

Abstract
Objective
To describe a phenotype caused by ATP1A3 mutations, which manifests as dystonia, dysmorphism of the face, encephalopathy with developmental delay, brain MRI abnormalities always including cerebellar hypoplasia, no hemiplegia (Ø) (D-DEMØ), and neonatal onset.
Methods
Review and analysis of clinical and genetic data.
Results
Patients shared the above traits and had whole-exome sequencing that showed de novo variants of the ATP1A3 gene, predicted to be disease causing and occurring in regions of the protein critical for pump function. Patient 1 (c.1079C>G, p.Thr360Arg), an 8-year-old girl, presented on day 1 of life with episodic dystonia, complex partial seizures, and facial dysmorphism. MRI of the brain revealed cerebellar hypoplasia. Patient 2 (c.420G>T, p.Gln140His), an 18-year-old man, presented on day 1 of life with hypotonia, tremor, and facial dysmorphism. He later developed dystonia. MRI of the brain revealed cerebellar hypoplasia and, later, further cerebellar volume loss (atrophy). Patient 3 (c.974G>A, Gly325Asp), a 13-year-old girl, presented on day 1 of life with tremor, episodic dystonia, and facial dysmorphism. MRI of the brain showed severe cerebellar hypoplasia. Patient 4 (c.971A>G, p.Glu324Gly), a 14-year-old boy, presented on day 1 of life with tremor, hypotonia, dystonia, nystagmus, facial dysmorphism, and later seizures. MRI of the brain revealed moderate cerebellar hypoplasia.
Conclusions
D-DEMØ represents an ATP1A3-related phenotype, the observation of which should trigger investigation for ATP1A3 mutations. Our findings, and the presence of multiple distinct ATP1A3-related phenotypes, support the possibility that there are differences in the underlying mechanisms.
The α3 subunit of the transmembrane Na+/K+ ATPase is the main contributor in rapid restoration of neuronal membrane potential after rapid depolarization. A reduction in the activity of this subunit leads to different neurologic diseases.1 The classical spectrum of disorders related to ATP1A3 mutations includes rapid-onset dystonia parkinsonism (RDP), alternating hemiplegia of childhood (AHC), cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss (CAPOS) syndrome, and multiple other phenotypes.1,2 Here, we report 4 cases of ATP1A3 mutations with a unique clinical phenotype consisting of the combination of 6 distinct features. These include dystonia, dysmorphism of the face, encephalopathy with developmental delay, brain MRI abnormalities always including cerebellar hypoplasia, no hemiplegia (Ø), and neonatal onset of symptoms.
Methods
To probe the potential effects of the variants, we performed structure and structure/function analyses using the PyMOL (pymol.org/2/) and Protein Data Bank databases (rcsb.org/).
Standard protocol approvals, registrations, and patient consent
Patients' guardians provided written consent, and their prospectively collected data from our center and retrospectively available data from previous centers were entered into our institutional review board–approved database.
Data availability
Anonymized data will be shared by request from any qualified investigator.
Results
Case series
Patient 1 is an 8-year-old girl born to healthy parents with no family history of neurologic disorders (table 1). On day 1 of life, limb and whole-body dystonia started and occurred multiple times per day, 2–3 days a week. Each episode lasted 5–10 minutes. Episodes were often triggered by overstimulation and were aborted by sleep (videos 1 and 2). Additional features that started on day 1 of life and continued throughout infancy and childhood include oculomotor abnormalities (nystagmus and eye deviation) that have occurred daily, episodes of autonomic dysfunction (flushing, sweating, and tachycardia) that have occurred 2–3 times per week, and complex partial seizures that have lasted a few minutes each and have occurred up to 10 times a day. Dysmorphic features included a high forehead with bitemporal narrowing, broad nasal bridge, narrow palpebral fissures, bulbous nasal tip, anteriorly facing nostrils, thickened alae nasi, long philtrum, micrognathia, thin upper lip, prominent lower lip, and incompletely formed antitragus and lower part of the antihelix in the ear pinnae (figure 1).3,4 Scoliosis was noted at 1 year of age. Brain MRI at 2 years of age revealed global cerebral hypoplasia, prominent parietal lobe hypoplasia, mild cerebellar hypoplasia, and pontine hypoplasia (figure 2A). She has had no episodes of hemiplegia, although she has quadriplegic episodes, occurring at most once a year, lasting for a few minutes, and generally only triggered by painful stimuli. She has continued to have daily complex partial and tonic seizures refractory to medications. Developmentally, she is nonverbal, nonambulatory, and hyperreflexic with clonus. Whole-exome sequencing revealed an ATP1A3 de novo missense heterozygous variant, c.1079C>G (p.Thr360Arg), predicted to be disease causing and involving the cytoplasmic domain, an area responsible for adenosine triphosphate (ATP) binding and subsequent phosphorylation-mediated mechanisms. Threonine is a polar, noncharged, and hydroxylic amino acid, whereas arginine is a positively charged and basic amino acid. The amino acid substitution is a nonconservative change in a well-conserved residue within the ATP binding site. In silico analysis predicts this variant likely affects the secondary structure of its respective protein, and thus, it is likely to be pathogenic. Mutations in the close residues, 358 and 363, have been reported to cause early infantile epileptic encephalopathy.5
Table 1.
