

Abstract
Patients with common variable immunodeficiency associated with autoimmune cytopenia (CVID+AIC) generate few isotype-switched B cells with severely decreased frequencies of somatic hypermutations (SHMs), but their underlying molecular defects remain poorly characterized. We identified a CVID+AIC patient who displays a rare homozygous missense M466V mutation in β-catenin–like protein 1 (CTNNBL1). Because CTNNBL1 binds activation-induced cytidine deaminase (AID) that catalyzes SHM, we tested AID interactions with the CTNNBL1 M466V variant. We found that the M466V mutation interfered with the association of CTNNBL1 with AID, resulting in decreased AID in the nuclei of patient EBV-transformed B cell lines and of CTNNBL1 466V/V Ramos B cells engineered to express only CTNNBL1 M466V using CRISPR/Cas9 technology. As a consequence, the scarce IgG+ memory B cells from the CTNNBL1 466V/V patient showed a low SHM frequency that averaged 6.7 mutations compared with about 18 mutations per clone in healthy-donor counterparts. In addition, CTNNBL1 466V/V Ramos B cells displayed a decreased incidence of SHM that was reduced by half compared with parental WT Ramos B cells, demonstrating that the CTNNBL1 M466V mutation is responsible for defective SHM induction. We conclude that CTNNBL1 plays an important role in regulating AID-dependent antibody diversification in humans.
Keywords: Autoimmunity, Immunology
Keywords: Autoimmune diseases, B cells
Introduction
Isotype-switched high-affinity antibodies are essential for protection against a vast range of pathogens. Activation-induced cytidine deaminase (AID) is the enzyme that mediates somatic hypermutation (SHM) and class-switch recombination (CSR), 2 processes resulting in the production of high-affinity, mutated isotype-switched antibodies (1, 2). As a consequence, AID deficiency in mice and humans is characterized by a susceptibility to bacterial infections associated with a lack of serum protective isotype-switched antibodies and memory B cells that are devoid of SHMs (1, 2). The absence of functional AID also prevents the establishment of central and peripheral B cell tolerance and leads to the emergence of autoreactive B cells that often produce autoantibodies targeting erythrocytes and platelets associated with the development of autoimmune cytopenias (AICs) or other self-antigens characteristic of arthritis and systemic lupus erythematosus (3–6).
Patients with common variable immunodeficiency (CVID) also suffer from recurrent infections resulting from decreased serum antibody production and low frequencies of isotype-switched B cells (7, 8). Paradoxically, about 20% of CVID patients display defective early B cell tolerance checkpoints and secrete autoantibodies targeting erythrocytes and/or platelets, which result in AIC development (7, 9). The analysis of the scarce circulating IgG+ B cells from CVID patients with autoimmune cytopenias (CVID+AIC) has revealed very low frequencies of SHM compared with counterparts in patients only affected by CVID, whereas CD27+IgA+ B cells are virtually absent in CVID+AIC patients (10). CVID+AIC patients are therefore unable to restrict gut microbiota to the intestine, exhibit hyperplastic germinal centers (GCs) also observed in AID deficiency, and produce autoreactive VH4-34–expressing IgG+ B cell clones that may recognize both commensal bacteria and I/i self-antigens expressed on erythrocytes, platelets, and other hematopoietic cells (6, 10–14). The genetic defects associated with a failure to properly induce SHM in B cells from most patients with CVID+AIC remain unknown.
Here we describe a CVID+AIC patient with progressive severe hypogammaglobulinemia who harbors a rare, homozygous, damaging missense variant allele in CTNNBL1, which encodes a single M466V amino acid replacement in the β-catenin–like protein 1 (CTNNBL1) protein. CTNNBL1 is a member of the armadillo-containing protein family and is part of the pre-mRNA processing factor 19 (Prp19) spliceosome through binding to the N-terminus of cell division cycle 5–like (CDC5L) (15–17). CTNNBL1 has also been shown to interact with AID, and AID mutations abolishing its interaction with CTNNBL1 reduce IgV diversification in chicken DT40 cells, suggesting that CTNNBL1 may play an important role in regulating AID function (18). Using patient-derived B cells and CRISPR/Cas9-engineered CTNNBL1 466V/V Ramos B cells, we provide evidence that the M466V mutation decreases CTNNBL1’s interaction with AID and its nuclear translocation, which results in defective SHM and CSR in human B cells.
Results
Sequencing the whole exome of a CVID patient with AIC identifies a CTNNBL1 homozygous mutation.
The patient is a 15-year-old Hispanic female born to nonconsanguineous parents who presented in early life with progressive hypogammaglobulinemia, AICs, and recurrent infections and was therefore diagnosed with CVID+AIC (Table 1, Supplemental Table 1, and Methods for detailed clinical presentation; supplemental material available online with this article; https://doi.org/10.1172/JCI131297DS1). Exome sequencing revealed a homozygous missense mutation at position Chr20(hg19):g.36488304A>G in exon 14 of the gene encoding CTNNBL1, resulting in a single amino acid change from methionine to valine at position 466 (NM_030877.4:c.1396A>G, M466V; Figure 1, A and B). This methionine 466 is near the C-terminal domain of CTNNBL1 and is conserved among species besides rodents that display an isoleucine, another bulky hydrophobic residue (Figure 1, C and D). The variant is very rare, with a minor allelic frequency of 7.97 × 10–6 and no homozygotes in the gnomAD database (19). At the time of this publication, no other human disease–causing mutation in CTNNBL1 has been reported to our knowledge. Because CTNNBL1 is part of the spliceosome complex, which associates with AID that catalyzes SHM in B cells, we investigated whether the CTNNBL1 M466V mutation could alter AID function and impair SHM and possibly CSR (18, 20–22).
Table 1. Immunological characteristics of the CTNNBL1 466V/V patient.
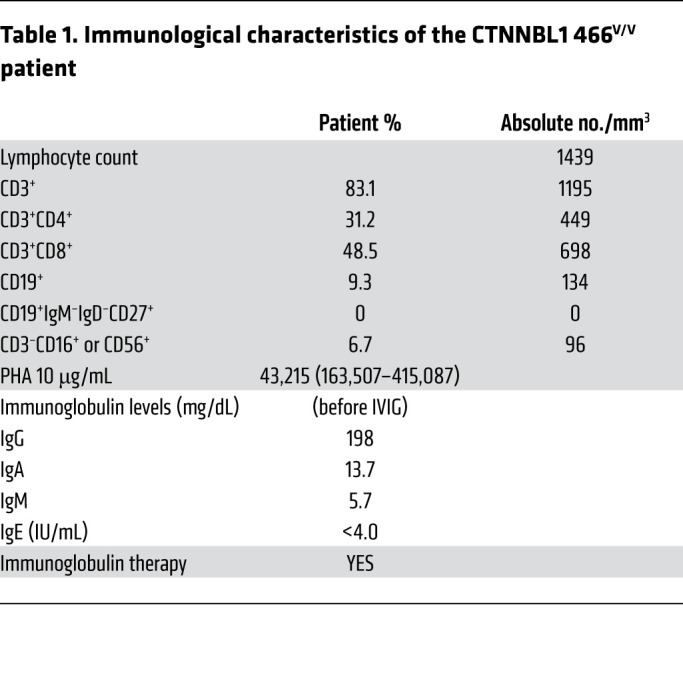
Figure 1. Homozygous CTNNBL1 mutation in a patient with CVID+AIC.

