

Abstract
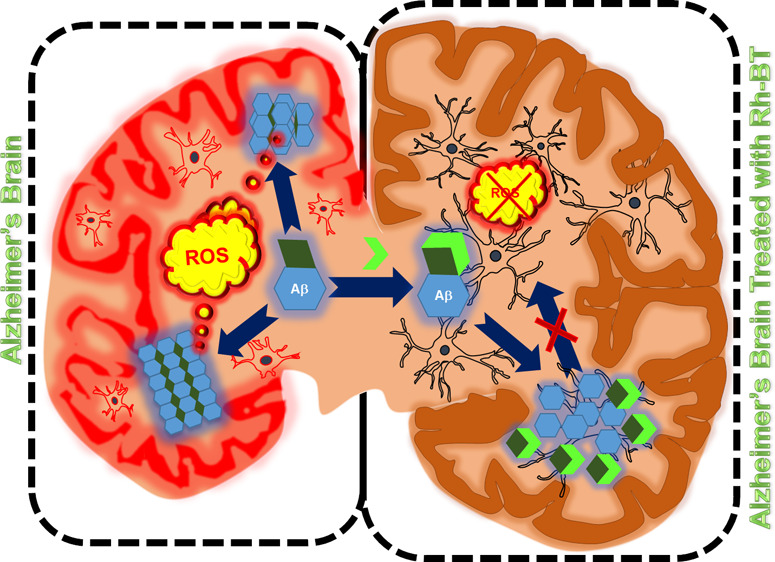
Alzheimer’s disease (AD) exhibits a multitude of syndromes which add up to its complex nature. In AD, amyloid plaques are deposited along with abnormal accumulation of transition-metal ions. These transition-metal ions are redox-active and help to induce the formation of various polymorphic forms of amyloid-β. Amyloid oligomeric and fibrillar aggregates are the main cause for neuronal toxicity. Another reason for neuronal toxicity arises from generation of reactive oxygen species (ROS) catalyzed by redox-active metal ions through Fenton’s reaction. In this direction, an Aβ inhibitor possessing the metal chelation property will be the most promising approach against multifaceted AD. Herein, a rhodamine-B-based compound (Rh-BT) has been designed and synthesized. Rhodamine was attached with benzothiazole as a recognition unit for amyloid-β aggregates. The molecule can effectively capture redox metal ions from the Aβ–Cu2+ complex as well as inhibit Aβ self-assembly such as toxic oligomeric and fibrillar aggregates. Various biophysical assays show that Rh-BT interacts with the Aβ peptide, is capable of decreasing metal-induced ROS generation, and inhibits Aβ–Cu2+-induced cytotoxicity. All these results support the multifunctional nature of Rh-BT, which has an Aβ-specific recognition unit. In addition to the above properties, Rh-BT also exhibits good serum stability in vivo and blood–brain barrier permeability. Therefore, Rh-BT can be considered as a potent multifunctional therapeutic for the treatment of AD.
1. Introduction
Alzheimer’s disease (AD) is a multifaceted, progressive neurodegenerative disorder characterized by the inabilities in thinking and memory, behavioral disorders, and deterioration of cognition, which finally lead to the death of neurons.1,2 Until now, the etiology of AD is not fully understood.3 Among all the neurodegenerative diseases, AD is known to be the most complicated disease.4 Several factors have been identified, which contribute to the development of AD, such as amyloid-β fibrillation and oligomerization,5−7 hyperphosphorylation of Tau protein,8−10 loss of acetylcholine function,11,12 oxidative stress, dyshomeostasis of biometals,13−15 and so forth.16−23 A different facet of amyloid toxicity is reflected in each of these hypotheses. Among all these factors, metal-ion-mediated amyloid toxicity has a significant role toward the development of neurodegenerative diseases.24−27 The presence of amyloid plaques in the brain is a trademark for all AD patients.28,29 Amyloid plaques consisting of amyloid-β (Aβ) peptides of 40–42 amino acids have been implicated to play an important role in most of the AD hypotheses.30,31 In the amyloid cascade hypothesis, polymorphic forms of Aβ peptide have been reported, such as the oligomeric and fibrillar aggregates.32,33 Among these polymorphic forms, oligomeric aggregates are the most toxic and the main culprit behind the neurotoxicity in the AD brain.34−36 The existence of Aβ oligomers depends on both the monomer and the fibril. This is due to the fact that after a certain concentration of amyloid fibril formation, they are catalyzed to form oligomeric aggregates known as secondary nucleation. Therefore, to develop inhibitors of amyloid toxicity, we have to target both oligomeric and fibrillar aggregates. In an AD brain, high concentration of biometals is also found in association with Aβ peptide, which forms the amyloid plaques. The metal ion accelerates the Aβ aggregation pathway and is also known for stabilizing the most toxic oligomeric aggregates.37,38 Amyloid-β contains one metal binding region Aβ1-16. Mainly, Cu(II) coordinates with His6 and the peptide’s N-terminal part. However, Cu(I) coordinates with His (13, 14). Additionally, Aβ-bound metal ions help to produce reactive oxygen species (ROS), which eventually induce oxidative stress finally leading to neuronal damage.39,40 In physiological conditions, in the presence of ascorbates, Aβ–Cu(II) is reduced to Aβ–Cu(I), and also in the presence of molecular oxygen, that is, biological oxidant, Aβ–Cu(I) can be oxidized back to Aβ–Cu(II). In this way, Aβ–Cu complexes start a copper-mediated Fenton-type reaction, which is mainly responsible for overproduction of ROS and slowly increases the oxidative strain in the neuronal cell.41 Therefore, copper ion is known to be the main culprit for AD. Currently, approved drugs are known to provide only temporary relief but do not act on the disease-causing pathway of AD. Keeping in mind the above arguments, the designed molecule causes disaggregation of Aβ aggregates and disruption of metal ions from the Aβ–metal complex and prevents generation of ROS.42−48 In literature, it has been previously reported that metal chelating peptides and small molecules are able to reduce metal-mediated toxicity. Briefly, Mirica et al. reported two bifunctional compounds, which contain both amyloid binding motifs and metal binding motifs. These compounds disrupted the metal–amyloid β interaction, disassembled the preformed amyloid fibrils, and reduced H2O2 formation.49 Mirica et al. also reported that small chelators reduced the toxicity of the cell but do not disaggregate the amyloid β fibrillar structure.50 Sharma et al. discovered that two bifunctional compounds, HL1 and HL2, bind with Cu and have antiamyloidogenic property.51 Govindaraju et al. reported that a peptidomimetic metal chelator (Gly-His-Lys-Sr-Val-Sr-Phe-Sr) prevented amyloid aggregation and metal-mediated toxicity.44 Faller et al. showed that Aβ12-20 was able to chelate metal ions and inhibit aggregation.45 Jiang et al. reported that peptide-conjugated cyclen was able to prevent metal-mediated amyloid toxicity.52 In this study, we have report a multifunctional molecule, Rh-BT, which is essentially a rhodamine-based metal chelator.
We have synthesized this molecule by attaching a benzothiazole moiety to the rhodamine molecule. The benzothiazole moiety serves as the recognition unit for the amyloid aggregates in AD, as well as an imaging agent for β-amyloid plaques as it has strong binding affinity with β-amyloid plaques.25 When tested in vivo in C57BL/6J mice, it showed considerable serum stability and blood–brain barrier (BBB) permeability, which makes it an even more attractive target for the development of future neurodiagnostics.
