

Abstract
Coronavirus disease 2019 (COVID-19) has affected almost every country in the world by causing a global pandemic with a high mortality rate. Lack of an effective vaccine and/or antiviral drugs against SARS-CoV-2, the causative agent, has severely hampered the response to this novel coronavirus. Natural products have long been used in traditional medicines to treat various diseases, and purified phytochemicals from medicinal plants provide a valuable scaffold for the discovery of new drug leads. In the present study, we performed a computational screening of an in-house database composed of ~1000 phytochemicals derived from traditional Saudi medicinal plants with recognised antiviral activity. Structure-based virtual screening was carried out against three druggable SARS-CoV-2 targets, viral RNA-dependent RNA polymerase (RdRp), 3-chymotrypsin-like cysteine protease (3CLpro) and papain like protease (PLpro) to identify putative inhibitors that could facilitate the development of potential anti-COVID-19 drug candidates. Computational analyses identified three compounds inhibiting each target, with binding affinity scores ranging from −9.9 to −6.5 kcal/mol. Among these, luteolin 7-rutinoside, chrysophanol 8-(6-galloylglucoside) and kaempferol 7-(6″-galloylglucoside) bound efficiently to RdRp, while chrysophanol 8-(6-galloylglucoside), 3,4,5-tri-O-galloylquinic acid and mulberrofuran G interacted strongly with 3CLpro, and withanolide A, isocodonocarpine and calonysterone bound tightly to PLpro. These potential drug candidates will be subjected to further in vitro and in vivo studies and may assist the development of effective anti-COVID-19 drugs.
Keywords: Antiviral, COVID-19, MD simulation, SARS-CoV-2, Virtual screening, Protease
1. Introduction
The global threat of the 2019 novel coronavirus disease (now officially named COVID-19 and the causative pathogen is SARS-CoV-2) is rapidly escalating with unprecedented international health and economic burden in the recent history. The whole-genome of SARS-CoV-2 had been sequenced and revealed that SARS-CoV-2 pathogen is the fifth strain of β-coronaviruses, which include OC43, HKU1, SARS-CoV-1 and Middle East respiratory syndrome coronavirus (MERS-CoV) (Wang et al., 2020). After the entry into the host cell through strong binding of β-coronaviruses’ protein spikes with the human angiotensin-converting enzyme 2 (ACE2) receptor (Xu et al., 2020), β-coronaviruses generate large polyproteins (PP1a and PP1ab) upon genome translation of open reading frame (ORF) 1a and ORF1ab by the host cell machinery. These polyproteins, also known as replicase polyproteins, are proteolytically cleaved by essential cysteine proteases encoded by the virus, explicitly 3-chymotrypsin-like protease (3CLpro; sometimes called main protease (Mpro)) and papain-like proteases (PLpro), to release 16 non-structural proteins (nsps) (Thiel et al., 2003, Ziebuhr, 2004). Among those nsps is the RNA-dependent RNA polymerase (RdRp or sometimes referred to as nsp12). RdRp catalyses the synthesis of a complementary RNA strand using the viral RNA and hence, it plays an essential role in directing the replication and transcription of SARS-CoV-2 genome (Chen et al., 2020, Lung et al., 2020, Zumla et al., 2016). The proteolysis of PP1a and PP1ab by 3CLpro occurs at 11 distinct sites and generates various nsps that are important for the viral replication (Anand et al., 2003). Therefore, 3CLpro is crucial for the virus particle replication and represent a valid target for the identification of coronavirus inhibitors (Needle et al., 2015). PLpro is another crucial cysteine protease, an enzyme that cleaves N-terminus of the replicase polyprotein to release several nsps, among them the nsp3, in which PLpro is encoded and is implicated not only in the viral replication but also in suppressing the host innate immune response (Chen et al., 2020, Lei et al., 2018, Yuan et al., 2015). In addition, PLpro possesses a nucleic acid-binding domain (NAB) with a nucleic acid chaperon function and is essential in the virus replication correction. Furthermore, PLpro has posttranslational modification activities, deubiquitination and deISGylating, on the host proteins, such as interferon regulatory factor 3(IRF3), which consequently lead to the removal of ubiquitin and human interferon-stimulated gene 15 (ISG15), the main signalling elements of the antiviral innate immune response (Báez-Santos et al., 2015, Chen et al., 2020, Lei et al., 2018, Liu et al., 2020). The indispensable role of the RdRp, 3CLpro and PLpro in orchestrating the RNA genome replication/transcription cycles of SARS-CoV-2 make them potential targets for designing antiviral candidates against COVID-19 (Báez-Santos et al., 2015, Ramajayam et al., 2011, Ren et al., 2013).
