

Collagen hydrolysate can stabilize Zn and Li electrodeposition in metal ion batteries.
Abstract
Lithium (Li) or zinc (Zn) metal anodes have attracted interest for battery research due to their high theoretical capacities and low redox potentials. However, uncontrollable dendrite growth, especially under high current (>4 mA cm−2), precludes reversable cycling in Li or Zn metal batteries with a high-loading (>4 mAh cm−2), precludes reversable cycling in Li or Zn metal batteries with high-loading (>4 mAh cm−2) cathode. We report a cation regulation mechanism to address this failure. Collagen hydrolysate coated on absorbed glass mat (CH@AGM) can simultaneously induce a deionization shock inside the separator and spread cations on the anode to promote uniform electrodeposition. Employing 24 mAh cm−2 cathodes, Li and Zn metal batteries with CH@AGM delivered 600 cycles with a Coulombic efficiency of 99.7%. In comparison, pristine Li and Zn metal batteries only survive for 10 and 100 cycles, respectively. This approach enabled 400 cycles in a 200 Ah-class Zn metal battery, which suggests a scalable method to achieve dendrite-free anodes in various batteries.
INTRODUCTION
The increasing demand for battery systems for portable electronics, electric vehicles, and other electric applications has motivated intensive research seeking high energy density, high unit capacity, and decently stable energy storage devices (1). Because of high theoretical capacity with low oxidation-reduction potential, lithium (Li) and zinc (Zn) metals are considered the most promising anode materials for rechargeable metal-ion batteries (2). However, the main obstacle for the practical application of Li and Zn metal batteries is the dendrite formation (3). Dendrites not only penetrate the separator to induce short circuit in the battery (4) but also generate high surface area on the anode to accelerate metal ion depletion, resulting in active material loss and subsequent battery failure (5). Great efforts have been made to stabilize Li and Zn anodes in lithium ion batteries in the last 10 years. Conductive hosts, such as graphene (6), carbon nanotubes (7), porous copper (8), and graphite fibers (9), have been developed to control the deposition of Li and Zn ions to achieve a uniform and stable metal anode. The large surface area of these materials decreases the local current density, which effectively improves battery safety and extends Sand’s time. Moreover, the ion distribution in the electrolyte can be regulated by surface functional groups and pore structures of these conductive materials. Simultaneously, electrolyte additives (10), porous separators (11), and artificial protective films (12) are used to manipulate ion diffusion to achieve a homogeneous Li and Zn deposition.
Although good cycling life and excellent specific capacity have been achieved in Li and Zn metal batteries (13, 14), these results were still lacking actual practicalities for a reliable battery system. For fundamental study, the batteries are normally tested with an extremely low mass loading of cathode material to minimize side reactions in the cathode and avoid metal ion consumption and electrolyte depletion. However, the low cathode loading (1 mAh cm−2 or less) used in most previous studies leads to very “shallow” stripping/deposition of metal in the same cell, which artificially leads to long cycle life that will not translate to high–energy density cell designs (15). For example, Li||nickel-cobalt-manganese batteries assembled under laboratory-scale, a low cathode mass setup delivers a low specific cathode capacity of 1.5 mAh cm−2, which leads to a unit capacity of only 1.4 mAh (per 0.95 cm2 cathode area). By considering the total weight of the cell unit, the energy density of such a battery would be less than 15 Wh kg−1 (15), which is far smaller than the energy density requirements for commercial batteries. It is well known that the cathode areal capacity should be above 4.0 mAh cm−2 to achieve the desired unit capacity and specific energy density (16). However, under such conditions, the high current density during charge-discharge due to the high mass loading of cathode materials always leads to fast heterogeneous nucleation and deposition of Li or Zn ions and, hence, dendritic metal growth, which results in an extremely short battery life span (17). Fundamental challenges associated with accelerated dendrite formation, fast metal depletion, and rapid electrode passivation under a high-loading intercalation cathode have so far presented formidable barriers to progress in achieving large-scale rechargeable batteries based on the Li and Zn metal anodes (16, 18).
Several models have been proposed to assess the stability of the electrodeposition in batteries using metal anodes, which provide feasible strategies to suppress the dendrites (19–21). Chazalviel (22) suggests that the planarity of the metal surface is destabilized at high current densities by a large electric field in the immediate vicinity of the metal electrode, caused by electrode anion depletion. Therefore, any protrusion of the electrode surface bears enhanced electrodeposition, leading to the growth of dendrite tips. On the basis of this model, Tikekar et al. (23) showed a weak effect of background charge theoretically on the linear stability of electrodeposition in steady state and proposed that a structured electrolyte with immobilized anions can stabilize the electrodeposition at large overpotentials. The group of M. Z. Bazant also investigated the influence of charged porous media in copper electrodeposition from copper sulfate solution. The negative surface charge in anodized aluminum oxide (AAO) membrane enables overlimiting current faster than diffusion. The surface conduction enhances the growth of copper metal along the nanopore surfaces, leading to an array of nanotubes. However, with a positive surface charge in AAO, the opposing surface conduction blocks dendrite penetration upon ion depletion. Dendrites are channeled along the pore centers, avoiding the electric double layers (24). In addition, it is also found that the positive separator (cellulose nitride or polyethylene) shows reduced flux of cation that is opposed by surface conduction, while the negative separator shows overlimiting current retained by surface conduction that leads to a deionization shock ahead of copper growth and substantially suppresses dendrites (25). The same effect of surface transport in charged porous media resulting in “deionization shockwaves” and stable shock electrodeposition have also been exploited for water desalination by “shock electrodialysis” (26, 27).
Inspired from the above fundamental studies, here we propose that collagen hydrolysate (CH) homogeneously coated on absorbed glass mat (CH@AGM) between the anode and the AGM separator can induce a similar shock electrodeposition as reported by Bazant’s group, which fundamentally stabilizes metal anodes and suppresses dendrite formation in lithium and zinc metal batteries with a high areal capacity cathode. Specifically, we found that CH shows a negative surface charge in both aqueous and organic electrolytes, which enables a surface conduction in CH@AGM and a deionization shock in random, charged porous media under overlimiting current, which promotes uniform Li+ and Zn2+ deposition. We also found that the binding of CH with Li+ or Zn2+ cations further spreads the distribution of cations over the whole surface of the metal anode, which navigates metal deposition along the surface/horizontal direction of the anode and significantly stabilizes Li and Zn growth. Such superimposed regulation effects in metal electrodeposition produce a dendrite-free metal deposition under high current density and enable the construction of stably cycled lithium and zinc metal batteries, where a Li or Zn metal anode is coupled with a high-loading cathode [LiMn2O4 (LMO)]. Compared with pristine batteries with a 24 mAh cm−2 cathode that can only survive for 10 and 100 cycles with Li and Zn anodes under 1 C, respectively, the assembled Li and Zn metal full batteries with CH@AGM deliver up to 600 cycles in both aqueous and organic electrolytes without dendrite piercing and a high cycling Coulombic efficiency of up to 99.7%. Such a biomolecule-based approach in dendrite suppression can be simply extended to commercial high-capacity battery systems. Six hundred and 400 cycles were realized in 5- and 200-Ah class aqueous Zn metal batteries at 100% depth of discharge, respectively.
RESULTS
Structure and morphology of CH and CH@AGM
CH originated from animal tissue was first used as glue as far back as 6000 BC. It is now widely applied in the photographic and food industries. CH consists of three α chains that form a triple-helix structure. The α chain is composed of repetitions of glycine-proline-hydroxyproline amino acid sequences, which are stabilized by hydrogen bonds between every three amino acids. These strong interactions are situated regularly on the chain and retain a triple-helical structure (Fig. 1A) (28). Typical CH consists of 19 amino acids, predominantly glycine, proline, and hydroxyproline, which together represent around 50% of the total amino acid content (Fig. 1B). Raman spectroscopy offers efficient characterization to investigate heterogeneous materials as it provides submicrometer spatial resolution with high sensitivity. As shown in Fig. 1C, the amide I band at 1654 cm−1 indicates the α helix structure (29). The amide I absorption is due to the stretching vibration of the C═O bond, which corresponds to the conformation of protein. Other characteristic peaks around 1440 and 1250 cm−1 are the amide III with C─N stretching and N─H bending vibrations of the peptide bond (30).
Fig. 1. Characterizations of CH and CH@AGM.
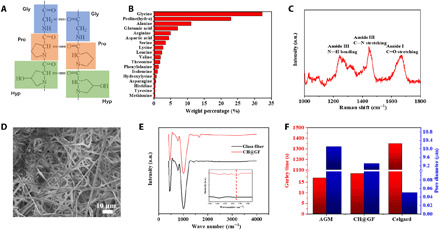
(A) Scheme of the CH structure. Dotted lines indicate the hydrogen bonds between CH chains. (B) Amino acid composition of a pig skin CH (28). (C) Raman spectra of CH. a.u., arbitrary units. (D) SEM images of CH@AGM. (E) FTIR spectra of CH@AGM and the pristine AGM. GF, glass fiber. (F) Air permeability and pore size diameter of the AGM separator, CH@AGM, and the Celgard separator.
The excellent solubility of CH in hot water leads to a facile one-step fabrication of CH@AGM, using porous AGM networks as supporting skeleton (see Materials and Methods). As characterized by scanning electron microscopy (SEM) (Fig. 1D) and corresponding energy-dispersive x-ray mapping (fig. S1), CH results in homogeneous deposition of thin adherent membranes on the immersed AGM. The obtained CH@AGM was further characterized by Fourier transform infrared spectra. While the pristine AGM exhibited only Si─O─Si and Si─O peaks corresponding to typical glass fibers, CH@AGM exhibited a newly appeared peak located at 1650 cm−1, which corresponds to the peptide bond from CH (Fig. 1E). The porosity of CH@AGM is investigated by measuring the air permeance using a Gurley densometer as well as by porosimetry. As shown in Fig. 1F, the air permeance and pore size of CH@AGM are superior to the Celgard separator and similar to those of the AGM separator. The electrolyte uptaking of CH@AGM is also investigated in electrolyte wetting test using a 1 M ZnSO4 and 2 M Li2SO4 aqueous electrolyte. As shown in fig. S2, CH@AGM exhibits superior electrolyte-uptaking performances in both vertical and horizontal electrolyte-wetting setups in comparison to the AGM separator. Given that the CH@AGM shows a similar porosity to the AGM separator (Fig. 1F), this result indicates an intrinsic physicochemical affinity of CH to aqueous solutions containing Li and Zn ions, which may be due to the surface charge of CH@AGM. To evaluate the mechanical property of CH@AGM, we performed puncture tests on CH@AGM with different thicknesses. As shown in fig. S3, the force required to penetrate the sample films depends on the film thickness. Increasing the film thickness resulted in a significant improvement in puncture resistance. By comprehensively considering the separator cost, electrical resistance, and the total weight of the final battery, we intentionally use a 1.3-mm-thick CH@AGM in both laboratory-scale and commercialized batteries in this work, which is strong enough for manufacturing operations.