2. Results and Discussion
2.1. Design and Synthesis of Rh-BT
Molecules capable of modulating Aβ aggregation are generally used to decrease the amyloid toxicity. However, the metal ion-mediated amyloid aggregation, that is, metal-induced amyloid toxicity, is more risky, whereas Aβ bound with metal ions, particularly Cu2+ ions, produce oxidative stress through ROS generation. For this reason, capturing copper from the Aβ–Cu2+ complex is the most important design strategy for effectively inhibiting the multifaceted AD. Here, we have attached rhodamine-B with the benzothiazole moiety such that Rh-BT can produce a metal binding site. Benzothiazole was used as an amyloid-β recognition moiety. Therefore, Rh-BT is expected to decrease the metal-mediated amyloid toxicity through metal ion chelation, as well as amyloid-β self-induced toxicity (Figure 1). For Rh-BT synthesis, rhodamine-B was first refluxed with phosphorus oxychloride (POCl3) in dichloroethane, and the resultant compound was refluxed with benzothiazole in dichloromethane (DCM). Then, the reaction mixture is extracted in chloroform and finally purified by column chromatography (Figure S1). Then, Rh-BT was characterized mass spectrometry, 1H NMR, and 13C NMR (Figure S2–S4).
Figure 1.
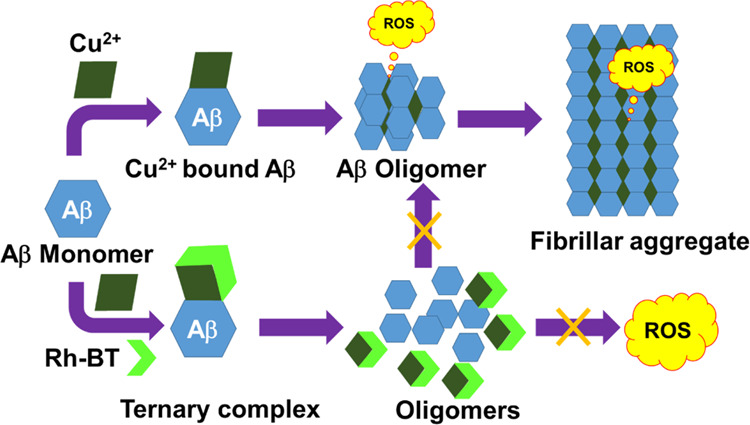
Mechanism for inhibition of metal-catalyzed amyloid toxicity.
2.2. UV–Vis Spectroscopic Studies of Rh-BT in the Presence of Different Metal Ions
To check the selectivity of Rh-BT toward various biologically relevant metals, UV–vis spectra were recorded for Rh-BT (10 μM in methanol) in the presence and absence of various biologically relevant metal ions (10 μM). Rh-BT shows absorption maximum at 313 nm, which may be due to the intramolecular π–π* charge-transfer transition. However, in the presence of Cu2+ and Zn2+, a new peak at 560 nm was observed because of the generation of xanthene through the ring opening of spirolactum via metal ligand binding. However, no such peak was observed in the presence of Ni2+ and Co2+ (Figure S5A,B). Therefore, it may be concluded that the Rh-BT molecule selectively binds with Cu2+ and Zn2+, which are mainly responsible for AD. During the sequential titration of Rh-BT with Cu2+ and Zn2+, the absorption peak at 560 nm increases gradually (Figure S5C,D). This study further supports the binding of Cu2+ and Zn2+ with Rh-BT.
2.3. Fluorescence Spectroscopic Studies
To further validate the quantitative analysis of four different ions, fluorescence spectroscopy was performed using excess of these metal ions. The excitation wavelength was 560 nm, and the emission range was 570–700 nm. For the excitation of only Rh-BT at 560 nm, no such emission peak was observed in the range from 570 to 700 nm, but in the presence of Cu2+ and Zn2+, a new peak was observed (Figure S6A, B). This is due to the fact that in the absence of metal ions, Rh-BT remains in the spirolactum form. However, in the presence of Cu2+ and Zn2+, opening of spirolactum ring triggers the highly conjugated xanthene form that is mainly responsible for fluorescence color. In the presence of Co2+ and Ni2+ ions, no spectral change was observed with respect to the only Rh-BT. From these studies, it is concluded that Rh-BT selectively binds with Cu2+ and Zn2+, which are mainly responsible for metal ion-mediated Alzheimer’s toxicity. Next, to get an insight into the binding pattern and metal coordination of metal ions with Rh-BT, fluorometric titration of 5 μM Rh-BT with varying concentrations of metals (Cu2+ and Zn2+) was performed until the saturation intensity was observed by exciting Rh-BT at 560 nm. The condition for Jobs plot is 1:1 binding, which is reflected for copper ions but not for zinc ions (Figure S6C,D). From the fluorescence titration data, the Jobs plot was drawn (Figure S7). For Cu2+, the jobs plot shows a 1:1 complex formation. However, for Zn2+, the Jobs plot shows a 1:2 complexation. The binding constant value from the nonlinear fit of the emission titration data was 4.5 × 104 M–1(Figure S8), which suggests a strong comparable binding affinity of Rh-BT with Cu2+ like other reported metal chelators.52 Further, sensitivity of Rh-BT toward Cu2+ was an important factor. Therefore, limits of detection (LOD) were calculated,53 and the LOD of Rh-BT for Cu2+ was 11 nM.
2.4. Inhibition of Aβ42 Fibrillar Aggregates
Polymorphic forms such as fibrillar and oligomeric aggregates are mainly responsible for neuronal toxicity in the AD brain. Therefore, inhibition of fibrillar and oligomeric aggregates is taken as one of the key approaches to develop therapeutics against AD. To check the ability of Rh-BT against fibrillar aggregates, thioflavin T (ThT) assay was performed. ThT results revealed that Rh-BT helped to inhibit aggregates. Increasing the compound ratio with the Aβ peptide improved the fibril inhibition ability. When we used Rh-BT with five molar excess of Aβ peptide, it inhibited the fibril formation up to 56% (Figure 2A). Therefore, these data support the idea of attaching benzothiazole with the rhodamine unit. Further confirmation was achieved from the dot blot assay. These data showed that Rh-BT reduces the fibrillar aggregate formation to ∼78% (50 μM), which further confirms its fibril inhibition ability (Figure 2B,C).
Figure 2.

(A) ThT assay of amyloid fibril inhibition in the presence of Rh-BT in different concentrations. (B) Dot blot assay for amyloid fibril inhibition. (C) Bar diagram of dot blot assay. (D) ThT assay of amyloid oligomer inhibition in the presence of Rh-BT in different concentrations. (E) Dot blot assay for inhibition of amyloid oligomerization. (F) Bar diagram of dot blot assay for amyloid oligomer inhibition. Error bar corresponds to the standard deviation (SD) of the value (*p ≤ 0.03 and **p ≤ 0.05).
2.5. Inhibition of Cu2+-Induced Aβ42 Fibrillar Aggregates
The effect of Rh-BT against Cu2+-mediated fibrillar aggregate formation was studied by the ThT assay. Thus, Aβ42 was incubated with Cu2+ independently and with two different concentrations of Rh-BT. The result showed that it inhibited the Cu2+-mediated aggregation up to 60% (Figure S9). These data support that Rh-BT inhibited not only Aβ self-fibrillar aggregate formation but also Cu2+-mediated fibrillar aggregate formation.