For ages, phytochemicals have been found to be a fruitful source of molecules with diverse therapeutic potentials and still are considered a valuable resource for the discovery of novel drug leads in other word, herbal medicines and purified phytochemicals can be used to develop more efficient drugs based on the structure of natural compounds (Mani et al., 2020). Given the time of its emergence, few studies related to the development of naturally-derived inhibitors of three main druggable targets of SARS-CoV-2 (RdRp, 3CLpro and PLpro) have already been reported using computer modelling for screening purposes. For instance, in silico or biological screening of a series of biologically active natural compounds have been demonstrated to directly inhibit these important proteins in pervious HCoVs such as SARS-CoV, MERS-CoV (Park et al., 2012, Shen et al., 2019) and the novel SARS-CoV-2 (Aanouz et al., 2020, Ul Qamar et al., 2020, Zhang et al., 2020). (Lung et al., 2020) virtually screened 83 compounds found in Chinese traditional medicines with potential activity against SARS-CoV-2 and identified theaflavin as a potential inhibitor of the RNA-dependent RNA polymerase. Likewise, (Zhang et al., 2020) screened 115 compounds that are commonly used in treating viral respiratory infection in China and highlighted 13 compounds for further studies to prove their activities against SARS-CoV-2. A virtual screening and molecular modelling study of marine natural products have also been investigated and proposed seventeen molecules with potent inhibitory action against 3CLpro (Gentile et al., 2020). In the present study, we conducted a computational study on a library of phytochemicals from Saudi medicinal plants with potential antiviral activities (Aati et al., 2019, Arbab et al., 2017) to identify potent inhibitors of RdRp, 3CLpro and PLpro, which eventually can be employed as novel lead molecules to accelerate the pace of designing anti-COVID-19 candidates.
2. Materials and methods
2.1. Preparation of protein structures
The X-ray crystal structures of three SARS-CoV-2 proteins including the RNA-dependent RNA polymerase (RdRp) with 2.50 Å resolution (accession number 7BV2) (Yin et al., 2020), 3-chymotrypsin-like cysteine protease (3CLpro) with 2.1 Å resolution (accession number 6 W63) (Mesecar, 2020) and papain like protease (PLpro) with 2.7 Å resolution (accession number 6W9C) (Osipiuk et al., 2020) were obtained from the Protein Data Bank. Discovery studio program (Studio, 2008) was used to prepare the proteins for virtual screening and molecular docking by removing co-crystalized ligands and water molecules and to prepare the proteins in monomer form in PDB formats.
2.2. Phytochemicals database
A library containing ~1000 plant-derived compounds obtained from 60 traditional Saudi medicinal plants with antiviral activity were designed. The phytochemicals were collected by comprehensive literature study. The 3D structures of compounds were obtained either from PubChem database (available online: https://pubchem.ncbi.nlm.nih.gov/) or were drown manually using ChemDraw Ultra 7.0 and saved in one SDF file.
2.3. Structure-based virtual screening
The virtual screening of phytochemical database against the structure of target proteins was performed individually using Autodock vina in PyRx 8.0 virtual screening tool (Dallakyan and Olson, 2015). At first, compounds were imported into OpenBabel program (Babel, 2011) implemented in PyRx for energy minimization using MMFF94 force field and for converting compounds into Autodock PDBQT format. The 3D grid box parameters were set to cover the active site cavity within each protein. The top three compounds with highest binding energy scores against each viral protein were selected as hit compounds for further assessment.
2.4. Molecular docking protocol
The molecular docking of candidate phytochemicals against their viral protein’s targets were carried out using Autodock vina 1.1.2 program (Trott and Olson, 2010). Autodock Tools 1.5.6 program (Huey and Morris, 2008) was employed to prepare the PDB structures of proteins for docking by adding polar hydrogen and to convert proteins and ligands PDB files into Autodock PDBQT format. The grid box parameters for RdRp were set to size 24 Å × 22 Å × 28 Å (x, y and z) and centre 113.78 Å × 118.474 Å × 132.008 Å (x, y and z). For 3CLpro, the grid box parameters were set to size 18 Å × 18 Å × 18 Å (x, y and z) and centre −20.318 Å × 19.141 Å × −28.343 Å (x, y and z). As for PLpro, the grid box parameters were set to size 21 Å × 22 Å × 22 Å (x, y and z) and centre −27.423 Å × 30.003 Å × 27.531 Å (x, y and z). The 2D ligand-protein interaction diagrams were generated by PoseView tool (available online: https://proteins.plus/). PyMOL (DeLano, 2002) was used to visualize and analyse the docking results.
2.5. Molecular dynamic simulation
All atom molecular dynamic (MD) simulation of ligand–protein complexes were performed with GROMACS 2018 software OPLS-AA/L force field. The high-resolution X-ray structures of proteins (PDB IDs: 6M71, 6W63 and 6W9C for RdRp, 3CLpro and PLpro, respectively) were used as initial structures for the MD simulation. The protein structures were further optimized for simulation by DockPerp tool in UCSF Chimera (Pettersen et al., 2004). The top-predicted docking pose of each ligand with the highest binding score was used as a starting point for the MD simulation. The ligands were parametrized using SwissParam webserver (Zoete et al., 2011). All systems were simulated for 20 ns using previously reported protocol (Alamri, 2020). Basic MD simulation parameters including real-mean-square-deviations (RMSD), real-mean-square-fluctuation (RMSF), radius of gyration (Needle et al., 2015), number of hydrogen bonds (H-bonds) were analysed for each complex.
