

Abstract
Colorectal cancer (CRC) is one of the most commonly diagnosed cancers with an estimated 1.8 million new cases worldwide and associated with high mortality rates of 881 000 CRC‐related deaths in 2018. Screening programs and new therapies have only marginally improved the survival of CRC patients. Immune‐related genes (IRGs) have attracted attention in recent years as therapeutic targets. The aim of this study was to identify an immune‐related prognostic signature for CRC. To this end, we combined gene expression and clinical data from the CRC data sets of The Cancer Genome Atlas (TCGA) into an integrated immune landscape profile. We identified a total of 476 IRGs that were differentially expressed in CRC vs normal tissues, of which 18 were survival related according to univariate Cox analysis. Stepwise multivariate Cox proportional hazards analysis established an immune‐related prognostic signature consisting of SLC10A2, FGF2, CCL28, NDRG1, ESM1, UCN, UTS2 and TRDC. The predictive ability of this signature for 3‐ and 5‐year overall survival was determined using receiver operating characteristics (ROC), and the respective areas under the curve (AUC) were 79.2% and 76.6%. The signature showed moderate predictive accuracy in the validation and GSE38832 data sets as well. Furthermore, the 8‐IRG signature correlated significantly with tumour stage, invasion, lymph node metastasis and distant metastasis by univariate Cox analysis, and was established an independent prognostic factor by multivariate Cox regression analysis for CRC. Gene set enrichment analysis (GSEA) revealed a relationship between the IRG prognostic signature and various biological pathways. Focal adhesions and ECM‐receptor interactions were positively correlated with the risk scores, while cytosolic DNA sensing and metabolism‐related pathways were negatively correlated. Finally, the bioinformatics results were validated by real‐time RT‑qPCR. In conclusion, we identified and validated a novel, immune‐related prognostic signature for patients with CRC, and this signature reflects the dysregulated tumour immune microenvironment and has a potential for better CRC patient management.
Keywords: bioinformatics, colorectal cancer, immunogenomic landscape, prognostic signature, TCGA
1. INTRODUCTION
Colorectal cancer (CRC) is the third most commonly diagnosed cancer worldwide and ranks second in terms of cancer‐related mortality. 1 An estimated 1.8 million new cases and 881 000 CRC‐related deaths were recorded in 2018 alone. 1 Recent screening programs have reported a decrease in both the incidence and mortality of CRC, 2 and new therapies have doubled the overall survival (OS) of patients with advanced CRC. 3 However, the OS of CRC patients is still low as they are usually diagnosed at the advanced‐stage. 4 , 5 Therefore, it is essential to identify novel therapeutic, prognostic and diagnostic biomarkers for CRC.
Immunotherapy is now part of the standard treatment regimen for many solid tumours, 6 as the immune system is frequently dysregulated in cancer patients and inextricably linked with tumour growth. 7 Anti‐cancer immunotherapeutic approaches are classified into six categories: oncolytic virus therapy, adjuvant immunotherapy and cytokines, adoptive cell therapy, therapeutic vaccines, checkpoint inhibitors and monoclonal antibodies. 8 However, insufficient immune response has long been a concern, especially for checkpoint inhibitors targeting the programmed death 1 (PD‐1) and PD‐1 ligands (PD‐L1s) in CRC. 9 Especially, use of immune checkpoint inhibitors (ICIs) has little or no clinical activity in most metastatic CRC patients. 10 The response to ICI therapy depends on several key factors, including mutation load (high levels of tumour neoantigen), tumour‐infiltrating lymphocytes and regulatory checkpoint receptors. 11 As seen in CRC patients with microsatellite stability (MSS), the main obstacle for ICI is low tumour immunogenicity 12 , 13 because of the presence of fewer neoantigens, 14 resulting in fewer infiltrating CD8 + T cells and fewer strongly positive for PD‐1. 15 This makes CRC become one of the tumours in which immunotherapy has been shown less effective, in relation to different classes. Therefore, it is essential to characterize the immune‐related genes (IRGs) in CRC to optimize treatment.
Gene microarrays and RNA‐sequencing have identified several prognostic biomarkers for human cancers in recent years. For instance, cytokines, 16 heat shock protein beta 3, 17 cyclin D1, 18 clusterin 19 and RBP7 20 are established prognostic markers for CRC. In addition, non‐coding RNAs such as microRNAs, 21 , 22 long non‐coding RNAs 23 , 24 and circRNAs 25 are increasingly reported to be associated with the survival of cancer patients. Several immune‐related prognostic signatures have also been established for multiple cancer types. 26 For example, an IRG prognostic signature consisting of SPAG11A, PTH2R, IL17C, IL11, FAM3B, CTGF and AGTR1 was constructed based on TCGA data for papillary thyroid cancer. 27 Wang et al 28 analysed the gene expression profiles of TCGA patients with renal papillary cell carcinoma to establish a risk signature of 15 IRGs. Similar IRG‐based prognostic signatures have been reported for gastric cancer, 29 invasive ductal carcinoma 30 and ovarian cancer 31 as well. Based on these studies, our aim was to establish an immune‐related prognostic signature for CRC.
Here, we systematically analysed the immunogenomic landscape of CRC based on the gene expression profiles in TCGA and identified 476 differentially expressed IRGs between tumour samples relative to normal tissues including 18 survival‐associated IRGs. An IRG prognostic signature including SLC10A2, FGF2, CCL28, NDRG1, ESM1, UCN, UTS2 and TRDC was constructed which showed moderate predictive ability for the overall survival of CRC patients in both the training and validation sets. Furthermore, this signature correlated with the tumour stage, invasion, lymph node metastasis and distant metastasis, and was identified as an independent prognostic indicator for CRC. This IRG signature may reflect the immune dysregulation in the tumour microenvironment and is a promising novel therapeutic target in addition to being an accurate prognostic biomarker for CRC.
2. MATERIALS AND METHODS
2.1. Data acquisition and IRG selection
RNA‐sequencing and clinical data of 568 CRC and 44 normal tissue samples were downloaded from TCGA database (https://portal.gdc.cancer.gov/) 32 as 15 August 2019. A total of 2,498 IRGs (Table S1) associated with human cancers were identified using the Immunology Database and Analysis Portal (ImmPort) database (https://www.immport.org/home). 33
2.2. Identification of differentially expressed IRGs
The limma R package 34 was used to identify IRGs that were differentially expressed between the tumour and normal tissue samples, with false discovery rate (FDR) of < 0.05 and log2‐fold change > 1 as the cut‐off values. Gene Ontology (GO) 35 and the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis 36 were conducted using the clusterProfiler R package 37 to identify the functionally enriched genes and classify the gene clusters. FDR < 0.01 was considered statistically significant.