2.6. Inhibition of Aβ Oligomeric Aggregates
The Aβ peptide undergoes a hydrophobic interaction to form the most toxic polymorphic structure, that is, Aβ oligomeric aggregates. The effectiveness of Rh-BT was studied through the ThT assay and through the dot blot assay against the Aβ oligomeric aggregates. At first, Aβ oligomers were prepared following previously reported protocols. The ThT assay showed that Rh-BT was able to inhibit the oligomeric formation in a concentration-dependent manner (Figure 2D). For further confirmation, immunohistochemistry was performed. Here, A11 primary antibody was used to detect the amyloid oligomer. Dot blot assay results showed that Rh-BT inhibited oligomerization through a concentration-dependent manner. Rh-BT inhibited oligomerization formation to ∼83% at 50 μM (Figure 2E,F). Rh-BT effectively inhibited fibrillar as well as oligomeric aggregate formation, which supports the multifunctional nature of Rh-BT.
2.7. Understanding the Interactions of Rh-BT with Aβ Peptide Using Molecular Docking
Blind docking of Rh-BT was performed with Aβ42 peptide. The result reveals that Rh-BT binds with Aβ10-16, which is the main copper binding region with a decent binding energy (−5.9 kcal/mol) through hydrophobic and hydrophilic attraction forces (Figure 3A,B). As Rh-BT has affinity toward the copper binding region of Aβ42, it can easily capture copper from the Aβ-bound copper complex, which also support that Rh-BT inhibited Cu2+-mediated amyloid toxicity.
Figure 3.
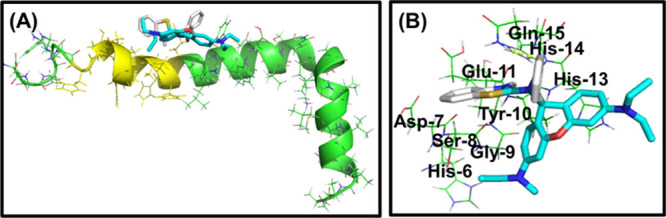
(A) Molecular docking experiment of Rh-BT with Aβ42, showing that it strongly binds with Aβ42. (B) Interacting partner of Rh-BT with different amino acids of amyloid-β.
2.8. Isothermal Titration Calorimetry Experiment for Understanding the Binding of Rh-BT with Aβ Peptide
To look into the interaction of Rh-BT with Aβ peptide experimentally, an isothermal titration calorimetry (ITC) experiment was performed. The titration curve was exothermic in nature, and all the corresponding thermodynamic parameters were calculated from the titration curve. The experiment showed that Rh-BT has the strong affinity (7.39 ± 1.54 × 105 M–1) for Aβ42 peptide (Figure S10).
2.9. Extraction of Metal Ions from the Aβ42–Cu2+ Complex
It is well reported that only Aβ peptide is unable to produce ROS in the AD brain, but the metal-bound Aβ complex helps to produce ROS through a Fenton-type reaction.54 Therefore, sequestration of copper ions from the Aβ-bound copper complex is an important strategy to suppress the metal-mediated Aβ toxicity through the prevention of redox process (Figure 4A). The metal extraction ability of Rh-BT from the Cu2+-bound Aβ complex was studied by tyrosine quenching experiment using the inherent Tyr10 fluorescence quenching in Aβ42.55 It is reported that the quenching of the Tyr10 fluorescence occurs because of the close proximity of Cu2+ to Aβ42, and the extent of regaining of fluorescence intensity by the metal chelator indicates the ability of the chelator to capture copper from the Aβ-bound copper complex. For this experiment, Aβ42 (10 mM), Aβ42–Cu2+ (10 mM), and Aβ42–Cu2+ + Rh-BT (10 mM) were incubated in phosphate-buffered saline (PBS; 10 mM, pH 7). Then, fluorescence intensity was measured (excitation: 285) (Figure 4B,C). Interestingly, Rh-BT was able to restore tyrosine fluorescence by ∼60%. Therefore, Rh-BT may be used as a metal chelator for inhibition of metal-induced Aβ toxicity.
Figure 4.

(A) Chelation of Cu2+ by Rh-BT, which leads to generate the fluorescence property of Rh-BT. (B) Extraction of Cu2+ from the Aβ–Cu2+ complex by Rh-BT through Tyr quenching experiment. (C) Bar diagram of Tyr quenching experiment. Error bar corresponds to the SD of the value (*p ≤ 0.03).
2.10. ROS Inhibition Properties of Rh-BT
In the AD brain, the Aβ-bound copper complex produces ROS through a Fenton-type reaction56 following the redox cycle of the metal ion (Figure 5A). From the previous study, it is well established that Rh-BT can remove Cu2+ from the Aβ-bound copper complex. As copper has a crucial role for producing ROS, we were interested to know that capturing copper from Aβ42 has any effect on ROS inhibition or not. Therefore, we investigated the redox silencing property of Rh-BT through ascorbate (reducing agent) assay.57 In this assay, Cu2+ is transferred to Cu+ by ascorbate, but Cu+ is unstable. It is easily oxidized back to Cu2+. One of the major constituents of ROS, hydroxyl radicals, is generated through this process, which causes toxicity in AD. These hydroxyl radicals were measured by measuring the fluorescence intensity of 7-hydroxycoumarin-3-carboxylic acid (7-OH-CCA) (λex = 395 nm and λem = 452 nm). Actually for this experiment, we used 50 μM coumarin-3-carboxylic acid (3-CCA), which is nonfluorescent. However, in the presence of hydroxyl radicals generated from the reaction of 5 μM Cu2+ and 150 μM ascorbate in PBS (pH 7.4), the nonfluorescent coumarin-3-carboxylic acid (3-CCA) transformed to the fluorescent 7-hydroxycoumarin-3-carboxylic acid (7-OH-CCA). Interestingly, in the presence of Rh-BT (10 μM), the fluorescence intensity of 7-OH-CCA is very negligible, which indicates that after complexation with Rh-BT, Cu2+ becomes unable to take part in redox cycle to produce ROS in highly reducing conditions (Figure 5B,C). Therefore, it may be concluded that Rh-BT inhibited the formation of hydroxyl radicals through copper capturing by subduing the redox cycle. Thus, the designing of Rh-BT for ROS inhibition by capturing metal from the Aβ–Cu2+ complex is fruitful.
Figure 5.
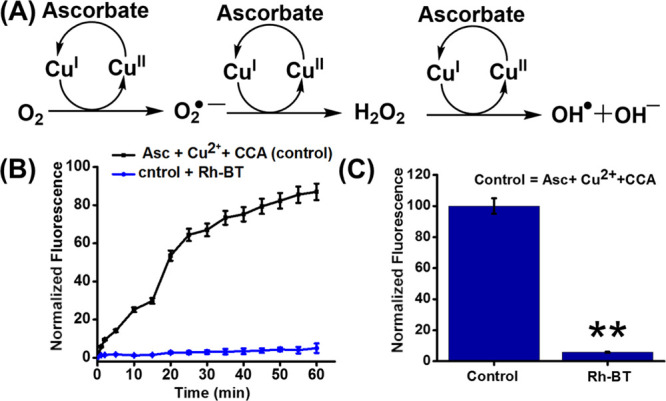
(A) Mechanism for ROS production through Fenton reaction cycle from molecular oxygen and ascorbate using copper. (B) Inhibition of copper-catalyzed ROS production through copper chelation via measuring the fluorescence intensity of 7-OH-CCA. (C) Bar diagram of fluorescence intensity of 7-OH-CCA after 1 h. Error bar corresponds to the SD of the value (**p ≤ 0.05).
2.11. MTT Assay of Rh-BT
So far, we were interested about the intriguing role of Rh-BT using varying in vitro experiments. Next, we were eager to examine the effect of Rh-BT for the inhibition of metal-induced Aβ toxicity in a cellular model. Therefore, we first checked the toxicity of Rh-BT in PC12-derived neurons. For that purpose, PC12-derived neurons were incubated with various concentrations of
Rh-BT for 24 h up to 200 μM. Using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) reduction method, cell viability was calculated. Interestingly, Rh-BT has shown no cytotoxic effect on the neuron viability even at a concentration of 200 μM (Figure 6A). Therefore, up to a concentration of 200 μM, Rh-BT may be used for various cell-based assays in PC12-differentiated neurons.
