

Abstract
Persistent viral infections require a host cell reservoir that maintains functional copies of the viral genome. To this end, several DNA viruses maintain their genomes as extrachromosomal DNA minichromosomes in actively dividing cells. These viruses typically encode a viral protein that binds specifically to viral DNA genomes and tethers them to host mitotic chromosomes, thus enabling the viral genomes to hitchhike or piggyback into daughter cells. Viruses that use this tethering mechanism include papillomaviruses and the gammaherpesviruses Epstein-Barr virus and Kaposi’s sarcoma-associated herpesvirus. This review describes the advantages and consequences of persistent extrachromosomal viral genome replication.
Keywords: human papillomavirus, Epstein-Barr virus, Kaposi’s sarcoma-associated herpesvirus, DNA replication, Partitioning, persistent infection
1. STRATEGIES OF PERSISTENT INFECTION OF VIRUSES WITH EXTRACHROMOSOMAL GENOMES
1.1. Introduction
Papillomaviruses (PVs) and gammaherpesviruses are ancient viruses that have coevolved with their human hosts (1, 2). For the most part, the healthy host immune system controls these infections so that they are asymptomatic or harmless. However, the longevity of the viral-host association, as well as the manipulation of host pathways to ensure that virally infected cells survive, can sometimes result in disease. The Achilles’ heel of these infections is their reliance on a single viral protein to tether the extrachromosomal viral genomes to host chromosomes in latent or persistently infected cells. Disruption of this tether should lead to viral genome loss and cure of infection.
1.2. Papillomaviridae
PVs are a ubiquitous group of viruses that infect and replicate in the epidermis of vertebrates. The persistent replication cycle is tightly linked to the differentiation process of this stratified epithelium; the basal layers of the epithelium must frequently divide and differentiate to replenish the outermost tissue layer that protects against injury, infection, and dehydration. PVs infect the basal layer of the epithelium through a microabrasion and establish a persistent infection in these dividing cells. The viral genome replicates at low levels within these cells and has very limited gene expression. This low viral activity, combined with immune evasion functions of viral proteins, results in long-term infection that escapes immune detection (3).
Basal epidermal cells divide either symmetrically (self-renewal) or asymmetrically. In the latter case, one daughter cell leaves the basement membrane and begins the process of differentiation, moving up through the epithelium until it is sloughed from the surface. PVs exploit this process as productive viral DNA replication, late gene expression, and viral particle assembly occur sequentially as cells progress toward terminal differentiation and viral-laden squames are shed from the tissue surface. This resourceful strategy results in long-term infection (without immune clearance) and continual dissemination of viral particles.
To date, almost 500 different animal and human papillomavirus (HPV) types have been identified (4), most of which give rise to benign lesions (warts or papillomas) or asymptomatic infection in their hosts. Infections are usually self-limiting, as the host immune system eventually clears the virus, but can be debilitating in immunocompromised hosts (e.g., organ transplant recipients, human immunodeficiency virus infection, or genetic disease). In addition, a handful of PVs are oncogenic, and persistent infection with these types sometimes leads to cancer. In fact, oncogenic HPVs are responsible for 5% of human cancers worldwide (e.g., cervical, anogenital, and oropharyngeal) (5).
Despite their complex infectious cycle, the small PV circular, dsDNA genomes (~7–8 kb) contain just four well-conserved genes; these encode for capsid proteins (L1 and L2) and nonstructural replication proteins (E1 and E2). Additional proteins (E4, E5, E6, and E7) encoded by some PVs influence cellular environments to optimize viral replication, virion release, and evasion of the host immune response and may be evolutionary adaptations to different niches in the host epithelium (1).
1.3. Gammaherpesvirinae: Human Herpesviruses-4 and −8
The gammaherpesviruses human herpesvirus 4 (HHV-4) (Epstein-Barr virus or EBV) and HHV-8 (Kaposi’s sarcoma-associated herpesvirus or KSHV) are also ancient viruses that coevolved with their hosts and give rise to lifelong infections (2). EBV infects 95% of the human population, but KSHV is more geographically restricted (2). In most individuals, EBV or KSHV infection occurs in early childhood, resulting in subclinical infection. Both viruses are transmitted in saliva and transferred from the oral cavity to circulating B cells where they establish persistent, latent infection (2, 6). This is best understood for EBV, which initially infects oral epithelial cells and is transferred to B cells in the oropharynx (6). EBV is relatively dormant in infected B cells but can be triggered to reactivate into the lytic phase to produce virions. Persistent EBV infection is rarely associated with disease in healthy individuals, but in rare cases, immunosuppression, cellular stress, or dysregulated viral gene expression give rise to EBV-associated lymphomas (Hodgkin disease, immunoblastic lymphoma, and Burkitt lymphoma). Similarly, KSHV infection can cause the B cell malignancies primary effusion lymphoma and multicentric Castleman disease and the endothelial cell–associated cancer Kaposi’s sarcoma (2, 6). In contrast to PVs, the gammaherpesviruses use two or more cell types to switch from latency to transmission modes. The EBV and KSHV genomes are also large (EBV is ~172 kbp, and KSHV is ~165–170 kbp). Both genomes are linear dsDNA in the virion but are circularized by ligation of their terminal ends shortly after infection and thereafter maintained as circular dsDNA (7). EBV and KSHV encode at least 85 gene products important for infection, immune evasion, B cell reprogramming, latency, viral DNA replication, and virion structure. Only a subset of genes is expressed in latency, but there is consistent expression of EBNA1 (Epstein-Barr virus nuclear antigen 1) and KSHV LANA (latency-associated nuclear antigen). A key function of these proteins is to maintain the viral genomes as extrachromosomal plasmids.
2. REPLICATION MECHANISMS OF PERSISTENT VIRUSES WITH EXTRACHROMOSOMAL GENOMES
2.1. Multiple Phases of Viral DNA Replication
The complex infectious cycles of the PVs and gammaherpesviruses require multiple phases and modes of viral genome replication (Figure 1). In the first phase (upon initial infection), the incoming viral genome undergoes limited DNA synthesis to amplify the genome to a low copy number. Next, these genomes are established in the host cell, a relatively rare event that requires evasion of antiviral restriction factors and epigenetic silencing and localization of the genomes to advantageous regions of the nucleus. Once established, viral genomes replicate at a constant copy number in dividing cells through synchronous replication with host DNA during S-phase, and these genomes are partitioned to daughter cells in association with host mitotic chromosomes. Infected cells can persist for many years (PVs) or for the life of the host (gammaherpesviruses), and a robust mechanism is required to retain the extrachromosomal viral genome in dividing cells. The final phase of viral DNA synthesis is productive DNA amplification that generates high numbers of genomes for packaging in progeny virions. This phase is usually induced by terminal differentiation of infected host lymphocytes or epithelial cells (2, 6, 8).
Figure 1.
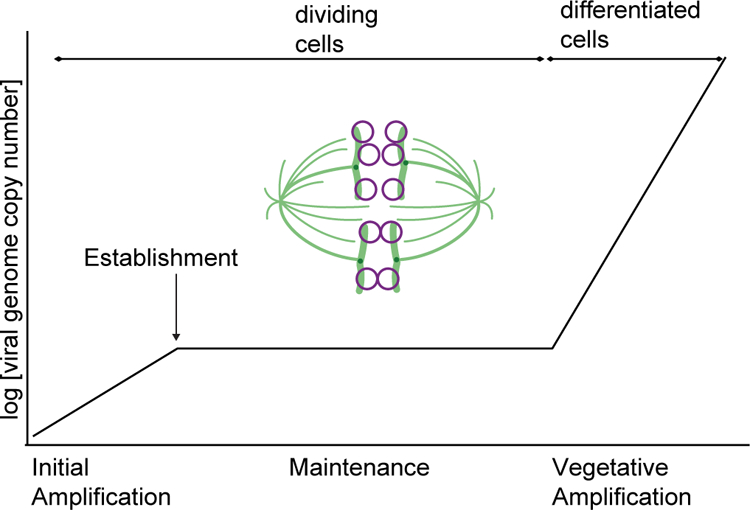
Phases of replication in gammaherpesvirus and papillomavirus infection. Upon infection, the virus undergoes limited DNA synthesis and becomes established as an extrachromosomal plasmid in the host nucleus. As the infected cells divide, viral genomes (purple circles) are maintained as stable copy number plasmids that are partitioned by binding to host chromosomes (green) in mitosis. Changes in the cell environment, such as differentiation, induce the productive or lytic phase, and large amounts of viral progeny genomes are synthesized.
2.2. Initiation of Viral DNA Synthesis
In theory, initiation of viral DNA synthesis and partitioning of viral genomes are two independent, separable processes. DNA synthesis is initiated at a replication origin bound by an initiator protein such as cellular origin recognition complex (ORC) or PV E1, and a cascade of cellular proteins assemble to synthesize DNA in a semiconservative manner. Viral genomes are partitioned by a virally encoded DNA binding protein that binds viral DNA and tethers the genomes to host chromosomes. In practice, these processes are tightly linked, as viral tethering proteins also recruit factors required for DNA replication.