2.3. Survival‐associated IRG analysis
The survival‐associated IRGs were screened using data from patients surviving at least 90 days and with known M stage (pM), tumour stage (pStage), T stage (pT) and N stage (pN) [according to American Joint Committee on Cancer (AJCC)]. Accordingly, the data set of 453 patients was randomly assigned to the training (362 patients, 80% of all samples) and validation (91 patients, 20% of all samples) groups, and the survival‐related IRGs were identified by univariate Cox analysis with the survival R package (P < .05).
2.4. Construction and verification of the immune signature
Based on the results of the univariate analysis, the immune‐related prognostic signature was generated using a stepwise multivariate Cox proportional hazards model with the survival package in R language. Here, a multivariate Cox proportional hazards regression model was used to solve the problem of multiple factors affecting the survival time of patients. In brief, we considered all the 18 IRGs that are significantly related to prognosis by univariate Cox analysis as influencing factors. After bringing them into multivariate Cox proportional hazards model, significant IRGs will be retained during multiple computing. The weighted coefficients based on individual gene expression levels were used to calculate the risk score as follows:
The patients in the training group were then stratified into the low‐ and high‐risk groups according to median risk score values, and their survival was analysed using the Kaplan‐Meier method and the log‐rank test. The specificity and sensitivity of the risk score in predicting 3‐ and 5‐year survival was determined by ROC analysis using the survival ROC R package, and the areas under curve (AUC) were calculated. The immune signature was further validated using the entire TCGA (n = 453) and validation (n = 91) data sets, as well as the GSE38832 data set 38 downloaded from the Gene Expression Omnibus (GEO) database. 39 The latter included 115 CRC samples and the survival information in accordance with the GPL570 (Affymetrix Human Genome U133 Plus 2.0 Array). Each data set was stratified into the low‐ and high‐risk groups as described.
2.5. Association of the immune signature and clinicopathological features
The correlation between patient survival and clinical factors including age, gender, pM, pN, pT, pStage and risk scores was determined by univariate Cox analysis. Multivariate Cox regression analysis was then used to identify the independent prognostic factors for CRC. Prognostic nomograms were generated using the Cox regression coefficients with the rms R package, and the calibration plots to assess the performance of these nomograms were drawn using the regplot R package.
2.6. Bioinformatics analyses
The biological relevance of the immune signature was determined by gene set enrichment analysis (GSEA) using the GSVA R package. FDR < 0.001 and a relationship coefficient of > 0.3 were used as the cut‐offs for the included KEGG pathways. The association between the immune‐related genes was analysed by the Spearman correlation coefficient. The protein‐protein interaction network was based on the STRING database (https://string‐db.org/cgi/input.pl) 40 and constructed using with an interaction confidence of 0.4 and 10 neighbouring nodes. The differentially expressed transcription factors (TFs) were identified from the Cistrome database (http://cistrome.org/), 41 with FDR < 0.05 and log2‐fold change > 1 as the criteria. TF correlation assay was performed using P < .001 and correlation coefficient > 0.3 as the cut‐offs. The association between CRC biomarkers like BRAF, 42 NRAS 42 and PIK3CA, 43 and the immune prognostic signature was then determined. Finally, a competing endogenous RNA (ceRNA) regulatory network based on eight biomarkers was constructed using the TarBase (version 8) 44 and LncBase (version 2) 45 databases, and visualized using Cytoscape software (version 3.7.2). 46
2.7. Clinical specimens
The expression of IRGs was clinically validated in 25 pairs of CRC and matched normal tissues surgically resected at the Second Affiliated Hospital of Zhejiang University School of Medicine from May 2018 to June 2018. These patients had received no preoperative chemotherapy, radiotherapy or targeted therapy. The tissue samples were collected within 30 minutes of surgical resection, cleaned and cryopreserved in liquid nitrogen. All patients provided informed consent, and the study was approved by The Human Research Ethics Committee of the hospital.
2.8. Real‐time quantitative polymerase chain reaction (RT‑qPCR)
Total RNA was extracted from the frozen tissue specimens using Trizol (#G3013, Servicebio) as per the manufacturer's instructions. The concentration and purity of the RNA were ascertained with a NanoDrop2000 spectrophotometer (ThermoFisher Scientific). Reverse transcription into complementary DNA (cDNA) was performed using a specific kit (#K1622, ThermoFisher Scientific) following the manufacturer's instructions. In brief, a total of 5 μg of total RNA (diluted to 12 μL using RNase‐free dH2O with 1 μL oligo(dT)18) were incubated at 65°C for 5 minutes and cooling on ice. Then, a 8 μL reaction mixture containing 4 μL of 5 × Reaction Buffer, 2 μL Of 10 mmol/L dNTP Mix, 1 μL of RiboLock RNAase inhibitor and 1 μL of RevertAi M‐MuLV reverse transcriptase was added in above RNA mixture (20 μL in total). The samples were finally incubated in a PCR thermocycler for 60 minutes at 42°C and 5 minutes at 70°C. RT‐qPCR was conducted in the StepOnePlus cycler (Applied Biosystems Inc) using FastStart Universal SYBR Green Master (#G3008, Servicebio). The PCR cycle parameters were as follows: pre‐denaturation at 95°C for 10 minutes, followed by 40 cycles at 95°C for 15 seconds and 60°C for 1 minute. Relative gene expression levels were measured using the comparative cycle threshold (ΔΔCt) method and normalized to GAPDH. The sequences and accession numbers for primers used in real‐time RT–PCR were shown in Table 1 (Servicebio). All samples were tested in triplicate.
TABLE 1.
The information of primers used in real‐time RT–PCR
| Gene | Sequences of primers | Accession No. | Tm (°C) | Amplicon (bp) |
|---|---|---|---|---|
| SLC10A2 | FOR: AGGTGCCGAACGGTTGCTT | NM_000452.2 | 60°C | 113 |
| REV: AGCGGGAAGGTGAATACGACA | 60°C | |||
| ESM1 | FOR: CTTGCTACCGCACAGTCTCA | NM_001135604.2 | 60°C | 124 |
| REV: GCCATGTCATGCTCTTTGCAG | 60°C | |||
| GAPDH | FOR: GGAAGCTTGTCATCAATGGAAATC | NM_002046 | 60°C | 168 |
| REV: TGATGACCCTTTTGGCTCCC | 60°C |
Abbreviations: FOR, forward; REV, reverse; Tm, Annealing temperatures.
2.9. Statistical analysis
All statistical analyses were conducted using R language (version 3.6.1). The Wilcoxon test was used to compare two independent non‐parametric samples, and the Kruskal‐Wallis test was used for multiple independent samples. The Kaplan‐Meier survival curves were compared with the log‐rank test. Independent prognostic factors related to survival were identified using the univariate and multivariate Cox proportional hazard regression analyses. The Spearman correlation coefficient was used to analyse the association among immune‐related genes. P‐value < .05 was considered statistically significant.