Suppression of Zn and Li dendrite by shock electrodeposition in charged porous media
Zeta potential experiments were performed to study the surface charge of CH@AGM and the pristine AGM. At a pH range of 2 to 6, which corresponds to the acidic electrolyte used in Zn and Li metal batteries, CH@AGM exhibited a negative surface charge derived from the oxygen functional groups in CH backbones (Fig. 2A) (31), while pristine AGM shows a positive surface charge (Fig. 2B) that may be attributed to the protective sizing used in fiber industries. On the basis of these surface charge results, it can be concluded that coating of CH on the AGM surface is realized via electrostatic self-assembling of the negatively charged CH and the positively charged AGM. Voltammetry measurements were further performed in symmetric Zn and Li cells with and without CH in the AGM separator, to fully understand the influence of background charge in metal electrodeposition. Figure 2C shows current-voltage curves of Zn|AGM + CH@AGM|Zn and Zn|AGM|Zn cells in the 1 M ZnSO4 electrolyte at a scan rate of 1 mV s−1. Although the current in Zn|AGM|Zn reaches −46 mA at −0.2 V and retains a current of −43 mA, Zn|AGM + CH@AGM|Zn exhibits a linear current increase starting from −0.34 V. Similar current-voltage profiles are also observed in the voltammetry of Li|AGM + CH@AGM|Li and Li|AGM|Li cells in 1 M LiPF6 in 1:1:1 dimethyl carbonate: ethyl carbamate: diethyl carbonate (Fig. 2D). These data clearly show that the sign of surface charge in porous media is responsible for either enhancement or suppression of overlimiting current between metal electrodes, which may control the metal electrodeposition behavior under high current (25). As determined from the plateau of voltammetry in Zn|AGM|Zn and Li|AGM|Li symmetric cells, the limiting current density of Zn and Li electrodeposition in this work is 11.3 and 2.06 mA cm−2, which is consistent to the reported value in similar battery systems (32, 33).
Fig. 2. Ion transportation behaviors in CH@AGM and the pristine AGM separator.
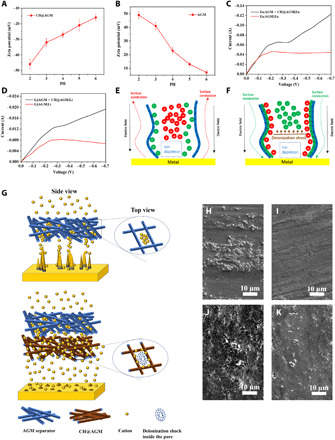
(A and B) Zeta potential of CH@AGM and the pristine AGM in different pH values. (C) Voltammetry of Zn|AGM + CH@AGM|Zn and Zn|AGM|Zn cells in the 1 M ZnSO4 electrolyte at a scan rate of 1 mV s−1. (D) Voltammetry of Li|AGM + CH@AGM|Li and Li|AGM|Li cells in 1 M LiPF6 in 1:1:1 DMC:EC:DEC. (E and F) Mechanistic sketches of the effect of surface conduction on metal electrodeposition in a charged pore. (G) Schematic diagrams of Li or Zn ion movements in CH@AGM (up) and the pristine AGM (down). (H and I) SEM analyses of the Zn anode after a rapid chronoamperometry experiment (10 min) in the pristine LMO|AGM|Zn battery (H) and the LMO|AGM + CH@AGM|Zn battery (I) at −135 mV overpotential. (J and K) SEM analyses of the Li anode after a rapid chronoamperometry experiment (10 min) of the pristine LMO|AGM|Li battery (J) and the LMO|AGM + CH@AGM|Li battery (K) with CH@AGM at −135-mV overpotential.
Figure 2 (E and F) shows mechanistic sketches of the effect of surface conduction on Zn or Li deposition in a charged random porous medium. It is well known that during metal electrodeposition, there is an ion depletion region as cations are continually reduced and deposited on the electrode surface (24, 25, 34). Owing to the reduced bulk conductivity, the axial electric field is amplified to sustain the current (34). With pristine AGM corresponding to a positive surface charge (Fig. 2E), the oppositely directed surface conduction greatly suppresses the overlimiting current and leads to a reduced cation flux (25). Meanwhile, the SO42− or PF6− counterions move away from the anode, further blocking the diffusion of Zn2+ or Li+ ions to maintain neutrality. Above a critical voltage, Zn or Li metal tends to grow avoiding the pore walls and forms large and irregular dendrites owing to the electro-osmotic flow returning to the pore centers (Fig. 2G, top). In comparison, with CH@AGM corresponding to a negative surface charge (Fig. 2F), Zn2+ or Li+ counterions provide surface conduction to maintain electrodeposition, and surface conduction eventually dominates bulk diffusion in carrying the current through the depleted region, leading to a significantly enhanced overlimiting current (25). Meanwhile, SO42− or PF6−counterions are repelled by the large electric field, thus further enhancing bulk depletion and resulting in a “deionization shock” analogous to pressure shocks in gases, leading to a uniform “shock electrodeposition” in the wake and suppressing dendritic growth (Fig. 2G, bottom). The mechanisms of surface-driven overlimiting current in a microchannel, which was shown to influence electrodeposition in this work, were predicted theoretically by Dydek and co-workers (35). Subsequently, Nam and co-workers performed in situ particle tracking and current-voltage measurements in a micro-/nanofluidic device to investigate the transition from surface conduction to electro-osmotic flow (36). All these reports provide strong evidence to support the surface conduction-controlled metal growth proposed in this work.
To investigate the electrochemical evidence for such deionization shock-controlled metal growth, we performed SEM analyses of the Zn and Li anode after a rapid chronoamperometry experiment (10 min) in Zn and Li metal batteries at −135 mV overpotential, which continuously sustain overlimiting current in the cell. To eliminate the effect of mechanical stress from CH@AGM on Zn and Li deposition, a hollow rubber film was stacked in the battery assembly. Owing to the short deposition time, no sharp dendrites can be seen on all the anodes. However, quantities of Zn and Li bushy-like deposits, which are the precursor of large dendrites, are observed on the anodes in pristine LMO|AGM|Zn and LMO|AGM|Li batteries (Fig. 2, H and J). In contrast, the surface of Zn and Li deposited beneath CH@AGM shows a dense layer with uniform metal hills (Fig. 2, I and K). Clearly, the positive surface charge in pristine AGM blocks cation penetration and induces the formation of dendrites avoiding the pore walls, while negative surface charge in CH@AGM leads to a uniform metal electrodeposition stabilized by a deionization shock. Khoo and co-workers have studied the linear stability of transient electrodeposition in a charged random porous medium through sophisticated modeling and concluded that after diffusion limitation is reached, a negative background charge significantly stabilizes the electrode surface instability, while a positive background charge destabilizes the system (37). This theory agrees well with our experimental phenomenon, which is a critical evidence to support the proposed mechanism in this work.
We performed galvanostatic electrochemical impedance spectroscopy (EIS) in symmetric Zn and Li cells at different dc biases to study how charged porous media affect electrodiffusion. As shown in fig. S4 (A and B), when applying a dc bias, the Warburg-like arc for Zn|AGM + CH@AGM|Zn in 1 M ZnSO4 shrinks as the current increases because of the surface conduction-driven overlimiting current as confirmed in Fig. 2C, while the low-frequency response of Zn|AGM|Zn diverges along with the increased current, which is due to the blocking of Zn2+. Consistent trend was also observed in the impedance spectra of Li|AGM + CH@AGM|Li and Li|AGM|Li in 1 M LiPF6 in 1:1:1 DMC:EC:DEC (fig. S4, C and D). These data demonstrate good agreement with galvanostatic impedance spectra of copper deposition using a negative or a positive porous separator as reported by Han et al. (24, 25), indicating the generality of background charge in cation diffusion.
We further used Song’s theory in electrochemical impedance under charged medium to interpret the galvanostatic EIS data in this work, which clearly describe the diffusion and electromigration of ions in a charged background (38). In Song’s model, a one-dimensional (1D) system from x = 0 to x = L is considered, which contains two oppositely charged mobile species and another charged immobile species (38). Two common cell configurations are considered for boundary conditions. In the reservoir configuration, an ideal reservoir maintains dimensionless concentration of charge carriers and electric potential , while in the symmetric configuration, the boundary at x = 0 has the same semi-blocking condition (dimensionless flux of negatively charged carriers = 0) identical to the boundary at x = 1 (38). After solving the boundary value problem and calculating the impedance for any combination of steady-state dimensionless direct current bias and charge density of immobile species , the limiting behaviors of dimensionless impedance as a function of in both reservoir and symmetric configurations are obtained. The calculation shows that both low-frequency resistance [] and high-frequency resistance [] increase exponentially under the diffusion-limiting regime and then converge to over the diffusion-limiting regime, when the background charge is negative (). When , and diverge before the diffusion-limiting bias.
To compare our experimental data to this theory, we fitted the impedance spectra of Zn and Li symmetrical cells through the equivalent circuit models (fig. S5) and obtained the solution resistance (Rs) and mass transfer resistance (Rm, corresponding to the sum of Rs, charge transfer resistance Rct, and bulk diffusion resistance Rbd). Figure S6 shows the Rs and Rm as a function of applied current in Zn and Li symmetrical cells. It is found that Rs in both Zn|AGM + CH@AGM|Zn and Li|AGM + CH@AGM|Li cells increases in the regime below limiting current (11.3 mA cm−2 for Zn and 2.06 mA cm−2 for Li metal batteries) and then converges in overlimiting current (fig. S6, A and B). As Rs corresponds to high-frequency resistance (ω → ∞), these results show close agreement with Song’s calculations in the trend of , demonstrating the transition in the impedance behavior around the diffusion-limiting bias with a charged immobile species. On the other hand, Rm decreases with increasing current. It can be attributed to the shrinking diffusion layer as the depletion zone expands (24). In the case of Zn|AGM |Zn and Li|AGM |Li cells, however, both Rs and Rm diverge as the current is increased (fig. S6, C and D), which is a strong evidence of severe ion depletion.