3. Results and discussion
3.1. Construction of potential phytochemical database
Initially, an in-house phytochemical database composed of ~1000 bioactive compounds derived from 60 traditional Saudi medicinal plants was designed. The extracts of the selected plants were reported to possess antiviral activity against a wide-range of viruses such as HIV, HCV and HCB at various concentrations (Aati et al., 2019, Arbab et al., 2017). The main constituents of each plant were identified by comprehensive literature study. This approach was applied to build a focused phytochemicals library with recognised antiviral activity, so it can be further screened in search for potential anti-COVID19 agents. The tested library composed of phytochemicals that represent major classes of natural products as shown in (Fig. 1 ). The natural occurrence and examples of the top five chemical classes are presented in Table S1.
Fig. 1.

The major classes of natural products in the tested library.
3.2. Structure-based virtual screening, molecular docking and MD simulation
A high-throughput virtual screening of a diverse plant-derived compounds from Saudi medicinal plants, followed by molecular docking and all atom 20 ns MD simulation studies were performed. The screening was conducted against three well-established drug discovery targets including SARS-CoV-2 RdRp, 3CLpro and PLpro enzymes.
3.2.1. RNA-dependent RNA polymerase (RdRp)
First, we screened the designed database against SARS-CoV-2 RdRp. The RdRp is a conserved enzyme in coronaviruses and essential for their replication and transcription of their genomes (Chen et al., 2020, Lung et al., 2020, Zumla et al., 2016). RdRp in its active form tends to form a heterotrimeric complex with nsp7 and nsp8 for maximum activity (Gao et al., 2020). The virtual screening of in-house phytochemical database was performed against the recently solved X-ray structure of the nsp12 protein. The analysis resulted in the identification of three compounds, namely chrysophanol 8-(6-galloylglucoside), luteolin 7-rutinoside and kaempferol 7-(6″-galloylglucoside), which have been reported to be isolated form Rumex dentatus (Mishra et al., 2018), Marrubium vulgare (Neamah et al., 2009) and Acacia species (El-Toumy et al., 2018), respectively. The natural source and docking scores of these compounds are presented in (Table 1 ).
Table 1.
Potential inhibitors of RdRp from in-house Saudi medicinal database.
| Name/ID | Chemical structure | Natural source | Docking score (kcal/mol) |
|---|---|---|---|
| Remdesivir monophosphate | 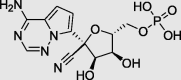 |
– | −7.0 |
|
Chrysophanol 8-(6-galloylglucoside) (PubChem ID:78384671) |
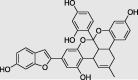 |
Rumex dentatus | −9.9 |
|
Luteolin 7-rutinoside (PubChem ID:44258082) |
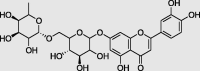 |
Marrubium vulgare | −9.8 |
|
Kaempferol 7-(6″-galloylglucoside) (PubChem ID:74978085) |
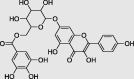 |
Acacia | −9.3 |
In order to gain more insight into the mechanism of interaction of these compounds with SARS-CoV-2 RdRp, a molecular docking was conducted using Autodock vina. Initially, Remdesivir which is the only approved drug to treat patients with severe COVID-19 was used to validate the docking approach (FDA, 12 April 2020). Remdesivir is a nucleotide analogue prodrug of (GS-441524) that converted into the active triphosphate form inside the cell. Recently, the structure of RdRp in complex with Remdesivir was solved (PDB: 7BV2). Remdesivir was found to interact covalently in its monophosphate form with the primer strand at the +1 position within the active site of SARS-CoV-2 RdRp (Yin et al., 2020). Fig. 2 a showed that the redocked Remdesivir monophosphate pose binds to the active site within RdRp as same as the co-crystalized ligand with RMSD = 2.969 Å. This result validates the ability of our docking protocol to predict the bioactive conformation of inhibitors. In addition, Remdesivir monophosphate exhibited lower binding free energy (−7.1 kcal/mol) in comparison to candidate compounds. This implies a strong possibility for these ligands to bind RdRp with higher affinity (Table 1). The molecular docking results of identified ligands illustrated that all compounds adapted binding modes similar to Remdesivir monophosphate within the active-site of RdRp (Fig. 2A). The chrysophanol 8-(6-galloylglucoside) was found to interact with the active site through six H-bonds (Tyr619, Asn691, Lys621, Asp623, Asp760 and Asp761) and one hydrophobic interaction (Pro621) (Fig. 2b). In the other hand, luteolin 7-rutinoside was involved in nine H-bonds with the active site (Arg553, Lys545, Arg555, Asp623, Asn691, Thr687, Asp760 and Ser759) (Fig. 2c). Fig. 2d, showed that kaempferol 7-(6″-galloylglucoside) formed seven H-bonds with RdRp (Arg553, Thr556, Tyr619, Trp617, Lys621, Asp760 and Asp761). Notably, the catalytic site of RdRp is composed of seven conserved motifs from A to G. Similar to Remdesivir, candidate compounds interact via H-bonds with several residues including the conserved Ser759-Asp60-Asp761 sequence within motif C of the RdRp polymerase domain (Fig. 2B) (Gao et al., 2020). These results suggested that the selected hits might act as non-nucleotide anti-polymerase agents.
Fig. 2.

