.45 mL, 3.80 mmol, 2.0 equiv) and TBSOTf (0.55 mL, 2.4 mmol, 1.25 equiv). 21b (0.71 g, 87%) was obtained as a yellow oil. TLC (EtOAc:hexanes = 1:10): Rf = 0.30 (UV, KMnO4). 1H NMR (400 MHz, CDCl3) δ 6.03 (ddd, J = 15.6, 1.7, 0.8 Hz, 1H), 5.67 (dtd, J = 15.5, 6.8, 6.2 Hz, 1H), 5.32 (ddd, J = 9.0, 1.3, 0.7 Hz, 1H), 5.10 (td, J = 8.9, 3.8 Hz, 1H), 5.00 (ddd, J = 7.6, 6.2, 1.1 Hz, 1H), 3.74 (dd, J = 16.1, 8.8 Hz, 1H), 3.46 (dd, J = 11.4, 7.8 Hz, 1H), 3.02 (dd, J = 11.4, 1.1 Hz, 1H), 3.00 (dd, J = 16.2, 3.8 Hz, 1H), 2.45 – 2.31 (m, 1H), 1.77 (d, J = 1.2 Hz, 3H), 1.76 (dd, J = 5.7, 1.2 Hz, 3H), 1.05 (d, J = 6.8 Hz, 3H), 0.97 (d, J = 7.0 Hz, 3H), 0.83 (s, 9H), 0.03 (s, 3H), 0.00 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 202.8, 171.2, 135.3, 133.3, 131.7, 124.6, 71.8, 66.9, 46.0, 30.9, 30.8, 25.8, 19.1, 18.2, 18.0, 17.8, 13.0, −4.3, −5.0. HRMS-ESI m/z calcd for C21H37NNaO2S2Si+ [M + Na]+450.1927, found 450.1911.
4.2.3.3. TBS ether 21c
TBS ether 21c was prepared according to the general procedure from 20c (2.30 g, 7.34 mmol, 1 equiv), 2,6-lutidine (1.70 mL, 14.70 mmol, 2.0 equiv) and TBSOTf (2.11 mL, 0.17 mmol, 1.25 equiv). 21c (3.08 g, 98%) was obtained as a yellow oil. TLC (EtOAc:hexanes = 1:10): Rf = 0.30 (UV, KMnO4). 1H NMR (400 MHz, CDCl3) δ 5.76 (dp, J = 11.6, 1.7 Hz, 1H), 5.47 (dq, J = 11.7, 7.2 Hz, 1H), 5.33 (dt, J = 8.9, 1.5 Hz, 1H), 5.12 – 4.93 (m, 2H), 3.76 (dd, J = 16.2, 8.7 Hz, 1H), 3.46 (dd, J = 11.4, 7.8 Hz, 1H), 3.11 – 2.96 (m, 2H), 2.46 – 2.32 (m, J = 6.9 Hz, 1H), 1.82 (d, J = 1.4 Hz, 3H), 1.78 (dd, J = 7.2, 1.8 Hz, 3H), 1.06 (d, J = 6.8 Hz, 3H), 0.97 (d, J = 6.9 Hz, 3H), 0.84 (s, 9H), 0.05 (s, 3H), 0.03 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 202.8, 171.2, 132.9, 132.6, 132.3, 125.1, 71.8, 66.9, 46.0, 31.0, 30.8, 25.8, 19.1, 18.0, 17.8, 17.3, 14.7, −4.4, −4.9. HRMS-ESI m/z calcd for C21H37NNaO2S2Si+ [M + Na]+ 450.1927, found 450.1911.
4.2.4. General procedures for preparation of the right halves 10a–c
A round-bottom flask charged with oxazolyl carboxylic acid 22 (2.0 equiv) was evacuated and flushed with nitrogen (this process was repeated a total of 3 times) and was sealed with a rubber septum. THF (10 mL per mmol 21) was added, resulting in a light yellow solution and the vessel and its contents were cooled to −78°C in a dry ice-acetone bath. A solution of n-butyllithium in hexanes (4.0 equiv) was added dropwise over 15 min, resulting in a deep red solution. After 30 min, a solution of 21 (1.0 equiv) in THF (10 mL per mmol 22) was added over 30 min by syringe pump. After an additional 30 min, water (100 mL) was added, followed by 1 M aqueous KHSO4 solution (8.0 equiv). The system was allowed to warm to 23 °C while the mixture was rapidly stirred. The biphasic mixture was transferred to a separatory funnel and the layers were separated. The aqueous layer was extracted with EtOAc (2 × 50 mL). The combined organic layers were washed with water (2 × 100 mL) and brine (100 mL) and the washed solution was dried (Na2SO4). The dried solution was filtered and the filtrate was concentrated. The resulting crude residue was purified by flash chromatography (silica gel, eluent: AcOH:EtOAc:hexanes = 0.5:50:50) to afford carboxylic acid 10.
