

Key Points
Free heme in hemolytic disorders induces rapid endothelial barrier dysfunction via the p38MAPK pathway.
MKK3 KO effectively protects from heme-induced endothelial barrier dysfunction in the lungs.
Abstract
Several studies demonstrate that hemolysis and free heme in circulation cause endothelial barrier dysfunction and are associated with severe pathological conditions such as acute respiratory distress syndrome, acute chest syndrome, and sepsis. However, the precise molecular mechanisms involved in the pathology of heme-induced barrier disruption remain to be elucidated. In this study, we investigated the role of free heme in the endothelial barrier integrity and mechanisms of heme-mediated intracellular signaling of human lung microvascular endothelial cells (HLMVECs). Heme, in a dose-dependent manner, induced a rapid drop in the endothelial barrier integrity of HLMVECs. An investigation into barrier proteins revealed that heme primarily affected the tight junction proteins zona occludens-1, claudin-1, and claudin-5, which were significantly reduced after heme exposure. The p38MAPK/HSP27 pathway, involved in the regulation of endothelial cytoskeleton remodeling, was also significantly altered after heme treatment, both in HLMVECs and mice. By using a knockout (KO) mouse for MKK3, a key regulator of the p38MAPK pathway, we showed that this KO effectively decreased heme-induced endothelial barrier dysfunction. Taken together, our results indicate that targeting the p38MAPK pathway may represent a crucial treatment strategy in alleviating hemolytic diseases.
Visual Abstract

Introduction
Elevated levels of circulating free heme have been shown to correlate with increased risk of several hemolytic disorders and are associated with endothelial barrier dysfunction. The pathological conditions associated with endothelial barrier dysfunction include sickle cell anemia, acute respiratory distress syndrome, myocardial infarction, hypertension, sepsis, and stroke.1 During hemolysis, free hemoglobin from ruptured red blood cells in the bloodstream undergoes rapid oxidation and releases free heme. Pathologically, high levels of free heme and iron release occur in acute conditions such as severe hemolytic crisis in sickle cell disease (up to 20 μM) or thalassemia (up to 280 μM).2,3 Therefore, understanding the mechanism of free heme–induced damage represents an important strategy in identifying appropriate treatment targets for a broad range of hemolytic diseases.
Although significant progress has been made in our understanding of signaling mechanisms and mediators that regulate endothelial permeability, important gaps still exist, and mouse models to explore heme-mediated damage are not clearly defined. We recently discovered that heme affects endothelial barrier function within minutes, and this is associated with activation of the p38/HSP27 pathway.4 In disorders with endothelial barrier dysfunction, the lungs are particularly vulnerable to vascular leak and fluid accumulation, with serious pathological consequences.5 Therefore, in this study, we used human lung microvascular endothelial cells (HLMVECs) to assess barrier dysfunction because they are most susceptible to injury with severe implications. We also took advantage of the feasibility of exploring endothelial damage in the lungs in vivo. Consequently, we demonstrate a strong association between the p38MAPK pathway and heme-induced barrier dysfunction by using a knockout (KO) mouse model of MKK3, a key regulator of the p38 pathway. Our findings provide a solid background for the development of novel therapeutic targets to treat hemolytic diseases with endothelial barrier dysfunction.
Methods
Detailed methods are provided in the supplemental Information, available on the Blood Web site.
Results and discussion
The endothelial cells form a monolayer lining the blood vessel, forming a semipermeable barrier that controls fluid homeostasis, nutrient transport, and cellular migration across the barrier.6 In continuation of our previous studies,4 based on the hypothesis that excess heme in blood potentially leads to endothelial barrier disruption, we studied the direct effect of heme on barrier function in HLMVECs. Indeed, treatment with free heme (50 µM) significantly increased intracellular heme content in HLMVECs (Figure 1A). We also observed a dose-dependent decrease in barrier function, which was a maximum between 15 and 25 minutes after heme treatment followed by a recovery phase (Figure 1B). When the barrier function was calculated, a significant decline in function was observed, as identified from the slope in Figure 1C. Recovery of the barrier function was proportional to the dose and did not revert to the control level. These data confirm that heme causes a rapid disruption of the endothelial barrier and could contribute to diseases involving endothelial barrier compromise.
Figure 1.
