

Abstract
Purpose of Review
West Nile virus (WNV) emerged from Central Africa in the 1990s and is now endemic throughout much of the world. Twenty years after its introduction in the USA, it is becoming apparent that neurological impairments can persist for years following infection. Here, we review the epidemiological data in support of such long-term deficits and discuss possible mechanisms that drive these persistent manifestations.
Recent Findings
Focusing on the recently discovered antimicrobial roles of amyloid and alpha-synuclein, we connect WNV late pathology to overlapping features encountered in neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease. We also summarize new research on microglial activation and engulfment of neural synapses seen in recovered WNV as well as in neurodegenerative diseases, and discuss how loss of integrity of the blood-brain barrier (BBB) may exacerbate this process.
Summary
Neuroinvasive viral infections such as WNV may be linked epidemiologically and mechanistically to neurodegeneration. This may open doors to therapeutic options for hitherto untreatable infectious sequelae; additionally, it may also shed light on the possible infectious etiologies of age-progressive neurodegenerative dementias.
Keywords: West Nile virus, Neurological sequelae, Neurodegenerative disease, Antimicrobial peptide, Amyloid beta, Alpha-synuclein, Alzheimer’s disease, Parkinson’s disease
Introduction
The neuroinvasive, mosquito-borne West Nile virus (genus Flavivirus, family Flaviviridae) is a small, positive-sense, single-stranded RNA virus originating from Central Africa. Its range has extended significantly in recent decades, and it now occurs throughout the Americas, Europe, Australia, Africa, Asia, and the Middle East [1]. Since its introduction to North America in 1999, it is estimated that 3 to 5 million people have been infected by WNV in the USA alone [2]. Approximately 1% of WNV infections result in neuroinvasive disease, including meningitis, encephalitis, or acute flaccid paralysis/poliomyelitis [3]. While clinically apparent neuroinvasive disease is relatively rare, even mild and asymptomatic WNV infections have been associated with a significant prevalence of sequelae such as memory loss, confusion, and fatigue years later [4, 5]. This has led to some intriguing epidemiological and animal-model studies of the acute and late CNS pathology of WNV, revealing features that overlap with neurodegenerative dementias such as Alzheimer’s disease (AD) and Parkinson’s disease (PD).
In this review, we discuss epidemiological studies on WNV neurological sequelae and delve into some key mechanisms that may be driving these long-term symptoms. The literature on neurological impairment, following neuroinvasive infections, and research on the etiology of neurodegenerative diseases are converging on the role of the innate immune system [6]. In both instances, production of antimicrobial peptides and activation of glial cells precede and may possibly underlie the ensuing CNS dysfunction. Comorbidities, such as diabetes mellitus and hypertension, appear to predispose individuals to both conditions, possibly by their vasculopathies or by weakening the blood-brain barrier (BBB). Considering these relationships and possibilities opens the door to many new questions. First, do these pathogens contribute to age-dependent neurodegeneration and if so, what are the molecular pathways affected? Which host factors—environmental as well as genetic—confer not only risk but also protection following infection by a neuroinvasive microbe? While we have definitely observed high levels of cognitive impairment following alphaviral infection in endemic regions (Carrera and Vittor, unpublished data), we will limit our discussion here to WNV since the literature is most robust for this pathogen. We will then link the WNV findings to recent developments in the two most common neurodegenerative dementias, namely AD and PD.
Neurological Sequelae of WNV
WNV infection results in a self-limited febrile illness in 25% of exposed individuals, while neuroinvasive disease (meningitis, encephalitis acute flaccid paralysis) occurs in less than 1% [3]. Following the initial waves of WNV outbreaks throughout North America in the early 2000s, it became increasingly clear to clinicians taking care of these patients that the acute phase of illness was often followed by a prolonged phase characterized by excessive fatigue, confusion, memory loss, muscle weakness, and occasionally, tremors. The first studies to systematically examine the possibility of such long-term sequelae demonstrated that indeed, 20 to 60% of patients reported difficulties with these symptoms even after 6 to 18 months after the initial infection [7–9]. While it was initially hypothesized that patients with neuroinvasive disease (encephalitis or meningitis) would report higher rates of ongoing symptoms, most studies have shown that even patients with non-neuroinvasive disease (WNV fever) reported similar sequelae at equal frequencies [10]. Due to a paucity of data on long-term outcomes in people asymptomatically infected with WNV, the fate of this population is unknown.
As more time elapsed since WNV first arrived in North America, it became possible to study these sequelae over longer periods of time. Though the studies are still few in number, it appears that significant portions of patients with WNV have ongoing symptoms years after the acute phase of illness [4, 11]. For example, Murray et al. [4] note that at 8 years from initial infection (neuroinvasive and febrile WNV), 40% of patients still reported sequelae [4]. Fatigue, weakness, and depression were most common, and interestingly, 9% of these patients reported memory loss and confusion. Poor recovery was associated with older age and diabetes.
These self-reported symptoms were further substantiated in a series of studies utilizing various batteries of neuropsychological tests [5, 11–13, 14••, 15, 16]. Most of these studies have included two groups of subjects—those who initially suffered from WNV neuroinvasive disease, and others who had WNV fever. The emerging themes from these studies are that psychomotor speed, memory, and executive functions are impacted significantly. In addition, as with the self-reported symptoms, the objective measures of cognitive impairment do not appear to differ significantly between the neuroinvasive and non-neuroinvasive groups [5, 12, 13]. Assessments are mostly performed 1 year after the onset of the initial illness. Samaan et al. [11], however, evaluated subjects whose time since infection ranged from less than 1 month up to 4 years. Not only did the authors demonstrate that a sizeable proportion of patients had ongoing motor function deficits (69%), executive function deficits (30%), and verbal learning and memory deficits (30%), but they showed that these impairments were more pronounced in subjects with longer intervals since their initial illness.
A caveat of these studies is that only few studies include healthy, or normative controls in their analyses [15, 17]. In a study that included age- and sex-matched controls, Balakrishnan et al. [17] followed up patients 1 year after illness due to WNV encephalitis. Cognitive impairment was determined using the Mini-Mental Status Exam (MMSE; probable dementia MMSE score < 21). The authors report a 57% (95% CI 22.5–76.1%) attributable risk of WNV encephalitis for probable dementia, with a crude relative risk of 2.3 (95% CI 1.3–4.2). Notably, of the 40 cases initially identified as having had WNV encephalitis, 10 had died within a year. On the other hand, Sejvar et al. [15] found that motor deficits persisted 1.5 years after illness onset, but did not find a significant difference in neurocognitive performance in WNV patients compared to normative controls using the Cambridge Neuropsychological Testing Automated Battery (CANTAB [18]).
A recent study extended these findings further by conducting magnetic resonance imaging (MRI) on a subset of a cohort of patients with WNV neuroinvasive disease or WNV fever [14••]. The population included subjects at 0 to 11 years following initial infection, 25–31% of whom demonstrated memory impairments on cognitive testing. Compared to age- and sex-matched controls, significant bilateral frontal and limbic cortical thinning was observed in WNV subjects. A review of MRI findings (Table 1) reveals that acute illness is characterized predominantly by abnormalities in the thalami and basal ganglia, though many other structures can be affected. Late findings include cortical thinning in structures within the frontal, temporal, and limbic cortices and regional atrophy in the thalami, basal ganglia, cerebellum, and brain stem. Interestingly, several of these affected structures are also atrophied in AD [39] (e.g. posterior cingulate cortex and insular cortex, and the hippocampus and entorhinal regions in the temporal lobe) and PD [40] (e.g., substantia nigra within the basal ganglia and thalamic connections).
Table 1.
MRI findings in acute and chronic WNV neuroinvasive disease. Italic structures are also abnormal in AD, and bold structures are abnormal in PD
| Acute vs chronic | Time after illness onset | MRI findings | Ref. |
|---|---|---|---|
| Acute | 1–30 days, 113 cases combined | T2 and FLAIR hyperintensities in the thalami, basal ganglia (i.e., substantia nigra, caudate nucleus, putamen), temporal lobes, parieto-occipital region, temporo-parietal region, fronto-parietal region, insula, hippocampus, periventricular white matter, corona radiata, internal capsule, corticospinal tracts, cerebellum, corpus callosum, pons | [7, 19–36] |
| Chronic | 2 months, 1 case | Temporal lobes, basal ganglia, internal capsule posterior limb, and corona radiata | [37] |
| Chronic | 5 months, 1 case | Periventricular and subcortical white matter of the bilateral frontal lobes, basal ganglia and thalami. Generalized volume loss all levels of cortex | [38] |
| Chronic | Average of 4 years, 30 subjects | Cortical thinning: frontal, temporal and limbic cortices (i.e., posterior cingulate cortex, superior and inferior frontal cortices, medial-orbito frontal region, anterior cingulate cortex, cuneus, parahippocampal region, middle and inferior temporal cortex, supramarginal region, insular cortex). Also atrophy of: cerebellum, brain stem, thalamus, basal ganglia. | [14] |
Taken together, the preponderance of epidemiological evidence suggests that cognitive impairment following WNV infection is common, and that its occurrence does not correlate with severity of the initial presentation (neuroinvasive cases and febrile WNV infections). On neuropsychological testing, the domains most affected in WNV patients include psychomotor, memory, and executive function. While these studies have definitely advanced our understanding of long-term clinical and neuropsychiatric outcomes of WNV, the lack of healthy controls and asymptomatically infected individuals in most studies hampers our ability to accurately define the extent of neurological sequelae following WNV exposure.