Figure 6.

(A) Toxicity of Rh-BT in PC12-derived neurons, showing that it was not toxic in neurons. (B) Cell rescue assay of Rh-BT in Aβ + Cu2+(1:1) (Cu2+ is 5 μM) mediated toxicity, showing that cell viability was increased upon treatment with Rh-BT. These experiments were repeated in triplicate (n = 3) and calculated the SD, which represent the error bars. Error bar corresponds to the SD of the value (*p ≤ 0.03, **p ≤ 0.05).
2.12. Cellular Uptake Rh-BT in the Presence of Cu2+
The in vitro assays so far have revealed the metal chelating property of Rh-BT. Inspired from the above results, we studied its metal chelating property in PC12-differentiated neurons. We incubated the PC12-derived neurons with 5 μM copper for 2 h. Then, we incubated the cells with 5 μM Rh-BT for another 2 h. When observed under a microscope, it showed red color, thereby proving the metal chelating property of Rh-BT. In the case of cells only incubated with Rh-BT or copper, it did not show any red color, thereby acting as negative controls (Figures S11–S13).
2.13. MTT Assay of Rh-BT against Cu2+-Triggered Aβ Toxicity
The above promising antiaggregation properties of Rh-BT led us to check whether Rh-BT could also exert neuroprotective effect against Cu2+-induced Aβ toxicity in a cellular model.25,58 To address this problem, PC12-derived neurons were treated with Aβ + Cu2+, and MTT assay was performed to analyze the cellular viability. After 24 h of incubation, the cell viability was decreased, but in the presence of a copper chelator, that is, Rh-BT, the cell viability increases. We also performed dose response analysis with higher concentrations of Rh-BT, which resulted in even more pronounced cell viability (Figure 6B). These data support the fact that Rh-BT can capture Cu2+ from Aβ–Cu2+ complex simultaneously, resulting in decreased amyloid toxicity in PC12-derived neurons. This result showed that Rh-BT not only inhibited Aβ oligomerization and fibrillation but also decreased Aβ–Cu2+-mediated cellular toxicity.
2.14. Microscopy Experiment for Cellular ROS Inhibition by Rh-BT
In order to scrutinize the ability of Rh-BT to aid the recovery of neurons against oxidative stress in an AD brain, the DCFDA assay was performed. ROS was generated by Aβ–Cu2+ in PC12-derived neurons. Then, the cellular ROS generation was monitored by measuring the fluorescence of DCF. PC12-derived neurons were treated with Aβ–Cu2+ in the presence and absence of Rh-BT. The microscopic images showed that when Rh-BT is present, the green fluorescence intensity of DCF was decreased compared to the control experiment (untreated) (Figure 7A,B).
Figure 7.

Live microscopy images of PC12-derived neurons stained with DCFDA. (A) Neurons were treated with Aβ + Cu2+ (1:1). (B) Neurons were treated with Aβ + Cu2+ (1:1) in the presence of Rh-BT. (C) Neurons were treated with only Rh-BT to check the autofluorescence of Rh-BT. Scale bars corresponding to 20 μm. (D) Quantitative analysis of green fluorescence intensity (by ImageJ software). (E) Bar diagram for the DCFDA assay. Error bar corresponds to the SD of the value (**p ≤ 0.05).
To check the autofluorescence of Rh-BT, PC12-derived neurons were treated with only Rh-BT, but we did not find any green or red fluorescence (Figure 7C). Further, we have measured the green fluorescence intensity of DCF to quantify the percentage of ROS inhibition by Rh-BT (Figure 7D). The bar diagram showed that there is a significant amount of ROS inhibition upon treatment with Rh-BT. This experiment further supports that Rh-BT inhibited cellular ROS generation induced by an Aβ42–Cu2+ redox cycle in PC12-derived neurons.
2.15. DCFDA Assay to Assess ROS Inhibition
Previously, it has been reported that the AD brain produces ROS through the electrochemical reaction of Aβ–Cu2+ complex.9,58 Therefore, the DCFDA assay was performed in PC12-derived neurons.25 This assay measures hydroxyl, peroxyl, and other various ROS within the cell. PC12-derived neurons seeded in black 96-well plates were treated with Aβ42–Cu2+ in the presence and absence of Rh-BT. Then, a fluorometric assay of DCFDA was performed. The fluorescence intensity measurement showed that in presence of Rh-BT, the fluorescence intensity was less than compared to untreated neurons (Figure 7E). Therefore, this result also supported that Rh-BT inhibited ROS production in the cellular model by capturing the Cu2+ ion from the Aβ42–Cu2+ complex.
2.16. Cellular Uptake of Rh-BT in Primary Cortical Neurons
Our prime target is to use Rh-BT in an in vivo model system. Therefore, we checked the toxicity and cellular uptake of Rh-BT in primary cortical neurons as a preliminary experiment. For this assay, we have taken Sprague-Dawley rat embryos to collect primary cortical neurons from their brains. This experiment was performed using the guidelines of institutional animal ethics committee. The freshly prepared primary cortical neurons were treated with Rh-BT in presence of Cu2+ to check their cellular uptake efficacy. The microscopic images showed that the cell body contains red color, which supports the cellular uptake of Rh-BT in presence of Cu2+ (Figure S14). The healthy morphology of the primary cortical neurons also supports the noncytotoxic nature of Rh-BT.
2.17. In Vivo BBB and Serum Stability in a Mouse Model
One of the key features of brain is that it is segregated completely from the spinal cord by the BBB. Therefore, it is mainly responsible for the limited transport of compounds from blood to brain. Therefore, we checked the BBB crossing ability of Rh-BT. Rh-BT was injected intraperitoneally, and after 4 h, the brain and plasma were collected from the mice. The brain was homogenized in acetonitrile. Then, we performed matrix-assisted laser desorption ionization time-of-flight mass spectrometry with the plasma as well as the mouse brain extract. The mass spectra of the mouse brain extract show the presence of molecular peak of Rh-BT, which suggests that Rh-BT was able to cross the BBB (Figure S15). A similar result was observed with the plasma, which proves its stability in serum (Figure S16). Therefore, it can be concluded that Rh-BT was also present in the brain as well as the plasma. This experiment demonstrates that the significant brain permeability of Rh-BT is sufficient to activate the different signal transduction cascades to prompt therapeutic effects.
3. Conclusions
In conclusion, we have successfully designed and synthesized a multifunctional compound against multifaceted AD. Rh-BT can capture metal ions from the Aβ–Cu2+ complex. Therefore, it inhibited the formation of metal-induced toxic polymorphic aggregates. In this way, Rh-BT suppressed the amyloidogenic pathway to keep the cell membrane integrity intact. As it can effectively sequester Cu2+ from the Aβ–Cu2+ complex, it successfully inhibited Fenton’s reaction, that is, ROS production is prevented in the AD brain. The antioxidant property of Rh-BT has been explored through the DCFDA assay in a cellular model and ascorbate assay in vitro. It is also found to be noncytotoxic in neurons derived from PC12 cells. Therefore, when we checked the ability of Rh-BT to reverse Aβ42–Cu2+-induced toxicity in neurons through the cell rescue assay, it effectively reduced the metal-mediated toxicity. All the experimental and docking results strongly agree with the designed mechanism of Rh-BT. Thus, the inhibition of metal-induced aggregation and fibrillation, selective capturing of metal ions from the metal Aβ complex, inhibition of metal-induced ROS production, and reduction of metal-induced toxicity make Rh-BT a multifunctional molecule against the multifactorial AD. Moreover, Rh-BT is stable in mouse plasma and crosses the BBB. This study opens the path for designing of more inhibitors of metal-mediated toxicity. Therefore, Rh-BT can be considered as a potential lead for the treatment of multifaceted AD.