4.2.4.1. Acid 10a
Acid 10a was prepared according to the general procedure from TBS ether 21a (0.83 g, 2.00 mmol, 1 equiv), acid 22 (0.80 g, 4.00 mmol, 2.0 equiv) and nBuLi (2.5 M, 3.20 mL, 8.00 mmol, 4.00 equiv). 10a (0.63 g, 70%) was obtained as a yellow solid. TLC (MeOH:DCM = 1:20): Rf = 0.30 (UV, KMnO4). 1H NMR (400 MHz, CDCl3) δ 6.30 (dd, J = 17.4, 10.7 Hz, 1H), 5.40 (d, J = 8.8 Hz, 1H), 5.18 (d, J = 17.4 Hz, 1H), 5.04 (d, J = 10.7 Hz, 1H), 4.99 (dt, J = 8.7, 4.3 Hz, 1H), 4.14 (d, J = 17.1 Hz, 1H), 4.06 (d, J = 17.1 Hz, 1H), 2.85 (dd, J = 14.9, 8.6 Hz, 1H), 2.51 (dd, J = 14.9, 4.2 Hz, 1H),1.76 (s, 3H), 0.84 (s, 9H), 0.36 (s, 9H), 0.03 (s, 3H), −0.01 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 201.1, 165.4, 165.3, 161.3, 140.7, 140.6, 134.0, 133.7, 113.3, 66.6, 50.1, 43.9, 25.8, 18.0, 12.1, −2.2, −4.5, −5.1. HRMS-ESI m/z calcd for C21H36O3Si2– [M – HCO2]– 406.2239, found 406.2339.
4.2.4.2. Acid 10b
Acid 10b was prepared according to the general procedure from TBS ether 21b (0.40 g, 0.94 mmol, 1.0 equiv), acid 22 (0.37 g, 1.87 mmol, 2.0 equiv) and nBuLi (2.5 M, 1.50 mL, 3.74 mmol, 4.00 equiv). 10b (0.30 g, 69%) was obtained as a yellow solid. TLC (MeOH:DCM = 1:20): Rf = 0.30 (UV, KMnO4). 1H NMR (400 MHz, CDCl3) δ 9.97 (br s, 1H), 5.99 (dd, J = 15.6, 0.9 Hz, 1H), 5.66 (dq, J = 15.6, 6.6 Hz, 1H), 5.25 (d, J = 8.8 Hz, 1H), 4.96 (td, J = 8.7, 4.2 Hz, 1H), 4.16 (d, J = 17.1 Hz, 1H), 4.07 (d, J = 17.2 Hz, 1H), 2.82 (dd, J = 14.8, 8.5 Hz, 1H), 2.49 (dd, J = 14.7, 4.3 Hz, 1H), 1.75 (dd, J = 6.7, 1.5 Hz, 3H), 1.73 (d, J = 1.3 Hz, 3H), 0.83 (s, 9H), 0.35 (s, 9H), 0.01 (s, 3H), −0.02 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 201.2, 165.4, 165.1, 161.4, 140.8, 135.1, 133.47, 131.2, 124.9, 66.7, 50.3, 43.9, 25.8, 18.2, 13.0, 12.8, −2.2, −4.4, −5.1. HRMS-ESI m/z calcd for C22H38O3Si2 [M – HCO2]– 420.2393.
4.2.4.3. Acid 10c
Acid 10c was prepared according to general procedure from TBS ether 21c (0.66 g, 1.54 mmol, 1.0 equiv), acid 22 (0.62 g, 3.09 mmol, 2.0 equiv) and nBuLi (2.5 M, 2.47 mL, 6.17 mmol, 4.00 equiv). 10c (0.48 g, 67%) was obtained as a yellow solid. TLC (MeOH:DCM = 1:20): Rf = 0.30 (UV, KMnO4). 1H NMR (400 MHz, CDCl3) δ 5.75 (dt, J = 11.7, 1.6 Hz, 1H), 5.48 (dq, J = 11.7, 7.2 Hz, 1H), 5.28 (dt, J = 8.7, 1.4 Hz, 1H), 4.93 (td, J = 8.6, 4.2 Hz, 1H), 4.15 (d, J = 17.2 Hz, 1H), 4.06 (d, J = 17.2 Hz, 1H), 2.86 (dd, J = 14.7, 8.5 Hz, 1H), 2.53 (dd, J = 14.7, 4.2 Hz, 1H), 1.78 (d, J = 1.4 Hz, 3H), 1.77 (dd, J = 7.2, 1.8 Hz, 3H), 0.85 (s, 9H), 0.36 (s, 9H), 0.05 (s, 3H), 0.02 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 201.2, 165.4, 165.3, 161.3, 140.6, 132.8, 132.7, 131.8, 125.3, 66.8, 50.4, 44.0, 25.8, 18.0, 17.3, 14.6, −2.2, −4.5, −5.0. HRMS-ESI m/z calcd for C22H38O3Si2– [M – HCO2]– 420.2396, found 420.2393.