CNS Infection in WNV Patients
Following peripheral infection, WNV accesses the CNS via one or more of four pathways: free virus crosses the BBB via passive transport through the endothelium [41], peripheral inflammation (e.g., due to tumor necrosis factor-α (TNF-α)) weakens the tight junctions of the BBB allowing the virus to “leak” in [42], infected virus leukocytes are trafficked across the BBB in a “trojan horse” fashion [41], or it infects peripheral nerves with retrograde axonal flow into the CNS [43]. Once the virus enters the brain, it can replicate in astrocytes, microglia, and neurons [44, 45], which produce pro-inflammatory cytokines such as type I interferons (IFN-α, IFN-β), type III interferons (IFN-λ), TNF-α, and interleukin-1β (IL-1β). Direct neuronal injury is incurred via apoptosis of infected cells [46] or by secondary insults due to the ensuing inflammation [47]. The innate immune response (i.e., pro-inflammatory cytokines, antimicrobial peptides, phagocytes, natural killer cells, and γδ T cells), complement, and the adaptive immune response are all critical for clearance of virus and survival [48]. WNV persistence in the CNS has been studied in mice, revealing that in immunecompetent mice, infectious virus is typically found up to 1 month after infection [49]. Thereafter, WNV RNA remains detectable for up to 6 months post infection (p.i.). However, Appler et al. demonstrated that infectious virus appears to reactivate beyond 1 month p.i. upon treatment with cyclophosphamide, suggesting that lymphocytes function to restrict the replication of WNV [49]. Detailed analysis of these lymphocyte subpopulations showed that activated, virus-specific CD4+ and CD8+ T cells as well as plasma cells remained in the CNS even at 4 months p.i. [50]. Importantly, regulatory T cells (Tregs) were also present at elevated levels, likely preventing neuronal injury due to ongoing inflammation.
Misfolded Protein or Antimicrobial Peptide?
As it has become clear that WNV is associated with long-term neurological deficits, a growing body of research indicates that neurodegenerative diseases may in turn be associated with an infectious trigger. The most prevalent neurodegenerative dementia, AD, currently affects 5.7 million Americans [51]. Hallmark pathological features of AD include amyloid (neuritic) plaques, which are extracellular β amyloid (Aβ) aggregates and intraneuronal neurofibrillary tangles (NFTs), which are comprised of hyperphosphorylated and aberrantly folded tau protein [52]. The progressive accumulation of these proteinopathies in selectively vulnerable brain regions, such as the hippocampus and cortex, are associated with increased brain inflammation, neuritic pathology, neurodegeneration, and brain atrophy. The prevailing paradigm for late-onset sporadic forms of AD (the most common manifestation), the Amyloid Cascade Hypothesis, posits that deposition of Aβ leads to harmful Aβ aggregates and to the pathological tau alterations that result in NFT which cause neuronal dysfunction and cell death [53]. Clinical trials targeting extracellular Aβ have successfully reduced levels of Aβ in the brain, but this has not correlated with an improvement or slowing of clinical progression of AD in most cases, albeit in already symptomatic subjects [54–58]. Though Aβ is posited to be the initial trigger for AD, most, if not all studies agree that Aβ is present even in brains of neurologically healthy aged individuals. Also interestingly, Aβ is conserved in organisms across kingdoms over 400 million years, including prokaryotes [59].
New studies suggest that Aβ may actually serve a protective, innate immune function role as an antimicrobial peptide (AMP) [60]. In vitro, the Aβ42 and Aβ40 (differently cleaved forms of Aβ peptide) oligomers were shown to potently inhibit the growth of the yeast Candida albicans, the gram-negative bacteria Escherichia coli, and gram-positive bacteria Staphylococcus epidermidis, Staphylococcus aureus, Streptococcus pneumoniae, Streptococcus agalactiae, Enterococcus faecalis, and Listeria monocytogenes. No inhibitory activity was detected against Streptococcus mitis, Streptococcus pyogenes, Streptococcus salivarius, or Pseudomonas aeruginosa [61]. The same group then examined the antimicrobial activity of Aβ in vivo in mouse and nematode models [62]. They found that Aβ oligomers bound to microbial cell wall carbohydrates and ultimately entrapped Salmonella typhimurium in the mouse model, and similarly interacted with Candida albicans in the nematode model.
A separate group went on to demonstrate that Aβ has antiviral properties as well [63, 64]. Infected neuroglioma H4 cells with herpes simplex virus-1 (HSV-1) resulted in the secretion of Aβ42 and the production of the inflammatory cytokines TNF-α and IL-1β. When they transferred the conditioned media from the HSV-1 infected cells to a de novo neuroglioma culture, the cells were refractory to HSV-1 infection. Subsequently, Eimer et al. went on to show that Aβ oligomers bound HSV-1 and human herpesvirus 6A and 6B surface glycoproteins, which caused the rapid seeding and deposition of additional Aβ [65]. Amyloid has been known to be elevated in HIV-infected brains as well and was recently shown to bind HIV-1 [66]. Interestingly, HIV-1 gag protein promotes secretase-dependent cleavage of the amyloid precursor protein (APP) into Aβ40 and Aβ42 isoforms. Viruses that are not known to establish latency in the CNS also are recognized by Aβ. In vitro, respiratory syncytial virus (RSV) was shown to bind amyloid [67], and Aβ42 was demonstrated to inhibit influenza A viral replication [68]. No studies on Aβ and WNV infection have been reported, but given the broad antimicrobial activity of Aβ and the neuroinvasive nature of WNV, it can be reasonably deduced that there may be increased Aβ production due to WNV infection, as a protective means for viral containment.
Evidence is also mounting for a potential antimicrobial role of α-synuclein (Asyn), the protein associated with PD. Asyn has been shown to have antimicrobial activity against bacteria (E. coli, S. aureus) and fungi (Aspergillusflavus, A.fumigatus, Rhizoctonia solani) [69], and infection of Asyn-knockout mice with WNV resulted in dramatically increased viral titers in the brain compared to wild-type control mice, with a 95% mortality rate (vs. 20% in controls) [70••]. The authors replicated these findings with Venezuelan equine encephalitis (VEE) virus (family Togaviridae, genus Alphavirus) vaccine strain TC-83 in the Asyn-knockout mice; TC-83 is not neuroinvasive in wild-type mice, demonstrating that Asyn restricts neuroinvasion. The authors further linked this to humans by showing increased Asyn expression in primary neuronal cell cultures infected with WNV.
CD-1 outbred mice, when infected with Western equine encephalitis virus (WEEV) and rescued with passive immunotherapy (WEEV E1 antibodies) [71], provided additional evidence of a link between viral infection and neurodegenerative proteinopathies. This served as a model of recovered alphaviral infection, with demonstration of complete viral clearance from the CNS 8 weeks p.i. The recovered mice had Asyn aggregates in the hippocampus, cortex, substantia nigra and mammillary bodies, and deficits consistent with parkinsonism (tremors, dyskinesia, rigidity) as well. The mice also had a 4- to 6-fold upregulation of genes linked to AD (APP, phospholipase D1 and D2, Akt1, and Pten) and PD (Park7, Aldh1a1, Irgm1). Chronic microglial and astrocyte activation, also seen in AD [72] and PD [73], were observed in the recovered brains.
While Aβ and Asyn may function as antimicrobial peptides, when produced in excess, or if clearance mechanisms fail, these proteins can trigger a neurotoxic cascade. Therapeutic interventions to clear these proteins have not yet produced clinical improvement or slowing of decline in AD, though some argue that patients need to undergo therapy earlier, in the presymptomatic state [74]. Anti-a-synuclein therapies (e.g., monoclonal antibodies) are currently in clinical trials [75, 76]; it remains to be seen if these will be successful.
Microglial Activation and Synaptic Engulfment
The cellular endpoint along the path of neurodegeneration constitutes the loss of neurons and synapses. Direct neuronal injury and death result from the acute phase of neuroinvasive West Nile disease [77]. Following viral clearance, however, progressive neurodegeneration is caused by complement-mediated microglial synaptic engulfment [78••] that shares similarity to the neurotoxic cascades seen in AD [72, 79–82] and PD [73]. Complement activation plays an important role in limiting WNV replication, though it can also lead to excessive inflammation [83]. All three complement pathways (classical, lectin, and alternative pathways) are engaged during WNV infection and lead to neutralization of virions and lysis of WNV-infected cells [84]. Mice depleted of microglia prior to WNV infection have increased mortality and elevated CNS viral titers following infection, demonstrating that microglia are crucial in limiting WNV disease [85]. However, Vasek et al. recently demonstrated that in mice infected with an attenuated WNV strain, complement C1q in concert with activated microglia led to neurodegeneration [78••]. The authors noted error-prone slower learning in the infected animals when submitted to spatial memory testing, which was attributed to persistent phagocytic microglia that drove complement-mediated (C1qA) CA3 synaptic terminal engulfment and remodeling in the hippocampus. An accompanying examination of postmortem brain tissue from patients with WNV neuroinvasive disease also showed reduced CA3 presynaptic terminals in the hippocampus and in the entorhinal cortex, compared to their age-matched controls [78••].