Binding mode and molecular interaction of compounds with RdRp. (A) Ribbon representation of RdRp (PDB: 7BV2) shown the binding mode of Remdesivir monophosphate (Liu et al., 2020), Chrysophanol8-(6-galloylglucoside) (yellow), Luteolin 7-rutinoside (orange) and Kaempferol 7-(6″-galloylglucoside) (blue). (B) The superimposition of co-crystalized (Liu et al., 2020) and docked (yellow) Remdesivir monophosphate (a). 2D ligand–protein interaction of (b) Chrysophanol8-(6-galloylglucoside), (c) Luteolin 7-rutinoside (Liu et al., 2020) and (d) Kaempferol 7-(6″-galloylglucoside).
In order to assess RdRp association and to determine the dynamic stability of screened compounds, all atom MD simulation at 20 ns were carried out by GROMACS. Analysis of RMSD from the starting structure revealed that most of complexes reach equilibrium within 20 ns (Fig. 3 A). This points to the high stability of protein–ligand complexes. Some fluctuation was observed within the first 10 ns in all systems. For the structure of RdRp bound to Remdesivir monophosphate and Chrysophanol8-(6-galloylglucoside), the RMSD values stabilized around ~0.32 nm. On the other hand, the RMSD values for the RdRp complex with Luteolin 7-rutinoside and Kaempferol 7-(6″-galloylglucoside), were equilibrated at around ~0.24 and ~0.20 nm, respectively. These results suggested that the binding of Remdesiver monophosphate and Chrysophanol8-(6-galloylglucoside) to RdRp may induce conformational changes (Fig. 3A). In consistent with this, the analysis of RMSF vs RdRp residue number showed that Remdesivir monophosphate and Chrysophanol8-(6-galloylglucoside) complexes showed higher oscillations in backbone residues in comparison to Luteolin 7-rutinoside and Kaempferol 7-(6″-galloylglucoside) systems (Fig. 3B).
Fig. 3.

(A) RMSD of backbone atoms (C, Cα, and N) for RdRp-Ligand complex systems. (B) RMSF of backbone atoms for RdRp-Ligand complex systems. (C) Radius of gyration of backbone atoms (D) Number of H-bonds over 20 ns simulation time.
The RMSF values were high in some residues located in the flexible loop as well as alpha helices regions such as Thr261, Asp336, Glu431 and Asp824. Radius of gyrate (Rg) is another parameter to assess the compactness changes of a ligand–protein complex. Remdesivir monophosphate displayed a high Rg fluctuation in comparison to other compounds. This is consistent with the docking results of Remdesiver monophosphate that showed the lowest binding free energy. In contrast, no significant changes were observed in Rg values of Chrysophanol8-(6-galloylglucoside) and Kaempferol 7-(6″-galloylglucoside) indicating sustained compactness of both systems throughout the simulation (Fig. 3C). The Luteolin 7-rutinoside complex was the most stable and compact system with Rg value of ~2.94 nm. Next, the formation and stability of H-bonds were investigated over the simulation time (Fig. 3D). The H-bond properties are essential parameter in drug design due to their role in drug specificity, metabolism and absorption (Bitencourt-Ferreira et al., 2019). The results illustrated that Remdesiver monophosphate, Chrysophanol8-(6-galloylglucoside), Luteolin 7-rutinoside and Kaempferol 7-(6″-galloylglucoside) could form up to 5, 10, 11 and 10H-bonds, respectively. Therefore, the stability of complexes was maintained by H-bonds formation with active site residues. According to the docking and MD simulation analyses, Luteolin 7-rutinoside showed the best affinity towards RdRp in comparison to other compounds. It showed a high docking score (−9.8 kcal/mol) and was able to form stable H-bonds with the essential Asp760 and Asp761 of SDD sequence of polymerase domain. Moreover, the results from RMSD, RMSF and Rg indicated that the Luteolin 7-rutinoside complex was stable during the simulation period. Therefore, it can be concluded that Luteolin 7-rutinoside is the most preferable compound for the inhibition of RdRp for further in vitro and in vivo studies.
3.2.2. 3-Chymotrypsin-like protease (3CLpro)
The 3CLpro, also known as the main protease (Mpro) is a cysteine protease. Its proteolytic activity is mediated by His41-Cys148 catalytic dyad (Anand et al., 2003, Needle et al., 2015). The structure based virtual screening of our phytochemical database led to identification of three compounds namely, 3,4,5-Tri-O-galloylquinic acid, chrysophanol 8-(6-galloylglucoside) and iso-mulbel-rochromene with docking scores of −9.7, −9.7- and −9.6 kcal/mol, respectively (Table 2 ). Interestingly, Chrysophanol 8-(6-galloylglucoside) was also identified as a potential RdRp inhibitor indicating that it might have a dual inhibitory mechanism.
Table 2.