Clinical features of D-DEMØ

Figure 1. Pictures of patients 1 and 4.
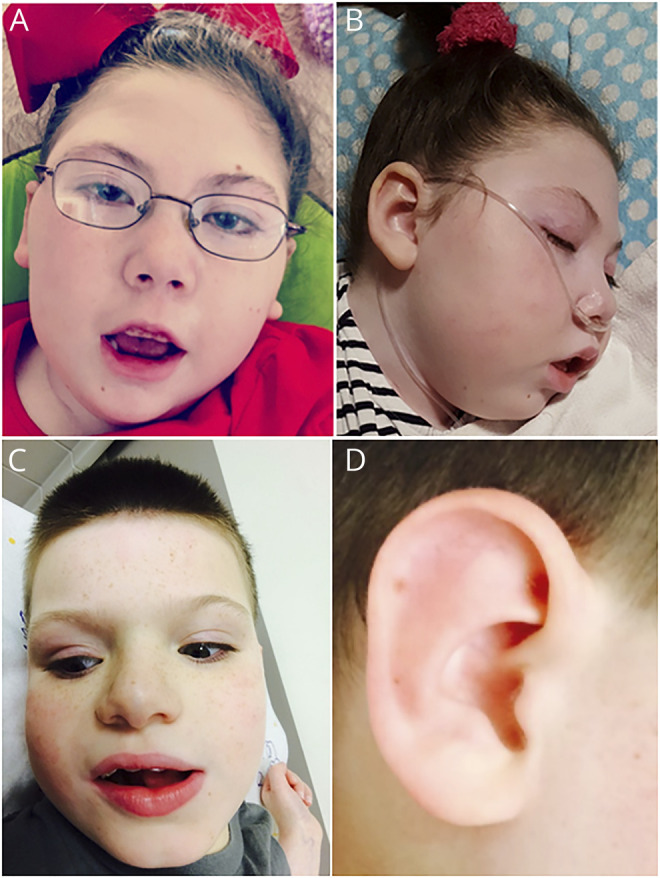
(A and B) Facial features of patient 1 demonstrating a high forehead with bitemporal narrowing, broad nasal bridge, narrow palpebral fissures, bulbous nasal tip, anteriorly facing nostrils, thickened alae nasi, long philtrum, micrognathia, thin upper lip, prominent lower lip, and incompletely formed antitragus and lower part of the antihelix in the ear pinnae. (C and D) Facial features of patient 4 demonstrating a high forehead with bitemporal narrowing, broad nasal bridge, slightly narrow palpebral fissure, bulbous nasal tip with anteriorly facing nostrils and thickened alae nasi, thin upper lip, prominent lower lip, and poorly folded ear pinnae.
Figure 2. Brain MRIs of patients 1–3.
