

Abstract
Over the past decade, advancements in high-throughput sequencing have greatly enhanced our knowledge of the mutational signatures responsible for hereditary hearing loss. In its present state, the field has a largely uncensored view of protein coding changes in a growing number of genes that have been associated with hereditary hearing loss, and many more that have been proposed as candidate genes. Sequencing data can now be generated using methods that have become widespread and affordable. The greatest hurdles facing the field concern functional validation of uncharacterized genes and rapid application to human diseases, including hearing and balance disorders. To date, over 30 hearing-related disease models exist in zebrafish. New genome editing technologies, including CRISPR/Cas9 will accelerate the functional validation of hearing loss genes and variants in zebrafish. Here, we discuss current progress in the field and recent advances in genome editing approaches.
Keywords: genetics of hearing loss, hereditary hearing loss, model organism, zebrafish, CRISPR/Cas9, base editing, genetic variants
Graphical Abstract
Hearing loss is a highly prevalent disorder, the incidence of which increases with age: At birth one to two of every 1,000 newborns are affected (Morton et al., 2006), yet by the eighth decade of life the disorder affects 60% of individuals (Davis, 1989). It has been long understood that hearing loss is associated with social isolation, delayed speech and language development, as well as increased risk of depression. However, the recent recognition that hearing loss is associated with dementia has recently uncovered another key risk factor for reduced quality of life (Livingston et al., 2017), and underscores the importance of understanding the molecular mechanisms of hearing loss and how they can be addressed by therapeutic treatment. In the past decade, an understanding of hidden hearing loss in seemingly normal hearing individuals has surfaced as another factor impacting quality of life. Although environmental contributions such as noise and drug exposures are long-recognized risk factors, a genetic susceptibility to hearing loss is thought to be responsible for over half of diagnoses (Morton et al., 2006). Particularly in developed countries, approximately 80% of congenital hearing loss is due to genetic causes and the remaining fraction is attributed to environmental factors (Shearer et al., 2017).
Genetic causes of hearing loss propagate several challenges: clinical heterogeneity manifests in the parameters used to characterize the type of hearing loss, including age-of-onset, severity, and appearance of additional clinical symptoms. Hearing loss apparent at birth is classified as congenital, whereas hearing loss with an onset before language development is called pre-lingual, and hearing loss starting after language acquisition is termed post-lingual. Presbycusis, or age-related hearing loss, is another category and is described as progressive. Hearing loss can be described as sensorineural, conductive, or mixed depending on the part of the auditory system that is affected. Severity of hearing loss is measured in decibels and is determined through a variety of physiologic and audiometric testing methods that are adapted to patient age. Hearing loss is further categorized based on the severity of the frequencies (high-, mid-, and low-frequency) that are affected. When hearing loss occurs as an isolated symptom, it is termed non-syndromic; if it presents as part of a constellation of symptoms, it is called syndromic. Adding to this complexity is the existence of syndromes that have a delayed clinical onset after hearing loss has been diagnosed; when these delays are present, it is initially impossible to make an accurate clinical diagnosis in the absence of molecular genetic data.
Descent into the genetics of hearing loss
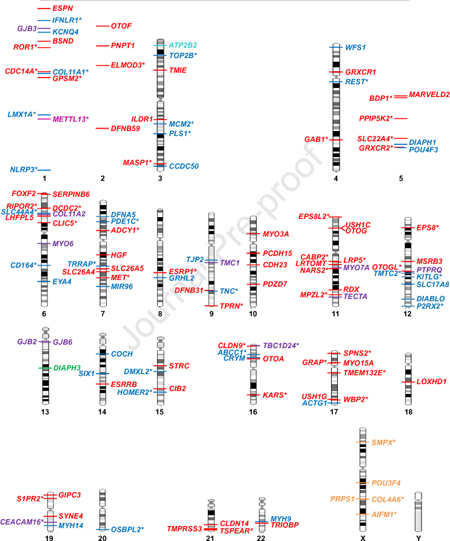
Given the complexity of the auditory system, it can be reasoned that hundreds of genes have essential roles in the auditory pathway. Presently, over 400 genes are associated with either non-syndromic or syndromic hearing loss based on searching the term “deafness” in the Human Gene Mutation Database (HGMD, version 2019.2). Of these, roughly 130 are associated with non-syndromic hearing loss (taken from the https://hereditaryhearingloss.org/, date last accessed, September 13, 2019 and recent literature) (Figure 1), and considerably more are associated with syndromic hearing loss. In some cases, different alleles of the same gene can underlie an autosomal dominant or recessive form of hearing loss, as well as either a syndromic or non-syndromic outcome. The most recent data from large-scale mouse phenotype screens puts the current estimate of the number of genes involved in normal hearing at roughly 1,000 (Ingham et al., 2019). Taking advantage of standardized nomenclature that has been assigned to gene loci, the Online Mendelian Inheritance in Man (https://www.omim.org/) database assigns gene locus identifiers to novel non-syndromic hearing loss genes as they are discovered depending on inheritance pattern: DFNA followed by a chronological number of discovery order denotes a locus that harbors a gene causing autosomal dominant non-syndromic hearing loss; DFNB is the locus identifier for autosomal recessive hearing loss; DFNX for X-linked; DFNY for Y-linked; and DFNM for genetic modifier loci. Other identifiers in use include AUNA for auditory neuropathy and OTSC for otosclerosis.
Figure 1.

Known non-syndromic hearing loss genes arranged according to cytogenetic location. Genes following autosomal dominant (blue), recessive (red), and those following both autosomal dominant and recessive patterns of inheritance (purple) are shown. Less common genes are X-linked (orange), associated with auditory neuropathy (green), modifiers of hearing loss (pink) and autosomal dominant non-syndromic deafness and a modifier of hearing loss (turquoise). Genes that were identified using high-throughput sequencing are marked with an asterisk. Updated and adapted from (Vona et al.) (Vona et al., 2016).
Gene identification in the time before the completion of the human genome project relied on large families and employed linkage studies based on microsatellite markers to map the chromosomal locus of hearing loss candidate genes. This type of analysis provided the first information about the chromosomal interval harboring the pathogenic variant, but often contained several dozens, if not hundreds, of genes. The genes in these intervals were either prioritized according to inner ear gene expression or were systematically Sanger sequenced until a putative pathogenic variant was identified. While positional gene mapping using linkage analysis has proven to be very successful, there still remain many linked deafness loci for which causative genes have not been identified. It is also worth considering that complex changes, such as copy number variation and chromosomal translocations, may be missed by conventional Sanger sequencing approaches.