4.2.5. General procedures for preparation of ring closing metathesis (RCM) precursors 8a–c
A 100-mL round-bottom flask was charged with amine 9 (1 equiv), carboxylic acid 10 (1.05 equiv) and iPr2EtN (2.0 equiv). DCM (10 mL per mmol 9) was added, resulting in a clear, colorless solution and HATU (1.25 equiv) was added to this solution in one portion at 23 °C. After stirring for 5 h, the mixture was diluted with DCM (100 mL). The solution was transferred to a separatory funnel and was washed with water (2 × 100 mL) and brine (100 mL). The washed solution was dried (Na2SO4). The dried solution was filtered and the filtrate was concentrated. The resulting crude residue was purified by flash chromatography (silica gel, eluent: EtOAc:hexanes = 1:6 to 1:4) to afford RCM precursor 8.
4.2.5.1. RCM precursor 8a
RCM precursor 8a was prepared according to the general procedure from amine 9 (0.40 g, 1.30 mmol, 1 equiv), acid 10a (0.62 g, 1.40 mmol, 1.05 equiv), iPr2EtN (0.45 mL, 2.60 mmol, 2.0 equiv), and HATU (0.62 g, 1.60 mmol, 1.25 equiv). 8a (0.86 g, 93%) was obtained as a yellow, waxy solid. TLC (EtOAc:hexanes = 1.2.5): Rf = 0.30 (UV, KMnO4). 1H NMR (400 MHz, CDCl3) δ 6.73 – 6.56 (m, 1H), 6.35 – 6.25 (m, 1H), 5.98 – 5.70 (m, 3H), 5.47 – 5.35 (m, 1H), 5.24 – 5.01 (m, 4H), 5.00 – 4.90 (m, 1H), 4.83 – 4.68 (m, 1H), 4.65 – 4.55 (m, 1H), 4.11 – 3.80 (m, 5H), 3.81 – 3.64 (m, 1H), 2.88 – 2.75 (m, 1H), 2.65 – 2.35 (m, 2H), 2.29 – 2.12 (m, 2H), 2.11 – 1.81 (m, 3H), 1.76 (s, 3H), 1.05 – 0.88 (m, 6H), 0.82 (m, 12H), 0.33 (s, 4.5H), 0.27 (s, 4.5H), 0.018 (s, 1.5H), 0.011 (s, 1.5H), −0.014 (s, 1.5H), −0.019 (s, 1.5H). 13C NMR (100 MHz, CDCl3) δ 201.7, 201.2, 172.35, 172.32, 165.7, 165.4, 163.1, 162.2, 161.57, 161.55, 159.32, 159.28, 145.34, 145.22, 145.17, 145.1, 140.56, 140.54, 134.12, 134.08, 134.06, 134.01, 133.6, 123.89, 123.72, 116.5, 116.3, 113.3, 80.9, 80.5, 66.8, 66.6, 60.4, 59.9, 50.01, 49.96, 48.8, 47.0, 44.4, 44.2, 41.91, 41.87, 38.6, 38.3, 38.0, 31.5, 29.9, 29.7, 28.9, 25.73, 25.66, 25.2, 21.5, 19.6, 19.4, 18.0, 17.2, 17.0, 14.7, 14.2, 12.1, −1.79, −1.81, −4.5, −5.10, −5.12. HRMS- ESI m/z calcd for C39H64N3O7Si2+ [M + H]+ 742.4277, found 742.4292.