(A) Family pedigree with homozygous CTNNBL1 M466V mutation. The patient is II.2. (B) Confirmation of single nucleotide substitution Chr20(hg19):36488304A>G by Sanger sequencing (highlighted). The CTNNBL1 region was amplified from gDNA from the patient and 3 relatives. Representative chromatograms are shown. (C) Schematic representation of the CTNNBL1 protein structure. Numbers indicate amino acid residue numbers. BLNS, bipartite nuclear localization sequence; NAM, N-terminal anchoring motive; NTD, N-terminal domain; ARM, armadillo repeats; CTD, C-terminal domain. (D) Multiple sequence alignment of human CTNNBL1 and its orthologues. The M466 residue of human CTNNBL1 (top row) and corresponding residues in other species are highlighted.
To determine whether the rare M466V variant is a pathogenic mutation, we first assessed potential functional consequences by examining its effect on CTNNBL1’s interaction with AID (Figure 2). Using CRISPR/Cas9 technology, we engineered Ramos B cells to carry the same biallelic A>G change in CTNNBL1 so that CTNNBL1 466V/V Ramos B cells only express the CTNNBL1 variant of the patient (Supplemental Figure 1). We then immunoprecipitated patient EBV-derived B lymphocyte cell lines (BLCLs) and CTNNBL1 466V/V Ramos B cells with an anti-CTNNBL1 antibody and tested by Western blot CTNNBL1 expression and interactions with AID and CDC5L, a spliceosome component that binds CTNNBL1 (Figure 2 and ref. 20). Comparisons were made by studying other EBV-immortalized B cell lines derived from 3 different healthy donors, an AID-deficient patient (AID–/–), and a uracil N-glycosylase–deficient (UNG-deficient) patient (UNG–/–), as well as unmodified CTNNBL1M/M Ramos B cells and CRISPR/Cas9-edited AID–/– Ramos B cells that lack AID expression (Figure 2, Supplemental Figures 1 and 2, and refs. 23, 24). We found that CTNNBL1 expression in both the patient EBV BLCLs and the CTNNBL1 466V/V Ramos B cells was similar to expression in control EBV-derived BLCLs and Ramos B cells, showing that the M466V missense mutation does not appear to alter the CTNNBL1 cellular pool (Figure 2). However, AID association with CTNNBL1 was severely decreased in patient EBV-derived BLCLs and only a quarter of AID-CTNNBL1 complexes were detected in these cells compared with EBV BLCLs from healthy donors (Figure 2, A and B, and Supplemental Figure 2). As anticipated, we did not observe an AID interaction with CTNNBL1 in AID–/– EBV BLCLs (Figure 2, A and B, and Supplemental Figure 2). Putative differences in AID expression in patient versus control EBV-derived BLCLs did not account for decreased AID binding to the CTNNBL1 466V variant because AID expression in the total lysates of patient EBV BLCLs was found to be similar to control B cell lines (Figure 2A). We also excluded the possibility that decreased AID-CTNNBL1 complexes in patient EBV BLCLs was due to differences in B cell development and the presence of isotype-switched B cells in EBV BLCLs derived from healthy donors because a similar conclusion was reached when comparing patient cells with EBV BLCLs derived from an UNG-deficient patient who also suffers from severe hypogammaglobulinemia due to a lack of isotype-switched B cells (Supplemental Figure 2 and ref. 23). In contrast, CDC5L’s interaction with CTNNBL1 was unaffected by the M466V missense mutation because equal amounts of CDC5L were detected after CTNNBL1 immunoprecipitation in EBV BLCLs from controls and patient (Figure 2, A and B, and Supplemental Figure 2). The analysis of Ramos B cell lines confirmed results obtained with EBV-derived BLCLs in that the M466V mutation decreased AID’s interaction with CTNNBL1 by half in CTNNBL1 466V/V Ramos B cells compared with unmodified CTNNBL1 466M/M control Ramos B cells, whereas AID was detected at approximately similar levels in the total lysates of both Ramos B cell lines (Figure 2, C and D). CDC5L binding to CTNNBL1 was found to be unaffected by the M466V mutation in Ramos B cell lines because similar amounts of CDC5L-CTNNBL1 complexes were detected in all Ramos B cell variants (Figure 2, C and D). Of note, immunoprecipitation of CDC5L showed its association with CTNNBL1 in the absence of AID, suggesting that CDC5L binding to CTNNBL1 does not require AID, which may not be part of CDC5L-CTNNBL1 complexes (Supplemental Figure 3, A and B). Our experiments therefore show that the M466V mutation decreases the ability of CTNNBL1 to interact with AID but does not alter CTNNBL1’s association with CDC5L.
Figure 2. CTNNBL1 M446V mutation decreases AID association with CTNNBL1.

(A) Lysates of EBV BLCLs from a healthy donor (HD), CTNNBL1 466V/V patient, or AID-deficient patient were immunoprecipitated with an anti-CTNNBL1 antibody; precipitates and total cell lysates were analyzed by Western blotting with the indicated antibodies (shown are representative blots). (B) Summary of densitometric quantification of at least 6 independent experiments. Represented are values relative to HDs, indicated by dashed lines; bars represent the mean. (C) Lysates of parental WT, CTNNBL1 466V/V, and AID–/– Ramos B cells were analyzed as in A (shown are representative blots). (D) Summary of densitometric quantification of 5 independent experiments. Represented are values relative to parental WT cells, indicated by dashed lines; bars represent the mean.
The CTNNBL1 M466V mutation decreases AID transport in the nucleus.
AID cellular localization is predominantly cytoplasmic, and AID needs to translocate to the nucleus to alter DNA and initiate molecular events resulting in SHM and CSR (25–27). Active nuclear transport of AID is mediated by a nuclear localization signal (NLS), and CTNNBL1 is a known NLS-binding and splicing-associated factor that has been shown to colocalize with AID to traffic and localize to the nucleus (21). We therefore investigated the impact of the CTNNBL1 M466V mutation on the nuclear localization of AID. We first assessed the stability of the CTNNBL1 variants by cellular thermal shift assay (CETSA). Ligand-bound proteins remain in solution when heat treated, whereas the unbound or poorly bound proteins denature (28). We found that the M466V mutation decreased the stability of CTNNBL1, as illustrated by the loss of CTNNBL1 in nuclear lysates of CTNNBL1 466V/V Ramos B cells at lower temperature than in wild-type (WT) CTNNBL1 cells (Figure 3A). CTNNBL1 stability was unaffected by the absence of AID expression in AID–/– Ramos B cells, suggesting that the decreased stability of CTNNBL1 466V is not the result of decreased AID binding by this variant (Figure 3A). We then analyzed CTNNBL1, AID, and CDC5L cellular localization in various Ramos B cell lines using Western blots with nuclear and cytosolic lysates characterized by the presence of lamin B1 and β-tubulin, respectively (Figure 3B). The CTNNBL1 M466V mutation impairs nuclear import of AID (Figure 3B). Both CTNNBL1 and CDC5L spliceosome proteins were enriched in the nucleus, whereas AID was found in the cytosol and to a lesser extent in the nucleus, as previously reported (20). Although CTNNBL1 and CDC5L nuclear pools were similar in all Ramos B cell lines, AID nuclear detection was decreased by half in CTNNBL1 466V/V Ramos B cells compared with parental WT CTNNBL1 466M/M counterparts and absent in AID–/– Ramos B cells, as expected (Figure 3, B and C). Hence, the CTNNBL1 M466V mutation specifically diminishes AID but not CDC5L nuclear localization.
Figure 3. CTNNBL1 M446V mutation interferes with CTNNBL1 thermal stability and reduces AID nuclear localization.

(A) Thermal treatment of parental WT, CTNNBL1 466V/V, and AID–/– Ramos B cells at indicated temperatures; nuclear lysates were analyzed by Western blotting with anti-CTNNBL1 antibody. (B) Nuclear and cytosolic lysates of WT, CTNNBL1 466V/V, and AID–/– Ramos B cells were analyzed by Western blotting with the indicated antibodies and (C) 4 independent experiments were quantified by densitometry. Represented are values relative to those of parental WT control indicated by dashed lines. Shown are representative blots from at least 3 independent experiments. Western blot of AID shows different exposures for nuclear and cytosolic fractions; bars represent the mean.