2.3. Partitioning of Extrachromosomal DNA in Dividing Cells
There are numerous ways in which extrachromosomal DNA can be effectively partitioned to daughter cells (Figure 2). In bacteria, low copy plasmids use specialized partitioning mechanisms, but high copy plasmids are partitioned by random diffusion without tethering to host structures (Figure 2a). In eukaryotes, extrachromosomal DNA must additionally be retained in the nucleus after cell division, and so it is advantageous to directly associate with the spindle apparatus through centromere-like elements (Figure 2b) or piggyback on host chromosomes (Figure 2c,d). DNA synthesis of extrachromosomal DNA can take place in close association with host DNA, with replicated viral genomes remaining associated with chromatids until they are faithfully separated in mitosis (Figure 2d). Alternatively, extrachromosomal DNA can replicate in a separate nuclear location and subsequently randomly associate with host chromosomes (Figure 2c). The viruses reviewed here distribute their genomes using the schemes shown in Figure 2c,d. Almost 90% of EBV genomes are observed associated with host chromosomes as pairs until anaphase and so partition in a quasi-faithful manner (9) (Figure 2d). While KSHV genomes are synthesized while tethered to host chromatin, they partition nonequally in clusters (Figure 2c), resulting in a larger number of genomes in fewer cells (10). The small PV genome size makes analogous experiments extremely challenging.
Figure 2.

Different models of plasmid partitioning. (a) Viral genomes or extrachromosomal plasmids (purple circles) randomly distributed throughout the mitotic cell (singly or in clusters), (b) attached to the microtubules of the spindle, (c) randomly attached to chromosomes, and (d) attached pairwise to sister chromatids. The pink bar represents a topological or protein link between daughter molecules.
2.4. Papillomavirus Replication
PV DNA synthesis requires two viral proteins and is dependent on cellular factors (11). E1 is a sequence-specific helicase that binds and unwinds the viral origin of replication. E2 is a multifunctional protein that regulates viral transcription, promotes initiation of DNA replication, and partitions the viral genomes by tethering them to mitotic chromosomes. The replication origin is in the upstream regulatory region (URR) of the genome and consists of overlapping E1 binding sites (E1BSs) flanked by E2 binding sites (E2BSs) (12) (Figure 3a). E1 binds to the origin cooperatively with E2 (13), E2 dissociates, and E1 converts into a double hexamer that unwinds the genome in a bidirectional manner (14). Cellular replication proteins are recruited to the replication fork to promote theta-mode replication of the viral DNA.
Figure 3.

Comparison of cis-elements required for stable genome maintenance. (a) The URRs from two PV types are shown. The consensus E2BSs are indicated by purple circles and the E1BS by a green rectangle. Minimal origins and MMEs are indicated. (b) EBV oriP contains a DS and an FR element. The DS element is required for initiation of replication and contains four EBNA1 BSs (green circles). The FR element contains multiple 30-bp tandem repeats (light purple box), each containing an EBNA1 BS, and is required for genome maintenance and partitioning. (c) Each repeat in the KSHV TR element (light purple box) contains three LANA binding sites (green circles), one of which (darker green) overlaps with an element required for initiation of DNA synthesis (42, 79). Abbreviations: BPV1, Bovine papillomavirus; DS, dyad symmetry; E1BS, E1 binding site; E2BS, E2 binding site; EBNA1 BS, Epstein-Barr virus nuclear antigen 1 binding site; FR, family of repeat; HPV16, human papillomavirus 16; KSHV TR, Kaposi’s sarcoma-associated herpesvirus terminal repeat; LANA, latency-associated nuclear antigen; MME, minichromosome maintenance element; MMEE; minichromosome maintenance enhancer element; oriP, viral replication origin; PV, papillomavirus; URR, upstream regulatory region.
Upon entry into the cell, the L1 capsid protein is dissociated from the virus particle, and the viral genome, in complex with the L2 minor capsid protein, accesses the nucleus encased in endocytic vesicles. After nuclear membrane breakdown in mitosis, the L2 genome vesicles bind to condensed host mitotic chromosomes (15, 16). After cell division, viral genomes associate with nuclear domain 10 bodies; paradoxically, these are important for antiviral defense, but many DNA viruses initiate the infectious cycle here by taking advantage of some components and disrupting or degrading others (17). E1 and E2 proteins then initiate low-level amplification of viral DNA. Successful establishment of viral genomes requires evasion of intrinsic immune defenses and association with regions of transcriptionally active host chromatin (18). In addition to its role in initiation of viral DNA synthesis, E2 binds and partitions the viral genomes in approximately equal numbers into daughter cells during the maintenance phase of replication by tethering them to mitotic chromosomes (19, 20).
Typically, both E1 and E2 are required at each stage of PV replication, but there is evidence that E1 might be dispensable during the maintenance phase (21, 22). E1 induces a DNA damage response (DDR) in host nuclei, and so its expression and nuclear localization are tightly regulated (23–25). Under certain circumstances, E1 is probably dispensable for maintenance replication, and viral DNA synthesis is initiated by cellular proteins. Cellular DNA is licensed so that it replicates once per cell cycle, and there is keen interest in whether viral genomes that replicate with a stable copy number are similarly restricted. Bovine papillomavirus (BPV1) replicates by a random choice mechanism whereby some molecules replicate more than once per cell cycle and others not at all (20, 26). For alpha-HPVs, the replication mode varies in different cell and HPV types, but overexpression of E1 results in unlicensed, random replication (27). Thus, we surmise that in the absence of E1, HPV replication is initiated by cellular replication proteins and is likely licensed, but in the presence of E1, random choice replication is more likely.
Vegetative (productive) replication of PV genomes occurs in differentiated keratinocytes. Activation of the late promoter generates high levels of E1 and E2 to rapidly amplify viral DNA. However, the dependence on cellular replication proteins is problematic in differentiated cells not in S-phase, and so PVs induce a cellular DDR (28). This response recruits cellular factors to replication foci to synthesize DNA and resolve replication intermediates, and replication switches from theta-mode to recombination-dependent replication (RDR) (29).
2.5. Epstein-Barr Virus Replication
The linear EBV genome is recircularized in the nucleus upon infection and amplified to a low copy number; successful genome establishment is relatively rare and requires association with beneficial regions of host chromatin (30). In the maintenance phase, viral genomes replicate once per cell cycle (licensed) in synchrony with the host DNA. Replication requires the viral replication origin (oriP) and EBNA1 protein (31). oriP contains two separable functional elements, the family of repeats (FRs) and the dyad symmetry (DS) (32), each of which contains multiple EBNA1 binding sites (Figure 3b). EBNA1 recruits cellular replication proteins to initiate DNA synthesis at the DS element and binds to FR to retain and partition the viral genomes. EBNA1 does this by tethering the viral genomes to host chromosomes, so that the genomes are faithfully or quasi-faithfully partitioned to daughter cells by association with sister chromatids (9, 33). The final phase of EBV DNA replication is initiated upon reactivation of latent EBV genomes in resting memory B cells. EBV encodes many additional proteins required for productive viral DNA synthesis, and replication switches to a rolling circle (RC)/RDR mode (34). As in many other viruses, DDR proteins are recruited to replication foci (35).
2.6. Kaposi’s Sarcoma-Associated Herpes Virus Replication
Similar to those of EBV, linear KSHV genomes are recircularized upon infection by ligation of the terminal repeats (TRs); however, unlike in EBV, these repeats also function as the latent replication origin and partitioning element (36) (Figure 3c). After initial genome amplification and establishment, viral genomes are maintained at a constant copy number through licensed DNA replication and partitioning by the viral LANA protein (37). LANA binds to the LANA binding sites (LBSs) in the TRs and tethers the genomes to host chromosomes (36, 38). Although KSHV genomes are partitioned in approximately equivalent numbers per cellular division, live cell imaging shows that viral genomes cluster at tethering sites and partition somewhat randomly, resulting in loss of KSHV genomes in some daughter cells (10). Finally, as in EBV, lytic reactivation leads to induction of virally encoded replication proteins, a switch from theta-mode DNA synthesis to RC/RDR replication (34) and recruitment of DDR factors to assist in DNA synthesis and resolution of replication intermediates (35).
3. VIRAL AND CELLULAR FACTORS INVOLVED IN TETHERING AND PARTITIONING VIRAL GENOMES
3.1. Viral Tethering Proteins
Tethering to host nuclear structures is critical for partitioning and nuclear retention of viral genomes in dividing cells. To this end, multi-functional viral proteins act as a physical tether between these nuclear structures and viral genomes. This section describes the viral tethering proteins.