3. RESULTS
3.1. Construction of an immune‐gene expression signature in CRC
Given the crucial role of the immune microenvironment in cancer development, we explored the prognostic value of IRGs in CRC (Figure 1). Screening of TCGA and ImmPort databases revealed 476 IRGs that were differentially expressed in CRC compared to normal tissues, of which 177 were up‐regulated and 299 down‐regulated (Table S2; Figure 2A,B). The differentially expressed IRGs were significantly enriched in GO terms related to complement activation, humoural immune response, protein activation, immunoglobulin‐mediated immune response, antigen binding, phagocytosis, immunoglobulin complex formation and cytokine activity (Figure 2C and Table S3), and in cytokine‐cytokine receptor interaction, IL‐17 signalling pathway, natural killer cell‐mediated cytotoxicity, chemokine signalling pathway, Rap1 signalling pathway and MAPK signalling pathway (Figure 2D and Table S4).
FIGURE 1.
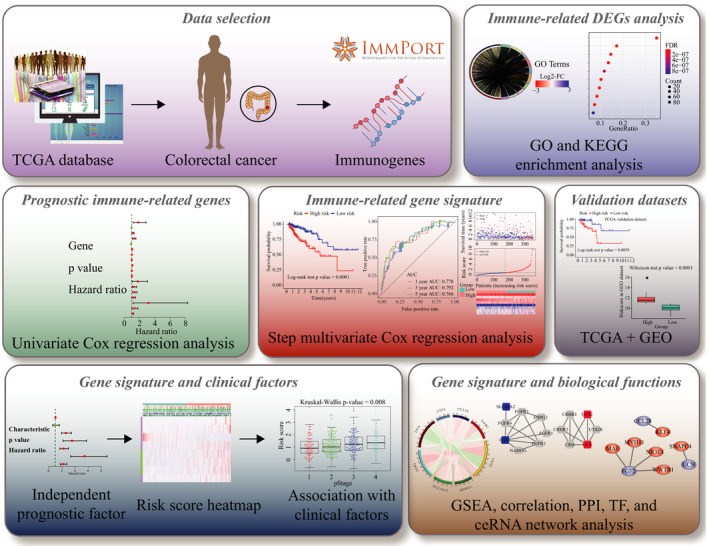
Flow chart for the development and verification of an immune‐related prognostic signature for CRC
FIGURE 2.
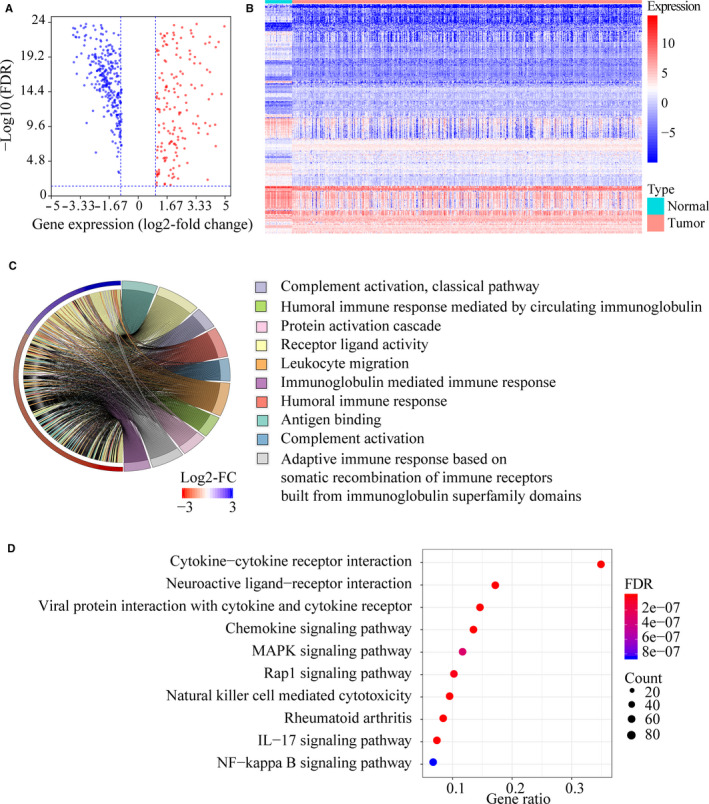
Screening of differentially expressed immune‐related genes (IRGs) in colorectal cancer (CRC). A, Volcano plot showing the differentially expressed IRGs in tumours vs normal tissue samples. Blue dots represent down‐regulated IRGs, and red dots represent up‐regulated IRGs. B, Gene expression heat map of differentially expressed IRGs in CRC. C, Results of the gene ontology (GO) term enrichment study. D, Results of the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment study
Eighteen survival‐associated IRGs were identified in the training group by univariate Cox analysis (Figure 3A), of which SLC10A2, FGF2, CCL28, NDRG1, ESM1, UCN, UTS2 and TRDC were further screened by the stepwise multivariate Cox proportional hazards model (Table 2). An eight‐gene immune signature was constructed using the independent regression coefficients of each gene, and the risk score was calculated as (0.639 * level of SLC10A2) + (0.387 * level of FGF2) + (−0.094 * level of CCL28) + (0.012 * level of NDRG1) + (0.124 * level of ESM1) + (0.378 * level of UCN) + (0.254 * level of UTS2) + (0.129 * level of TRDC). The risk scores were calculated for each patient in the training group from 0.02 and 24.80, and the patients were assigned to the high‐risk or low‐risk group based on the median risk score of 0.95. As shown in Figure 3B, patients with high‐risk scores had significantly worse survival outcome than those with low‐risk scores (log‐rank test, P < .0001). Furthermore, the AUC of the risk score for 3‐year and 5‐year OS were 79.2% and 76.6%, respectively (Figure 3C). The survival status, risk scores and gene expression data of CRC patients in the training group are illustrated in Figure 3D.
FIGURE 3.
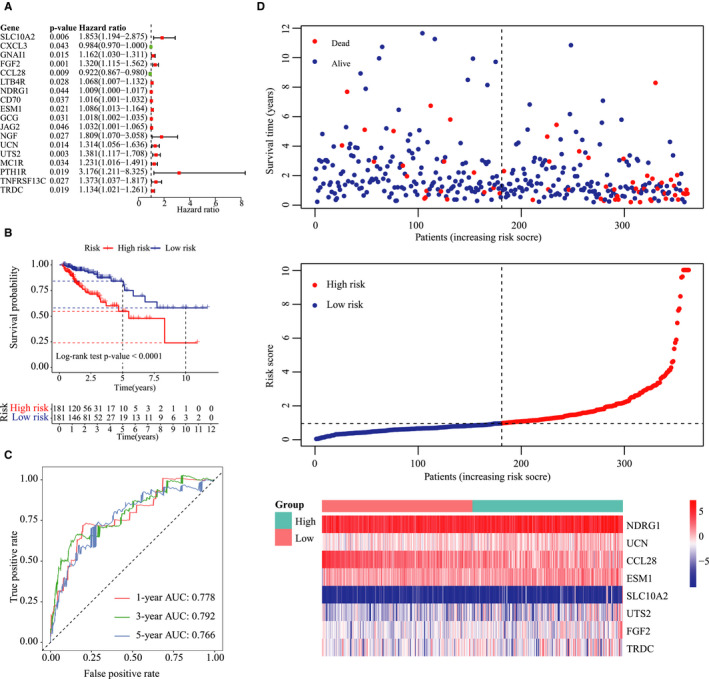
Construction of the immune‐related prognostic signature in CRC. A, Forest plot of immune‐related prognostic genes based on univariate Cox regression analysis. B, Kaplan‐Meier plots of the immune‐related signature showing worse survival in the high‐risk group compared to the low‐risk group (log‐rank P‐value < .0001). C, Time‐dependent (ROC) curve of the immune‐related signature for 1‐, 3‐ and 5‐year overall survival. D, Distribution of the survival status, risk score and gene expression data of CRC patients in the training group
TABLE 2.