We also used Khoo’s model to interpret the voltammetry data in Fig. 2 (C and D). Khoo and Bazant (39) proposed a system of a charged nanoporous medium filled with a binary electrolyte flanked on the left by anode and on the right by cathode. Through numerically solving the steady-state equations under different boundary conditions, steady-state current-voltage relations are obtained (39). With negative surface charge, I varies linearly with V for a sufficiently large V and the overlimiting conductance σOLC is the gradient of this linear relationship. At a positive surface charge, I tends to a finite maximum value as →∞. Experimental voltammetry data in symmetrical Zn and Li cells (Fig. 2, C and D) agree well with this model, indicating the sensitivity of surface conduction to the sign of the surface charge. By performing a linear fit of the linear portion of the current-voltage relation, the experimental σOLC of Zn|AGM + CH@AGM|Zn and Li|AGM + CH@AGM|Li cells can be obtained, which shows a value of 0.164 and 0.188 ohm−1, respectively. Moreover, Yan and co-workers also simulated an increasing trend of current in response to a voltage ramp in a liquid electrolyte with one electrode and negative background charge (40), which further supports the proposed mechanism in this work.
On the basis of the surface conduction-driven theory, for a current smaller than the limiting current, dendrites will not occur anyway, and there is little effect of charge surface on metal electrodeposition. To verify this, we performed a 72-hour galvanostatic deposition of Zn and Li in symmetric cells at a current density of −1.5 mA cm−2, which is below the limiting current density from Fig. 2 (C and D). No sharp dendrite can be found from the SEM images of Zn and Li electrode after deposition, regardless of the CH in the separator, demonstrating that stable growth of Zn and Li is possible below the limiting current (fig. S7). However, root-growing mossy structures are still formed during electrodeposition in both Zn and Li electrodes (fig. S7). Similar to dendrites, mossy structures are mechanically instable and may also affect the battery life, which appears below the limiting current in Zn and Li metal batteries (41, 42). It should be noted that the moss-structured area in the Zn and Li electrode with CH in the separator (fig. S7, A and C) is much smaller than the ones in the sample without CH (fig. S7, B and D). Clearly, there should be another mechanism to stabilize the anode in Zn and Li metal batteries using CH@AGM, which will be discussed in the next section.
CH-guided Zn and Li electrodeposition on the anode surface
It is reported that during electrochemical tests, the extra heat generated from the side reactions accumulates in the electrode and results in large temperature rise within the cell (43). It is also well known that CH can be slowly dissolved in water and organic solvents when the temperature is above 40°C (44). As a result, there should be some CH fine particles stripped from CH@AGM, dispersed in both aqueous and organic electrolytes in CH@AGM-based batteries, and then absorbed on the anode surface during the long-term chronoamperometry test. This assumption was confirmed by the chemical compositions of the Zn and Li metal anodes with CH@AGM in the separator through x-ray photoelectric spectroscopy (XPS), where the N component from CH was detected after 10- and 30-min chronoamperometry at −135 mV overpotential in the Zn and Li anode, respectively (fig. S8 and tables S1 and S2). However, such CH dispersing in the electrolyte is only partial from this electrochemical condition. As evidenced in fig. S9, even after 1-hour chronoamperometry, a large amount of CH coatings can still be observed on the AGM separator.
The interaction between Zn2+ and Li+ cations with CH was first studied using isothermal titration calorimetry (ITC), which is the most direct and quantitative approach for characterizing the thermodynamic properties of protein-ligand interactions. In CH, glycine (Gly), proline (Pro), and alanine (Ala) hold more than 50% of the total amino acids (Fig. 1B). As a result, an aqueous solution of Gly-Pro-Ala peptide (10−4 M) is used as titrate to represent CH in ITC, while 10−3 M aqueous ZnSO4 or Li2SO4 acts as titrant. Strong interaction between Gly-Pro-Ala and Zn2+/Li+ is confirmed through the exothermicity of the calorimetry peaks in Fig. 3 (A and B), which indicates the released heat in one injection of titrant into the amino acids. As the sites available on Gly-Pro-Ala to interact Zn2+/Li+ are occupied during titration, the exothermicity of the peaks decreases and finally saturates. For comparison, aqueous solution of 10−4 M colloidal SiO2 is used as titrate to investigate the Zn2+/Li+ cations binding to AGM, as SiO2 is the main component of glass fiber in AGM. Figure 3 (C and D) shows the calorimetric response of colloidal SiO2 with the reaction of ZnSO4 or Li2SO4 at the pH 7.4. The data show a significant scatter with no obvious trend. A comparison of the heats evolved in the injection of Zn2+/Li+ to Gly-Pro-Ala (Fig. 3, A and B) with that in the reaction of Zn2+/Li+ with SiO2 (Fig. 3, C and D) shows that the interaction of Zn2+/Li+ with SiO2 is quite weak.
Fig. 3. The binding of Zn2+ and Li+ cations with CH.
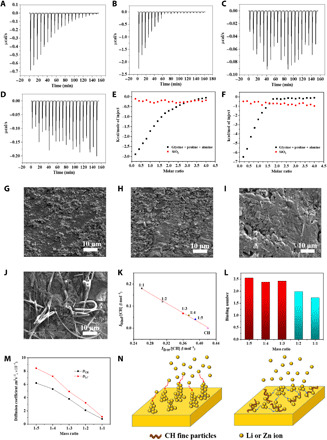
(A and B) ITC during injection of 10−3 M ZnSO4 and Li2SO4 in calorimetric cell containing 10−4 M Gly-Pro-Ala solution at pH 7.4, respectively. (C and D) ITC during injection of 10−3 M ZnSO4 and Li2SO4 in calorimetric cell containing 10−4 M colloidal SiO2 at pH 7.4, respectively. (E and F) The integrated heat response from ITC against molar ratio of titrant solution. (E) Titration of 10−3 M ZnSO4 into 10−4 M Gly-Pro-Ala solution. (F) Titration of 10−3 M Li2SO4 into 10−4 M colloidal SiO2. (G and H) SEM images of the Zn anode in Zn|AGM|Zn cell after 10 min of chronoamperometry with and without CH in the aqueous electrolyte. (I and J) SEM images of the Li anode in the Li|AGM|Li cell after 30 min of chronoamperometry with and without CH in the organic electrolyte. (K) Ibind/[CH] versus Ifree/[CH] in ZnSO4-CH electrolyte with various ZnSO4-CH mass ratios. (L) Binding numbers in different ZnSO4-CH mass ratio electrolytes derived from Raman spectra in fig. S10. Red columns indicate the binding number of the stable Zn2+-CH complex. (M) Diffusion coefficients of CH (DCH) and the Li+ ions (DLi+) with different ZnSO4-CH mass ratios. (N) Schematic diagrams of Li or Zn nucleation on the anode with CH dispersed (left) and the pristine electrolyte (right). The blue and red arrows demonstrate the transverse interaction imposed from CH and the vertical electrostatic force, respectively.
Figure 3 (E and F) shows plots of the integrated heat response from calorimetry data in Fig. 3 (A to D) against the molar ratio of Zn2+/Li+ cations to amino acids in the reaction vessel. The heat data, combined with the known quantity of titrant and titrate, are fit using a nonlinear least squares method to a binding model, which can yield the best fit values of the stoichiometry (n), binding constant (K), and heat of binding (enthalpy, ΔH). As shown in table S3, the binding constant from the injection of Zn2+ and Li+ cations to Gly-Pro-Ala solution is 1.13 × 106 M−1 and 4.38 × 106 M−1, respectively, which is two orders of magnitude higher than the values of SiO2 with Li+ and Zn2+. These characterizations thus exhibit an obvious interaction between amino acids and Zn2+ or Li+ cations, which is consistent with previous theoretical studies (45).
Inspired by the strong interactions between CH and Zn2+/Li+ cations as quantified from ITC characterization, we speculate that the CH fine particles absorbed on the anode surface can also bind to Zn2+/Li+ cations, which may affect the dendrite growth on the anode surface as well. To verify this hypothesis, we intentionally designed a battery for chronoamperometry experiment, in which CH fine particles was dispersed in the aqueous and organic electrolyte [2% (w/w)] and absorbed on the anode surface, using the pristine AGM as separator. Figure 3 (G and H) shows the SEM images of the Zn anode after 10-min chronoamperometry, which enables continuous overlimiting current to induce dendrite, with and without CH in the aqueous electrolyte, respectively. Without the dispersing effect from CH@AGM on cation transport through the separator, adding only CH into the electrolyte cannot guarantee a uniform electrodeposition in the cell. Dendritic growth is evident on both Zn anodes. However, there are still big differences in their dendrite morphology. In the aqueous electrolyte with 2% CH, the anode surface was only partially covered with Zn coating, with a grain diameter smaller than 1 μm. In comparison, continuous granular microstructure with grains up to 10 μm in diameter was formed on the anode surface in the pristine aqueous electrolyte. An analogous phenomenon is also observed after 30-min chronoamperometry for the Li anode (Fig. 3, I and J). The surface of Li deposits in the electrolyte with 2% CH exhibits a densely packed Li metal flake with smooth morphology, while abundance of uneven and dead Li deposits embedded with AGM residues is detected on the Li anode in the pristine electrolyte. Obviously, CH fine particles absorbed on the anode surface can significantly refine grain size, obtain uniform structure, and affect the texture and dendrite growth direction.
