

Abstract
Purpose
This study aims to develop an impedance-based drug screening platform that will help identify drugs that can enhance the vascular barrier function by stabilizing vascular endothelial cell junctions.
Methods
Changes in permeability of cultured human retinal microvascular endothelial cells (HRMECs) monolayer were monitored in real-time with the xCELLigence RTCA system. Using this platform, we performed a primary screen of 2100 known drugs and confirmed hits using two additional secondary permeability assays: the transwell permeability assay and the XPerT assay. The cellular and molecular mechanisms of action and in vivo therapeutic efficacy were also assessed.
Results
Eleven compounds blocked interleukin 1 beta (IL-1β) induced hyperpermeability in the primary screen. Two of 11 compounds, apigenin and ethaverine hydrochloride, reproducibly blocked multiple cytokines induced hyperpermeability. In addition to HRMEC monolayers, the two compounds stabilized three other types of primary vascular endothelial cell monolayers. Preliminary mechanistic studies suggest that the two compounds stabilize the endothelium by blocking ADP-ribosylation factor 6 (ARF6) activation, which results in enhanced VE-cadherin membrane localization. The two compounds showed in vivo efficacy in an animal model of retinal permeability.
Conclusions
We developed an impedance-based cellular phenotypic drug screening platform that can identify drugs that enhance vascular barrier function. We found apigenin and ethaverine hydrochloride stabilize endothelial cell junctions and enhance the vascular barrier by blocking ARF6 activation and increasing VE-cadherin membrane localization.
Translational Relevance
The drugs identified from the phenotypic screen would have potential therapeutic efficacy in retinal vascular diseases regardless of the underlying mechanisms that promote vascular leak.
Keywords: drug screen, impedance, vascular barrier, VE-cadherin, ARF6
Introduction
The retina is one of the most metabolically active tissues, resulting in high demand for oxygen and nutrients.1 There are two vascular layers that provide nutrients and oxygen to the retina: retinal blood vessels and choroidal blood vessels. One-third to one-half of the retina relies on choroidal blood vessels for its supply of oxygen and nutrients, thus reducing the need for high densities of blood vessels in the anterior segment of the retina. Because photoreceptors are located in the posterior retina, a high density of retinal blood vessels can interfere with light transmission. The blood-retinal barrier (BRB) is a strictly regulated permeable system.2 The BRB consists of the inner BRB (tight junction between retinal capillary endothelial cell) and the outer BRB (tight junction between retinal epithelial cell). Any pathological alteration can break the equilibrium of the BRB and cause angiogenesis or vascular leakage that interferes with the recycling of metabolic waste. This can cause damage to photoreceptors and other retinal neuronal cells, eventually impairing the visual system.
Diabetic retinopathy (DR) and wet age-related macular degeneration (AMD) are two retinal diseases that are caused by vascular leakage and angiogenesis.3 Breakdown of inner BRB is a common pathologic feature in patients with diabetes and experimental animal models. DR is a common microvascular complication of diabetes mellitus and is one of the fastest growing causes of blindness and visual impairment in the working age populations, whereas AMD mainly affects aged populations. AMD is characterized by visual disability, including blindness in elderly people.4 There are about 14 million people affected by AMD worldwide.5 There are two primary types of AMD: wet and dry. Wet AMD is characterized by choroidal neovascularization (CNV), in which immature and fragile blood vessels cross the retinal pigment epithelium (RPE), breakdown the outer BRB, and enter the retina. Breakdown of the BRB causes macular edema through multiples ways and results in damage of retinal neuron cells, which eventually impair vision. Dry AMD is the most common form of AMD (85%–90%) and is caused by small yellow deposits (drusen) under the macula. In contrast to wet AMD, there is no neovascularization or edema in dry AMD. It has been commonly accepted that AMD can be caused by multiple pathogenic factors.6,7
Blocking the vascular endothelial growth factor (VEGF) signaling pathway is the current standard pharmacological therapy for both DR and wet AMD.3 However, anti-VEGF treatments require frequent and long-term pharmacologic administration to maintain vision gains, and although many patients respond well to anti-VEGF therapy, some have only a moderate or poor response and patients may lose their initial vision gains over time.8–10 Therefore, the development of new drugs for DR and AMD is vital.
Herein, we developed a label-free, phenotypic drug screening platform to discover drugs that could potentially be used to treat DR and wet AMD by enhancing the vascular barrier function. This is a high throughput compatible phenotypic screen that monitors changes in the endothelial cell monolayer barrier function. We utilized the xCELLigence RTCA system (Supplementary Fig. S1), a method for label-free, real-time monitoring of electrical impedance of endothelial cell monolayers, which can be used to assess changes in permeability over time (Supplementary Fig. 2 shows an overview of the screen). Because we did not screen against a single target, but rather screened for endothelial cell monolayer barrier function enhancement, the drugs we identified in this screen should be useful for the treatment of DR and wet AMD regardless of the pathogenic factors promoting the disease. To facilitate the transition of the identified hits to clinical use, we utilized a repurposing drug strategy and chose to screen a known drug compound library.
Materials and Methods
Cell Culture
Human retinal microvascular endothelial cells (HRMECs), human dermal microvascular endothelial cells (HMVECs-d), and human umbilical vein endothelial cells (HUVECs) were purchased from Lonza (Portsmouth, NH, USA) and cultured in the EBM-2 media that supplemented with reagents from EGM-2MV kit (for HRMEC and HMVEC-d) or EGM-2 SingleQuots kit (for HUVEC). Human synovial microvascular endothelial cells (HSMECs) were purchased from Cell Systems (Kirkland, WA, USA) and cultured in the EBM-2 media supplemented with EGM-2 SingleQuots kit. All the primary endothelial cells were counted as passage 0 upon thawing of the original vial from the vendor. After three expansions (passage 3), cells were frozen at 1.5 × 106 cell/mL/vial. The frozen cells were thawed and expanded one more time for all screening experiments.