4.2.5.2. RCM precursor 8b
RCM precursor 8b was prepared according to the general procedure from amine 9 (0.26 g, 0.84 mmol, 1 equiv), acid 10b (0.41 g, 0.89 mmol, 1.05 equiv), iPr2EtN (0.29 mL, 1.70 mmol, 2.0 equiv), and HATU (0.40 g, 1.10 mmol, 1.25 equiv). 8b (0.51 g, 80%) was obtained as a yellow, waxy solid. TLC (EtOAc:hexanes = 1.2.5): Rf = 0.30 (UV, KMnO4). 1H NMR (400 MHz, CDCl3) δ 6.74 – 6.54 (m, 1H), 6.04 – 5.95 (m, 1H), 5.95 – 5.57 (m, 4H), 5.29 – 5.05 (m, 3H), 5.00 – 4.90 (m, 1H), 4.81 – 4.56 (m, 1H), 4.12 – 3.81 (m, 5H), 3.81 – 3.63 (m, 1H), 2.85 – 2.71 (m, 1H), 2.68 – 2.48 (m, 1H), 2.48 – 2.38 (m, 1H), 2.29 – 2.10 (m, 2H), 2.11 – 1.81 (m, 3H), 1.82 – 1.65 (m, 6H), 1.07 – 0.87 (m, 6H), 0.87 – 0.67 (m, 12H), 0.33 (s, 4H), 0.27 (s, 5H), 0.01 (s, 1.5H), 0.01 (s, 1.5H), −0.01 (s, 1.5H), −0.02 (s, 1.5H). 13C NMR (100 MHz, CDCl3) δ 201.9, 201.4, 172.4, 172.3, 165.7, 165.4, 163.0, 162.2, 161.6, 159.39, 159.35, 145.34, 145.22, 145.16, 145.07, 135.04, 135.02, 134.13, 134.02, 133.5, 131.27, 131.24, 125.03, 125.02, 123.9, 123.7, 116.5, 116.3, 80.8, 80.5, 67.9, 66.9, 66.8, 60.4, 59.9, 50.3, 50.2, 48.8, 47.0, 44.4, 44.2, 41.91, 41.87, 38.6, 38.3, 38.0, 31.5, 29.9, 29.7, 28.9, 25.8, 25.7, 25.6, 25.2, 21.5, 19.6, 19.4, 18.2, 18.0, 17.2, 17.0, 14.7, 14.2, 12.9, −1.79, −1.80, −4.4, −5.09, −5.11. HRMS- ESI m/z calcd for C40H66N3O7Si2+ [M + H]+ 756.4434, found 756.4435.
4.2.5.3. RCM precursor 8c
RCM precursor 8c prepared according to the general procedure from amine 9 (0.50 g, 1.60 mmol, 1 equiv), acid 10c (0.79 g, 1.70 mmol, 1.05 equiv), iPr2EtN (0.57 mL, 3.200 mmol, 2.0 equiv), and HATU (0.77 g, 2.0 mmol, 1.25 equiv). 8c (1.10 g, 90%) was obtained as a yellow, waxy solid. TLC (EtOAc:hexanes = 1.2.5): Rf = 0.30 (UV, KMnO4). 1H NMR (400 MHz, CDCl3) δ 6.74 – 6.56 (m, 1H), 5.97 – 5.65 (m, 4H), 5.55 – 5.40 (m, 1H), 5.35 – 4.99 (m, 3H), 4.98 – 4.58 (m, 3H), 4.17 – 3.84 (m, 5H), 3.84 – 3.63 (m, 1H), 2.90 −2.75 (m, 1H), 2.70 – 2.38 (m, 2H), 2.32 – 2.13 (m, 2H), 2.13 – 1.83 (m, 3H), 1.83 – 1.72 (m, 6H), 1.07 – 0.89 (m, 6H), 0.89 – 0.79 (m, 12H), 0.34 (s, 4H), 0.29 (s, 5H), 0.05 (s, 1.5H), 0.04 (s, 1.5H), 0.03 (s, 1.5H), 0.02 (s, 1.5H). 13C NMR (100 MHz, CDCl3) δ 201.9, 201.4, 172.4, 165.7, 165.4, 163.1, 162.2, 161.6, 159.38, 159.35, 145.5, 145.3, 145.2, 145.1, 134.1, 134.0, 132.7, 131.90, 131.88, 125.3, 123.9, 123.7, 116.5, 116.4, 80.9, 80.5, 67.0, 66.8, 65.8, 60.5, 59.9, 50.3, 50.2, 48.8, 47.0, 44.4, 44.2, 41.93, 41.89, 38.3, 38.0, 31.6, 29.9, 29.8, 28.9, 25.8, 25.7, 25.24, 25.18, 22.6, 21.5, 19.6, 19.4, 18.0, 17.2, 17.02, 15.2, 14.7, 14.2, 14.1, 11.4, −1.78, −1.79, −4.5, −4.6, −5.04, −5.06. HRMS-ESI m/z calcd for C40H66N3O7Si2+ [M + H]+ 756.4434, found 756.4435.