The binding structures of Zn2+-CH fine particles in the aqueous electrolyte at various mass ratios were investigated using Raman spectroscopy in 1550 to 1780 cm−1 (Supplementary note 2). The pure CH in water shows a band around 1590 to 1670 cm−1 that is associated with amide I C═O stretching (46). After adding ZnSO4, a new band at 1630 to 1740 cm−1 is probably due to the binding of CH with the Zn2+ ions. By deconvoluting into two Gaussian-Lorentzian bands, the Raman bands at 1650 cm−1 (Ifree) and 1680 cm−1 (Ibind) can be attributed to the concentration of free CH molecules ([CH]free) and CH molecules bonded to Zn2+ ([CH]bind; fig. S10). The plot of Ibind/[CH] versus Ifree/[CH] in the ZnSO4-CH electrolyte with various ZnSO4-CH mass ratios shows a linear feature, and the molar scattering coefficient ratio of bonded and free CH (Jbind/Jfree) can be obtained from the slope of the plot (Fig. 3K). By knowing Ifree/Ibind and Jbind/Jfree, the binding number (nbind) of CH with Zn2+ can be calculated from eqs. S2 to S7 in Supplementary note 1 (47). For mass ratio between 1:5 and 1:3, the CH molecules form a stable Zn2+-CH complex with a binding number of 2.5 (Fig. 3L). As mass ratio increases, the concentration of CH decreases, and its binding number with Zn2+ decreases, probably due to the depletion of the CH.
We further used 15N and 7Li nuclear magnetic resonance (NMR) with pulsed-field gradient (PFG) to study the association of CH with Li+ ions (Supplementary note 2) (48). For an ideal NMR sample, all the compounds should be fully dissolved in the solvent to obtain a reliable result. Unfortunately, the solubility of CH in 1:1:1 DMC:EC:DEC mixed solvent is very low. Therefore, it is not possible to prepare a clear NMR sample in the mixture of Li salt and CH under an organic electrolyte. To overcome this challenge, here we studied the binding behavior of Li2SO4 with collagen in aqueous solution to simulate the association of Li+ with CH in an organic electrolyte, by reasonably assuming that solvent will not affect the interaction between Li+ and CH. Figure S11 shows that along with Li2SO4-CH mass ratio increases, the 15N NMR peak from CH, δ(15N), shifts upfield due to the screening of Li+ ions on the lone electron pair density of the oxygen in C═O groups from amino acids in CH (49). Owing to the Li+ ion complexation by CH, the upfield shift of the 7Li NMR peak, δ(7Li), is proportional to mass ratio. Diffusion coefficients of CH (DCH) and Li+ ions (DLi+) were obtained using 15N and 7Li NMR, respectively. As shown in Fig. 3M, diffusion coefficients decrease along with the mass ratio increase. In all mass ratios, DCH is higher than DLi+, and both DCH and DLi+ decrease with the mass ratio increases. The values of DLi+ and DCH become the same after the mass ratio reaches 1, indicating the firm binding of Li+ with CH.
The above molecular-level analysis suggests a clear binding mechanism between CH fine particles and Zn2+ or Li+ cations in aqueous solution and indicates a possible guiding effect of CH in cation movement and nucleation on the anode surface. As shown in Fig. 3N (right), Zn or Li will be preferentially deposited around the tips of protuberances on the CH-free anode rather than on smooth regions to form large dendrites, due to the strong electrical field on the sharp edge. In contrast, the strong binding of CH fine particles absorbed on the anode to Zn2+ or Li+ cations imposes a competing effect to the deposition of Zn2+ or Li+ cations on the protuberances, which spreads Zn or Li to adjacent regions of the initial growth tips of protuberances and prevents the formation of large dendrites (Fig. 3N, left). The computer simulation (density functional theory) also indicates that Zn tends to grow into 2D clusters around -NH─C═O in CH due to the more favorable energy, which facilitates Zn deposition along the surface/horizontal direction of the anode and leads to flake-shaped small dendrites (fig. S12). However, on the pristine anode surface, Zn favors the vertical direction of growth to minimize the surface area exposed to the electrolyte (fig. S12), which results in the large dendrites on the flat anode (Fig. 3N, right). Of course, the actual electrochemical cation deposition is much more complicated than the nucleation phenomena discussed here and deserves in-depth study in the future.
Long-term electrochemical cycles
The long-term interfacial stability of the Zn anode was first investigated through symmetric Zn|AGM + CH@AGM|Zn and Zn|AGM|Zn cells using an aqueous 1 M ZnSO4 and 2 M Li2SO4 electrolyte. During cycling, Zn ions are transported back and forth between the two Zn metal electrodes at 1.0 mA cm−2, and the corresponding voltage versus time plot is shown in Fig. 4A. Amplified voltage profiles in Fig. 4A for 0 to 4 hours (16,000 s) and 36 hours (130,000 s) to 43 hours (155,000 s) of cycling are provided in Fig. 4 (C and D). Lower mass transfer–controlled potential (μmtc) values and better potential retentions are realized for the Zn|AGM + CH@AGM|Zn cell than for the Zn|AGM|Zn cell, indicating a homogeneous Zn metal electrode with a stable electrolyte/metal interface. The electrochemical impedances of Zn symmetrical cells were measured at the end of 0-, 20-, and 40-hour deposition/dissolution cycles in the 1 M ZnSO4 and 2 M Li2SO4 electrolyte at 1.0 mA cm−2. As shown in Fig. 4G, the initial charge transfer resistance (Rct) of Zn|AGM + CH@AGM|Zn is equal to that of Zn|AGM|Zn (29.3 versus 27.8 ohms, after fitting the Nyquist plots through equivalent circuit in fig. S13), indicating the well-retained pore structure after CH coating. The Rct remains stable at around 23.6 ohms in 40-hour cycling, indicating the stabilized Zn growth and little consumption in the electrolyte. However, with pristine AGM separators, the Rct exhibits large fluctuations during cycling. The value significantly drops to 12.9 ohms after 20-hour cycling and further reduces to 6.7 ohms in 40 hours (Fig. 4H). Such large fluctuations in Rct suggest unstable electrochemical environments during Zn depositing and stripping, which induces breakage of native oxide on the surface and exposes the fresh Zn to the electrolyte.
Fig. 4. Long-term interfacial stability of the Zn and Li anode.
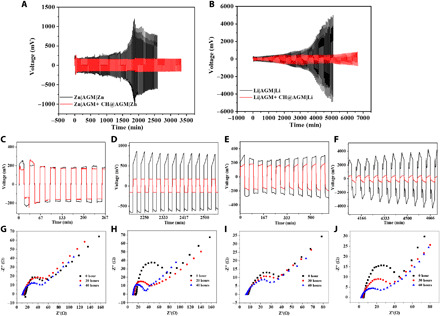
(A) Galvanostatic Zn plating/stripping voltage profiles for symmetric Zn|AGM + CH@AGM|Zn and Zn|AGM|Zn cells at a capacity of 1 mAh cm−2 and a current density of 1 mA cm−2. (B) Galvanostatic Li plating/stripping voltage profiles for symmetric Li|AGM + CH@AGM|Li and Li|AGM|Li cells at a capacity of 1 mAh cm−2 and a current density of 1 mA cm−2. (C and D) Amplified voltage profiles from 0 to 16,000 s (C) and 130,000 to 155,000 s (D) of cycling from (A), respectively. (E and F) Magnified voltage profiles from 0 to 35,000 s (E) and 255,000 to 280,000 s (F) of cycling from (B), respectively. (G and H) Nyquist plots of Zn|AGM + CH@AGM|Zn and Zn|AGM|Zn cells at the end of 0-, 20-, and 40-hour deposition/dissolution cycles in the 1 M ZnSO4 and 2 M Li2SO4 electrolyte. (I and J) Nyquist plots of Li|AGM + CH@AGM|Li and Li|AGM|Li cells at the end of 0-, 30-, and 60-hour deposition/dissolution cycles in the organic electrolyte (1 M LiPF6 in 1:1:1 DMC:EC:DEC mixed solvent).
Furthermore, the Li migrations for the symmetrical Li|AGM + CH@AGM|Li and Li|AGM|Li cells using the organic electrolyte (1 M LiPF6 in 1:1:1 DMC:EC:DEC mixed solvent) are measured at 1 mA cm−2 (Fig. 4B). Magnified voltage profiles in Fig. 4B for 0 to 9 hours (35,000 s) and 70 hours (255,000 s) to 78 hours (280,000 s) of cycling are shown in Fig. 4 (E and F). Compared with the cell using routine AGM separators, the Li|AGM + CH@AGM|Li cell renders a tighter voltage hysteresis (200 to 500 mV versus >5000 mV for the pristine AGM) and much more extended cycle life (111 hours versus <85 hours for AGM), indicating the depletion of Li ions in the electrolyte resulting from the dead Li. Figure 4 (I and J) shows typical impedance spectra measured at the end of 0-, 30-, and 60-hour cycle of Li deposition/dissolution in Li symmetrical cells. Consistent with the Zn symmetrical cell, the Li|AGM + CH@AGM|Li cell shows similar initial Rct to the Li|AGM|Li cell and exhibits less reduction of Rct (from 14.6 to 12.8 ohms; see fig. S13 for equivalent circuit) during cycling compared to the value (from 18.2 to 7.3 ohms) in the Li|AGM|Li cell, implying the stable Li growth. Clearly, the CH in Li and Zn significantly stabilizes Zn and Li growth, which affords new insights into the realization of stable cycling in Zn and Li metal batteries using both aqueous and organic electrolytes.
We also assembled coin-type Li|AGM + CH@AGM|Cu and Li|AGM|Cu half-cells using 1 M LiPF6 in 1:1:1 DMC:EC:DEC as electrolyte, to further evaluate the long-term stability of Li plating/stripping behavior on a metal substrate. High Li reversibility and CE (~99%) are achieved in the Li|AGM + CH@AGM|Cu cell at a current density of 1 mA cm−2, while the pristine Li|AGM|Cu cell fails after about the 80th cycle (fig. S14). The polarization profiles in different cycles were also recorded to gain the kinetic behavior during Li stripping/plating (fig. S15). As shown in fig. S15A, Li|AGM + CH@AGM|Cu exhibited a similar Li migration potential at the 10th and 80th cycle from the charge/discharge curves in 1 mA cm−2 (126 mV versus 139 mV), indicating a stable Li electrodeposition. In comparison, large polarization was observed in the Li|AGM|Cu cell, which resulted in a much larger Li migration potential at the 80th cycle (182 mV; fig. S15B).
