

Publisher's Note: There is a Blood Commentary on this article in this issue.
Key Points
Trf-LKO mice display iron-deficiency anemia, iron overload in tissues, and susceptibility to liver damage via ferroptosis.
Ferroptosis inhibition or deletion of Slc39a14 attenuated iron overload and CCl4-induced liver fibrosis in Trf-LKO.
Abstract
Although the serum-abundant metal-binding protein transferrin (encoded by the Trf gene) is synthesized primarily in the liver, its function in the liver is largely unknown. Here, we generated hepatocyte-specific Trf knockout mice (Trf-LKO), which are viable and fertile but have impaired erythropoiesis and altered iron metabolism. Moreover, feeding Trf-LKO mice a high-iron diet increased their susceptibility to developing ferroptosis-induced liver fibrosis. Importantly, we found that treating Trf-LKO mice with the ferroptosis inhibitor ferrostatin-1 potently rescued liver fibrosis induced by either high dietary iron or carbon tetrachloride (CCl4) injections. In addition, deleting hepatic Slc39a14 expression in Trf-LKO mice significantly reduced hepatic iron accumulation, thereby reducing ferroptosis-mediated liver fibrosis induced by either a high-iron diet or CCl4 injections. Finally, we found that patients with liver cirrhosis have significantly lower levels of serum transferrin and hepatic transferrin, as well as higher levels of hepatic iron and lipid peroxidation, compared with healthy control subjects. Taken together, these data indicate that hepatic transferrin plays a protective role in maintaining liver function, providing a possible therapeutic target for preventing ferroptosis-induced liver fibrosis.
Visual Abstract

Introduction
Iron homeostasis is essential for maintaining the function of many tissues, particularly the liver, which serves as the major organ for iron metabolism. Transferrin (Trf), a serum-abundant metal-binding protein, is primarily synthesized in the liver.1 In the circulation, Trf binds to ferric iron (Fe3+) in a soluble, nontoxic form to deliver iron to the bone marrow and other tissues. At physiological pH, Trf binds Fe3+ with high affinity, and cellular uptake of Trf is mediated by the Trf receptor 1 (Tfr1), a ubiquitously expressed membrane protein. After Trf binds to Tfr1, the resulting Trf-Tfr1 complex is internalized, followed by endosomal acidification, which causes the release of iron from Trf and the transfer of iron to the cytosol.2 Trf binds and releases iron via the N lobe and/or the C lobe. Parrow et al recently reported that mice carrying mutations in Trf that prevent iron binding at either lobe have hepatocellular iron overload and decreased liver expression of the iron regulatory hormone hepcidin.3 These findings provide further support that Trf plays a key role in iron homeostasis and erythropoiesis.
In the clinic, reduced Trf levels have been associated with poor prognosis in patients with severe conditions such as liver cirrhosis.4-7 Under normal conditions, the majority of Trf-bound iron (TBI) is delivered to the bone marrow,2,8,9 and the majority of non-TBI (NTBI), which can be toxic, is cleared by the liver.8 When serum Trf levels decrease, however, the level of NTBI increases, resulting in an accumulation of NTBI in hepatocytes.10,11 Accordingly, approximately one-third of patients with end-stage liver disease present with hepatic iron overload,12 which suggests that Trf likely plays a pathogenic role in liver fibrosis.
Consistent with this notion, a growing body of data suggests that excessive iron is associated with liver damage. For example, we previously reported that iron overload induces liver damage via ferroptosis, a recently identified iron-dependent form of nonapoptotic cell death.13-16 Moreover, the high redox potential of iron may underlie its toxicity, as excessive iron leads to an accumulation of toxic reactive oxygen species, increased oxidative stress, and lipid peroxidation in the cell membrane, resulting in the cellular damage that ultimately triggers cell death.17 However, the molecular mechanisms by which iron overload and ferroptosis contribute to liver fibrosis and cirrhosis remain unclear. Moreover, the role that Trf plays in iron homeostasis in various tissues is poorly understood.
To address these clinically relevant questions and study the physiological role of hepatic Trf, we generated a hepatocyte-specific Trf knockout mouse (referred to hereafter as Trf-LKO). We found that although Trf-LKO mice are both viable and fertile, they have impaired erythropoiesis and iron-deficiency anemia. Moreover, Trf-LKO mice are more sensitive to phenylhydrazine (PHZ)-induced hemolysis and are more susceptible to iron overload‒induced ferroptosis upon consuming a high-iron diet (HID). Importantly, we found that treating Trf-LKO mice with the ferroptosis inhibitor ferrostatin-1 (Fer-1) and hepatocyte-specific knockout of the metal transporter Slc39a14 both significantly reduced iron overload‒induced liver ferroptosis. These results support the notion that hepatic Trf plays a protective role with respect to maintaining iron homeostasis, promoting erythropoiesis and liver function. In addition, these findings suggest that both the ferroptosis pathway and Slc39a14 are possible therapeutic targets for ferroptosis-induced liver damage.
Materials and methods
Patients and liver biopsies
Serum samples were obtained from 48 patients with liver cirrhosis and 52 healthy control subjects at the First Affiliated Hospital of Zhejiang University School of Medicine. Individual serum Trf levels were measured by using the immunoturbidimetric method (Roche Diagnostics), and serum alanine transaminase (ALT) levels were measured by using a Hitachi 704 Analyzer (Roche Diagnostics). Liver fibrosis was estimated with the Fibrosis-4 Index, a fluorescence immunoassay that includes hyaluronic acid, laminin, procollagen III, and collagen IV. The demographic and clinical characteristics in each group are summarized in supplemental Table 1 (available on the Blood Web site).
The use of biopsy material was approved by the Ethics Committee of the First Affiliated Hospital of Zhejiang University School of Medicine. Liver biopsy specimens were collected from 20 patients with liver cirrhosis and 20 noncancerous sections from patients with hepatocellular carcinoma (as a control group). Immunohistochemistry (IHC) was performed by using the following primary antibodies: anti-Transferrin (ab82411; Abcam), anti-Slc39a14 (TB2519862D; Thermo Fisher Scientific), and anti-Malondialdehyde (ab6463; Abcam).
Animal models
The gene-targeting strategy used to delete exons 4 and 5 in the mouse Trf gene is shown in supplemental Figure 1A, producing Trffl/+ offspring (Shanghai Biomodel Organism). Trffl/+ mice were then backcrossed to the C57BL/6J background for >10 generations. The resulting Trffl/+ mice were then crossed with either CMV-Cre or Alb-Cre transgenic mice to generate global Trf knockout (Trf-KO) and hepatocyte-specific Trf knockout (Trf-LKO) mice, respectively. The generation and characterization of hepatocyte-specific Slc39a14 knockout (Slc39a14-LKO) mice were previously described.18 Hepatocyte-specific Trf/Slc39a14 double-knockout (DKO) mice were generated by crossing Trf-LKO mice with Slc39a14-LKO mice.
All mice were fed a standard AIN-76A diet containing 50 mg/kg of iron (Research Diets, Inc.) unless stated otherwise. Age- and sex-matched mice were used in each treatment group. All animal experiments were approved by the Institutional Animal Care and Use Committee of Zhejiang University.
Animal treatments and related measurements
The following were performed as previously described: PHZ-induced hemolysis; iron dextran injection; HID treatment; Fer-1 treatment; carbon tetrachloride (CCl4)-induced fibrosis murine model; flow cytometry analysis; serum iron, tissue non-heme iron, and serum NTBI testing; Perls Prussian blue, ALT, Sirius Red, Masson’s trichrome, and 4-hydroxynonenal (4-HNE) staining; analysis of malondialdehyde (MDA), thiol, and superoxide dismutase (SOD) enzyme activity; reverse transcription polymerase chain reaction (RT-PCR); western blot analysis; IHC; and measurement of serum Trf and serum erythropoietin (Epo).15,19-26 Detailed information about these methods is available in the supplemental Methods.
Statistical analysis
All summary data are presented as the mean ± standard error of the mean. Groups were compared by using the Student t test or one-way analysis of variance with Tukey’s post hoc test, where appropriate. Differences with a P value <.05 were considered statistically significant. Categorical variables were compared by using the χ2 test. Pearson’s correlation coefficient (r) and P values were used to analyze correlations, and correlations were considered statistically significant at P < .05. The log-rank test was used to analyze the survival curve. All statistical analyses were performed by using SPSS version 21.0 (IBM SPSS Statistics, IBM Corporation).
Results
Trf-LKO mice have impaired erythropoiesis and altered iron metabolism
Given that Trf is expressed predominantly in the liver (supplemental Figure 1B), we generated Trf-LKO mice by crossing Trffl/fl mice with Albumin-Cre (Alb-Cre) transgenic mice. Loss of hepatic Trf expression was confirmed by using RT-PCR (supplemental Figure 1C), western blot analysis (Figure 1A), and Trf IHC (supplemental Figure 1D). We found that Trf-LKO mice are both viable (supplemental Figure 1E) and fertile but have significantly lower serum Trf levels (only 6% of normal) compared with control (Trffl/fl) littermates (Figure 1A). The Trf-LKO mice also have significantly lower levels of serum iron (Figure 1B), reduced red blood cell counts, reduced total hemoglobin, lower hematocrit values, and microcytic hypochromic anemia with reticulocytosis and thrombocytosis (supplemental Figure 2A-B). The fact that the platelet count increases in the circulation indicates iron-restricted hematopoiesis. The presence of these typical features associated with iron-deficient anemia in the Trf-LKO mice underscores the important role that liver-derived Trf plays in erythropoiesis.