Methodological advancements in sequencing
Considering the extreme genetic heterogeneity in hereditary hearing loss, one gene in particular, GJB2 (which maps to the DFNB1 and DFNA3A loci), is involved in a disproportionate fraction of genetic diagnoses. Pathogenic variants of GJB2 are associated with both autosomal recessive and autosomal dominant forms of hearing loss, although the autosomal recessive form prevails. Systematic reviews of the literature estimate a world-wide GJB2 diagnostic yield of 17.3% (Chan et al., 2014). GJB2-associated hearing loss is most prevalent in Spain and Eastern Europe, where it is reported to be involved in 40 to 50% of genetic diagnoses (Chan et al., 2014). Due to the relative ease of sequencing the single coding and non-coding exons that comprise this gene, direct sequencing of GJB2 is the most logical starting point for molecular genetic testing. Exclusionary testing of GJB2 and ethnic-specific deafness-associated pathogenic variants was identified as a valuable starting strategy (albeit with a limited total diagnostic yield), before working toward positional cloning of other genes in undiagnosed patients. However, the strategy became less promising as it became clear that the sheer number of genes involved was beyond what was feasible to test in a molecular genetic diagnostic setting at that time. The combination of milestone achievements in genomics, including the completion of the Human Genome Project in 2003, and advances in bioinformatics and computational power, now permit hypothesis-free genetic analysis that may range from a subset of genes with a strong gene-disease association to all coding genes in the human genome. This has revolutionized the possibilities and approaches used in molecular genetics research and diagnostic settings.
The past decade has witnessed a transformative shift with the advent and widespread accessibility of high-throughput sequencing approaches. This is reflected by the surge of genes that have been identified using these newer sequencing approaches (Figure 1, genes marked with asterisks, Figure 2). The most notable outcomes from the continued development and application of these technologies is the tremendous reduction in data generation cost and subsequent increase in sequencing data output. The largest bottleneck in sequencing data generation in molecular genetic diagnostics and novel gene identification studies is removed. However, it comes at the expense of increasingly complex demands for data interpretation because of the large number of novel variants identified across many genes.
Figure 2.

Non-syndromic hearing loss gene discovery over time. Black shows the number of genes identified per year since 1995. Grey shows the cumulative number of genes that have been identified. Genes that are associated with both autosomal recessive and autosomal dominant modes of inheritance are counted twice for each individual discovery.
These technologies have been seamlessly integrated into the diagnostic testing setting in the form of diagnostic testing of gene panels for targeted sequencing of over 100 genes or a much boarder approach, as with exome or genome sequencing and targeted gene analysis of known or candidate genes. The advantage of the latter is that analysis of existing exome or genome datasets for newly discovered genes is possible in undiagnosed patients, which opens potential avenues for continued diagnostic analysis. This will be increasingly important, as understanding of the genes involved in hearing loss becomes more complete with rapid gene discovery.
The mutational signatures of hereditary hearing loss
The high prevalence of hereditary hearing loss due to a single gene like GJB2 is the exception rather than the rule. In one of the largest molecular genetic epidemiology studies of hearing impaired patients to date, DNA samples from 1,119 patients were sequenced using various versions of hearing loss gene panels (Sloan-Heggen et al., 2016). The results indicated that 39% of patients received a diagnosis that involved genetic changes in 49 genes. The most frequently implicated gene was GJB2 (involved in 22% of diagnoses), followed by STRC (16%), SLC26A4 (7%), and TECTA (5%). Although 49 genes were implicated in the diagnoses of this hearing loss cohort, nearly three-fourths of the diagnoses were attributed to pathogenic and copy number variations in only 10 genes, with the remaining 39 genes making up a very small proportion of diagnoses (Sheffield et al., 2019; Sloan-Heggen et al., 2016). However, individually sequencing each of the 10 most frequently involved genes from this study with an alternative widespread technique such as Sanger sequencing is unfeasible in most molecular genetic diagnostic settings.
The classification of genetic variants represents a tremendous hurdle in genetic diagnostics and novel gene discovery. The Deafness Variation Database provides the best glimpse into the molecular landscape of 152 deafness-associated genes (Azaiez et al., 2018). In a recent study that described the interpretation and prioritization of genetic variants in 152 genes implicated in non-syndromic and syndromic deafness, the authors evaluated 876,139 variants and classified >8,100 as pathogenic or likely pathogenic, >172,000 variants as benign or likely benign, and >695,000 variants as having uncertain significance. The authors also re-categorized 1,270 variants (Azaiez et al., 2018). The take-home message of this study clearly highlights two main challenges in the genetics of deafness: (1) the vast majority of variants uncovered, even in well-studied genes, are variants with unknown significance, and (2) the notion of a secure genetic analysis and diagnosis in these genes is in flux, as illustrated by the re-categorization of so many variants.
Although the majority of molecular genetic diagnoses involve single nucleotide exchanges, insertion/deletion (indels), stop mutations (nonsense variants) and duplications at a single nucleotide resolution, genetic changes identifiable at lower resolutions are quite common and are involved in a significant fraction of diagnoses. The burden of copy number variation (deletion or duplication of genetic material comprising regions of 1 kilobase or larger) in the diagnostic yield of patients has proven to be substantial. Copy number variations were implicated in the molecular genetic diagnosis of approximately 18.7% patients from a large-scale study that included 686 hearing loss patients (Shearer et al., 2014). Additionally, chromosomal translocations, although apparently rare, may also contribute to the molecular spectrum of hereditary hearing loss through the interruption of critical gene sequences.
As many laboratories shift to genome sequencing technologies that provide information about non-protein coding sequences as possible contributors to inherited disease in humans, interpretation of genetic variants is likely to encounter a wave of complexity unlike we have seen before. Our current knowledge, largely restricted to coding sequences and adjacent non-coding sequences that coordinate splicing, has already uncovered hundreds of thousands of variants with uncertain significance, conflicting interpretations of pathogenicity, and many falsely classified variants and false gene-disease associations. This is a prime example of the fallout caused by post-genomics “growing pains” and is a major deterrent to the precision medicine movement, for which achieving a secure genetic diagnosis is crucial.
Although our understanding of the genes and variants involved in hearing loss has significantly advanced in the past 20 years (Vona et al., 2015), there is an urgent need to develop and utilize robust experimental systems to understand the function of newly identified genes. The last couple of decades have seen tremendous progress in identifying hearing loss genes thanks to inexpensive sequencing technologies that facilitated an increasing number of hereditary hearing loss screening projects. However, validation of genetic loci or genes that underlie the disease and determination of the pathological mechanisms of disease genes has been unacceptably slow, and has delayed progress toward the development of targeted therapies for hearing loss.