The CTNNBL1 M466V patient displays severely decreased CSR and SHM.
We have established that the CTNNBL1 M466V mutation interferes with CTNNBL1’s interaction with AID and its nuclear localization, but its impact on CSR and SHM remained undetermined. We evaluated the production of isotype-switched IgG+ and IgA+ B cells generated by CSR and SHM frequency in IgG+ B cells isolated from the blood of the CTNNBL1 466V/V patient compared with her relatives who were all heterozygous for the CTNNBL1 variant allele (Figure 1, A and B) and unrelated healthy donors (Figure 4). We found markedly reduced frequencies of CD19+CD27+ memory B cells in the patient’s blood compared with healthy donors and relatives who displayed memory B cell proportions similar to controls, revealing the recessive nature of the CTNNBL1 M466V mutation (Figure 4, A and B). Among memory B cells, the homozygous CTNNBL1 466V/V mutation resulted in a decrease in CD19+CD27+IgG+ B cells and a virtual absence of CD19+CD27+IgA+ isotype-switched B cells, as previously observed for other CVID+AIC patients with uncharacterized genetic defects (Figure 4, A and B, and ref. 10). In agreement with impaired CSR processes in the CTNNBL1 466V/V patient, most of her CD19+CD27+ B cells expressed IgM (Figure 4B). We then investigated whether SHM was also affected by the CTNNBL1 M466V mutation by evaluating the frequency of mutations in her scarce IgG+ B cells and comparing them with those in counterparts from 14 healthy donors and 6 previously analyzed CVID+AIC patients (10). The average number of mutations in VH transcripts from CD27+IgG+ B cells from the CTNNBL1 466V/V patient was very low (6.7 mutations) and similar to those in other CVID+AIC patients (7.6 ± 2.2 mutations) compared with 18.2 ± 3.0 mutations per VH segment in healthy donors (Figure 4, C and D). Many of the CD27+IgG+ B cells from the CTNNBL1 466V/V patient appeared to display VH transcripts with even fewer SHMs than those in other CVID+AIC patients, whereas a few clones managed to accumulate large numbers of mutations potentially induced by recurrent exposure to a specific pathogen (Figure 4D). Similarly, SHM frequencies were also significantly decreased in CD27+IgM+ B cells from the CTNNBL1 466V/V patient compared with counterparts in healthy donors (Supplemental Figure 4). The analysis of the VH repertoire also revealed biased VH gene usage associated with the homozygous CTNNBL1 variant. Although VH3 is the most-used gene segment in CD27+IgG+ B cells from healthy donors, the CTNNBL1 466V/V patient’s CD27+IgG+ B cells favored clones expressing VH4-encoded antibodies like other CVID+AIC patients (Figure 4E). Among VH4 family members, VH4-34, which is known to encode autoreactive antibodies that bind the conserved I/i self-antigen on hematopoietic cells (11, 14), has recently been reported to be enriched in IgG+ B cells in patients that may fail to contain gut microbiota and which include CVID+AIC patients likely due to their defects in producing IgA antibodies (10, 13). In agreement with these observations, we found that the CD27+IgG+ B cells from the CTNNBL1 466V/V patient were also enriched in VH4-34–expressing clones (5.4%), as in other CVID+AIC patients (8.1% ± 5.4%), and contrasted with the very low frequencies (1.5% ± 2.4%) of these rare clones in healthy donors (Figure 4F).
Figure 4. CTNNBL1 466V/V patient shows defective CSR, SHM, and skewed VH4 gene segment usage in her scarce IgG+ B cell compartment.

(A) Surface CD19 and CD27 expression on PBMCs (upper panels) and IgA and IgG expression on CD19+CD27+ memory B cells (lower panels). Shown are representative flow cytometric plots from a healthy donor (HD) and a patient with the CTNNBL1 466V/V mutation. (B) Frequency of total CD19+CD27+, and IgM+, IgG+, or IgA+ memory B cells. (C) Mutations in VH transcripts from CD27+IgG+ B cells from 14 HDs, 6 CVID patients with AICs, and 1 patient with the CTNNBL1 466V/V mutation are displayed as averaged mutation number per subject. Bars represent the mean. (D) Distribution of number of mutations per sequence. (E) Pie charts represent VH family gene segment usage from pooled IgG transcripts of 14 HDs, 6 CVID patients with AICs, and 1 patient with the CTNNBL1 466V/V mutation. Number of transcripts is indicated in pie’s center. (F) Frequency of VH4-34 gene segment usage in transcripts from 14 HDs, 6 CVID patients with AICs, and 1 patient with the CTNNBL1 466V/V mutation. Bars represent the mean. **P ≤ 0.01; ****P ≤ 0.0001 by unpaired 2-tailed Student’s t test; single measurements from the CTNNBL1 466V/V patient were excluded from statistical analysis.
To further assess whether CTNNBL1 466V/V–associated SHM defects correlated with an alteration in GC development in secondary lymphoid organs, as recently reported for patients with CVID+AIC (10), we evaluated a lymph node obtained through an excisional biopsy from our patient (Supplemental Figure 5). This lymph node displayed distorted, irregularly shaped follicles (Supplemental Figure 5A), follicular hyperplasia (Supplemental Figure 5B), and many GCs that appeared to have attenuation of the mantle zones and some coalescent follicles (Supplemental Figure 5C) with some prominent tingible body macrophages and some peripheral epithelioid histiocytes (Supplemental Figure 5D). These findings were consistent with the lymph node analysis of patients with CVID+AIC (10). The expanded GC also correlated with the presence of an expanded peripheral autoimmune/autoreactive CD19hiCD21–/loCD10–CD27– B cell subset, as reported in other CVID+AIC patients (Supplemental Figure 5E and refs. 10, 29). Additional immunological analysis of the CTNNBL1 466V/V patient revealed a decreased frequency of CD3+CD4+CD25hiCD127loFoxP3+ regulatory T cells (Tregs) compared with healthy donors and her asymptomatic relatives and resembled those in CVID+AIC patients (Supplemental Figure 6, A and B, and ref. 10). However, Tregs from the CTNNBL1 466V/V patient suppressed the proliferation of stimulated T responder cells in vitro, unlike Tregs from other CVID+AIC patients (Supplemental Figure 6C and ref. 10). Finally, the hyperplastic GC reactions in both AID-deficient and CVID+AIC patients were associated with an increase in circulating CD3+CD4+CXCR5+PD-1hi T follicular helper–like (Tfh-like) T cells, a feature also identified in the CTNNBL1 466V/V patient (Supplemental Figure 6, D and E, and refs. 6, 10). Thus, the CTNNBL1 466V/V patient demonstrates defective CSR and SHM processes that are associated with hyperplastic GC reactions and a dysregulated T cell compartment characterized by decreased Treg and increased circulating Tfh-like cell frequencies previously reported in other CVID+AIC patients. In addition, her asymptomatic heterozygous relatives who display normal memory B and T cell populations reveal that CSR and SHM defects in the patient likely follow an autosomal recessive inheritance pattern.
The CTNNBL1 466V mutation is responsible for defective SHMs.