Figure 1.
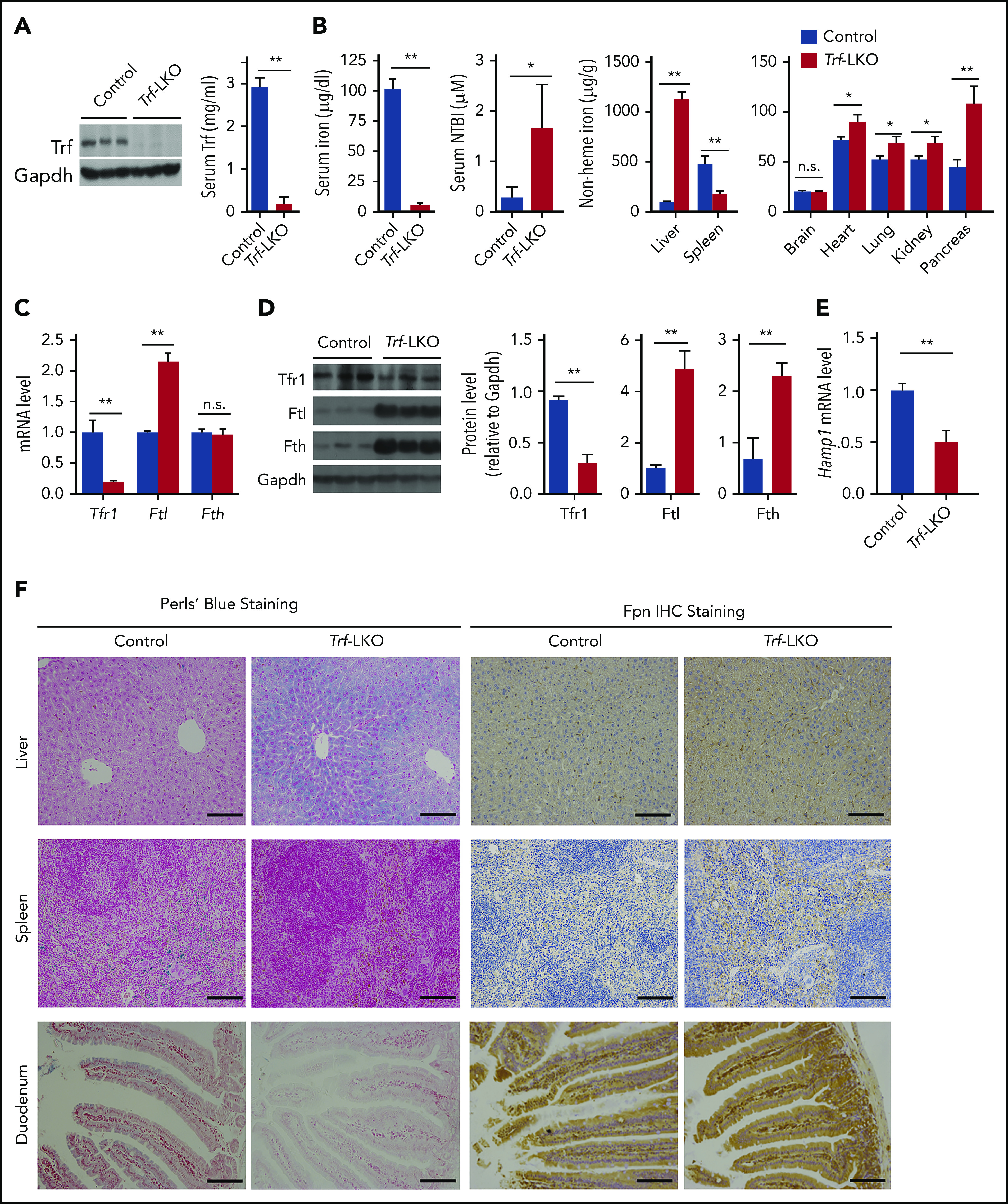
Trf-LKO mice have impaired iron metabolism. (A) Hepatic Trf protein and serum Trf levels were measured in 8-week-old control (Trffl/fl) and Trf-LKO (n = 6-7 mice/group) mice. (B) Serum iron levels, serum NTBI levels, and tissue non-heme iron concentrations were measured in 8-week-old control and Trf-LKO mice (n = 8-10 mice/group). (C) Hepatic Tfr1, Ftl, and Fth mRNA levels were measured in control and Trf-LKO mice using RT-PCR (n = 6 mice/group). (D) Western blot analysis of hepatic Tfr1, ferritin-L (Ftl), and ferritin-H (Fth) proteins in control and Trf-LKO mice (n = 3 mice/group). (E) RT-PCR analysis of hepatic Hamp mRNA in control and Trf-LKO mice (n = 6 mice/group). (F) Perls Prussian blue staining for iron and Fpn1 IHC were performed in liver, spleen, and duodenum sections obtained from control and Trf-LKO mice; the scale bars represent 100 μm. *P < .05, **P <. 01, Student t test. n.s., not significant.
To examine the role of hepatic Trf in the development of proerythroblasts into mature red blood cells, flow cytometry was performed by using the erythroid lineage markers Ter119 and CD44.27 We found that the cell number of polychromatophilic erythroblasts (R3) increased significantly, whereas orthochromatophilic erythroblasts (R4) and mature erythrocytes (R5) were significantly decreased in the bone marrow of Trf-LKO mice compared with control mice (supplemental Figure 2C). The reticulocyte count decreased in the bone marrow and increased in the circulation, which is consistent with normal compensatory responses to release reticulocytes into the circulation earlier in conditions of more severe anemia. In the spleen, mature erythrocytes decreased significantly but early-stage erythrocytes increased in Trf-LKO mice (supplemental Figure 2D), suggesting the impairment of erythropoiesis upon loss of hepatic Trf.
We also examined the role of hepatic Trf in iron recycling using PHZ-induced hemolytic anemia. Notably, even 8 days after PHZ injections, the Trf-LKO mice continued to have severe hypoferric anemia, whereas control mice had nearly fully recovered from the effects of PHZ by this time (supplemental Figure 3A). We also used the iron-sensitive fluorophore calcein-AM to measure intracellular iron levels and found that 8 days after PHZ treatment, the Trf-LKO mice had significantly lower levels of intracellular iron in the bone marrow compared with control mice (supplemental Figure 3B). This outcome is consistent with the notion that hepatic Trf serves as the principal source of systemic Trf for delivering iron to meet erythropoietic requirements in the bone marrow.
We next examined the putative role of hepatic Trf in iron metabolism. Trf-LKO mice had higher levels of serum NTBI (Figure 1B) and developed iron overload in a variety of tissues, including the liver, heart, lungs, kidneys, and pancreas. Strikingly, hepatic non-heme iron levels were ∼10-fold higher in Trf-LKO mice compared with control mice at 2 months of age. Consistent with hepatic iron overload, we found reduced expression of Tfr1 at both the messenger RNA (mRNA) and protein levels, increased ferritin-L expression at the mRNA and protein levels, and increased ferritin-H protein levels in the liver of Trf-LKO mice compared with control mice (Figure 1C-D).
Hepcidin, the master regulator of iron homeostasis, mediates iron through internalization and degradation of ferroportin (Fpn), the sole iron exporter. Because Trf-LKO mice exhibited hypoferric anemia, we measured Epo in the kidney and serum, and erythroferrone (Erfe) in bone marrow and spleen, and found significantly higher levels of Epo and Erfe in 8-week-old Trf-LKO mice (supplemental Figure 4A-B); this suggests decreased Epo-responsiveness in Trf-LKO mice. As a result, increased Erfe could suppress expression of hepcidin.28-30 As shown in Figure 1E, Hamp mRNA levels were significantly lower in Trf-LKO mice. Despite higher levels of iron accumulated in the liver of Trf-LKO mice compared with control mice (Figure 2A), Hamp mRNA levels in Trf-LKO mice remained significantly downregulated (supplemental Figure 4C), accompanied by higher levels of Epo mRNA levels in the kidney (supplemental Figure 4D) and higher Erfe mRNA levels in the bone marrow and spleen (supplemental Figure 4E).
Figure 2.