The results of stepwise multivariate Cox proportional hazards model
| Gene symbol | Coef | HR | HR.95L | HR.95H | P‐value | Regulation |
|---|---|---|---|---|---|---|
| SLC10A2 | 0.638812 | 1.89423 | 1.202826 | 2.983063 | .005834 | Down |
| FGF2 | 0.386611 | 1.471984 | 1.234656 | 1.754931 | 1.63E‐05 | Down |
| CCL28 | −0.09426 | 0.910051 | 0.851113 | 0.973069 | .005796 | Down |
| NDRG1 | 0.011524 | 1.011591 | 1.003183 | 1.020069 | .006806 | Down |
| ESM1 | 0.124352 | 1.132414 | 1.057736 | 1.212365 | .000354 | Up |
| UCN | 0.377663 | 1.458871 | 1.157773 | 1.838275 | .001364 | Up |
| UTS2 | 0.254362 | 1.289639 | 1.028137 | 1.617652 | .02781 | Up |
| TRDC | 0.129341 | 1.138078 | 1.015503 | 1.275449 | .026111 | Down |
Coef represents regression coefficient of each gene.
Abbreviations: HR, hazard ratio; HR.95H, high 95% confidence interval of HR; HR.95L, low 95% confidence interval of HR.
3.2. Validation of the immune‐related signature
To validate the immune‐related signature, its prognostic accuracy was further assessed in three independent cohorts, including the validation group, TCGA data set and the GSE38832 data set. The OS was significantly longer for patients in the low‐risk vs the high‐risk group in the validation cohort (n = 91; log‐rank test, P = .006, Figure 4A), and the predicted 3‐year and 5‐year OS was 75.5% and 82.2%, respectively (Figure 4B). The TCGA data set (n = 453) also validated the prognostic accuracy of the immune‐related signature (log‐rank test, P < .0001, Figure 4C), with respective AUCs of 77.7% and 77.5% for 3‐year and 5‐year OS. Thus, the 8‐IRG immune signature predicted OS of CRC patients with superior accuracy (Figure 4D). Consistent with this, significantly longer OS was also seen for patients in the low‐risk group of the GSE38832 data set (n = 115) compared to those in the high‐risk group (log‐rank test, P = .016, Figure 4E). Furthermore, the risk scores were significantly different in both groups (Wilcoxon test, P < .0001, Figure 4F).
FIGURE 4.

Verification of the immune‐related signature in three independent cohorts. A, Kaplan‐Meier plots of the immune‐related signature in the validation group. B, Time‐dependent receiver operating characteristics (ROC) curve of the immune‐related signature in the validation group. C, Kaplan‐Meier plots of the immune‐related signature in The Cancer Genome Atlas (TCGA) data set. D, Time‐dependent ROC curve of the immune‐related signature in TCGA data set. E, Kaplan‐Meier plots of the immune‐related signature in the GSE38832 database. F, Risk score distribution in low‐ and high‐risk groups
The expression level of the IRGs was validated in 25 matched tissue pairs by qRT‐PCR ESM1 and SLC10A2 showed the maximum differential expression between CRC and normal tissues, and consistent with the results of the bioinformatics analysis, ESM1 was significantly elevated and SLC10A2 was significantly down‐regulated (P < .05) in the tumours (Figure S1A,B).
3.3. The IRG signature confers additional prognostic power for CRC patients
As shown in Figure 5A, clinical factors including age, pStage, pT, pN, pM and the immune risk score were closely associated with patient survival. Multivariate Cox regression analysis further showed that the IRG signature is an independent prognostic indicator for OS (P < .001, Figure 5B and Table 3). Expression profiles of the eight IRGs are shown in Figure 5C, and the risk score increased with advanced tumour parameters (Figure 5D). Furthermore, the expression level of CCL28 was significantly associated with pT (P = .048), that of ESM1 with pT (P = .008) and pStage (P = .01), UTS2 with pM (P = .039), and TRDC with pN (P = .012), pM (P < .0001) and pStage (P < .0001, Figure 6A). We next created a nomogram that integrated clinicopathological characteristics with the IRG signature as a quantitative tool for predicting OS of CRC patients (Figure 6B). As shown in the calibration plots in Figure 6C, the nomogram performed with moderate accuracy compared to an ideal model.
FIGURE 5.
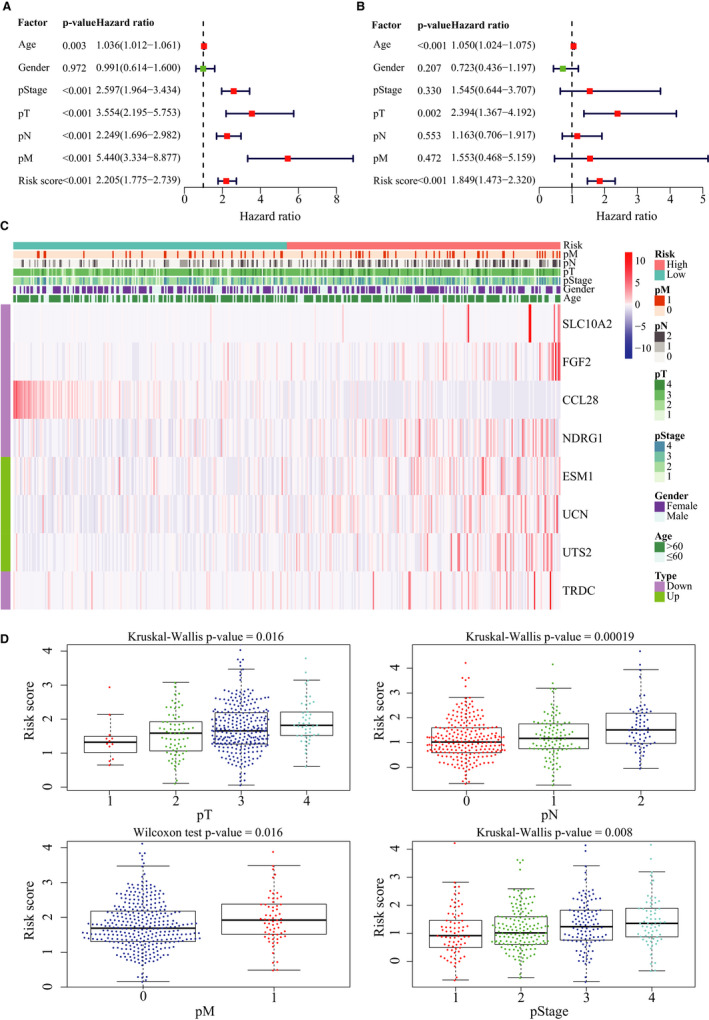
Association of the immune‐related signature with clinicopathological characteristics. A, Forest plot of risk scores and other clinical factors based on a univariable Cox regression analysis. B, Forest plot of risk scores and other clinical factors based on a multivariate Cox regression analysis. C, Expression profiles of the eight immune‐related genes. D, Box plots showing risk score distribution of different clinical factors
TABLE 3.
