

Abstract
An asymmetric total synthesis of [13C4]-anatoxina ([13C4]-1) has been developed from commercially available ethyl [13C4]-acetoacetate ([13C4]-15). The unique requirements associated with isotope incorporation inspired a new, robust, and highly scalable route, providing access to 0.110 g of this internal standard for use in the detection and precise quantification of anatoxin-a in freshwater. A highlight of the synthesis is a method that leverages a cyclic iminium ion racemization to achieve dynamic kinetic resolution in an enantioselective Morita–Baylis–Hillman (MBH) cyclization.
Keywords: cyclization, isotopes, natural products, rearrangements, total synthesis
Anatoxin-a (1) is a cyanobacterial toxin produced by several genera of blue-green algae, including Anabaena, Aphanizomenon, and Oscillatoria.[1] This potent agonist of nicotinic acetylcholine receptors (nAChRs) causes prolonged excitation at the neuromuscular junction and a blockage of further electrical transmission, which can lead to muscular paralysis and death by asphyxiation.[2] It has been associated with the poisoning and deaths of livestock, domestic animals, and waterfowl after exposure to cyanobacteria-contaminated water.[3]
Cyanobacterial algal blooms have occurred with increasing frequency in recent years, likely caused by factors such as rising water temperatures and fertilizer runoff.[4] These blooms pose a significant threat to public safety because of the production of cyanobacterial toxins, such as anatoxin-a. As such, methods for the detection and quantification of anatoxin-a in fresh water are critically important to human health.[1b] The analytical method currently adopted by the Environmental Protection Agency for anatoxin-a quantification in finished drinking water involves a combination of liquid chromatography, electrospray ionization, and tandem mass spectrometry (LC/ESI-MS/MS) using deuterated phenylalanine (L-phenylalanine-d5) as an internal standard.[5] While this method is useful, its accuracy hinges on the assumption that L-phenylalanine-d5 and anatoxin-a will have identical behavior during sample preparation and analysis.[6] A more ideal standard, which would account for potential sample loss and ion suppression from other components present in the sample matrix, would be an isotopically labelled variant of anatoxin-a. However, no such standard is available.
Since its isolation, a plethora of synthetic approaches to the natural isotope of anatoxin-a have been developed and extensively reviewed.[7] Its popularity as a synthetic target has inspired several innovative routes (Figure 1). Elegant work has been described by the groups of Trost,[8] Aggarwal,[9] Seitz,[10] and Martin,[11] relying on a variety of creative strategies including asymmetric allylic amination, asymmetric deprotonation, tropanone ring expansion, and enyne meta-thesis. These impressive syntheses proceeded in as few as eight steps (Seitz) and up to 27% overall yield (Martin), truly setting a high bar for future approaches to this important target.