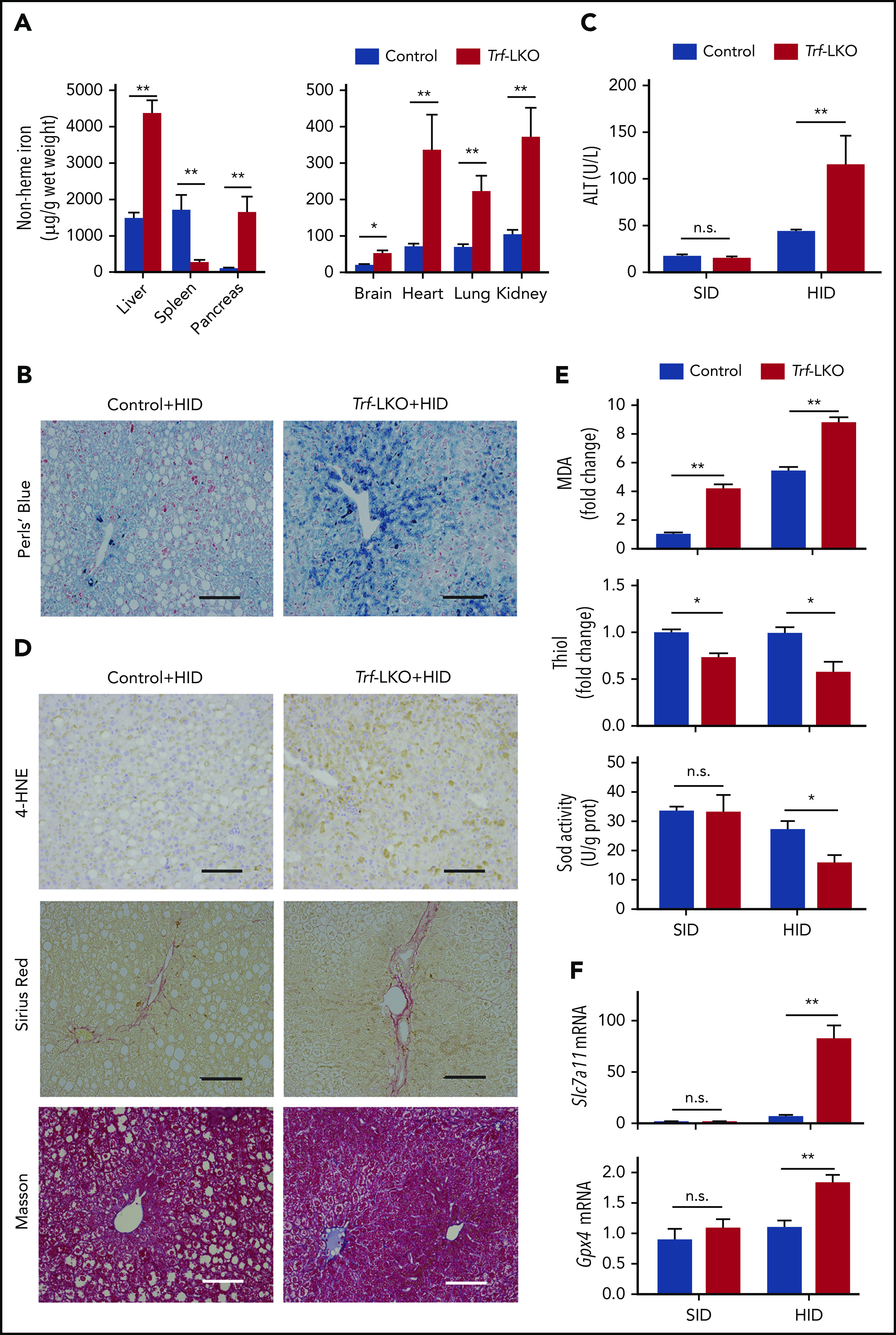
Loss of hepatic Trf exacerbates liver damage induced by an HID. (A) Non-heme iron was measured in the indicated tissues of control and Trf-LKO mice (n = 4 mice/group) after 8 weeks on an HID. (B) Perls Prussian blue staining of liver sections obtained from HID-fed control and Trf-LKO mice; the scale bars represent 100 μm. (C) Serum ALT levels were measured in control and Trf-LKO mice fed either an SID or an HID (n = 4 mice/group). (D) 4-HNE, Sirius Red, and Masson’s trichrome staining were performed on liver sections obtained from HID-fed control and Trf-LKO mice; the scale bars represent 100 μm. Hepatic MDA, thiol, and SOD activity (E) and hepatic Slc7a11 and Gpx4 mRNA (F) were measured in SID-fed and HID-fed control mice and Trf-LKO mice (n = 4 mice/group). *P < .05, **P < .01, Student t test. n.s., not significant.
In addition, we found that Fpn levels (measured by using both IHC and western blot analysis) were increased in the duodenum and spleen of Trf-LKO mice (Figure 1F; supplemental Figure 4F). This increased level of Fpn1 in the duodenum resulted in an increased absorption of dietary iron and subsequent efflux of iron into the blood, leading to iron accumulation in the liver of Trf-LKO mice; this action was confirmed by using Perls Prussian blue staining. Moreover, the increased levels of splenic Fpn1 in Trf-LKO mice accounts for the decrease in splenic iron measured by using Perls Prussian blue staining. Taken together, these data indicate that loss of hepatic Trf leads to iron overload in several organs, particularly the liver.
Trf-LKO mice have increased liver damage via iron overload‒induced ferroptosis
To further study the putative role of hepatic Trf in iron overload‒induced liver damage, we measured non-heme iron, MDA, and ALT levels in Trf-LKO mice and control mice fed a standard iron diet (SID group) or intraperitoneally injected with iron dextran, respectively. Hepatic non-heme iron levels (∼1200 μg/g tissue weight) and MDA changes were comparable between iron dextran loading control mice and Trf-LKO mice, but ALT levels did not differ among these 3 groups (supplemental Figure 5A-C). However, compared with age-matched (2, 6, and 12 months of age) control mice, Trf-LKO mice had significant hepatic iron overload and increased MDA levels (supplemental Figure 5D-E), and a significant, although slight, increase in ALT levels was noted in 12-month-old Trf-LKO mice (supplemental Figure 5F). Thus, loss of hepatic Trf eventually leads to liver damage due to chronic iron accumulation.
We recently reported that feeding mice an HID induces liver fibrosis and hepatic ferroptosis.15 We therefore examined the role of hepatic Trf in mice that were fed an HID for 8 weeks. Compared with HID-fed control mice, HID-fed Trf-LKO mice developed more severe iron overload in a variety of tissues, including the liver, pancreas, brain, heart, lungs, and kidneys (Figure 2A); in contrast, HID-fed Trf-LKO mice had significantly reduced iron levels in the spleen. The approximately threefold higher level of hepatic non-heme iron in HID-fed Trf-LKO mice compared with HID-fed control mice was supported by more intense Perls Prussian blue staining of liver sections (Figure 2B). Notably, compared with HID-fed control mice, HID-fed Trf-LKO mice developed more severe liver damage, reflected by increased levels of serum ALT (Figure 2C) and stronger staining of liver fibrosis markers, including Sirius Red and Masson’s trichrome staining (Figure 2D; supplemental Figure 6A-B).
We previously reported that ferroptosis underlies liver damage in mouse models of hemochromatosis.15 We therefore measured several putative biomarkers of ferroptosis, including lipid peroxidation‒derived 4-HNE staining,16,31 MDA levels,15,32 hepatic thiol levels,33,34 SOD enzyme activity,35 and glutathione peroxidase 4 (Gpx4)36,37 and Slc7a1115,38 mRNA levels, in both control and Trf-LKO mice fed either an HID or SID. We found that in addition to having more severe iron overload, HID-fed Trf-LKO mice had higher levels of 4-HNE compared with HID-fed control mice (Figure 2D). Moreover, both SID-fed and HID-fed Trf-LKO mice had higher hepatic MDA levels and lower hepatic thiol levels compared with their respective control groups (Figure 2E). In addition, SOD activity was significantly decreased in HID-fed Trf-LKO mice compared with HID-fed control mice but was similar between SID-fed Trf-LKO mice and SID-fed control mice. Finally, the levels of both Gpx4 mRNA and Slc7a11 mRNA were significantly increased in HID-fed Trf-LKO mice compared with HID-fed control mice but were similar between SID-fed Trf-LKO mice and SID-fed control mice (Figure 2F). Similarly, we previously showed that Slc7a11 expression could be upregulated by iron.15 Taken together, these results indicate that loss of hepatic Trf contributes to ferroptosis-induced liver fibrosis.
Blocking ferroptosis reduces HID-induced liver fibrosis in Trf-LKO mice
We next examined whether Fer-1, a specific inhibitor of ferroptosis,13 could reduce liver fibrosis in HID-fed Trf-LKO mice. We found that treating both HID-fed control mice and HID-fed Trf-LKO mice with Fer-1 (1 mg/kg per day) had no effect on hepatic non-heme iron levels (Figure 3A). Nevertheless, treating HID-fed Trf-LKO mice with Fer-1 reduced ALT levels to control (ie, nontreated) levels (Figure 3B). In addition, treating Trf-LKO mice with Fer-1 significantly decreased MDA levels (Figure 3C), increased thiol levels (Figure 3D), decreased both 4-HNE and liver fibrosis measured by using Sirius Red and Masson’s trichrome staining (Figure 3E-F), and decreased hepatic mRNA levels of the fibrotic genes Col1a1 and PDGF (Figure 3G) compared with the respective nontreated HID-fed Trf-LKO groups. These results suggest that due to their increased susceptibility to iron accumulation, Trf-LKO mice are more vulnerable to ferroptosis-induced liver fibrosis, and Fer-1 can significantly reduce these in vivo effects.
Figure 3.