4. Experimental Section
4.1. Chemicals
Rhodamine-B, 2-aminobenzothiazole, and phosphorus(V) oxychloride were purchased from Sigma-Aldrich. Triethyl amine, ThT, dry DCM, hexane, chloroform, ethyl acetate, and nitrocellulose membrane were purchased from Merck (Germany). Silica gel was taken from SRL. 1,1,1,3,3,3-Hexafluoroisopropanol (HFIP) was procured from Spectrochem. 2-[4-(2-Hydroxyethyl)piperazin-1-yl]ethanesulfonic acid and nickel chloride hexahydrate were purchased from Himedia. Copper(II) chloride, MTT, zinc nitrate hexahydrate, cobalt chloride, 4-piperazinediethanesulfonic acid, Dulbecco’s modified Eagle’s medium (DMEM), ethylenediaminetetraacetic acid, and cell culture-grade dimethylsulfoxide (DMSO) were purchased from Sigma-Aldrich. Various culture media and serum were procured from Invitrogen. β-Amyloid (1–42) was procured from Alxotec (Sweden). Primary antibody 6E10 was taken from Bio Legend, A11 was taken from Thermo Scientific, and OC were taken from Millipore. All these chemicals were used in various experiments without further purification.
4.2. Synthesis and Characterization of Rhodamine-Based Compound Rh-BT
Rh-BT was synthesized from rhodamine-B. In brief, 400 mg of rhodamine-B was taken in a two-neck round-bottom flask. To that round-bottom flask, 20 mL of 1,2 dichloroethane and 750 μL of phosphorus oxychloride were added. Then, the reaction mixture was refluxed for 6 h to obtain acid chloride from rhodamine-B. After 6 h, the reaction mixture was evaporated out and 20 mL of DCM along with 150 mg of 2-aminobenzothiazole were added to the round-bottom flask. After adding 1 mL of triethylamine, the reaction mixture was refluxed for another 6 h. Next, the solvent was removed under reduced pressure, and the residue was extracted in chloroform. Then, the purple solid obtained was dried and purified by column chromatography using ethyl acetate and hexane as an eluent. Finally, Rh-BT was characterized by mass spectrometry and NMR.
4.3. Preparation Aβ42 Peptide Solution
The Aβ42 peptide stock solution was prepared in 1,1,1,3,3,3-hexafluoroisopropanol (HFIP) and stored in a −20 °C freezer. For various experiments, a required amount of stock solution was taken out. Then, the isopropanol solution was evaporated out using a nitrogen gas flow to separate out the Aβ42 peptide residue. Finally, the Aβ42 peptide solution was prepared in PBS buffer.
4.4. Preparation of ThT Solution
ThT solution was prepared in PBS buffer and stored in a 4 °C freezer. The solution was stored in a dark place to avoid degradation. The freshly prepared ThT solution was used for the experiment.
4.5. Preparation of Oligomers and Fibrils of Aβ42 Peptide59
For the preparation of oligomeric aggregates, Aβ42 peptide was dissolved in molecular biology-grade DMSO to obtain a final concentration of 1 μM. Then, the solution was further diluted with PBS buffer to obtain a final concentration of 200 μM with a final DMSO concentration of 1%. The solution was incubated at 37 °C for 1 h, and the incubation was further continued for 24 h at 4 °C. After incubation, the peptide solution was centrifuged to obtain the oligomers in the solution. To obtain the confirmation of oligomer formation, we performed dot blot analysis. For the preparation of fibrillar aggregates of Aβ42 peptide, the peptide solution was incubated at 37 °C with constant agitation for 48 h. To obtain the confirmation of fibril formation, the ThT fluorescence intensity was measured (excitation: 435 nm, emission: 450–600 nm) with a slit width of 1.25 nm in a Quanta Master Spectrofluorometer (QM-40). Finally, the data were plotted using Origin Pro 8.5 software.
4.6. Dot Blot Analysis
Aβ42 oligomeric and fibrillar aggregates were prepared following the previous procedure in PBS buffer. Then, on a nitrocellulose, membrane spotting was performed from this solution. For blocking the nonspecific sites, a blocking buffer was used. The blocking buffer was prepared in TBST buffer containing 5% bovine serum albumin. After that, the membranes were soaked in primary antibodies 6E10, A11, and OC to detect Aβ peptide, oligomer, and fibril, respectively, and then washed with TBST buffer. A secondary antimouse antibody was added and incubated with the membrane for 1 h. To capture the image of the dot blot, electrochemical luminescence was performed to the membranes and incubated for 2 min. The image appears through a phenomenon called chemiluminescence.
4.7. Analysis through Isothermal Titration Calorimetric Study
For this isothermal experiment, the temperature was kept fixed at 298 K. The freshly prepared Aβ42 peptide (20 μM) was used for this experiment. Twenty-eight injections were made with each solution containing 10 μL of Rh-BT solution. The mixing of Rh-BT with Aβ42 peptide was performed for 5 min by a computer control programming. The heat was measured after every addition of Rh-BT. To subtract the heat of dilution, a blank experiment with Aβ42 peptide was performed. Finally, the heat released against the molar ratio of Rh-BT was plotted through origin pro 8.5 software.
4.8. Cultivation of PC12 Cells
PC12 cells (rat adrenal pheochromocytoma cell line) were procured from NCCS Pune, India. The cells were grown in DMEM containing horse serum (10%) as well as fetal bovine serum (5%) under 5% CO2 at 37 °C. The PC12 cells were differentiated into neurons using nerve growth factor (NGF) (100 ng/mL) in 1% horse serum. The differentiated neurons were used for various cell-based assays.
4.9. Cell Cytotoxicity Assay with Rh-BT
The cytotoxic response of Rh-BT was assessed in PC12-derived neurons through the MTT reduction method. Generally, MTT is reduced in live cells into formazan through reductase. The color of the formazan is purple. However, dead cells are unable to perform this type of reduction step. From this reduction assay, we easily separate the healthy cells from the dead cells. Before the treatment of Rh-BT, the PC12-derived neurons were seeded in a 96-well plate. After the treatment of 24 h, the MTT solution (10 mg/mL) was added and incubated for 4 h at 37 °C. After that, the formazan was dissolved in DMSO/MeOH (1:1). Then, the absorbance of the solution was measured using a microplate ELISA reader at 570 nm. Finally, the cell viability of the cells was measured using the following formula:
4.10. Microscopy Study for Cellular Uptake of Rh-BT with Cu2+
Microscopy study of cellular uptake was performed in PC12-derived neurons with Cu2+ chelating Rh-BT. For differentiation of neurons from PC12 cells, 100 ng/mL of NGF in serum-free media was used for 7 days. Then, the neurons were treated with Rh-BT (10 μM) and Cu2+ (10 μM) for 2 h. After washing the neurons, they were fixed with 4% formaldehyde. Washing was performed with PBS. The nucleus of the neurons was stained with Hoechst 33258. The images of the neurons were captured using an Olympus (IX83) microscope equipped with an Andor iXon3 897 EMCCD camera.