Figure 1.
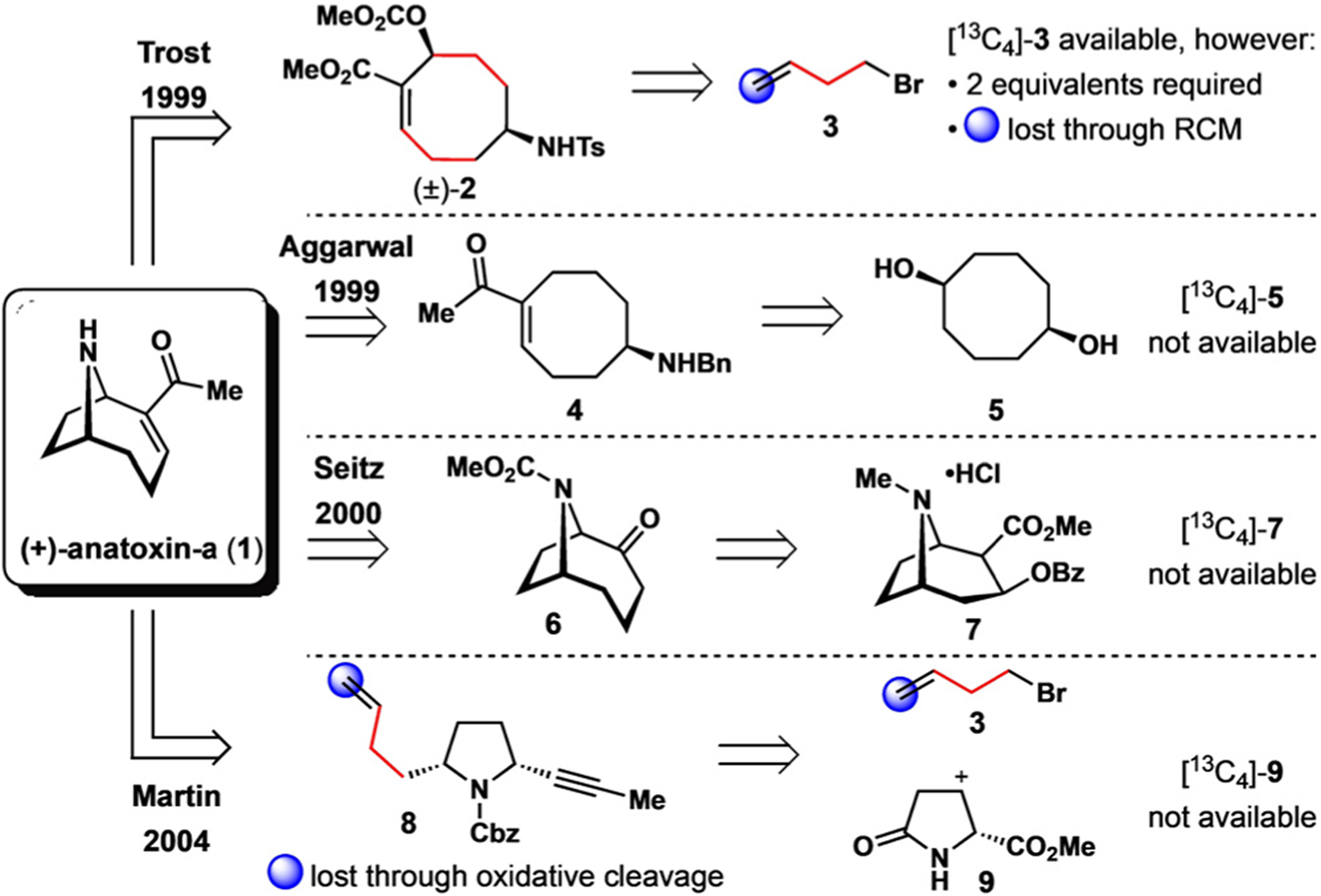
Previous synthetic approaches to (+)-anatoxin-a and consideration of their ability to produce an isotopically labelled internal standard.
Our goal was to provide practical synthetic access to an isotopically labelled anatoxin-a that could be used as an internal standard for highly sensitive and precise freshwater quantification. In the initial stages of synthesis planning, there were several important criteria that had to be taken into consideration.[12] First, isotope incorporation at non-exchangeable positions throughout the synthesis is essential to preventing isotope loss and maintaining a uniform molecular mass in the final product. Second, to prevent any signal overlap during mass spectral analysis, we sought to prepare a standard with a mass difference of 4 atomic units compared to the natural anatoxin-a. Finally, given the high cost of isotopically labelled starting materials, the synthesis itself must be as concise as possible, highly scalable, and rely on robust chemical transformations to ensure reproducibility.
Inspired by the past approaches described above, we attempted to identity a suitable route to isotopically labelled anatoxin-a that would satisfy these criteria. Previous syntheses, while succinct and high yielding, appeared to be unfeasible in this regard because of a lack of either commercially available isotopically labelled starting materials or synthetic transformations that would result in isotope loss (Figure 1). We therefore decided to develop a de novo synthesis of [13C4]-anatoxin-a ([13C4]-1) from a suitable source of heavy isotopes. We envisioned a synthetic route in which the final C—C bond of anatoxin-a would be constructed from an iminium Morita–Baylis–Hillman (MBH) cyclization of the enone [13C4]-10 (Scheme 1).[13,14] The cyclization precursor [13C4]-10 would be derived from the acid [13C4]-11, which in turn would arise from an Ireland–Claisen rearrangement of the allylic ester [13C4]-12.[15] Disconnection of the ester C—O bond identified the chiral alcohol 14 and carboxylic acid 15 as potential materials for the introduction of isotopes into the synthesis. After a thorough survey of commercially available isotopically labelled compounds, ethyl [13C4]-acetoacetate ([13C4]-15) was determined to be an excellent precursor to alcohol [13C4]-14. This material serves as the sole source of all heavy isotopes in the synthesis. More importantly, these isotopes are placed at non-exchangeable positions throughout the designed route. Herein, we describe our successful approach to [13C2,13C3,13C11]-anatoxin-a ([13C4]-1) from [13C4]-15 and the development of a dynamic kinetic resolution process through enantioselective MBH cyclization.