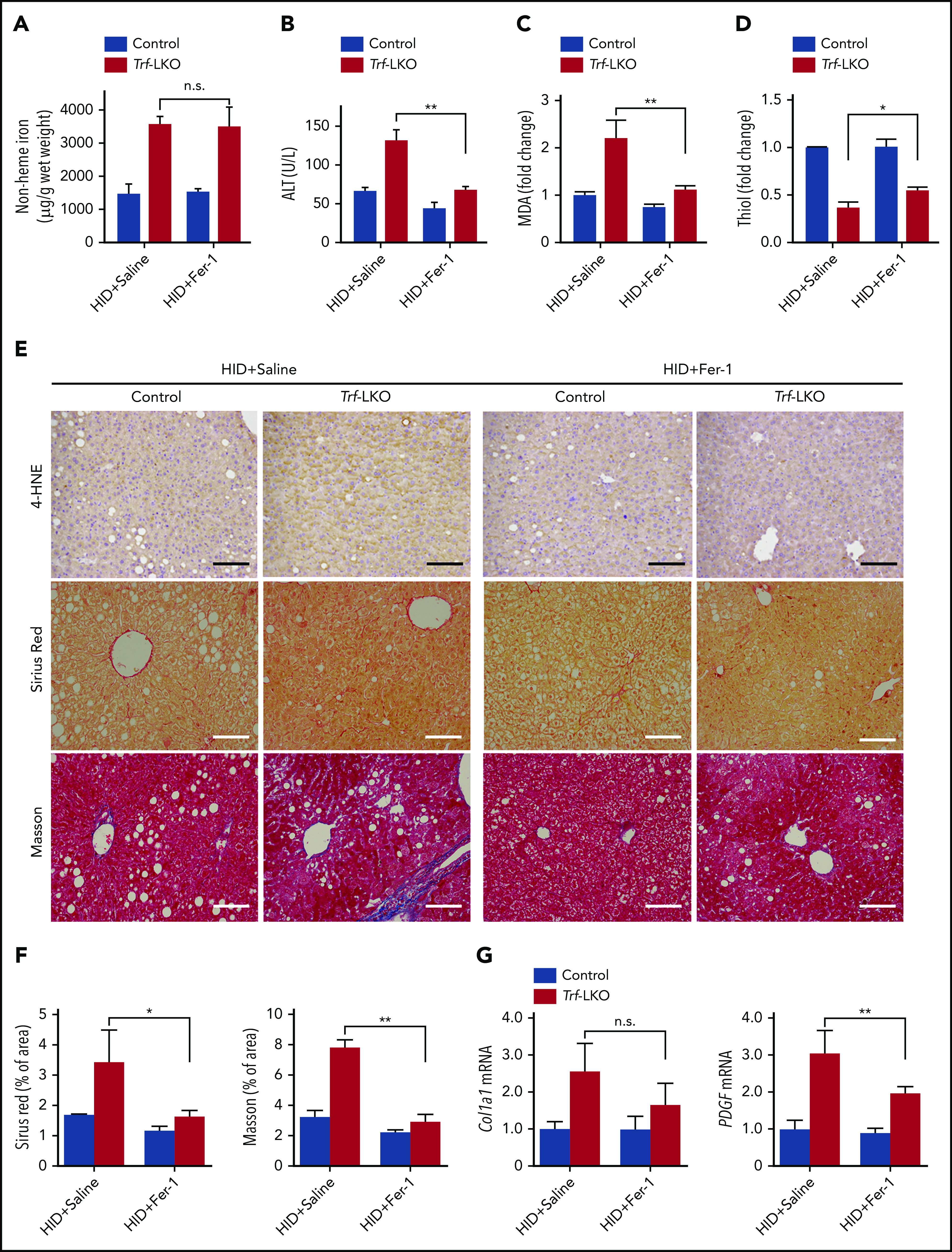
Fer-1 treatment rescues iron overload‒induced ferroptosis in Trf-LKO mice. Hepatic non-heme iron (A), serum ALT (B), hepatic MDA (C), and hepatic thiol (D) levels were measured in HID-fed control mice and Trf-LKO mice treated with saline or Fer-1 (n = 3-6 mice/group). (E) Liver sections obtained from HID-fed control and Trf-LKO mice treated with saline or Fer-1 were stained with 4-HNE, Sirius Red, and Masson’s trichrome; the scale bars represent 100 μm. (F) Quantitative analyses for Sirius red and Masson’s trichrome staining in the indicated groups. (G) Hepatic mRNA levels of the fibrotic genes Col1a1 and PDGF were measured in the indicated groups. *P < .05, **P < .01, one-way analysis of variance with Tukey’s post hoc test. n.s., not significant.
Loss of hepatic Slc39a14 protects against ferroptosis-induced liver fibrosis in Trf-LKO mice
Having shown that iron accumulation in the liver causes ferroptosis-induced fibrosis in Trf-LKO mice, we next investigated the route through which iron enters hepatocytes. Jenkitkasemwong et al39 previously suggested that Slc39a14 might serve as a possible hepatic transporter for NTBI in mouse models of hereditary hemochromatosis. To test whether hepatic Slc39a14 serves as the transporter of NTBI and accelerates iron-induced liver damage in Trf-LKO mice, we crossed Trf-LKO mice with Slc39a14-LKO mice, thereby generating offspring that lack both Trf and Slc39a14 specifically in their hepatocytes (ie, Trf/Slc39a14 double-knockout, or simply DKO, mice). Consistent with our previous findings,18 Slc39a14-LKO mice had tissue iron levels similar to those in control mice, suggesting Slc39a14 is not a major iron transporter under physiological conditions (supplemental Figure 7A-G). However, we found that hepatic iron levels were significantly lower in DKO mice compared with Trf-LKO mice when fed an SID (Figure 4A-B), indicating that Slc39a14 functions as the hepatic transporter of NTBI in the absence of Trf. In addition, DKO mice showed higher iron levels in the brain, heart, lung, kidney, and pancreas compared with Trf-LKO mice (supplemental Figure 7B-C, and E-G), which suggests that NTBI entered in extrahepatic tissues. Interestingly, HID-fed DKO mice had lower levels of hepatic non-heme iron (Figure 4C) and higher levels of hepatic thiol (Figure 4D) compared with HID-fed Trf-LKO mice. Moreover, HID-fed DKO mice had lower levels of hepatic MDA (Figure 4E) and 4-HNE (Figure 4F) compared with HID-fed Trf-LKO mice. Thus, the loss of hepatic Slc39a14 in the context of Trf-LKO mice reduces iron accumulation, alleviates ferroptosis-triggered liver fibrosis when fed an HID (supplemental Figure 8A-B), and reduces hepatic Col1a1 and PDGF expression (supplemental Figure 8C-D). In summary, absence of hepatic Slc39a14 may protect HID-fed Trf-LKO mice from ferroptosis-induced liver fibrosis, suggesting that hepatic Slc39a14 might serve as a possible therapeutic target for preventing ferroptosis-induced liver damage in the absence of hepatic Trf.
Figure 4.
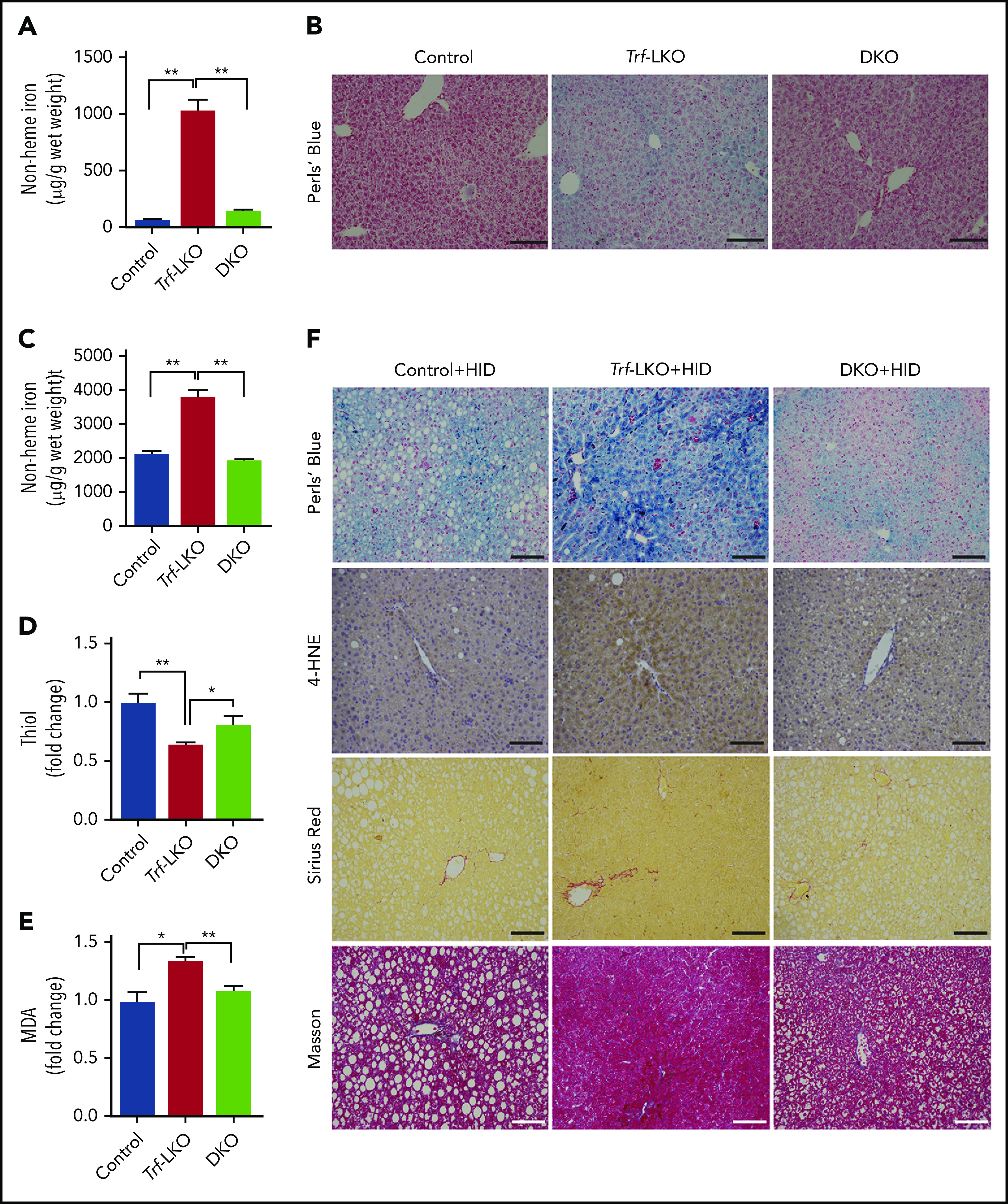
Loss of hepatic Slc39a14 protects against ferroptosis in Trf-LKO mice. (A) Hepatic non-heme iron was measured in control (Trffl/fl), Trf-LKO, and Trf/Slc39a14 double-knockout (DKO) mice fed an SID (n = 4 mice/group). (B) Representative images of Perls Prussian blue‒stained liver sections obtained from SID-fed control, Trf-LKO, and DKO mice; the scale bars represent 100 μm. Hepatic non-heme iron (C), thiol (D), and MDA (E) levels were measured in HID-fed control, Trf-LKO, and DKO mice (n = 4 mice/group). (F) Representative images of liver sections obtained from HID-fed control, Trf-LKO, and DKO mice and stained with Perls Prussian blue, 4-HNE, Sirius Red, and Masson’s trichrome; the scale bars represent 100 μm. *P < .05 and **P < .01, one-way analysis of variance with Tukey’s post hoc test.