To determine whether the CTNNBL1 M466V missense mutation interferes with the acquisition of SHMs, we analyzed the emergence of SHMs in single, sorted, unmodified CTNNBL1 466M/M Ramos B cells that spontaneously undergo SHM in culture and compared it to SHM in CTNNBL1 466v/v and AID–/– counterparts. Because SHM can generate stop codons or other deleterious mutations in the immunoglobulin heavy and light chain genes, we first monitored the loss of IgM expression by flow cytometry 12 weeks after seeding sorted IgM+ single B cell clones from the 3 Ramos B cell lines (Figure 5, A and B). Although about 10% of the unmodified CTNNBL1 466M/M Ramos B cell clones lost IgM expression during the 12-week culture, over 97% of CTNNBL1 466v/v Ramos B cell clones retained IgM expression, suggesting that SHM is affected by the CTNNBL1 M466V mutation (Figure 5, A and B). We then sequenced VH transcripts amplified from a total of 572 sorted single B cells from the seeded clones cultivated for 12 weeks from the 3 Ramos B cell lines and assessed the number of unique mutations per IgM sequence. We found that the CTNNBL1 466v/v Ramos mutants displayed a significant, approximately 2-fold reduction in SHMs as compared with unmodified CTNNBL1 466M/M Ramos B cell clones (P = 0.0064; Figure 5, C and D). In addition, the measure of BrdU incorporation that quantifies cycling B cells demonstrated that proliferation rates were similar in all Ramos B cell lines, documenting that defective SHM induction resulting from the CTNNBL1 M466V mutation is not due to decreased B cell division (Figure 5E). As expected, virtually none of the AID–/– Ramos B cell clones lost IgM expression or acquired mutations (Figure 5, A–D). We conclude that the CTNNBL1 M466V mutation is likely responsible for defective SHMs.
Figure 5. The CTNNBL1 466V mutation impairs SHM in Ramos B cells.

(A) Representative flow cytometric plots of surface IgM expression on single-cell-derived colonies of WT, CTNNBL1 466V/V, and AID–/– Ramos B cells 12 weeks after seeding. (B) Summary of IgM+ cell frequencies in single-cell-derived Ramos colonies. Bars represent the mean. **P ≤ 0.01; ***P ≤ 0.001 by 1-way ANOVA followed by Tukey’s post hoc test. (C) Pie charts represent the number of newly acquired mutations per VH4-34 sequence after single-cell seeding. Number of analyzed sequences indicated in the center of the chart. (D) Summary of newly acquired mutations per sequence originating from a single cell. Each data point represents the average mutations per sequence of 1 single-cell-derived culture; bars indicate the mean. **P ≤ 0.01 comparing CTNNBL1 genotypes, obtained by 2-tailed, unpaired t test. (E) Proliferation rate of randomly selected single-cell-derived colonies measured by BrdU incorporation at indicated time points. Represented are means and SD. Significance was tested by 2-way ANOVA followed by Tukey’s post hoc test. NS, not significant.
Lentivirus-driven expression of WT CTNNBL1 restores SHM induction in CTNNBL1 466V/V Ramos B cells.
To exclude the possibility that decreased SHMs associated with the engineered CTNNBL1 M466V missense mutation in Ramos cells may be due to a CRISPR/Cas9-mediated off-target activity that might alter another gene involved in SHM regulation, we reintroduced either WT CTNNBL1 or the or 466V variant in CTNNBL1 466v/v Ramos B cells using a GFP-tagged lentiviral strategy and assessed loss of IgM expression and SHM induction in comparison with WT Ramos cells transduced with a lentivirus expressing WT CTNNBL1 (Figure 6A and ref. 30). GFP+ CTNNBL1 WT and 466v/v Ramos B cells that express lentivirus-driven CTNNBL1 variants were batch sorted 1 week after transduction and single GFP+IgM+ Ramos cells were subsequently plated the following week and cultivated for 12 weeks before assessment of IgM cell surface expression and SHM frequencies, as described above (Figure 6A). We found that the lentivirus-driven expression of WT CTNNBL1 in CTNNBL1 466v/v Ramos clones induced loss of IgM expression during the 12-week culture, similarly to that in WT Ramos clone counterparts, whereas CTNNBL1 466v/v Ramos B cells expressing the lentivirus-driven CTNNBL1 466V variant retained significantly higher IgM cell surface expression, suggesting that the reintroduction of WT CTNNBL1 in 466v/v Ramos B cells favored the generation of deleterious mutations that abrogate IgM production (CTNNBL1 466v/v Ramos + lentivirus-driven WT vs. 466V CTNNBL1, P = 0.0257; Figure 6, B and C). In agreement with these observations, sequencing VH transcripts amplified from 1,060 sorted single B cells from these seeded clones revealed that the lentivirus-driven expression of WT CTNNBL1 in WT and CTNNBL1 466v/v Ramos B cells induced an average of 0.458 and 0.694 mutation per VH transcript, respectively, compared with only 0.266 mutation per VH transcript in CTNNBL1 466v/v Ramos B cells transduced with lentiviruses encoding the CTNNBL1 466V variant and which therefore displayed a significant, approximately 2- to 3-fold reduction in SHM (WT Ramos + lentivirus-driven WT CTNNBL1 vs. CTNNBL1 466v/v Ramos + lentivirus-driven 466V CTNNBL1, P = 0.0199; CTNNBL1 466v/v Ramos + lentivirus-driven WT vs. 466V CTNNBL1, P < 0.0001; Figure 6, D and E). Thus, the WT CTNNBL1 variant rescues SHM impairments in CTNNBL1 466v/v Ramos B cells, which is consistent with the CTNNBL1 M466V mutation decreasing SHMs in humans.
Figure 6. Lentivirus-driven expression of WT CTNNBL1 restores SHM induction in CTNNBL1 466V/V Ramos B cells.

(A) WT and CTNNBL1 466V/V Ramos B cells were transduced with lentiviruses allowing the coexpression of GFP and either WT or 466V variants of CTNNBL1. GFP+ Ramos B cells were batch sorted 1 week after lentiviral transduction and single GFP+IgM+ Ramos cells were plated the following week and cultivated for 12 weeks before assessment of IgM cell surface expression and SHM frequencies. (B) Representative flow cytometric plots of cell surface IgM expression on GFP+ single-cell-derived colonies of WT and CTNNBL1 466V/V Ramos B cells transduced with the indicated lentivirus expressing either WT or 466V CTNNBL1 variants 12 weeks after seeding. (C) Summary of IgM+ cell frequencies in single-cell-derived Ramos colonies. (D) Pie charts represent the number of newly acquired mutations per VH4-34 sequence after single-cell seeding. Number of analyzed sequences indicated in the center of the chart. (E) Summary of newly acquired mutations per sequence originating from a single cell. Each data point represents the average mutations per sequence of 1 single-cell-derived culture; bars indicate the mean. *P ≤ 0.05; **P ≤ 0.01; ****P ≤ 0.0001 by 1-way ANOVA followed by Tukey’s post hoc test. NS, not significant.
Discussion
We report herein that a pathogenic homozygous allele in the CTNNBL1 gene impairs SHM and CSR in human B cells. Indeed, the c.1396A>G (NM_030877.4) CTNNBL1 gene change, which induces a single methionine to valine amino acid replacement at position 466, interfered with SHM in Ramos B cells and resulted in progressive hypogammaglobulinemia associated with defective isotype-switched B cell production, a failure to secrete serum IgG and IgA, and immune-mediated thrombocytopenia in the first years of the patient’s life. Because the father, mother, and brother who all carry a heterozygous CTNNBL1 variant allele are unaffected and display normal isotype-switched B cell numbers and function, we conclude that the patient has a potentially novel autosomal recessive primary immunodeficiency caused by the biallelic CTNNBL1 defect. Our patient showed striking similarities to other CVID+AIC of unknown genetic origin and AID-deficient patients because they all display hyperplastic GCs in their lymph nodes likely resulting from extended immune reactions induced by impaired high-affinity antibody production associated with defective SHMs (2, 6, 10). Although we cannot rule out the possibility that abnormal cell cycle regulation associated with defective CTNNBL1 function might contribute to this phenotype (31, 32), defective SHM and CSR in these patients led to severely reduced IgA production and a failure to control gut microbiota, which may also result in systemic immune responses targeting commensal bacteria that will also favor GC formation (6, 10, 33). This pathobiological scenario is supported by the detection of increased endotoxin plasma concentrations in IgA-deficient patients with CVID+AIC and AID deficiency and the emergence of nontolerized commensal reactive VH4-34–expressing clones in their IgG memory B cell compartment (10, 13). Because these VH4-34–encoded IgG antibodies still recognize I/i self-antigen expressed on red blood cells and platelets, they may be responsible for the elimination of these cells, thereby resulting in autoimmune cytopenia that developed in all of these patients, including our CTNNBL1 466V/V patient (10).