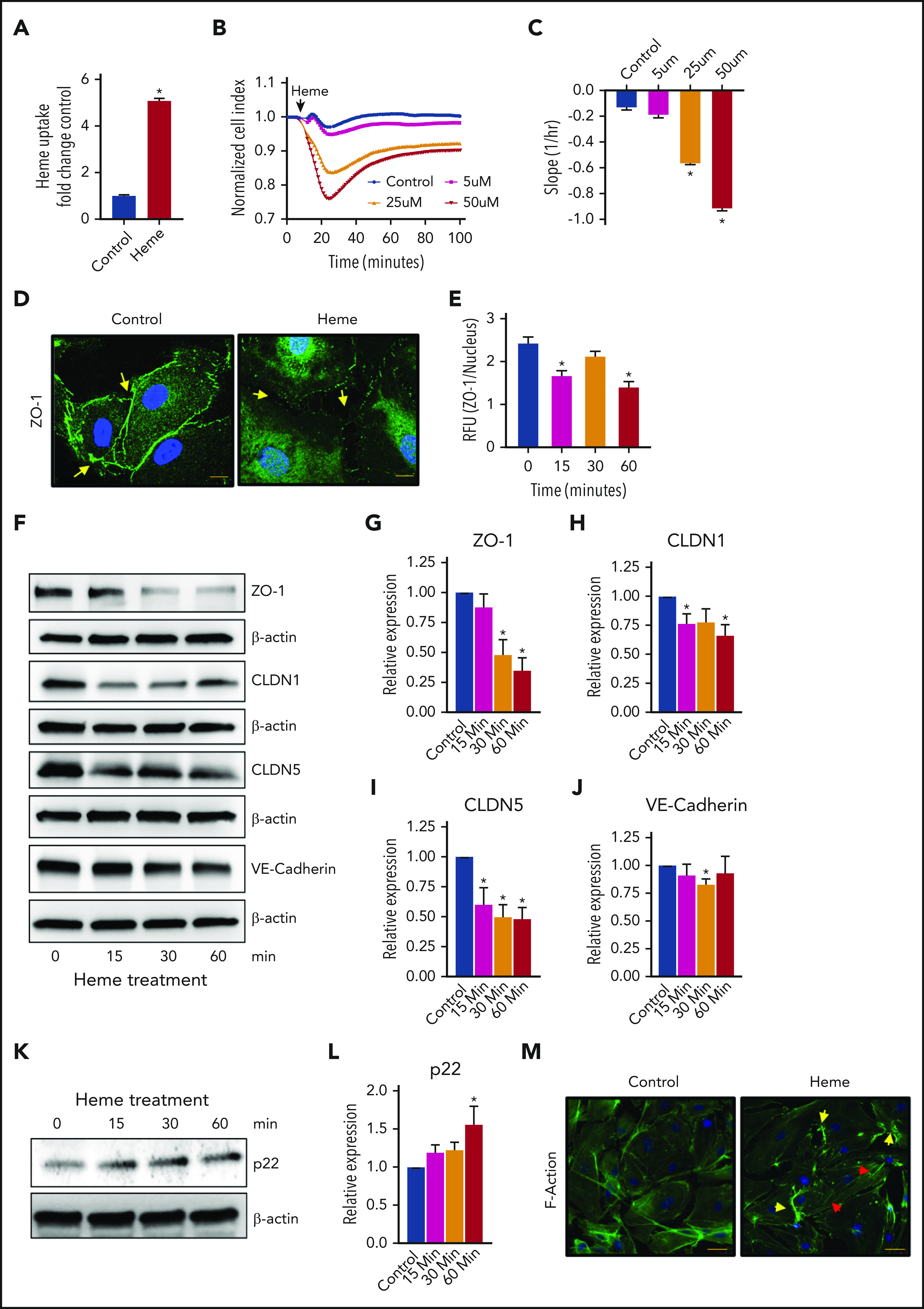
Heme causes endothelial cell barrier disruption. (A) Heme entry into HLMVECs was quantified using the Hemin assay kit (Abcam; ab65332). HLMVECs treated with heme (50 μM, 15 minutes) had significantly increased intracellular heme content. (B) HLMVECs were seeded in ECIS plates, and after resistance measurements attained a stable level, heme was added to the media. Readings were recorded every minute after heme treatment and plotted as normalized cell index. (C) Rate of decline in resistance, reflective of barrier disruption, was calculated as slope using xCELLigence software. (D) Heme affects tight junction proteins. HLMVECs were treated with heme (50 μM, 60 minutes), and cells were fixed, immunostained for zona occludens-1 (ZO-1, green), and imaged using a confocal microscope (original magnification ×63). Arrows indicate the cellular organization of ZO-1. (E) Similarly, HLMVECs were treated with heme for the given amount of time and imaged at 20× and quantified for ZO-1 levels using Image J software. Images were quantified using at least 5 random images. (F-J) HLMVECs were treated with heme (50 µM) for different time points, and cell lysate was prepared in RIPA buffer with protease and phosphatase inhibitors. Endothelial junction proteins were detected using western blotting. Representative blots of ZO-1, claudin-1 [CLDN-1], CLDN-5, and VE-cadherin are shown (F), quantified using BioRad software (G-J). (K-L) Protein levels were normalized to β-actin. Stress fiber formation was seen after heme exposure. p22, the EF-hand protein required for actin polymerization, was found significantly increased. (M) HLMVECs were treated with heme (50 µM, 60 minutes), and cells were fixed, stained for F-actin using phalloidin (green), and imaged using a fluorescence microscope (original magnification ×63). Yellow arrows indicate apical stress fibers, and red arrows indicate stress fibers. Experiments were repeated at least 3 times. Values are mean ± standard error of the mean. *P < .05 compared with control. RFU, relative fluorescence unit.
To investigate whether heme disrupts tight junction (TJ) proteins, we studied endothelial junctions such as TJs and adherens junctions, which are prominent and regulate vascular permeability, mediate cell adhesion, and transfer intracellular signals.7 TJs are composed of transmembrane proteins (eg, CLDNs) and intracellular proteins (eg, ZO). Disruption in interactions between these proteins perturbs the TJs affecting permeability of the endothelium. Adhesion in adherens junctions is mediated by cadherins, and among them, VE-cadherin is the major component. These junctions are continuously remodeled in response to mechanical and chemical cues in physiological settings.8 We