4.11. Cell Rescue Assay through the MTT Reduction Method
Cell rescue assay was performed in neurons derived from PC12 cells using neurite growth factor. Dulbecco’s modified Eagle’s medium (DMEM) containing horse serum (10%) and fetal bovine serum (5%) was used to culture the neurons. For this assay, neurons were seeded on a 96-well plate. After 24 h of seeding, the neurons were treated with Aβ42 (5 μM) with Cu2+(5 μM) in the absence or presence of Rh-BT for 24 h in a 1:1 ratio. Then, the MTT reduction method was performed to examine the cell rescue ability of Rh-BT.
4.12. Cellular Uptake and Toxicity of Rh-BT in Primary Cortical Neurons
We have followed previous published literature to culture the primary cortical neurons.60,61 For this experiment, we have taken pregnant Sprague-Dawley rats to collect E18 embryos. Next, the brains were collected from the embryos. After performing microdissection and digestion, the brains were dissolved in MEM (minimum essential medium), which contains horse serum (10%). Poly-d-lysine-coated confocal dishes were used to culture the cells at normal cell culture conditions at 37 °C with a 5% CO2 environment. Then, the culture medium was taken out and the neurobasal medium was added. Rh-BT (5 μM) and Cu2+ (5 μM) were treated for 4 h to check the toxicity and cellular uptake.
4.13. In Vivo BBB Permeability and Serum Stability of Rh-BT62,63
In order to understand the in vivo functioning of Rh-BT, we tested its efficacy through BBB permeability and serum stability in C57BL/6J mice (n = 3) of an average weight of 25–35 g. The compound prepared in saline solution was injected intraperitoneally. The dosage of the experiment was 10 mL/kg body weight of the mice. After 4 h of injection, avertin (i.p) was used to anesthetize the mice. Around 0.5 mL of blood from each mouse was collected in heparinized tubes through cardiac puncture with a 23 G needle. After 30 min, this tube was centrifuged for 10 min at 5000 rpm at 4 °C. Then, the supernatant was taken as plasma and stored in −30 °C for mass analysis. The mice were thereafter sacrificed through transcardial perfusion and the brain was taken out. All the blood vessels as well as the meninges were removed from the brain and collected in PBS buffer. The cortex was collected and homogenized under liquid nitrogen using a pestle. Finally, it was extracted in high-performance liquid chromatography-grade acetonitrile. Then, the solution was centrifuged at 10,000 rpm at 4 °C for 10 min. After centrifugation, the supernatant was collected and stored at −20 °C for mass analysis. Finally, mass spectrometry was performed with mouse brain extract as well as plasma.
4.14. Docking
To find the molecular interaction between Rh-BT and Aβ42 peptide, blind docking was performed in AutoDock-Vina software (version 1.1.2). The receptor Aβ42 peptide was PDB ID: 1IYT.
4.15. Data Analysis
Origin 8.5 pro software was used to plot and calculate all the spectroscopic data. For the analysis of microscopic study, we have used ImageJ software. To perform statistical analysis, we have performed t-test and one-way analysis of variance. For all the experiments, the statistical values are *p ≤ 0.03 and **p ≤ 0.05.
Acknowledgments
K.P. and S.B. thank UGC, G.D. thanks ICMR, C.K. thanks SERB, India (PDF/2017/000074), T.M. thanks CSIR, J.K. thanks DST Inspire, and N.M. thanks IIT Jodhpur for awarding their fellowships. We thank Dr. P. Jaisankar, CSIR IICB, Kolkata, for his kind and generous support and help. S.G. kindly acknowledges SERB India (CRG/2019/000670) for financial assistance. S.G. also thanks CSIR-IICB, Kolkata, Animal House Facility, and IIT Jodhpur for financial and infrastructural support.
Glossary
Abbreviations
- Aβ
amyloid beta
- AD
Alzheimer’s disease
- NGF
nerve growth factor
- MTT
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
- PDB
protein data bank
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.0c02235.
Scheme of Rh-BT preparation, mass chromatogram, NMR chromatogram, ThT assay, various fluorometric and UV–vis spectra and microscopic images of PC12-derived neurons, and mass spectra of mouse brain extract and plasma (PDF)
Author Present Address
∥ Department Of Chemistry, Amity University, Kolkata, India.
Author Contributions
K.P. and S.G. designed the project. K.P. carried out the synthesis, purification, and characterization of Rh-BT; various in vitro assays such as the ThT assay; ITC; fluorometric study; and molecular docking study. G.D. helped K.P. to perform the dot blot assay. G.D. performed cell-based assays such as the MTT assay and the DCFDA assay. S.B., N.M., and T.M. supported K.P. and G.D. for performing various experiments. K.P. and S.G. wrote the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Scheltens P.; Blennow K.; Breteler M. M. B.; de Strooper B.; Frisoni G. B.; Salloway S.; Van der Flier W. M. Alzheimer’s disease. Lancet 2016, 388, 505–517. 10.1016/s0140-6736(15)01124-1. [DOI] [PubMed] [Google Scholar]
- Rajasekhar K.; Chakrabarti M.; Govindaraju T. Function and toxicity of amyloid beta and recent therapeutic interventions targeting amyloid beta in Alzheimer’s disease. Chem. Commun. 2015, 51, 13434–13450. 10.1039/c5cc05264e. [DOI] [PubMed] [Google Scholar]
- Brettschneider J.; Tredici K. D.; Lee V. M.-Y.; Trojanowski J. Q. Spreading of pathology in neurodegenerative diseases: a focus on human studies. Nat. Rev. Neurosci. 2015, 16, 109–120. 10.1038/nrn3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamley I. W. The Amyloid Beta Peptide: A Chemist’s Perspective. Role in Alzheimer’s and Fibrillization. Chem. Rev. 2012, 112, 5147–5192. 10.1021/cr3000994. [DOI] [PubMed] [Google Scholar]