Drug Treatment
The compound library (Spectrum collection) was purchased from MicroSource Discovery System (Gaylordsville, CT, USA) at 2 mM in 100% DMSO. At the time of treatment, compounds were diluted to 1 mM with complete culture media, then 1 µl of the appropriate diluted compound was added to each well for a final concentration of 10 µM in 0.5% DMSO (vehicle) in complete cell culture media (100 µl). The hit compounds from the primary screen were ordered from MicroSource Discovery System individually and dissolved in 100% DMSO for a stock concentration of 20 mM for the following dose response, transwell permeability assay, XPerT assay, and in vivo animal experiments. For the in vitro dose response and other cellular assays, compounds were serially diluted with 100% DMSO and then diluted 1:200 into cell culture media to get the final treatment concentration with 0.5% DMSO.
Impedance-Based Primary Screen
HRMEC were cultured to 80% confluency in EBM-2 media that was supplemented with EGM-2MV kit (Lonza, USA), treated with trypsin, and then collected for permeability assay. The 96-well E-plate (ACEA Biosciences, Inc., San Diego, CA, USA) was coated with human fibronectin (10 µg/mL, 100 µl/well, 37°C for 30 minutes) before seeding the HRMEC. The HRMEC single cell suspension was diluted to 1 × 105 cells/mL, then seeded 100 µl/well (10,000 cell/well) onto a 96-well E-plate. The 96-well E-plate was loaded onto xCELLigence RTCA system (ACEA Biosciences, Inc.) inside a tissue culture incubator (37°C, 5% CO2). Cell growth and monolayer formation were monitored in real-time. The culture media was replaced with fresh complete media with interleukin 1 beta (IL-1β; 20 ng/mL) plus compound (10 µM) or vehicle control (DMSO = 0.5%) only once the cells formed a monolayer (determined by impedance plateau). Changes in impedance were monitored for another 12 hours. The impedance was converted to cell index by the xCELLigence RTCA software, and so all the graph's Y-axis were labeled as cell index (normalized to the time point before treatment, and so called “relative cell index”) instead of impedance.
Transwell Permeability Assay
Ninety-six-well transwell insert (Corning, NY, USA) was coated with human fibronectin (10 µg/mL, 100 µl/well, at 4°C overnight) and equilibrated with complete media (50 µl/well in insert, 100 µl/well in bottom receiving plate, 37°C tissue culture incubator overnight) before seeding HRMEC. The HRMEC single cell suspension was diluted to 1 × 105 cells/mL and then 100 µl/well (10,000 cell/well) of cells was seeded onto the top insert. The bottom receiving plate was filled with complete media (100 µl/well). Cells were cultured for 2 days and media was changed daily before the induction of permeability and compound treatment. Both top insert and bottom wells in the receiving plate were changed to fresh complete media with IL-1β (20 ng/mL), or IL-6 (20 ng/mL) + soluble IL-6 receptor (sIL-6r; 40 ng/mL) plus compound (1:200 dilution to get the final treatment concentration) or vehicle control (0.5% DMSO in media). No induction control wells had vehicle control only. FITC-dextran (70 KD; Sigma, USA) stock solution was 100 mg/mL, which was diluted to 50 mg/mL with PBS before adding 1 µl/well to the top insert at 8 hours post-treatment. After 2 hours of incubation, 100 µl of media was removed from each bottom well of the receiving plate and transferred to a 96-well black wall plate to measure fluorescence intensity using a plate reader (Ex 485 nm, Em 528 nm).
XPerT Assay
Clear bottom black 96-well plates (Corning) were coated with homemade biotinylated gelatin (0.25 mg/mL, 50 µl/well) at 4°C for overnight and then washed twice with PBS before being seeded with HRMEC (10,000 cell/well). The cells were cultured for 2 days after seeding and media was changed daily. The culture media was changed to fresh complete media with IL-1β (20 ng/mL) plus compound (1:200 dilution into culture media to the final treatment concentration in 0.5% DMSO) or 0.5% DMSO only once the cells formed a monolayer. The uninduced control wells had media only with 0.5% DMSO. FITC-avidin (5 mg/mL in 50% PBS + 50% glycerol; Molecular Probes, Eugene, OR, USA) was added to the media (5 µl/well, final concentration was 0.25 mg/mL) at 8 hours post IL-1β treatment and incubated for 10 minutes at 37°C. The plate was then washed three times with 100 µl PBS and fluorescent intensity was read using a plate reader (Ex 490 nm, Em 520 nm).
Immunocytochemistry
Twenty-four well chambered cover glass was coated with human fibronectin (10 µg/mL, 100 µl/well at 4°C for overnight) before seeding HRMEC (30,000 cell/well). Cells were treated with IL-1β (20 ng/mL) or BSA control for 30 minutes at 24 hours post-seeding and then compound (1:200 dilution into culture media to get the final treatment concentration) or 0.5% DMSO only was added to the corresponding treatment wells and the cells were incubated for an additional 2 hours at 37°C. Cells were washed once with PBS and were fixed with 4% paraformaldehyde for 20 minutes at room temperature. Cells were blotted with VE-cadherin antibody (Cell Signaling Technology, Danvers, MA, USA) overnight at 4°C after blocking with 1% BSA, thoroughly washed, and then blotted with Alexa 488 labeled anti-Rabbit secondary antibody 2 hours at room temperature. The nucleus was stained with 4,6-diamidino-2-phenylindole (DAPI). Confocal microscopy was used to take images.
VE-Cadherin Membrane Localization
HRMECs were treated with the compound or DMSO only in EBM-2 + 1% fetal bovine serum (FBS) media for 3 hours at 37°C after the cells formed a monolayer on a 60-mm dish. The cells were then treated with IL-1β (20 ng/mL) and primaquine (0.6 mM) in EBM-2 + 1% FBS for 2 hours at 37°C. The media was aspirated and the dish was immediately placed on ice. Cells were washed with ice-cold PBS and labeled with EZ-link Sulfo-NHS-SS-Biotin (0.5 mg/mL; Thermo Fisher Scientific) for 2 hours at 4°C in the dark box. The unbound free biotin was washed off with PBS, and the cells were lysed in NP-40 buffer supplemented with protease inhibitor. The cell lysate was clarified by centrifugation. Biotin labeled protein was pulled-down from cleared lysate using streptavidin agarose resin (Thermo Fisher Scientific). Protein was eluted from resin using 2X sample buffer supplemented with 5% β-mercaptoethanol and then boiled for 5 minutes. Western blotting was performed using the anti-VE-cadherin antibody.