IFN-γ also plays an important role in hippocampal neurogenesis and synapse formation [86, 87]. Garber et al. demonstrated that hippocampus-resident memory WNV-specific T cells secrete IFN-γ, which activate microglia; this led to a loss ofpresynaptic terminals and spatial learning defects in the infected mice [88•]. These studies build on the mounting evidence of the importance of microglia, the brain’s resident macrophages, in AD and PD [89–91]. Indeed, it is now appreciated that there is a complex interplay between the immune system and neurodegeneration [92]. Microglia are crucial for pathogen host defense as well as neural circuit pruning [79]. Microglial activation, mediated by the pro-inflammatory cytokines IL-6, TNF-α, and IFN-γ, is also critical for clearance of Aβ deposits [93–95], whereas anti-inflammatory cytokines IL-4 and IL-10 actually promote the deposition of Aβ [96, 97]. Proper microglial function is therefore protective in AD, but prolonged activation may lead to synaptoxicity and neuronal injury [92]. Based on murine models, excessive accumulation of Aβ andhyperphosphorylated tau may induce microglia into this pathological activation [98]. Furthermore, in murine AD models, microglia can induce astrocytes into a neurotoxic state via release of IL-1α, TNF-α, and C1q complement [98]. In WNV, reactive changes in microglia and astrocytes persist for weeks after viral clearance, which may result in induction of such a pathological state as well, [78••, 99•]. Interestingly, in mice, antiviral CD8+ T cells have been shown to persist for months in the CNS in clusters; these are presumed to be prior infection hot spots, irrespective of persisting antigen [100]. Though controversial in AD, T cells and antigen presentation have been thought to play an important role in the pathogenesis of PD [101].
In PD, pro-inflammatory microglia have also been implicated in dopaminergic neuron loss, mediated by aberrant activation of the complement system [73, 102]. Bodea et al. [102] repeatedly injected mice with bacterial LPS, resulting in elevated plasma levels of TNF-α and IL-113. This, in turn, elevated the levels of these cytokines in the brain and stimulated microglia into a prolonged activated state. Expression of complement C1q, C3, and C4b was upregulated, and there was a 40% loss of dopaminergic neurons in the substantia nigra. In C3-deficient mice, no loss of dopaminergic neurons occurred, suggesting that neuron loss was mediated by complement-mediated glial phagocytosis. This is strikingly similar to the process described in AD and in WNV-infected mice above. Microglia have also been shown to engulf Asyn, though aggregated forms of Asyn may actually inhibit phagocytosis by blocking FcyR signaling leading to accumulation of extracellular Asyn [103]. In addition, the importance of microglia in PD as well as AD has emerged from genomewide association studies (GWAS), in which the triggering receptor expressed on myeloid cells 2 (TREM2) gene has been identified as a genetic risk factor for both diseases [104, 105]. TREM2, a cell surface receptor expressed on myeloid cells, promotes microglial survival, activation, cytokine release, and phagocytosis [106]. It plays a crucial role in synaptic pruning during postnatal development, but TREM2 signaling may also serve as a switch between a homeostatic and neurodegenerative microglial phenotype [107]. While research on TREM2 in WNV is nascent, Garber et al. noted increased microglial expression of TREM2 isolated from mouse brain infected with an attenuated strain of WNV [99•].
Healthy brain function has been compared to the planetary science concept of a “Goldilocks zone,” in that it may also require an immune environment that is balanced and just right [92]. CNS infections such as WNV cause a sizeable inflammatory response [108], and as such, are highly likely to disturb this delicate balance.
Disruption of the BBB
The BBB is comprised of vascular endothelial cells and basal lamina lining the microvasculature of the CNS and the pericytes and astrocytic foot processes that surround them. Inter-endothelial tight junctions, lack of fenestration, and specialized transport mechanisms are key features of the BBB [109]. These serve to minimize the entry of pathogens, fluid, and entrained molecules into the CNS. A compromised BBB, therefore, can lead to an influx of pathogens and neurotoxic agents with trigger CNS inflammation and possibly neurodegeneration [110, 111]. Murine studies have demonstrated that amyloid protein upregulates the gelatinase matrix metalloproteinase-9 (MMP-9), which in turn degrades the basal lamina and the tight junction in acute injury (though MMPs also play a role in angiogenesis and remodeling) [112]. A detailed examination was provided in a recent study using a 3D AD BBB model with neurons expressing mutations in the amyloid precursor protein (APP) and PSEN1 genes [113]. This model demonstrated increased BBB permeability due to diminished expression of tight junction proteins, resulting from increased levels of matrix metalloproteinase-2, reactive oxygen species (ROS), interferon-γ (IFN-γ), and aggregation of Aβ onto the endothelium [113].
The BBB plays a central role in WNV infection as well. Early host responses are triggered when WNV activates pattern recognition receptors (PRRs), including Toll-like receptors (TLR 3, TLR 7), RIG-I-like receptors (RLRs), and NODlike receptors [114]. This results in the production of type I and III interferons (IFNs) as well as other antiviral cytokines. In mice, type 1 and III IFNs promote the integrity of the BBB by enhancing tight junction formation, thereby restricting the neuroinvasive capacity of WNV [115]. However, type IIIFN (IFN-γ) secreted by T cells during WNV infection compromises the BBB by promoting transendothelial migration of CD4+ T cells [116] as well as by causing junctional breakdown and cell-cell separation [117]. This is a double-edged sword, in that T cells are crucial for WNV clearance in the CNS [118].
In addition to IFNs, TNF-α mediates BBB permeability in WNV infection. In a murine model of WNV CNS infection, γδ T cells proliferate by day 2 after WNV infection. These cells reduce viral burden and inhibit viral CNS infiltration by secretion of IFN-γ and TGF-β, respectively, but simultaneously increase BBB permeability by producing TNF-α [119]. These pro-inflammatory and anti-inflammatory effector functions are mediated by different γδ T cell subsets. In aged mice, Vγ4+ cells comprise a larger proportion compared to younger mice, and it is these cells that secrete TNF-α, which compromises the BBB [119]. TNF-α induces BBB endothelial cells to increase expression of adhesion molecules (i.e., selectins, vascular endothelial cell adhesion molecule-1 (VCAM-1), intercellular cell adhesion molecule-1 (ICAM-1)) [120]. This leads to endothelial adhesion of circulating leukocytes, which in turn produce reactive oxygen species and proteases that damage the microvessels [121]. Activated microglia also produce TNF-α, as well as IL-1|3, reactive oxygen species, and nitric oxide, all of which contribute to enhancing BBB permeability [122].
Thus, WNV infection leads to the production of cytokines that promote BBB integrity as well as permeability, and host factors, such as age, modulate the balance of the two. Indeed, normal aging in itself appears to be associated with BBB breakdown [123]. Compared to young mice, aged mice demonstrated significantly higher levels of inflammation (TNF-α), leading to extravasation of circulating IgG in the cortex and hippocampus—mostly attributable to diminished tight junction complex expression [123]. Hypertension is also a significant factor in BBB breakdown, mediated by angiotensin II-driven downregulation of key BBB protein constituents (endothelial barrier antigen, transferrin receptor) [124]. Diabetes mellitus also impairs the BBB integrity [125]; in vitro models of the BBB under hyperglycemic conditions demonstrated the expression of numerous pro-inflammatory cytokines (TNF-α, IL-6, IL-1, and IL-4) in astrocytes [125], which may also act to compromise the tight junction complex. It is no coincidence, then, that WNV neuroinvasive disease occurs predominantly in the elderly, and that hypertension and diabetes mellitus are prominent risk factors as well [126–128].
From this, we can surmise that a compromised BBB will be more permissive to the entry of WNV as well as other microbes and injurious molecules, which in turn activate microglia and lead to the upregulation of Aβ and/or Asyn as antimicrobial peptides. Persistent microglial activation and excess production, or lack of clearance of Aβ and/or Asyn may lead to complement-mediated synaptoxicity, and ultimately neurodegeneration.
Conclusion
The frequency with which cognitive and psychomotor deficits persist long after WNV exposure, coupled with the continued transmission of WNV in the USA and worldwide, underscores the importance of understanding the pathways underlying these impairments. Furthermore, the world is experiencing the emergence of ongoing outbreaks due to other neuroinvasive arboviruses such as Zika virus, eastern equine encephalitis virus, VEE virus, and Japanese encephalitis virus, to name just a few. Many of these are also associated with long-term neurological sequelae [129,130]. While there are no therapies nor prognoses available at present, critical progress has been made in recent years to assess the relationship between neuroinvasive viral infection and neurodegenerative diseases.