- Viola K. L.; Klein W. L. Amyloid β oligomers in Alzheimer’s disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 2015, 129, 183–206. 10.1007/s00401-015-1386-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerasoli E.; Ryadnov M. G.; Austen B. M. The elusive nature and diagnostics of misfoldedA beta oligomers. Front. Chem. 2015, 3, 17. 10.3389/fchem.2015.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G.-f.; Xu T.-h.; Yan Y.; Zhou Y.-r.; Jiang Y.; Melcher K.; Xu H. E. Amyloid beta: structure, biology and structure-based therapeutic development. ActaPharmacol. Sin. 2017, 38, 1205–1235. 10.1038/aps.2017.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šimić G.; Leko M. B.; Wray S.; Harrington C.; Delalle I.; Jovanov-Milosevic N.; Bazadona D.; Buee L.; de Silva R.; Di Giovanni G.; Wischik C.; Hof P. R. Tau Protein Hyperphosphorylation and Aggregation in Alzheimer’s Disease and Other Tauopathies, and Possible Neuroprotective Strategies. Biomolecules 2016, 6, 6. 10.3390/biom6010006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iqbal K.; Liu F.; Gong C.-X.; Grundke-Iqbal I. Tau in Alzheimer Disease and Related Tauopathies. Curr. Alzheimer Res. 2010, 7, 656–664. 10.2174/156720510793611592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao J.; Shi R. R.; Li L. F.; Chen F.; Zhou Y.; Tung Y. C.; Hu W.; Gong C. X.; Iqbal K.; Liu F. Pathological Tau From Alzheimer’s Brain Induces Site-Specific Hyperphosphorylation and SDS- and Reducing Agent-Resistant Aggregation of Tau in vivo. Front. Aging Neurosci. 2019, 11, 34. 10.3389/fnagi.2019.00034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis P. T.; Palmer A. M.; Snape M.; Wilcock G. K. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J. Neurol. Neurosurg. Psychiatry 1999, 67, 558. 10.1136/jnnp.67.4.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferreira-Vieira T. H.; Guimaraes I. M.; Silva F. R.; Ribeiro F. M. Alzheimer’s Disease: Targeting the Cholinergic System. Curr. Neuropharmacol. 2016, 14, 101–115. 10.2174/1570159X13666150716165726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatta P.; Drago D.; Bolognin S.; Sensi S. L. Alzheimer’s disease, metal ions and metal homeostatic therapy. Trends Pharmacol. Sci. 2009, 30, 346–355. 10.1016/j.tips.2009.05.002. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Nguyen M.; Robert A.; Meunier B. Metal Ions in Alzheimer’s Disease: A Key Role or Not?. Acc. Chem. Res. 2019, 52, 2026–2035. 10.1021/acs.accounts.9b00248. [DOI] [PubMed] [Google Scholar]
- Adlard P. A.; Bush A. I. Metals and Alzheimer’s Disease: How Far Have We Come in the Clinic?. J. Alzheimer’s Dis 2018, 62, 1369–1379. 10.3233/jad-170662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattson M. P. Pathways towards and away from Alzheimer’s disease. Nature 2004, 430, 631–639. 10.1038/nature02621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekhar K.; Govindaraju T. Current progress, challenges and future prospects of diagnostic and therapeutic interventions in Alzheimer’s disease. RSC Adv. 2018, 8, 23780–23804. 10.1039/c8ra03620a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciechanover A.; Kwon Y. T. Degradation of misfolded proteins in neurodegenerative diseases: therapeutic targets and strategies. Exp. Mol. Med. 2015, 47, e147 10.1038/emm.2014.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teplow D. B. On the subject of rigor in the study of amyloid β-protein assembly. Alzheimer’s Res. 2013, 5, 39. 10.1186/alzrt203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauk A. The chemistry of Alzheimer’s disease. Chem. Soc. Rev. 2009, 38, 2698–2715. 10.1039/b807980n. [DOI] [PubMed] [Google Scholar]
- Roychaudhuri R.; Yang M.; Hoshi M. M.; Teplow D. B. Amyloid β-Protein Assembly and Alzheimer Disease. J. Biol. Chem. 2009, 284, 4749–4753. 10.1074/jbc.r800036200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y.; Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell 2012, 148, 1204–1222. 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles T. P. J.; Vendruscolo M.; Dobson C. M. The amyloid state and its association with protein misfolding diseases. Nat. Rev. Mol. Cell Biol. 2014, 15, 384–396. 10.1038/nrm3810. [DOI] [PubMed] [Google Scholar]
- Axelsen P. H.; Komatsu H.; Murray I. V. J. Oxidative Stress and Cell Membranes in the Pathogenesis of Alzheimer’s Disease. Physiology 2011, 26, 54–69. 10.1152/physiol.00024.2010. [DOI] [PubMed] [Google Scholar]
- Geng J.; Li M.; Wu L.; Ren J.; Qu X. Liberation of Copper from Amyloid Plaques: Making a Risk Factor Useful for Alzheimer’s Disease Treatment. J. Med. Chem. 2012, 55, 9146–9155. 10.1021/jm3003813. [DOI] [PubMed] [Google Scholar]
- Bush A. I. The metallobiology of Alzheimer’s disease. Trends Neurosci. 2003, 26, 207–214. 10.1016/s0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- Karran E.; Mercken M.; Strooper B. D. The amyloid cascade hypothesis for Alzheimer’s disease: an appraisal for the development of therapeutics. Nat. Rev. Drug Discovery 2011, 10, 698–712. 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- Hardy J.; Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol. Sci. 1991, 12, 383–388. 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- Selkoe D. J.; Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradhan K.; Das G.; Mondal P.; Khan J.; Barman S.; Ghosh S. Genesis of Neuroprotective Peptoid from Aβ30-34 Inhibits Aβ Aggregation and AChE Activity. ACS Chem. Neurosci. 2018, 9, 2929–2940. 10.1021/acschemneuro.8b00071. [DOI] [PubMed] [Google Scholar]
- Buxbaum J. N.; Linke R. P. A molecular history of the amyloidoses. J. Mol. Biol. 2012, 421, 142. 10.1016/j.jmb.2012.01.024. [DOI] [PubMed] [Google Scholar]
- Jankowska A.; Wesolowska A.; Pawlowski M.; Chlon-Rzepa G. Multi-Target-Directed Ligands Affecting Serotonergic Neurotransmission for Alzheimer’s Disease Therapy: Advances in Chemical and Biological Research. Curr. Med. Chem. 2018, 25, 2045–2067. 10.2174/0929867324666170529122802. [DOI] [PubMed] [Google Scholar]
- Kantham S.; Chan S.; McColl G.; Miles J. A.; Veliyath S. K.; Deora G. S.; Dighe S. N.; Khabbazi S.; Parat M.-O.; Ross B. P. Effect of the Biphenyl Neolignan Honokiol on Aβ42-Induced Toxicity inCaenorhabditis elegans, Aβ42Fibrillation, Cholinesterase Activity, DPPH Radicals, and Iron(II) Chelation. ACS Chem. Neurosci. 2017, 8, 1901–1912. 10.1021/acschemneuro.7b00071. [DOI] [PubMed] [Google Scholar]
- Paul A.; Nadimpally K. C.; Mondal T.; Thalluri K.; Mandal B. Inhibition of Alzheimer’s amyloid-β peptide aggregation and its disruption by a conformationally restricted α/β hybrid peptide. Chem. Commun. 2015, 51, 2245–2248. 10.1039/c4cc09063b. [DOI] [PubMed] [Google Scholar]
- Mckenzie-Nickson S.; Bush A. I.; Barnham K. J. Bis(thiosemicarbazone) metal complexes as therapeutics for neurodegenerative diseases. Curr. Top. Med. Chem. 2016, 16, 3058–3068. 10.2174/1568026616666160216155746. [DOI] [PubMed] [Google Scholar]
- Caballero A. B.; Terol-Ordaz L.; Espargaró A.; Vázquez G.; Nicolás E.; Sabaté R.; Gamez P. Histidine-Rich Oligopeptides To Lessen Copper-Mediated Amyloid-β Toxicity. Chem. - Eur. J. 2016, 22, 7268–7280. 10.1002/chem.201600286. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Hu X.; Wang W.; Yuan Z. Study of a Bifunctional Aβ Aggregation Inhibitor with the Abilities of Antiamyloid-β and Copper Chelation. Biomacromolecules 2016, 17, 661–668. 10.1021/acs.biomac.5b01603. [DOI] [PubMed] [Google Scholar]
- Toyoshima I. Toxic effect of clioquinol on vesicular transport in axons of dorsal root ganglion cells. J. Neurol. Sci. 2017, 381, 502. 10.1016/j.jns.2017.08.3623. [DOI] [Google Scholar]