Scheme 1.
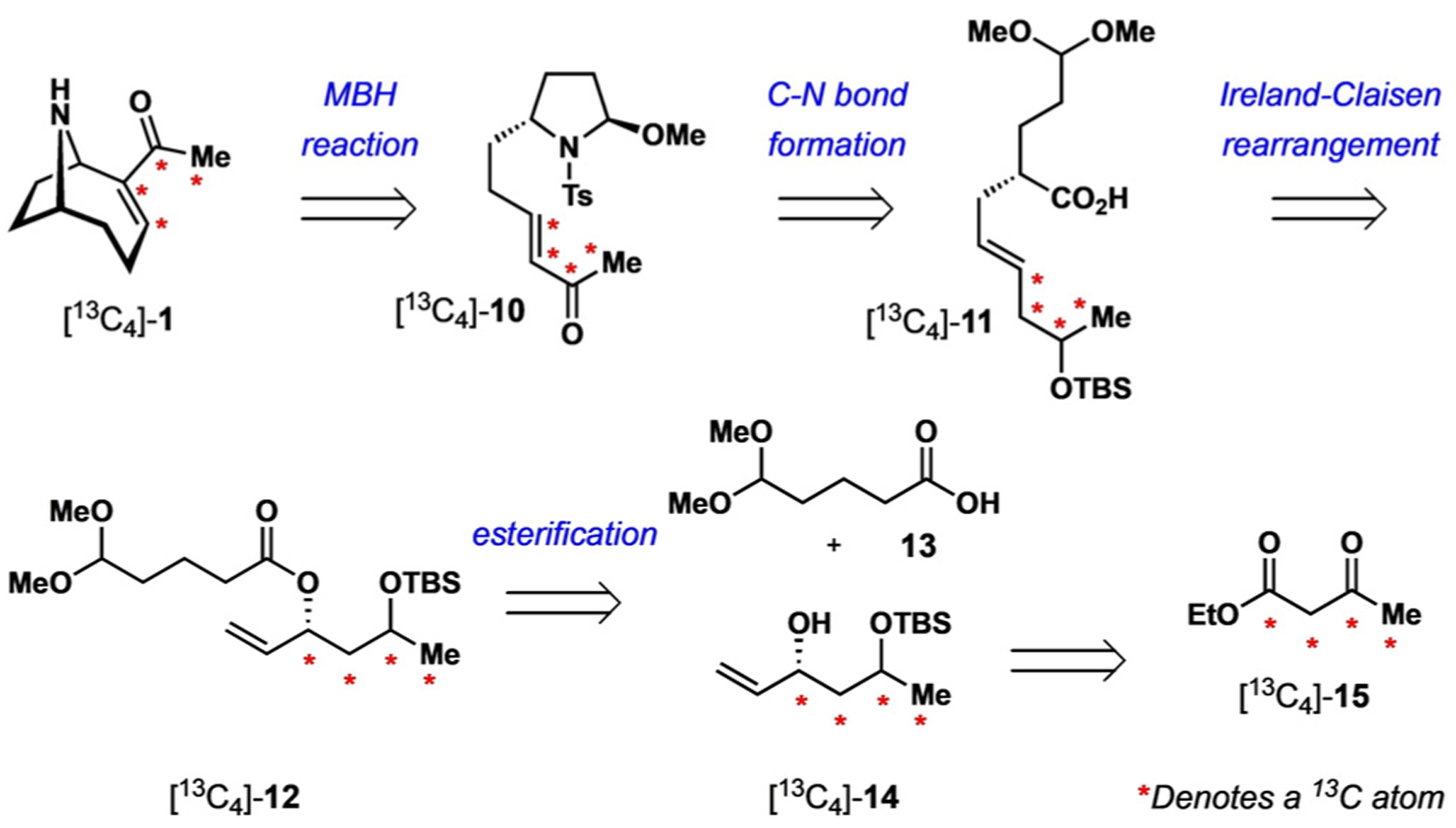
Synthesis plan for [13C4]-anatoxin-a. TBS=tert-butyldimethylsilyl.