A proven approach to address these issues is to make use of animal models: mutated genes (designed to recapitulate mutations identified in human patients) are introduced into the model and the resulting phenotypes are examined. For decades, the mouse has been the most popular model organism to validate human gene function, and it has provided an invaluable tool for studying the mechanisms of hearing that would not be possible in human studies alone. However, the large-scale identification and validation of candidate genes in mice is limited by many factors including the small number of progeny, the in utero lethality of many knockouts, the labor involved in targeting the genes, and the relatively expensive husbandry.
Zebrafish as an animal model
The zebrafish (Danio rerio) is a small tropical fish that belongs to the teleost clade, originates in Southeast Asia and was first used to study developmental biology by George Streisinger in the early 1980s (Chakraborty et al., 2017). Zebrafish is a vertebrate model organism with several advantages over mice: zebrafish have a large number of progeny, undergo external fertilization and relatively fast embryonic development, produce transparent larvae, and are particularly suitable for high-throughput mutagenesis and drug screening approaches (Varshney et al., 2015a). As a result of the number of genetic tools and techniques available to manipulate the zebrafish genome, several large-scale mutagenesis projects have generated the second biggest mutant collection in a vertebrate model (Kettleborough et al., 2013; Varshney et al., 2013b). Three different approaches are being used to study gene function in zebrafish.
Random mutagenesis approaches
In the early 1990s, chemical mutagenesis using N-ethyl-N-nitrosourea (ENU) as a mutagen was used to perform large-scale random mutagenesis screens (Driever et al., 1996; Haffter et al., 1996; Solnica-Krezel et al., 1994), and later retroviruses were used for random insertional mutagenesis (Amsterdam et al., 1997; Gaiano et al., 1996; Golling et al., 2002). Both screens identified thousands of mutations affecting embryonic development, including genes involved in ear development. Mutations affecting ear development were identified based on ear morphology (e.g. otolith development and the shape of the ear) and simple behavioral assays (e.g. aberrant swimming pattern) (Driever et al., 1996; Granato et al., 1996; Malicki et al., 1996; Nicolson et al., 1998; Whitfield et al., 1996). Malicki et al characterized 20 mutants affecting ear development; identifying a variety of phenotypes such as absence of otolith, abnormal ear morphology, and abnormal otic placode development (Malicki et al., 1996). Similarly, another screen identified 58 mutants with a variety of phenotypes including abnormal development of sensory patches, otoliths, and semicircular canals (Whitfield et al., 1996). Many of these mutants with ear and otolith defects also displayed behavioral phenotypes and failed to balance properly. Another class of mutants in this screen displayed balance defects without any defects in the ear. A group of mutants show ear phenotypes as secondary phenotypes i.e. associated with a different phenotype. In another independent screen, Teresa Nicolson identified seven mutants based on abnormal swimming behavior of the larvae; these were called circler mutants (sputnik, mariner, orbiter, mercury, gemini, skylab, astronaut, and cosmonaut). All of them had abnormal vestibular function, and none of these mutants showed any obvious morphological defects (Granato et al., 1996; Nicolson et al., 1998) (Driever et al., 1996; Haffter et al., 1996; Solnica-Krezel et al., 1994).
Over the years, many labs have identified a number of mutants affecting ear development; however, only a small number of these mutants are linked to non-syndromic hearing loss (Bang et al., 2002; Hardison et al., 2005; Nissen et al., 2003; Schibler et al., 2007). Primarily, the circler class of mutants has an association with non-syndromic hearing loss genes (Sollner et al., 2004). The Sanger Institute in the UK carried out a large scale ENU mutagenesis study to identify mutations in all protein-coding genes (Kettleborough et al., 2013), while Shawn Burgess’ and Shuo Lins’ labs used retroviruses to generate genome-wide insertional mutations (Varshney et al., 2013b). Both projects generated a large number of mutants across thousands of genes; thousands of mutants from both ENU and retroviral mutagenesis projects are currently available to the community (Kettleborough et al., 2013; Varshney et al., 2013a).
An alternative approach to random insertional mutagenesis in zebrafish involves transposable elements such as Tol2, or Ac/Ds from heterologous hosts (Varshney et al., 2015a). These transposable elements are easy to use and can carry complex DNA transgenes. The Tol2 transposon from medaka is the most popular genetic tool for creating transgenic zebrafish, and many transgenic lines expressing fluorescent proteins in the inner ear or lateral line are available to the community. A list of hearing related transgenic lines can be found in Baxendale et al (Baxendale et al., 2016). Tol2-based gene or protein traps, used to generate conditional mutants with inactivated gene function in a tissue (spatial) or time point (temporal) specific manner are also available (Varshney et al., 2015a).
Targeted mutagenesis approaches
While there are a number of mutants available from both ENU and retroviral screens, mutants for all hearing loss genes are not available. Targeted mutagenesis approaches provide an opportunity to target the genes of interest at will. Moreover, mutants generated by random mutagenesis might carry additional background mutations that could complicate the phenotypic analysis (San et al., 2019). During the time when random mutagenesis-based forward genetic screens identified a number of mutants related to ear development and function, no tools existed for targeted mutagenesis in reverse genetic screens. However, the past ten years have seen an enormous development of reverse genetic tools including Zinc Finger Nucleases (ZFNs), TALENs, and CRISPR/Cas9. ZFNs and TALENs are engineered proteins, and CRISPR/Cas9 is an RNA-guided endonuclease, but all function in a similar manner by binding to the target DNA sequence and inducing a double-stranded (DSB) break. The DSB can either be repaired by the error-prone non-homologous end joining repair pathway (NHEJ) or, when a donor template is provided, by the homology-directed repair (HDR) pathway (Varshney et al., 2015a). NHEJ repair is being exploited to generate loss-of-function alleles. The CRISPR/Cas9 technique has revolutionized reverse genetics approaches because it is inexpensive, simple, easy to use, and allows multiple genes to be targeted at the same time. Multiplexing is particularly useful in zebrafish because one-third of the genome is duplicated, and targeting two paralogs is now possible (Jao et al., 2013). CRISPR/Cas9 has two components - a guide RNA (sgRNA) that binds to the target site, and the Cas9 enzyme that induces the DSB. Any DNA sequence can be targeted using this technique, the only requirement being that a 20-nucleotide target sequence must be followed by a specific protospacer sequence motif (PAM). Different Cas9 enzymes have different PAM requirements, and the most popular Cas9 from Streptococcus pyogens (SpCas9) requires NGG PAM (i.e. the 20-nucleotide target sequence must be followed by NGG (e.g. AGG/CGG, TGG or GGG)). Cas9 from Streptococcus aureus (SaCas9) with NNGRRT (R = A or G) PAM and Cas12a, another class of enzyme, with TTTV PAM (V = A or C or G) have also shown to work efficiently in zebrafish (Feng et al., 2016; Moreno-Mateos et al., 2017). There are many other orthologous enzymes and engineered versions of SpCas9 that can be used as an alternate (Liu et al., 2019). Using CRISPR/Cas9 technology, mutations in more than 50 zebrafish orthologs of genes involved in human hearing loss have been generated. It is projected that this resource will accelerate the functional validation of hearing loss genes in zebrafish (Varshney et al., 2015b).