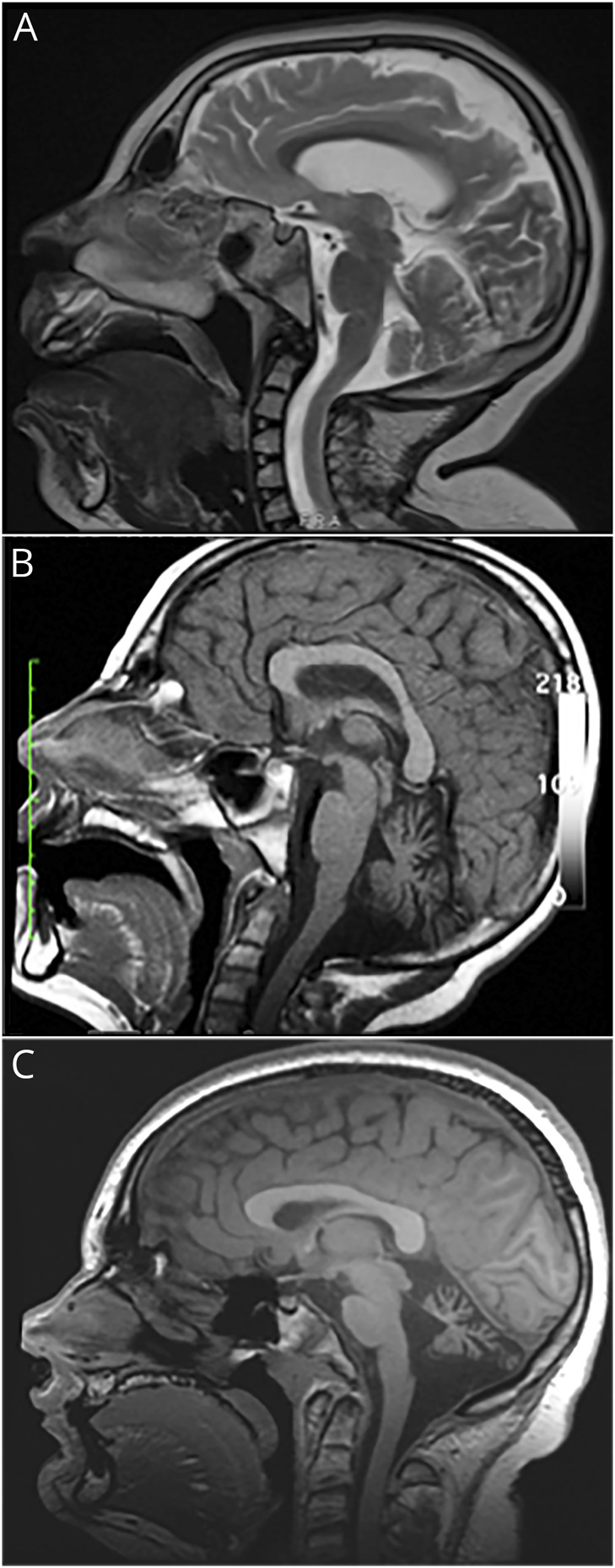
(A) Brain MRI of patient 1, taken at 8 years of age, showing mild cerebellar hypoplasia and pontine hypoplasia. (B) Brain MRI of patient 2, taken at 8 years of age, showing moderate to severe cerebellar atrophy. (C) Brain MRI of patient 3, taken at 13 years of age, showing severe cerebellar hypoplasia with a smaller-than-expected pons.
Eight year old female with dystonia of the left arm.Download Supplementary Video 1 (30MB, mp4) via http://dx.doi.org/10.1212/000466_Video_1
Eight year old female with dystonia of the left arm and hand.Download Supplementary Video 2 (44MB, mp4) via http://dx.doi.org/10.1212/000466_Video_2
Patient 2 is an 18-year-old man born to healthy parents with no family history of neurologic disorders. On day 1 of life, hypotonia and tremor were noted. Intermittent episodes of flushing of extremities with, on examination, tactile coldness and 1 complex partial seizure were noted at 2 months of age. Alternating esotropia was noted at 5 months of age. Dysmorphic features included long palpebral fissures and mildly thick eyebrows. Brain MRI at 1 year of age revealed mild cerebellar hypoplasia. Developmentally, the patient was slow in gaining milestones. Ambulation started at 23 months of age with a wide-based gait. He had hyperreflexia with clonus. Fine motor movements have always been slow, and speech has always been dysarthric. Progressive ataxia was noted beginning at 6 years of age. Cognitively, he made some progress during childhood but never progressed beyond the first-grade level of function. Repeat brain MRI at 8 years of age noted further cerebellar volume loss, indicating progressive cerebellar atrophy (figure 2B). Episodes of cervical dystonia, lasting minutes to several hours, began in adolescence and were often triggered by excitement or nervousness. The patient also manifested mild kyphoscoliosis, which was noted at 16 years of age. He has no history of hemiplegic or quadriplegic episodes. Whole-exome sequencing revealed an ATP1A3 de novo heterozygous missense variant, c.420G>T (p.Gln140His), predicted to be disease causing and involving the M2 transmembrane domain. Glutamine is a polar molecule with a high tendency for hydrogen bonding, whereas histidine is a bulkier, positively charged amino acid that forms less hydrogen bonds with surrounding elements. The amino acid substitution is a nonconservative change in a well-conserved residue within the M2 domain. In silico analysis predicts this variant likely affects the secondary structure of its respective protein, and thus, it is likely to be pathogenic. A different mutation at the same site, Gln140Leu, has been reported to cause AHC.5
Patient 3 is a 13-year-old girl born to healthy parents with no family history of neurologic disorders. On day 1 of life, a continuous tremor was noted as well as dystonic episodes that involved tensing of the feet and curling of the toes. The dystonic episodes would last several minutes. Dysmorphic features included broad nasal bridge and tip, hypoplastic alae nasi, slightly large mouth, and a thin upper lip. Developmentally, ambulation and speech occurred at 15 months of age. She acquired and subsequently lost the ability to write her name. Diagnostic evaluation included a brain MRI at 9 years of age revealing diffuse cerebellar hypoplasia. Repeat brain MRI at 13 years of age showed stable cerebellar hypoplasia and a small pontine volume that was not captured by previous brain MRI (figure 2C). She exhibits lower extremity spasticity, hyperreflexia, clonus, and a wide-based gait with ataxia. She has no history of hemiplegia, abnormal eye movements, autonomic dysfunction, or seizures. Whole-exome sequencing revealed an ATP1A3 de novo heterozygous variant, c.974G>A (Gly325Asp), predicted to be disease causing and involving the M4 transmembrane domain. Glycine is a small nonpolar amino acid, whereas aspartate is a large polar amino acid with a negative charge. The amino acid substitution is a nonconservative change in a well-conserved residue within the M4 domain. In silico analysis predicts this variant likely affects the secondary structure of its respective protein, and thus, it is likely to be pathogenic. This variant has been reported in the ClinVar database to cause the CAPOS phenotype.6