tested the effect of heme on these junctions in HLMVECs by immunostaining and western blotting. We observed that heme caused attenuation of peripheral ZO-1, as evident from confocal images (Figure 1D). Quantification of ZO-1 levels over nuclear staining showed a significant decrease, even at 15 minutes after heme treatment (Figure 1E), indicating that ZO-1, upon losing its cellular organization and being a high molecular weight protein, intercalates with other cytoskeleton proteins. Indeed, ZO-1 has been shown to bind to F-actin and has been linked to regulation of the actomyosin cytoskeleton.9 An investigation into the other junction proteins by western blotting revealed that ZO-1, CLDN-1, and CLDN-5 were significantly decreased at all time points tested, with a slight drop in VE-cadherin seen only at 30 minutes after heme treatment (Figure 1F-J). Interestingly, we found that heme activated p22, an EF-hand Ca2+-binding protein, which facilitates microtubule-membrane interactions and could potentiate stress fiber formations (Figure 1K-L).10 Furthermore, we also found that heme promoted actin stress fiber formation in HLMVECs (Figure 1M).
Heme is known to activate multiple signaling pathways, including AKT-ERK, lipid oxidation, and the canonical TLR4-NFΚβ inflammatory pathway.11,12 However, we recently discovered that heme-mediated endothelial barrier disruption took place 10 times faster and occurred independent of TLR4 signaling.4 We also demonstrated an early activation of the p38/HSP27 pathway during hemolysis.4 Interestingly, the p38/HSP27 pathway is known to rapidly modulate the cytoskeleton and affects junction organization in response to stress.13 Investigating the p38 pathway proteins showed that heme caused p38 phosphorylation at early time points (Figure 2A-B). Phosphorylation of p38 is known to activate the downstream cascade of MAPK-activated protein kinase 2 (MK2) and HSP27.13 Correspondingly, we observed significant activation of MK2 by phosphorylation at Thr334 residue but only a trending increase in the phosphorylation of the Thr222 residue (Figure 2A,C-D). Activation of MK2 has been shown to phosphorylate HSP27.14 In addition, MK2 has also been shown to phosphorylate HSP27 in response to oxidative stress.13 Indeed, free heme promotes the formation of reactive oxygen species, which induces direct cellular damage via the oxidation of lipids, proteins, and DNA, thus activating the p38/MAPK pathway.13,15 In this study, we observed phosphorylation of HSP27 from early time points (Figure 2A,E). This activation also corresponded to the activation of MKK3 (Figure 2A,F), a key regulator upstream of the p38/MAPK pathway, indicating that heme potentially targets the MKK3/p38/MAPK axis in disrupting the endothelial barrier.