ADP-Robosylation Factor 6-GTP Pull-Down Assay
HRMECs were cultured in complete media in 6-well plates until 80% confluency was reached. Cells were treated with compound or 0.5% DMSO only in EBM-2 media with 1% FBS for 4 hours at 37°C. Treated cells were then stimulated with IL-1β (20 ng/mL) or BSA control for 10 minutes to activate ADP-ribosylation factor 6 (ARF6). The plate was placed on ice immediately after aspirating the treatment media and washed with ice cold PBS for one time. Cells were lysed on ice for 15 minutes with ice cold lysis buffer from ARF6-GTP pull-down kit (Cell Biolabs, San Diego, CA, USA) supplemented with protease inhibitor. The lysates were collected into ice cold 1.5 mL tubes and centrifuged at 13,000 rpm for 10 minutes in 4°C. Supernatants were transferred to new ice cold 1.5 mL tubes. Pull-down was performed according to the manufacturer's directions (all the experimental procedures were performed in the 4°C cold room).
Quantitative Real-Time PCR
HRMECs were cultured in complete media in 10 cm dish until 80% confluency was reached. Cells were treated with compound or 0.5% DMSO in EBM-2 media with 1% FBS for 4 hours in 37°C thermal conductivity (TC) incubator. The IL-1β (final concentration was 20 ng/mL) or BSA control were added to the dishes and incubated for 10 minutes in 37°C TC incubator at 4 hours post compound treatment. Following treatment, the media was quickly aspirated from the dish and cells were washed once with PBS before lysing them for total RNA isolation. Total RNA was extracted from the HRMECs using Trizol reagent and converted into cDNA using TransStart All-in-One First-Strand cDNA Synthesis SuperMix for qPCR (One-Step gDNA removal) (Transgen, China). Quantitative real-time polymerase chain reaction (qRT-PCR) was performed on 7500 Fast Real-Time PCR system using TransStart Tip Green qPCR SuperMix (+DyeI/+DyeII; TransGen Biotech Co., Beijing, China). Primers used for PCR: VE-cadherin-F (5’-TTGGAACCAGATGCACATTGAT-3’) and VE-cadherin-R (5’-TCTTGCGACTCACGCTTGAC-3’), GAPDH-F (5’-AGGTCGGAGTCAACGGATTT-3’) and GAPDH-R (5’-TGACGGTGCCATGGAATTTG-3’).
Retinal Vascular Permeability Assay
Retinal vascular permeability assay was performed using 6 to 8 weeks old C57BL/6 mice as described previously.11–13 All the animals were provided by and experimental protocols were reviewed and approved by The Institution of Animal Medical Research of Sichuan Provincial People's Hospital in accordance with the ARVO STATEMENT for the use of animals in ophthalmic and vision research. For intravitreal injection of IL-1β (or BSA control; 2 µl of 100 ng/µl) and compound (1 µl of 200 ng/µl) or DMSO control (1 µl of 100% DMSO), the mice were sedated by 4% chloral hydrate (8 µl/g, intraperitoneal), then carefully injected with the mixture of IL-1β (or BSA control) with compound (or DMSO control) from the side of the eye into the vitreous without touching the retina. Evans blue (45 mg/kg; Sigma Aldrich, USA) was injected via the tail vein at 2 hours post intravitreal injection. The mice were sacrificed by CO2 10 hours after receiving Evans blue. Mouse eyes and retina were collected for Evans blue quantification. The Evans blue that penetrated into retina was extracted using formamide (Sigma Aldrich) for 20 hours at 70°C. The absorbance of extracts was measured at 620 nm and 740 nm after clarification by centrifugation (13,000 rpm, 5 minutes). Data are presented as mean ± SEM.
Statistical Analysis
All the data were analyzed using GraphPad Prism 6.0. One-way analysis of variance (ANOVA) with Tukey's multiple comparisons test was performed to assess statistical significance. When a P value is <0.05 it was counted as statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001.
Results
Development of Impedance-Based Primary Screen
The xCELLigence RTCA system allows us to monitor cell growth and monolayer formation on 96-well E-plates (the bottom is coated with microelectrode) in real-time and label free (Supplementary Fig. S1 shows an image of the system that was used in this study). The impedance plateaued once the cells formed a monolayer on the E-plates, meaning transcellular resistance had hit a maximum. We added IL-1β to destabilize the monolayer and to induce hyperpermeability, which is measured as a reduction in impedance on the xCELLigence RTCA system and converted to cell index by its software. By optimizing cell's seeding number, culture time, and IL-1β dose, we consistently decreased HRMEC monolayer barrier function. The positive control compound, Forskolin, blocks IL-1β induced hyperpermeability (Fig. 1A).
Figure 1.