- Muthuraj B.; Layek S.; Balaji S. N.; Trivedi V.; Iyer P. K. Multiple Function Fluorescein Probe Performs Metal Chelation, Disaggregation, and Modulation of Aggregated Aβ and Aβ-Cu Complex. ACS Chem. Neurosci. 2015, 6, 1880–1891. 10.1021/acschemneuro.5b00205. [DOI] [PubMed] [Google Scholar]
- Wu W.-h.; Lei P.; Liu Q.; Hu J.; Gunn A. P.; Chen M.-s.; Rui Y.-f.; Su X.-y.; Xie Z.-p.; Zhao Y.-F.; Bush A. I.; Li Y.-m. Sequestration of Copper from β-Amyloid Promotes Selective Lysis by Cyclen-Hybrid Cleavage Agents. J. Biol. Chem. 2008, 283, 31657–31664. 10.1074/jbc.m804722200. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Zhao B. Oxidative stress and the pathogenesis of Alzheimer’s disease. Oxid. Med. Cell. Longevity 2013, 2013, 316523. 10.1155/2013/316523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassagnes L.-E.; Hervé V.; Nepveu F.; Hureau C.; Faller P.; Collin F. The catalytically active copper-amyloid-beta state: Coordination site responsible for reactive oxygen species production. Angew. Chem., Int. Ed. 2013, 52, 11110–11113. 10.1002/anie.201305372. [DOI] [PubMed] [Google Scholar]
- Folch J.; Petrov D.; Ettcheto M.; Abad S.; Sánchez-López E.; García M. L.; Olloquequi J.; Beas-Zarate C.; Auladell C.; Camins A. Current Research Therapeutic Strategies for Alzheimer’s Disease Treatment. Neural Plast. 2016, 2016, 8501693. 10.1155/2016/8501693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajasekhar K.; Madhu C.; Govindaraju T. Natural Tripeptide-Based Inhibitor of Multifaceted Amyloid β Toxicity. ACS Chem. Neurosci. 2016, 7, 1300–1310. 10.1021/acschemneuro.6b00175. [DOI] [PubMed] [Google Scholar]
- Jensen M.; Canning A.; Chiha S.; Bouquerel P.; Pedersen J. T.; Østergaard J.; Cuvillier O.; Sasaki I.; Hureau C.; Faller P. Inhibition of Cu-Amyloid-β by using Bifunctional Peptides with β-Sheet Breaker and Chelator Moieties. Chem.—Eur. J. 2012, 18, 4836–4839. 10.1002/chem.201103546. [DOI] [PubMed] [Google Scholar]
- Rana M.; Sharma A. K. Cu and Zn interactions with Aβ peptides: consequence of coordination on aggregation and formation of neurotoxic soluble Aβ oligomers. Metallomics 2019, 11, 64–84. 10.1039/c8mt00203g. [DOI] [PubMed] [Google Scholar]
- IUPAC Compendium of Analytical Nomenclature, Definitive Rules, 1997, web edition, Lnczédy J.; Lengyel T.; Ure A. M., Eds. IUPAC, 2002. [Google Scholar]
- Gamez P.; Caballero A. B. Copper in Alzheimer’s disease: Implications in amyloid aggregation and neurotoxicity. AIP Adv 2015, 5, 092503. 10.1063/1.4921314. [DOI] [Google Scholar]
- Sharma A. K.; Pavlova S. T.; Kim J.; Finkelstein D.; Hawco N. J.; Rath N. P.; Kim J.; Mirica L. M. Bifunctional Compounds for Controlling Metal-Mediated Aggregation of the Aβ42Peptide. J. Am. Chem. Soc. 2012, 134, 6625–6636. 10.1021/ja210588m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma A. K.; Kim J.; Prior J. T.; Hawco N. J.; Rath N. P.; Kim J.; Mirica L. M. Small Bifunctional Chelators That Do Not Disaggregate Amyloid β Fibrils Exhibit Reduced Cellular Toxicity. Inorg. Chem. 2014, 53, 11367–11376. 10.1021/ic500926c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rana M.; Cho H.-J.; Roy T. K.; Mirica L. M.; Sharma A. K. Azo-dyes Based Small Bifunctional Molecules for Metal Chelation and Controlling Amyloid Formation. Inorganica Chim Acta 2018, 471, 419–429. 10.1016/j.ica.2017.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang D.; Zhang L.; Grant G. P. G.; Dudzik C. G.; Chen S.; Patel S.; Hao Y.; Millhauser G. L.; Zhou F. The Elevated Copper Binding Strength of Amyloid-β Aggregates Allows the Sequestration of Copper from Albumin: A Pathway to Accumulation of Copper in Senile Plaques. Biochemistry 2013, 52, 547–556. 10.1021/bi301053h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigoni F.; Prosdocimi T.; Mollica L.; De Gioia L.; Zampella G.; Bertini L. Copper reduction and dioxygen activation in Cu-amyloid beta peptide complexes: insight from molecular modelling. Metallomics 2018, 10, 1618–1630. 10.1039/c8mt00216a. [DOI] [PubMed] [Google Scholar]
- Gomes L. M. F.; Mahammed A.; Prosser K. E.; Smith J. R.; Silverman M. A.; Walsby C. J.; Gross Z.; Storr T. A catalytic antioxidant for limiting amyloid-beta peptide aggregation and reactive oxygen species generation. Chem. Sci. 2019, 10, 1634–1643. 10.1039/c8sc04660c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Z.; Yu D.; Du X.; Scott P.; Ren J.; Qu X. Self-triggered click reaction in an Alzheimer’s disease model:in situbifunctional drug synthesis catalyzed by neurotoxic copper accumulated in amyloid-β plaques. Chem. Sci. 2019, 10, 10343–10350. 10.1039/c9sc04387j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arredondo M.; Núñez M. T. Iron and copper metabolism. Mol. Aspects Med. 2005, 26, 313–327. 10.1016/j.mam.2005.07.010. [DOI] [PubMed] [Google Scholar]
- Guilloreau L.; Combalbert S.; Sournia-Saquet A.; Mazarguil H.; Faller P. Redox Chemistry of Copper-Amyloid-β: The Generation of Hydroxyl Radical in the Presence of Ascorbate is Linked to Redox-Potentials and Aggregation State. ChemBioChem 2007, 8, 1317–1325. 10.1002/cbic.200700111. [DOI] [PubMed] [Google Scholar]
- Geng J.; Zhao C.; Ren J.; Qu X. Alzheimer’s disease amyloid beta converting left-handed Z-DNA back to right-handed B-form. Chem. Commun. 2010, 46, 7187–7189. 10.1039/c0cc02049d. [DOI] [PubMed] [Google Scholar]
- Klein W. L. Abeta Toxicity in Alzheimer’s Disease: Globular Oligomers (ADDLs) as New Vaccine and Drug Targets. Neurochem. Int. 2002, 41, 345–352. 10.1016/s0197-0186(02)00050-5. [DOI] [PubMed] [Google Scholar]
- Xu S.-Y.; Wu Y.-M.; Ji Z.; Gao X.-Y.; Pan S.-Y. A Modified Technique for Culturing Primary Fetal Rat Cortical Neurons. J. Biomed. Biotechnol. 2012, 2012, 803930. 10.1155/2012/803930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaudoin G. M. J.; Lee S.-H.; Singh D.; Yuan Y.; Ng Y.-G.; Reichardt L. F.; Arikkath J. Culturing Pyramidal Neurons From the Early Postnatal Mouse Hippocampus and Cortex. Nat. Protoc. 2012, 7, 1741–1754. 10.1038/nprot.2012.099. [DOI] [PubMed] [Google Scholar]
- Fortuna A.; Alves G.; Soares-da-Silva P.; Falcão A. Pharmacokinetics, brain distribution and plasma protein binding of carbamazepine and nine derivatives: new set of data for predictive in silico ADME models. Epilepsy Res 2013, 107, 37–50. 10.1016/j.eplepsyres.2013.08.013. [DOI] [PubMed] [Google Scholar]
- Singh A. K.; Gothwal A.; Rani S.; Rana M.; Sharma A. K.; Yadav A. K.; Gupta U. Dendrimer Donepezil Conjugates for Improved Brain Delivery and Better in Vivo Pharmacokinetics. ACS Omega 2019, 4, 4519–4529. 10.1021/acsomega.8b03445. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.