It is becoming clear that there are convergent mechanisms resulting in neuronal loss [131]. The data thus far suggest that hosts who are older, have diabetes, hypertension, or other disorders associated with BBB-damage are more susceptible to neuroinvasive WNV infection and subsequent neurological sequelae, as well as to neurodegenerative diseases. In the case of WNV, this may be mediated by enhanced viral entry into the CNS, but additional factors play an important role in residual cognitive and psychomotor impairments. Abnormal BBB permeability may lead to CNS entry of peripheral antigens that activate microglia, which then precipitates a neurodegenerative phenotype that results in synaptotoxicity [132, 133]. Infection stimulates production of Aβ and Asyn, which aggregate in the setting of a viral or bacterial brain infection [59, 60]. The inciting infection and initial host response lead to an aberrant inflammatory response, excess elevation of Aβ and/or Asyn, and in time, neurodegenerative changes [59] (Box 1, Fig. 1).
Box 1. Proposed pathways underlying WNV-mediated reduction in neural resilience and cognitive damage leading to neurodegenerative dementias:
Neuroinvasion → complement mediated neuronal damage and synaptotoxicity
White matter injury → impaired cognition
Chronic inflammation → dysfunctional immunity → reduced Aβ clearance increased hyperphosphorylated tau
Impaired BBB → vascular instability → impaired brain metabolism
Selective neuronal vulnerability and progressive brain organ failure
Fig. 1.
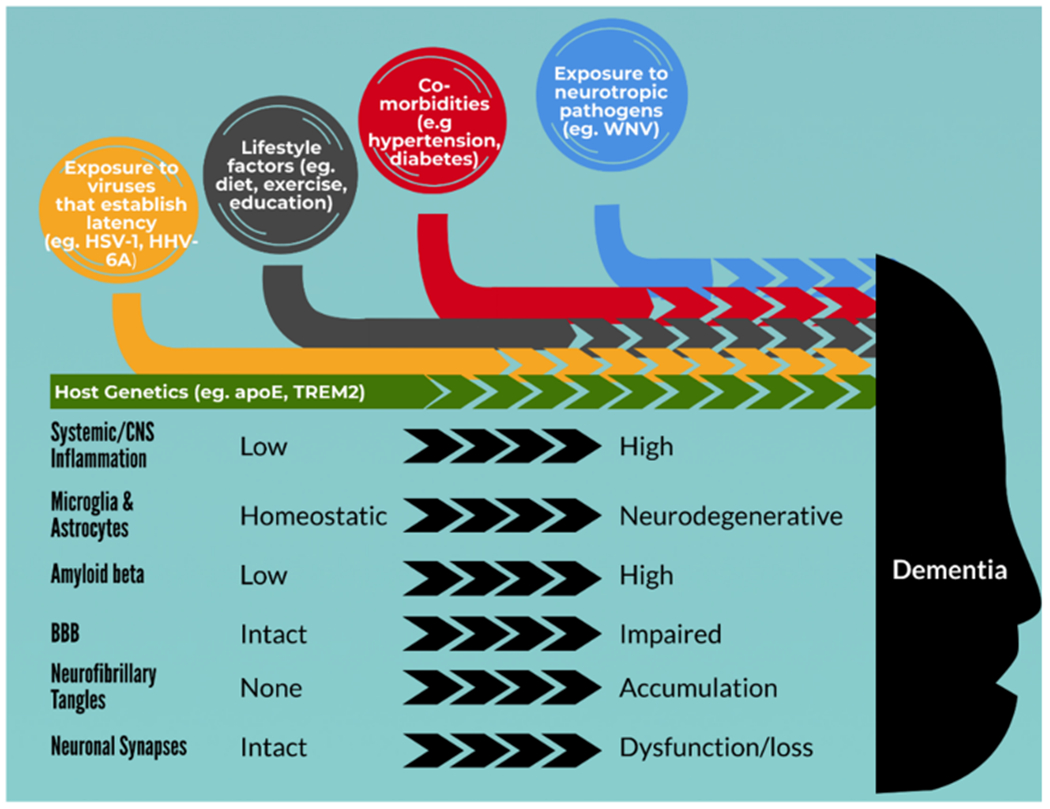
Conceptual model of the role of WNV and the multifactorial cascade leading to neurodegenerative dementia
This proposed sequence is not limited to WNV. Early work connecting an infectious agent to AD was performed by Itzhaki et al. in the 1990s, who examined the role of herpes simplex virus-1 (HSV-1) in AD [134, 135]. Her group found that the presence of HSV-1 DNA, coupled with the presence of an apolipoprotein E4 allele (apoE; apoE4 is strongly associated with AD), was correlated with AD. A subsequent population-based study examined the association between HSV-1 and AD, reporting twice the risk for AD in reactivated HSV-1 (defined by the presence of HSV-1 IgM) as well as an interaction with ApoE4 [136, 137]. An expanding suite of pathogens has since been identified that promotes this pattern of neurodegeneration, including spirochetes [138], HIV [139–141], Chlamydia pneumoniae [142], Porphyromonas gingivalis [143], human herpesvirus-6A (HHV-6A), and HHV-7 [144]. Post-encephalitic parkinsonism and encephalitis lethargica, conditions that significantly overlap with PD, have been associated with infectious etiologies for a century [145]. In this sense, an infectious association with PD has long been entertained. Specific pathogens that have been considered include influenza A virus [146], Porphyromonas gingivalis [147], Helicobacter pylori [148], hepatitis C virus [149] , and gram-negative bacterial gastrointestinal infections [150] .
Moving forward, studies that further our mechanistic understanding are crucial. ApoE is ofparticular interest in its potential ability to modify the effect of a neurotropic virus. Apolipoprotein E (ApoE) is a cholesterol carrier that binds to cell surface receptors to deliver lipids as well as the Aβ peptide in the CNS [151]. The apolipoprotein E E4 allele, which confers a strong risk for AD, has also been associated with higher CNS viral loads [152]. In a murine model of acute HSV-1 infection, apoE4 mice had a 10-fold increased number of HSV-1 genomic copies compared to apoE3 mice, and apoE3 mice had in turn a 10-fold higher number of genomic copies than mice lacking apoE [152]. The HSV-1 genomic copy number is directly linked to the risk of reactivation [153], thus explaining how apoE may influence HSV-1 reactivation. While no such connection has been made to WNV, the capsid protein of another flavivirus, dengue virus, has been shown to bind to the apoE component of very low-density lipoprotein (VLDL) and thought to thus “hitch a ride” to distant organs [154].
Looking to modify disease outcomes, could we learn about neuroprotective factors and resilience from people fully recovered from neuroinvasive pathogens? What mechanisms promote a return to microglial homeostasis in such individuals? And while there is epidemiological and MRI evidence for cognitive and other neurological deficits in WNV-exposed patients suggesting syndromes similar to those seen in neurodegenerative diseases, data on WNV exposure in people with known AD or PD are lacking. In AD as well as psychiatric diseases, efforts to study the “brain microbiome” are expanding with the increasing recognition that microbes are permanent residents ofthe CNS and can also distally influence a wide range of neurological functions [155]. Such studies will help to establish the role played by latent microbes such as HSV-1, HHV-6A/B, and HHV-7. However, these studies will not address infections that are transient but leave a lasting imprint, as described for WNV. To understand the impact of these types of pathogens, carefully designed epidemiological studies will prove important in assessing the magnitude of the link between neuroinvasive viral exposure and subsequent neurodegeneration. Bringing together experts in infectious disease, neuroscience and neurology will help deepen our understanding of the intricacies of how pathogens may trigger neurodegenerative cascades, ultimately with the aim of improving outcomes for people living with chronic sequelae of infectious diseases as well as patients devastated by dementias.
Footnotes
This article is part of the Topical Collection on Emerging Tropical Diseases
Conflict of Interest The authors declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent This article does not contain any studies with human or animal subjects performed by any of the authors.