Patient 4 is a 14-year-old boy born to healthy parents with no family history of neurologic disorders. The mother's pregnancy was uncomplicated, other than increased amniotic fluid that was noted toward the end of the pregnancy. On day 1 of life, episodes of bilateral lower extremity dystonia and hypotonia were noted, in addition to bilateral tremor and atypical nystagmus. Complex partial seizures started on day 2 of life and consisted of circumoral cyanosis accompanied by unilateral arm stiffness and lip smacking. Seizure frequency increased at around 6–7 years of age, occurring up to twice a day and lasting for several minutes each. He has had several episodes of status epilepticus. Seizures decreased after starting clobazam to biweekly with a duration of less than 1 minute each. He has had no episodes of hemiplegia. Brain MRI at 7 years of age displayed moderate hypoplasia of the cerebellum. The cerebral hemispheres also had mildly prominent perivascular spaces but no focal parenchymal signal abnormality. Dysmorphic features include a high forehead with bitemporal narrowing, broad nasal bridge, slightly narrow palpebral fissure, bulbous nose with anteriorly facing nostrils and thickened alae nasi, thin upper lip, prominent lower lip, and poorly folded pinnae. Developmentally, he remains unable to sit independently, hyperreflexic with clonus, uses a wheelchair, and never developed speech. Whole-exome sequencing revealed an ATP1A3 de novo heterozygous mutation, c.971A>G (p.Glu324Gly), predicted to be disease causing and involving the M4 transmembrane domain. Glutamate is a negatively charged amino acid with high tendency for interactions, whereas glycine is nonpolar, smaller amino acid and contains no side chains, thus carries no steric hindrance. The amino acid substitution is a nonconservative change in a well-conserved residue within the M4 domain. In silico analysis predicts this variant likely affects the secondary structure of its respective protein, and thus, it is likely to be pathogenic. A different mutation at the same site, Glu324Gln, has been reported to cause AHC.5
Structure/function relationship analysis of D-DEMØ mutations
Table 2 shows results for the following parameters: allele frequency in gnomAD, binary predictions from various programs, Combined Annotation-Dependent Depletion scores, and the interpretation of the American College of Medical Genetics and Genomics criteria with the categories we used to make the call. The Na+/K+-ATPases are responsible for maintaining sodium and potassium gradients across the cell membrane. The Na+/K+-ATPases are composed of α, β, and FXYD subunits. The α-subunit is encoded by ATP1A for which there are 4 isoforms, ATP1A1–4. The primary sequences of the α-isoforms are highly conserved, especially between isoforms 1–3. ATP1A3 is primarily expressed in the brain, whereas the other isoforms are more widely expressed across various cells (including muscle, brain, and sperm cells).
Table 2.
Variant classification and analyses

ATP1A can be divided into a membrane domain composed of 10 transmembrane helices, M1–M10, and 3 cytoplasmic domains, N, P, and A domains.5,7 The N domain is the kinase responsible for ATP binding, the P domain contains the conserved aspartate residue that gets phosphorylated during the catalytic cycle, and the A domain is responsible for phosphatase activity.5,7 In the reaction cycle, 3 intracellular Na+ ions are exchanged with 2 extracellular K+ ions. During the reaction cycle, a conserved aspartate in the P domain, Asp366, is phosphorylated when the α-subunit binds 3 Na+ ions, which is later dephosphorylated with the binding of 2 K+ ions. Thus, the P-cytoplasmic domain is central to the activity of the pump.