3.1.1. Papillomavirus: E2 protein.
The full-length PV E2 protein (E2-TA or E2) plays roles in replication and transcription but also tethers viral and host genomes together during maintenance replication. E2 contains two conserved domains linked by a flexible hinge (Figure 4). The N-terminal transcriptional activation domain forms a cashew-shaped structure important for transcriptional regulation and interaction with E1. This domain interacts with multiple cellular proteins that influence replication, transcription, and tethering of E2 to host chromatin (39). The C-terminal domain of E2 binds specifically to 12-bp E2BS motifs in the viral genome. This DNA binding domain (DBD) dimerizes to form a stable, antiparallel beta-barrel core with recognition alpha helices on the surface that insert into the major groove of DNA and specifically interact with nucleotides of the conserved E2BS sequence (40). This structure is well conserved in E2 proteins from all PV genera and has a strong similarity to the dimeric DBDs of LANA and EBNA1, despite little sequence similarity among the three proteins (40–42). The conserved E2 domains are connected by a flexible and unstructured hinge region that is not required for transcription or replication functions. The hinge has low sequence complexity and is not well conserved, in sequence or length, among the E2 proteins of different PV genera. However, the hinges contain sites of post-translational modification that are conserved within each genus and regulate E2 half-life, nuclear location, protein-protein interactions, and tethering to host chromatin (39).
Figure 4.

Functional domains of the viral tethering proteins. Although having unrelated sequences, the tethering proteins have a comparable modular organization and the DNA binding dimerization domains (purple) have similar structures. The regions important for host chromatin association are shown in green. These domains are linked by repetitive sequences (light purple). Abbreviations: EBNA1, Epstein-Barr virus nuclear antigen 1; LANA, latency-associated nuclear antigen; PV, papillomavirus.
3.1.2. Epstein-Barr virus: EBNA1.
The N-terminal half of EBNA1 contains two regions important for replication, transcriptional regulation, and tethering (domains A and B, or linking regions 1 and 2) separated by a long glycine-alanine repeat region (Figure 4). The dimeric, sequence-specific DBD is located at the C terminus. EBNA1 chromatin association is mediated by two glycine-arginine-rich regions within domains A and B (43, 44). These regions function as AT hooks that bind AT-rich regions of DNA (45) and also to cellular proteins important for tethering to host chromatin (43, 46). The two domains are relatively equivalent in function, and two copies of either can mediate binding to host chromatin (44, 47, 48). EBNA1 binds to conserved motifs in the FR and DS region of oriP, the latent origin of replication. This is mediated by the C-terminal DBD that, akin to E2, forms a dimeric eight-stranded beta-barrel structure (41, 49). Site-specific DNA recognition is mediated by a surface alpha helix and an extended chain that extends into the DNA minor groove (41, 49, 50). During latent infection, EBNA1 is required to replicate and maintain the extrachromosomal viral genomes, and this provides a potential target for antigen recognition by circulating immune cells. The glycine-alanine repeat region is encoded by purine-rich sequences that inhibit antigen presentation. This is due to the formation of G-quadruplex structures that inhibit translation and reduce the formation of EBNA1-derived T cell epitopes (51).
3.1.3. Kaposi’s sarcoma-associated herpesvirus: LANA.
LANA has three distinct regions: a small N-terminal domain, a large central region containing multiple interspersed repeat and unique sequences, and a C-terminal DNA binding/dimerization domain (Figure 4). The 23 residue N-terminal domain contains a bipartite nuclear localization signal and a short chromatin binding domain (CBD) (52). The N-terminal domain is important for LANA-mediated transactivation, and residues 5–13 are necessary for tethering to chromatin (53–55). As in E2 and EBNA, the C-terminal domain of LANA is the specific DNA binding/dimerization domain that forms a beta-barrel core with surface recognition alpha helices that contact residues in the LBS motif (42, 56, 57). On the face of the domain opposite from the recognition helix is a basic patch that interacts with cellular factors and impacts KSHV plasmid maintenance (58, 59). The N-terminal CBD is primarily responsible for host chromosome binding, but sequences in the C-terminal DBD can also mediate binding to pericentromeric and telomeric regions of host chromosomes (60–62). The N- and C-terminal domains of LANA are separated by long stretches of repetitive sequences that, like EBNA1, inhibit antigen presentation (63), and computational analysis indicates that they may also form G-quadruplex structures that inhibit translation (51).
3.2. Viral Cis-Elements Required for Viral Plasmid Maintenance
Viral tethering proteins regulate replication, transcription and plasmid maintenance by binding to distinct cis elements located within the regulatory regions of PV, KSHV and EBV genomes. The cis-elements required for initiation of viral DNA synthesis and plasmid maintenance are often separable.
3.2.1. Papillomavirus E2 binding sites and enhancer elements.
E2 proteins bind consensus sites (ACCN6GGT) located within the URR (64) (Figure 3a). Two or three E2BSs flank an E1BS to form the replication origin (12, 13), and these E2BSs also regulate expression of the adjacent early promoter. There are additional E2BSs upstream in the URR, but the number varies greatly among different genera of PV (39). The deltapapillomavirus, BPV1, contains 12 E2BSs in the URR. A minichromosome maintenance element (MME), containing a transcriptional enhancer and six of the E2BSs, and the minimal replication origin are required for maintenance replication (20, 65). However, multimerized E2BSs can substitute for the MME. In contrast, most alphapapillomaviruses contain four E2BSs, and only the two to three adjacent to the origin are required for genome maintenance (66, 67). In addition to the minimal replication origin, the URR transcriptional enhancer is necessary for maintenance of an alphapapillomavirus HPV18 replicon (66). This minichromosome maintenance enhancer element contains binding sites for several cellular factors, which may impact the maintenance of the HPV18 genome (68).
3.2.2. Epstein-Barr virus: oriP (dyad symmetry and family of repeats).
EBV oriP, the latent origin of replication, is the only cis-element required to maintain the EBV genome; it contains multiple 30-bp repeats, each of which contains an EBNA1 binding site (69). oriP contains two separate elements, FR and DS (32, 70, 71). DS contains four 30-bp repeats and is necessary for initiation of DNA synthesis but unnecessary for genome maintenance (72, 73). FR contains 20 tandem repeats and is required for genome maintenance and partitioning (69, 70, 74, 75). At least seven EBNA1 BSs are required for EBV plasmid maintenance (76).
3.2.3. Kaposi’s sarcoma-associated herpesvirus: terminal repeat.
In contrast to that of EBV, the KSHV latent replication origin is formed by recircularization of the 801-bp TR repeats. On average, there are 35–40 repeats, each of which contains three 18–20-bp LANA BS motifs (one overlaps the site of replication initiation) (42). As in EBV oriP, the LBSs are spaced optimally apart on the same DNA helix face, thus facilitating cooperative binding of adjacent LANA dimers (42, 77). The KSHV TR cis-element is both necessary and sufficient for initiation of DNA synthesis and genome maintenance (77–79). However, little is known about how many of the individual TR repeats initiate DNA synthesis on a single viral genome.
3.3. Cellular Partners of the Viral Tethering Proteins
E2, EBNA1, and LANA interact with many cellular proteins to facilitate viral transcriptional regulation, DNA synthesis, and partitioning. Some of these are proposed to directly mediate, or assist, in tethering viral genomes to host chromosomes, but it is difficult to separate their tethering role from functions related to viral transcription and replication. The following section lists cellular targets that may be involved in chromatin tethering.
3.3.1. Papillomavirus E2 protein.
Several cellular proteins have been postulated to be the anchor of the E2 tethering protein, but the strength and nature of these interactions vary among viral types, and often occurs only during discrete stages of mitosis. It is probable that multiple cellular factors participate in the tethering complex.
3.3.1.1. Transcriptional and epigenetic regulators: Brd4.
Brd4 (bromodomain and extraterminal domain protein 4) is the best-studied candidate for PV chromosomal attachment. It binds acetylated histones in cellular enhancers and superenhancers, recruits transcription factors, and promotes transcriptional elongation (80, 81). Brd4 both activates and represses PV transcription and binds and colocalizes with many different E2 proteins on mitotic chromosomes (82–85). Two conserved residues in the E2 transactivation domain interact with the C terminus of Brd4 (82, 83, 86). The Brd4-E2 protein association modulates viral transcription in all PVs studied, but the importance of Brd4 in tethering and partitioning viral genomes is more complex. E2 proteins that bind Brd4 with high affinity colocalize in punctate foci with Brd4 on mitotic chromosomes (84, 85). However, Brd4 binding to E2 proteins from the alpha genus (HPV11, HPV16, HPV18, HPV31) is weak and difficult to detect on mitotic chromosomes except in late telophase and under specific fixation conditions (85, 87). Figure 5 shows example of different PV E2 proteins associated with host mitotic chromosomes. Although Brd4 is the most-studied E2 chromosomal partner, differences in this association among different PV types indicate that additional cellular factors are required.
Figure 5.

Variations in the pattern of E2 proteins binding to mitotic chromosomes. Mitotic cells were briefly extracted before fixation to stabilize HPV31 E2 binding. Figure reproduced from Reference 92a and reused with permission under a CC-BY license.