Electrochemical performance of full cells
It is well known that for currents beyond diffusion limitation, the concentration of salt at the electrode surface decreases to zero at a certain time. This time is known as “Sand’s time,” after which the cations preferentially deposit on the protrusions of the surface, resulting in a self-amplifying process of dendritic growth (42). By multiplying with the current density, we converted Sand’s time to Sand’s capacity and plot the current-dependent Sand’s capacity in Zn and Li metal anodes (fig. S16), which clearly shows the transition from mossy to dendritic metal. Because of the much higher limiting current in Zn deposition (11.3 mA cm−2) than that in Li deposition (2.06 mA cm−2), the Zn metal electrode exhibits a harsher experimental condition to form dendrites than Li metal. Moreover, the results in the Li metal electrode (fig. S16B) show close agreement with the previous literature revealing the conditions for dendritic versus mossy growth of lithium (42). To demonstrate the feasibility of CH@AGM in Zn metal batteries, we first investigate the cycling performances of LMO|AGM + CH@AGM|Zn and LMO|AGM|Zn laboratory-scale cells, in which a low-capacity LMO cathode (3.6 mAh cm−2), a thick Zn metal, and a much flooded electrolyte [electrolyte weight versus cathode capacity (E/C) of 30 g(Ah)−1] were used (table S4 and fig. S17). Owing to the low cathode capacity, these cells fall into the mossy region of fig. S16. As shown in Fig. 5A and fig. S18, the LMO|AGM + CH@AGM|Zn cell exhibited a good rate capability and stable cycling over 1200 cycles at 1 C with an initial capacity retention above 90% and efficiency above 98%. When using the regular AGM as separator, the capacity retention of the cell starts to decline after 400 cycles with an obvious efficiency decay. The cycling performance in Fig. 5A is further analyzed by observing the charge-discharge curves of LMO|AGM + CH@AGM|Zn and LMO|AGM|Zn at a specific cycle. Compared to the voltage profile of LMO|AGM|Zn that shows a rapid potential spike at first charge and gradually leftward and up movement of the charge-discharge intersection (Fig. 5B), the steady potential spike at the beginning of the first charge and leftward horizontal movement of the traced intersection points are observed in the LMO|AGM + CH@AGM|Zn cell (Fig. 5C). It implies that the Zn anode is stably operated during cycling and the decrease in capacity of this cell is mainly due to the unavoidable degradation of the cathode.
Fig. 5. Electrochemical performance of laboratory-scale full cells.
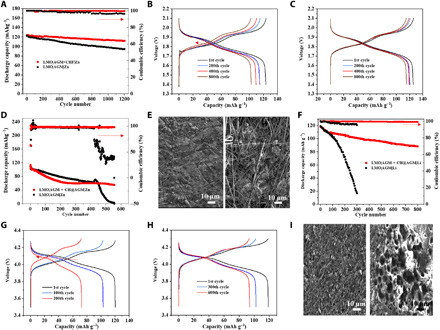
(A) Cycling performance of the discharge capacity and coulombic efficiency of LMO|AGM + CH@AGM|Zn and LMO|AGM|Zn cells with the 3.6 mAh cm−2 cathode at a rate of 1 C. (B and C) Voltage profiles of LMO|AGM|Zn (B) and LMO|AGM + CH@AGM|Zn (C) cells in different cycles. The dashed red arrow indicates the trace of the intersection between charge/discharge curves. (D) Cycling performance coulombic efficiency of LMO|AGM + CH@AGM|Zn and LMO|AGM|Zn cells with the 3.6 mAh cm−2 cathode at a rate of 1 C under 60°C. (E) SEM image of LMO|AGM + CH@AGM|Zn (left) and LMO|AGM|Zn (right) cells after 400 cycles at a rate of 1 C under 60°C. (F) Cycling performance of the discharge capacity and coulombic efficiency of LMO|AGM + CH@AGM|Li and LMO|AGM|Li cells with the 1.8 mAh cm−2 cathode at a rate of 1 C. (G and H) Voltage profiles of LMO|AGM|Li (G) and the LMO|AGM + CH@AGM|Li (H) cells in different cycles. The dashed red arrow indicates the same trace as (B). (I) SEM image of LMO|AGM + CH@AGM|Li (left) and LMO|AGM|Li (right) cells after 200 cycles at a rate of 1 C.
In addition, we further evaluated the 60°C cycling performance of an aqueous Zn metal battery under the same C rate as in room temperature. As shown in Fig. 5D, the LMO|AGM + CH@AGM|Zn cell exhibits more than 600 stable cycles with above 99% Coulombic efficiency in such harsh conditions. In contrast, the pristine LMO|AGM|Zn cell shows a sharp capacity and efficiency decay after 400 cycles. To reveal the morphology changes of Zn metal anodes during charge-discharge interaction, ex situ SEM of Zn anodes was conducted after 400 cycles under 60°C. A uniform Zn metal layer is retained during cycling in the cells with the CH@AGM separator [Fig. 5E (left) and fig. S19]. In contrast, irregular Zn deposits are observed beneath the AGM residues in the cell using the pristine AGM separator [Fig. 5E (right) and fig. S20]. The much better cycling performance in Zn|AGM + CH@AGM|Zn than in Zn|AGM|Zn under 60°C mainly comes from the binding of dissolved CH to the Zn2+ on the anode surface, which spreads the distribution of cations over the whole surface of the metal anode to stabilize Zn growth (Fig. 3N, right). Previous reports revealed that larger total surface areas resulted in more metal ion consumptions in electrolytes. Therefore, stabilizing Zn growth through CH@AGM contributes to the prolonged life span, which is consistent with the long-term plating/stripping performances obtained from symmetrical cells.
Moreover, we have also investigated the cycling properties of LMO|AGM + CH@AGM|Li and LMO|AGM|Li cells, using a low-capacity LMO cathode (1.8 mAh cm−2) and a flooded electrolyte (table S5) that also fall into the mossy regime similar to Zn metal batteries (fig. S16). As expected, such a LMO|AGM + CH@AGM|Li full cell shows stable cycling over 800 cycles (Fig. 5F) with more stable voltage profiles (Fig. 5H). When using the regular AGM separator, the pristine LMO|AGM|Li cell (Fig. 5, F and G) shows a quick capacity drop after 300 cycles. The ex situ SEM images of the Li anode in LMO|AGM + CH@AGM|Li and LMO|AGM|Li cells after 600 and 200 cycles are shown in Fig. 5I. Rather than forming disordered Li mosses in the LMO|AGM|Li cell, the Li surface in the LMO|AGM + CH@AGM|Li cell exhibits a uniform and densely packed Li deposition layer. This phenomenon suggests a unique effect of CH in mosses’ formation on the Li and Zn anode, which is derived from the interaction between dissolved CH and Li and Zn ions in the electrolyte as discussed in the previous section. The mass of dissolved CH in aqueous and organic electrolytes during cycling can be obtained by calculating the mass of CH@AGM before and after battery cycling. As plotted in fig. S21, the highest content of CH in aqueous (13%) and organic (9%) electrolytes was reached after 200 and 400 cycles, respectively, demonstrating the maximum solubility of CH.
Although stable cyclability of the LMO cathode is verified in both Zn metal and Li metal full batteries by adding CH@AGM between anode and separator, the result is still lacking actual practicalities for a real battery as the cathode areal capacity in the above cells are extremely low (3.6 mAh cm−2 for the Zn metal battery and 1.8 mAh cm−2 for the Li metal battery), which greatly reduces the total capacity in a full battery. However, it is highly challenging to construct cycling stable Zn and Li metal batteries using high–mass loading cathode materials. The higher area capacity actually involves a higher current density, and a large fraction of Zn and Li metal was reacted in each charge-discharge cycle, which generally produces a fast Zn and Li consumption that causes an accelerated depletion of metal ions in the electrolyte and premature cell failure. For instance, under conventional condition with low-capacity cathode (3.6 mAh cm−2; table S4), after 1200 cycles, the LMO|AGM|Zn cell still shows a high capacity of 95 mAh g−1. However, when tested under constrained conditions with a cathode capacity of 24 mAh cm−2, this cell only retains 65% of its original capacity after 400 cycles (Fig. 6A). At the 450th cycle, the battery encountered an immediate short circuit, which is due to the piercing of obvious large dendrites formed on the anode surface as this battery falls into the dendritic Zn regime in fig. S16. This performance discrepancy from cell configurations is even larger in the LMO|AGM|Li cell, which can only survive 10 cycles with high-capacity cathode (24 mAh cm−2) as evidenced in Fig. 6B, in comparison to the results of the LMO|AGM|Li cell in conventional testing conditions with a cathode capacity of 1.8 mAh cm−2 (Fig. 5F). It should be noted that within this high-loading cathode, both ion transportation and mechanical property will be poor, and the changes in cathode structure also lead to the fading in Zn and Li metal batteries. It is reported that the diffusion of Li+ inside the electrolyte is faster than that inside the cathode (50). Abundant Li+ tends to accumulate on the surface of Li transition metal oxides and induce a partial transformation in the crystalline structure (51). This structural distortion, the so-called Jahn-Teller distortion, severely blocks the Li+ diffusion pathways and reduces the capacity retention (52). Moreover, the electrochemical oxidation of the conductive carbon in the cathode is responsible for the capacity loss of Li-ion batteries. It is induced by contacting water at low potentials and/or by oxygen evolved from the unstable delithiated materials (53, 54). As a result, we attribute the capacity fading of Zn and Li metal batteries in Fig. 6 (A and B) to the combination of structure instabilities in cathode materials, and the metal depletion in the anode, both of which can be greatly amplified under a high-loading cathode.
Fig. 6. Electrochemical performance of large-scale full cells with high–mass loading cathode materials.