How does the CTNNBL1 M466V mutation interfere with SHM and CSR? CTNNBL1 is a part of Prp19-CDC5L spliceosome complexes in distinct nuclear structures called Cajal bodies (15–17). AID, which initiates SHM and CSR, has been identified as a CTNNBL1 binding partner (18, 34). Our studies confirm that CTNNBL1 binds CDC5L and AID but we found that CDC5L appears to associate with CTNNBL1 in the absence of AID. CDC5L binds the C-terminal armadillo domain VII (ARM VII) of CTNNBL1 (32, 34–37). A structural model of the AID-CTNNBL1 interaction suggests that AID binds both N- and C-terminal domains of CTNNBL1 (34). Hence, CDC5L and AID may compete for docking to the protein binding groove of CTNNBL1, resulting in the formation of CTNNBL1 complexes containing either CDC5L or AID. However, analysis of the CTNNBL1 mutation in our patient revealed that the M466V amino acid change decreases CTNNBL1’s association with AID but not CDC5L. M466 is located in the ARM VII domain of CTNNBL1 that is predicted to interact with AID residues 39–43, which are important for CTNNBL1 interaction (34). Indeed, mutations in amino acid stretch A39TSFS43 of AID abrogate the formation of AID-CTNNBL1 complexes (18). Hence, the M466V mutation likely disrupts the interaction of the C-terminal domain of CTNNBL1 with the A39TSFS43 AID motif and therefore weakens the formation of CTNNBL1-AID complexes when AID can only bind the N-terminal domain of CTNNBL1. Our data also reveal that the C-terminal domain of CTNNBL1 that binds CDC5L differs from the region that interacts with AID and which is perturbed by the M466V mutation. Of note, mutations of AID residues 39–43 important for binding CTNNBL1 do not affect AID phosphorylation at nearby position S38 by the catalytic subunit of cAMP-dependent protein kinase A (PKA), which enables binding to replication protein A (RPA) and strongly stimulates substrate deamination by AID and SHM (38–41). In addition, the S38 mutation does not alter AID binding to CTNNBL1, suggesting that AID’s interaction with RPA and CTNNBL1 can occur simultaneously (18, 39, 42). In line with this hypothesis, AID, CTNNBL1, and PKA all colocalize to Cajal bodies and nuclear speckles, the site of maturation and storage of splicing machinery (22). Hence, the analysis of the CTNNBL1 M466V variant confirms that the predicted ARM VII region of CTNNBL1 is important for interactions with AID.
Studies in human cell lines suggest that CTNNBL1 plays a role in nuclear translocation and subnuclear localization of AID into spliceosome-associated compartments and that AID nuclear shuttling occurs through an NLS-dependent import mechanism (21, 22, 27). We found that the CTNNBL1 M466V mutation decreases the AID nuclear pool without affecting global AID cellular expression, suggesting that decreased AID binding to the CTNNBL1 466V variant impairs AID shuttling to the nucleus. Decreased AID in the nucleus is likely to result in reduced AID-mediated function, i.e., SHM and CSR, as observed in our CTNNBL1 466V/V patient. In agreement with this hypothesis, it has been shown that AID haploinsufficiency in both mice and humans results in decreased AID expression and suboptimal SHM and CSR processes (5, 6, 43–45). However, previous studies investigating the role of CTNNBL1 in AID-mediated antibody diversification have yielded conflicting conclusions. Loss of CTNNBL1 expression in chicken DT40 B cells reduced AID-dependent IgV gene conversion, supporting a role for CTNNBL1 in regulating this antibody diversification processes (18). However, B cell–specific knockout of Ctnnbl1 in mice only showed a mild decrease in CSR when B cells were activated in vitro, and Ctnnbl1 knockout in the CH12F3 mouse B cell line had no effect on CSR (18, 33). In humans, the S43P missense mutation in AID identified in a patient with hyper-IgM syndrome abrogated CSR and was shown to interfere with AID binding to CTNNBL1, consistent with an important role for CTNNBL1 in regulating AID function in human B cells (18, 46). Because methionine 466 is conserved among many vertebrates, with the notable exception of rodents, where it is replaced by an isoleucine which is structurally more similar to valine (Figure 1D), this amino acid replacement at CTNNBL1 position 466 in mice may potentially account for some of the observed differences between results obtained in murine B cells compared with avian and human B cells. Analysis of CTNNBL1 466v/v Ramos B cells engineered by CRISPR/Cas9 to express only the CTNNBL1 variant with the patient’s mutation demonstrated that the M466V amino acid change impairs SHM in these human B cells without altering B cell proliferation rates. In addition, the rescue of defective SHM processes in CTNNBL1 466v/v Ramos B cells by the reintroduction of WT but not 466V CTNNBL1 strongly suggests that decreased SHM and CSR in the CTNNBL1 466v/v patient are the result of her homozygous CTNNBL1 gene defect and that impaired CTNNBL1 function causes an autosomal recessive immunodeficiency characterized by SHM and CSR impediment associated with severe CVID with AIC.
Methods
Patient and donor controls.
A Hispanic female with a history of sinopulmonary infections first presented at 2 years of age with idiopathic thrombocytopenia purpura and was treated with i.v. steroids and discharged (Supplemental Table 1). Born to nonconsanguineous parents in Columbia, she developed some growth delay, 2 other episodes of immune thrombocytopenic purpura, vitiligo, and by 8 years of age lymphadenopathy. As a part of the lymphadenopathy evaluation, she was found to have severe progressive hypogammaglobulinemia and no specific antibody responses with low B cells and absence of switched memory B cells (Table 1). The lymph node biopsy showed benign reactive follicular and intrafollicular hyperplasia with some intra- and perifollicular histiocytic infiltrates with ill-defined epithelioid cell granulomas, and no evidence of malignancy. She had persistent lymphopenia with low absolute numbers of T and B cells and poor T cell proliferation in response to phytohemagglutinin (PHA). She continues to require romiplostim to treat her steroid-resistant thrombocytopenia and IVIG for progressive severe hypogammaglobulinemia; however, the vitiligo and growth delay have resolved. The unusual nature of disease and clinical presentation prompted a genetic evaluation. Whole-exome sequencing revealed a rare homozygous missense variant NM_030877.4(CTNNBL1):c.[1396A>G];[1396A>G], which results in a single amino acid exchange: M466V in CTNNBL1. The study also includes comparison with 6 CVID+AIC patients and 15 healthy donors previously reported (10).
Generation of EBV lymphoblastoid cells.
BLCLs were generated from healthy donors and patient PBMCs by addition of EBV as previously described (47). PBMCs were transduced with B95-8 EBV supernatant and cultured for 3 weeks in RPMI medium with 10% FBS and 1 μg/mL cyclosporine A. Cell lines were confirmed mycoplasma negative before use.
Cell staining and sorting, cDNA, RT-PCR, and VH sequence analysis.