CRISPR-based approaches to study genetic variants
Since most of the mutations in hearing loss genes are point mutations, investigating their role in disease pathology has been challenging. With the advancement of CRISPR/Cas9 technology, it is possible to mimic these mutations in zebrafish if the sequences are well conserved in a targeted manner. CRISPR/Cas9 mediated knock-in approaches (via HDR with a donor template including the desired mutation) are being used successfully (Irion et al., 2014; Prykhozhij et al., 2018). In zebrafish, the HDR-based targeted knock-in approaches are more challenging than generating knock-out alleles. The very first use of integrating an exogenous DNA in zebrafish was done by a TALEN-mediated knock-in approach using 900bp long homologous arms. However, the germline transmission frequency was less than 2%, limiting its use on a large-scale (Zu et al., 2013). Chang et al used CRISPR/Cas9 for the first time to integrate LoxP sequences using single-stranded oligonucleotide (ssODN) donor templates for HDR (Chang et al., 2013). Using the same strategy of ssODN donor template, single nucleotide changes were introduced in zebrafish genes fus (R536H) and tardbp (A379T), mimicking amyotrophic lateral sclerosis (ALS)-causing missense mutations in FUS (R521H) and TARDBP (A328T) genes (Armstrong et al., 2016). Two different template sizes were tested, at 23bp and 100bp in length, identifying 1/46 (~2%) and 3/77(~4%) F0 founders transmitting the mutations in the germline (Armstrong et al., 2016). In another strategy using donor oligonucleotides, an HA tag at C9t3 locus was introduced with 1.7% germline transmission efficiency (Hruscha et al., 2013). In a different strategy, plasmids were used as a donor to correct a premature stop codon in the albino locus (slc45a2) that prematurely truncates the protein and the germline transmission efficiencies were comparable to an earlier study by Hruscha et al (Hruscha et al., 2013; Irion et al., 2014). While many knock-in alleles have been reported mimicking disease-causing nonsense mutations in zebrafish, there is only one example related to hearing loss. A knock-in allele was generated where 670bp of ush2a intron 40 were replaced with 557 bp of the corresponding human USH2A sequence. In patients, the c.7595–2144A>G mutation in USH2A intron 40 introduces a splice donor site resulting in the incorporation of a pseudoexon predicted to terminate USH2A translation prematurely (Slijkerman et al., 2018). Over the years, several strategies are being used for knock-in of exogenous DNA or mimicking point mutations in zebrafish with low germline efficiencies. More recently, newer approaches are being developed that are more efficient and promising, but come with their own challenges (Wierson et al., 2018). Nonetheless, these new approaches need to be tested on a large-scale to effectively address the flood of variants being identified. As of today, the most common approach to study variants in zebrafish has been injecting mRNA with a specific variant into one-cell stage embryos in a null background. A wild-type mRNA should rescue the phenotype, while mRNA with a variant may not rescue the phenotype if the variant is critical to the protein function. If the phenotypes appear at later stages of animal development, then phenotype rescue or variant testing is done using a transgene. This approach is time consuming, labor-intensive, and not amenable to high-throughput testing of variants. In 2016–17, David Liu’s group at the Broad Institute repurposed the CRISPR/Cas9 based system to change the single bases at a target site (Gaudelli et al., 2017; Komor et al., 2016). Catalytically inactive Cas9 is fused with enzymes such as cytidine deaminase or adenine base editor (a combination of wild-type TadA (tRNA specific adenosine deaminase) and evolved TadA enzyme) that can convert C.G base pair to T.A or A.T to G.C, respectively. The conversion of the nucleotide occurs without creating a DSB and thus does not generate indels (or fewer indels) via DSB repair mechanisms. This base editing approach can be used to investigate pathogenic vs non-pathogenic single nucleotide mutations wherever a targeting window is available, which is usually ~5bp and PAM dependent. The PAM requirement can be relaxed by using a different enzyme recognizing a different PAM.
Morpholino-mediated knockdown of mRNA
Since early 2000, the use of chemically synthesized morpholinos (MOs) has been a popular method among zebrafish researchers for the transient knockdown of target mRNAs to study loss-of-function phenotypes (Nasevicius et al., 2000). MOs work either by binding to the translation start site to inhibit translation (ATG blocker) or by interfering with the splicing mechanism of mRNA (splice blocker). One of the reasons that MOs became the most popular antisense knockdown tool in zebrafish is their ease of delivery in early stage embryos. MOs can knockdown the expression of both maternal and zygotic transcripts to create complete loss-of-function phenotypes. Soon after, MOs became a popular tool for targeted reverse genetic studies in zebrafish. These studies made it possible to quickly and inexpensively study genes from various exome- and genome-based studies, as well as genes and variants that were identified as interesting targets by genome-wide association studies. Using MO-mediated knockdown, ~ 25 hearing related genes have been studied (Table 1) and their roles in hearing and balance validated. MOs were also used to phenocopy mutants with otic defects identified in a random mutagenesis screen (Whitfield, 2002). The first hearing loss gene analyzed by MO was for the DFNA5 locus. In humans, DFNA5-associated hearing loss is caused by pathogenic variants in the gene GSDME that result in a non-syndromic, autosomal dominant form of progressive hearing loss. The MO-mediated knockdown of dfna5a in zebrafish showed disorganization of the developing semicircular canals (Busch-Nentwich et al., 2004).
Table 1.