Co-expression of alpha-PV E1 and E2 results in nuclear foci that recruit Brd4, and Brd4 localizes on the surface of HPV31 late replication foci (88). This localization could represent transcriptionally active regions of viral DNA or the interface between viral and host chromatin. Brd4 is required for foci formation but is not absolutely required for E1-E2-dependent replication or HPV31 genome maintenance (88–91).
3.3.1.2. Chromosomal architectural proteins.
Replication, repair, condensation, and faithful partitioning of cellular chromosomes require the concerted efforts of the structural maintenance of chromosome (SMC architectural proteins), condensin (SMC2/4), cohesin (SMC1/3), and SMC5/6. Although these proteins have not yet been demonstrated to be directly involved in the PV genome partitioning mechanism, they do influence maintenance replication. SMC1/CTCF complexes bind the late region of HPV genomes and promote maintenance and vegetative replication (92). SMC5/6 bind to PV E2 proteins, localize to replication foci, and are required for maintenance replication (93). Another E2 interacting protein, the DNA helicase ChlR1, links replication to chromatid cohesion (94). ChlR1 promotes the establishment of E2 and viral genomes with chromatin in the early stages of mitosis, but this interaction does not persist through mitosis and thus ChlR1 does not directly partition viral DNA (95). Similarly, topoisomerase ll binding protein 1 localizes with E2 on mitotic chromosomes only in telophase (87, 90).
3.3.1.3. Mitotic checkpoint proteins, mitotic spindles, and Mklp2.
When overexpressed, HPV11, −16, and −18 E2 proteins colocalize with mitotic spindle fibers (96). Similarly, some E2s interact with spindle assembly checkpoint proteins BubR1, Mad2, and Cdc20 (97) and mitotic kinesin-like protein 2 (Mklp2) (98). These interactions have not been shown to mediate partitioning of viral DNA and may reflect the promiscuity of E2 binding.
3.3.2. EBNA1.
EBNA1 interacts with both cellular proteins and nucleic acids. As described for PV E2, it is probable that multiple cellular factors participate in the EBV tethering complex.
3.3.2.1. Interaction with AT-rich host DNA by EBNA1 AT hooks.
The GR-rich domains A/B of EBNA1 have AT hook DNA binding properties and bind to AT-rich regions of host chromatin to maintain EBV plasmids (45). Inhibiting AT hook binding blocks this association and reduces EBV plasmid maintenance (99). Furthermore, the AT hooks from high motility group 1a, when fused to the EBNA1 DBD, support EBV oriP-dependent plasmid maintenance (100, 101).
3.3.2.2. EBP2.
EBNA1 binding protein 2 (EBP2) is a nucleolar protein involved in rRNA processing that binds the GR-rich CBDs of EBNA1 (43). This interaction was originally thought to mediate EBNA1 mitotic chromosome association and EBV plasmid maintenance (43). However, EBNA1 binds mitotic chromosomes throughout mitosis while EBP2 colocalizes with EBNA1 from late prophase onward (48). Therefore, EBP2 might stabilize EBNA1 chromosomal association but does not directly mediate attachment.
3.3.2.3. Transcriptional and epigenetic regulators: Brd4.
EBNA1 interacts with Brd4 through a region close to CB1 (residues 61–83) and localizes to the FR element of oriP (102). This interaction is important for EBNA1-mediated transcriptional activation.
3.3.2.4. G-quadruplex RNA.
EBNA1 binds to G-quadruplex RNA through RGG motifs in the chromatin binding regions; this is required for ORC recruitment for viral DNA synthesis (103). Small molecules that disrupt G-quadruplexes inhibit the ORC-EBNA1 association, viral DNA replication, and EBNA1 binding to mitotic chromosomes (104). It remains to be determined whether this interaction is a primary, additive, or alternative tethering mechanism.
3.3.2.5. Chromatin binding protein: RCC1.
Regulator of chromosome condensation 1 (RCC1) is an interaction partner of EBNA1 (105). RCC1 interacts directly with cellular histones, and EBNA1 binding to RCC1 is mediated through its CBDs; both proteins colocalize during interphase and mitosis, and inhibition of this interaction disrupts EBNA1 chromatin association (105). Further studies are required to determine the precise role of this essential protein in EBV genome partitioning.
3.3.3. Kaposi’s sarcoma-associated herpesvirus LANA.
Similar to E2 and EBNA1, KSHV LANA associates with numerous cellular factors and many have been proposed to be relevant for viral genome maintenance.
3.3.3.1. Nucleosomes: histone H2A/H2B interface.
The N-terminal LANA CBD residues form a hairpin structure that associates with the H2A/H2B interface of nucleosomes and mediates LANA’s association with mitotic chromosomes (52). This interaction is the dominant chromosome attachment but could be modulated by the factors described below.
3.3.3.2. Chromatin binding proteins: MeCP2 and DEK.
The C-terminal domain of LANA localizes to the heterochromatin at pericentromeric regions of chromosomes, and this correlates with binding to methyl CpG binding protein 2 (MeCP2) (106). The abundant chromatin architectural protein DEK also interacts with the C-terminal LANA domain but does not display the same punctate patterning on mitotic chromosomes (60).
3.3.3.3. Mitotic checkpoint proteins, spindle, and kinetochore.
The nuclear mitotic apparatus interacts with the LANA C-terminal domain during interphase, but this interaction is lost at the onset of mitosis (107). LANA also forms a complex with Bub1 and CENP-F, but only Bub1 and LANA remain colocalized throughout mitosis (108). Further study is needed to determine the role of Bub1 in KSHV genome maintenance.
3.3.3.4. Transcriptional and epigenetic regulators: Brd2/Brd4.
The Bromodomain and Extra-Terminal Domain proteins Brd2 and Brd4 interact with the electrostatic patch on the LANA DBD (38, 109). Brd4 and LANA colocalize with KSHV genomes on cellular chromosomes throughout mitosis, while Brd2 partially binds chromosomes and only in the presence of KSHV TR plasmids (109). Notably, Brd4 is one of the few cellular target interactions shared by all three viral tethering proteins.
4. CHROMOSOME REGIONS BOUND BY VIRAL TETHERING COMPLEX
4.1. Specific Chromosome Regions
Viral tethering proteins are detected on mitotic chromosomes either alone or in complex with their respective viral genome. Full-length LANA and EBNA1 show a diffuse binding pattern in the absence of viral DNA, likely reflecting the ubiquitous nature of their histone- or AT-rich DNA targets (55). In the presence of viral DNA, the protein-DNA complexes are observed as punctate dots across all chromosomes (33).
PV E2 proteins that bind with high affinity to Brd4 are observed colocalized with Brd4 in punctate speckles distributed randomly over mitotic chromosomes, and PV genomes are distributed in a similar pattern (85). As described below, this binding may be related to regions of chromosomes undergoing replication stress. Betapapillomavirus E2 (HPV8) proteins bind to the rDNA loci located on acrocentric chromosomes; this is mediated by the E2 DBD and a chromatin binding peptide from the hinge region. When this peptide is mutated, HPV8 E2 binds to chromosomes with Brd4 as observed for other E2 proteins (110, unpublished data).
The alphapapillomavirus E2 proteins bind Brd4 with low affinity and are not easily detected on mitotic chromosomes (111). However, when analyzed by immunofluorescence after a pre-extraction technique, these E2s are also observed bound to the short arms of acrocentric chromosomes (85). Notably, the LANA DBD also binds pericentromeric/telomeric regions of specific mitotic chromosomes through the secondary C-terminal CBD (60, 61).
4.2. Euchromatin and Transcriptionally Active Regions
All three viral tethering proteins bind to transcriptionally active host chromatin, although this might be associated with their role in viral transcriptional regulation. For example, E2 binds with Brd4 to transcriptionally active cellular promoters but does not alter gene expression, and binding does not persist through mitosis (18, 89). E2 binding to these promoters is Brd4 dependent and not due to direct DNA binding; it likely reflects the importance of associating with euchromatic, active chromatin that is unlikely to be silenced (18).
LANA also associates with sites of host active transcription and does not alter the transcriptional profile (112, 113). In contrast, EBNA1 binds to both active and repressed host chromatin, including Long interspersed nuclear element-1 retrotransposons (114). EBNA1 also upregulates transcription of many associated cellular genes and relieves compaction at targeted sites through H1 displacement (115).
In situ Hi-C analysis of interaction between viral and host DNA revealed differences between the chromosomal interactions of the gammaherpesviruses and PVs. As predicted, HPV plasmids are tethered to gene-rich, euchromatic regions, but EBV interacted with gene-poor, heterochromatic regions (116). This could be because HPVs undergo low-level, persistent infection in the cell types analyzed while gammaherpesvirus establish a more silent, latent infection. Upon reactivation, the EBV genomes move toward active euchromatin, presumably to facilitate the productive phase of the viral replication cycle. Thus, the host chromatin tethering site may not be static through the viral replication cycle.