Potential inhibitors of 3CLpro from in-house Saudi medicinal database.
| Name/ID | Chemical structure | Natural source | Docking score (kcal/mol) |
|---|---|---|---|
|
X77 (PubChem ID: 145998279) |
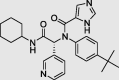 |
– | −8.6 |
|
3,4,5-Tri-O-galloylquinic acid (PubChem ID: 127406) |
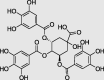 |
Guiera senegalensis | −9.7 |
|
Chrysophanol 8-(6-galloylglucoside) (PubChem ID: 78384671) |
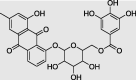 |
Rumex dentatus | −9.7 |
|
Iso-mulbel-rochromene (PubChem ID: 196583) |
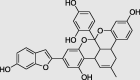 |
Chenopodium ambrosioides, Morus alba L | −9.6 |
To predict these inhibitors conformation and orientation inside the active site of 3CLpro, a molecular docking was carried out. All selected compounds were found to fit into the active site with similar binding mode (Fig. 4 A). The co-crystalized non-covalent ligand, X77, was re-docked to assess the capability of our docking protocol to re-produce the bioactive conformation. As shown in Fig. 4a, the docked pose adapted similar binding mode as the co-crystalized ligand with RMSD = 0.789 Å indicating the robustness of our docking procedure. 3,4,5-Tri-O-galloylquinic acid binds with the active site of 3CLpro through several H-bonds (Met49, Leu141, Ser144, Glu166, His163 and Asp187) (Fig. 4b). The interaction between chrysophanol 8-(6-galloylglucoside) and 3CLpro was essentially mediated through H-bonds with four residues (Glu166, Arg188, Thr190 and Gln192) (Fig. 4c). Iso-mulbel-rochromene formed six H-bonds with the active site of 3CLpro (Thr25, Leu141, Ser144, His163, Arg188 and Gln189). These compounds block most of the essential residues involved in substrate recognition through H-bonds interaction indicating that these compounds may have potential to inhibit SARS-CoV-2 3CLpro activity.
Fig. 4.

Binding mode and molecular interaction of compounds with 3CLpro. (A) Ribbon representation of 3CLpro (PDB: 6w63) shown the binding mode of co-crystalized (Pettersen et al., 2004) and docked (pink) X77, 3,4,5-Tri-O-galloylquinic acid (orange), chrysophanol8-(6-galloylglucoside) (cyan), iso-mulbel-rochromene (blue). (B) The superimposition of co-crystalized (Pettersen et al., 2004) and docked (pink) X77 (a). His41-Cys145 catalytic dyad is shown in cyan sticks. 2D ligand–protein interaction of (b) 3,4,5-Tri-O-galloylquinic acid, (c) chrysophanol8-(6-galloylglucoside) and (d) iso-mulbel-rochromene with 3CLpro.
Next, we characterized the stability of dynamic behavior and binding conformation of 3CLpro in complex with co-crystalized ligand, X77 as well as the identified compounds by 20 ns all-atom MD simulations. Fig. 5 A illustrated the plot of RMSD (nm) vs. time (ns) for apo-3CLpro and three 3CLpro-ligand complexes (3,4,5-Tri-O-galloylquinic acid, chrysophanol8-(6-galloylglucoside) and iso-mulbel-rochromene). All complexes attained the equilibration state during the 20 ns MD simulation period. The 3CLpro-X77 complex showed higher RMSD value as compared to identified ligand complexes during the last 10 ns, whereas 3,4,5-Tri-O-galloylquinic acid showed the least RMSD value, i.e., a greater dynamic stability compared to the other compounds. The RMSF values showed normal fluctuation with all ligand–protein complexes with respect to the RMSF value of co-crystalized ligand (Fig. 5B). The Rg profiles were quite similar for all ligand-complexes as compared to X77-3CLpro complex. Thus, protein-ligands complexes were stable complexes and well folded (Fig. 5C).
Fig. 5.

(A) RMSD of backbone atoms (C, Cα, and N) for 3CLpro-Ligand complex systems. (B) RMSF of backbone atoms for 3CLpro-Ligand complex systems. (C) Radius of gyration (Rg) of backbone atoms (D) Number of H-bonds over 20 ns simulation time.
The number of H-bonds for ligand–protein complexes was calculated over 20 ns simulation time. Fig. 5D showed that 3,4,5-Tri-O-galloylquinic acid, chrysophanol8-(6-galloylglucoside) and iso-mulbel-rochromene had an average of eight, eleven and seven H-bonds, respectively, whereas an average of four H-bonds are formed in the X77-3CLpro complex. This explains the high binding energy scores of identified ligands compared to co-crystallized ligand, X77. These findings offer an extensive opportunity for these natural compounds, especially 3,4,5-Tri-O-galloylquinic acid, to contradict 3CLpro for the treatment of COVID-19.
3.2.3. Papain-like protease (PLpro)
The PLpro is another key cysteine protease enzyme for coronaviruses life cycle. In addition to its function in virus replication, it is involved in facilitating the virus entry by antagonizing the host cell immunity (Báez-Santos et al., 2015, Chen et al., 2020, Lei et al., 2018, Liu et al., 2020). The virtual screening against PLpro revealed that withanolide A, isocodonocarpine and calonysterone exhibited the highest binding affinity scores of −7.4, −7.0 and −6.9 kcal/mol, respectively (Table 3 ).
Table 3.