Mononuclear cells were isolated from peripheral blood using Ficoll density gradient centrifugation, and B cells were enriched using anti-CD20 microbeads (Miltenyi Biotec). B cells were stained with Pacific Blue anti–human CD19 (HIB19), PerCPCy5.5 anti–human CD27 (M-T271), APC anti–human CD21 (Bu32), and PE anti–human IgG (HP6017) (all from BioLegend). Single CD19+CD21+CD27+IgG+ and CD19+CD21+CD27+IgM+ B cells from the CTNNBL1 466V/V patient were sorted on FACSAria (Becton Dickinson) flow cytometers into 96-well PCR plates that were immediately frozen on dry ice. RNA from single cells was reverse transcribed in the original 96-well plate in 12.5-μL reactions containing Superscript II RT (Gibco BRL) for 45 minutes at 42°C. RT-PCR conditions, including primer sequences, were as described previously (5). In brief, IgG gene transcripts were amplified in 96-well plates with 2 rounds of nested PCRs using HotStart Taq DNA polymerase (QIAGEN) and 3.5 μL of cDNA as template for the first PCRs and 3.5 μL of the first PCRs as templates for the second PCRs (5). VH sequences were analyzed using the National Center for Biotechnology Information (NCBI) IGBLAST software.
Flow cytometric analysis.
The following antibodies were used for flow cytometric staining: APC-Cy7 anti-CD19 (HIB19), PerCP-Cy5.5 anti-CD27 (M-T271), Pacific Blue anti-CD21 (B-ly4), APC-Cy7 anti-CD4 (OKT4), PE-Cy7, PE or PE Dazzle anti-CD25 (BC96), PerCP-Cy5.5 or APC anti-CD127 (A019D5), Alexa Fluor 700 anti-CD45RO (UCHL1), Pacific Blue anti-CXCR5 (J252D4), PE-Cy7 anti–PD-1 (EH12.2H7) (all from BioLegend); eFluor 605NC anti-CD3 (OKT3), FITC or PerCP-Cy5.5 anti-IgM (MHM-88) (eBioscience); and APC anti-IgG (G18-145) (Becton Dickinson). Intracellular staining with Alexa Fluor 488 anti-FOXP3 (PCH101) (eBioscience) or Alexa Fluor 647 anti-FOXP3 (150D) (BioLegend) was performed using the Foxp3/Transcription Factor Staining Buffer Set (eBioscience) in accordance with the manufacturer’s instructions.
In vitro Treg suppression assay.
CD4+ T cells were enriched using the EasySep Human CD4+ T cell enrichment kit (STEMCELL). CD4+CD25hiCD127lo/– Tregs and CD3+CD4+CD25– responder T (Tresp) cells were sorted by flow cytometry and labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) (Thermo Fisher Scientific). Treg and Tresp cells were cocultured at a 1:1 ratio with beads loaded with anti-CD2, anti-CD3, and anti-CD28 (Treg Suppression Inspector, human; Miltenyi Biotec). Proliferation of viable Tresp cells was analyzed by CFSE dilution at 3.5–4.5 days.
Lymph node staining and analysis.
Hematoxylin and eosin–stained lymph node sections were analyzed at low power (×12.5) on a DM4000B microscope (Leica Biosystems), to capture the nodes’ entire 2-dimensional area with a Spot RT/SE Slider camera (Spot Imaging). All pathology images were reviewed by one or more hematopathologists. Immunohistochemical staining was performed with anti-CD21 (1:100; Dako, clone IF8), anti-CD3 (Ventana, clone SP7 [prediluted]), anti-CD20 (1:200; Dako, clone L26), and anti-BCL6 (1:50; Dako, clone PG-B6p) antibodies.
Generation of CTNNBL1 466V/V Ramos cell lines.
Guide RNAs targeting human AID CTNNBL1 were designed using the Broad Institute’s sgRNA designer (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). Guide RNAs were then subcloned into px458 (Addgene, 48138) and transiently transfected with Gene Pulser Electroporation Buffer (Bio-Rad, 1652676) into Ramos cells with an ssODN. Cells were single-cell sorted for the GFPhi population into 96-well plates 24–36 hours after transfection. After 2–3 weeks, colonies were expanded and genomic DNA prepared. To determine potential CTNNBL1 466V/V mutants, genomic DNA collected from individual colonies was analyzed by PCR for a region surrounding exon 14 of human CTNNBL1. Because successful repair from the ssODN results in the introduction of a BmgBI/Ajil site, PCR products were digested with Ajil (Thermo Fisher Scientific, ER1941). The CTNNBL1 466V/V clone was further validated by TA cloning and Sanger Sequencing.
Lentivirus-driven expression of WT and 466V CTNNBL1 variants in Ramos cells.
The pTRIP-Ubi-GFP lentiviral vector has been used for expression of WT and 466V CTNNBL1 variants. Vector constructions have been described previously (30). Lentiviral particles were produced by transient transfection of 293T cells, as previously described (48). Viruses were then used to transduce either WT or 466V/V Ramos cells in the presence of protamine sulfate (MilliporeSigma).
IgM fluctuation assay and SHM frequency assessment.
IgM-positive single Ramos cells were FACS sorted into round-bottom 96-well cell culture plates. After 12 weeks in culture, surface IgM expression was assessed on individual Ramos subclones by flow cytometry using FITC–anti–human IgM monoclonal antibody (BioLegend). Subsequently, single cells from each Ramos subclone were sorted into 96-well PCR plates as described above and immediately frozen on dry ice. VH sequences were amplified by RT-PCR and analyzed using the NCBI IGBLAST software; VH4-34 transcripts were aligned to the germ-line consensus VH4-34*01 sequence. Proliferation rates were assessed by BrdU incorporation following the manufacturer’s instructions (FITC BrdU Flow Kit, Becton Dickinson).
Cytosol-nucleus fractionation.
Cells were lysed in hypotonic buffer (20 mM Tris-HCl, pH 7.4; 10 mM NaCl, and 3 mM MgCl2). After centrifugation, the supernatant was collected as the cytosolic fraction. The nuclei were lysed in cell extraction buffer (100 mM Tris-HCl, pH 7.4; 100 mM NaCl, 1 mM EDTA, 1 mM EGTA, 10% glycerol, 1% Triton X-100, 0.1% SDS, and 0.5% sodium deoxycholate). After centrifugation, the supernatant was collected as the nuclear fraction.
CETSA.
The CETSA protocol was previously described (28). In brief, cells were distributed into PCR tubes for each temperature step. A PCR machine (Eppendorf) was preheated to the desired temperatures and cell aliquots were heated for precisely 3 minutes and then kept on ice until further processing.
Western blot analysis.
Cells were lysed in 0.1% Triton X-100 buffer (diluted in 50 mM Tris, pH 7.4; 150 mM NaCl, 1 mM sodium orthovanadate, 25 mM β-glycerophosphate, and protease and phosphatase inhibitor cocktail [Thermo Fisher Scientific, 78443]). Immunoprecipitations were performed using anti–human CTNNBL-1 [EP2669Y] (ab76243, Abcam), anti–human AID [30F12] (4949S, Cell Signaling Technology), anti–human AID [ZA001] (39-2500, Thermo Fisher Scientific), anti–human CDC5L [2136C1a] (sc-81220, Santa Cruz Biotechnology), and anti–human actin [13E5] (4970S, Cell Signaling Technology) as control. Prepared samples were electrophoresed in Mini-Protean SDS-PAGE gels (Bio-Rad) following protein quantitation by BCA Protein Assay (Thermo Fisher Scientific) according to the manufacturer’s protocol and equivalent protein amounts were loaded for each condition. Proteins were blotted onto PVDF membranes (Roche) by wet transfer. The membranes were probed with the appropriate primary and secondary antibodies, developed using enhanced chemiluminescence reagent, and images were acquired using a ChemiDoc gel documentation system. Densitometry was performed using Image Lab v6 (all Bio-Rad).
Statistics.
Statistical analysis was performed using Prism v8.4.2 (GraphPad). Data are reported as means ± SD. Differences between 2 groups of research subjects were analyzed for statistical significance with unpaired 2-tailed Student’s t test. Differences between 3 groups were analyzed by 1- or 2-way ANOVA followed by Tukey’s post hoc test. A P value of 0.05 or less was considered significant.