4.3. Common Fragile Sites
The punctate PV E2-Brd4 chromosomal target sites mapped by chromatin immunoprecipitation techniques have properties characteristic of common fragile sites (CFSs) (117). CFSs are regions of the host genome undergoing replication stress and could represent an advantageous location for a virus that uses the DDR for productive DNA replication. Late replication foci form more frequently adjacent to these sites (117). One consequence of viral genome tethering at CFSs is the risk of integration into the host genome, as observed in many HPV-associated cancers (118).
4.4. rDNA
Betapapillomavirus E2s associate with the repetitive ribosomal DNA genes on the short arms of acrocentric chromosomes (110, 119). This interaction is mitosis specific, as rDNA resides in the nucleolus during interphase, and E2 binds only after nucleolar disassembly in mitosis (120). Under specific fixation conditions, alpha-PV E2s also bind this location (85, 120). The repetitive rDNA genes are prone to genetic instability and could serve as a DNA damage sensor (121), making them an attractive target for a virus that requires the DDR to facilitate viral DNA amplification.
5. REGULATION OF VIRAL GENOME PARTITIONING
5.1. Post-Translational Modification
Post-translational modifications of the viral tethering proteins impact stability, replication and transactivation functions, nuclear localization, and chromatin interaction. Multiple phosphorylation sites have been identified within the hinges of the PV E2 proteins. In BPV1, CK2 phosphorylation induces proteosomal degradation and regulates BPV1 genome copy number during maintenance replication (122, 123). In contrast, phosphorylation of the HPV8 E2 hinge by PKA at a conserved RXXS motif stabilizes E2 and is essential for E2 host chromosome association (110, 120). In alphapapillomaviruses, phosphorylation at a nonconserved RXXS motif in the HPV16 E2 hinge increases protein stability and promotes interaction with Brd4 and host chromosomes (124).
EBNA1 is modified by serine/threonine phosphorylation and arginine methylation, particularly in the RXXS motif in GR-rich domain A. Phosphorylation regulates EBNA1 functions in plasmid maintenance and interaction with cellular proteins (i.e., EBP2) (125, 126). EBNA1 methylation is important for proper nuclear distribution of EBNA1; loss of EBNA1 methylation results in EBNA1 localization to the nucleolar periphery (125).
LANA has a multitude of post-translational modifications. RSK3 phosphorylates serine/threonine residues 13/14 within the CBD to stabilize the protein and promote H2B interaction (127). DNA-PK phosphorylates LANA between residues 31–52 and 91–340, and a kinase recruited by Brd2 phosphorylates the C-terminal domain. LANA is also arginine methylated, acetylated, sumoylated, and poly ADP-ribosylated (38). These modifications impact protein interactions and viral genome copy number.
Notably, all three tethering proteins contain RXXS motifs (often located in the chromosomal attachment domains) targeted by arginine methylation and/or serine phosphorylation. Post-translational modifications of these motifs impact protein stability and protein function.
5.2. Truncated Viral Repressor Proteins
All PVs encode a second E2 protein (E8Ê2) that consists of a short basic peptide (E8) fused to the hinge and DBD of E2 (128). E8Ê2 antagonizes the function of E2-TA in transcription and replication by forming heterodimers and competitively binding to E2BSs. The E8 moiety also recruits transcriptional repressor complexes to viral genomes (129). In the absence of E8Ê2 expression, HPV genomes cannot stably maintain extrachromosomal viral genomes because of the propensity to enter unrestricted, productive DNA replication (130–132).
5.3. Higher-Order Multimerization
The formation of higher-order structures of the viral tethering proteins is central to genome tethering and partitioning. The DNA binding function of BPV1 E2 is not required for its association with Brd4 and host chromatin, but the dimerization function greatly enhances this activity (133). Accordingly, a dimeric E2 protein with a single transactivation domain cannot support maintenance replication (134). Dimerization of E2 through the C-terminal domain allows two transactivation domains to interact with chromosomal targets, promoting the formation of multimeric complexes consistent with the punctate E2 speckles on mitotic chromosomes. The CBDs of E2 and EBNA also self-associate to form DNA loops between individual DNA-bound dimers, although the significance of this is not known (47, 135, 136). The dimeric DBDs of LANA and EBNA1 form higher-order oligomeric structures (42, 57, 58, 137). This oligomerization is required for cooperative DNA binding and maintenance replication and is responsible for the nuclear speckles formed by the proteins. (47, 135, 136). LANA bound to KSHV DNA binds the histone H2A/B interface on cellular chromatin but can also interact with histones on viral chromatin, promoting the observed clusters of viral genomes associated with mitotic chromosomes (10).
5.4. Epigenetic Modifications of Host and Viral DNA
During persistent/latent infection, viral DNA is packaged in cellular nucleosomes that are subject to a wide range of epigenetic modifications (138). The viral tethering proteins directly influence these modifications to modulate the viral transcriptional program as well as the efficiency of genome maintenance and partitioning (139).
Supplementary Material
SUMMARY POINTS.
Several dsDNA oncogenic viruses cause persistent infection that can last the lifetime of the host.
These viruses often have viral genomes that replicate as stable copy number, extrachromosomal dsDNA circular plasmids in the nucleus of infected cells.
A robust mechanism is required to maintain extrachromosomal viral genomes in dividing cells; a viral tethering protein binds to viral DNA and links it to host chromosomes to partition the genomes to daughter cells.
FUTURE ISSUES.
Disruption of binding of the viral tethering complex to host chromosomes is an attractive therapeutic target that could cure viral infection.
Novel, more sensitive technologies will allow a more detailed understanding of the tethering and partitioning process in living cells and tissues.
Understanding how extrachromosomal viral DNA escapes detection has important implications for gene therapy.
ACKNOWLEDGMENTS
Research of the DNA Tumor Virus Section is supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Van Doorslaer K 2013. Evolution of the papillomaviridae. Virology 445:11–20 [DOI] [PubMed] [Google Scholar]
- 2.Mariggio G, Koch S, Schulz TF. 2017. Kaposi sarcoma herpesvirus pathogenesis. Philos. Trans. R. Soc. B Biol. Sci 372:20160275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stanley MA. 2012. Epithelial cell responses to infection with human papillomavirus. Clin. Microbiol. Rev 25:215–22 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Doorslaer K, Li Z, Xirasagar S, Maes P, Kaminsky D, et al. 2017. The Papillomavirus Episteme: a major update to the papillomavirus sequence database. Nucleic Acids Res 45:D499–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McBride AA. 2017. Oncogenic human papillomaviruses. Philos. Trans. R. Soc. B Biol. Sci 372:20160273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Thorley-Lawson DA. 2015. EBV persistence—introducing the virus. Curr. Top. Microbiol. Immunol 390:151–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zimmermann J, Hammerschmidt W. 1995. Structure and role of the terminal repeats of Epstein-Barr virus in processing and packaging of virion DNA. J. Virol 69:3147–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kranjec C, Doorbar J. 2016. Human papillomavirus infection and induction of neoplasia: a matter of fitness. Curr. Opin. Virol 20:129–36 [DOI] [PubMed] [Google Scholar]
- 9.Nanbo A, Sugden A, Sugden B. 2007. The coupling of synthesis and partitioning of EBV’s plasmid replicon is revealed in live cells. EMBO J 26:4252–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chiu YF, Sugden AU, Fox K, Hayes M, Sugden B. 2017. Kaposi’s sarcoma-associated herpesvirus stably clusters its genomes across generations to maintain itself extrachromosomally. J. Cell Biol 216:2745–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ustav M, Stenlund A. 1991. Transient replication of BPV-1 requires two viral polypeptides encoded by the E1 and E2 open reading frames. EMBO J 10:449–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ustav M, Ustav E, Szymanski P, Stenlund A. 1991. Identification of the origin of replication of bovine papillomavirus and characterization of the viral origin recognition factor E1. EMBO J 10:4321–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mohr IJ, Clark R, Sun S, Androphy EJ, MacPherson P, Botchan MR. 1990. Targeting the E1 replication protein to the papillomavirus origin of replication by complex formation with the E2 transactivator. Science 250:1694–99 [DOI] [PubMed] [Google Scholar]