The univariate and multivariate Cox analysis for risk signature and clinical factors
| Factors | Univariate Cox analysis | Multivariate Cox analysis | ||
|---|---|---|---|---|
| Hazard ratio (95% CI) | P‐value | Hazard ratio (95% CI) | P‐value | |
| Age | 1.036 (1.012‐1.061) | .003 | 1.050 (1.024‐1.075) | <.0001 |
| Gender | 0.991 (0.614‐1.600) | .972 | 0.723 (0.436‐1.197) | .207 |
| pStage | 2.597 (1.964‐3.434) | <.0001 | 1.545 (0.644‐3.707) | .330 |
| pT | 3.554 (2.195‐5.753) | <.0001 | 2.394 (1.367‐4.192) | .002 |
| pN | 2.249 (1.696‐2.982) | <.0001 | 1.163 (0.706‐1.917) | .553 |
| pM | 5.440 (3.334‐8.877) | <.0001 | 1.553 (0.468‐5.159) | .472 |
| Risk score | 2.205 (1.775‐2.739) | <.0001 | 1.849 (1.473‐2.320) | <.0001 |
Abbreviations: 95% CI, 95% confidence interval; pM: pathological metastasis stage; pN, pathological lymph node stage; pT, pathological tumour stage.
FIGURE 6.
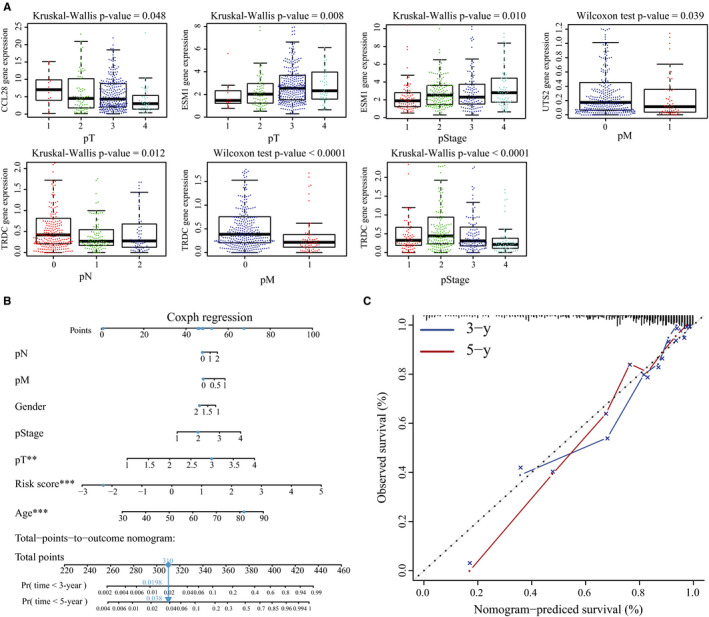
Expression of immune‐related genes (IRGs) associated with clinicopathological features, and construction of a nomogram for survival assessment. A, Associations between the different IRGs and clinicopathological features. B, Nomogram integrating the immune‐related signature to clinicopathological characteristics. C, Plots displaying the calibration of each model comparing predicted and actual 3‐ and 5‐year overall survival. The graph relative to the 45° line showing the model relative to perfect prediction
3.4. Biological revenant and regulatory network of the IRG signature in CRC
The potential association between the IRG signature and biological functions was assessed by GSEA using samples from TCGA data set. We found 28 KEGG pathways that were significantly correlated to either the low‐ or high‐risk CRC patient group (Figure S2A and Table S5). Pathways modulating cancer‐related functions such as focal adhesions, the actin cytoskeleton, ECM‐receptor interactions and endocytosis were positively correlated with the risk scores, while cytosolic DNA sensing, linoleic acid metabolism, arachidonic acid metabolism and ether lipid metabolism pathways were negatively correlated.
The potential correlation between the IRGs and other cancer‐related genes was also evaluated. As shown in Figure S2B, there was a significant negative correlation between UCN and FGF2 (Spearman's correlation coefficient = −0.31), while the IRGs were weakly correlated with other genes. The interactions of the proteins encoded by these genes were next analysed (Figure S2C), and the Cistrome database further showed that UCN, FGF2 and CCL28 were regulated by transcription factors (Figure S2D). Finally, the expression of several CRC biomarkers was assessed in the tumours from the low‐ and high‐risk groups. BRAF, NRAS and PIK3CA were differentially expressed, indicating that the IRG signature is closely related to CRC progression (Figure 7A–C). Based on the above, a complex ceRNA network regulating IRGs was constructed using the TarBase and LncBase databases (Figure 7D).
FIGURE 7.