The 4 mutations identified in this study in ATP1A3 are in residues that are conserved between the 4 isoforms of ATP1A and also with other species, including porcine and shark ATP1A. Three of the mutant residues are within 2 transmembrane helices, M2 (p.Gln140His) and M4 (p.Glu324Gly and p.Gly325Asp). The fourth mutation, p.Thr360Arg, is in the cytoplasmic domain that proceeds off of the transmembrane domain M4 (figure 3). These mutations all lie in regions where other disease mutations have been identified with a plethora of disease mutations identified in M4 including a different disease allele in Glu324, while the same Gly325 allele has been reported to cause CAPOS syndrome.5–7 Disease mutations have also been identified in M2, and an alternate disease allele has been identified in residue Gln140. Two other disease mutations have been identified around the Thr360 site of our first patient—those in Gly358 and Ile363.
Figure 3. Model showing the location of the disease mutations in ATP1A.

(A) The crystal structure of the Na+/K+-ATPase is shown with subunits α (green), β (blue), and FXYD (purple). (B) This is an expanded image from “A” that shows the positions of the mutated residues. K+ is colored as a dark purple sphere, whereas the atoms of the diseased residues are spheres colored as orange, red, and blue for carbon, oxygen, and nitrogen, respectively. The atoms for Asp366, the key residue that is phosphorylated during pump function, are colored as red, light red, and blue, for carbon, oxygen, and nitrogen, respectively. The crystal structure is determined from porcine, PDB, and 4RES (rcsb.org/structure/4RES). (C) Similar to “B,” except that it shows the crystal structure of the Na+/K+-ATPase with Na+ ions (dark purple spheres) bound, determined from porcine, PDB, and 3WGU (rcsb.org/structure/3wgu). The primary sequence of ATP1A from the porcine protein in this structure is identical to that of ATP1A3 from the human protein. This figure was made using PyMOL.32 Note, only the Na+/K+ ions bound to the transmembrane helices are transported during the pumping. The Na+/K+ cations in close proximity to Thr360 play a structural role and are not pumped across the cell membrane. Their primary role is to facilitate the phosphorylation process. PDB = Protein Data Bank.
Glu324 is involved in binding Na+/K+ ion along with a number of residues in transmembrane helices, M5, M6, and M8. As such, it is no surprise that p.Glu324His is causative for the disease. Likewise, p.Gly325Asp places a second negative charge near the Na+/K+ binding site, and it is adjacent to M6. Although it is difficult to predict the rotamer form of Asp325, the extra charge is expected to disrupt the Na+/K+ binding site by either directly interacting with the Na+/K+ or altering the conformation of the Na+/K+ binding site.
Thr360 is in the P domain at the interface of the transmembrane domain and just 13 Å from a Na+/K+ binding site in the P domain and about 14 Å from Asp366, which is phosphorylated during the reaction cycle. The Na+/K+ binding site at this location has been shown to be involved in the activation of dephosphorylation of Asp366 during the reaction cycle, but the bound ions are not transported.8 Thr360 is at the end of the helix, which abuts M5 in which the largest number of disease alleles has been identified.5,7 Thus, it is likely that substitution of the large side chain of Arg for that of Thr elicits structural changes of the Na+/K+ binding sites.
Gln140 is positioned in the transmembrane helix, M2, which abuts M4 and M3, and is close to the cytoplasmic side of the pump. The Na+/K+ binding site in the P domain is about 13 Å from Gln140. Thus, p.Gln140His can affect both the ion binding sites within the membrane domain and that in the cytoplasmic domain.