4.2.6. TBS-TMS-viginiamycin M2 (23)
A 50-mL round-bottom flask was charged with RCM precursor 8c (30 mg, 40 μmol, 1 equiv) and Grela II catalyst (2.1 mg, 3.2 μmol, 0.08 equiv). The vessel was evacuated and filled with nitrogen (this process was repeated a total of 3 times) and was sealed with a rubber septum. Benzentrifluoride (8 mL) was added, resulting in a green solution. A stream of argon was passed through the solution for 30 min. After 12 hours, another portion of Grela II catalyst (2.1 mg, 3.2 μmol, 0.08 equiv) was added. The mixture was stirred for another 12 hours and concentrated under vacuum. The resulting residue was purified by flash chromatography (silica gel, eluent: EtOAc: Hexanes = 1:3 to 1:1.5) to afford TBS-TMS- viginiamycin M2 (23, 14 mg, 49%) as a white solid. TLC (EtOAc:hexanes = 1:2): Rf = 0.20 (UV). 1H NMR (400 MHz, CDCl3) δ 6.49 (dd, J = 16.3, 4.2 Hz, 1H), 6.19 – 6.10 (m, 1H), 6.07 (dd, J = 9.2, 3.2 Hz, 1H), 5.77 (dd, J = 16.4, 2.0 Hz, 1H), 5.57 (ddd, J = 15.5, 9.4, 4.2 Hz, 1H), 5.42 (d, J = 8.9 Hz, 1H), 5.00 (ddd, J = 8.9, 7.0, 5.9 Hz, 1H), 4.85 – 4.72 (m, 2H), 4.57 – 4.43 (m, 1H), 3.89 (d, J = 17.2 Hz, 1H), 3.78 – 3.69 (m, 3H), 3.39 (ddd, J = 14.8, 9.5, 3.3 Hz, 1H), 2.92 (dd, J = 15.9, 7.0 Hz, 1H), 2.79 – 2.68 (m, 2H), 2.18 – 2.04 (m, 1H), 1.90 (dddd, J = 24.9, 15.9, 11.3, 6.8 Hz, 3H), 1.77 – 1.68 (m, 1H), 1.66 (d, J = 1.2 Hz, 3H), 1.08 (d, J = 6.9 Hz, 3H), 0.99 (d, J = 6.5 Hz, 3H), 0.94 (d, J = 6.8 Hz, 3H), 0.85 (s, 9H), 0.30 (s, 9H), 0.05 (s, 3H), 0.02 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 201.0, 172.1, 166.4, 161.8, 161.3, 159.6, 145.1, 144.8, 136.7, 134.7, 132.4, 124.9, 123.7, 81.1, 65.4, 58.7, 50.6, 48.4, 43.7, 41.3, 36.7, 29.3, 28.2, 25.7, 24.8, 19.9, 18.6, 18.1, 12.67, 9.9, −1.8, −4.5, −5.0. HRMS-ESI m/z calcd for C37H60N3O7Si2 [M + H]+ 714.3964, found 714.3968.
4.2.7. viginiamycin M2 (1)
A 100-mL round-bottom flask charged with 23 (58 mg, 81 μmol, 1 equiv) was evacuated and filled with nitrogen (this process was repeated a total of 3 times) and was sealed with a rubber septum. THF (1.6 mL) was added, resulting in a light yellow solution. In a separate flask, Im·HCl (84 mg, 0.81 mmol, 10.0 equiv) was added to a solution of TBAF in THF (1 M, 0.81 mL, 0.81 mmol, 10.0 equiv). The resulting colorless solution was added dropwise to the solution of 23. After 12 h, the mixture was concentrated and the residue was dissolved in DCM (50 mL). The resulting solution was transferred to a separatory funnel and was washed with water (3 × 30 mL) and brine (30 mL). The washed solution was dried (Na2SO4). The dried solution was filtered and the filtrate was concentrated. The resulting crude residue was purified by flashed chromatography (silica gel, eluent: MeOH:DCM = 1:40) to afford virginiamycin M2 (1, 35 mg, 82%) as a light yellow solid. m. p. 120 – 125 °C (DCM). TLC (MeOH:DCM = 1:20): Rf = 0.30 (UV). [α]25D = - 67.4 (c = 0.3, DCM). 