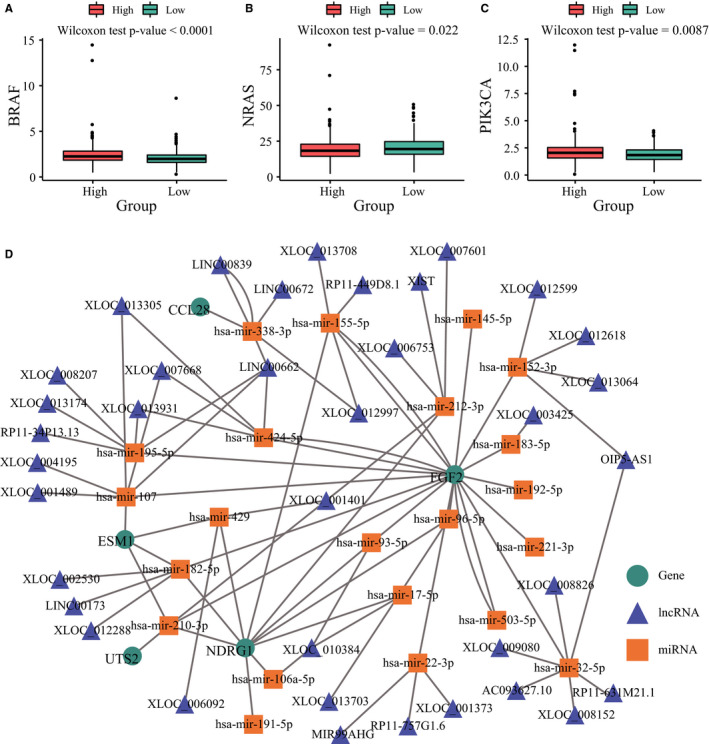
Correlation of immune‐related genes with biomarkers and construction of a competing endogenous RNA (ceRNA) network for colorectal cancer. A, BRAF gene expression in risk groups. B, NRAS gene expression in risk groups. C, PIK3CA gene expression in risk groups. D, The regulatory network of ceRNAs based on immune‐related genes
4. DISCUSSION
The tumour microenvironment includes immune cells that are key players in tumour progression, 47 and variously modulate inflammation 48 and metastasis. 49 Thus, both immunology‐cancer interface and the microenvironment have a major impact on the diagnosis and treatment of cancers, including CRC. 50 Resistance to CRC immunotherapy is the result of poorly antigenic tumour cells displaying PD‐1 and PD‐L1. 14 We systematically evaluated the immunogenomic landscape of CRC tumours based on gene expression profiles in TCGA database and constructed an immune‐related prognostic signature including SLC10A2, FGF2, CCL28, NDRG1, ESM1, UCN, UTS2 and TRDC. The AUC values for 3‐year and 5‐year OS of this prognostic signature were 79.2% and 76.6%, respectively, indicating moderate predictive accuracy. Furthermore, the signature correlated with tumour stage, invasion, lymph node metastasis and distant metastasis, and is an independent predictor for the OS of CRC patients. This IRG signature likely reflects immune dysregulation in the tumour microenvironment and is a novel prognostic biomarker of CRC.
High throughput molecular analyses, gene expression databases and bioinformatic tools have enabled systematic profiling of immune signatures in cancers. For example, Yang et al 29 built a prognostic model for gastric cancer consisting of immune‐related genes including TNFRSF18, PBK, MICB, ITGA6, TLR5, PNMA1, LBP, CXCR4, C6 and NRP1, which accurately distinguished between patients with poor and satisfactory OS. Similarly, Bao et al identified an independent prognostic signature for invasive ductal carcinoma 30 that consisted of FLT3LG, SPIB, KLRB1, BATF, IGHA1, TIMM8A and QRSL1. In this study, we identified an immune‐related signature comprising SLC10A2, FGF2, CCL28, NDRG1, ESM1, UCN, UTS2 and TRDC for CRC. N‐myc downstream‐regulated gene 1 (NDRG1) was reported that it can play a key regulatory role in signalling pathways related to tumour progression, especially in tumour metastasis. Mi et al 51 determined that NDRG1 inhibits epithelial‐mesenchymal transition (EMT), migration and invasion by interacting and promoting caveolin‐1 ubiquitination in human CRC cells. Another study also proved that knockdown of NDRG1 can promote EMT progress of CRC via NF‐κB signalling. 52 ESM1 that can be secreted into saliva, blood and urine is considered a candidate biomarker for CRC. 53 The ceRNA‐ and TF‐mediated regulatory networks further identified FGF2, CCL28 and UCN in CRC progression. FGF2 is associated with increased CRC responsiveness to irinotecan 54 and is also a member of a gene hub associated with cetuximab insensitivity in CRC. 55 In addition, CCL28 was previously identified as part of a prognostic signature that accurately predicted the OS of patients with CRC. 56 Sun et al 57 also identified CCL28 as a critical prognostic factor for CRC using GEO gene expression data and the rank aggregation method.
In human cancer studies, none of the single biomarkers can be used to detect cancers achieved the required specificity and sensitivity. 58 When analysing one or two typical biomarkers, conflicting results are often obtained, leading to incorrect cancer diagnosis and unsuccessful treatment. Because it is known that several pathways and biological processes in tumour cells have changed, the concept of ‘single marker’ in cancer is incorrect. 59 Some studies have reported extensive combinations of serum biomarkers in various cancers. The combination of serum biomarkers with nucleic acids including free DNA, mRNA, microRNA and circulating tumour DNA (ctDNA) is becoming a diagnostic tool for malignant tumours. 60 In breast cancer, several recurrence prediction models including multi‐gene panels, such as Oncotype DX, EndoPredict, BCI and Curebest 95GC, were established. 61 Therefore, above results may give us a hint: the combination of several biomarkers from different biological pathways can lead to better understanding of cancer progression and prognostic significance in solid cancers. However, the toxic side effects and other adverse reactions of multiple target genes therapy are uncertain, thus it is worth our serious considerations.
The prognostic IRGs in our study were enriched in 28 KEGG pathways, including cancer‐related pathways such as focal adhesions, the actin cytoskeleton, ECM‐receptor interactions and endocytosis. The actin cytoskeleton of cytotoxic lymphocytes is a major mediator of immune synapse formation and maturation, 62 cell migration and immune surveillance. 63 Focal adhesion is an essential step in cell migration, and the extracellular matrix (ECM) and its secreted cytokines play key roles in the immune escape of human tumours. 64 The activation of several metabolism‐related pathways such as cytosolic DNA sensing, linoleic acid metabolism, arachidonic acid metabolism and ether lipid metabolism negatively correlated with the CRC risk scores. Thus, the IRG gene signature is biologically significant in CRC.
Our study has some limitations that should be addressed in future studies. First, transcriptomic analysis cannot reflect the molecular mechanisms underlying the immunobiology of CRC, which may be better elucidated by proteomics and/or metabolomics. Second, the robustness of our IRG signature must be verified in a large prospective clinical study. Finally, our findings have to be validated by in vitro and in vivo functional assays to further our understanding of the biological role of this IRG signature in CRC.
In conclusion, we identified and validated a novel immune‐related prognostic signature for patients with CRC, which likely reflects the immune dysregulation in the tumour microenvironment and is a potential prognostic biomarker and therapeutic target.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
AUTHOR CONTRIBUTION
Jun Wang: Conceptualization (lead); Formal analysis (lead); Writing‐original draft (lead); Writing‐review & editing (equal). Shaojun Yu: Methodology (lead); Writing‐review & editing (equal). Guofeng Chen: Conceptualization (supporting); Writing‐original draft (supporting). Muxing Kang: Conceptualization (supporting); Writing‐original draft (supporting). Xiaoli Jin: Methodology (supporting); Writing‐review & editing (equal). Yi Huang: Formal analysis (supporting); Methodology (supporting). Lele Lin: Formal analysis (supporting); Methodology (supporting). Dan Wu: Conceptualization (supporting); Writing‐original draft (supporting). Lie Wang: Writing‐review & editing (equal). Jian Chen: Writing‐review & editing (equal).