References
Papers of particular interest, published recently, have been highlighted as:
• Of importance
•• Of major importance
- 1.Chancey C, Grinev A, Volkova E, Rios M. The global ecology and epidemiology of West Nile virus. Biomed Res Int. 2015;2015: 376230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petersen LR, Carson PJ, Biggerstaff BJ, Custer B, Borchardt SM, Busch MP. Estimated cumulative incidence of West Nile virus infection in US adults, 1999-2010. Epidemiol Infect. 2013;141 (3):591–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petersen LR, Brault AC, Nasci RS. West Nile virus: review of the literature. JAMA. 2013;310(3):308–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Murray KO, Garcia MN, Rahbar MH, Martinez D, Khuwaja SA, Arafat RR, et al. Survival analysis, long-term outcomes, and percentage of recovery up to 8 years post-infection among the Houston West Nile virus cohort. PLoS One. 2014;9(7):e102953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carson PJ, Konewko P, Wold KS, Mariani P, Goli S, Bergloff P, et al. Long-term clinical and neuropsychological outcomes of West Nile virus infection. Clin Infect Dis. 2006;43(6):723–30. [DOI] [PubMed] [Google Scholar]
- 6.Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer’s disease. Nat Immunol. 2015;16(3):229–36. [DOI] [PubMed] [Google Scholar]
- 7.Sejvar JJ, Haddad MB, Tierney BC, Campbell GL, Marfin AA, Van Gerpen JA, et al. Neurologic manifestations and outcome of West Nile virus infection. JAMA. 2003;290(4):511–5. [DOI] [PubMed] [Google Scholar]
- 8.Klee AL, Maidin B, Edwin B, Poshni I, Mostashari F, Fine A, et al. Long-term prognosis for clinical West Nile virus infection. Emerg Infect Dis. 2004;10(8):1405–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ou AC, Ratard RC. One-year sequelae in patients with West Nile Virus encephalitis and meningitis in Louisiana. J La State Med Soc. 2005;157(1):42–6. [PubMed] [Google Scholar]
- 10.Watson JT, Pertel PE, Jones RC, Siston AM, Paul WS, Austin CC, et al. Clinical characteristics and functional outcomes of West Nile fever. Ann Intern Med. 2004;141(5):360–5. [DOI] [PubMed] [Google Scholar]
- 11.Samaan Z, McDermid Vaz S, Bawor M, Potter TH, Eskandarian S, Loeb M. Neuropsychological impact of West Nile virus infection: an extensive neuropsychiatric assessment of 49 cases in Canada. PLoS One. 2016;11(6):e0158364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kathleen Y, Haaland JS, Pergam S, Echevarria LA, Davis LE, Goade D, et al. Mental status after West Nile virus infection. Emerg Infect Dis. 2006;12(8):1260–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sadek JR, Pergam SA, Harrington JA, Echevarria LA, Davis LE, Goade D, et al. Persistent neuropsychological impairment associated with West Nile virus infection. J Clin Exp Neuropsychol. 2010;32(1):81–7. [DOI] [PubMed] [Google Scholar]
- 14.••.Murray KO, Nolan MS, Ronca SE, Datta S, Govindarajan K, Narayana PA, et al. The neurocognitive and MRI outcomes of West Nile Virus infection: preliminary analysis using an external control group. Front Neurol. 2018;9:111. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study examines neuropsychological testing and MRI findings in patients an average of 4 years after their initial illness onset. Normative controls are used as a comparison group for the MRI portion of the study. The authors report significant frontal, temporal, and limbic cortical thinning in the WNV participants.
- 15.Sejvar JJ, Curns AT, Welburg L, Jones JF, Lundgren LM, Capuron L, et al. Neurocognitive and functional outcomes in persons recovering from West Nile virus illness. J Neuropsychol. 2008;2(Pt 2):477–99. [DOI] [PubMed] [Google Scholar]
- 16.Sheppard DP, Woods SP, Hasbun R, Salazar L, Nolan MS, Murray KO. Does intra-individual neurocognitive variability relate to neuroinvasive disease and quality of life in West Nile Virus? J Neuro-Oncol. 2018;24(4):506–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Balakrishnan A, Thekkekara RJ, Tandale BV. Outcomes of West Nile encephalitis patients after 1 year of West Nile encephalitis outbreak in Kerala, India: a follow-up study. J Med Virol. 2016;88(11):1856–61. [DOI] [PubMed] [Google Scholar]
- 18.Fray PJ, Robbins TW. CANTAB battery: proposed utility in neurotoxicology. Neurotoxicol Teratol. 1996;18(4):499–504. [DOI] [PubMed] [Google Scholar]
- 19.Rabadi MH. Brought down by a mosquito? West Nile Virus encephalitis. Am J Med. 2018;131(9):1064–6. [DOI] [PubMed] [Google Scholar]
- 20.Pradhan S, Anand S, Choudhury SS. Cognitive behavioural impairment with irreversible sensorineural deafness as a complication of West Nile encephalitis. J Neurovirol. 2019;25(3):429–33. [DOI] [PubMed] [Google Scholar]
- 21.Lyons JL, Schaefer PW, Cho TA, Azar MM. Case 34-2017. A 76-year-old man with fever, weight loss, and weakness. N Engl J Med. 2017;377(19):1878–86. [DOI] [PubMed] [Google Scholar]
- 22.Guth JC, Futterer SA, Hijaz TA, Liotta EM, Rosenberg NF, Naidech AM, et al. Pearls & oy-sters: bilateral thalamic involvement in West Nile virus encephalitis. Neurology. 2014;83(2):e16–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brilla R, Block M, Geremia G, Wichter M. Clinical and neuroradiologic features of 39 consecutive cases of West Nile Virus meningoencephalitis. J Neurol Sci. 2004;220(1–2):37–40. [DOI] [PubMed] [Google Scholar]
- 24.Racsa L, Gander R, Chung W, Southern P, Le J, Beal S, et al. Clinical features of West Nile virus epidemic in Dallas, Texas, 2012. Diagn Microbiol Infect Dis. 2014;78(2):132–6. [DOI] [PubMed] [Google Scholar]
- 25.Ocal M, onder H, Arsava EM, Alp S, Ozkul A, Ergunay K. A case of central nervous system infection due to West Nile Virus lineage-1 in Ankara province, Turkey. Mikrobiyol Bul. 2013;47(1):164–72. [DOI] [PubMed] [Google Scholar]
- 26.Mainali S, Afshani M, Wood JB, Levin MC. The natural history of West Nile virus infection presenting with West Nile virus meningoencephalitis in a man with a prolonged illness: a case report. J Med Case Rep. 2011;5:204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abraham A, Ziv S, Drory VE. Posterior reversible encephalopathy syndrome resulting from Guillain-Barre-like syndrome secondary to West Nile virus infection. J Clin Neuromuscul Dis. 2011;12(3): 113–7. [DOI] [PubMed] [Google Scholar]
- 28.Jain N, Fisk D, Sotir M, Kehl KS. West Nile encephalitis, status epilepticus and West Nile pneumonia in a renal transplant patient. Transpl Int. 2007;20(9):800–3. [DOI] [PubMed] [Google Scholar]
- 29.Bosanko CM, Gilroy J, Wang AM, Sanders W, Dulai M, Wilson J, et al. West Nile Virus encephalitis involving the substantia nigra: neuroimaging and pathologic findings with literature review. Arch Neurol. 2003;60(10):1448–52. [DOI] [PubMed] [Google Scholar]
- 30.DeBiasi RL, Parsons JA, Grabert BE. West Nile virus meningoencephalitis in an immunocompetent adolescent. Pediatr Neurol. 2005;33(3):217–9. [DOI] [PubMed] [Google Scholar]
- 31.Ali M, Safriel Y, Sohi J, Llave A, Weathers S. West Nile virus infection: MR imaging findings in the nervous system. AJNR Am J Neuroradiol. 2005;26(2):289–97. [PMC free article] [PubMed] [Google Scholar]
- 32.Rosas H, Wippold FJ 2nd. West Nile virus: case report with MR imaging findings. AJNR Am J Neuroradiol. 2003;24(7):1376–8. [PMC free article] [PubMed] [Google Scholar]
- 33.Brener ZZ, et al. Acute renal failure in a patient with West Nile viral encephalitis. Nephrol Dial Transplant. 2007;22(2):662–3. [DOI] [PubMed] [Google Scholar]
- 34.Shepherd JC, Subramanian A, Montgomery RA, Samaniego MD, Gong G, Bergmann A, et al. West Nile virus encephalitis in a kidney transplant recipient. Am J Transplant. 2004;4(5):830–3. [DOI] [PubMed] [Google Scholar]
- 35.Farnaes L, Schiff D, McElroy A, Coufal NG, Crawford JR, Cannavino C. Encephalitis and thalamic injury from neuroinvasive West Nile Virus in children on treatment for acute lymphoblastic leukemia. Pediatr Neurol. 2018;80:84–7. [DOI] [PubMed] [Google Scholar]
- 36.Petropoulou KA, Gordon SM, Prayson RA, Ruggierri PM. West Nile virus meningoencephalitis: MR imaging findings. AJNR Am J Neuroradiol. 2005;26(8):1986–95. [PMC free article] [PubMed] [Google Scholar]
- 37.Hwang J, Ryu H-S, Kim H, Lee S-A. The first reported case ofWest Nile encephalitis in Korea. J Korean Med Sci. 2015;30(3):343–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kadkhoda K, Embil JM, McKibbin LR, McEachern J, Michael A. Drebot West Nile Virus infection in a renal transplant recipient resulting in polioencephalomylelitis, quadriplegia, and global brain atrophy. IDCases. 2019;17:e00551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ledig C, Schuh A, Guerrero R, Heckemann RA, Rueckert D. Structural brain imaging in Alzheimer’s disease and mild cognitive impairment: biomarker analysis and shared morphometry database. Sci Rep. 2018;8(1):11258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Helmich RC, Vaillancourt DE, Brooks DJ. The future of brain imaging in Parkinson’s disease. J Park Dis. 2018;8(s1):S47–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Verma S, Lo Y, Chapagain M, Lum S, Kumar M, Gurjav U, et al. West Nile virus infection modulates human brain microvascular endothelial cells tight junction proteins and cell adhesion molecules: transmigration across the in vitro blood-brain barrier. Virology. 2009;385(2):425–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Diamond MS, Shrestha B, Mehlhop E, Sitati E, Engle M. Innate and adaptive immune responses determine protection against disseminated infection by West Nile encephalitis virus. Viral Immunol. 2003;16(3):259–78. [DOI] [PubMed] [Google Scholar]