Zebrafish orthologs of human non-syndromic hearing loss genes
| Human deafness locus | Human gene | Zebrafish gene(s) | Zebrafish model | Type of mutant | References |
|---|---|---|---|---|---|
| DFNA20/26 | ACTG1 | - | - | (van Wijk et al., 2003; Zhu et al., 2003) | |
| DFNB44 | ADCY1 | adcy1a, adcy1b | Yes | Morpholino | (Santos-Cortez et al., 2014) |
| DFNX5 | AIFM1 | aifm1 | - | (Zong et al., 2015) | |
| - | ATP2B2 | atp2b2 | - | (Smits et al., 2019) | |
| DFNB93 | CABP2 | cabp2a, cabp2b | - | (Schrauwen et al., 2012) | |
| DFNA44 | CCDC50 | ccdc50 | - | (Modamio-Hoybjor et al., 2007) | |
| DFNB32/105 | CDC14A | cdc14aa, cdc14ab | Yes | Morpholino CRISPR | (Delmaghani et al., 2016; Imtiaz et al., 2018) |
| DFNA66 | CD164 | cd164 | - | (Nyegaard et al., 2015) | |
| DFNB12 | CDH23 | cadherin-like 23 | Yes | ENU Morpholino | (Bork et al., 2001; Nicolson et al., 1998; Söllner et al., 2004) |
| DFNA4B, DFNB113 | CEACAM16 | si:dkey-11o1.7 | - | (Booth et al., 2018) (Zheng et al., 2011) | |
| DFNB48 | CIB2 | cib2 | Yes | Morpholino | (Riazuddin et al., 2012) |
| - | CLDN9 | - | - | (Sineni et al., 2019) | |
| DFNB29 | CLDN14 | - | - | (Wilcox et al., 2001) | |
| DFNB103 | CLIC5 | clic5a, clic5b | - | (Seco et al., 2015) | |
| DFNA9 | COCH | coch | - | (Robertson et al., 1998) | |
| DFNX6 | COL4A6 | col4a6 | - | (Rost et al., 2014) | |
| DFNA37 | COL11A1 | col11a1a, col11a1b | - | (Booth et al., 2019) | |
| DFNB53, DFNA13 | COL11A2 | col11a2 | - | (Chen et al., 2005; McGuirt et al., 1999) | |
| DFNA40 | CRYM | crym | - | (Abe et al., 2003) | |
| DFNB66 | DCDC2 | dcdc2b | Yes | Morpholino | (Grati et al., 2015) |
| DFNA5 | GSDME | dfna5a, dfna5b | Yes | Morpholino | (Busch-Nentwich et al., 2004; Van Laer et al., 1998) |
| DFNA1 | DIAPH1 | - | - | (Lynch et al., 1997) | |
| DFNA71 | DMXL2 | dmxl2 | - | (Chen et al., 2017) | |
| DFNB88 | ELMOD3 | elmod3 | - | (Jaworek et al., 2013; Li et al., 2018) | |
| DFNB102 | EPS8 | eps8a, eps8b | - | (Behlouli et al., 2014) | |
| DFNB106 | EPS8L2 | eps8l2 | - | (Dahmani et al., 2015) | |
| DFNB36 | ESPN | espn, espnl | - | (Naz et al., 2004) | |
| DFNB109 | ESRP1 | esrp1 | - | (Rohacek et al., 2017) | |
| DFNB35 | ESRRB | esrrb | - | (Collin et al., 2008) | |
| DFNA10 | EYA4 | eya4 | Yes | Morpholino | (Wayne et al., 2001) (Schonberger et al., 2005) |
| DFNB104 | RIPOR2 | ripor2 | Yes | Morpholino | (Diaz-Horta et al., 2014) |
| - | FOXF2 | foxf2a, foxf2b | - | (Bademci et al., 2019) | |
| DFNB26 | GAB1 | gab1 | Yes | Morpholino | (Yousaf et al., 2018b) |
| DFNB15/72/95 | GIPC3 | gipc3 | - | (Ain et al., 2007; Charizopoulou et al., 2011; Rehman et al., 2011) | |
| DFNB1A, DFNA3A | GJB2 | cx30.3 | Yes | Morpholino | (Alvarez et al., 2005; Barashkov et al., 2011; Chang-Chien et al., 2014; Kelsell et al., 1997) |
| DFNB91, DFNA2B | GJB3 | cx35.4 | - | (Xia et al., 1998) | |
| DFNB1B, DFNA3B | GJB6 | cx30.3 | Yes | Morpholino | (Chang-Chien et al., 2014; del Castillo et al., 2002; Grifa et al., 1999) |
| DFNB82 | GPSM2 | gpsm2 | - | (Yousaf et al., 2018a) | |
| DFNA114 | GRAP | grapa, grapb | - | (Li et al., 2019) | |
| DFNA28 | GRHL2 | grhl2a, grhl2b | Yes | Tol2 | (Vona et al., 2013) (Han et al., 2011) |
| DFNB25 | GRXCR1 | grxcr1 | Yes | CRISPR TALEN | (Blanco-Sánchez et al., 2018; Schraders et al., 2010b) |
| DFNB101 | GRXCR2 | - | - | (Imtiaz et al., 2014) | |
| DFNB39 | HGF | hgfa, hgfb | - | (Schultz et al., 2009) | |
| DFNA68 | HOMER2 | homer2 | - | (Azaiez et al., 2015) | |
| DFNA2C | IFNLR1 | crfb2 | Yes | Morpholino | (Gao et al., 2018) |
| DFNB42 | ILDR1 | ildra1, ildr1b | Yes | Morpholino | (Borck et al., 2011; Sang et al., 2014) |
| DFNB89 | KARS | kars | - | (Santos-Cortez et al., 2013) | |
| DFNA2A | KCNQ4 | kcnq4 | - | (Kubisch et al., 1999) | |
| DFNA69 | KITLG | kitlga, kitlgb | - | (Zazo Seco et al., 2015) | |
| DFNB66/67 | LHFPL5 | lhfpl5a, lhfpl5b | Yes | ENU | (Kalay et al., 2006; Obholzer et al., 2012; Shabbir et al., 2006; Tlili et al., 2005) |
| DFNA7 | LMX1A | lmx1aI | - | (Wesdorp et al., 2018a) | |
| DFNB77 | LOXHD1 | loxhd1a, loxhd1b | - | (Grillet et al., 2009) | |
| - | LRP5 | - | - | (Xia et al., 2017) | |
| DFNB63 | LRTOMT/COMT2 | tomt | Yes | Tol2 | (Ahmed et al., 2008b; Du et al., 2008; Erickson et al., 2017) |
| DFNB49 | MARVELD2 | marveld2a, marveld2b | - | (Riazuddin et al., 2006a) | |
| DFNA70 | MCM2 | mcm2 | - | (Gao et al., 2015) | |
| DFNB97 | MET | met | Yes | Morpholino | (Mujtaba et al., 2015) |
| DFNM1 | METTL13 | mettl13 | - | (Yousaf et al., 2018b) | |
| DFNA50 | MIR96 | mir96 | - | (Mencia et al., 2009) | |
| DFNB111 | MPZL2 | mpzl2b | - | (Bademci et al., 2018; Wesdorp et al., 2018b) | |
| DFNB74 | MSRB3 | msrb3 | Yes | Morpholino | (Ahmed et al., 2011; Shen et al., 2015) |
| DFNA4A | MYH14 | myh14 | - | (Donaudy et al., 2004) | |
| DFNA17 | MYH9 | myh9a, myh9b | - | (Lalwani et al., 2000) | |
| DFNB3 | MYO15A | myosin XVAa, myosin XVAb | - | (Wang et al., 1998) | |
| DFNB30, DFNA | MYO3A | myosin IIIA | - | (Grati et al., 2016; Walsh et al., 2002) | |
| DFNB37, DFNA22 | MYO6 | myosin VIa, myosin VIb | Yes | ENU | (Ahmed et al., 2003c; Melchionda et al., 2001; Seiler et al., 2004) |
| DFNB2, DFNA11 | MYO7A, MYO7A | myosin VIIAa, myosin VIIAb | Yes | ENU | (Ernest et al., 2000; Liu et al., 1997a; Wasfy et al., 2014; Weil et al., 1997) |
| DFNB94 | NARS2 | nars2 | - | (Simon et al., 2015) | |