1H NMR (400 MHz, CDCl3) δ 8.08 (s, 1H), 6.47 (dd, J = 16.4, 5.0 Hz, 1H), 6.39 (dd, J = 9.0, 3.7 Hz, 1H), 6.11 (m, J = 15.6 Hz, 1H), 5.78 (dd, J = 16.4, 1.9 Hz, 1H), 5.69 (ddd, J = 15.6, 9.2, 4.6 Hz, 1H), 5.41 (d, J = 8.8 Hz, 1H), 4.90 (dt, J = 8.9, 5.6 Hz, 1H), 4.73 (dd, J = 10.1, 2.0 Hz, 1H), 4.70 (dd, J = 8.9, 3.2 Hz, 1H), 4.45 (ddd, J = 13.9, 8.9, 4.6 Hz, 1H), 4.00 – 3.92 (m, 1H), 3.82 (s, 2H), 3.79 – 3.70 (m, 1H), 3.39 (ddd, J = 14.0, 9.2, 3.6 Hz, 1H), 3.05 (dd, J = 17.0, 6.0 Hz, 1H), 2.89 (dd, J = 17.0, 5.2 Hz, 1H), 2.74 (ddt, J = 6.9, 4.9, 2.0 Hz, 1H), 2.60 (br s, 1H), 2.24 – 2.08 (m, 1H), 2.01 – 1.88 (m, 3H), 1.88 – 1.75 (m, 1H), 1.71 (d, J = 1.2 Hz, 3H), 1.03 (d, J = 6.9 Hz, 3H), 0.98 (d, J = 6.5 Hz, 3H), 0.95 (d, J = 6.8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 202.1, 171.6, 166.5, 160.2, 156.9, 144.5, 143.9, 136.92, 136.86, 134.3, 132.7, 125.2, 124.0, 81.4, 65.0, 59.6, 48.9, 48.4, 43.3, 40.9, 36.6, 29.4, 28.3, 25.0, 19.7, 18.7, 12.6, 10.4. HRMS-ESI m/z calcd for C28H38N3O7 [M + H]+ 528.2704, found 528.2703.
4.2.8. RCM precursor 8d
An 100-mL round-bottom flask charged with 8c (0.38 g, 0.50 mmol, 1 equiv) was evacuated and filled with nitrogen (this process was repeated a total of 3 times) and was sealed with a rubber septum. THF (20 mL) was added, resulting in a light yellow solution. In a separate flask, Im·HCl (0.52 mg, 5.00 mml, 10.0 equiv) was added to a solution of TBAF (5.0 mL, 5.00 mml, 10.0 equiv). The resulting colorless solution was added dropwise to the above solution of 8c. After 12 h, the mixture was concentrated and the residue was dissolved in DCM (100 mL). The resulting solution was transferred to a separatory funnel and was washed with water (4 × 100 mL) and brine (100 mL). The washed solution was dried (Na2SO4). The dried solution was filtered and the filtrate was concentrated. The resulting crude residue was purified by flashed chromatography (silica gel, eluent: acetone:hexanes = 1:3 to 1:1.5) to RCM precursor 8d (0.23 g, 83%) as a light yellow solid. TLC (acetone:hexanes = 1:1.5): Rf = 0.40 (UV, KMnO4).1H NMR (400 MHz, CDCl3) δ 8.21 (s, 0.4H), 8.16 (s, 0.6H), 6.65 – 6.50 (m, 1H), 6.35 (t, J = 5.8 Hz, 0.6H), 5.96 – 5.69 (m, 3.4H), 5.60 – 5.40 (m, 2H), 5.35 – 5.26 (m, 1H), 5.23 – 5.06 (m, 2H), 5.00 – 4.85 (m, 1H), 4.81 – 4.59 (m, 2H), 4.05 (t, J = 6.5 Hz, 1H), 4.01 – 3.84 (m, 4H), 3.83 – 3.62 (m, 1H), 2.88 – 2.73 (m, 1H), 2.73 – 2.45 (m, 2H), 2.33 – 2.19 (m, 2H), 2.13 – 1.85 (m, 3H), 1.84 – 1.71 (m, 6H), 1.00 – 0.81 (m, 9H). 13C NMR (100 MHz, CDCl3) δ 202.74, 202.72, 172.2, 171.7, 166.1, 165.3, 160.08, 160.05, 157.2, 157.1, 145.3, 144.7, 144.5, 144.2, 137.3, 137.1, 135.4, 135.3, 134.3, 134.0, 132.6, 132.5, 130.0, 129.9, 125.7, 125.6, 124.3, 123.6, 116.4, 116.2, 81.2, 80.8, 64.74, 64.71, 60.5, 60.2, 49.4, 49.3, 48.74 47.1, 43.2, 43.1, 41.91, 41.88, 38.2, 37.6, 31.5, 29.9, 29.8, 28.8, 25.3, 21.4, 19.6, 19.3, 17.6, 17.4, 17.3, 14.70, 14.2, 13.9. HRMS-ESI m/z calcd for C31H44N3O7+[M + H]+ 570.3174, found 570.3181.