Trf-LKO mice are susceptible to CCl4-induced fibrosis
Approximately one-third of patients with end-stage liver disease present with hepatic iron overload.12 Nevertheless, even though the toxicity of excess iron could be attributed to oxidative stress, its precise role in the pathway of liver fibrogenesis remains unclear. To investigate further the role of hepatic Trf in the classical pathogenesis of liver fibrosis, we used a mouse model in which liver fibrosis was induced by repeated injections of CCl4. Interestingly, we found that Trf-LKO mice chronically exposed to CCl4 were more susceptible to fibrosis than both vehicle-treated Trf-LKO mice and CCl4-injected control littermates (Figure 5A-B; supplemental Figure 9). In addition, we found that apo-transferrin supplementation could partially rescue liver fibrosis (supplemental Figures 10 and 11), anemia, and accumulated NTBI in Trf-LKO mice upon CCL4 treatment.
Figure 5.
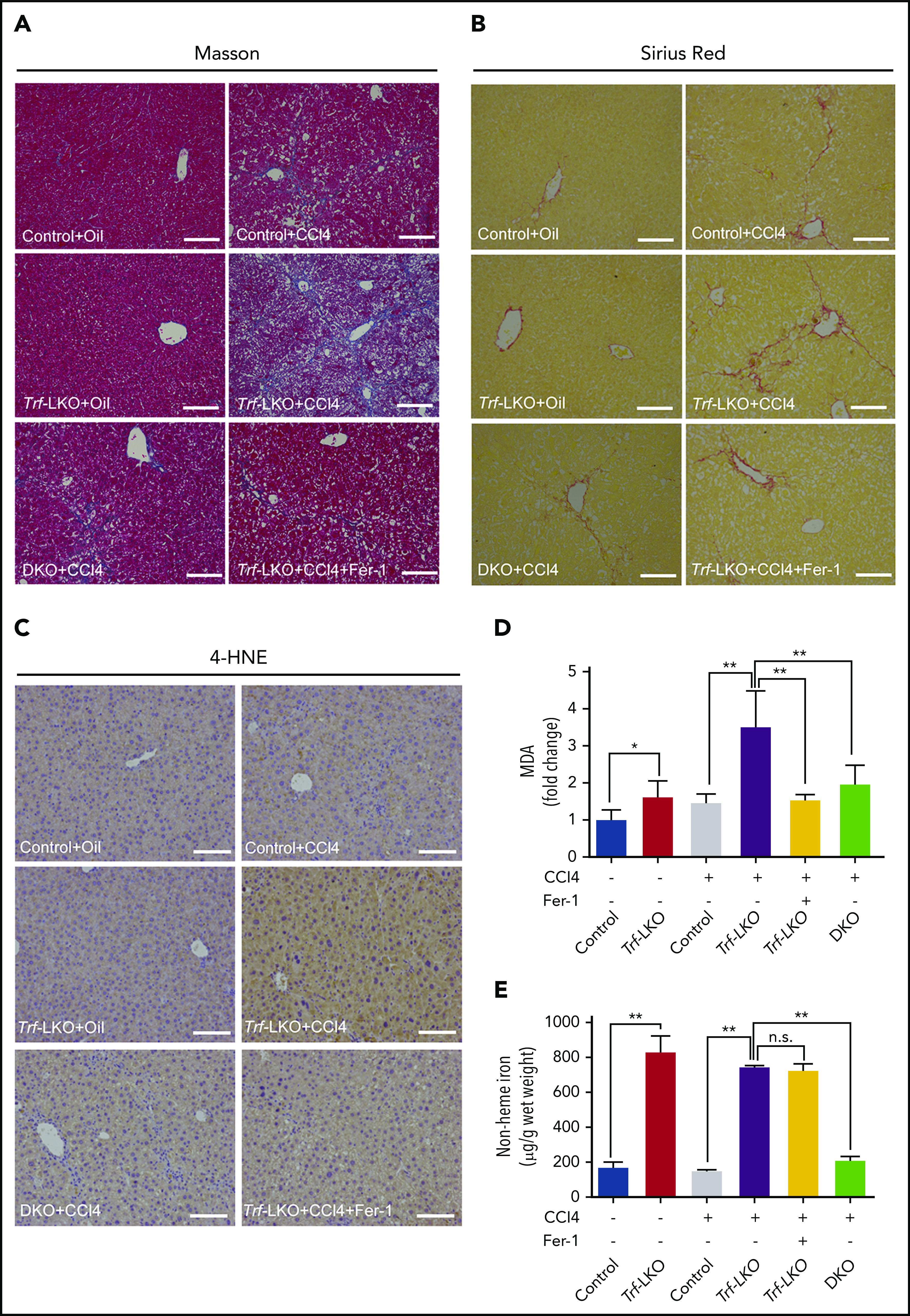
Both inhibiting ferroptosis and deleting hepatic Slc39a14 alleviates CCl4-induced fibrosis in Trf-LKO mice. Control, Trf-LKO, and DKO mice were treated with vehicle (oil) or CCl4, and liver sections were obtained and stained with Masson’s trichrome (A), Sirius Red (B), or 4-HNE (C); the scale bars represent 100 μm. Hepatic MDA (D) and non-heme iron (E) were measured in the indicated mice (n = 4-5 mice/group). *P < .05, **P < .01, one-way analysis of variance with Tukey’s post hoc test. n.s., not significant.
Notably, hepatic ferroptosis was significantly increased in CCl4-injected Trf-LKO mice and rescued by treating these mice with Fer-1 (Figure 5C-D). Consistent with decreased levels of hepatic iron (Figure 5E), the loss of hepatic Slc39a14 in DKO mice also reduced liver fibrosis compared with Trf-LKO mice. Taken together, these results indicate that Trf-LKO mice are highly susceptible to CCl4-induced liver fibrosis, and both Fer-1 treatment and loss of hepatic Slc39a14 can reduce this effect.
Serum Trf levels are significantly lower in patients with liver cirrhosis
To investigate the clinical relevance of our results obtained with animal models, we next collected serum samples from 48 adult patients (40-83 years of age) with nonviral liver cirrhosis and from 52 age- and sex-matched healthy control subjects. We found that serum Trf levels were significantly lower in the patient group compared with the control group (Figure 6A), consistent with previous reports based on virus-infected patients with cirrhosis.40-44 Moreover, 4 clinical indicators of hepatic fibrosis (hyaluronic acid, laminin, type III collagen, and type IV collagen) were measured in the patient group, and all 4 indicators were found to be inversely correlated with Trf levels (Figure 6B-E). Of note, the correlation of low Trf with poor outcome could be a consequence of worsening disease because Trf is primarily synthesized by the liver. However, together with the recent report by Viveiros et al7 that Trf deficiency is associated with poor prognosis in patients with liver cirrhosis, these data suggest that serum Trf likely plays a role in this disease.
Figure 6.

Patients with liver cirrhosis have reduced serum Trf levels. (A) Trf was measured in serum samples obtained from 48 patients with cirrhosis and 52 healthy control subjects. (B-E) The indicated indices of liver fibrosis were measured in the serum of 48 patients with liver cirrhosis and are plotted against serum Trf concentration. The data in panel A were analyzed by using a Student t test; the data in panels B-E were analyzed by using Pearson’s correlation coefficient (r).