Discussion
The cases we describe, summarized in table 1, do not fit the AHC nor the RDP diagnostic criteria but do have some shared features with each syndrome.2 Features shared with AHC include early onset of symptoms (4/4 patients), presence of dystonia that is triggered by hyperexcitation and/or physiologic or psychological stressors (4/4 patients), global developmental delay (4/4 patients), seizures (3/4 patients), episodes of quadriplegia (2/4 patients), and symptoms of autonomic dysfunction (2/4 patients).2,9 However, none of these cases have the cardinal feature of AHC, which is episodes of alternating hemiplegia. Furthermore, all 4 patients have cerebellar hypoplasia, which is found in only the minority of patients with AHC. When comparing our patients with RDP, dystonia is a common feature (table e-1, links.lww.com/NXG/A307). Hyperreflexia, not a feature of AHC, is also shared with RDP. However, RDP symptoms do not occur in the neonatal period or in childhood, whereas in D-DEMØ, symptoms start at birth.10 These patients have facial dysmorphism and early severe developmental delay not present in RDP.10 Our patients are different from all the other atypical cases in the literature that we could find previously reported due to ATP1A3 mutations. As seen in table 3, all the atypical cases reported previously lacked 3–5 of the 6 cardinal D-DEMØ manifestations.11–15
Table 3.
D-DEMØ vs spectrum of atypical cases

Mutations of ATP1A3 cause reduction in enzyme activity and in sodium-potassium transfer across the neuronal membranes.16–18 They also lead to increased excitability in the brain and abnormal cerebellar Purkinje cell and deep nuclei neuronal firing.19–21 Most ATP1A3 mutations causing AHC affect an amino acid located within a transmembrane domain, changing the configuration of the protein, and therefore affecting several factors such as binding to Na+ and K+, or transport of these ions in and out of the cell.5,16,22 In RDP, which has a phenotype milder than AHC, mutations are more evenly distributed across the ATPA13 protein.16 In 3/4 of our patients, the mutations occurred in a transmembrane domain (M2 in case 2 and M4 in cases 3 and 4). In only 1 of the 4 patients, case 1, the mutation occurred in the intracellular domain just outside the transmembrane domains. It should be noted that the transmembrane domains contain only about 19% of the total number of amino acids in ATP1A3 (194/1,013).5,23 This is consistent with previous observations that mutations in the transmembrane domain are more likely to cause more severe phenotypes.16
Sites of sodium and potassium binding are predominantly clustered around transmembrane helices M4–M6 of the alpha subunit. The first sodium ion is localized to M4–M6, the second localized to M4 and M6, and the third localized to M5, M6, and M8. For potassium, the first ion is localized to M5 and M6, whereas the second is localized to M4–M6 segments. Analysis of the potential relationship of the positions of these variants, and their effect on the polarity and other characteristics of the amino acid residues, support pathogenicity as discussed in the Structure/function relationship analysis of D-DEMØ mutations section above.
The resulting clinical features in all 4 cases probably reflect a shared pathophysiologic phenomenon at the level of the Na+/K+-ATPase pump. This pump plays an essential role in maintaining the excitatory-inhibitory balance in various brain regions, which potentially could account for dystonia and seizures manifested in our patients.18–21,24 ATP1A3 is highly expressed in cerebellar Purkinje cells.25 This could explain the cerebellar hypoplasia observed in our patients. Although we recognize that the MRI calls of “cerebellar hypoplasia” and “pontine hypoplasia” are nonquantitative, the images provided in figure 2 support these findings. ATP1A3 is expressed in utero in the human brain in multiple areas.26 This could potentially explain the neonatal onset in all 4 patients and the brain malformation in the first patient. ATP1A3 is also highly expressed in brainstem nuclei.25 Given that congenital cranial nerve pathology has been shown to result in facial dysmorphism,27 and that ATP1A3 is also expressed in utero,26 this could potentially be the underlying mechanism behind the facial dysmorphic features observed in our patients. Also, given the increasing availability of the gene panel and whole-exome sequencing testing coupled with increasing awareness of phenotypes caused by ATP1A3 mutations, it is evident that the spectrum of manifestations secondary to these mutations will continue to grow.2 D-DEMØ appears to be not only a different but also a more severe phenotype than AHC, RDP, and CAPOS.10,22,28–30 The different manifestations between these syndromes suggest that their underlying mechanisms may differ, thus supporting the investigation of their underlying pathophysiologies.
We must consider the possibility of digenic or higher-order polygenic/oligogenic etiologies. The unique features present in our patients may not necessarily only be due to the mutation itself but may be related to the interaction between genetic background and the ATP1A3 mutations. This may explain why some ATP1A3 mutations can cause different phenotypes in different individuals such as AHC and RDP,1 or AHC and D-DEMØ. It is known that there are many loci capable of modifying the Na+/K+-ATPase–related manifestations,31 and future investigations may provide additional insights for the basis of multiple distinct syndromes and symptoms associated with ATP1A3-related disease.
The persistent unique features coupled with the absence of hemiplegia indicate the existence of a stand-alone phenotype in our patients. Observation of this phenotype, with its neonatal onset and above distinct findings without hemiplegia, should trigger investigation for ATP1A3 mutations.