The synthesis commenced with a sodium borohydride reduction of [13C4]-15 (Scheme 2). Silylation of the nascent alcohol with TBSCl (96% yield over 2 steps) followed by partial reduction of the ethyl ester gave aldehyde [13C4]-16. Homologation with triethyl phosphonoacetate followed by reduction of the intermediate α,β-unsaturated ester furnished the allylic alcohol [13C4]-17 in 84% yield over two steps. Sharpless asymmetric epoxidation of [13C4]-17 gave epoxide [13C4]-18, which was then immediately subjected to iododehydroxylation conditions.[16] Reduction with zinc occurred with concomitant epoxide opening to deliver chiral alcohol [13C4]-14 in 88% yield and 86% ee over three steps. It is worth mentioning that we did attempt to access [13C4]-14 directly through enantioselective addition of divinyl zinc to aldehyde [13C4]-16.[17] However, both the yield and enantioselectivity for this one-step transformation were lower compared to the route outlined in Scheme 2 (see the Supporting Information for more details).
Scheme 2.
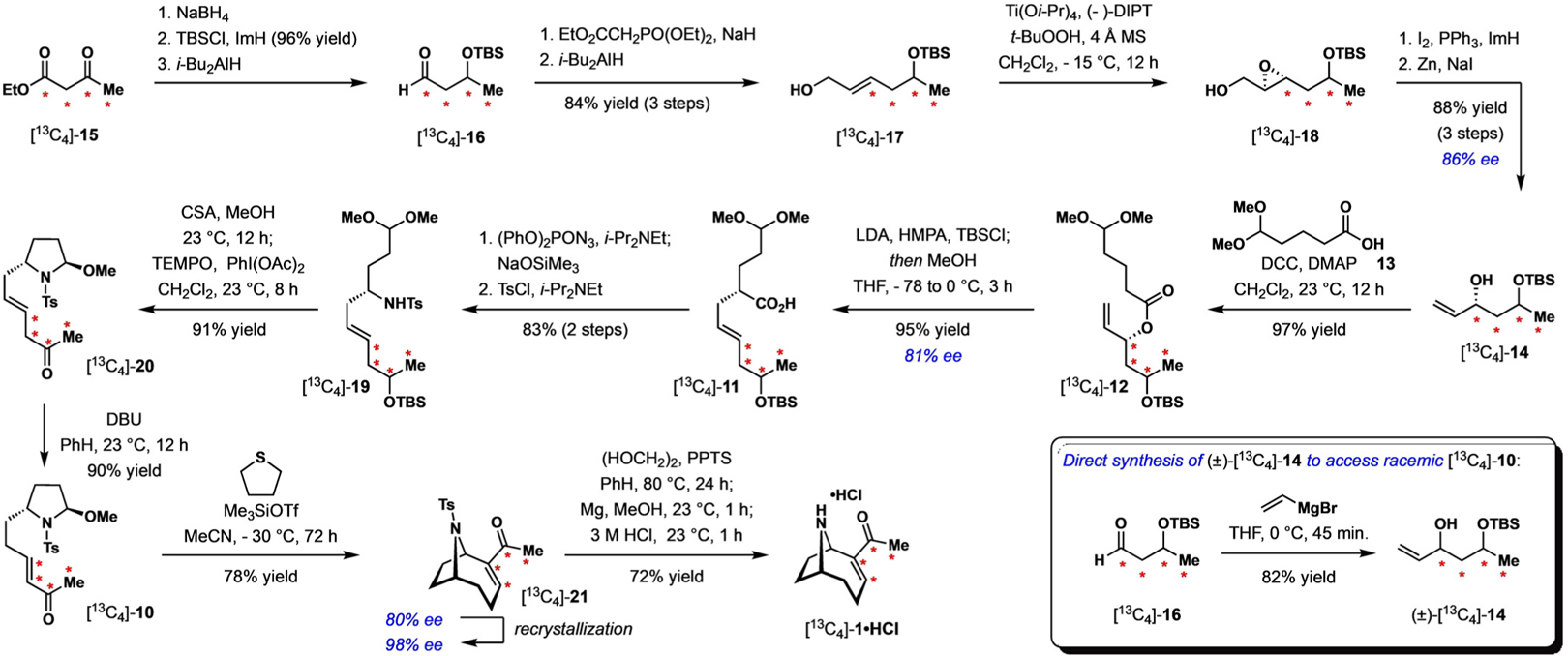
Total synthesis of [13C4]-anatoxin-a. CSA=10-camphorsulfonic acid, DBU=1,8-diazabicyclo[5.4.0]undec-7-ene, DCC=N,N’-dicyclohexylcarbodiimide, DIPT=diisopropyl tartrate, DMAP=4-(N,N-dimethyl)aminopyridine, HMPA=hexamethylphosphoric triamide, LDA=lithium diisopropylamide, MS=molecular sieves, PPTS=pyridinium p-toluenesulfonate, TEMPO=, Tf=trifluoromethanesulfonyl, THF=tetrahydrofuran, Ts = 4-toluenesulfonyl.