Patients with hepatic cirrhosis have increased levels of hepatic iron and lipid peroxidation
Lastly, we measured hepatic iron levels and ferroptosis markers in liver biopsy samples obtained from 20 patients with liver cirrhosis; as a control, healthy liver tissue samples were also obtained from patients who underwent hepatocellular carcinoma resection. Consistent with our results obtained with Trf-LKO mice shown in Figure 1, we found lower levels of hepatic Trf in biopsy samples from patients with cirrhosis compared with control samples (Figure 7A). Moreover, the biopsy specimens from patients with cirrhosis contained higher levels of iron deposits and stronger staining for MDA (Figure 7B-C). Notably, the biopsy samples from patients with cirrhosis also had significantly stronger hepatic SLC39A14 staining, which is consistent with our finding of strong hepatic Slc39a14 staining in Trf-LKO mice compared with control mice and in both HID-fed control mice and HID-fed Trf-LKO mice (Figure 7D-E).
Figure 7.
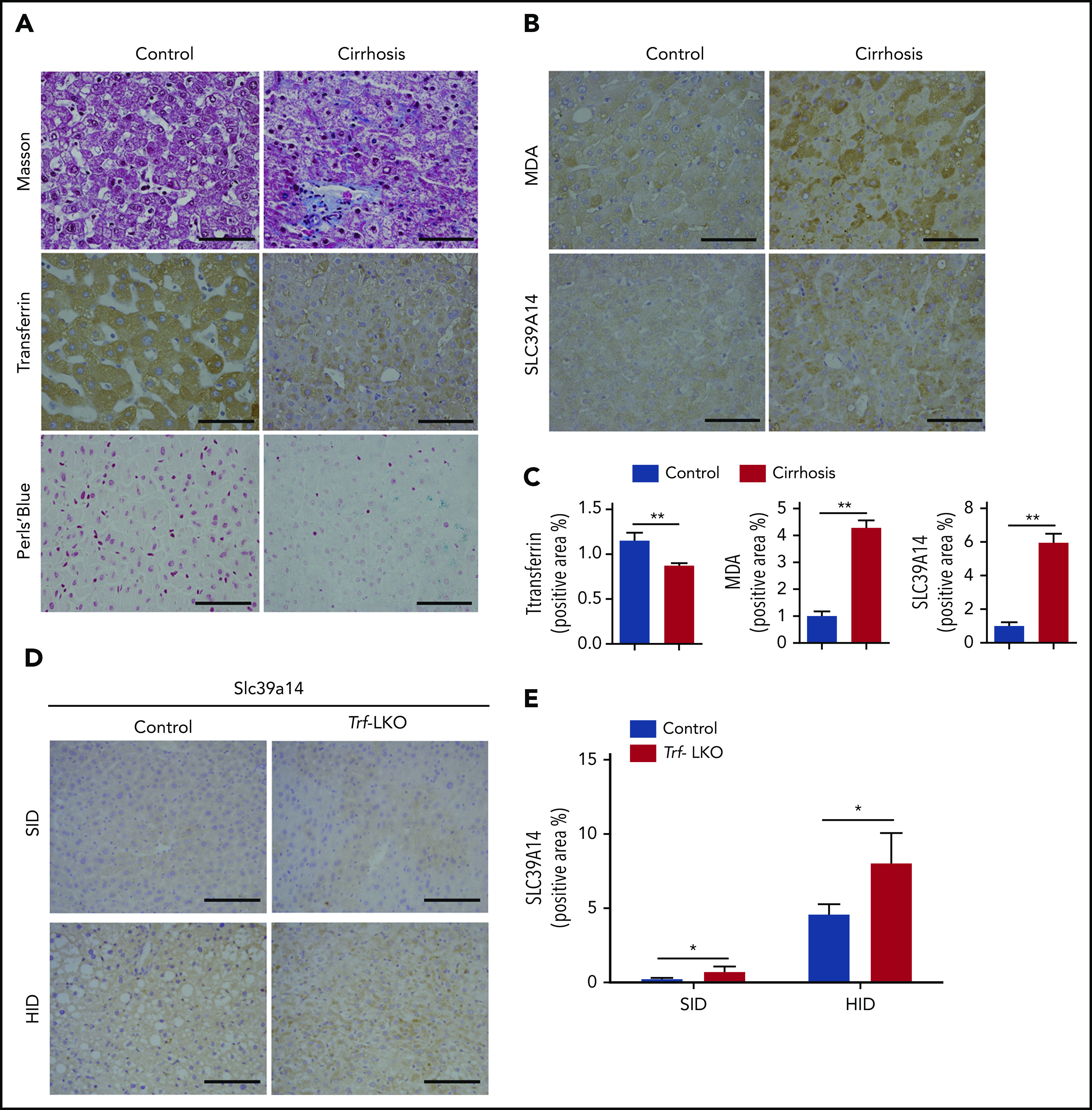
Hepatic Trf, iron, MDA, and SLC39A14 levels in liver biopsy samples obtained from patients with liver cirrhosis and in Trf-LKO mice. Liver biopsy samples were obtained from 20 patients with liver cirrhosis and 20 control subjects who underwent resection for hepatocellular carcinoma. (A,B) Representative images of liver sections stained with Masson’s trichrome, Trf IHC, Perls Prussian blue, MDA IHC, and SLC39A14 IHC. (C) Quantitative analyses of Trf, MDA, and SLC39A14 staining of liver sections obtained from control and cirrhotic biopsy samples. (D) Representative images of murine liver sections stained with Slc39a14 obtained from 12-week-old SID-fed and HID-fed control mice and Trf-LKO mice. The scale bars represent 100 μm. (E) Quantitative analyses of hepatic Slc39a14 staining from 12-week-old SID-fed and HID-fed control mice and Trf-LKO mice. *P < .05, **P < .01, Student t test.
Discussion
Excessive iron has long been postulated as a risk factor for developing liver fibrosis and cirrhosis. However, the precise role of iron regulatory proteins in liver fibrosis has not been defined. Here, we characterized the functional role of hepatic Trf in iron metabolism and its implications for the iron overload–induced pathogenesis of liver fibrosis and cirrhosis.
The hypotransferrinemic (Hpx) mouse model, which was first described in 1987, carries a point mutation in the Trf gene.1,8,45,46 However, despite their clear value in studying Trf, there are several major different characteristics between our newly generated conditional Trf- KO mice (eg, Trf-LKO) and Hpx mice. First, the murine models are genetically different. Hpx mice presented more severe serum Trf deficiency (<1% of the normal levels), whereas our Alb-Cre–driven Trf depletion mouse model specially deletes Trf in hepatocytes, resulting in ∼6% of circulating Trf of the normal levels. Our data suggest that the liver is the principal source of Trf in mice. Second, the 2 models are phenotypically different. The Hpx mice presented severe anemia, and they cannot survive unless Trf is exogenously administered during the first 3 weeks following birth. In comparison, our hepatocyte-specific Trf null mice are viable even without Trf supplementation. Notably, our Trf-LKO mice displayed less severe anima, suggesting that extrahepatic-produced Trf could at least partially compensate for loss of hepatic Trf. Moreover, expression levels of the iron regulatory hormone hepcidin significantly differ between these 2 mouse lines. Hepcidin deficiency is one of the major characteristics of the Hpx mouse,47 whereas our hepatocyte-specific Trf null mice (8 weeks old) had 50% of hepcidin compared with their littermate controls (Figure 1E). Most importantly, our conditional Trf-KO mouse model allows precise dissection of tissue-specific production and function of Trf.
Considering that the global Trf-deficient (Trf-KO) mice died within 1 day of birth (supplemental Figure 12), whereas Trf-LKO mice are viable, it is interesting to further explore extrahepatic source of Trf. Previous reports suggest Trf could be produced by brain48,49 and mammary gland.1,50 Our qPCR results indicate that Trf is mainly expressed in the liver, white adipose, and lung in mice. Future studies are needed for dissecting the role of extrahepatic sources of Trf.
In the normal physiological condition, serum iron binds to Trf. When serum iron levels exceed the buffering capacity of Trf (∼75% of Trf saturation), NTBI accumulates.51 Given the almost complete Trf (94%) deficiency in Trf-LKO mice, the TBI in serum is low, and thus the predominant form of circulating iron in Trf-LKO mice is NTBI. Previous studies have shown that NTBI in the plasma is rapidly and efficiently taken up mostly by the liver, and to a lesser extent by the pancreas, kidney, and heart.10 The profound tissue iron overload observed in Trf-LKO mice could be attributed to tissue uptake of NTBI.
Phenotypes observed in Trf-LKO mice could be mechanistically attributed to Trf deficiency and hepcidin suppression. Liver-selective loss of Trf leads to microcytic anemia because of insufficient serum iron delivery to the bone marrow for erythropoiesis, resulting in iron-restricted erythropoiesis and decreased Epo-responsiveness. Moreover, the reticulocyte count decreasing in the bone marrow and increasing in the circulation is consistent with normal compensatory responses to release reticulocytes into the circulation earlier in more severe anemia. As a result, Erfe expression is upregulated, which in turn suppresses hepcidin expression,28,29,52 followed by increased dietary iron absorption from small intestinal enterocytes via ferroportin-1.2 Eventually, iron accumulates in the liver of Trf-LKO mice, which in turn leads to downregulation of Tfr1 and upregulation of protein levels of ferritin. This scenario is mainly because both Tfr1 and ferritin genes are posttranscriptionally regulated by the iron regulatory protein/iron-responsive element system through the iron-responsive elements at the 3′-untranslated region of the Tfr1 gene and the 5′-untranslated region of the ferritin gene in an iron-dependent manner.