Acknowledgment
The authors thank all members of the Duke AHC Multidisciplinary Program. M. McLean is deceased.
Glossary
- AHC
alternating hemiplegia of childhood
- ATP
adenosine triphosphate
- CAPOS
cerebellar ataxia, areflexia, pes cavus, optic atrophy, and sensorineural hearing loss
- RDP
rapid-onset dystonia parkinsonism
Appendix. Authors
Study funding
This study was supported by Duke Fund Nos. 4410161 and 3912247, by a donation by the Cure AHC Foundation (M.A.M.), and by NIH R35GM131731 to D.M.M.
Disclosure
L. Prange, M. Pratt, K. Herman, R. Schiffmann, and D. M. Mueller report no disclosures. M. McLean is deceased; disclosures are not included for this author. M. Moya Mendez, N. Walley, E.L. Heinzen, D. Goldstein, V. Shashi, A. Hunanyan, and V. Pagadala report no disclosures. M.A. Mikati reports a pending patent application for the treatment of alternating hemiplegia of childhood. Go to Neurology.org/NG for full disclosures.
References
- 1.Heinzen EL, Arzimanoglou A, Brashear A, et al. Distinct neurological disorders with ATP1A3 mutations. Lancet Neurol 2014;13:503–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fernandes C, Mikati M. The expanding spectrum of ATP1A3 related disease. Eur J Paediatr Neurol 2019;23:345–346. [DOI] [PubMed] [Google Scholar]
- 3.Hennekam RC, Cormier-Daire V, Hall J, Méhes K, Patton M, Stevenson R. Elements of morphology: standard terminology for the nose and philtrum. Am J Med Genet A 2008;149A:61–76. [DOI] [PubMed] [Google Scholar]
- 4.Cox TC, Camci ED, Vora S, Luquetti DV, Turner EE. The genetics of auricular development and malformation: new findings in model systems driving future directions for microtia research. Eur J Med Genet 2014;57:394–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Holm R, Toustrup-Jensen MS, Einholm AP, Schack VR, Andersen JP, Vilsen B. Neurological disease mutations of alpha3 Na+,K+-ATPase: structural and functional perspectives and rescue of compromised function. Biochim Biophys Acta 2016;1857:1807–1828. [DOI] [PubMed] [Google Scholar]
- 6.Landrum MJ, Lee JM, Benson M, et al. ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res 2018;46:D1062–D1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clausen MV, Hilbers F, Poulsen H. The structure and function of the Na,K-ATPase isoforms in health and disease. Front Physiol 2017;8:371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schack VR, Morth JP, Toustrup-Jensen MS, et al. Identification and function of a cytoplasmic K+ site of the Na+, K+ -ATPase. J Biol Chem 2008;283:27982–27990. [DOI] [PubMed] [Google Scholar]
- 9.Masoud M, Prange L, Wuchich J, Hunanyan A, Mikati MA. Diagnosis and treatment of alternating hemiplegia of childhood. Curr Treat Options Neurol 2017;19:8. [DOI] [PubMed] [Google Scholar]
- 10.Brashear A, Dobyns WB, de Carvalho Aguiar P, et al. The phenotypic spectrum of rapid-onset dystonia-parkinsonism (RDP) and mutations in the ATP1A3 gene. Brain 2007;130:828–835. [DOI] [PubMed] [Google Scholar]
- 11.Nicita F, Travaglini L, Sabatini S, et al. Childhood-onset ATP1A3-related conditions: report of two new cases of phenotypic spectrum. Parkinsonism Relat Disord 2016;30:81–82. [DOI] [PubMed] [Google Scholar]
- 12.Paciorkowski AR, McDaniel SS, Jansen LA, et al. Novel mutations in ATP1A3 associated with catastrophic early life epilepsy, episodic prolonged apnea, and postnatal microcephaly. Epilepsia 2015;56:422–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rosewich H, Baethmann M, Ohlenbusch A, Gärtner J, Brockmann K. A novel ATP1A3 mutation with unique clinical presentation. J Neurol Sci 2014;341:133–135. [DOI] [PubMed] [Google Scholar]
- 14.Sasaki M, Ishii A, Saito Y, Hirose S. Intermediate form between alternating hemiplegia of childhood and rapid-onset dystonia-parkinsonism. Mov Disord 2014;29:153–154. [DOI] [PubMed] [Google Scholar]