Coupling of [13C4]-14 with 13 in the presence of DCC gave [13C4]-12 in 97% yield. Deprotonation with LDA followed by addition of TBSCl and Ireland–Claisen rearrangement of the resulting silyl ketene acetal delivered [13C4]-11 in 95% yield and 81% ee. [13C4]-11 was transformed into the corresponding acyl azide with (PhO)2PON3, which then underwent Curtius rearrangement upon heating. Hydrolysis of the intermediate isocyanate with NaOSiMe3 gave a primary amine, which was tosylated to give [13C4]-19 in 83% yield over two steps.[18] Treatment with CSA resulted in pyrrolidine formation and simultaneous removal of the TBS group. In the same pot, addition of TEMPO and PhI(OAc)2 gave [13C4]-20, and subsequent treatment with DBU led to migration of the double bond into the α,β-position, delivering [13C4]-10 in 83% yield over two steps. [13C4]-10 served as a precursor for the penultimate MBH cyclization. After extensive experimentation, it was found that the use of tetrahydrothiophene as the nucleophile in the presence of Me3SiOTf in MeCN at −30°C gave [13C4]-21 in 78% yield and 80% ee, which could be enriched to 98% ee following recrystallization.[19] Heating [13C4]-21 with ethylene diol and PPTS generated an acetal, tosyl-group removal was successfully accomplished through reduction with magnesium, and in situ hydrolysis of the acetal gave [13C4]-anatoxin-a in 72% yield as its hydrochloride salt. High-resolution mass spectral analysis revealed the final product had 99.1% 13C incorporation at all four positions.
During optimization of the iminium MBH cyclization, we noticed an unexpected relationship between reaction temperature and enantiopurity of the cyclized product. At temperatures above −30°C, significant racemization was observed. This racemization caught us by surprise, given the apparent lack of an epimerizable carbon center in the molecule. Tanner and co-workers observed the same phenomenon in their synthesis of anatoxin-a, in which they used an MBH cyclization on a similar precursor.