Potential inhibitors of PLpro from in-house Saudi medicinal database.
| Name/ID | Chemical structure | Natural source | Docking score (kcal/mol) |
|---|---|---|---|
|
GRL0617 (PubChem ID: 24941262) |
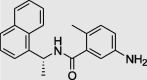 |
– | −6.5 |
|
Withanolide A (PubChem ID: 11294368) |
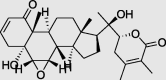 |
Datura innoxia | −7.4 |
| Isocodonocarpine | 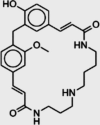 |
Capparis decidua | −7.0 |
|
Calonysterone (PubChem ID: 101281312) |
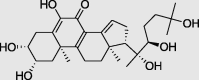 |
Senna obtusifolia | −6.9 |
Next, we performed a molecular docking to determine the molecular interactions of these inhibitors with PLpro. At first, the docking protocol was validated by docking a known SARS-CoV inhibitor; GRL0617 (PDB ID: 3E9S) into the same site within SRS-CoV-2 PLpro (PDB ID: 6W9C) (Ratia et al., 2008). This site is a conserved in PLpro for both SARS viruses. It is located in a proximity to the catalytic site which is composed of catalytic triad residues (Cys112-His273-Asp287) of PLpro (Fig. 6 A). Notably, the initial virtual screening was carried out against this pocket. Fig. 6A showed that GRL0617 as well as all identified ligands fit into the binding pocket with similar binding modes. A closer view into the binding of GRL0617 to PLpro of SRAS-CoV-2 showed that it adapted a similar binding mode and conformational arrangement for SARS-CoV (PDB ID: 3E9S), which demonstrated the validity of docking approach (Fig. 6a). Withanolide A interacted with the active site of PLpro through three H-bonds with Lys157, Asp164 and Glu167 (Fig. 6b). In the other hand, the cyclic compound, isocodonocarpine formed hydrophobic interactions with Tyr264 and Tyr268 and π-π stacking with Tyr264 (Fig. 6c). The calonysterone’s binding was found to involve in three H-bonds with Glu161, Asp164 and Tyr246 and one hydrophobic interaction with Pro248 (Fig. 6d).
Fig. 6.

Binding mode and molecular interaction of compounds with PLpro. (A) Ribbon representation of PLpro (PDB: 6w9c) showing the binding mode of docked GRL0617 (Pettersen et al., 2004), withanolide A (Liu et al., 2020), isocodonocarpine (blue) and calonysterone (orange). The active site and catalytic triad Cys112-His273-Asp287 is shown in red sticks. (B) The superimposition of binding mode of docked GRL0617 (Pettersen et al., 2004) to SRAR-CoV-2 (cyan) (PDB: 6w9c) with the binding mode of co-crystalized GRL0617 (yellow) in complex with SARS-CoV (gold) (PDB: 3E9S). (a). 2D ligand–protein interaction of (b) withanolide A (c) isocodonocarpine and (d) calonysterone with PLpro.
Similarly, to ensure the stability of ligands within the active site of PLpro, 20 ns all-atom MD simulation using GROMAS 2018 was performed. The RMSD results of withanolide A-PLpro, isocodonocarpine-PLpro and calonysterone-PLpro complexes reached a stable state throughout the simulation period. The fluctuation in the RMSD values of withanolide A- and isocodonocarpine-PLpro complexes were less compared to the apo-protein and calonysterone-PLpro complex (Fig. 7 A). The RMSF values for isocodonocarpine showed less RMS fluctuation than other complexes, with respect to the RMSF values of apo-protein (Fig. 7B).
Fig. 7.

(A) RMSD of backbone atoms (C, Cα, and N) for PLpro-Ligand complex systems. (B) RMSF of backbone atoms for PLpro-Ligand complex systems. (C) Radius of gyration (Rg) of backbone atoms (D) Number of H-bonds over 20 ns simulation time.
Fig. 7C illustrated that all ligand–protein complexes showed similar Rg behaviour, with respect to the apo-protein suggesting stable ligand–protein complexes in general. The H-bonds were also measured for each complex (Fig. 7D). Withanolide A, isocodonocarpine and calonysterone showed an average of two, three and five H-bonds. Although, isocodonocarpine did not show any H-bonds by molecular docking, three H-bonds were predicted by MD simulation. In addition, the number of H-bonds formed by withanolide A were almost stable, while the number of H-bonds formed by calonysterone was decreased over the MD simulation. This could explain the low docking score of calonysterone. Overall, Withanolide A showed the best results with respect to docking score as well as the MD simulation analyses suggesting that it might be a potential PLpro inhibitor candidate to combat COVID-19 infection.
4. Conclusion
In the presented study, we employed computational screening approach for the discovery of potential anti-COVID-19 against the novel coronavirus SARS-CoV-2 infection. A total of ~1000 phytochemicals derived from 60 Saudi medicinal plants with recognised antiviral activities have been investigated and lead to the identification of nine potential inhibitors for three major druggable targets of SARS-CoV-2. The obtained candidates from the virtual screening were subjected to MD simulation to confirm their binding and dynamic stability at the target site. It has been demonstrated that chrysophanol 8-(6-galloylglucoside), luteolin 7-rutinoside and kaempferol 7-(6″-galloylglucoside) have the potential for further optimisation as an RdRp inhibitors. Moreover, 3,4,5-tri-O-galloylquinic acid, chrysophanol 8-(6-galloylglucoside) and iso-mulbel-rochromene showed potential to efficiently bind and inhibit the viral 3CLpro. Withanolide A, isocodonocarpine and calonysterone have also shown promising results in targeting the PLpro. Future in vitro and in vivo evaluations of these compounds can confirm their antiviral activity and subsequently their potential to serve as drug candidates for further designing of target-specific anti-COVID-19 agents.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
Authors would like to thank Prince Sattam Bin Abdulaziz University for their support and providing the necessary computational tools to conduct this study.