| DFNA34 | NLRP3 | - | - | (Nakanishi et al., 2017) | |
| DFNA67 | OSBPL2 | osbpl2a, osbpl2b | Yes | ENU | (Thoenes et al., 2015; Wang et al., 2019; Xing et al., 2015) |
| DFNB22 | OTOA | - | - | (Zwaenepoel et al., 2002) | |
| DFNB9 | OTOF | otoferlin-a, otoferlin-b, | Yes | Morpholino | (Chatterjee et al., 2015; Yasunaga et al., 1999) |
| DFNB18B | OTOG | otogelin | Yes | ENU | (Schraders et al., 2012) (Stooke-Vaughan et al., 2015) |
| DFNB84 | OTOGL | otogelin-like | Yes | Morpholino | (Yariz et al., 2012) |
| DFNA41 | P2RX2 | p2rx2 | - | (Yan et al., 2013) | |
| DFNB23 | PCDH15 | pcdh15a, pcdh15b | Yes | Tol2 | (Ahmed et al., 2003a; Seiler et al., 2005) |
| DFNA74 | PDE1C | pde1ca, pde1cb | - | (Wang et al., 2018) | |
| DFNB57 | PDZD7 | pdzd7a, pdzd7b | Yes | Morpholino | (Ebermann et al., 2010; Schneider et al., 2009) |
| DFNB59 | PJVK | dfnb59 | - | (Delmaghani et al., 2006) | |
| - | PLS1 | pls1 | - | (Schrauwen et al., 2019) | |
| DFNB70 | PNPT1 | - | - | (von Ameln et al., 2012) | |
| DFNX2 (DFN3) | POU3F4 | - | - | (de Kok et al., 1995) | |
| DFNA15 | POU4F3 | pou4f3 | - | (Vahava et al., 1998) | |
| DFNB100 | PPIP5K2 | ppip5k2 | - | (Yousaf et al., 2018a) | |
| DFNX1 (DFN2) | PRPS1 | prps1a, prps1b | Yes | Morpholino | (DeSmidt et al., 2019; Liu et al., 2010; Pei et al., 2016) |
| DFNB84, DFNA73 | PTPRQ | ptprq | - | (Eisenberger et al., 2018; Schraders et al., 2010a) | |
| DFNB24 | RDX, | moesin a, moesin b | - | (Khan et al., 2007) | |
| DFNA27 | REST | rest | - | (Nakano et al., 2018) | |
| DFNB108 | ROR1 | ror1 | - | (Diaz-Horta et al., 2016) | |
| DFNB68 | S1PR2 | s1pr2 | Yes | Morpholino | (Santos-Cortez et al., 2016) |
| DFNA91 | SERPINB6 | - | - | (Sirmaci et al., 2010) | |
| DFNA23 | SIX1 | six1a, six1b | - | (Mosrati et al., 2011) | |
| DFNA25 | SLC17A8 | slc17a8/vglut3 | Yes | Morpholino | (Obholzer et al., 2008; Ruel et al., 2008) |
| DFNB60 | SLC22A4 | slc22a4 | - | (Ben Said et al., 2016) | |
| DFNB4 | SLC26A4 | slc26a4 | - | (Li et al., 1998) | |
| DFNB61 | SLC26A5 | slc26a5 | - | (Liu et al., 2003) | |
| DFNA64 | SMAC/DIABLO | diabloa diablob | - | (Cheng et al., 2011) | |
| DFNX4 (DFN6) | SMPX | smpx | - | (Huebner et al., 2011; Schraders et al., 2011) | |
| - | SPATC1L | - | - | (Morgan et al., 2019) | |
| DFNB115 | SPNS2 | spns2 | - | (Ingham et al., 2019) | |
| DFNB16 | STRC | strca strcb | - | (Verpy et al., 2001) | |
| DFNB76 | SYNE4 | - | - | (Horn et al., 2013) | |
| DFNB86, DFNA65 | TBC1D24 | tbc1d24 | - | (Azaiez et al., 2014; Rehman et al., 2014) | |
| DFNB21, DFNA8/12 | TECTA | tecta | Yes | ENU | (Mustapha et al., 1999; Stooke-Vaughan et al., 2015; Verhoeven et al., 1998) |
| DFNA51 | TJP2 | tjp2a, tjp2b | - | (Walsh et al., 2010) | |
| DFNB7/11, DFNA36 | TMC1 | tmc1 | - | (Kurima et al., 2002) | |
| DFNB99 | TMEM132E | tmem132e | Yes | Morpholino | (Li et al., 2015) |
| DFNB6 | TMIE | tmie | Yes | Morpholino | (Gleason et al., 2009; Naz et al., 2002) |
| DFNB8/10 | TMPRSS3 | tmprss3a, tmprss3b | - | (Scott et al., 2001) | |
| - | TMTC2 | Tmtc2a, tmtc2b | (Runge et al., 2016) | ||
| DFNA56 | TNC | tenascin C | - | (Zhao et al., 2013) | |
| - | TOP2B | top2b | (Xia et al., 2019b) | ||
| DFNB79 | TPRN | tprn | - | (Li et al., 2010; Rehman et al., 2010) | |
| DFNB28 | TRIOBP | triobpa triobpb | - | (Riazuddin et al., 2006b; Shahin et al., 2006) | |
| - | TRRAP | trrap | Yes | CRISPR Morpholino | (Xia et al., 2019a) |
| DFNB98 | TSPEAR | tspeara tspearb | - | (Delmaghani et al., 2012) | |
| DFNB18 | USH1C | ush1c | Yes | ENU Morpholino | (Ahmed et al., 2002; Ouyang et al., 2002; Phillips et al., 2011) |
| DFNB107 | WBP2 | wbp2 | - | (Buniello et al., 2016) | |
| DFNA6/14/38 | WFS1 | wfs1a, wfs1b | - | (Bespalova et al., 2001; Young et al., 2001) | |
| DFNB31 | WHRN | dfnb31a, dfn31b | - | (Mburu et al., 2003) |
While MOs are an effective knockdown tool, their utility can be limited by off target toxicity and the fact that their knockdown effect is transient (typically 2–4 days post-fertilization (dpf)). The off-target toxicity can be excluded by injecting wild-type mRNA from the target gene together with MO, specific phenotypes should be rescued by wild-type mRNAs either partially or fully. As targeted mutagenesis methods like CRISPR/Cas9 and TALENS became available, a few studies demonstrated that genetic mutants did not recapitulate the MO-induced phenotype (Kok et al., 2014; Law et al., 2014). For example, phenotypes induced by targeting the mRNA of the non-syndromic hearing loss gene, CDC14A, ortholog by MO-mediated knockdown were not replicated in a genetic mutant (Delmaghani et al., 2016) (Imtiaz et al., 2018). These findings led to the introduction of more stringent guidelines for MO-mediated knockdown experiments; the community now recommends using multiple MOs, or MOs in combination with other approaches such as genetic mutants or CRISPR interference mediated knockdown (CRISPRi) (Stainier et al., 2017). Since the MO-based phenotypes are dose-dependent, it is recommended to have a dose response curve, and to perform additional experiments when a dose higher than 5 ng is required to observe a phenotype. In conclusion, MOs are not recommended as a reverse genetic tool. In cases where a genetic mutant is not available, appropriate controls must be used to rule out phenotypes caused by off-target effects (Stainier et al., 2017). Alternatively, CRISPR/Cas9 are highly efficient in inducing biallelic mutations, and phenotypes can be observed in the F0 generation (akin to morphants) (Varshney et al., 2015a). Recently, a study adopted this method to screen for phenotypes in the F0 generation by utilizing multiple sgRNAs together with Cas9 (Wu et al., 2018). Furthermore, a recent publication showed that the mutagenesis efficiencies can be increased by using a dual gRNA approach where CRISPR RNA (crRNA) and trans-activating CRISPR RNA (tracrRNA) are synthesized separately and injected together with Cas9 protein (Hoshijima et al., 2019).