[11] The authors propose that racemization occurs through a hydride shift of the initially formed cyclic iminium ion. However, this mechanism seems improbable given that [1,3] hydride shifts are symmetry-forbidden.[20] It is conceivable that this racemization involves a simple trace-acid-catalyzed isomerization of the initially-formed C-monosubstituted imine to the more stable C-disubstituted imine (i to ii, Scheme 3a). However, at present the exact origin of epimerization remains unknown.
Scheme 3.
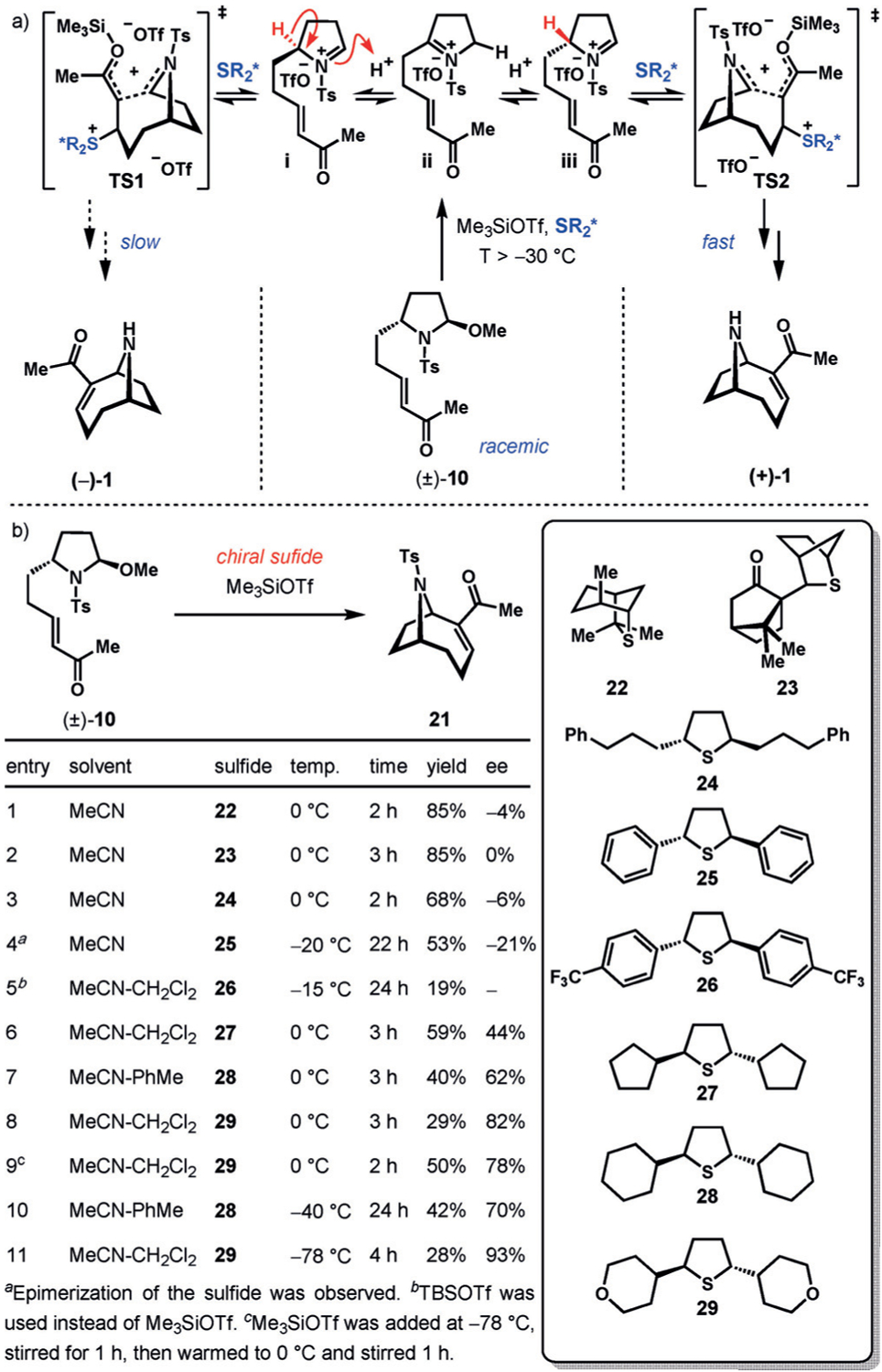
a) Proposed dynamic kinetic resolution process through an enantioselective aza-MBH reaction. b) Optimization of the chiral sulfide used for enantioselective cyclization.
We became intrigued by the practical implications of the cyclic imine undergoing facile racemization. At temperatures above −30°C, the rate of imine isomerization begins to outcompete the rate of cyclization. We hypothesized that this phenomenon could be leveraged to achieve dynamic kinetic resolution through use of a chiral nucleophile to induce enantioselectivity. Treatment of the racemic cyclization precursor (±)-10 with Me3SiOTf produces a series of rapidly equilibrating cyclic imines (i, ii, and iii, Scheme 3a). If reaction parameters are adjusted such that equilibration between i and iii occurs rapidly relative to cyclization, the product distribution should reflect the difference in energy between the two diastereomeric transition states TS1 and TS2, in accordance with the Curtin–Hammett principle.[21] The ability to use (±)-10 would obviate the need to introduce chirality into 14. Racemic (±)-[13C4]-14 can be accessed directly through addition of vinylmagnesium bromide to (±)-[13C4]-16 in 82% yield (Scheme 2 inset), shortening the synthesis by four steps.