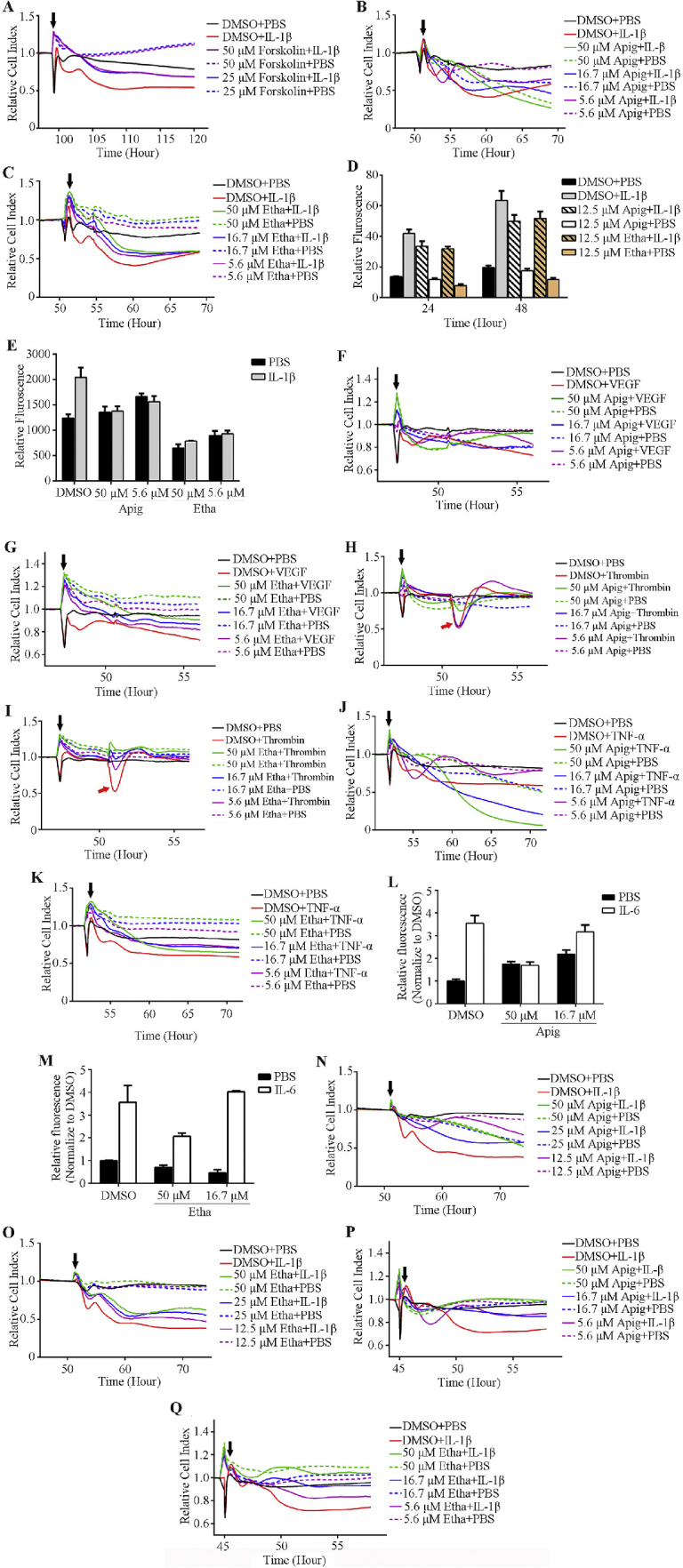
Identification of compounds that enhance endothelial cell monolayer barrier function and block cytokines induced hyperpermeability. (A–C) Positive control compound Forskolin (A) and two hits, Apigenin (Apig) and Ethaverine Hydrochloride (Etha) (B, C), block IL-1β induced hyperpermeability in an impedance-base permeability assay. (D, E) Apigenin and ethaverine hydrochloride rescue IL-1β induced permeability in two secondary permeability assays: transwell permeability assay (D) and XPerT (E). (F–K) Apigenin and ethaverine hydrochloride block the hyperpermeability induced by multiple cytokines in the impedance assay. (L, M) Compounds enhance HSMEC monolayer barrier function and block IL-6 induced permeability in transwell permeability assay. (N–Q) Compounds enhance HMVEC-d (N and O) and HUVEC (P and Q) monolayer barrier function in impedance-based permeability assay. Black arrowhead indicates the time of starting compound and/or cytokine treatment in the impedance-based permeability assay.
Execution of Primary Screen
To further evaluate the impedance-based screen platform, we screened a collection of 2100 small molecules (Spectrum collection, MicroSource Discovery System, USA). These 2100 compounds include known drugs or compounds with known biological activity and greatly facilitates the translation of hits to clinical use. Repurposing of existing drugs (known as drug repurposing, drug repositioning, or drug rediscovery) has many advantages, and there are many successful examples.14 We chose to screen this library consisting of many known drugs to validate our platform in the hope that this would accelerate clinical usage of identified drugs. Eleven compounds blocked IL-1β induced hyperpermeability in our screen (Table). We re-ordered these 11 hits individually and tested dose responses in the primary screen. Two out of 11 hits (apigenin and ethaverine hydrochloride) consistently rescued IL-1β induced hyperpermeability (Figs. 1B, 1C).
Table.