Iron-dependent ferroptosis is characterized by a reduced glutathione level and increased lipid peroxidation.14,16,53,54 A large number of studies have shown that ferroptosis plays a role in neurotoxicity,13,55 cancer,38,56 ischemia-reperfusion injury,16,36,57 renal damage,55,58 and iron metabolism‒related diseases15; however, the role of ferroptosis in liver fibrosis and cirrhosis is unclear. Here, we suggest 3 features that are specific to the iron overload‒induced form of ferroptosis in the liver. First, iron overload can initiate ferroptosis, as we found that 8 weeks on an HID is sufficient to induce ferroptosis-induced liver injury in control mice. Second, Trf protects against liver fibrosis by buffering ferric iron (Fe3+), as Trf-LKO mice are more susceptible to HID-induced liver damage. Nevertheless, our in vivo results differ from those of a previous in vitro study, which found that Trf and the amino acid glutamine induce ferroptosis in an in vitro system.57 In contrast, our in vivo mouse results suggest that Trf protects against excessive NTBI-triggered ferroptosis in the liver. Third, in the absence of Trf, Slc39a14 mediates ferroptosis-induced liver fibrosis by transporting massive amounts of NTBI into hepatocytes under high dietary iron conditions and CCl4 injections, supporting the notion that Slc39a14 promotes NTBI-induced ferroptosis.
Ferroptosis-induced liver fibrosis is initiated by the accumulation of free non-heme iron in liver tissue. In the circulation, Trf carries Fe3+ to the tissues for utilization. When Trf saturation exceeds 75%, NTBI progressively accumulates in the tissues, causing tissue damage and giving rise to pathological conditions such as β-thalassemia and hemochromatosis.51 However, precisely how NTBI enters various cell types remains unclear. Thus, our tissue-specific Trf knockout mice provide an ideal animal model for addressing this question. Previously, our group and others reported that hepatic Slc39a14 functions as a manganese transporter under physiological conditions.18,59,60 Here, we report that Slc39a14 can transport iron into hepatocytes in the absence of Trf, as deleting hepatic Slc39a14 in Trf-LKO mice reduced liver fibrosis in Trf-LKO mice induced by either an HID or CCl4 injections. These findings suggest that Slc39a14 might serve as a possible therapeutic target for preventing ferroptosis-induced liver fibrosis in the absence of hepatic Trf.
From a clinical perspective, 4 clinical indicators of hepatic fibrosis (hyaluronic acid, laminin, type III collagen, and type IV collagen) were found to be inversely correlated with Trf levels, with intermediate strength, in a cohort of liver cirrhosis patients using multiple linear regression analysis; this finding suggests that serum Trf might have additional value for better predicting the prognosis of liver cirrhosis. Nevertheless, the correlation of low Trf with poor outcome could likely be a consequence of worsening disease. Future studies are warranted for clinically testing the implication of Trf in the progress of liver diseases. It is interesting to note that liver biopsy specimens from patients with congenital atransferrinemia often reveal hepatic iron deposits and liver fibrosis.61,62 Conversely, we found that patients with liver cirrhosis have reduced levels of Trf. In addition, low serum Trf levels have been associated with poor prognosis in patients with cirrhosis,4,7 suggesting that Trf might serve as a prognostic marker for liver cirrhosis. Importantly, we found that patients with cirrhosis and low Trf levels have more iron deposits in the liver, as well as higher levels of lipid peroxidation‒derived MDA, which suggests that ferroptosis-induced fibrosis might play a pathogenic role in the development of liver cirrhosis. Interestingly, we also found that patients with cirrhosis have significantly higher levels of hepatic SLC39A14 compared with control subjects. Although the underlying molecular mechanism warrants further investigation, one possibility is that excessive hepatic iron due to reduced hepatic Trf levels may cause an increase in hepatic SLC39A14. Consistent with this hypothesis, Zhao et al63 reported that the plasma membrane metal transporter ZIP14 (also known as SLC39A14) is regulated by iron via the glycosylation-dependent proteasomal degradation pathway.
In conclusion, we report that hepatic Trf plays an essential role in regulating systemic iron metabolism and liver function. This conclusion is based on 3 key findings. First, hepatic Trf maintains iron homeostasis. Second, we identified 3 characteristic features of iron-induced ferroptosis associated with the pathogenesis of liver fibrosis. Finally, we found that using the ferroptosis inhibitor Fer-1 and deleting hepatic Slc39a14 can rescue ferroptosis-induced liver fibrosis in hepatic Trf knockout mice. Given that excessive hepatic iron deposits in patients with cirrhosis have been associated with a significantly increased risk of liver failure, cardiac complications, and the development of liver cancer, further clinical studies are warranted to better understand the protective role that Trf plays in the pathogenesis of ferroptosis-induced liver fibrosis and cirrhosis, and to test the feasibility of targeting ferroptosis and/or iron overload for treating liver cirrhosis.
Supplementary Material
The online version of this article contains a data supplement.
Acknowledgments
The authors thank Zhaohui Yu (The First Affiliated Hospital, Zhejiang University School of Medicine) and the members of the Wang and Min laboratories for helpful discussions.
This study was supported by research grants from the National Natural Science Foundation of China (31530034 and 31930057, F.W.; 31570791, J.M.; 91542205, S.Z.; 31701034, Q.W.; and 31701035, H.W.), the National Key Research and Development Program of China (2018YFA0507802, F.W., 2018YFA0507801, J.M.), and the China Postdoctoral Science Foundation (2019M650139, X.F.; 2019M662028, L.J.).
Footnotes
Original data requests should be submitted via e-mail to the corresponding author Fudi Wang (fwang@zju.edu.cn).
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: Y.Y., L.J., S.Z., J.M., and F.W. designed the experiments and drafted the manuscript; Y.Y. and L.J. performed the statistical analyses; Y.Y., L.J., H.W., S.Z., J.M., and F.W. revised the manuscript; F.W., J.M., and S.Z. obtained funding and supervised the study. All authors acquired and analyzed the data, and all authors approved the final version of the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Fudi Wang, 866 Yuhangtang Rd, Zhejiang University School of Medicine, Hangzhou 310058, China; e-mail: fwang@zju.edu.cn or Junxia Min, 866 Yuhangtang Rd, Zhejiang University School of Medicine, Hangzhou 310058, China; e-mail: junxiamin@zju.edu.cn; or Shusen Zheng, 866 Yuhangtang Rd, Zhejiang University School of Medicine, Hangzhou 310058, China; e-mail: zyzss@zju.edu.cn.