- 43.Samuel MA, Wang H, Siddharthan V, Morrey JD, Diamond MS. Axonal transport mediates West Nile virus entry into the central nervous system and induces acute flaccid paralysis. Proc Natl Acad Sci U S A. 2007;104(43):17140–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shrestha B, Gottlieb D, Diamond MS. Infection and injury of neurons by West Nile encephalitis virus. J Virol. 2003;77(24): 13203–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hayes EB, Sejvar JJ, Zaki SR, Lanciotti RS, Bode AV, Campbell GL. Virology, pathology, and clinical manifestations of West Nile virus disease. Emerg Infect Dis. 2005;11(8):1174–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Diniz JA, da Rosa AP, Guzman H, Xu F, Xiao SY, Popov VL, et al. West Nile virus infection of primary mouse neuronal and neuroglial cells: the role of astrocytes in chronic infection. Am J Trop Med Hyg. 2006;75(4):691–6. [PubMed] [Google Scholar]
- 47.Lesteberg KE, Beckham JD. Immunology of West Nile virus infection and the role of alpha-synuclein as a viral restriction factor. Viral Immunol. 2019;32(1):38–47. [DOI] [PubMed] [Google Scholar]
- 48.Suthar MS, Diamond MS, Gale M Jr. West Nile virus infection and immunity. Nat Rev Microbiol. 2013;11(2):115–28. [DOI] [PubMed] [Google Scholar]
- 49.Appler KK, Brown AN, Stewart BS, Behr MJ, Demarest VL, Wong SJ, et al. Persistence of West Nile virus in the central nervous system and periphery of mice. PLoSOne. 2010;5(5):e10649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stewart BS, Demarest VL, Wong SJ, Green S, Bernard KA. Persistence of virus-specific immune responses in the central nervous system of mice after West Nile virus infection. BMC Immunol. 2011;12:6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Matthews KA, Xu W, Gaglioti AH, Holt JB, Croft JB, Mack D, et al. Racial and ethnic estimates of Alzheimer’s disease and related dementias in the United States (2015-2060) in adults aged >/=65 years. Alzheimers Dement. 2019;15(1):17–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nelson PT, Alafuzoff I, Bigio EH, Bouras C, Braak H, Cairns NJ, et al. Correlation of Alzheimer disease neuropathologic changes with cognitive status: a review of the literature. J Neuropathol Exp Neurol. 2012;71(5):362–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1991;6(4):487–98. [DOI] [PubMed] [Google Scholar]
- 54.Doody RS, Farlow M, Aisen PS, Alzheimer’s Disease Cooperative Study Data Analysis and Publication Committee. Phase 3 trials of solanezumab and bapineuzumab for Alzheimer’s disease. N Engl J Med. 2014;370(15):1460. [DOI] [PubMed] [Google Scholar]
- 55.Doody RS, Raman R, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Alzheimer’s Disease Cooperative Study Steering Committee, Siemers E, Sethuraman G, Mohs R; Semagacestat Study Group. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N Engl J Med. 2013;369(4):341–50. [DOI] [PubMed] [Google Scholar]
- 56.Doody RS, Thomas RG, Farlow M, Iwatsubo T, Vellas B, Joffe S, et al. Phase 3 trials of solanezumab for mild-to-moderate Alzheimer’s disease. N Engl J Med. 2014;370(4):311–21. [DOI] [PubMed] [Google Scholar]
- 57.LaMotte S Another promising Alzheimer’s drug trial ends in failure: This one hurts: CNN Health; 2019. [Google Scholar]
- 58.Salloway S, Sperling R, Gilman S, Fox NC, Blennow K, Raskind M, et al. A phase 2 multiple ascending dose trial of bapineuzumab in mild to moderate Alzheimer disease. Neurology. 2009;73(24): 2061–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gosztyla ML, Brothers HM, Robinson SR. Alzheimer’s amyloid-beta is an antimicrobial peptide: a review of the evidence. J Alzheimers Dis. 2018;62(4):1495–506. [DOI] [PubMed] [Google Scholar]
- 60.Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement. 2018;14(12):1602–14. [DOI] [PubMed] [Google Scholar]
- 61.Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5(3):e9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, et al. Moir RD Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016;8(340):340–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bourgade K, Dupuis G, Frost EH, Fulop T. Anti-viral properties of amyloid-beta peptides. J Alzheimers Dis. 2016;54(3):859–78. [DOI] [PubMed] [Google Scholar]
- 64.Bourgade K, Le Page A, Bocti C, Witkowski JM, Dupuis G, Frost EH, et al. Protective effect of amyloid-beta peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J Alzheimers Dis. 2016;50(4):1227–41. [DOI] [PubMed] [Google Scholar]
- 65.Eimer WA, Vijaya Kumar DK, Navalpur Shanmugam NK, Rodriguez AS, Mitchell T, Washicosky KJ, et al. Alzheimer’s disease-associated beta-amyloid is rapidly seeded by herpesviridae to protect against brain infection. Neuron. 2018;100(6):1527–32. [DOI] [PubMed] [Google Scholar]
- 66.Chai Q, Jovasevic V, Malikov V, Sabo Y, Morham S, Walsh D, et al. HIV-1 counteracts an innate restriction by amyloid precursor protein resulting in neurodegeneration. Nat Commun. 2017;8(1):1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ezzat K, Pernemalm M, Palsson S, Roberts TC, Jarver P, Dondalska A, et al. The viral protein corona directs viral pathogenesis and amyloid aggregation. Nat Commun. 2019;10(1): 2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.White MR, Kandel R, Tripathi S, Condon D, Qi L, Taubenberger J, et al. Alzheimer’s associated beta-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS One. 2014;9(7):e101364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Park SC, Moon JC, Shin SY, Son H, Jung YJ, Kim NH, et al. Functional characterization of alpha-synuclein protein with antimicrobial activity. Biochem Biophys Res Commun. 2016;478(2): 924–8. [DOI] [PubMed] [Google Scholar]
- 70.••.Beatman EL, Massey A, Shives KD, Burrack KS, Chamanian M, Morrison TE, et al. Alpha-synuclein expression restricts RNA viral infections in the brain. J Virol. 2015;90(6):2767–82 [DOI] [PMC free article] [PubMed] [Google Scholar]; This murine study shows that neuronal alpha-synuclein inhibited WNV infection in the CNS, and that deletion of the gene for alpha-synuclein gene resulted in decreased survival and increased CNS viral burden and neuronal injury. This article speaks directly to the hypothesis that asyn is an antimicrobial peptide.
- 71.Bantle CM, Phillips AT, Smeyne RJ, Rocha SM, Olson KE, Tjalkens RB. Infection with mosquito-borne alphavirus induces selective loss of dopaminergic neurons, neuroinflammation and widespread protein aggregation. NPJ Parkinsons Dis. 2019;5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Hong S, Beja-Glasser VF, Nfonoyim BM, Frouin A, Li S, Ramakrishnan S, et al. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 2016;352(6286):712–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tremblay ME, Cookson MR, Civiero L. Glial phagocytic clearance in Parkinson’s disease. Mol Neurodegener. 2019;14(1):16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Golde TE, DeKosky ST, Galasko D. Alzheimer’s disease: the right drug, the right time. Science. 2018;362(6420):1250–1. [DOI] [PubMed] [Google Scholar]
- 75.Jankovic J, Goodman I, Safirstein B, Marmon TK, Schenk DB, Koller M, et al. Safety and tolerability of multiple ascending doses of PRX002/RG7935, an anti-alpha-synuclein monoclonal antibody, in patients with Parkinson disease: a randomized clinical trial. JAMA Neurol. 2018;75(10):1206–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brys M, Fanning L, Hung S, Ellenbogen A, Penner N, Yang M, et al. Randomized phase I clinical trial of anti-alpha-synuclein antibody BIIB054. Mov Disord. 2019;34(8):1154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Chambers TJ, Diamond MS. Pathogenesis of flavivirus encephalitis. Adv Virus Res. 2003;60:273–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.••.Vasek MJ, Garber C, Dorsey D, Durrant DM, Bollman B, Soung A, et al. A complement-microglial axis drives synapse loss during virus-induced memory impairment. Nature. 2016;534(7608):538–43 [DOI] [PMC free article] [PubMed] [Google Scholar]; This murine study found impaired spatial learning, attributable to persistent phagocytic microglia that drove complement-mediated synaptic loss in the hippocampus following infection with an attenuated WNV strain. An accompanying examination of human postmortem brain tissue showed reduced CA3 presynaptic terminals in the hippocampus and entorhinal cortex associated with WNV neuroinvasive disease, compared to age-matched controls.