The Name and Structure of 11 Hits from the Primary Screen
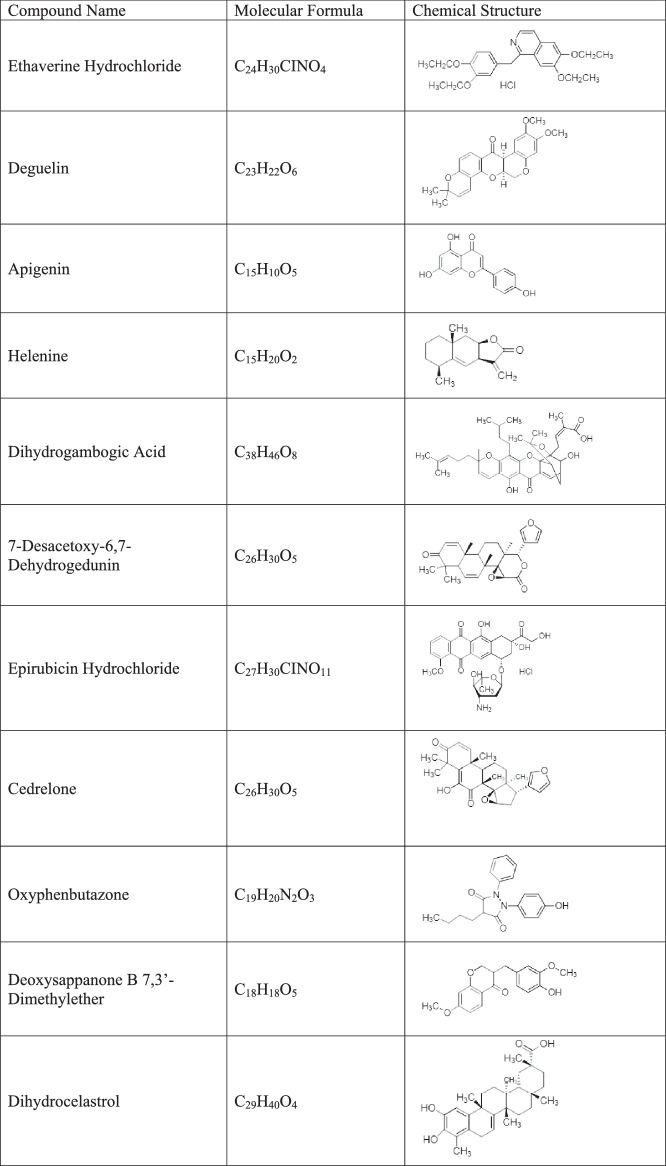
|
Validating Hits in Secondary Screen
We validated apigenin and ethaverine hydrochloride in two different secondary screen assays: the transwell permeability assay and the XPerT assay. Impedance-based screens monitor permeability changes through the change in electrical current of the microelectrode in a 96-well E-plate on which the HRMEC monolayer formed. The transwell permeability assay is a more direct measure of permeability changes at a specific time point because it monitors the permeability tracer that passes through the cell monolayer and enters the bottom chamber in the receiving plate. We were able to generate a very high signal to background ratio in transwell permeability assay by optimizing cell seeding numbers, culture time, and the size of FITC-dextran. The XPerT assay was originally developed by Dubrovskyi et al. and later modified by Rokhzan et al.15,16 In this assay, the paracellular permeability changes can be detected either by reading the FITC fluorescent intensity or by imaging FITC-avidin that crosses paracellular gaps and binds to the pre-coated biotin on the plate. Both apigenin and ethaverine hydrochloride inhibited IL-1β induced hyperpermeability in these two secondary permeability screening assays (Figs. 1D, 1E).
Apigenin and Ethaverine Hydrochloride Rescue Multiple Cytokines Induced Hyperpermeability
Currently, blocking VEGF signaling is the gold standard treatment for retinal vascular diseases that are caused by leaking blood vessels or angiogenesis, including DR and wet AMD. However, it has been commonly accepted that multiple factors/signaling pathways can cause hyperpermeability, thus likely accounting for the existence of nonresponders to anti-VEGF treatment. The purpose of our study is to develop a screening platform for the discovery of drugs that stabilize vascular endothelium and inhibit leaky blood vessels or angiogenesis regardless of the initiating factors. Therefore, we tested apigenin and ethaverine hydrochloride for efficacy in reducing the hyperpermeability induced by other cytokines, including thrombin (1 U/mL), VEGF (20 ng/mL), TNFα (0.2 nΜ), and IL-6 + sIL-6r (thrombin, VEGF, and TNFα induce hyperpermeability were performed in the primary screen. IL-6 + sIL-6r induced permeability was performed in transwell assay.). Thrombin induced hyperpermeability lasts a very short time, which is indicated by the red arrowhead in Figures 1H and 1I. Both apigenin and ethaverine hydrochloride were effective in reducing the hyperpermeability induced by these four cytokines (Figs. 1F–M).
Apigenin and Ethaverine Hydrochloride Enhance Barrier Function of Monolayers of Endothelial Cells Isolated from Different Tissues (organs)
Pathogenic vascular leak can happen anywhere in the body, not just the eyes. We wanted to identify drugs through our screening platform that not only could be used for retinal vascular diseases but also diseases caused or exacerbated by leakage in other vascular beds. Vascular endothelial cells isolated from three other vascular beds HMVEC-d, HUVEC, and HSMEC were cultured on 96-well E-plates (HMVEC-d and HUVEC) or transwell inserts (HSMEC) and hyperpermeability was induced with IL-1β or IL-6 + sIL-6r once a monolayer was formed. Both apigenin and ethaverine hydrochloride blocked IL-1β- or IL-6-induced hyperpermeability in these three types of endothelial cell monolayers (Figs. 1L–Q).
Apigenin and Ethaverine Hydrochloride Enhance VE-Cadherin Membrane Localization by Blocking ARF6 Activation
We next explored the mechanism by which these two compounds enhance vascular barrier function. VE-cadherin is a cell surface protein that is a key component of endothelial cell adherens junctions. Published studies also suggest that VE-cadherin can promote the formation of tight junctions by upregulating claudin-5 expression.17
We checked VE-cadherin mRNA levels in IL-1β- and compound-treated HRMEC by RT-PCR. Neither IL-1β nor the compound affected VE-cadherin transcription (Fig. 2A). However, Western blots of membrane-associated proteins (biotinylated) showed that IL-1β significantly reduced surface VE-cadherin without affecting the total VE-cadherin protein level (Figs. 2B, 2C). This suggested that IL-1β induced VE-cadherin internalization and resulted in hyperpermeability. Both apigenin and ethaverine hydrochloride blocked IL-1β-induced VE-cadherin internalization and enhanced VE-cadherin membrane localization (Figs. 2B–D).
Figure 2.

Compounds stabilize VE-cadherin membrane localization by blocking IL-1β-induced ARF6 activation. (A) RT-PCR analysis of VE-cadherin mRNA levels. (B, C) Western blot analysis of membrane-bound VE-cadherin. (D) Immunofluorescence images of HRMEC. Scale bar = 100 µm. (E, F) Activated ARF6 (ARF6-GTP) pull-down assays of HRMEC following treatment with compounds and/or IL-1β.
Our recent studies suggested that ARF6 might be a convergence point in regulating vascular barrier function.18,19 ARF6 is a small GTPase, cycling between GDP-bound inactive and GTP-bound active forms. This cycling procedure regulates membrane protein traffic, including VE-cadherin. We performed active ARF6 pull-downs from IL-1β and compound treated HRMECs. Apigenin and ethaverine hydrochloride blocked IL-1β-induced ARF6 activation (Figs. 2E, 2F), suggesting that these compounds act by inhibiting ARF6.