- 14.Sanders CM, Stenlund A. 1998. Recruitment and loading of the E1 initiator protein: an ATP-dependent process catalysed by a transcription factor. EMBO J 17:7044–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DiGiuseppe S, Luszczek W, Keiffer TR, Bienkowska-Haba M, Guion LG, Sapp MJ. 2016. Incoming human papillomavirus type 16 genome resides in a vesicular compartment throughout mitosis. PNAS 113:6289–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aydin I, Villalonga-Planells R, Greune L, Bronnimann MP, Calton CM, et al. 2017. A central region in the minor capsid protein of papillomaviruses facilitates viral genome tethering and membrane penetration for mitotic nuclear entry. PLOS Pathog 13:e1006308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.McBride AA. 2017. Mechanisms and strategies of papillomavirus replication. Biol. Chem 398:919–27 [DOI] [PubMed] [Google Scholar]
- 18.Jang MK, Kwon D, McBride AA. 2009. Papillomavirus E2 proteins and the host BRD4 protein associate with transcriptionally active cellular chromatin. J. Virol 83:2592–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skiadopoulos MH, McBride AA. 1998. Bovine papillomavirus type 1 genomes and the E2 transactivator protein are closely associated with mitotic chromatin. J. Virol 72:2079–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Piirsoo M, Ustav E, Mandel T, Stenlund A, Ustav M. 1996. Cis and trans requirements for stable episomal maintenance of the BPV-1 replicator. EMBO J 15:1–11 [PMC free article] [PubMed] [Google Scholar]
- 21.Kim K, Lambert PF. 2002. E1 protein of bovine papillomavirus 1 is not required for the maintenance of viral plasmid DNA replication. Virology 293:10–14 [DOI] [PubMed] [Google Scholar]
- 22.Egawa N, Nakahara T, Ohno S, Narisawa-Saito M, Yugawa T, et al. 2012. The E1 protein of human papillomavirus type 16 is dispensable for maintenance replication of the viral genome. J. Virol 86:3276–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fradet-Turcotte A, Bergeron-Labrecque F, Moody CA, Lehoux M, Laimins LA, Archambault J. 2011. Nuclear accumulation of the papillomavirus E1 helicase blocks S-phase progression and triggers an ATM-dependent DNA damage response. J. Virol 85:8996–9012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fradet-Turcotte A, Moody C, Laimins LA, Archambault J. 2010. Nuclear export of human papillomavirus type 31 E1 is regulated by Cdk2 phosphorylation and required for viral genome maintenance. J. Virol 84:11747–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakakibara N, Mitra R, McBride AA. 2011. The papillomavirus E1 helicase activates a cellular DNA damage response in viral replication foci. J. Virol 85:8981–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gilbert DM, Cohen SN. 1987. Bovine papilloma virus plasmids replicate randomly in mouse fibroblasts throughout S phase of the cell cycle. Cell 50:59–68 [DOI] [PubMed] [Google Scholar]
- 27.Hoffmann R, Hirt B, Bechtold V, Beard P, Raj K. 2006. Different modes of human papillomavirus DNA replication during maintenance. J. Virol 80:4431–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Moody CA, Laimins LA. 2009. Human papillomaviruses activate the ATM DNA damage pathway for viral genome amplification upon differentiation. PLOS Pathog 5:e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sakakibara N, Chen D, McBride AA. 2013. Papillomaviruses use recombination-dependent replication to vegetatively amplify their genomes in differentiated cells. PLOS Pathog 9:e1003321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leight ER, Sugden B. 2001. Establishment of an oriP replicon is dependent upon an infrequent, epigenetic event. Mol. Cell. Biol 21:4149–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yates JL, Guan N. 1991. Epstein-Barr virus-derived plasmids replicate only once per cell cycle and are not amplified after entry into cells. J. Virol 65:483–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yates J, Warren N, Reisman D, Sugden B. 1984. A cis-acting element from the Epstein-Barr viral genome that permits stable replication of recombinant plasmids in latently infected cells. PNAS 81:3806–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanda T, Kamiya M, Maruo S, Iwakiri D, Takada K. 2007. Symmetrical localization of extrachromosomally replicating viral genomes on sister chromatids. J. Cell Sci 120:1529–39 [DOI] [PubMed] [Google Scholar]
- 34.Chiu YF, Sugden B. 2018. Plasmid partitioning by human tumor viruses. J. Virol 92:e02170–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weitzman MD, Fradet-Turcotte A. 2018. Virus DNA replication and the host DNA damage response. Annu. Rev. Virol 5:141–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ballestas ME, Chatis PA, Kaye KM. 1999. Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen. Science 284:641–44 [DOI] [PubMed] [Google Scholar]
- 37.Stedman W, Deng Z, Lu F, Lieberman PM. 2004. ORC, MCM, and histone hyperacetylation at the Kaposi’s sarcoma-associated herpesvirus latent replication origin. J. Virol 78:12566–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Juillard F, Tan M, Li S, Kaye KM. 2016. Kaposi’s Sarcoma herpesvirus genome persistence. Front. Microbiol 7:1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.McBride AA. 2013. The papillomavirus E2 proteins. Virology 445:57–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hegde RS, Grossman SR, Laimins LA, Sigler PB. 1992. Crystal structure at 1.7 A of the bovine papillomavirus-1 E2 DNA-binding domain bound to its DNA target. Nature 359:505–12 [DOI] [PubMed] [Google Scholar]
- 41.Bochkarev A, Barwell JA, Pfuetzner RA, Furey W Jr., Edwards AM, Frappier L 1995. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein EBNA 1. Cell 83:39–46 [DOI] [PubMed] [Google Scholar]
- 42.Hellert J, Weidner-Glunde M, Krausze J, Lunsdorf H, Ritter C, et al. 2015. The 3D structure of Kaposi sarcoma herpesvirus LANA C-terminal domain bound to DNA. PNAS 112:6694–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Shire K, Ceccarelli DFJ, Avolio-Hunter TM, Frappier L. 1999. EBP2, a human protein that interacts with sequences of the Epstein-Barr virus nuclear antigen 1 important for plasmid maintenance. J. Virol 73:2587–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Marechal V, Dehee A, Chikhi-Brachet R, Piolot T, Coppey-Moisan M, Nicolas JC. 1999. Mapping EBNA-1 domains involved in binding to metaphase chromosomes. J. Virol 73:4385–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sears J, Ujihara M, Wong S, Ott C, Middeldorp J, Aiyar A. 2004. The amino terminus of Epstein-Barr virus (EBV) nuclear antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV genomes by tethering them to cellular chromosomes. J. Virol 78:11487–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kapoor P, Lavoie BD, Frappier L. 2005. EBP2 plays a key role in Epstein-Barr virus mitotic segregation and is regulated by aurora family kinases. Mol. Cell. Biol 25:4934–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mackey D, Sugden B. 1999. The linking regions of EBNA1 are essential for its support of replication and transcription. Mol. Cell. Biol 19:3349–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nayyar VK, Shire K, Frappier L. 2009. Mitotic chromosome interactions of Epstein-Barr nuclear antigen 1 (EBNA1) and human EBNA1-binding protein 2 (EBP2). J. Cell Sci 122:4341–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bochkarev A, Barwell JA, Pfuetzner RA, Bochkareva E, Frappier L, Edwards AM. 1996. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein, EBNA1, bound to DNA. Cell 84:791–800 [DOI] [PubMed] [Google Scholar]
- 50.Cruickshank J, Shire K, Davidson AR, Edwards AM, Frappier L. 2000. Two domains of the Epstein-Barr virus origin DNA-binding protein, EBNA1, orchestrate sequence-specific DNA binding. J. Biol. Chem 275:22273–77 [DOI] [PubMed] [Google Scholar]
- 51.Murat P, Zhong J, Lekieffre L, Cowieson NP, Clancy JL, et al. 2014. G-quadruplexes regulate Epstein-Barr virus-encoded nuclear antigen 1 mRNA translation. Nat. Chem. Biol 10:358–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, et al. 2006. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science 311:856–61 [DOI] [PubMed] [Google Scholar]
- 53.Wong LY, Matchett GA, Wilson AC. 2004. Transcriptional activation by the Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen is facilitated by an N-terminal chromatin-binding motif. J. Virol 78:10074–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Barbera AJ, Ballestas ME, Kaye KM. 2004. The Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen 1 N terminus is essential for chromosome association, DNA replication, and episome persistence. J. Virol 78:294–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Piolot T, Tramier M, Coppey M, Nicolas JC, Marechal V. 2001. Close but distinct regions of human herpesvirus 8 latency-associated nuclear antigen 1 are responsible for nuclear targeting and binding to human mitotic chromosomes. J. Virol 75:3948–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Correia B, Cerqueira SA, Beauchemin C, Pires de Miranda M, Li S, et al. 2013. Crystal structure of the gamma-2 herpesvirus LANA DNA binding domain identifies charged surface residues which impact viral latency. PLOS Pathog 9:e1003673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Domsic JF, Chen HS, Lu F, Marmorstein R, Lieberman PM. 2013. Molecular basis for oligomeric-DNA binding and episome maintenance by KSHV LANA. PLOS Pathog 9:e1003672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hellert J, Weidner-Glunde M, Krausze J, Richter U, Adler H, et al. 2013. A structural basis for BRD2/4-mediated host chromatin interaction and oligomer assembly of Kaposi sarcoma-associated herpesvirus and murine gammaherpesvirus LANA proteins. PLOS Pathog 9:e1003640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li S, Tan M, Juillard F, Ponnusamy R, Correia B, et al. 2015. The Kaposi sarcoma herpesvirus latency-associated nuclear antigen DNA binding domain dorsal positive electrostatic patch facilitates DNA replication and episome persistence. J. Biol. Chem 290:28084–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krithivas A, Fujimuro M, Weidner M, Young DB, Hayward SD. 2002. Protein interactions targeting the latency-associated nuclear antigen of Kaposi’s sarcoma-associated herpesvirus to cell chromosomes. J. Virol 76:11596–604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kelley-Clarke B, Ballestas ME, Komatsu T, Kaye KM. 2007. Kaposi’s sarcoma herpesvirus C-terminal LANA concentrates at pericentromeric and peri-telomeric regions of a subset of mitotic chromosomes. Virology 357:149–57 [DOI] [PubMed] [Google Scholar]