Supporting information
Fig S1
Fig S2
Table S1
Table S2
Table S3
Table S4
Table S5
Supplementary Material
ACKNOWLEDGEMENTS
We greatly thank the Department of Surgery, the Second Affiliated Hospital, Zhejiang University School of Medicine for technical advice. We thank the Department of Surgical Oncology, the Second Affiliated Hospital, Zhejiang University School of Medicine for providing human CRC tissue samples. We also thank Elsevier (https://webshop.elsevier.com/language‐editing‐services/) for language editing services (#CDUNW_1).
Wang J, Yu S, Chen G, et al. A novel prognostic signature of immune‐related genes for patients with colorectal cancer. J Cell Mol Med. 2020;24:8491–8504. 10.1111/jcmm.15443
Jun Wang and Shaojun Yu are contributed equally to this work.
Funding information
This work was supported by Zhejiang Provincial Key Project of Research and Development (2019C03043), the National Natural Science Foundation of China (81830006, 31670887, 31870874 and 31800734).
Contributor Information
Lie Wang, Email: wanglie@zju.edu.cn.
Jian Chen, Email: zrchenjian@zju.edu.cn.
DATA AVAILABILITY STATEMENT
All data generated or analysed during this study are included in this published article and its supplementary information files.
REFERENCES
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2018;68:394‐424. [DOI] [PubMed] [Google Scholar]
- 2. Hissong E, Pittman ME. Colorectal carcinoma screening: established methods and emerging technology. Crit Rev Clin Lab Sci. 2020;57(1):22–36. [DOI] [PubMed] [Google Scholar]
- 3. Dekker E, Tanis PJ, Vleugels JLA, Kasi PM, Wallace MB. Colorectal cancer. Lancet. 2019;394:1467‐1480. [DOI] [PubMed] [Google Scholar]
- 4. Pawa N, Arulampalam T, Norton JD. Screening for colorectal cancer: established and emerging modalities. Nat Rev Gastroenterol Hepatol. 2011;8:711‐722. [DOI] [PubMed] [Google Scholar]
- 5. Ladabaum U, Dominitz JA, Kahi C, Schoen RE. Strategies for colorectal cancer screening. Gastroenterology. 2020;158(2):418–432. [DOI] [PubMed] [Google Scholar]
- 6. Pagni F, Guerini‐Rocco E, Schultheis AM, et al. Targeting immune‐related biological processes in solid tumors: we do need biomarkers. Int J Mol Sci. 2019;20(21):5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gessani S, Belardelli F. Immune dysfunctions and immunotherapy in colorectal cancer: the role of dendritic cells. Cancers (Basel). 2019;11(10):1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Abakushina EV, Gelm YV, Pasova IA, Bazhin AV. Immunotherapeutic approaches for the treatment of colorectal cancer. Biochemistry (Mosc). 2019;84:720‐728. [DOI] [PubMed] [Google Scholar]
- 9. Yaghoubi N, Soltani A, Ghazvini K, Hassanian SM, Hashemy SI. PD‐1/ PD‐L1 blockade as a novel treatment for colorectal cancer. Biomed Pharmacother. 2019;110:312‐318. [DOI] [PubMed] [Google Scholar]
- 10. Ciardiello D, Vitiello PP, Cardone C, et al. Immunotherapy of colorectal cancer: challenges for therapeutic efficacy. Cancer Treat Rev. 2019;76:22‐32. [DOI] [PubMed] [Google Scholar]
- 11. Lichtenstern CR, Ngu RK, Shalapour S, Immunotherapy KM. Inflammation and colorectal cancer. Cells. 2020;9(3):618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gutting T, Burgermeister E, Hartel N, Ebert MP. Checkpoints and beyond ‐ immunotherapy in colorectal cancer. Semin Cancer Biol. 2019;55:78‐89. [DOI] [PubMed] [Google Scholar]
- 13. Morse MA, Hochster H, Benson A. Perspectives on treatment of metastatic colorectal cancer with immune checkpoint inhibitor therapy. Oncologist. 2020;25:33‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kather JN, Halama N, Jaeger D. Genomics and emerging biomarkers for immunotherapy of colorectal cancer. Semin Cancer Biol. 2018;52:189‐197. [DOI] [PubMed] [Google Scholar]
- 15. Zhang Y, Sun Z, Mao X, et al. Impact of mismatch‐repair deficiency on the colorectal cancer immune microenvironment. Oncotarget. 2017;8:85526‐85536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gunawardene A, Dennett E, Larsen P. Prognostic value of multiple cytokine analysis in colorectal cancer: a systematic review. J Gastrointest Oncol. 2019;10:134‐143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kalioraki MA, Artemaki PI, Sklirou AD, et al. Heat shock protein beta 3 (HSPB3) is an unfavorable molecular biomarker in colorectal adenocarcinoma. Mol Carcinog. 2019;59(1):116‐125. [DOI] [PubMed] [Google Scholar]
- 18. Palaiologos P, Chrysikos D, Theocharis S, Kouraklis G. The prognostic value of G1 Cyclins, p21 and Rb protein in patients with colon cancer. Anticancer Res. 2019;39:6291‐6297. [DOI] [PubMed] [Google Scholar]
- 19. Artemaki PI, Sklirou AD, Kontos CK, et al. High clusterin (CLU) mRNA expression levels in tumors of colorectal cancer patients predict a poor prognostic outcome. Clin Biochem. 2020;75:62–69. [DOI] [PubMed] [Google Scholar]
- 20. Elmasry M, Brandl L, Engel J, Jung A, Kirchner T, Horst D. RBP7 is a clinically prognostic biomarker and linked to tumor invasion and EMT in colon cancer. J Cancer. 2019;10:4883‐4891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bottani M, Banfi G, Lombardi G. Circulating miRNAs as diagnostic and prognostic biomarkers in common solid tumors: focus on lung, breast, prostate cancers, and osteosarcoma. J Clin Med. 2019;8(10):1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Fadaka AO, Klein A, Pretorius A. In silico identification of microRNAs as candidate colorectal cancer biomarkers. Tumour Biol. 2019;41:1010428319883721. [DOI] [PubMed] [Google Scholar]
- 23. Jiang X, Zhu Q, Wu P, Zhou F, Chen J. Upregulated long noncoding RNA LINC01234 predicts unfavorable prognosis for colorectal cancer and negatively correlates with KLF6 expression. Ann Lab Med. 2020;40:155‐163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yao F, Wang Q, Wu Q. The prognostic value and mechanisms of lncRNA UCA1 in human cancer. Cancer Manag Res. 2019;11:7685‐7696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hao S, Cong L, Qu R, Liu R, Zhang G, Li Y. Emerging roles of circular RNAs in colorectal cancer. Onco Targets Ther. 2019;12:4765‐4777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kwon MJ. Emerging immune gene signatures as prognostic or predictive biomarkers in breast cancer. Arch Pharm Res. 2019;42(11):947–961. [DOI] [PubMed] [Google Scholar]