Figure 2.
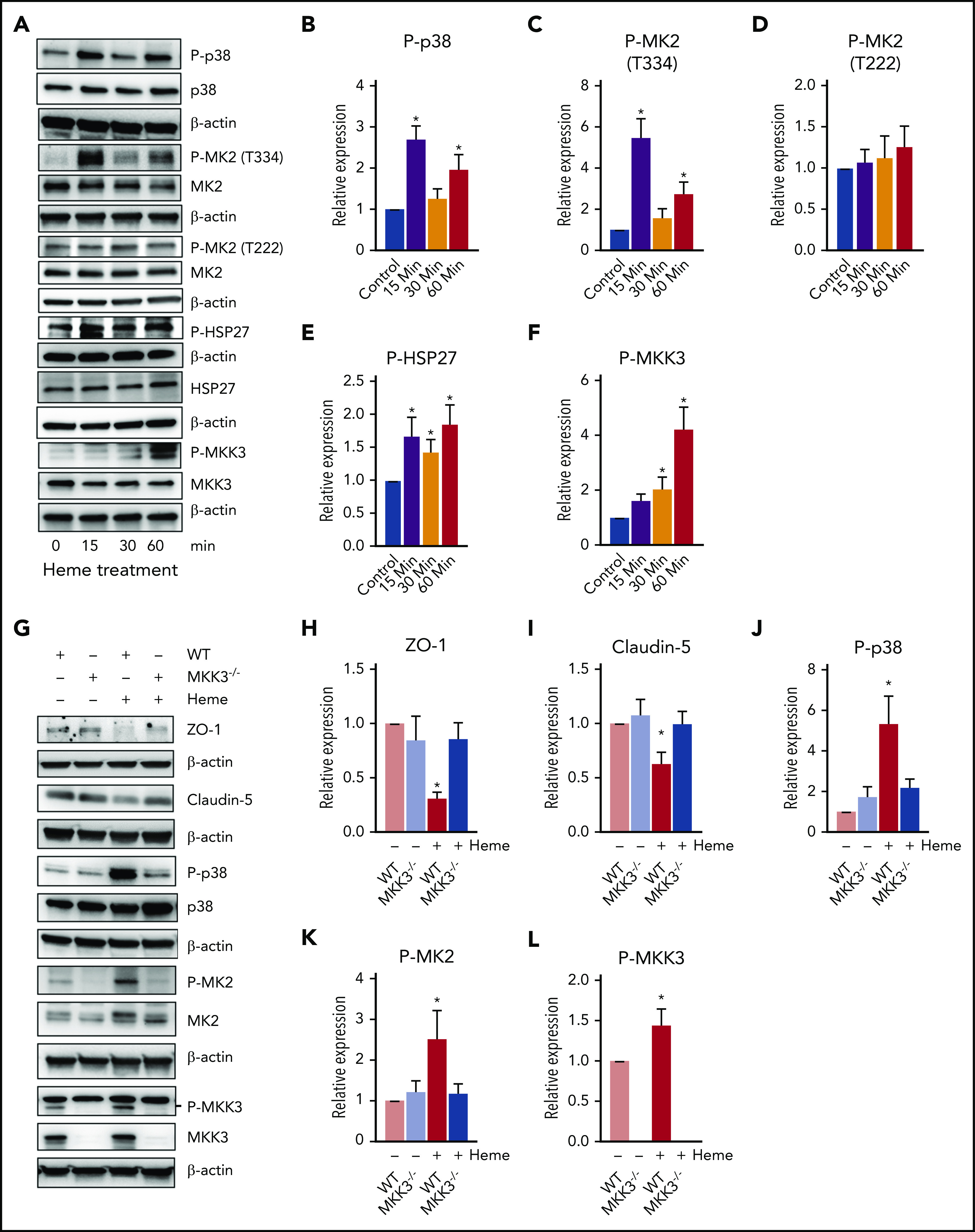

Intracellular MKK3/P38 pathway affected by heme. (A-F) HLMVECs were treated with heme (50 µM) for different time points, and cell lysate was prepared in RIPA buffer with protease and phosphatase inhibitors. The MKK3/p38/HSP27 axis proteins were found to be activated after heme treatment. Representative blots of proteins and corresponding proteins phosphorylation levels for P-p38, p38, P-MK2 (T334), P-MK2 (T222), P-HSP27, HSP27, P-MKK3 (S189), and MKK3 (A), quantified using BioRad software (B-F). (H-L) MKK3−/− mice had attenuated barrier disruption and diminished p38 pathway activation. Wild-type (WT) and MKK3−/− mice were treated with heme (250 µM IV) or with vehicle for 6 hours, their were lungs excised, and lysates were prepared in RIPA buffer with protease and phosphatase inhibitors. Our data indicate activation of MKK3/P38 on heme treatment that was blunted in MKK3−/− lungs. TJ proteins showed a decrease only in WT mice treated with heme. Representative blots of proteins ZO-1, CLDN-5, P-p38, p38, P-MK2 (T334), MK2, P-MKK3(S189), and MKK3 (K), quantified using BioRad software (H-L). (M-P) Heme mildly activated adhesion molecules, specifically ICAM-1. Representative blots of E-selectin, ICAM-1, and VCAM-1, and quantifications are shown. All protein levels were normalized to β-actin. (Q) Assessment of heme-induced barrier dysfunction in mouse lungs. Mice were injected with heme or vehicle (6 hours) and, in the end, injected with fluorescein isothiocyanate (FITC)–dextran to visualize FITC-dependent fluorescence in the lungs. Our data indicated a high level of extravasation of FITC-dextran into the lungs in WT rats treated with heme, whereas MKK3−/− rats showed protection against heme-mediated lung barrier disruption. (R) Quantification of FITC-dextran extravasation into the lungs showed that MKK3−/− mice were significantly protected from heme-induced barrier damage. Experiments were repeated at least 3 times. Values are mean ± standard error of the mean. (S) Heme injected into mice activates the MKK3/p38MAPK/HSP27 pathway, leading to disruption of TJs, causing cytoskeletal derangements and ultimately causing barrier disruption (red). Compromised endothelial barrier is reflected by flow of FITC-dextran from bloodstream and into lung tissue. MKK3−/− mice are highly protected (green) from heme-induced endothelial barrier disruption. *P < .05 compared with WT, **P < .05 compared with MKK3 vehicle, ***P < .05 compared with WT heme group.