Morpholinos Vs Genetic Mutants
More than 50% of the zebrafish models used in hearing research have been generated using MO-mediated knockdown that only permit analysis of effects during the first 4–5 days of embryonic development and makes the study of genes involved in delayed-onset hearing loss challenging. Some studies have suggested that MO-mediated knockdown phenotypes may not be repeatable by genetic mutants (Kok et al., 2014). The suggestion has fueled a debate about whether MO-mediated phenotypes are generated by off-target effects and strengthened the argument in favor of genetic mutants as superior. However, another study showed that the absence of phenotypes in genetic mutants could be caused by genetic compensation (Rossi et al., 2015). Since most of the mutations generated by genome editing technology are small indels that introduce mediated decay of mRNA, the decayed mRNA enters the nucleus and upregulates genes similar to ones targeted in mutants (Rossi et al., 2015). The activity of the upregulated gene compensates for the loss of the target gene and eliminates the phenotype that would otherwise be observed with loss of the target. The mechanisms of genetic compensation in mutants have been described in two recent, independent publications (El-Brolosy et al., 2019; Ma et al., 2019). These studies have identified that smaller indels cause nonsense-mediated decay of the mutant mRNA, which in turn triggers the recruitment of the COMPASS complex to the regulatory regions of closely related genes. This results in epigenetic changes that activate the expression of the closely related genes. It has been suggested that genetic compensation can be avoided if RNA-less alleles (in which the promoter and first exon have been deleted), which do not create a premature termination codon are employed (El-Brolosy et al., 2019). An alternative approach could be generating missense mutations in the functional domains of the target protein that could make the protein non-functional but will not induce nonsense-mediated decay of mRNA; however, this approach has not yet been tested. It has also been shown that CRISPR/Cas9-mediated indels might induce altered mRNA processing such as exon skipping that could escape nonsense-mediated decay (Anderson et al., 2017). However, a large-scale mutant analysis showed only 4/36 alleles have altered mRNA processing, and only two of them generated in-frame mutations at low-levels suggesting that this process may not be very common in CRISPR generated alleles but cDNA sequencing must be done as a quality control step for functional genomics studies (Ramanagoudr-Bhojappa et al., 2018). The gold standard approach to verify null alleles is to perform western blot or immunohistochemistry experiments in cases where antibodies are available.
Zebrafish as a model to study hearing and balance disorders
Numerous studies have shown that zebrafish and mammalian genes are highly conserved, and ~82% of human disease genes listed in the OMIM database have an ortholog in zebrafish (Howe et al., 2013). Thus far, 129 loci, and 130 genes have been identified in humans that are associated with non-syndromic hearing loss as per the hereditary hearing loss database (http://hereditaryhearingloss.org). Out of 130 human hearing loss genes, 103 have a matching ortholog in zebrafish, suggesting that hearing loss genes are highly conserved in zebrafish and humans. Since ~20% of the zebrafish genome is duplicated, there are many genes with a paralog. Of 103 non-syndromic hearing loss genes, 36 have known/annotated paralogs in zebrafish. Classical forward genetic screens performed in the early 1990s for identifying hearing related mutants did not identify many genes linked to non-syndromic hearing loss genes.
Zebrafish have two related sensory organs that utilize mechanosensory hair cell receptors: the inner ear, similar to mammalian ears, is used to detect sound, gravity, and motion; and the lateral line, a fish- and amphibian-specific organ used to detect water flow over the surface of the body and the head (Elepfandt, 1988; Nicolson, 2005) (Figure 3 A–D). The lateral line consists of neuromasts. Each neuromast is composed of support cells surrounding hair cells. The lateral line hair cells are similar to the hair cells found in the inner ear. Their development and differentiation follow the similar developmental regime (Ghysen et al., 2004; Nicolson, 2005) (Figure 3 B). Many non-syndromic deafness-related genes are expressed in lateral line hair cells (Erickson et al., 2015) (Matern et al., 2018), as well as the inner ear of zebrafish (Yao et al., 2019) (Barta et al., 2018).
Figure 3.