Apigenin and Ethaverine Hydrochloride Enhance Retinal Vascular Barrier Function In Vivo
Next, we investigated whether these two compounds could enhance retinal vascular barrier function in vivo. We performed a retinal permeability assay by intravitreal injection of IL-1β with or without the compounds. Evans blue was injected via the tail vein as a permeability tracer. IL-1β alone increased the amount of Evans blue that penetrated into the surrounding tissues (Fig. 3). Both apigenin and ethaverine hydrochloride significantly reduced the extravasation of Evans blue into surrounding retinal tissues. These results suggest that both compounds enhance blood vessel barrier function and block IL-1β-induced hyperpermeability in vivo.
Figure 3.
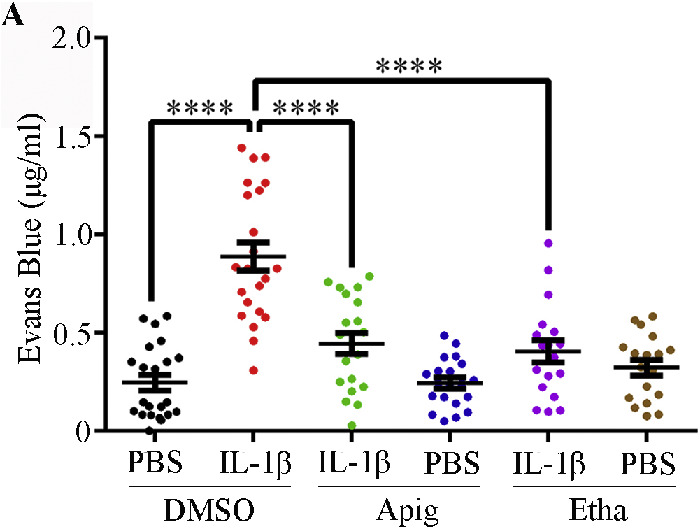
Compounds enhance retinal vascular barrier function in vivo. Retinal vascular permeability assays were performed on 6 to 8 week old C57BL/6 mice. IL-1β (or BSA control) and compound (or DMSO control) were administered by intravitreal injection, and Evans blue was injected via tail vein at 2 hours post intravitreal injection. The mice were euthanized by CO2 inhalation 10 hours after receiving Evans blue. Mouse eyes and retinas were collected for Evans blue quantification. Data are presented as mean ± SEM.
Discussion
Retinal blood vessels are a good model to study vascular systems because the structures can be viewed directly and noninvasively using a digital fundus camera.20 Retinal blood vessel structure changes can be associated with vascular diseases of other tissues or organs, such as cardiovascular disease and hypertension.21 Therefore, the retinal blood vessel system can suggest drug therapeutic efficacy for vascular diseases of other tissues or organs that cannot be viewed directly.
The traditional label-based cellular assay for studying drug effects and cellular functions requires the use of labeled either drug or target or staining the whole cell with a fluorescent tracer. The advantages of xCELLigence RTCA system is that it is label free and can monitor multiple physiological factors of the cell in real-time. This phenotypic assay allows us to study complex biology process in a physiologic relevant environment.22 Since the advent of this label free electrical biosensor system in 1993, it has been widely used in many different types of research and has been compared to the transwell permeability assay.23 Like the study BRB of the human induced pluripotent stem cells (hiPSC)-RPE from patients with AMD and sibling control,24 and the role of S1P signaling pathway in regulation of blood-brain barrier (BBB).25 In the Wiltshire et al. paper, they studied S1P's effect on the barrier function of human brain microvascular endothelia cells (hCMVECs; mimic BBB) using the xCELLigene RTCA system. They showed that S1P temporarily affect the BBB and quickly rebound back to normal and enhanced BBB. They also found S1P promoted release of proinflammatory cytokines like IL-6 and IL-8.25 However, our study suggested that IL-6 + sIL-6r decreased the HSMEC monolayer barrier function. This inconsistency might be due to barrier difference (BBB versus HSMEC) or lack sIL-6r in their assay since sIL-6r is necessary for IL-6 signaling, in addition to our IL-6 + sIL-6r induced hyperpermeability was performed in transwell permeability assay.
Target-based drug screens identify drugs that target a specific signaling pathway associated with a disease. But some diseases, such as DR and wet AMD, can be caused by multiple pathogenic factors. In these cases, phenotypic screens can be used to identify drugs that give the desired effect in cultured cells, tissues/organs, or animals although the mechanism underlying a drug's effect may take time to determine.26,27
We chose HRMEC as the primary model of our screen and changes in endothelium barrier function as our cellular phenotypic readout. We reasoned that this screening system could help us identify a drug that blocks the effect of multiple factors that induce vascular leakage in the retina. Our data show that apigenin and ethaverine hydrochloride each blocked IL-1β-, ΙL-6-, thrombin-, VEGF-, and TNFα-induced hyperpermeability, which verified our hypothesis that a single compound can block leakage induced by multiple factors and suggested that these two compounds might be potential drug candidates for DR and wet AMD.
Pathological capillary leakage can take place in different diseases and different regions of our body.28 We designed this cellular phenotypic screen platform with the hope that it would also identify compounds that would be useful for the treatment of diseases caused or exacerbated by leak in other vascular beds, so we tested permeability of endothelial monolayers from three other type of tissues: umbilical cord (HUVEC), dermis (HMVEC-d), and synovial tissue (HSMEC). Apigenin and ethaverine hydrochloride successfully blocked IL-1β or IL-6 induced hyperpermeability in vascular endothelial cell monolayers from all three tissues. This result suggests that our screening platform has broad applications.