Next, to test whether blocking the p38 pathway would alleviate heme-mediated endothelial barrier dysfunction in the lungs, we targeted its regulator, MKK3, which controls the p38-mediated actin cytoskeleton rearrangements.13 Therefore, we tested whether disrupting this pathway by knocking out MKK3 would protect from heme-mediated action on the endothelial barrier in mouse lungs. The wild-type mice injected with heme considerably supported the in vitro findings and demonstrated a significant decrease in barrier proteins ZO-1 and CLDN-5 (Figure 2G-I). Also, we found heme-induced activation of MK2 and MKK3 and, correspondingly, significant activation of p38 in wild-type mice (Figure 2G,J-L). In contrast, both barrier disruption and activation of the p38MAPK pathway were significantly attenuated in the MKK3−/− mice (Figure 2G-L). We also observed that heme significantly activated the inflammatory adhesion protein, ICAM1, but only showed a trending activation in E-selectin and no change in VCAM1 (Figure 2M-P), indicating that inflammation could be a later outcome of heme-induced damage. Interestingly, the MKK3−/− mice did not show any increase in these inflammatory markers but showed lower basal expression of ICAM1 (Figure 2M-P). This could signify that MKK3−/− mice are protected from later onset of heme-induced inflammation. To assess the lung barrier disruption, we evaluated lung leakage by FITC-dextran extravasation. Strikingly, the MKK3−/− mice injected intravenously with heme (250 μM) and FITC-dextran (1 mg per 30 g of body weight) in circulation for 6 hours showed a dramatic decrease in lung extravasation of FITC-dextran through the endothelial barrier as compared with controls (Figure 2Q-R). Interestingly, the in vivo circulating concentration of heme used in this study was highly comparable to hemolytic pathological conditions,2,3 and the MKK3−/− mice circumvented the heme-induced damage, indicating that targeting the p38MAPK pathway effectively attenuates hemolytic endothelial barrier dysfunction (Figure 2S). In summary, our data suggest that the MKK3/p38 axis regulates heme-mediated barrier disruption in pulmonary endothelial cells. Understanding intracellular heme signaling is important in the pathobiology of hemolytic conditions. This knowledge could be applied to the development of treatment options that target the MKK3 pathway for resolution of hemolysis-related lung complications (eg, acute chest syndrome, acute respiratory distress syndrome, and pulmonary edema).