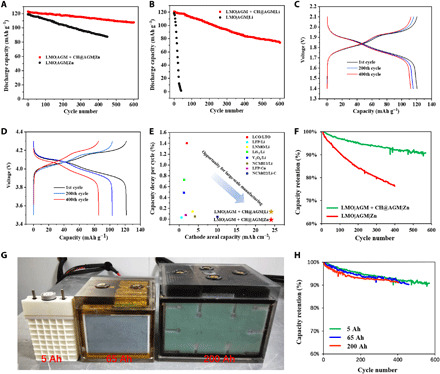
(A) Cycling performance of the discharge capacity of LMO|AGM + CH@AGM|Zn and LMO|AGM|Zn cells with a cathode capacity of 24 mAh cm−2 at a rate of 1 C. (B) Cycling performance of the discharge capacity of LMO|AGM + CH@AGM|Li and LMO|AGM|Li cells with a cathode capacity of 24 mAh cm−2 at a rate of 1 C. (C and D) Corresponding voltage profiles of LMO|AGM + CH@AGM|Zn (C) and LMO|AGM|Li (D) cells at various cycles. (E) Cathode areal capacities and reported capacity decay per cycle of other state-of-the-art Li-ion electrochemistry systems: LiCoO2/Li4Ti5O12 (LCO/LTO) (55), LiFePO4/Li (LFP/Li) (56), LiNi0.5Mn1.5O4/Li (LNMO/Li) (57), TiS2/Li (58), V2O5/Li (59), LiNi0.8Mn0.1Co0.1O2/Li (NCM811/Li) (16), LiFePO4/Cu (LFP/Cu) (60), and LiNi0.6Mn0.2Co0.2O2/Li-C(NCM622/Li-C) (15). (F) Cycling performance of the discharge capacity of 5-Ah LMO|AGM + CH@AGM|Zn and LMO|AGM|Zn cells under constrained cell parameters at a rate of 1 C. (G) Photos of 5-, 65-, and 200-Ah cells. (H) Cycling performance of the discharge capacity of 5-, 65-, and 200-Ah LMO|AGM + CH@AGM|Zn cells at a rate of 1 C. Photo credit: Yunfeng Luo, Rehab Energy Co. Ltd.
Unexpectedly, when CH@AGM was added in the Zn metal battery (LMO|AGM + CH@AGM|Zn) with a 24 mAh cm−2 cathode, a stable cycling over 600 cycles was realized with a capacity retention of 92% at a rate of 1 C (Fig. 6A). In addition, when CH@AGM was used, the Li metal battery using the same 24 mAh cm−2 cathode can maintain an excellent cycling stability of 93% capacity retention until the 100th cycle and even 68% capacity retention at the 600th cycle (Fig. 6B). Moreover, the charge/discharge curves of both batteries were well retained during cycling without an obvious overpotential increase (Fig. 6, C and D), due to the relatively stable interface between the metal anode and the electrolyte. The superimposed effects of CH-enabled cation regulation in the separator and on the anode surface are crucial to achieve such a highly stable cycling in Zn or Li metal batteries under realistic conditions. Figure S22 shows the cycling performance of the LMO|AGM|Zn cell, using CH as electrolyte additives and pristine AGM as separator under the same battery conditions in Fig. 6A. This cell can be charged-discharged over 500 cycles without short circuit. However, compared to the cell with CH@AGM that exhibits more than 100 mAh g−1 capacity at the 600th cycle (Fig. 6A), this cell only shows a low capacity of 81 mAh g−1 after 500 cycles and a rapid capacity decrease after the 500th cycle. As evidenced from the SEM image of the Zn anode at the 500th cycle, although no bushy dendrites are found, large amounts of accumulated Zn grains inside the pores of the AGM separator are still formed on the surface of the anode, which can seriously affect the cycle life of a rechargeable battery (fig. S23). Obviously, the key function of absorbed CH on the anode is to suppress the formation of large dendrites by spreading the cations to a homogeneous distribution, and CH coating on AGM is still necessary to realize a complete dendrite-free anode.
A comprehensive comparative summary of cathode areal capacities and reported cycle decay per cycle of representative materials in Li-ion batteries is presented in Fig. 6E. It can be seen that both LMO|AGM + CH@AGM|Zn and LMO|AGM + CH@AGM|Li batteries are standouts for either cathode loading or cycling life. Clearly, CH@AGM enables the high stability and rechargeability of Zn and Li metal batteries with realistic, high-loading cathode, which suggests a significant promise for the industrial fabrication in high-capacity Li-ion batteries. For the purpose of commercialization, we have also fabricated a 5-Ah commercial high-capacity battery based on a single LMO|AGM + CH@AGM|Zn cell (1.056 Ah, with a 80-cm2 cathode sheet; see fig. S24) with constrained conditions (cathode capacity of 13.2 mAh cm−2; table S4). Note that to seek the ease of quality assurance and industrial manufacturing, we intentionally choose a less constrained cell parameter (relatively lower cathode areal capacity compared to the battery in Fig. 6A) in a single cell. However, this battery still falls into the dendritic regime of Zn metal (fig. S16) due to the much higher cathode loading than the laboratory-scale battery. As shown in Fig. 6F, a 5-Ah LMO|AGM + CH@AGM|Zn full cell can realize over 600 stable cycles under 100% depth of discharge. We have also fabricated 65-Ah and 200-Ah high-capacity Zn metal batteries (Fig. 6G), both of which show an excellent cyclability over 400 cycles with >90% capacity retention (Fig. 6H). We have also tried to further decrease the amount of electrolyte to increase the energy density of the commercial battery. When an extremely constrained E/C ratio (3 g Ah−1) was used, the single battery unit from the 200-Ah cell, even with CH@AGM in the separator, can only survive 80 cycles (fig. S25). Obviously, the electrolyte consumption becomes a limiting factor when an extremely low amount of electrolyte was used. More optimization in electrolyte formulation is needed in this system.
DISCUSSION
In this contribution, we report a facile and general approach to resolving the stability issue in Zn and Li meal batteries with a practical, high-loading intercalation cathode, where a cation-regulating layer CH@AGM between anode and separator can effectively suppress metal dendrite formation. The surface charge of CH and the binding between CH and cations (Li+ or Zn2+) can simultaneously induce a deionized shock inside the separator and realize a homogeneous distribution of cations on the anode surface, which leads to efficient dendrite suppression and control in both Zn and Li metal batteries. Benefiting from superimposed effects of CH in cation regulation in electrolyte and on the anode surface, Zn and Li metal full cells coupled with CH@AGM and a 3.6 mAh cm−2 cathode delivered up to 1200 dendrite-free cycles at 1 C and exhibit over 600 stable cycles with above 99% Coulombic efficiency under 60°C. At a cathode loading as high as 24 mAh cm−2, Li and Zn metal batteries using CH@AGM can still deliver 600 dendrite-free cycles at 1 C in both organic and aqueous electrolytes, with a Coulombic efficiency of up to 99.7%. In contrast, the same batteries with the pristine AGM separator could only survive for 10 and 100 cycles, respectively. In addition, such CH-based dendrite control technology can be readily applied in large-scale, Ah class battery systems. By adding CH@AGM, 600 and 400 cycles were realized in 5-Ah class and 200-Ah class batteries at 100% depth of discharge, respectively, which show more than 30% improvement compared to the pristine Zn metal batteries. The proposed cation-biomolecule interaction for uniform metal deposition and nucleation suggests a scalable process to realize dendrite-free charge/discharge in various metal anodes, which can be facilely translated to commercial-level battery designs.
MATERIALS AND METHODS
Preparation of CH@AGM
The CH solutions are prepared by 5% [weight % (wt %)] CH (Sigma-Aldrich) mixed with deionized water with constant stirring for 2 hours at 80°C. The fabrication of CH@AGM used in the Zn metal battery is slightly different to the fabrication of CH@AGM used in Li metal batteries. For the Zn metal battery, AGM was fully immersed into CH solution for a few seconds and followed by air drying at 110°C for 12 hours to obtain CH@AGM. For the Li metal battery, 1 wt % LiOH (Sigma-Aldrich) was added into 5% aqueous CH solution. Then, AGM was immersed into the solution for a few seconds and followed by air drying at 110°C for 12 hours to obtain CH@AGM. The mass loading of absorbed CH in AGM is about 38 mg cm−2 for the Zn metal battery and 43 mg cm−2 for the Li metal battery.
Battery assembly
For both Li and Zn metal batteries, the cathodes are the same, which were prepared by casting slurries of LMO (Wuxi Jewel Power & Materials Co., Ltd.), conductive carbon (KS-6, MTI, Co.) with styrene-butadiene rubber/carboxymethylcellulose as binding material (83:10:7 wt.%) in N-methyl-2-pyrrolidinone (Sigma-Aldrich Co.) on a conductive polyethylene film. The mass loadings on the cathode of Zn and Li metal batteries under unconstrained testing conditions are 30 and 15 mg cm−2, corresponding to a 3.6 and 1.8 mAh cm−2 cathode, respectively. For batteries under constrained conditions, the mass loading on the cathode of Zn and Li metal batteries is 200 mg cm−2.
To assemble a LMO|AGM + CH@AGM|Zn battery under conventional, unconstrained conditions, a 3.6 mAh cm−2 LMO cathode (4 cm2, square) was coupled with the conventional AGM separator. Then, CH@AGM was attached on the top of the separator before assembling a 147 mAh cm−2 Zn anode (250-μm thickness); 0.42 g of 2 M Li2SO4 and 1M ZnSO4 aqueous solution (pH 4) was used as the electrolyte, to make the E/C ratio equal to 30 g(Ah)−1. The full battery pack is shown in fig. S17.
To assemble a LMO|AGM + CH@AGM|Li battery under unconstrained conditions, a 1.8 mAh cm−2 LMO cathode (0.95 cm−2, disk) was coupled with the AGM separator, and then CH@AGM and a 51.5 mAh cm−2 Li anode (250-μm thickness) were subsequently assembled on top of the separator to obtain the full cell; 0.09 g of mixed solution of 1 M LiPF6 in dimethyl carbonate, ethyl carbonate, and diethyl carbonate (1:1:1 by weight) was used as the electrolyte. The E/C ratio is 60 g(Ah)−1, and the battery is tested under a CR2032 coin-cell kit.
To assemble a LMO|AGM + CH@AGM|Zn battery under constrained conditions, a 24 mAh cm−2 LMO cathode (9 cm−2, square) was coupled with the AGM separator, CH@AGM, and a 58 mAh cm−2 Zn anode (100-μm thickness); 2.16 g of 2 M Li2SO4 and 1 M ZnSO4 aqueous solution (pH 4) was used as the electrolyte, to make the E/C ratio equal to 10 g(Ah)−1. The battery pack is the same as the LMO|AGM + CH@AGM|Zn battery under unconstrained conditions (fig. S17), but the cathode, anode, and separator are differently sized.