REFERENCES
- 1.Huggenvik JI, Craven CM, Idzerda RL, Bernstein S, Kaplan J, McKnight GS. A splicing defect in the mouse transferrin gene leads to congenital atransferrinemia. Blood. 1989;74(1):482-486. [PubMed] [Google Scholar]
- 2.Muckenthaler MU, Rivella S, Hentze MW, Galy B. A red carpet for iron metabolism. Cell. 2017;168(3):344-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Parrow NL, Li Y, Feola M, et al. Lobe specificity of iron binding to transferrin modulates murine erythropoiesis and iron homeostasis. Blood. 2019;134(17):1373-1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bruns T, Nuraldeen R, Mai M, et al. Low serum transferrin correlates with acute-on-chronic organ failure and indicates short-term mortality in decompensated cirrhosis. Liver Int. 2017;37(2):232-241. [DOI] [PubMed] [Google Scholar]
- 5.Cho HJ, Kim SS, Ahn SJ, et al. Serum transferrin as a liver fibrosis biomarker in patients with chronic hepatitis B. Clin Mol Hepatol. 2014;20(4):347-354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maras JS, Maiwall R, Harsha HC, et al. Dysregulated iron homeostasis is strongly associated with multiorgan failure and early mortality in acute-on-chronic liver failure. Hepatology. 2015;61(4):1306-1320. [DOI] [PubMed] [Google Scholar]
- 7.Viveiros A, Finkenstedt A, Schaefer B, et al. Transferrin as a predictor of survival in cirrhosis. Liver Transpl. 2018;24(3):343-351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Craven CM, Alexander J, Eldridge M, Kushner JP, Bernstein S, Kaplan J. Tissue distribution and clearance kinetics of non-transferrin-bound iron in the hypotransferrinemic mouse: a rodent model for hemochromatosis. Proc Natl Acad Sci U S A. 1987;84(10):3457-3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ganz T. Systemic iron homeostasis. Physiol Rev. 2013;93(4):1721-1741. [DOI] [PubMed] [Google Scholar]
- 10.Bradbury MW, Raja K, Ueda F. Contrasting uptakes of 59Fe into spleen, liver, kidney and some other soft tissues in normal and hypotransferrinaemic mice. Influence of an antibody against the transferrin receptor. Biochem Pharmacol. 1994;47(6):969-974. [DOI] [PubMed] [Google Scholar]
- 11.Wang CY, Knutson MD. Hepatocyte divalent metal-ion transporter-1 is dispensable for hepatic iron accumulation and non-transferrin-bound iron uptake in mice. Hepatology. 2013;58(2):788-798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ludwig J, Hashimoto E, Porayko MK, Moyer TP, Baldus WP. Hemosiderosis in cirrhosis: a study of 447 native livers. Gastroenterology. 1997;112(3):882-888. [DOI] [PubMed] [Google Scholar]
- 13.Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060-1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stockwell BR, Friedmann Angeli JP, Bayir H, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171(2):273-285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang H, An P, Xie E, et al. Characterization of ferroptosis in murine models of hemochromatosis. Hepatology. 2017;66(2):449-465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fang X, Wang H, Han D, et al. Ferroptosis as a target for protection against cardiomyopathy. Proc Natl Acad Sci U S A. 2019;116(7):2672-2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bacon BR, Britton RS. The pathology of hepatic iron overload: a free radical—mediated process? Hepatology. 1990;11(1):127-137. [DOI] [PubMed] [Google Scholar]
- 18.Xin Y, Gao H, Wang J, et al. Manganese transporter Slc39a14 deficiency revealed its key role in maintaining manganese homeostasis in mice. Cell Discov. 2017;3(1):17025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang Z, Zhang F, An P, et al. Ferroportin1 deficiency in mouse macrophages impairs iron homeostasis and inflammatory responses. Blood. 2011;118(7):1912-1922. [DOI] [PubMed] [Google Scholar]
- 20.Muto Y, Nishiyama M, Nita A, Moroishi T, Nakayama KI. Essential role of FBXL5-mediated cellular iron homeostasis in maintenance of hematopoietic stem cells. Nat Commun. 2017;8(1):16114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang Z, Zhang F, Guo X, An P, Tao Y, Wang F. Ferroportin1 in hepatocytes and macrophages is required for the efficient mobilization of body iron stores in mice. Hepatology. 2012;56(3):961-971. [DOI] [PubMed] [Google Scholar]
- 22.Gosriwatana I, Loreal O, Lu S, Brissot P, Porter J, Hider RC. Quantification of non-transferrin-bound iron in the presence of unsaturated transferrin. Anal Biochem. 1999;273(2):212-220. [DOI] [PubMed] [Google Scholar]
- 23.Delima RD, Chua AC, Tirnitz-Parker JE, et al. Disruption of hemochromatosis protein and transferrin receptor 2 causes iron-induced liver injury in mice. Hepatology. 2012;56(2):585-593. [DOI] [PubMed] [Google Scholar]
- 24.Li H, Rybicki AC, Suzuka SM, et al. Transferrin therapy ameliorates disease in beta-thalassemic mice. Nat Med. 2010;16(2):177-182. [DOI] [PubMed] [Google Scholar]
- 25.An P, Wang H, Wu Q, et al. Elevated serum transaminase activities were associated with increased serum levels of iron regulatory hormone hepcidin and hyperferritinemia risk. Sci Rep. 2015;5(1):13106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Magdaleno F, Arriazu E, Ruiz de Galarreta M, et al. Cartilage oligomeric matrix protein participates in the pathogenesis of liver fibrosis. J Hepatol. 2016;65(5):963-971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang S, He X, Wu Q, et al. Transferrin receptor 1-mediated iron uptake plays an essential role in hematopoiesis[published online ahead of print 10 October 2019] . Haematologica. doi:10.3324/haematol.2019.224899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kautz L, Jung G, Valore EV, Rivella S, Nemeth E, Ganz T. Identification of erythroferrone as an erythroid regulator of iron metabolism [published correction appears in Nat Gene. 2020;52(4):463]. Nat Genet. 2014;46(7):678-684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kautz L, Jung G, Du X, et al. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of β-thalassemia. Blood. 2015;126(17):2031-2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Arezes J, Foy N, McHugh K, et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood. 2018;132(14):1473-1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Breitzig M, Bhimineni C, Lockey R, Kolliputi N. 4-Hydroxy-2-nonenal: a critical target in oxidative stress? Am J Physiol Cell Physiol. 2016;311(4):C537-C543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425-1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carlson BA, Tobe R, Yefremova E, et al. Glutathione peroxidase 4 and vitamin E cooperatively prevent hepatocellular degeneration. Redox Biol. 2016;9:22-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Telorack M, Meyer M, Ingold I, Conrad M, Bloch W, Werner S. A glutathione-Nrf2-thioredoxin cross-talk ensures keratinocyte survival and efficient wound repair. PLoS Genet. 2016;12(1):e1005800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Galadari S, Rahman A, Pallichankandy S, Thayyullathil F. Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radic Biol Med. 2017;104:144-164. [DOI] [PubMed] [Google Scholar]
- 36.Friedmann Angeli JP, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180-1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ingold I, Berndt C, Schmitt S, et al. Selenium utilization by GPX4 is required to prevent hydroperoxide-induced ferroptosis. Cell. 2018;172(3):409-422.e21. [DOI] [PubMed] [Google Scholar]
- 38.Jiang L, Kon N, Li T, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520(7545):57-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jenkitkasemwong S, Wang CY, Coffey R, et al. SLC39A14 is required for the development of hepatocellular iron overload in murine models of hereditary hemochromatosis. Cell Metab. 2015;22(1):138-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mao W, Hu Y, Lou Y, Chen Y, Zhang J. Abnormal serum iron markers in chronic hepatitis B virus infection may be because of liver injury. Eur J Gastroenterol Hepatol. 2015;27(2):130-136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Lv Q, Gao J, et al. Coinfection with HIV-1 alleviates iron accumulation in patients with chronic hepatitis C virus infection. PLoS One. 2014;9(6):e98039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin D, Ding J, Liu JY, et al. Decreased serum hepcidin concentration correlates with brain iron deposition in patients with HBV-related cirrhosis. PLoS One. 2013;8(6):e65551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vagu C, Sultana C, Ruta S. Serum iron markers in patients with chronic hepatitis C infection. Hepat Mon. 2013;13(10):e13136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gao YH, Wang JY, Liu PY, et al. Iron metabolism disorders in patients with hepatitis B-related liver diseases. World J Clin Cases. 2018;6(13):600-610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bernstein SE. Hereditary hypotransferrinemia with hemosiderosis, a murine disorder resembling human atransferrinemia. J Lab Clin Med. 1987;110(6):690-705. [PubMed] [Google Scholar]
- 46.Trenor CC III, Campagna DR, Sellers VM, Andrews NC, Fleming MD. The molecular defect in hypotransferrinemic mice. Blood. 2000;96(3):1113-1118. [PubMed] [Google Scholar]
- 47.Bartnikas TB, Andrews NC, Fleming MD. Transferrin is a major determinant of hepcidin expression in hypotransferrinemic mice. Blood. 2011;117(2):630-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Levin MJ, Tuil D, Uzan G, Dreyfus JC, Kahn A. Expression of the transferrin gene during development of non-hepatic tissues: high level of transferrin mRNA in fetal muscle and adult brain. Biochem Biophys Res Commun. 1984;122(1):212-217. [DOI] [PubMed] [Google Scholar]
- 49.Hoshi K, Matsumoto Y, Ito H, et al. A unique glycan-isoform of transferrin in cerebrospinal fluid: a potential diagnostic marker for neurological diseases. Biochim Biophys Acta, Gen Subj. 2017;1861(10):2473-2478. [DOI] [PubMed] [Google Scholar]
- 50.Yan J, Gong X, Pan S, Guo X, Ren Z, Zeng Y. Expression of human transferrin can be regulated effectively by rabbit transferrin regulatory elements in transgenic mice. Biotechnol Lett. 2014;36(6):1209-1216. [DOI] [PubMed] [Google Scholar]
- 51.Le Lan C, Loréal O, Cohen T, et al. Redox active plasma iron in C282Y/C282Y hemochromatosis. Blood. 2005;105(11):4527-4531. [DOI] [PubMed] [Google Scholar]
- 52.Arezes J, Foy N, McHugh K, et al. Erythroferrone inhibits the induction of hepcidin by BMP6. Blood. 2018;132(14):1473-1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Doll S, Proneth B, Tyurina YY, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91-98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wenzel SE, Tyurina YY, Zhao J, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171(3):628-641.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Alvarez SW, Sviderskiy VO, Terzi EM, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature. 2017;551(7682):639-643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gao M, Monian P, Quadri N, Ramasamy R, Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Mol Cell. 2015;59(2):298-308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Linkermann A, Skouta R, Himmerkus N, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci U S A. 2014;111(47):16836-16841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tuschl K, Meyer E, Valdivia LE, et al. Mutations in SLC39A14 disrupt manganese homeostasis and cause childhood-onset parkinsonism-dystonia. Nat Commun. 2016;7(1):11601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Aydemir TB, Kim MH, Kim J, et al. Metal transporter Zip14 (Slc39a14) deletion in mice increases manganese deposition and produces neurotoxic signatures and diminished motor activity. J Neurosci. 2017;37(25):5996-6006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hamill RL, Woods JC, Cook BA. Congenital atransferrinemia. A case report and review of the literature. Am J Clin Pathol. 1991;96(2):215-218. [DOI] [PubMed] [Google Scholar]
- 62.Beutler E, Gelbart T, Lee P, Trevino R, Fernandez MA, Fairbanks VF. Molecular characterization of a case of atransferrinemia. Blood. 2000;96(13):4071-4074. [PubMed] [Google Scholar]
- 63.Zhao N, Zhang AS, Worthen C, Knutson MD, Enns CA. An iron-regulated and glycosylation-dependent proteasomal degradation pathway for the plasma membrane metal transporter ZIP14. Proc Natl Acad Sci U S A. 2014;111(25):9175-9180. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.