- 79.Schafer DP, Lehrman EK, Kautzman AG, Koyama R, Mardinly AR, Yamasaki R, et al. Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 2012;74(4):691–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wu T, Dejanovic B, Gandham VD, Gogineni A, Edmonds R, Schauer S, et al. Complement C3 is activated in human AD brain and is required for neurodegeneration in mouse models of amyloidosis and tauopathy. Cell Rep. 2019;28(8):2111–23 e6. [DOI] [PubMed] [Google Scholar]
- 81.Lian H, Yang L, Cole A, Sun L, Chiang AC, Fowler SW, et al. NFκB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron. 2015;85(1):101–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Canchi S, Raao B, Masliah D, Rosenthal SB, Sasik R, Fisch KM, et al. Integrating gene and protein expression reveals perturbed functional networks in Alzheimer’s disease. Cell Rep. 2019;28(4):1103–16 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Conde JN, Silva EM, Barbosa AS, Mohana-Borges R. The complement system in Flavivirus infections. Front Microbiol. 2017;8:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mehlhop E, Whitby K, Oliphant T, Marri A, Engle M, Diamond MS, et al. Complement activation is required for induction of a protective antibody response against West Nile virus infection. J Virol. 2005;79(12):7466–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Seitz S, Clarke P, Tyler KL. Pharmacologic depletion of microglia increases viral load in the brain and enhances mortality in murine models of flavivirus-induced encephalitis. J Virol. 2018;92(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kim IJ, Beck HN, Lein PJ, Higgins D. Interferon gamma induces retrograde dendritic retraction and inhibits synapse formation. J Neurosci. 2002;22(11):4530–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Li L, Walker TL, Zhang Y, Mackay EW, Bartlett PF. Endogenous interferon gamma directly regulates neural precursors in the noninflammatory brain. J Neurosci. 2010;30(27):9038–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.•.Garber C, Soung A, Vollmer LL, Kanmogne M, Last A, Brown J, et al. Tcells promotemicroglia-mediated synaptic elimination and cognitive dysfunction during recovery from neuropathogenic flaviviruses. NatNeurosci. 2019;22(8):1276–88 [DOI] [PMC free article] [PubMed] [Google Scholar]; In a murine model of West Nile neuroinvasive disease (WNND)-induced cognitive dysfunction, the authors demonstrate that T cell-derived IFN-γ signaling in microglia gives rise to spatial-learning defects and presynaptic termini elimination with lack of repair.
- 89.Nott A, Holtman IR, Coufal NG, CM SJ, Yu M, Hu R, et al. Brain cell type-specific enhancer-promoter interactome maps and disease risk association. Science. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Labzin LI, Heneka MT, Latz E. Innate immunity and neurodegeneration. Annu Rev Med. 2018;69:437–49. [DOI] [PubMed] [Google Scholar]
- 91.Salter MW, Stevens B. Microglia emerge as central players in brain disease. Nat Med. 2017;23(9):1018–27. [DOI] [PubMed] [Google Scholar]
- 92.Golde TE. Harnessing immunoproteostasis to treat neurodegenerative disorders. Neuron. 2019;101(6):1003–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chakrabarty P, Ceballos-Diaz C, Beccard A, Janus C, Dickson D, Golde TE, et al. IFN-gamma promotes complement expression and attenuates amyloid plaque deposition in amyloid beta precursor protein transgenic mice. J Immunol. 2010;184(9):5333–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chakrabarty P, Herring A, Ceballos-Diaz C, Das P, Golde TE. Hippocampal expression of murine TNFα results in attenuation of amyloid deposition in vivo. Mol Neurodegener. 2011;6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chakrabarty P, Jansen-West K, Beccard A, Ceballos-Diaz C, Levites Y, Verbeeck C, et al. Massive gliosis induced by interleukin-6 suppresses Aβ deposition in vivo: evidence against inflammation as a driving force for amyloid deposition. FASEB J. 2010;24(2):548–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chakrabarty P, Li A, Ceballos-Diaz C, Eddy JA, Funk CC, Moore B, et al. IL-10 alters immunoproteostasis in APP mice, increasing plaque burden and worsening cognitive behavior. Neuron. 2015;85(3):519–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Chakrabarty P, Tianbai L, Herring A, Ceballos-Diaz C, Das P, Golde TE. Hippocampal expression of murine IL-4 results in exacerbation of amyloid deposition. Mol Neurodegener. 2012;7:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hansen DV, Hanson JE, Sheng M. Microglia in Alzheimer’s disease. J Cell Biol. 2018;217(2):459–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.•.Garber C, Vasek MJ, Vollmer LL, Sun T, Jiang X, Klein RS. Astrocytes decrease adult neurogenesis during virus-induced memory dysfunction via IL-1. Nat Immunol. 2018;19(2):151–61 [DOI] [PMC free article] [PubMed] [Google Scholar]; In a murine model of WNND-induced cognitive dysfunction, the authors demonstrate that there are fewer neuroblasts and increased astrogenesis without recovery of hippocampal neurogenesis at 30 days, mediated by IL-1 secreted predominantly by pro-inflammatory astrocytes.
- 100.Wakim LM, Woodward-Davis A, Bevan MJ. Memory T cells persisting within the brain after local infection show functional adaptations to their tissue of residence. Proc Natl Acad Sci U S A. 2010;107(42):17872–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sulzer D, Alcalay RN, Garretti F, Cote L, Kanter E, Agin-Liebes J, et al. T cells from patients with Parkinson’s disease recognize alpha-synuclein peptides. Nature. 2017;546(7660):656–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bodea LG, Wang Y, Linnartz-Gerlach B, Kopatz J, Sinkkonen L, Musgrove R, et al. Neurodegeneration by activation of the microglial complement-phagosome pathway. J Neurosci. 2014;34(25):8546–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Choi YR, Kang SJ, Kim JM, Lee SJ, Jou I, Joe EH, et al. FcyRIIB mediates the inhibitory effect of aggregated α-synuclein on microglial phagocytosis. Neurobiol Dis. 2015;83:90–9. [DOI] [PubMed] [Google Scholar]
- 104.Lill CM, Rengmark A, Pihlstrom L, Fogh I, Shatunov A, Sleiman PM, et al. The role of TREM2 R47H as a risk factor for Alzheimer’s disease, frontotemporal lobar degeneration, amyotrophic lateral sclerosis, and Parkinson’s disease. Alzheimers Dement. 2015;11(12):1407–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW, et al. TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson’s disease. Mol Neurodegener. 2013;8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Colonna M, Wang Y. TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat Rev Neurosci. 2016;17(4): 201–7. [DOI] [PubMed] [Google Scholar]
- 107.Krasemann S, Madore C, Cialic R, Baufeld C, Calcagno N, el Fatimy R, et al. The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity. 2017;47(3):566–81 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Schafer A, Brooke CB, Whitmore AC, Johnston RE. The role of the blood-brain barrier during Venezuelan equine encephalitis virus infection. J Virol. 2011;85(20):10682–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Obermeier B, Verma A, Ransohoff RM. The blood-brain barrier. Handb Clin Neurol. 2016;133:39–59. [DOI] [PubMed] [Google Scholar]
- 110.Sweeney MD, Sagare AP, Zlokovic BV. Blood-brain barrier breakdown in Alzheimer disease and other neurodegenerative disorders. Nat Rev Neurol. 2018;14(3):133–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sweeney MD, Kisler K, Montagne A, Toga AW, Zlokovic BV. The role of brain vasculature in neurodegenerative disorders. Nat Neurosci. 2018;21(10):1318–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Seo JH, Guo S, Lok J, Navaratna D, Whalen MJ, Kim KW, et al. Neurovascular matrix metalloproteinases and the blood-brain barrier. Curr PharmDes. 2012;18(25):3645–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Shin Y, Choi SH, Kim E, Bylykbashi E, Kim JA, Chung S, et al. Blood-brain barrier dysfunction in a 3D in vitro model of Alzheimer’s disease. Adv Sci (Weinh). 2019;6(20):1900962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Luo H, Wang T. Recent advances in understanding West Nile virus host immunity and viral pathogenesis. F1000Res. 2018;7:338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Daniels BP, Holman DW, Cruz-Orengo L, Jujjavarapu H, Durrant DM, Klein RS. Viral pathogen-associated molecular patterns regulate blood-brain barrier integrity via competing innate cytokine signals. MBio. 2014;5(5):e01476–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sonar SA, Shaikh S, Joshi N, Atre AN, Lal G. IFN-gamma promotes transendothelial migration of CD4(+) T cells across the blood-brain barrier. Immunol Cell Biol. 2017;95(9):843–53. [DOI] [PubMed] [Google Scholar]