- 27. Lin P, Guo YN, Shi L, et al. Development of a prognostic index based on an immunogenomic landscape analysis of papillary thyroid cancer. Aging (Albany NY). 2019;11:480‐500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wang Z, Song Q, Yang Z, Chen J, Shang J, Ju W. Construction of immune‐related risk signature for renal papillary cell carcinoma. Cancer Med. 2019;8:289‐304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Yang W, Lai Z, Li Y, et al. Immune signature profiling identified prognostic factors for gastric cancer. Chin J Cancer Res. 2019;31:463‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bao X, Shi R, Zhang K, et al. Immune landscape of invasive ductal carcinoma tumor microenvironment identifies a prognostic and immunotherapeutically relevant gene signature. Front Oncol. 2019;9:903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Shen S, Wang G, Zhang R, et al. Development and validation of an immune gene‐set based prognostic signature in ovarian cancer. EBioMedicine. 2019;40:318‐326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cancer Genome Atlas Research N , Weinstein JN, Collisson EA, et al. The cancer genome atlas pan‐cancer analysis project. Nat Genet. 2013;45:1113‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Bhattacharya S, Andorf S, Gomes L, et al. ImmPort: disseminating data to the public for the future of immunology. Immunol Res. 2014;58:234‐239. [DOI] [PubMed] [Google Scholar]
- 34. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gene OC. The gene ontology project in 2008. Nucleic Acids Res. 2008;36:D440‐D444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yu G, Wang LG, Han Y, He QY. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284‐287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tripathi MK, Deane NG, Zhu J, et al. Nuclear factor of activated T‐cell activity is associated with metastatic capacity in colon cancer. Cancer Res. 2014;74:6947‐6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Edgar R, Domrachev M, Lash AE. Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002;30:207‐210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Szklarczyk D, Morris JH, Cook H, et al. The STRING database in 2017: quality‐controlled protein‐protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362‐D368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu T, Ortiz JA, Taing L, et al. Cistrome: an integrative platform for transcriptional regulation studies. Genome Biol. 2011;12:R83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cicenas J, Tamosaitis L, Kvederaviciute K, et al. KRAS, NRAS and BRAF mutations in colorectal cancer and melanoma. Med Oncol. 2017;34:26. [DOI] [PubMed] [Google Scholar]
- 43. Lech G, Slotwinski R, Slodkowski M, Krasnodebski IW. Colorectal cancer tumour markers and biomarkers: recent therapeutic advances. World J Gastroenterol. 2016;22:1745‐1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Karagkouni D, Paraskevopoulou MD, Chatzopoulos S, et al. DIANA‐TarBase v8: a decade‐long collection of experimentally supported miRNA‐gene interactions. Nucleic Acids Res. 2018;46:D239‐D245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Paraskevopoulou MD, Vlachos IS, Karagkouni D, et al. DIANA‐LncBase v2: indexing microRNA targets on non‐coding transcripts. Nucleic Acids Res. 2016;44:D231‐D238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shannon P, Markiel A, Ozier O, et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003;13:2498‐2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu T, Dai Y. Tumor microenvironment and therapeutic response. Cancer Lett. 2017;387:61‐68. [DOI] [PubMed] [Google Scholar]
- 48. Gonzalez H, Hagerling C, Werb Z. Roles of the immune system in cancer: from tumor initiation to metastatic progression. Genes Dev. 2018;32:1267‐1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Janssen LME, Ramsay EE, Logsdon CD, Overwijk WW. The immune system in cancer metastasis: friend or foe? J Immunother Cancer. 2017;5:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Koi M, Carethers JM. The colorectal cancer immune microenvironment and approach to immunotherapies. Future Oncol. 2017;13:1633‐1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mi L, Zhu F, Yang X, et al. The metastatic suppressor NDRG1 inhibits EMT, migration and invasion through interaction and promotion of caveolin‐1 ubiquitylation in human colorectal cancer cells. Oncogene. 2017;36:4323‐4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ma J, Gao Q, Zeng S, Shen H. Knockdown of NDRG1 promote epithelial‐mesenchymal transition of colorectal cancer via NF‐kappaB signaling. J Surg Oncol. 2016;114:520‐527. [DOI] [PubMed] [Google Scholar]
- 53. Ding D, Han S, Zhang H, He Y, Li Y. Predictive biomarkers of colorectal cancer. Comput Biol Chem. 2019;83:107106. [DOI] [PubMed] [Google Scholar]
- 54. Makondi PT, Chu CM, Wei PL, Chang YJ. Prediction of novel target genes and pathways involved in irinotecan‐resistant colorectal cancer. PLoS One. 2017;12:e0180616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Yu C, Hong H, Lu J, et al. Prediction of target genes and pathways associated with cetuximab insensitivity in colorectal cancer. Technol Cancer Res Treat. 2018;17:1533033818806905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen L, Lu D, Sun K, et al. Identification of biomarkers associated with diagnosis and prognosis of colorectal cancer patients based on integrated bioinformatics analysis. Gene. 2019;692:119‐125. [DOI] [PubMed] [Google Scholar]
- 57. Sun G, Li Y, Peng Y, et al. Identification of differentially expressed genes and biological characteristics of colorectal cancer by integrated bioinformatics analysis. J Cell Physiol. 2019;234(9):15215–15224. [DOI] [PubMed] [Google Scholar]
- 58. Muinao T, Deka Boruah HP, Pal M. Multi‐biomarker panel signature as the key to diagnosis of ovarian cancer. Heliyon. 2019;5:e02826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Rodriguez‐Enriquez S, Pacheco‐Velazquez SC, Gallardo‐Perez JC, et al. Multi‐biomarker pattern for tumor identification and prognosis. J Cell Biochem. 2011;112:2703‐2715. [DOI] [PubMed] [Google Scholar]
- 60. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med. 2014;6:224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Naoi Y, Noguchi S. Multi‐gene classifiers for prediction of recurrence in breast cancer patients. Breast Cancer. 2016;23:12‐18. [DOI] [PubMed] [Google Scholar]
- 62. Wurzer H, Hoffmann C, Al Absi A, Thomas C. Actin cytoskeleton straddling the immunological synapse between cytotoxic lymphocytes and cancer cells. Cells. 2019;8(5):463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Paluch EK, Aspalter IM, Sixt M. Focal adhesion‐independent cell migration. Annu Rev Cell Dev Biol. 2016;32:469‐490. [DOI] [PubMed] [Google Scholar]
- 64. Gao S, Yang D, Fang Y, et al. Engineering nanoparticles for targeted remodeling of the tumor microenvironment to improve cancer immunotherapy. Theranostics. 2019;9:126‐151. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1
Fig S2
Table S1
Table S2
Table S3
Table S4
Table S5
Supplementary Material
Data Availability Statement
All data generated or analysed during this study are included in this published article and its supplementary information files.