Our success using tetrahydrothiophene as a nucleophile in our original route inspired us to explore a variety of chiral sulfur-containing nucleophiles for enantioselective iminium MBH cyclization. We began our experiments by employing the commercially available chiral sulfide 22 (Scheme 3b, entry 1).[22] In this case, 21 was isolated in high yield, but virtually no enantioselectivity was achieved. We then screened the camphor-derived sulfide 23 developed by Aggarwal and co-workers.[23] We were excited by the potential of this sulfide to induce selectivity given its previous application in enantioselective MBH reactions.[24] Unfortunately, while the cyclized product was isolated in excellent yield, no enantioselectivity was observed (entry 2). Our attention was then turned toward a series of C2-symmetric trans-2,5-disubstituted thiolanes. The bis-primary alkyl substituted thiolane 24 gave the product in 68% yield with an unimpressive ee value of −6% (entry 3). The diphenyl thiolane 25 gave 21 in 22% ee with a drop in yield to 53%.[25] In this case, the recovered sulfide had partially epimerized to a mixture of cis- and trans-2,5-diphenylthiolane (dr 14:86; 89% ee for the recovered trans isomer), likely because of formation of a benzylic carbocation under the reaction conditions. To prevent epimerization, we sought to install suitable electron-withdrawing groups on the phenyl rings. To this end, the sulfide 26 was synthesized and applied in our optimization. The para-CF3 groups did indeed prevent racemization. However, the yield and purity of the product declined drastically compared to 26 (Scheme 3b, entry 5). The use of the dicyclopentyl sulfide 27 gave the product in reasonable yield with an improved ee value of 44%. We then tested the dicyclohexyl and ditetrahydropyranyl thiolanes 28 and 29, respectively, postulating that increasing the steric bulk of the thiolane substituents could potentially lead to greater selectivity. In both cases, improved enantioselectivity was observed, with the cyclized products being isolated in 40% yield, 62% ee (entry 7) and 29% yield, 82% ee (entry 8), respectively. For the sulfide 29, adding Me3SiOTf at −78°C, stirring for 1 hour, and then stirring the mixture at 0°C for 1 hour led to a substantial increase in the yield (50%) with a similar ee value (78%, entry 9). Finally, we also explored conditions in which the reaction temperature was kept low enough to prevent imine rearrangement (below −30°C) in an attempt to achieve simple kinetic resolution. Performing the cyclization at −40°C using 28 gave the cyclized product in a slightly improved yield of 42% and 70% ee (entry 10). Using 29 and performing the reaction at −78°C, the cyclized product was isolated in 28% yield, this time with a remarkable enantioselectivity of 93% ee. These observations indicate that increased steric encumbrance surrounding sulfur leads to higher selectivity. In contrast, this comes at the expense of decreased yield, perhaps because of a lower rate of conjugate addition for more hindered tetrahydrothiophenes, resulting in a greater prevalence of side reactions. In entries 1–9 in Scheme 3b, the remaining mass balance appears to be a complex mixture of products caused by decomposition or oligomerization. No spirocyclic cyclization products arising from cyclization of ii were isolated under any of the surveyed conditions, likely owing to its decreased reactivity.
Our approach to [13C4]-anatoxin-a was framed by a need to develop a route which commenced from a commercially feasible isotopically labeled starting material and utilized transformations that would retain all heavy isotopes. These constraints inspired the first synthesis of [13C4]-1, relying on a Sharpless asymmetric epoxidation as the enantiodetermining step, and an MBH cyclization to forge the final bicyclic scaffold of the anatoxin-a skeleton. The synthesis was completed in 12 steps from [13C4]-15 and 14% overall yield through the kinetic resolution strategy. As a testament to the scalability of this route, 0.110 grams of [13C4]-1 have been prepared to date. The ability to access substantial quantities of [13C4]-1 will enable the development of a new analytical method for its precise and highly accurate quantification in samples of fresh water. Finally, an observation made during the penultimate MBH cyclization in our original route prompted an investigation into a unique dynamic kinetic resolution process that leveraged a cyclic iminium ion rearrangement. Highly enantioselective cyclization was achieved starting from (±)-10 through the use of the chiral thiolanes 28 and 29. Our results suggest that iminium ions derived from 2-alkoxy-5-alkylpyrrolidines undergo facile rearrangement under acidic conditions, and that this process can be taken advantage of to achieve dynamic kinetic resolution. This finding has potential application in future enantioselective syntheses of 2,5-disubstituted pyrrolidines.
Supplementary Material
Acknowledgements
This work was supported by the NIH (NIGMS, R01-077379 and NIEHS, R03 ES025345-01). J.J.L. thanks the Natural Sciences and Engineering Research Council of Canada (NSERC) for a postgraduate scholarship (PGS-D) and UCSB for a Chancellor’s Fellowship. Dr. Hongjun Zhou is acknowledged for assistance with NMR spectroscopy. Dr. Dmitriy Uchenik and the UCSB mass spectroscopy facility are thanked for assistance with mass spectral analysis.
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].a) Devlin JP, Edwards OE, Gorham PR, Hunter NR, Pike RK, Stavric B, Can. J. Chem 1977, 55, 1367; [Google Scholar]; b) Osswald J, Rellán S, Gago A, Vasconcelos V, Environ. Int 2007, 33, 1070. [DOI] [PubMed] [Google Scholar]
- [2].a) Carmichael WW, Biggs DF, Gorham PR, Science 1975, 187, 542; [DOI] [PubMed] [Google Scholar]; b) J. Harris in Encyclopedia of Neuroscience (Ed.: Squire LR), Elsevier, Amsterdam, 2009, pp. 539 – 549. [Google Scholar]
- [3].Health Effects Support Document for the Cyanobacterial Toxin Anatoxin-A. EPA-820R15104. https://www.epa.gov/sites/production/files/2017-2006/documents/anatoxin-a-report-2015.pdf.