4.2.9. viginiamycin M2 (1) from RCM precursor 8d
An 50-mL round-bottom flask was charged with RCM precursor 8d (30 mg, 53 mol, 1 equiv) and Grela II catalyst (2.8 mg, 4.2 mmol, 0.08 equiv). The vessel was evacuated and filled with nitrogen (this process was repeated a total of 3 times) and was sealed with a rubber septum. DCM (10 mL) was added, resulting in a green solution. A stream of argon was passed through the solution for 30 min at 0 °C, then the reaction mixture was warmed to 23 °C. After 12 h, another portion of Grela II catalyst (Grela II catalyst (2.8 mg, 4.2 mmol, 0.08 equiv) was added. After the next 12 hours, the solvent was removed under vacuum. The resulting residue was purified by flash chromatography (silica gel, eluent: acetone:hexanes = 1:2 to 1:1) to afford virginianmycin M2 (1, 20 mg, 72%) as a white solid. The spectral data were consistent with those reported above.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of General Medical Sciences of the National Institutes of Health (R35GM128656). Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH. We are grateful to Dr. Ziyang Zhang (Shokat laboratory, UCSF) for assistance with high-resolution mass spectral analysis.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Aminov RI Frontiers Microbiol. 2010, 1, article 134, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.World Health Organization. ANTIBACTERIAL AGENTS IN CLINICAL DEVELOPMENT An analysis of the antibacterial clinical development pipeline, including tuberculosis. Geneva; 2017. https://www.who.int/medicines/areas/rational_use/antibacterial_agents_clinical_development/en/ [Google Scholar]
- 3. (a).Charney J; Fisher WP; Curran C; Machlowitz RA; Tytell AA Antibiotics and Chemotherapy 1953, 3, 1283–1286. [PubMed] [Google Scholar]; (b) Vazquez D. In Antibiotics; Gottlieb D, Shaw PD, Eds.; Springer: New York, 1967; Vol. 1, pp 387–403. [Google Scholar]
- 4. (a).Mukhtar TA; Wright GD Chem. Rev 2005, 105, 529–542; [DOI] [PubMed] [Google Scholar]; (b) Lee VZ, In Comprehensive Medicinal Chemistry II; Taylor JD, Triggle DJ, EDs.; Elsevier: Oxford, 2007; Vol. 7, pp 653–671. [Google Scholar]
- 5. (a).Politano AD; Sawyer RG Curr. Opin. Invest. Drugs 2010, 11, 225–236. [PMC free article] [PubMed] [Google Scholar]; (b) Pankuch GA; Lin G; Clark C; Appelbaum PC Antimicrob. Agents Chemother. 2011, 55, 1787–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. (a).Delgado G Jr.; Neuhauser MM; Bearden DT; Danziger LH Pharmacotherapy 2000, 20, 1469–1485; [DOI] [PubMed] [Google Scholar]; (b) Pavan B Curr. Opin. Invest. Drugs 2000, 1, 173–180; [PubMed] [Google Scholar]; (c) Bonfiglio G; Furneri PM Expert Opin. Invest. Drugs 2001, 10, 185–198. [DOI] [PubMed] [Google Scholar]; (d) Allington DR; Rivey MP Clin. Ther 2001, 23, 24–44. [DOI] [PubMed] [Google Scholar]
- 7. (a).Schlessinger RH; Li Y-JJ Am. Chem. Soc 1996, 118, 3301–3302; [Google Scholar]; (b) Tavares F; Lawson JP; Meyers AI J. Am. Chem. Soc 1996, 118, 3303–3304. [Google Scholar]
- 8.Dvorak CA; Schmitz WD; Poon DJ; Pryde DC; Lawson JP; Amos RA; Meyers AI Angew. Chem., Int. Ed 2000, 39, 1664–1666. [DOI] [PubMed] [Google Scholar]
- 9. (a).Wu J; Panek JS Angew. Chem., Int. Ed 2010, 49, 6165–6168. [DOI] [PubMed] [Google Scholar]; (b) Wu J; Panek JS J. Org. Chem 2011, 76, 9900–9918. [DOI] [PubMed] [Google Scholar]