Apigenin is a flavonoid natural product from plants. It has therapeutic effects in many diseases, including neuroprotective, anticancer, and anti-inflammatory effects.29–31 Apigenin has been shown to inhibit angiogenesis by suppressing VEGF, erythropoietin expression, and blocking other signaling pathways.32 Our study suggests that apigenin enhances vascular barrier function by blocking ARF6 activation, which leads to increased VE-cadherin membrane localization. Earlier studies have shown that apigenin inhibits histamine release and this might be another potential mechanism by which apigenin enhances the vascular barrier.33,34 In addition, apigenin has been shown to inhibit choroidal angiogenesis and retinal vascular leakage in laser-induced choroidal neovascularization animal models.35
Vascular barrier integrity can be disrupted by multiple pathogenic factors in the body.36 Shear stress, which is caused by constant blood flow, is one of the factors that can affect vascular barrier integrity.37,38 Physiologic shear stress helps maintain normal vascular barrier function. Either excessive shear stress or static flow, which is caused by impaired circulation, can compromise vascular integrity. There is clinical evidence suggesting that retinal circulation is altered in both patients with DR and AMD.39,40 The elevated expression of proinflammatory cytokines caused by abnormal shear stress might be one of the reasons for the existence of anti-VEGF treatment nonresponders in DR and wet AMD.41 Our previous studies have indicated that ARF6 is a convergence point in the signaling pathways of these proinflammatory cytokines.18,19 We have shown that apigenin and ethaverine hydrochloride blocked ARF6 activation. All of these results suggest that our screening platform was effective in identifying drugs that could be repurposed for the treatment of DR and wet AMD.
In summary, we have developed a cellular drug screening platform designed to identify known drugs that can enhance vascular barrier function by stabilizing endothelium. We found two compounds, apigenin and ethaverine hydrochloride, that blocked IL-1β-induced hyperpermeability both in vitro and in vivo. The two compounds blocked cytokines induced hyperpermeability of monolayers of endothelial cells isolated from four different tissues. Cellular studies suggest that these two compounds stabilize adherens junctions by blocking ARF6 activity, which promotes VE-cadherin membrane localization. These results indicate that this drug screening platform can be used to identify drugs that can prevent vascular leakage in multiple tissues regardless of the underlying factors that contribute to the hyperpermeability.
Supplementary Material
Acknowledgments
The authors thank Shannon Jay Odelberg for the helpful discussions and suggestions.
Supported by the National Natural Science Foundation of China (81371053).
Disclosure: W. Jiang, None; H. Chen, None; Z. Tai, None; T. Li, None; L. Luo, None; Z. Tong, University of Utah (F), Navigen, Inc. (F); W. Zhu, None
References
- 1. Eshaq RS, Wright WS, Harris NR. Oxygen delivery, consumption, and conversion to reactive oxygen species in experimental models of diabetic retinopathy. Redox Biology. 2014; 2: 661–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cunha-Vaz J, Bernardes R, Lobo C. Blood-retinal barrier. Eur J Ophthalmol. 2011; 21: S3–S9. [DOI] [PubMed] [Google Scholar]
- 3. Das UN. Diabetic macular edema, retinopathy and age-related macular degeneration as inflammatory conditions. Arch Med Sci. 2016; 12: 1142–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tomany SC, Wang HJ, van Leeuwen R, et al.. Risk factors for incident age-related macular degeneration - pooled findings from 3 continents. Ophthalmology. 2004; 111: 1280–1287. [DOI] [PubMed] [Google Scholar]
- 5. Chappelow AV, Kaiser PK. Neovascular age-related macular degeneration - potential therapies. Drugs. 2008; 68: 1029–1036. [DOI] [PubMed] [Google Scholar]
- 6. Wong T, Chakravarthy U, Klein R, et al.. The natural history and prognosis of neovascular age-related macular degeneration. Ophthalmology. 2008; 115: 116–126. [DOI] [PubMed] [Google Scholar]
- 7. Gallenga CE, Parmeggiani F, Costagliola C, Sebastiani A, Gallenga PE. Inflammaging: should this term be suitable for age related macular degeneration too? Inflamm Res. 2014; 63: 105–107. [DOI] [PubMed] [Google Scholar]
- 8. Ashraf M, Souka A, Adelman R. Predicting outcomes to anti-vascular endothelial growth factor (VEGF) therapy in diabetic macular oedema: a review of the literature. Br J Ophthalmol. 2016; 100: 1596–1604. [DOI] [PubMed] [Google Scholar]
- 9. Duh EJ, Sun JK, Stitt AW. Diabetic retinopathy: current understanding, mechanisms, and treatment strategies. JCI Insight. 2017;2:pii: 93751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Simo R, Sundstrom JM, Antonetti DA. Ocular anti-VEGF therapy for diabetic retinopathy: the role of VEGF in the pathogenesis of diabetic retinopathy. Diabetes Care. 2014; 37: 893–899. [DOI] [PubMed] [Google Scholar]
- 11. Scheppke L, Aguilar E, Gariano RF, et al.. Retinal vascular permeability suppression by topical application of a novel VEGFR2/Src kinase inhibitor in mice and rabbits. J Clin Investig. 2008; 118: 2337–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Maharjan S, Lee S, Agrawal V, et al.. Sac-0601 prevents retinal vascular leakage in a mouse model of diabetic retinopathy (vol 657, pg 35, 2011). Eur J Pharmacol. 2011; 659: 302. [DOI] [PubMed] [Google Scholar]
- 13. Kim JH, Kim JH, Yu YS, Cho CS, Kim KW. Blockade of angiotensin II attenuates VEGF-mediated blood-retinal barrier breakdown in diabetic retinopathy. J Cereb Blood Flow Metab. 2009; 29: 621–628. [DOI] [PubMed] [Google Scholar]
- 14. Simsek M, Meijer B, van Bodegraven AA, de Boer NKH, Mulder CJJ. Finding hidden treasures in old drugs: the challenges and importance of licensing generics. Drug Discov Today. 2018; 23: 17–21. [DOI] [PubMed] [Google Scholar]
- 15. Dubrovskyi O, Birukova AA, Birukov KG. Measurement of local permeability at subcellular level in cell models of agonist- and ventilator-induced lung injury. Lab Invest. 2013; 93: 254–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Rokhzan R, Ghosh CC, Schaible N, et al.. Multiplexed, high-throughput measurements of cell contraction and endothelial barrier function. Lab Invest. 2019; 99: 138–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Taddei A, Giampietro C, Conti A, et al.. Endothelial adherens junctions control tight junctions by VE-cadherin-mediated upregulation of claudin-5. Nat Cell Biol. 2008; 10: 923–934. [DOI] [PubMed] [Google Scholar]
- 18. Zhu WQ, London NR, Gibson CC, et al.. Interleukin receptor activates a MYD88-ARNO-ARF6 cascade to disrupt vascular stability. Nature. 2012; 492: 252–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Davis CT, Zhu WQ, Gibson CC, et al.. ARF6 inhibition stabilizes the vasculature and enhances survival during endotoxic shock. J Immunol. 2014; 192: 6045–6052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miri M, Amini Z, Rabbani H, Kafieh R. A comprehensive study of retinal vessel classification methods in fundus images. J Med Signals Sens. 2017; 7: 59–70. [PMC free article] [PubMed] [Google Scholar]
- 21. Fraz MM, Rudnicka AR, Owen CG, Strachan DP, Barman SA. Automated arteriole and venule recognition in retinal images using ensemble classification. Proceedings of the 2014 9th International Conference on Computer Vision, Theory and Applications (Visapp 2014), Vol 3 2014;194–202.