Study approval.
The present human studies were reviewed and approved by the Yale University institutional review board in New Haven (IRB protocol 0906005336) and by the Baylor College of Medicine institutional review board in Houston (protocol numbers H-29697 and H-21453); subjects provided written informed consent before their participation in the study.
Author contributions
MK, EC, AVL, VK, RKD, SG, DGS, and EM designed the experiments and discussed the data with LRF, ICH, and JSO who provided patient samples and clinical data. JRL and RAG performed exome sequencing and ASP and IKC analyzed the results. ZHCA, SNJ, and DMM performed bioinformatic analysis on the variant. AEP and AVH generated EBV-derived B cell lines and processed patient samples. MK performed immunoprecipitations and Western blot experiments and evaluated SHM in various Ramos B cell clones. EC and AVL generated lentivirus-transduced Ramos cell lines and evaluated SHM in these B cells. SG analyzed the patient’s immunological phenotype and SHM in the patient’s B cells. MLX analyzed the patient’s lymph node biopsy. VK and RKD generated CTNNBL1 466V/V and AID–/– Ramos B cell lines using CRISPR/Cas9 technology under the supervision of DGS. LRF and EM wrote the manuscript with substantial edits by MK, JRL, and DGS.
Supplementary Material
Acknowledgments
We thank L. Devine and C. Wang for cell sorting. We also thank Alison Bertuch and Jennifer Despotovic for the care of the patient. This work was supported by grant funding from the NIH NIAID AI061093 to EM and NIH NHGRI UM1 HG006542 to the Baylor-Hopkins Center for Mendelian Genomics.
Version 1. 06/02/2020
In-Press Preview
Version 2. 07/20/2020
Electronic publication
Version 3. 08/03/2020
Print issue publication
Footnotes
Conflict of interest: EM is an advisor for and receives funding from AbbVie, Inc.
Copyright: © 2020, American Society for Clinical Investigation.
Reference information: J Clin Invest. 2020;130(8):4411–4422.https://doi.org/10.1172/JCI131297.
Contributor Information
Marcel Kuhny, Email: marcelkuhny@googlemail.com.
Lisa R. Forbes, Email: Lisa.Forbes@bcm.edu.
Elif Çakan, Email: Elif.cakan@yale.edu.
Andrea Vega-Loza, Email: andreavegaloza25@outlook.com.
Valentyna Kostiuk, Email: Valentyna.kostiuk@yale.edu.
Ravi K. Dinesh, Email: Ravi.dinesh@yale.edu.
Salomé Glauzy, Email: s.glauzy@gmail.com.
Asbjorg Stray-Pedersen, Email: astrayped@gmail.com.
Ashley E. Pezzi, Email: aepezzi@gmail.com.
I. Celine Hanson, Email: hansonceline20@gmail.com.
Alexander Vargas-Hernandez, Email: vargashe@bcm.edu.
Mina LuQuing Xu, Email: mina.xu@yale.edu.
Shalini N. Jhangiani, Email: jhangian@bcm.edu.
Donna M. Muzny, Email: donnam@bcm.edu.
Richard A. Gibbs, Email: agibbs@bcm.edu.
James R. Lupski, Email: jlupski@bcm.edu.
Ivan K. Chinn, Email: ivan.chinn@bcm.edu.
David G. Schatz, Email: david.schatz@yale.edu.
Jordan S. Orange, Email: jso2121@cumc.columbia.edu.
Eric Meffre, Email: eric.meffre@yale.edu.
References
- 1.Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, Honjo T. Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell. 2000;102(5):553–563. doi: 10.1016/S0092-8674(00)00078-7. [DOI] [PubMed] [Google Scholar]
- 2.Revy P, et al. Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2) Cell. 2000;102(5):565–575. doi: 10.1016/S0092-8674(00)00079-9. [DOI] [PubMed] [Google Scholar]
- 3.Meyers G, et al. Activation-induced cytidine deaminase (AID) is required for B-cell tolerance in humans. Proc Natl Acad Sci U S A. 2011;108(28):11554–11559. doi: 10.1073/pnas.1102600108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kuraoka M, et al. Activation-induced cytidine deaminase mediates central tolerance in B cells. Proc Natl Acad Sci U S A. 2011;108(28):11560–11565. doi: 10.1073/pnas.1102571108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cantaert T, et al. Activation-induced cytidine deaminase expression in human B cell precursors is essential for central B cell tolerance. Immunity. 2015;43(5):884–895. doi: 10.1016/j.immuni.2015.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cantaert T, et al. Decreased somatic hypermutation induces an impaired peripheral B cell tolerance checkpoint. J Clin Invest. 2016;126(11):4289–4302. doi: 10.1172/JCI84645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bonilla FA, et al. International Consensus Document (ICON): common variable immunodeficiency disorders. J Allergy Clin Immunol Pract. 2016;4(1):38–59. doi: 10.1016/j.jaip.2015.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Warnatz K, et al. Severe deficiency of switched memory B cells (CD27(+)IgM(-)IgD(-)) in subgroups of patients with common variable immunodeficiency: a new approach to classify a heterogeneous disease. Blood. 2002;99(5):1544–1551. doi: 10.1182/blood.V99.5.1544. [DOI] [PubMed] [Google Scholar]
- 9.Romberg N, et al. CVID-associated TACI mutations affect autoreactive B cell selection and activation. J Clin Invest. 2013;123(10):4283–4293. doi: 10.1172/JCI69854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romberg N, et al. Patients with common variable immunodeficiency with autoimmune cytopenias exhibit hyperplastic yet inefficient germinal center responses. J Allergy Clin Immunol. 2019;143(1):258–265. doi: 10.1016/j.jaci.2018.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sabouri Z, et al. Redemption of autoantibodies on anergic B cells by variable-region glycosylation and mutation away from self-reactivity. Proc Natl Acad Sci U S A. 2014;111(25):E2567–E2575. doi: 10.1073/pnas.1406974111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reed JH, Jackson J, Christ D, Goodnow CC. Clonal redemption of autoantibodies by somatic hypermutation away from self-reactivity during human immunization. J Exp Med. 2016;213(7):1255–1265. doi: 10.1084/jem.20151978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schickel JN, et al. Self-reactive VH4-34-expressing IgG B cells recognize commensal bacteria. J Exp Med. 2017;214(7):1991–2003. doi: 10.1084/jem.20160201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Potter KN, Hobby P, Klijn S, Stevenson FK, Sutton BJ. Evidence for involvement of a hydrophobic patch in framework region 1 of human V4-34-encoded Igs in recognition of the red blood cell I antigen. J Immunol. 2002;169(7):3777–3782. doi: 10.4049/jimmunol.169.7.3777. [DOI] [PubMed] [Google Scholar]
- 15.Jabbour L, Welter JF, Kollar J, Hering TM. Sequence, gene structure, and expression pattern of CTNNBL1, a minor-class intron-containing gene--evidence for a role in apoptosis. Genomics. 2003;81(3):292–303. doi: 10.1016/S0888-7543(02)00038-1. [DOI] [PubMed] [Google Scholar]
- 16.Makarova OV, et al. A subset of human 35S U5 proteins, including Prp19, function prior to catalytic step 1 of splicing. EMBO J. 2004;23(12):2381–2391. doi: 10.1038/sj.emboj.7600241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grote M, et al. Molecular architecture of the human Prp19/CDC5L complex. Mol Cell Biol. 2010;30(9):2105–2119. doi: 10.1128/MCB.01505-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Conticello SG, Ganesh K, Xue K, Lu M, Rada C, Neuberger MS. Interaction between antibody-diversification enzyme AID and spliceosome-associated factor CTNNBL1. Mol Cell. 2008;31(4):474–484. doi: 10.1016/j.molcel.2008.07.009. [DOI] [PubMed] [Google Scholar]