- 62.Kelley-Clarke B, Ballestas ME, Srinivasan V, Barbera AJ, Komatsu T, et al. 2007. Determination of Kaposi’s sarcoma-associated herpesvirus C-terminal latency-associated nuclear antigen residues mediating chromosome association and DNA binding. J. Virol 81:4348–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zaldumbide A, Ossevoort M, Wiertz EJ, Hoeben RC. 2007. In cis inhibition of antigen processing by the latency-associated nuclear antigen I of Kaposi sarcoma herpes virus. Mol. Immunol 44:1352–60 [DOI] [PubMed] [Google Scholar]
- 64.Androphy EJ, Lowy DR, Schiller JT. 1987. Bovine papillomavirus E2 trans-activating gene product binds to specific sites in papillomavirus DNA. Nature 325:70–73 [DOI] [PubMed] [Google Scholar]
- 65.Li R, Knight J, Bream G, Stenlund A, Botchan M. 1989. Specific recognition nucleotides and their DNA context determine the affinity of E2 protein for 17 binding sites in the BPV-1 genome. Genes Dev 3:510–26 [DOI] [PubMed] [Google Scholar]
- 66.Van Doorslaer K, Chen D, Chapman S, Khan J, McBride AA. 2017. Persistence of an oncogenic papillomavirus genome requires cis elements from the viral transcriptional enhancer. mBio 8:e01758–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ustav M Jr., Castaneda FR, Reinson T, Mannik A, Ustav M 2015. Human papillomavirus type 18 cis-elements crucial for segregation and latency. PLOS ONE 10:e0135770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.O’Connor M, Chan SY, Bernard HU. 1995. Transcription factor binding sites in the long control region of genital HPVs In Human Papillomaviruses, pp. III21–40, Los Alamos, NM: Los Alamos Natl. Lab. [Google Scholar]
- 69.Rawlins DR, Milman G, Hayward SD, Hayward GS. 1985. Sequence-specific DNA binding of the Epstein-Barr virus nuclear antigen (EBNA-1) to clustered sites in the plasmid maintenance region. Cell 42:859–68 [DOI] [PubMed] [Google Scholar]
- 70.Reisman D, Yates J, Sugden B. 1985. A putative origin of replication of plasmids derived from Epstein-Barr virus is composed of two cis-acting components. Mol. Cell. Biol 5:1822–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yates JL, Warren N, Sugden B. 1985. Stable replication of plasmids derived from Epstein-Barr virus in various mammalian cells. Nature 313:812–15 [DOI] [PubMed] [Google Scholar]
- 72.Norio P, Schildkraut CL, Yates JL. 2000. Initiation of DNA replication within oriP is dispensable for stable replication of the latent Epstein-Barr virus chromosome after infection of established cell lines. J. Virol 74:8563–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ott E, Norio P, Ritzi M, Schildkraut C, Schepers A. 2011. The dyad symmetry element of Epstein-Barr virus is a dominant but dispensable replication origin. PLOS ONE 6:e18609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lupton S, Levine AJ. 1985. Mapping genetic elements of Epstein-Barr virus that facilitate extrachromosomal persistence of Epstein-Barr virus-derived plasmids in human cells. Mol. Cell. Biol 5:2533–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Harrison S, Fisenne K, Hearing J. 1994. Sequence requirements of the Epstein-Barr virus latent origin of DNA replication. J. Virol 68:1913–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wysokenski DA, Yates JL. 1989. Multiple EBNA1-binding sites are required to form an EBNA1-dependent enhancer and to activate a minimal replicative origin within oriP of Epstein-Barr virus. J. Virol 63:2657–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Garber AC, Hu J, Renne R. 2002. Latency-associated nuclear antigen (LANA) cooperatively binds to two sites within the terminal repeat, and both sites contribute to the ability of LANA to suppress transcription and to facilitate DNA replication. J. Biol. Chem 277:27401–11 [DOI] [PubMed] [Google Scholar]
- 78.Komatsu T, Ballestas ME, Barbera AJ, Kelley-Clarke B, Kaye KM. 2004. KSHV LANA1 binds DNA as an oligomer and residues N-terminal to the oligomerization domain are essential for DNA binding, replication, and episome persistence. Virology 319:225–36 [DOI] [PubMed] [Google Scholar]
- 79.Hu J, Renne R. 2005. Characterization of the minimal replicator of Kaposi’s sarcoma-associated herpesvirus latent origin. J. Virol 79:2637–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dey A, Chitsaz F, Abbasi A, Misteli T, Ozato K. 2003. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. PNAS 100:8758–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. 2005. The bromodomain protein Brd4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol. Cell 19:523–34 [DOI] [PubMed] [Google Scholar]
- 82.You J, Croyle JL, Nishimura A, Ozato K, Howley PM. 2004. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 117:349–60 [DOI] [PubMed] [Google Scholar]
- 83.Baxter MK, McPhillips MG, Ozato K, McBride AA. 2005. The mitotic chromosome binding activity of the papillomavirus E2 protein correlates with interaction with the cellular chromosomal protein, Brd4. J. Virol 79:4806–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.McPhillips MG, Ozato K, McBride AA. 2005. Interaction of bovine papillomavirus E2 protein with Brd4 stabilizes its association with chromatin. J. Virol 79:8920–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Oliveira JG, Colf LA, McBride AA. 2006. Variations in the association of papillomavirus E2 proteins with mitotic chromosomes. PNAS 103:1047–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Abbate EA, Voitenleitner C, Botchan MR. 2006. Structure of the papillomavirus DNA-tethering complex E2:Brd4 and a peptide that ablates HPV chromosomal association. Mol. Cell 24:877–89 [DOI] [PubMed] [Google Scholar]
- 87.Donaldson MM, Boner W, Morgan IM. 2007. TopBP1 regulates human papillomavirus type 16 E2 interaction with chromatin. J. Virol 81:4338–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Sakakibara N, Chen D, Jang MK, Kang DW, Luecke HF, et al. 2013. Brd4 is displaced from HPV replication factories as they expand and amplify viral DNA. PLOS Pathog 9:e1003777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang X, Helfer CM, Pancholi N, Bradner JE, You J. 2013. Recruitment of Brd4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J. Virol 87:3871–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gauson EJ, Donaldson MM, Dornan ES, Wang X, Bristol M, et al. 2015. Evidence supporting a role for TopBP1 and Brd4 in the initiation but not continuation of human papillomavirus 16 E1/E2-mediated DNA replication. J. Virol 89:4980–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Stubenrauch F, Lim HB, Laimins LA. 1998. Differential requirements for conserved E2 binding sites in the life cycle of oncogenic human papillomavirus type 31. J. Virol 72:1071–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mehta K, Laimins L. 2018. Human papillomaviruses preferentially recruit DNA repair factors to viral genomes for rapid repair and amplification. mBio 9:e00064–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bentley P, Tan MJA, McBride AA, White EA, Howley PM. 2018. The SMC5/6 complex interacts with the papillomavirus E2 protein and influences maintenance of viral episomal DNA. J. Virol 92:e00356–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Samora CP, Saksouk J, Goswami P, Wade BO, Singleton MR, et al. 2016. Ctf4 links DNA replication with sister chromatid cohesion establishment by recruiting the Chl1 helicase to the replisome. Mol. Cell 63:371–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Harris L, McFarlane-Majeed L, Campos-Leon K, Roberts S, Parish JL. 2017. The cellular DNA helicase ChlR1 regulates chromatin and nuclear matrix attachment of the human papillomavirus 16 E2 protein and high-copy-number viral genome establishment. J. Virol 91:e01853–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Van Tine BA, Dao LD, Wu SY, Sonbuchner TM, Lin BY, et al. 2004. Human papillomavirus (HPV) origin-binding protein associates with mitotic spindles to enable viral DNA partitioning. PNAS 101:4030–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Tan CL, Teissier S, Gunaratne J, Quek LS, Bellanger S. 2015. Stranglehold on the spindle assembly checkpoint: The human papillomavirus E2 protein provokes BUBR1-dependent aneuploidy. Cell Cycle 14:1459–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yu T, Peng YC, Androphy EJ. 2007. Mitotic kinesin-like protein 2 binds and colocalizes with papillomavirus E2 during mitosis. J. Virol 81:1736–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chakravorty A, Sugden B. 2015. The AT-hook DNA binding ability of the Epstein