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
Certain elements in Figure 2S and the visual abstract were derived from the image bank of Servier Medical Art (http://smart.servier.com/) under a Creative Commons Attribution 3.0 Unported License. This work was supported by National Heart, Lung, and Blood Institute, National Institutes of Health, grants R01HL133085 (O.R.), R01HL132918 (R.R.) and R01HL151447 (R.R.), and by Arizona Area Heath and Education Center grant RG2017-11 (A.S.).
Footnotes
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Contact the corresponding author for original data.
Authorship
Contribution: R.R., O.R., and A.S. were responsible for conception and design; J.J., A.S., C.A.E., M.V.V., and M.Z. analyzed and interpreted the data; and O.R., R.R., J.J., and A.S. drafted the manuscript for important intellectual content.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Ruslan Rafikov, Department of Medicine, Division of Endocrinology, University of Arizona College of Medicine, 1501 N. Campbell Ave, Tucson, AZ 85721; e-mail: ruslanrafikov@deptofmed.arizona.edu.
REFERENCES
- 1.Kayyali US, Pennella CM, Trujillo C, Villa O, Gaestel M, Hassoun PM. Cytoskeletal changes in hypoxic pulmonary endothelial cells are dependent on MAPK-activated protein kinase MK2. J Biol Chem. 2002;277(45):42596-42602. [DOI] [PubMed] [Google Scholar]
- 2.Alayash AI. Oxidative pathways in the sickle cell and beyond. Blood Cells Mol Dis. 2018;70:78-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schaer DJ, Buehler PW, Alayash AI, Belcher JD, Vercellotti GM. Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood. 2013;121(8):1276-1284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rafikova O, Williams ER, McBride ML, et al. . Hemolysis-induced lung vascular leakage contributes to the development of pulmonary hypertension. Am J Respir Cell Mol Biol. 2018;59(3):334-345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maniatis NA, Kotanidou A, Catravas JD, Orfanos SE. Endothelial pathomechanisms in acute lung injury. Vascul Pharmacol. 2008;49(4-6):119-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Komarova YA, Kruse K, Mehta D, Malik AB. Protein interactions at endothelial junctions and signaling mechanisms regulating endothelial permeability. Circ Res. 2017;120(1):179-206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778(3):660-669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Barry AK, Wang N, Leckband DE. Local VE-cadherin mechanotransduction triggers long-ranged remodeling of endothelial monolayers. J Cell Sci. 2015;128(7):1341-1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tornavaca O, Chia M, Dufton N, et al. . ZO-1 controls endothelial adherens junctions, cell-cell tension, angiogenesis, and barrier formation. J Cell Biol. 2015;208(6):821-838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Andrade J, Pearce ST, Zhao H, Barroso M. Interactions among p22, glyceraldehyde-3-phosphate dehydrogenase and microtubules. Biochem J. 2004;384(Pt 2):327-336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang JY, Lee YT, Chang PF, Chau LY. Hemin promotes proliferation and differentiation of endothelial progenitor cells via activation of AKT and ERK. J Cell Physiol. 2009;219(3):617-625. [DOI] [PubMed] [Google Scholar]
- 12.Higdon AN, Benavides GA, Chacko BK, et al. . Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: the protective role of autophagy. Am J Physiol Heart Circ Physiol. 2012;302(7):H1394-H1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Corre I, Paris F, Huot J. The p38 pathway, a major pleiotropic cascade that transduces stress and metastatic signals in endothelial cells. Oncotarget. 2017;8(33):55684-55714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Soni S, Anand P, Padwad YS. MAPKAPK2: the master regulator of RNA-binding proteins modulates transcript stability and tumor progression. J Exp Clin Cancer Res. 2019;38(1):121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rafikov R, James J, McClain N, Tofovic SP, Rafikova O. Role of gender in regulation of redox homeostasis in pulmonary arterial hypertension. Antioxidants. 2019;8(5):135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.