Footnotes
Peer review under responsibility of King Saud University.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.arabjc.2020.08.004.
Appendix A. Supplementary material
The following are the Supplementary data to this article:
References
- Aanouz I., Belhassan A., El Khatabi K., Lakhlifi T., El Idrissi M., Bouachrine M. Moroccan medicinal plants as inhibitors of COVID-19: Computational investigations. J. Biomol. Struct. Dyn. 2020:1–12. doi: 10.1080/07391102.2020.1758790. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aati H., El-Gamal A., Shaheen H., Kayser O. Traditional use of ethnomedicinal native plants in the Kingdom of Saudi Arabia. J. Ethnobiol. Ethnomed. 2019;15(1):2. doi: 10.1186/s13002-018-0263-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alamri M.A. Pharmacoinformatics and molecular dynamic simulation studies to identify potential small-molecule inhibitors of WNK-SPAK/OSR1 signaling that mimic the RFQV motifs of WNK kinases. Arab. J. Chem. 2020;13(4):5107–5117. [Google Scholar]
- Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science. 2003;300(5626):1763–1767. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Arbab A.H., Parvez M.K., Al-Dosari M.S., Al-Rehaily A.J. In vitro evaluation of novel antiviral activities of 60 medicinal plants extracts against hepatitis B virus. Exp. Therapeut. Med. 2017;14(1):626–634. doi: 10.3892/etm.2017.4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babel O. An open chemical toolbox. J. Cheminf. 2011;3:33. doi: 10.1186/1758-2946-3-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Báez-Santos Y.M., John S.E.S., Mesecar A.D. The SARS-coronavirus papain-like protease: structure, function and inhibition by designed antiviral compounds. Antiviral Res. 2015;115:21–38. doi: 10.1016/j.antiviral.2014.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bitencourt-Ferreira G., Veit-Acosta M., de Azevedo W.F., Jr. Hydrogen bonds in protein-ligand complexes. Methods Mol. Biol. (Clifton, N.J.) 2019;2053:93–107. doi: 10.1007/978-1-4939-9752-7_7. [DOI] [PubMed] [Google Scholar]
- Chen Y., Liu Q., Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J. Med. Virol. 2020;92(4):418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallakyan S., Olson A.J. Chemical Biology. Springer; 2015. Small-molecule library screening by docking with PyRx; pp. 243–250. [DOI] [PubMed] [Google Scholar]
- DeLano W.L. Pymol: An open-source molecular graphics tool. CCP4 Newslett. Protein Crystallogr. 2002;40(1):82–92. [Google Scholar]
- El-Toumy S.A., Salib J.Y., El-Kashak W.A., Marty C., Bedoux G., Bourgougnon N. Antiviral effect of polyphenol rich plant extracts on herpes simplex virus type 1. Food Sci. Hum. Wellness. 2018;7(1):91–101. [Google Scholar]
- FDA, U. 12 April 2020. Coronavirus (COVID-19) update: FDA issues emergency use authorization to decontaminate millions of N95 respirators.
- Gao Y., Yan L., Huang Y., Liu F., Zhao Y., Cao L., Wang T., Sun Q., Ming Z., Zhang L. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science. 2020 doi: 10.1126/science.abb7498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gentile D., Patamia V., Scala A., Sciortino M.T., Piperno A., Rescifina A. Putative inhibitors of SARS-CoV-2 main protease from a library of marine natural products: a virtual screening and molecular modeling study. Mar. Drugs. 2020;18(4):225. doi: 10.3390/md18040225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huey R., Morris G.M. The Scripps Research Institute; USA: 2008. Using AutoDock 4 with AutoDocktools: a tutorial; pp. 54–56. [Google Scholar]
- Lei J., Kusov Y., Hilgenfeld R. Nsp3 of coronaviruses: Structures and functions of a large multi-domain protein. Antiviral Res. 2018;149:58–74. doi: 10.1016/j.antiviral.2017.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W., Morse J.S., Lalonde T., Xu S. Learning from the past: possible urgent prevention and treatment options for severe acute respiratory infections caused by 2019-nCoV. ChemBioChem. 2020 doi: 10.1002/cbic.202000047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lung J., Lin Y.S., Yang Y.H., Chou Y.L., Shu L.H., Cheng Y.C., Liu H.T., Wu C.Y. The potential chemical structure of anti-SARS-CoV-2 RNA-dependent RNA polymerase. J. Med. Virol. 2020;92(6):693–697. doi: 10.1002/jmv.25761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani J.S., Johnson J.B., Steel J.C., Broszczak D.A., Neilsen P.M., Walsh K.B., Naiker M. Natural product-derived phytochemicals as potential agents against coronaviruses: A review. Virus Res. 2020;284:197989. doi: 10.1016/j.virusres.2020.197989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesecar A. A taxonomically-driven approach to development of potent, broad-spectrum inhibitors of coronavirus main protease including SARS-CoV-2 (COVID-19) Be Publ. 2020 [Google Scholar]
- Mishra A.P., Sharifi-Rad M., Shariati M.A., Mabkhot Y.N., Al-Showiman S.S., Rauf A., Salehi B., Župunski M., Sharifi-Rad M., Gusain P. Bioactive compounds and health benefits of edible Rumex species-A review. Cell. Mol. Biol. 2018;64(8):27–34. [PubMed] [Google Scholar]
- Neamah, S.I., Sarhan, I.A., Al-Shaye’a, O.N., 2009. Bioactive compounds from Marrubium Vulgare L. based on in vitro antioxidant activity.