- 10. (a).Entwistle DA; Jordan SI; Montgomery J; Pattenden GJ Chem. Soc., Perkin Trans 1 1996, 1315–1317; [Google Scholar]; (b) Ghosh AK; Liu WJ Org. Chem 1997, 62, 7908–7909; [DOI] [PubMed] [Google Scholar]; (c) Entwistle DA; Jordan SI; Montgomery J; Pattenden G Synthesis 1998, 1998, 603–612; [Google Scholar]; (d) Breuilles P; Uguen D Tetrahedron Lett 1998, 39, 3149–3152. [Google Scholar]
- 11.Li Q, Seiple IB J. Am. Chem. Soc 2017, 139,13304–13307. [DOI] [PubMed] [Google Scholar]
- 12.Villemin D Tetrahedron Lett. 1980, 21, 1715–1718. [Google Scholar]
- 13.For representative books on RCM:; (a) Cossy J; Arseniyadis S; Meyer C Metathesis in Natural Product Synthesis: Strategies, Substrates, and Catalysts.; Wiley-VCH: Weinheim, Germany, 1st ed, 2010; [Google Scholar]; (b) van Lierop BJ, Limmiss JAM, Fogg DE In Olefin Metathesis: Theory and Practice; Grela K, Ed.; John Wiley & Sons: Hoboken, NJ, 2014; chapter 3, pp 85–153; [Google Scholar]; (c) Grubbs RH, Handbook of Metathesis, 3 volume set; Wiley-VCH: Weinheim, 2nd ed., 2015. [Google Scholar]
- 14.For representative reviews on RCM:; (a) Grubbs RH; Miller SJ Miller Fu, G. C. Acc. Chem. Res 1995, 28, 446–452; [Google Scholar]; (b) Nicolaou KC, Bulger PG, Sarlah D, Angew. Chem. Int. Ed 2005, 44, 4490–4527; [DOI] [PubMed] [Google Scholar]; (c) Dhambri CL, Sanogo LAY; Zeghbib R Othman RB, Lannou M-I, Sprom G; Ardisson J Nat. Prod. Rep 2018, 35, 105–124; [DOI] [PubMed] [Google Scholar]; (d) Ogba OM, Waner NC, O’Leary DJ, Grubbs RH Chem. Soc. Rev 2018, 47, 4510–4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Simsek S; Kalesse M Tetrahedron Lett. 2009, 50, 3485–3488. [Google Scholar]
- 16.González A; Aiguadé J; Urpı´ F; Vilarrasa J Tetrahedron Lett. 1996, 37, 8949–8952. [Google Scholar]
- 17.Nagao Y, Hagiwara Y; Kumagai T; Ochiai M, Inoue T, Hashimoto K; Fujita EJ Org. Chem 1986, 51, 2391–2393. [Google Scholar]
- 18.Agency for Toxic Substances and Disease Registry (ATSDR) 2005. https://www.atsdr.cdc.gov/ToxProfiles/TP.asp?id=543&tid=98 (Accessed February, 2019).
- 19.Hong SH; Day MW; Grubbs RH J. Am. Chem. Soc 2004, 126, 7414–7415. [DOI] [PubMed] [Google Scholar]
- 20.Love JA; Morgan JP; Trnka TM; Grubbs RH Angew. Chem., Int. Ed 2002, 41, 4035–4037. [DOI] [PubMed] [Google Scholar]
- 21.Ung T; Hejl A; Grubbs RH; Schrodi Y Organometallics 2004, 23, 5399–5401. [Google Scholar]
- 22.Stewart IC; Ung T; Pletnev AA; Berlin JM; Grubbs RH; Schrodi Y Org. Lett 2007, 9, 1589–1592. [DOI] [PubMed] [Google Scholar]
- 23. (a).Courchay FC; Sworen JC; Wagener KB Macromolecules 2003, 36, 8231–8239; [Google Scholar]; (b) Romero PE; Piers WE; McDonald R Angew. Chem., Int. Ed 2004, 43, 6161; [DOI] [PubMed] [Google Scholar]; (c) Zannan catalyst 1-B (CAS: 918870-76-5): Patents CN1907992A, US 2007/0043180 A1, PCT WO 2007/003135 A1. [Google Scholar]
- 24.Ru-; (a) Grela K; Harutyunyan S; Michrowska A Angew. Chem., Int. Ed 2002, 41, 4038–4040. [DOI] [PubMed] [Google Scholar]; (b) Michrowska A; Bujok R; Harutyunyan S; Sashuk V; Dolgonos G; Grela KJ Am. Chem. Soc 2004, 126, 9318–9325. [DOI] [PubMed] [Google Scholar]
- 25.Lin YA; Davis BG Beilstein J. Org. Chem 2010, 6, 1219–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. (a).Ahmed TS; Grubbs RH Angew. Chem. Int. Ed 2017, 56, 11213–11216; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Müller DS; Baslé O; Mauduit M Beilstein J. Org. Chem 2018, 14, 2999–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.