- 117.Bonney S, Seitz S, Ryan CA, Jones KL, Clarke P, Tyler KL, et al. Gamma interferon alters junctional integrity via Rho kinase, resulting in blood-brain barrier leakage in experimental viral encephalitis. MBio. 2019;10(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sitati EM, Diamond MS. CD4+ T-cell responses are required for clearance of West Nile virus from the central nervous system. J Virol. 2006;80(24):12060–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wang T, Welte T. Role ofnatural killer and Gamma-delta T cells in West Nile virus infection. Viruses. 2013;5(9):2298–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Frijns CJ, Kappelle LJ. Inflammatory cell adhesion molecules in ischemic cerebrovascular disease. Stroke. 2002;33(8):2115–22. [DOI] [PubMed] [Google Scholar]
- 121.Wang F, Zou Z, Gong Y, Yuan D, Chen X, Sun T. Regulation of human brain microvascular endothelial cell adhesion and barrier functions by memantine. J Mol Neurosci. 2017;62(1):123–9. [DOI] [PubMed] [Google Scholar]
- 122.Dudvarski Stankovic N, Teodorczyk M, Ploen R, Zipp F, Schmidt MHH. Microglia-blood vessel interactions: a double-edged sword in brain pathologies. Acta Neuropathol. 2016;131(3):347–63. [DOI] [PubMed] [Google Scholar]
- 123.Elahy M, Jackaman C, Mamo JC, Lam V, Dhaliwal SS, Giles C, et al. Blood-brain barrier dysfunction developed during normal aging is associated with inflammation and loss of tight junctions but not with leukocyte recruitment. Immun Ageing. 2015;12:2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Biancardi VC, Son SJ, Ahmadi S, Filosa JA, Stern JE. Circulating angiotensin II gains access to the hypothalamus and brain stem during hypertension via breakdown of the blood-brain barrier. Hypertension. 2014;63(3):572–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Prasad S, Sajja RK, Naik P, Cucullo L. Diabetes mellitus and blood-brain barrier dysfunction: an overview. Aust J Pharm. 2014;2(2):125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Badawi A, Velummailum R, Ryoo SG, Senthinathan A, Yaghoubi S, Vasileva D, et al. Prevalence of chronic comorbidities in dengue fever and West Nile virus: a systematic review and meta-analysis. PLoS One. 2018;13(7):e0200200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Hart J Jr, Tillman G, Kraut MA, Chiang HS, Strain JF, Li Y, et al. NIAID Collaborative Antiviral Study Group West Nile Virus 210 Protocol Team. West Nile virus neuroinvasive disease: neurological manifestations and prospective longitudinal outcomes. BMC Infect Dis. 2014;14:248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Jean CM, Honarmand S, Louie JK, Glaser CA. Risk factors for West Nile virus neuroinvasive disease, California, 2005. Emerg Infect Dis. 2007;13(12):1918–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ronca SE, Dineley KT, Paessler S. Neurological sequelae resulting from encephalitic alphavirus infection. Front Microbiol. 2016;7:959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lebov JF, Brown LM, MacDonald P, Robertson K, Bowman NM, Hooper SR, et al. Review: evidence of neurological sequelae in children with acquired Zika virus infection. Pediatr Neurol. 2018;85:16–20. [DOI] [PubMed] [Google Scholar]
- 131.Liddelow SA, Barres BA. Reactive astrocytes: production, function, and therapeutic potential. Immunity. 2017;46(6):957–67. [DOI] [PubMed] [Google Scholar]
- 132.Clarke LE, Liddelow SA, Chakraborty C, Munch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci USA. 2018;115(8):E1896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Itzhaki RF, Lin WR, Shang D, Wilcock GK, Faragher B, Jamieson GA. Herpes simplex virus type 1 in brain and risk of Alzheimer’s disease. Lancet. 1997;349(9047):241–4. [DOI] [PubMed] [Google Scholar]
- 135.Jamieson GA, Maitland NJ, Wilcock GK, Craske J, Itzhaki RF. Latent herpes simplex virus type 1 in normal and Alzheimer’s disease brains. J Med Virol. 1991;33(4):224–7. [DOI] [PubMed] [Google Scholar]
- 136.Lovheim H, Gilthorpe J, Adolfsson R, Nilsson LG, Elgh F. Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimers Dement. 2015;11(6):593–9. [DOI] [PubMed] [Google Scholar]
- 137.Lovheim H, Norman T, Weidung B, Olsson J, Josefsson M, Adolfsson R, et al. Herpes simplex virus, APOEvarepsilon4, and cognitive decline in old age: results from the Betula cohort study. J Alzheimers Dis. 2019;67(1):211–20. [DOI] [PubMed] [Google Scholar]
- 138.Miklossy J, Khalili K, Gem L, Ericson RL, Darekar P, Bolle L, et al. Borrelia burgdorferi persists in the brain in chronic lyme neuroborreliosis and may be associated with Alzheimer disease. J Alzheimers Dis. 2004;6(6):639–49 discussion 673–81. [DOI] [PubMed] [Google Scholar]
- 139.Stanley LC, Mrak RE, Woody RC, Perrot LJ, Zhang S, Marshak DR, et al. Glial cytokines as neuropathogenic factors in HIV infection: pathogenic similarities to Alzheimer’s disease. J Neuropathol Exp Neurol. 1994;53(3):231–8. [DOI] [PubMed] [Google Scholar]
- 140.Esiri MM, Biddolph SC, Morris CS. Prevalence of Alzheimer plaques in AIDS. J Neurol Neurosurg Psychiatry. 1998;65(1):29–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Green DA, Masliah E, Vinters HV, Beizai P, Moore DJ, Achim CL. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. AIDS. 2005;19(4):407–11. [DOI] [PubMed] [Google Scholar]
- 142.Balin BJ, Gérard HC, Arking EJ, Appelt DM, Branigan PJ, Abrams JT, et al. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med Microbiol Immunol. 1998;187(1):23–42. [DOI] [PubMed] [Google Scholar]
- 143.Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, et al. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5(1):eaau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Readhead B, Haure-Mirande JV, Funk CC, Richards MA, Shannon P, Haroutunian V, et al. Multiscale analysis of independent Alzheimer’s cohorts finds disruption of molecular, genetic, and clinical networks by human herpesvirus. Neuron. 2018;99(1): 64–82 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vilensky JA, Gilman S, McCall S. Does the historical literature on encephalitis lethargica support a simple (direct) relationship with postencephalitic Parkinsonism? Mov Disord. 2010;25(9):1124–30. [DOI] [PubMed] [Google Scholar]
- 146.Takahashi M, Yamada T. Viral etiology for Parkinson’s disease-a possible role of influenza a virus infection. Jpn J Infect Dis. 1999;52(3):89–98. [PubMed] [Google Scholar]
- 147.Adams B, Nunes JM, Page MJ, Roberts T, Carr J, Nell TA, et al. Parkinson’s disease: a systemic inflammatory disease accompanied by bacterial inflammagens. Front Aging Neurosci. 2019;11:210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Shen X, Yang H, Wu Y, Zhang D, Jiang H. Meta-analysis: association of Helicobacter pylori infection with Parkinson’s diseases. Helicobacter. 2017;22(5). [DOI] [PubMed] [Google Scholar]
- 149.Wijarnpreecha K, Chesdachai S, Jaruvongvanich V, Ungprasert P. Hepatitis C virus infection and risk of Parkinson’s disease: a systematic review and meta-analysis. Eur J Gastroenterol Hepatol. 2018;30(1):9–13. [DOI] [PubMed] [Google Scholar]
- 150.Matheoud D, Cannon T, Voisin A, Penttinen AM, Ramet L, Fahmy AM, et al. Intestinal infection triggers Parkinson’s disease-like symptoms in Pink1(−/−) mice. Nature. 2019;571(7766):565–9. [DOI] [PubMed] [Google Scholar]
- 151.Liu CC, Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013;9(2):106–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Burgos JS, Ramirez C, Sastre I, Valdivieso F. Effect of apolipoprotein E on the cerebral load of latent herpes simplex virus type 1 DNA. J Virol. 2006;80(11):5383–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Hoshino Y, Pesnicak L, Cohen JI, Straus SE. Rates of reactivation of latent herpes simplex virus from mouse trigeminal ganglia ex vivo correlate directly with viral load and inversely with number of infiltrating CD8+ T cells. J Virol. 2007;81(15):8157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Martins AS, Carvalho FA, Faustino AF, Martins IC, Santos NC. West Nile virus capsid protein interacts with biologically relevant host lipid systems. Front Cell Infect Microbiol. 2019;9:-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Mayer EA, Knight R, Mazmanian SK, Cryan JF, Tillisch K. Gut microbes and the brain: paradigm shift in neuroscience. J Neurosci. 2014;34(46):15490–6. [DOI] [PMC free article] [PubMed] [Google Scholar]