Barr virus EBNA1 protein is necessary for the maintenance of viral genomes in latently infected cells. Virology 484:251–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sears J, Kolman J, Wahl GM, Aiyar A. 2003. Metaphase chromosome tethering is necessary for the DNA synthesis and maintenance of oriP plasmids but is insufficient for transcription activation by Epstein-Barr nuclear antigen 1. J. Virol 77:11767–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hung SC, Kang MS, Kieff E. 2001. Maintenance of Epstein-Barr virus (EBV) oriP-based episomes requires EBV-encoded nuclear antigen-1 chromosome-binding domains, which can be replaced by high-mobility group-I or histone H1. PNAS 98:1865–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lin A, Wang S, Nguyen T, Shire K, Frappier L. 2008. The EBNA1 protein of Epstein-Barr virus functionally interacts with Brd4. J. Virol 82:12009–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Norseen J, Thomae A, Sridharan V, Aiyar A, Schepers A, Lieberman PM. 2008. RNA-dependent recruitment of the origin recognition complex. EMBO J 27:3024–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Norseen J, Johnson FB, Lieberman PM. 2009. Role for G-quadruplex RNA binding by Epstein-Barr virus nuclear antigen 1 in DNA replication and metaphase chromosome attachment. J. Virol 83:10336–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Deschamps T, Bazot Q, Leske DM, MacLeod R, Mompelat D, et al. 2017. Epstein-Barr virus nuclear antigen 1 interacts with regulator of chromosome condensation 1 dynamically throughout the cell cycle. J. Gen. Virol 98:251–65 [DOI] [PubMed] [Google Scholar]
- 106.Matsumura S, Persson LM, Wong L, Wilson AC. 2010. The latency-associated nuclear antigen interacts with MeCP2 and nucleosomes through separate domains. J. Virol 84:2318–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Si H, Verma SC, Lampson MA, Cai Q, Robertson ES. 2008. Kaposi’s sarcoma-associated herpesvirus-encoded LANA can interact with the nuclear mitotic apparatus protein to regulate genome maintenance and segregation. J. Virol 82:6734–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Xiao B, Verma SC, Cai Q, Kaul R, Lu J, et al. 2010. Bub1 and CENP-F can contribute to Kaposi’s sarcoma-associated herpesvirus genome persistence by targeting LANA to kinetochores. J. Virol 84:9718–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.You J, Srinivasan V, Denis GV, Harrington WJ Jr., Ballestas ME, et al. 2006. Kaposi’s sarcoma-associated herpesvirus latency-associated nuclear antigen interacts with bromodomain protein Brd4 on host mitotic chromosomes. J. Virol 80:8909–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sekhar V, Reed SC, McBride AA. 2010. Interaction of the betapapillomavirus E2 tethering protein with mitotic chromosomes. J. Virol 84:543–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.McPhillips MG, Oliveira JG, Spindler JE, Mitra R, McBride AA. 2006. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J. Virol 80:9530–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hu J, Yang Y, Turner PC, Jain V, McIntyre LM, Renne R. 2014. LANA binds to multiple active viral and cellular promoters and associates with the H3K4methyltransferase hSET1 complex. PLOS Pathog 10:e1004240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Mercier A, Arias C, Madrid AS, Holdorf MM, Ganem D. 2014. Site-specific association with host and viral chromatin by Kaposi’s sarcoma-associated herpesvirus LANA and its reversal during lytic reactivation. J. Virol 88:6762–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lu F, Wikramasinghe P, Norseen J, Tsai K, Wang P, et al. 2010. Genome-wide analysis of host-chromosome binding sites for Epstein-Barr virus nuclear antigen 1 (EBNA1). Virol. J 7:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Coppotelli G, Mughal N, Callegari S, Sompallae R, Caja L, et al. 2013. The Epstein-Barr virus nuclear antigen-1 reprograms transcription by mimicry of high mobility group A proteins. Nucleic Acids Res 41:2950–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Moquin SA, Thomas S, Whalen S, Warburton A, Fernandez SG, et al. 2017. The Epstein-Barr virus episome maneuvers between nuclear chromatin compartments during reactivation. J. Virol 92:e01413–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jang MK, Shen K, McBride AA. 2014. Papillomavirus genomes associate with BRD4 to replicate at fragile sites in the host genome. PLOS Pathog 10:e1004117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Thorland EC, Myers SL, Persing DH, Sarkar G, McGovern RM, et al. 2000. Human papillomavirus type 16 integrations in cervical tumors frequently occur in common fragile sites. Cancer Res 60:5916–21 [PubMed] [Google Scholar]
- 119.Poddar A, Reed SC, McPhillips MG, Spindler JE, McBride AA. 2009. The human papillomavirus type 8 E2 tethering protein targets the ribosomal DNA loci of host mitotic chromosomes. J. Virol 83:640–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Sekhar V, McBride AA. 2012. Phosphorylation regulates binding of the human papillomavirus type 8 E2 protein to host chromosomes. J. Virol 86:10047–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kobayashi T 2008. A new role of the rDNA and nucleolus in the nucleus-rDNA instability maintains genome integrity. Bioessays 30:267–72 [DOI] [PubMed] [Google Scholar]
- 122.Penrose KJ, McBride AA. 2000. Proteasome-mediated degradation of the papillomavirus E2-TA protein is regulated by phosphorylation and can modulate viral genome copy number. J. Virol 74:6031–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.McBride AA, Howley PM. 1991. Bovine papillomavirus with a mutation in the E2 serine 301 phosphorylation site replicates at a high copy number. J. Virol 65:6528–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Chang SW, Liu WC, Liao KY, Tsao YP, Hsu PH, Chen SL. 2014. Phosphorylation of HPV-16 E2 at serine 243 enables binding to Brd4 and mitotic chromosomes. PLOS ONE 9:e110882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shire K, Kapoor P, Jiang K, Hing MN, Sivachandran N, et al. 2006. Regulation of the EBNA1 Epstein-Barr virus protein by serine phosphorylation and arginine methylation. J. Virol 80:5261–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Duellman SJ, Thompson KL, Coon JJ, Burgess RR. 2009. Phosphorylation sites of Epstein-Barr virus EBNA1 regulate its function. J. Gen. Virol 90:2251–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Woodard C, Shamay M, Liao G, Zhu J, Ng AN, et al. 2012. Phosphorylation of the chromatin binding domain of KSHV LANA. PLOS Pathog 8:e1002972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Dreer M, van de Poel S, Stubenrauch F. 2017. Control of viral replication and transcription by the papillomavirus E8Ê2 protein. Virus Res 231:96–102 [DOI] [PubMed] [Google Scholar]
- 129.Dreer M, Fertey J, van de Poel S, Straub E, Madlung J, et al. 2016. Interaction of NCOR/SMRT repressor complexes with papillomavirus E8Ê2C proteins inhibits viral replication. PLOS Pathog 12:e1005556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Straub E, Dreer M, Fertey J, Iftner T, Stubenrauch F. 2014. The viral E8Ê2C repressor limits productive replication of human papillomavirus 16. J. Virol 88:937–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Lace MJ, Anson JR, Thomas GS, Turek LP, Haugen TH. 2008. The E8Ê2 gene product of human papillomavirus type 16 represses early transcription and replication but is dispensable for viral plasmid persistence in keratinocytes. J. Virol 82:10841–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Stubenrauch F, Hummel M, Iftner T, Laimins LA. 2000. The E8Ê2C protein, a negative regulator of viral transcription and replication, is required for extrachromosomal maintenance of human papillomavirus type 31 in keratinocytes. J. Virol 74:1178–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cardenas-Mora J, Spindler JE, Jang MK, McBride AA. 2008. Dimerization of the papillomavirus E2 protein is required for efficient mitotic chromosome association and Brd4 binding. J. Virol 82:7298–305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kurg R, Uusen P, Vosa L, Ustav M. 2010. Human papillomavirus E2 protein with single activation domain initiates HPV18 genome replication, but is not sufficient for long-term maintenance of virus genome. Virology 408:159–66 [DOI] [PubMed] [Google Scholar]
- 135.Sim J, Ozgur S, Lin BY, Yu JH, Broker TR, et al. 2008. Remodeling of the human papillomavirus type 11 replication origin into discrete nucleoprotein particles and looped structures by the E2 protein. J. Mol. Biol 375:1165–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Avolio-Hunter TM, Frappier L. 1998. Mechanistic studies on the DNA linking activity of Epstein-Barr nuclear antigen 1. Nucleic Acids Res 26:4462–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Deakyne JS, Malecka KA, Messick TE, Lieberman PM. 2017. Structural and functional basis for an EBNA1 hexameric ring in Epstein-Barr virus episome maintenance. J. Virol 91:e01046–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Lieberman PM. 2016. Epigenetics and genetics of viral latency. Cell Host Microbe 19:619–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Chen HS, Lu F, Lieberman PM. 2013. Epigenetic regulation of EBV and KSHV latency. Curr. Opin. Virol 3:251–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.