To assemble a LMO|AGM + CH@AGM|Li battery under constrained conditions, a 24 mAh cm−2 LMO cathode (4 cm−2, square) was coupled with the AGM separator, CH@AGM, and a 51.5 mAh cm−2 Li anode (250-μm thickness); 0.98 g of mixed solution of 1 M LiPF6 in dimethyl carbonate, ethyl carbonate, and diethyl carbonate (1:1:1 by weight) was used as the electrolyte with the E/C ratio of 10 g(Ah)−1. The battery is assembled in an argon glove box, and the testing pack used in this battery is the same as that of the LMO|AGM + CH@AGM|Zn battery under constrained conditions (fig. S17).
To assemble an industrial-level LMO|AGM + CH@AGM|Zn battery, 1-Ah single-cell unit was first prepared. Briefly, a 13.2 mAh cm−2 LMO cathode (80 cm−2 rectangular sheet; fig. S24) was coupled with the AGM separator, CH@AGM, and a 147 mAh cm−2 Zn anode (250 μm thickness). Five, 65, and 200 repeated single-cell units were stacked together in a factory-made battery bay to assemble 5-, 65-, and 200-Ah batteries. After adding 150 g of 2 M Li2SO4 and 1 M ZnSO4 aqueous solution (pH 4) as electrolyte, the battery pack was sealed and aged for 24 hours before charge-discharge tests.
To prepare a 2 M Li2SO4 and 1 M ZnSO4 aqueous electrolyte with CH for LMO|AGM|Zn cells, 0.5% (weight %) CH (Sigma-Aldrich) was mixed with deionized water with constant stirring for 12 hours at 80°C. In this process, the CH was fully swollen by water and decomposed into CH. After cooling to room temperature, Li2SO4 and ZnSO4 were added to the liquid to make 2 M Li2SO4 and 1 M ZnSO4 aqueous solution (pH 4).
Electrochemical test
Cycling and rate tests were performed using a Neware series 3000 battery tester and maintained at 25° or 60°C under the working voltage of 1.4 to 2.1 V versus Zn2+/Zn for the Zn metal battery or the working voltage of 3.5 to 4.3 V versus Li+/Li for the Li metal battery. Chronoamperometry measurements were performed on a Bio-Logic VMP3 electrochemical workstation at −135 mV overpotential for Zn and Li metal batteries. Chronopotentiometry and linear sweep voltammetry were also performed on the same instruments for symmetric Zn and Li cells. The cycling performance of symmetric Zn and Li cells was tested by a multichannel battery tester (BTS-5V10mA, Neware) at a 1 mA cm−2 current density. Potentiostatic and galvanostatic EIS were conducted on a Biologic VMP3 electrochemical workstation using the symmetric cells, at an amplitude of 10 mV or various dc biases.
Morphology and structure characterization
The SEM images of the separator, lithium, anode and zinc anode were taken on UltraPlus field-emission scanning electron microscopes equipped with energy-dispersive spectroscopy for elemental analysis. Fourier transform infrared and Raman spectroscopy spectra were obtained using a Bruker Optics Vertex 70 Spectrometer and a Jobin Yvon Raman microspectrometer (HR 800), respectively. A Thermo-VG Scientific ESCALab 250 microprobe was used to record XPS spectra. A mercury porosimeter (Autopore IV9500 model, Micromeritics Inc.) and a Gurley density meter (4340 N) were used to investigate the porosity of various separators. PFG NMR was performed on a 600-MHz NMR spectrometer (Agilent) equipped with a 5-mm z-gradient probe.
Isothermal titration calorimetry
ITC experiments were conducted using a MicroCal VP-ITC instrument, wherein 300 μl of an aqueous solution of 10−3 M ZnSO4 or Li2SO4 was injected in equal steps into the cell containing 10−4 M Gly-Pro-Ala peptide. For comparison, aqueous solution of colloidal SiO2 is used as titrate to investigate the Zn2+/Li+ cations binding to AGM. Each negative peak shown in the heat signal curves represents an exothermic process, which denotes the heat released in one injection. After integrating heat response obtained from the raw signals, the calorimetric data against the total volume can be obtained. Ligand and receptor solutions were prepared with the same buffer stock solution, 20 mM tris(hydroxymethyl)aminomethane (tris) at pH 7.4.
Supplementary Material
Acknowledgments
We thank W. Wu in Rehab Energy Co. Ltd. for the support of battery commercialization and X. Luo and Y. Luo in Rehab Energy Co. Ltd. for the assembly and testing of 5-, 65-, and 200-Ah industrial-level Zn metal batteries. Funding: This research was supported by Rehab Energy Co. Ltd., the Canadian Foundation for Innovation (CFI), the Canada Research Chairs (CRC) program, and Mitacs (IT04444). Author contributions: J.Z. and P.C. proposed the idea. J.Z. conducted part of materials characterizations and battery testing, performed density functional theory simulations, and wrote the manuscript. S.L. performed part of electrochemical characterizations and assisted in microscopic and spectroscopic characterizations. M.H. performed part of material characterizations and assisted in revising the manuscript. P.C. arranged industrial-level batteries assembled in Rehab Energy Co. Ltd., oversaw all research phases, and revised the manuscript. Competing interests: J.Z. and P.C. are authors on a pending patent application related to this work (CN 202010258213.X, provisional patent filed on 23 April 2020). S.L. and P.C. are authors on another pending patent application related to this work (CN 201910355540.4, provisional patent filed on 29 April 2019). The authors declare that they have no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/32/eabb1342/DC1
REFERENCES AND NOTES
- 1.M. Yoshio, R. J. Brodd, A. Kozawa, Lithium-Ion Batteries (Springer, 2009), vol. 1. [Google Scholar]
- 2.Choudhury S., Mangal R., Agrawal A., Archer L. A., A highly reversible room-temperature lithium metal battery based on crosslinked hairy nanoparticles. Nat. Commun. 6, 10101 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Higashi S., Lee S. W., Lee J. S., Takechi K., Cui Y., Avoiding short circuits from zinc metal dendrites in anode by backside-plating configuration. Nat. Commun. 7, 11801 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ding F., Xu W., Graff G. L., Zhang J., Sushko M. L., Chen X., Shao Y., Engelhard M. H., Nie Z., Xiao J., Liu X., Sushko P. V., Liu J., Zhang J.-G., Dendrite-free lithium deposition via self-healing electrostatic shield mechanism. J. Am. Chem. Soc. 135, 4450–4456 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Harry K. J., Hallinan D. T., Parkinson D. Y., MacDowell A. A., Balsara N. P., Detection of subsurface structures underneath dendrites formed on cycled lithium metal electrodes. Nat. Mater. 13, 69–73 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Zhang R., Chen X.-R., Chen X., Cheng X.-B., Zhang X.-Q., Yan C., Zhang Q., Lithiophilic sites in doped graphene guide uniform lithium nucleation for dendrite-free lithium metal anodes. Angew. Chem. Int. Ed. 56, 7764–7768 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Zuo T.-T., Yin Y.-X., Wang S.-H., Wang P.-F., Yang X., Liu J., Yang C.-P., Guo Y.-G., Trapping lithium into hollow silica microspheres with a carbon nanotube core for dendrite-free lithium metal anodes. Nano Lett. 18, 297–301 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Li Q., Zhu S., Lu Y., 3D porous Cu current collector/Li-metal composite anode for stable lithium-metal batteries. Adv. Funct. Mater. 27, 1606422 (2017). [Google Scholar]
- 9.Wang L.-P., Li N.-W., Wang T.-S., Yin Y.-X., Guo Y.-G., Wang C.-R., Conductive graphite fiber as a stable host for zinc metal anodes. Electrochim. Acta 244, 172–177 (2017). [Google Scholar]
- 10.Sun K. E. K., Hoang T. K. A., Doan T. N. L., Yu Y., Zhu X., Tian Y., Chen P., Suppression of dendrite formation and corrosion on zinc anode of secondary aqueous batteries. ACS Appl. Mater. Interfaces 9, 9681–9687 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Ryou M. H., Lee D. J., Lee J.-N., Lee Y. M., Park J.-K., Choi J. W., Excellent cycle life of lithium-metal anodes in lithium-ion batteries with mussel-inspired polydopamine-coated separators. Adv. Energy Mater. 2, 645–650 (2012). [Google Scholar]
- 12.Liu Y., Lin D., Yuen P. Y., Liu K., Xie J., Dauskardt R. H., Cui Y., An artificial solid electrolyte interphase with high Li-ion conductivity, mechanical strength, and flexibility for stable lithium metal anodes. Adv. Mater. 29, 1605531 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Fan L., Wei S., Li S., Li Q., Lu Y., Recent progress of the solid-state electrolytes for high-energy metal-based batteries. Adv. Energy Mater. 8, 1702657 (2018). [Google Scholar]
- 14.Parker J. F., Chervin C. N., Nelson E. S., Rolison D. R., Long J. W., Wiring zinc in three dimensions re-writes battery performance—Dendrite-free cycling. Energ. Environ. Sci. 7, 1117–1124 (2014). [Google Scholar]
- 15.Niu C., Pan H., Xu W., Xiao J., Zhang J.-G., Luo L., Wang C., Mei D., Meng J., Wang X., Liu Z., Mai L., Liu J., Self-smoothing anode for achieving high-energy lithium metal batteries under realistic conditions. Nat. Nanotechnol. 14, 594–601 (2019). [DOI] [PubMed] [Google Scholar]
- 16.Kim M. S., Ryu J.-H., Deepika, Lim Y. R., Nah I. W., Lee K.-R., Archer L. A., Cho W. I., Langmuir–Blodgett artificial solid-electrolyte interphases for practical lithium metal batteries. Nat. Energy 3, 889–898 (2018). [Google Scholar]