- 15.Termsarasab P, Yang AC, Frucht SJ. Intermediate phenotypes of ATP1A3 mutations: phenotype-genotype correlations. Tremor Other Hyperkinet Mov (NY) 2015;5:336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinzen EL, Swoboda KJ, Hitomi Y, et al. De novo mutations in ATP1A3 cause alternating hemiplegia of childhood. Nat Genet 2012;44:1030–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li M, Jazayeri D, Corry B, et al. A functional correlate of severity in alternating hemiplegia of childhood. Neurobiol Dis 2015;77:88–93. [DOI] [PubMed] [Google Scholar]
- 18.Simmons C, Thompson C, Cawthon B, et al. Direct evidence of impaired neuronal Na/K-ATPase pump function in alternating hemiplegia of childhood. Neurobiol Dis 2018;115:29–38. [DOI] [PubMed] [Google Scholar]
- 19.Hunanyan A, Helseth A, Abdelnour E, et al. Mechanisms of increased hippocampal excitability in the Mashl(+/-) mouse model of Na(+)/K(+)-ATPase dysfunction. Epilepsia 2018;59:1455–1468. [DOI] [PubMed] [Google Scholar]
- 20.Hunanyan A, Fainberg N, Linabarger M, et al. Knock-in mouse model of alternating hemiplegia of childhood: behavioral and electrophysiologic characterization. Epilepsia 2015;56:82–93. [DOI] [PubMed] [Google Scholar]
- 21.Helseth A, Hunanyan A, Adil S, et al. Novel E815K knock-in mouse model of alternating hemiplegia of childhood. Neurobiol Dis 2018;119:100–112. [DOI] [PubMed] [Google Scholar]
- 22.Panagiotakaki E, De Grandis E, Stagnaro M, et al. Clinical profile of patients with ATP1A3 mutations in alternating hemiplegia of childhood—a study of 155 patients. Orphanet J Rare Dis 2015;10:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Holm R, Einholm AP, Andersen JP, Vilsen B. Rescue of na+ affinity in aspartate 928 mutants of na+,k+-ATPase by secondary mutation of glutamate 314. J Biol Chem 2015;290:9801–9811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Isaksen TJ, Kros L, Vedovato N, et al. Hypothermia-induced dystonia and abnormal cerebellar activity in a mouse model with a single disease-mutation in the sodium-potassium pump. PLoS Genet 2017;13:e1006763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bøttger P, Tracz Z, Heuck A, Nissen P, Romero-Ramos M, Lykke-Hartmann K. Distribution of Na/K-ATPase alpha 3 isoform, a sodium-potassium P-type pump associated with rapid-onset of dystonia parkinsonism (RDP) in the adult mouse brain. J Comp Neurol 2011;519:376–404. [DOI] [PubMed] [Google Scholar]
- 26.BrainSpan: Atlas of the Developing Human Brain. Available at: brainspan.org/lcm/search?exact_match=false&search_term=atp1a3&search_type=gene. Accessed November 2, 2019. [Google Scholar]
- 27.ten Donkelaar HJ, Lammens M, Cruysberg JRM, Cremers CWJR. Development and developmental disorders of the brain stem. In: ten Donkelaar HJ, Lammens M, Hori A, eds. Clinical Neuroembryology. Berlin, Germany: Springer; 2006:269–308. [Google Scholar]
- 28.Masoud M, Gordon K, Hall A, et al. Motor function domains in alternating hemiplegia of childhood. Dev Med Child Neurol 2017;59:822–828. [DOI] [PubMed] [Google Scholar]
- 29.Jasien JM, Bonner M, D'alli R, et al. Cognitive, adaptive, and behavioral profiles and management of alternating hemiplegia of childhood. Dev Med Child Neurol 2019;61:547–554. [DOI] [PubMed] [Google Scholar]
- 30.Uchitel J, Helseth A, Prange L, et al. The epileptology of alternating hemiplegia of childhood. Neurology 2019;93:e1248–e1259. [DOI] [PubMed] [Google Scholar]
- 31.Talsma AD, Chaves JF, LaMonaca A, et al. Genome-wide screen for modifiers of Na (+)/K (+) ATPase alleles identifies critical genetic loci. Mol Brain 2014;7:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeLano WL. The PyMOL Molecular Graphics System. San Carlos, CA: Delano Scientific; 2002. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Eight year old female with dystonia of the left arm.Download Supplementary Video 1 (30MB, mp4) via http://dx.doi.org/10.1212/000466_Video_1
Eight year old female with dystonia of the left arm and hand.Download Supplementary Video 2 (44MB, mp4) via http://dx.doi.org/10.1212/000466_Video_2
Data Availability Statement
Anonymized data will be shared by request from any qualified investigator.