- 19.Lek M, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536(7616):285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ganesh K, Adam S, Taylor B, Simpson P, Rada C, Neuberger M. CTNNBL1 is a novel nuclear localization sequence-binding protein that recognizes RNA-splicing factors CDC5L and Prp31. J Biol Chem. 2011;286(19):17091–17102. doi: 10.1074/jbc.M110.208769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hu Y, et al. A combined nuclear and nucleolar localization motif in activation-induced cytidine deaminase (AID) controls immunoglobulin class switching. J Mol Biol. 2013;425(2):424–443. doi: 10.1016/j.jmb.2012.11.026. [DOI] [PubMed] [Google Scholar]
- 22.Hu Y, Ericsson I, Doseth B, Liabakk NB, Krokan HE, Kavli B. Activation-induced cytidine deaminase (AID) is localized to subnuclear domains enriched in splicing factors. Exp Cell Res. 2014;322(1):178–192. doi: 10.1016/j.yexcr.2014.01.004. [DOI] [PubMed] [Google Scholar]
- 23.Durandy A, Cantaert T, Kracker S, Meffre E. Potential roles of activation-induced cytidine deaminase in promotion or prevention of autoimmunity in humans. Autoimmunity. 2013;46(2):148–156. doi: 10.3109/08916934.2012.750299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dinesh RK, et al. Transcription factor binding at Ig enhancers is linked to somatic hypermutation targeting. Eur J Immunol. 2020;50(3):380–395. doi: 10.1002/eji.201948357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rada C, Di Noia JM, Neuberger MS. Mismatch recognition and uracil excision provide complementary paths to both Ig switching and the A/T-focused phase of somatic mutation. Mol Cell. 2004;16(2):163–171. doi: 10.1016/j.molcel.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 26.Di Noia JM, Williams GT, Chan DT, Buerstedde JM, Baldwin GS, Neuberger MS. Dependence of antibody gene diversification on uracil excision. J Exp Med. 2007;204(13):3209–3219. doi: 10.1084/jem.20071768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Patenaude AM, et al. Active nuclear import and cytoplasmic retention of activation-induced deaminase. Nat Struct Mol Biol. 2009;16(5):517–527. doi: 10.1038/nsmb.1598. [DOI] [PubMed] [Google Scholar]
- 28.Jafari R, et al. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat Protoc. 2014;9(9):2100–2122. doi: 10.1038/nprot.2014.138. [DOI] [PubMed] [Google Scholar]
- 29.Isnardi I, et al. Complement receptor 2/CD21– human naive B cells contain mostly autoreactive unresponsive clones. Blood. 2010;115(24):5026–5036. doi: 10.1182/blood-2009-09-243071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schickel JN, et al. PTPN22 inhibition resets defective human central B cell tolerance. Sci Immunol. 2016;1(1):aaf7153. doi: 10.1126/sciimmunol.aaf7153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chandra A, van Maldegem F, Andrews S, Neuberger MS, Rada C. Deficiency in spliceosome-associated factor CTNNBL1 does not affect ongoing cell cycling but delays exit from quiescence and results in embryonic lethality in mice. Cell Cycle. 2013;12(5):732–742. doi: 10.4161/cc.23594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Maldegem F, et al. CTNNBL1 facilitates the association of CWC15 with CDC5L and is required to maintain the abundance of the Prp19 spliceosomal complex. Nucleic Acids Res. 2015;43(14):7058–7069. doi: 10.1093/nar/gkv643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Han L, Masani S, Yu K. Cutting edge: CTNNBL1 is dispensable for Ig class switch recombination. J Immunol. 2010;185(3):1379–1381. doi: 10.4049/jimmunol.1001643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang X, Wang G, Wu Y, Du Z. The structure of full-length human CTNNBL1 reveals a distinct member of the armadillo-repeat protein family. Acta Crystallogr D Biol Crystallogr. 2013;69(pt 8):1598–1608. doi: 10.1107/S0907444913011360. [DOI] [PubMed] [Google Scholar]
- 35.Ganesh K, et al. Structural and mutational analysis reveals that CTNNBL1 binds NLSs in a manner distinct from that of its closest armadillo-relative, karyopherin α. FEBS Lett. 2014;588(1):21–27. doi: 10.1016/j.febslet.2013.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ahn JW, Kim S, Kim EJ, Kim YJ, Kim KJ. Structural insights into the novel ARM-repeat protein CTNNBL1 and its association with the hPrp19-CDC5L complex. Acta Crystallogr D Biol Crystallogr. 2014;70(pt 3):780–788. doi: 10.1107/S139900471303318X. [DOI] [PubMed] [Google Scholar]
- 37.Ahn JW, Jin KS, Son HF, Chang JH, Kim KJ. Small angle X-ray scattering studies of CTNNBL1 dimerization and CTNNBL1/CDC5L complex. Sci Rep. 2015;5:14251. doi: 10.1038/srep14251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mu Y, Zelazowska MA, McBride KM. Phosphorylation promotes activation-induced cytidine deaminase activity at the Myc oncogene. J Exp Med. 2017;214(12):3543–3552. doi: 10.1084/jem.20170468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Basu U, et al. The AID antibody diversification enzyme is regulated by protein kinase A phosphorylation. Nature. 2005;438(7067):508–511. doi: 10.1038/nature04255. [DOI] [PubMed] [Google Scholar]
- 40.Chaudhuri J, Khuong C, Alt FW. Replication protein A interacts with AID to promote deamination of somatic hypermutation targets. Nature. 2004;430(7003):992–998. doi: 10.1038/nature02821. [DOI] [PubMed] [Google Scholar]
- 41.Mu Y, McBride KM. Targeting mutagenesis in B cells: Phosphorylation goes beyond AID association. Mol Cell Oncol. 2018;5(5):e1432259. doi: 10.1080/23723556.2018.1432259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pasqualucci L, Kitaura Y, Gu H, Dalla-Favera R. PKA-mediated phosphorylation regulates the function of activation-induced deaminase (AID) in B cells. Proc Natl Acad Sci U S A. 2006;103(2):395–400. doi: 10.1073/pnas.0509969103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McBride KM, Barreto V, Ramiro AR, Stavropoulos P, Nussenzweig MC. Somatic hypermutation is limited by CRM1-dependent nuclear export of activation-induced deaminase. J Exp Med. 2004;199(9):1235–1244. doi: 10.1084/jem.20040373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Takizawa M, et al. AID expression levels determine the extent of cMyc oncogenic translocations and the incidence of B cell tumor development. J Exp Med. 2008;205(9):1949–1957. doi: 10.1084/jem.20081007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kracker S, Imai K, Gardès P, Ochs HD, Fischer A, Durandy AH. Impaired induction of DNA lesions during immunoglobulin class-switch recombination in humans influences end-joining repair. Proc Natl Acad Sci U S A. 2010;107(51):22225–22230. doi: 10.1073/pnas.1012591108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhu Y, Nonoyama S, Morio T, Muramatsu M, Honjo T, Mizutani S. Type two hyper-IgM syndrome caused by mutation in activation-induced cytidine deaminase. J Med Dent Sci. 2003;50(1):41–46. [PubMed] [Google Scholar]
- 47.Tosato G, Cohen JI. Generation of Epstein-Barr virus (EBV)-immortalized B cell lines. Curr Protoc Immunol. 2007;Chapter 7:Unit 7.22. doi: 10.1002/0471142735.im0722s76. [DOI] [PubMed] [Google Scholar]
- 48.Schickel JN, et al. Carabin deficiency in B cells increases BCR-TLR9 costimulation-induced autoimmunity. EMBO Mol Med. 2012;4(12):1261–1275. doi: 10.1002/emmm.201201595. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.