- Needle D., Lountos G.T., Waugh D.S. Structures of the Middle East respiratory syndrome coronavirus 3C-like protease reveal insights into substrate specificity. Acta Crystallogr. D Biol. Crystallogr. 2015;71(5):1102–1111. doi: 10.1107/S1399004715003521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipiuk, J., Jedrzejczak, R., Tesar, C., Endres, M., Stols, L., Babnigg, G., Kim, Y., Michalska, K., Joachimiak, A., 2020. The crystal structure of papain-like protease of SARS CoV-2. RSCB PDB.
- Park J.-Y., Kim J.H., Kim Y.M., Jeong H.J., Kim D.W., Park K.H., Kwon H.-J., Park S.-J., Lee W.S., Ryu Y.B. Tanshinones as selective and slow-binding inhibitors for SARS-CoV cysteine proteases. Bioorg. Med. Chem. 2012;20(19):5928–5935. doi: 10.1016/j.bmc.2012.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25(13):1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Ramajayam R., Tan K.-P., Liang P.-H. Portland Press Ltd.; 2011. Recent Development of 3C and 3CL Protease Inhibitors for Anti-coronavirus and Anti-picornavirus Drug Discovery. [DOI] [PubMed] [Google Scholar]
- Ratia K., Pegan S., Takayama J., Sleeman K., Coughlin M., Baliji S., Chaudhuri R., Fu W., Prabhakar B.S., Johnson M.E. A noncovalent class of papain-like protease/deubiquitinase inhibitors blocks SARS virus replication. Proc. Natl. Acad. Sci. 2008;105(42):16119–16124. doi: 10.1073/pnas.0805240105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren Z., Yan L., Zhang N., Guo Y., Yang C., Lou Z., Rao Z. The newly emerged SARS-like coronavirus HCoV-EMC also has an“ Achilles' heel”: current effective inhibitor targeting a 3C-like protease. Protein Cell. 2013;4(4):248. doi: 10.1007/s13238-013-2841-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L., Niu J., Wang C., Huang B., Wang W., Zhu N., Deng Y., Wang H., Ye F., Cen S. High-throughput screening and identification of potent broad-spectrum inhibitors of coronaviruses. J. Virol. 2019;93(12):e00023–00019. doi: 10.1128/JVI.00023-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studio D. Accelrys [2.1]; 2008. Discovery Studio. [Google Scholar]
- Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weißbrich B., Snijder E.J., Rabenau H., Doerr H.W. Mechanisms and enzymes involved in SARS coronavirus genome expression. J. Gen. Virol. 2003;84(9):2305–2315. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- Trott O., Olson A.J. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010;31(2):455–461. doi: 10.1002/jcc.21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ul Qamar M.T., Alqahtani S.M., Alamri M.A., Chen L.-L. Structural basis of SARS-CoV-2 3CLpro and anti-COVID-19 drug discovery from medicinal plants. J. Pharm. Anal. 2020 doi: 10.1016/j.jpha.2020.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020;30(3):269–271. doi: 10.1038/s41422-020-0282-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X., Chen P., Wang J., Feng J., Zhou H., Li X., Zhong W., Hao P. Evolution of the novel coronavirus from the ongoing Wuhan outbreak and modeling of its spike protein for risk of human transmission. Sci. China Life Sci. 2020;63(3):457–460. doi: 10.1007/s11427-020-1637-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin W., Mao C., Luan X., Shen D.-D., Shen Q., Su H., Wang X., Zhou F., Zhao W., Gao M. Structural basis for inhibition of the RNA-dependent RNA polymerase from SARS-CoV-2 by remdesivir. Science. 2020 doi: 10.1126/science.abc1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L., Chen Z., Song S., Wang S., Tian C., Xing G., Chen X., Xiao Z.-X., He F., Zhang L. p53 degradation by a coronavirus papain-like protease suppresses type I interferon signaling. J. Biol. Chem. 2015;290(5):3172–3182. doi: 10.1074/jbc.M114.619890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D.H., Wu K.L., Zhang X., Deng S.Q., Peng B. In silico screening of Chinese herbal medicines with the potential to directly inhibit 2019 novel coronavirus. J. Integr. Med. 2020;18(2):152–158. doi: 10.1016/j.joim.2020.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziebuhr J. Molecular biology of severe acute respiratory syndrome coronavirus. Curr. Opin. Microbiol. 2004;7(4):412–419. doi: 10.1016/j.mib.2004.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoete V., Cuendet M.A., Grosdidier A., Michielin O. SwissParam: a fast force field generation tool for small organic molecules. J. Comput. Chem. 2011;32(11):2359–2368. doi: 10.1002/jcc.21816. [DOI] [PubMed] [Google Scholar]
- Zumla A., Chan J.F., Azhar E.I., Hui D.S., Yuen K.-Y. Coronaviruses—drug discovery and therapeutic options. Nat. Rev. Drug Discovery. 2016;15(5):327. doi: 10.1038/nrd.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.