- [4].Cheung MY, Liang S, Lee J, J. Microbiol 2013, 51, 1. [DOI] [PubMed] [Google Scholar]
- [5].Method 545: Determination of Cylindrospermopsin and Anatoxin-a in Drinking Water by Liquid Chromatography Electro-spray Ionization Tandem Mass Spectrometry (LC/ESI-MS/MS). EPA 815-R-15–009. https://www.epa.gov/sites/production/files/2017-10/documents/epa_815-r-15-009_method_545.pdf.
- [6].a) Annesley TM, Clin. Chem 2003, 49, 1041; [DOI] [PubMed] [Google Scholar]; b) Stevens DK, Krieger RI, Toxicon. 1991, 29, 167. [DOI] [PubMed] [Google Scholar]
- [7].a) Fonseca C, Aureliano M, Abbas F, A. Furey in Phycotoxins: Chemistry and Biochemistry Second Edition (Eds.: Botana LM, Alfanso A), Wiley, Hoboken, 2015, pp. 137 – 180; [Google Scholar]; b) Mansell HL, Tetrahedron 1996, 52, 6025. [Google Scholar]
- [8].Trost BM, Oslob JD, J. Am. Chem. Soc 1999, 121, 3057. [Google Scholar]
- [9].Aggarwal VK, Humphries PS, Fenwick A, Angew. Chem. Int. Ed 1999, 38, 1985; Angew. Chem. 1999, 111, 2178. [DOI] [PubMed] [Google Scholar]
- [10].Wegge T, Schwarz S, Seitz G, Tetrahedron: Asymmetry 2000, 11, 1405. [Google Scholar]
- [11].a) Brenneman JB, Martin SF, Org. Lett 2004, 6, 1329; [DOI] [PubMed] [Google Scholar]; b) Brenneman JB, Machauer R, Martin SF, Tetrahedron 2004, 60, 7301. [Google Scholar]
- [12].Mailyan AK, Chen JL, Li W, Keller AA, Sternisha SM, Miller BG, Zakarian A, J. Am. Chem. Soc 2018, 140, 6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Hjelmgaard T, Søtofte I, Tanner D, J. Org. Chem 2005, 70, 5688. [DOI] [PubMed] [Google Scholar]
- [14].Somfai P, Åhman J, Tetrahedron Lett. 1992, 33, 3791. [Google Scholar]
- [15].Ireland RE, Mueller RH, Willard AK, J. Am. Chem. Soc 1976, 98, 2868. [Google Scholar]
- [16].Katsuki T, Sharpless KB, J. Am. Chem. Soc 1980, 102, 5974. [Google Scholar]
- [17].Brubaker JD, Myers AG, Org. Lett 2007, 9, 3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Gluyas JBG, Burschka C, Dörrich S, Vallet J, Gronemeyer H, Tacke R, Org. Biomol. Chem 2012, 10, 6914. [DOI] [PubMed] [Google Scholar]
- [19].Kitulagoda JE, Palmelund A, Aggarwal VK, Tetrahedron 2010, 66, 6293. [Google Scholar]
- [20].Woodward RB, Hoffmann R, Angew. Chem. Int. Ed. Engl 1969, 8, 781; Angew. Chem. 1969, 81, 797. [Google Scholar]
- [21].Seeman JI, Chem. Rev 1983, 83, 83. [Google Scholar]
- [22].Illa O, Arshad M, Ros A, McGarrigle EM, Aggarwal VK, J. Am. Chem. Soc 2010, 132, 1828. [DOI] [PubMed] [Google Scholar]
- [23].a) Aggarwal VK, Alonso E, Hynd G, Lydon KM, Palmer MJ, Porcelloni M, Studley JR, Angew. Chem. Int. Ed 2001, 40, 1430; Angew. Chem. 2001, 113, 1479; [DOI] [PubMed] [Google Scholar]; b) Aggarwal VK, Fang GY, Kokotos CG, Richardson J, Unthank MG, Tetrahedron 2006, 62, 11297. [Google Scholar]
- [24].Myers EL, de Vries JG, Aggarwal VK, Angew. Chem. Int. Ed 2007, 46, 1893; Angew. Chem. 2007, 119, 1925. [DOI] [PubMed] [Google Scholar]
- [25].Periasamy M, Ramani G, Muthukumaragopal GP, Synthesis 2009, 1739. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.