- 17.Schaefer J. L., Lu Y., Moganty S. S., Agarwal P., Jayaprakash N., Archer L. A., Electrolytes for high-energy lithium batteries. App. Nanosci. 2, 91–109 (2012). [Google Scholar]
- 18.Lu W., Xie C., Zhang H., Li X., Inhibition of zinc dendrite growth in zinc-based batteries. ChemSusChem 11, 3996–4006 (2018). [DOI] [PubMed] [Google Scholar]
- 19.Lu Y., Tu Z., Shu J., Archer L. A., Stable lithium electrodeposition in salt-reinforced electrolytes. J. Power Sources 279, 413–418 (2015). [Google Scholar]
- 20.Tikekar M. D., Choudhury S., Tu Z., Archer L. A., Design principles for electrolytes and interfaces for stable lithium-metal batteries. Nat. Energy 1, 16114 (2016). [Google Scholar]
- 21.Lu Y., Tu Z., Archer L. A., Stable lithium electrodeposition in liquid and nanoporous solid electrolytes. Nat. Mater. 13, 961–969 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Chazalviel J.-N., Electrochemical aspects of the generation of ramified metallic electrodeposits. Phys. Rev. A 42, 7355–7367 (1990). [DOI] [PubMed] [Google Scholar]
- 23.Tikekar M. D., Archer L. A., Koch D. L., Stability analysis of electrodeposition across a structured electrolyte with immobilized anions. J. Electrochem. Soc. 161, A847–A855 (2014). [Google Scholar]
- 24.Han J.-H., Khoo E., Bai P., Bazant M. Z., Over-limiting current and control of dendritic growth by surface conduction in nanopores. Sci. Rep. 4, 7056 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Han J.-H., Wang M., Bai P., Brushett F. R., Bazant M. Z., Dendrite suppression by shock electrodeposition in charged porous media. Sci. Rep. 6, 28054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deng D., Dydek E. V., Han J.-H., Schlumpberger S., Mani A., Zaltzman B., Bazant M. Z., Overlimiting current and shock electrodialysis in porous media. Langmuir 29, 16167–16177 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Schlumpberger S., Lu N. B., Suss M. E., Bazant M. Z., Scalable and continuous water deionization by shock electrodialysis. Environ. Sci. Technol. Lett. 2, 367–372 (2015). [Google Scholar]
- 28.Duconseille A., Astruc T., Quintana N., Meersman F., Sante-Lhoutellier V., Gelatin structure and composition linked to hard capsule dissolution: A review. Food Hydrocoll. 43, 360–376 (2015). [Google Scholar]
- 29.Liu X., Zhang N., Yu L., Zhou S., Shanks R., Zheng J., Imaging the phase of starch–gelatin blends by confocal Raman microscopy. Food Hydrocoll. 60, 7–10 (2016). [Google Scholar]
- 30.Sun W., Zhao Q., Zhao M., Yang B., Cui C., Ren J., Structural evaluation of myofibrillar proteins during processing of Cantonese sausage by Raman spectroscopy. J. Agric. Food Chem. 59, 11070–11077 (2011). [DOI] [PubMed] [Google Scholar]
- 31.Kaul G., Amiji M., Cellular interactions and in vitro DNA transfection studies with poly (ethylene glycol)-modified gelatin nanoparticles. J. Pharm. Sci. 94, 184–198 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Banik S. J., Akolkar R., Suppressing dendritic growth during alkaline zinc electrodeposition using polyethylenimine additive. Electrochim. Acta 179, 475–481 (2015). [Google Scholar]
- 33.Li G., Liu Z., Huang Q., Gao Y., Regula M., Wang D., Chen L.-Q., Wang D., Stable metal battery anodes enabled by polyethylenimine sponge hosts by way of electrokinetic effects. Nat. Energy 3, 1076–1083 (2018). [Google Scholar]
- 34.Mani A., Bazant M. Z., Deionization shocks in microstructures. Phys. Rev. E 84, 061504 (2011). [DOI] [PubMed] [Google Scholar]
- 35.Dydek E. V., Zaltzman B., Rubinstein I., Deng D. S., Mani A., Bazant M. Z., Overlimiting current in a microchannel. Phys. Rev. Lett. 107, 118301 (2011). [DOI] [PubMed] [Google Scholar]
- 36.Nam S., Cho I., Heo J., Lim G., Bazant M. Z., Moon D. J., Sung G. Y., Kim S. J., Experimental verification of overlimiting current by surface conduction and electro-osmotic flow in microchannels. Phys. Rev. Lett. 114, 114501 (2015). [DOI] [PubMed] [Google Scholar]
- 37.Khoo E., Zhao H., Bazant M. Z., Linear stability analysis of transient electrodeposition in charged porous media: Suppression of dendritic growth by surface conduction. J. Electrochem. Soc. 166, A2280–A2299 (2019). [Google Scholar]
- 38.Song J., Khoo E., Bazant M. Z., Electrochemical impedance of electrodiffusion in charged medium under dc bias. Phys. Rev. E 100, 042204 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Khoo E., Bazant M. Z., Theory of voltammetry in charged porous media. J. Electroanal. Chem. 811, 105–120 (2018). [Google Scholar]
- 40.Yan D., Bazant M. Z., Biesheuvel P., Pugh M. C., Dawson F. P., Theory of linear sweep voltammetry with diffuse charge: Unsupported electrolytes, thin films, and leaky membranes. Phys. Rev. E 95, 033303 (2017). [DOI] [PubMed] [Google Scholar]
- 41.Riede J.-C., Turek T., Kunz U., Critical zinc ion concentration on the electrode surface determines dendritic zinc growth during charging a zinc air battery. Electrochim. Acta 269, 217–224 (2018). [Google Scholar]
- 42.Bai P., Li J., Brushett F. R., Bazant M. Z., Transition of lithium growth mechanisms in liquid electrolytes. Energ. Environ. Sci. 9, 3221–3229 (2016). [Google Scholar]
- 43.Zhi J., Bertens K., Yazdi A. Z., Chen P., Acrylonitrile copolymer/graphene skinned cathode for long cycle life rechargeable hybrid aqueous batteries at high-temperature. Electrochim. Acta 268, 248–255 (2018). [Google Scholar]
- 44.Biscarat J., Galea B., Sanchez J., Pochat-Bohatier C., Effect of chemical cross-linking on gelatin membrane solubility with a non-toxic and non-volatile agent: Terephthalaldehyde. Int. J. Biol. Macromol. 74, 5–11 (2015). [DOI] [PubMed] [Google Scholar]
- 45.Talmard C., Bouzan A., Faller P., Zinc binding to amyloid-β: Isothermal titration calorimetry and Zn competition experiments with Zn sensors. Biochemistry 46, 13658–13666 (2007). [DOI] [PubMed] [Google Scholar]
- 46.Frushour B. G., Koenig J. L., Raman scattering of collagen, gelatin, and elastin. Biopolymers 14, 379–391 (1975). [DOI] [PubMed] [Google Scholar]
- 47.Deng Z., Irish D. E., Raman spectral studies of ion association and solvation in solutions of LiAsF6–acetone. J. Chem. Soc. Faraday Trans. 88, 2891–2896 (1992). [Google Scholar]
- 48.Wang S., Sun P., Zhang R., Lu A., Liu M., Zhang L., Cation/macromolecule interaction in alkaline cellulose solution characterized with pulsed field-gradient spin-echo NMR spectroscopy. Phys. Chem. Chem. Phys. 19, 7486–7490 (2017). [DOI] [PubMed] [Google Scholar]
- 49.Zeng Z., Murugesan V., Han K. S., Jiang X., Cao Y., Xiao L., Ai X., Yang H., Zhang J.-G., Sushko M. L., Liu J., Non-flammable electrolytes with high salt-to-solvent ratios for Li-ion and Li-metal batteries. Nat. Energy 3, 674–681 (2018). [Google Scholar]
- 50.Thackeray M., Structural considerations of layered and spinel lithiated oxides for lithium ion batteries. J. Electrochem. Soc. 142, 2558 (1995). [Google Scholar]
- 51.Li X., Xu Y., Wang C., Suppression of Jahn–Teller distortion of spinel LiMn2O4 cathode. J. Alloys Compd. 479, 310–313 (2009). [Google Scholar]
- 52.Li B., Xing L., Xu M., Lin H., Li W., New solution to instability of spinel LiNi0.5 Mn1.5O4 as cathode for lithium ion battery at elevated temperature. Electrochem. Commun. 34, 48–51 (2013). [Google Scholar]
- 53.Gallagher K. G., Fuller T. F., Kinetic model of the electrochemical oxidation of graphitic carbon in acidic environments. Phys. Chem. Chem. Phys. 11, 11557–11567 (2009). [DOI] [PubMed] [Google Scholar]
- 54.Wang H., Tang A., Huang K., Oxygen evolution in overcharged LixNi1/3Co1/3Mn1/3O2 electrode and its thermal analysis kinetics. Chin. J. Chem. 29, 1583–1588 (2011). [Google Scholar]
- 55.Cheng Q., Song Z., Ma T., Smith B. B., Tang R., Yu H., Jiang H., Chan C. K., Folding paper-based lithium-ion batteries for higher areal energy densities. Nano Lett. 13, 4969–4974 (2013). [DOI] [PubMed] [Google Scholar]
- 56.Luo J., Fang C.-C., Wu N.-L., High polarity poly (vinylidene difluoride) thin coating for dendrite-free and high-performance lithium metal anodes. Adv. Energy Mater. 8, 1701482 (2018). [Google Scholar]
- 57.Patoux S., Sannier L., Lignier H., Reynier Y., Bourbon C., Jouanneau S., Cras F. L., Martinet S., High voltage nickel manganese spinel oxides for Li-ion batteries. Electrochim. Acta 53, 4137–4145 (2008). [Google Scholar]
- 58.Yoo H. D., Liang Y., Li Y., Yao Y., High areal capacity hybrid magnesium–lithium-ion battery with 99.9% coulombic efficiency for large-scale energy storage. ACS Appl. Mater. Interfaces 7, 7001–7007 (2015). [DOI] [PubMed] [Google Scholar]
- 59.Chen X., Zhu H., Chen Y.-C., Shang Y., Cao A., Hu L., Rubloff G. W., MWCNT/V2O5 core/shell sponge for high areal capacity and power density Li-ion cathodes. ACS Nano 6, 7948–7955 (2012). [DOI] [PubMed] [Google Scholar]
- 60.Qian J., Adams B. D., Zheng J., Xu W., Henderson W. A., Wang J., Bowden M. E., Xu S., Hu J., Zhang J.-G., Anode-free rechargeable lithium metal batteries. Adv. Funct. Mater. 26, 7094–7102 (2016). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/6/32/eabb1342/DC1