- 22. Hillger JM, Lieuw WL, Heitman LH, AP IJ. Label-free technology and patient cells: from early drug development to precision medicine. Drug Discov Today. 2017; 22: 1808–1815. [DOI] [PubMed] [Google Scholar]
- 23. Bischoff I, Hornburger MC, Mayer BA, Beyerle A, Wegener J, Furst R. Pitfalls in assessing microvascular endothelial barrier function: impedance-based devices versus the classic macromolecular tracer assay. Sci Rep. 2016; 6: 23671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gamal W, Borooah S, Smith S, et al.. Real-time quantitative monitoring of hiPSC-based model of macular degeneration on electric cell-substrate impedance sensing microelectrodes. Biosens Bioelectron. 2015; 71: 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Wiltshire R, Nelson V, Kho DT, Angel CE, O'Carroll SJ, Graham ES. Regulation of human cerebro-microvascular endothelial baso-lateral adhesion and barrier function by S1P through dual involvement of S1P1 and S1P2 receptors. Sci Rep. 2016; 6: 19814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Swinney DC. Phenotypic vs. target-based drug discovery for first-in-class medicines. Clin Pharmacol Ther. 2013; 93: 299–301. [DOI] [PubMed] [Google Scholar]
- 27. Eder J, Sedrani R, Wiesmann C. The discovery of first-in-class drugs: origins and evolution. Nat Rev Drug Discov. 2014; 13: 577–587. [DOI] [PubMed] [Google Scholar]
- 28. Bruni C, Frech T, Manetti M, et al.. Vascular leaking, a pivotal and early pathogenetic event in systemic sclerosis: should the door be closed? Front Immunol. 2018; 9: 2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Nabavi SF, Khan H, D'onofrio G, et al.. Apigenin as neuroprotective agent: of mice and men. Pharmacol Res. 2018; 128: 359–365. [DOI] [PubMed] [Google Scholar]
- 30. Madunic J, Madunic IV, Gajski G, Popic J, Garaj-Vrhovac V. Apigenin: a dietary flavonoid with diverse anticancer properties. Cancer Lett. 2018; 413: 11–22. [DOI] [PubMed] [Google Scholar]
- 31. Zhang XX, Wang GJ, Gurley EC, Zhou HP. Flavonoid apigenin inhibits lipopolysaccharide-induced inflammatory response through multiple mechanisms in macrophages. Plos One. 2014; 9: e107072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kashyap D, Sharma A, Tuli HS, et al.. Apigenin: a natural bioactive flavone-type molecule with promising therapeutic function. J Funct Foods. 2018; 48: 457–471. [Google Scholar]
- 33. Ogasawara H, Fujitani T, Drzewiecki G, Middleton E. The role of hydrogen-peroxide in basophil histamine-release and the effect of selected flavonoids. J Allergy Clin Immunol. 1986; 78: 321–328. [DOI] [PubMed] [Google Scholar]
- 34. Middleton E, Drzewiecki G.. Flavonoid inhibition of human basophil histamine-release stimulated by various agents. Biochem Pharmacol. 1984; 33: 3333–3338. [DOI] [PubMed] [Google Scholar]
- 35. Zou YH, Chiou GCY. Apigenin inhibits laser-induced choroidal neovascularization and regulates endothelial cell function. J Ocul Pharmacol Ther. 2006; 22: 425–430. [DOI] [PubMed] [Google Scholar]
- 36. Park-Windhol C, D'Amore PA.. Disorders of vascular permeability. Annu Rev Pathol. 2016; 11: 251–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lu D, Kassab GS.. Role of shear stress and stretch in vascular mechanobiology. J R Soc Interface. 2011; 8: 1379–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Paszkowiak JJ, Dardik A. Arterial wall shear stress: observations from the bench to the bedside. Vasc Endovascular Surg. 2003; 37: 47–57. [DOI] [PubMed] [Google Scholar]
- 39. Nagaoka T, Sato E, Takahashi A, Yokota H, Sogawa K, Yoshida A. Impaired retinal circulation in patients with type 2 diabetes mellitus: retinal laser Doppler velocimetry study. Invest Ophthalmol Vis Sci. 2010; 51: 6729–6734. [DOI] [PubMed] [Google Scholar]
- 40. Burgansky-Eliash Z, Barash H, Nelson D, et al.. Retinal blood flow velocity in patients with age-related macular degeneration. Curr Eye Res. 2014; 39: 304–311. [DOI] [PubMed] [Google Scholar]
- 41. Molins B, Mora A, Romero-Vazquez S, et al.. Shear stress modulates inner blood retinal barrier phenotype. Exp Eye Res. 2019; 187: 107751. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.