A) Schematic showing the inner ear of adult zebrafish with semi-circular canal, three sensory epithelia and otoliths. B) Schematic showing the anterior (aLL) and posterior (pLL) lateral lines of a larval zebrafish. Neuromasts are shown by dots. C & D) The maturing inner ear during embryonic development at 20 hours post fertilization (hpf) and 48 hpf, respectively. E & F) Larvae at 5-day post fertilization, stained with FM1–43 live dye, in wild-type and cdh23 mutant animals. Absence of staining in cdh23 mutants is due to dysfunctional mechanotransduction channels.
In addition, zebrafish offer unique advantages for studying defects in the inner ear. Their external fertilization and optically transparent larvae enable direct observation of the sensory epithelium of the inner ear, while observation or manipulation of sensory hair cells in mice requires the dissection of the inner ear, which is technically challenging. Unlike mice, the embryonic inner ear is accessible for imaging. As the development of zebrafish hearing and vestibular system has been covered in greater detail in many excellent reviews and research articles, we aim to summarize the important steps of inner ear and lateral line development here (Nicolson, 2005; Whitfield, 2002; Whitfield et al., 2002).
In zebrafish, the otic vesicle starts to form within 16 hours post fertilization (hpf) in the form of otic placodes (which result from thickening of the ectoderm), and by 24 hpf, the anterior and posterior sensory maculae start to form at ends of the otic vesicle (Nicolson, 2005; Whitfield et al., 2002). The inner ear has multiple sensory patches required for different functions. The vestibular system is required to sense gravity and head movement, and is composed of the semicircular canal, and three macular endorgans, such as saccule, utricle, and lagena in zebrafish. These endorgans are composed of neuroepithelium attached to specialized structures made up of mostly aggregates of calcium carbonate crystals called otoliths (Figure 3 A) (Lundberg et al., 2015). During the first seven days of embryonic development, only the saccule and utricle develop; the lagena develops at a later stage (~11 days post fertilization (dpf)) (Lu et al., 2013). The saccule is the main hearing organ, while the utricle is important for balance, motor co-ordination and gravity sensing. The function of the lagena is still not clear, though some reports suggest that the lagena might also play a role in hearing function (Lu et al., 2013; Yao et al., 2016). The hair cells begin to function by 4 dpf, and by 5 dpf, larvae can respond to vibrational reflexes and vestibular reflexes (Raible et al., 2000) (Metcalfe et al., 1985).
Genetic tools to study hearing and vestibular disorders in zebrafish
To investigate the roles of candidate disease genes in hearing loss, a growing number of tools including transgenic lines, antibodies, and live dyes that can label hair cells, the lateral line system, and other inner ear structures in zebrafish are available (Baxendale et al., 2016) (Kindt et al., 2018). A number of transgenic lines such as Tg(pou4f3:GAP-GFP) (Xiao et al., 2005) and Tg(myo6b:GFP) label hair cells (Obholzer et al., 2008), Tg(pvalb3b:ribeyea-mCherry) labels hair cell synapses (Kindt et al., 2018; Obholzer et al., 2008), Tg(atoh1a:tdTomato) labels immature hair cells (Wibowo et al., 2011) or transgenic lines that can detect neuronal activity using a genetically encoded calcium indicator (GECI), such as GCaMP6 (Lukasz et al., 2018). There are many live dyes such as DASPEI (2-(4-dimethylaminostyryl)-N-ethylpyridinium iodide) that labels hair cells, and DiASP (4-(4-diethylaminostyryl)-N-methylpyridinium iodide) that labels hair cells, as well as afferent fibers. FM1–43, FM4–64 labels hair cells, and Yo-Pro1 can label hair cell nuclei which is useful for counting hair cells (Figure 3 G&H). FM1–43/Yo-Pro1 labeling is being used to investigate the mechanotransduction defects in inner ear and lateral line hair cells in many zebrafish mutants. Other live dyes can also be used but FM1–43 and Yo-Pro1 are easy to use and have strong fluorescence signals. Other stains such as Phalloidin, a phallotoxin from the poisonous mushroom, Amanita phalloides, binds to filamentous actin and, thus, can provide a snapshot of the size, shape or splaying of the hair bundles. Phalloidin is available coupled with different fluorophores such as FITC or TRITC and other dyes. A number of antibodies are available such as Myosin VI or Otoferlin that are used for hair cell staining, as well as Ribeye a & b for staining ribbon synapses. A list of antibodies to study hair cell development and function are described in Kindt et al (Kindt et al., 2018).
Zebrafish disease models for hearing loss
As of December 2019, only 34 models for non-syndromic hearing loss-related genes in zebrafish have been published. Twenty-five of them have been generated using MO-mediated mRNA knockdown, followed by 8 models generated by ENU mutagenesis, 3 models by Tol2 insertional mutagenesis, and only 4 models by targeted mutagenesis approaches such as CRISPR/Cas9 and TALEN. Mutants for non-syndromic hearing loss can be grouped in many categories, such as genes affecting hair cell function, synapse function, morphogenesis of otic vesicles, and epithelial integrity. Here, we discussed selected genes from each category. Many of these genes have been discussed in reviews by Blanco-Sánchez et al and Nicolson T. in detail (Blanco-Sanchez et al., 2017; Nicolson, 2017).
Myosin 7AA (MYO7AA)
Myo7aa was one of the earliest genes linked to hearing disease pathology. In humans, MYO7A has been involved in both syndromic (Usher syndrome) and non-syndromic (DFNB2, DFNA11) deafness (Bharadwaj et al., 2000; Hildebrand et al., 2010; Liu et al., 1997b; Riazuddin et al., 2008; Weil et al., 1997). In zebrafish mariner, a circler mutant was identified with mutations in its myo7aa gene (Ernest et al., 2000). myo7aa is expressed in hair cells of the inner ear and the lateral line in zebrafish. Zebrafish mariner mutants showed abnormal stereocilia of the hair cells in the inner ear, and unbalanced swimming behavior. These phenotypes were also observed in CRISPR/Cas9-generated alleles of myo7aa (Zou et al., 2019).
Myosin 6 (MYO6)
Another gene from the myosin family, MYO6, is involved in both non-syndromic autosomal recessive (DFNB37) and autosomal dominant (DFNA22) deafness (Ahmed et al., 2003b; Melchionda et al., 2001). In zebrafish, myo6a and myo6b are both paralogs, but only myo6b is expressed in sensory hair cells of the inner ear and lateral line. One of the mutants (satellite) from earlier forward genetic screens with vestibular defects was later mapped to the myo6b gene (Seiler et al., 2004). The myo6b mutants have abnormal stereocilia growth and showed splaying of the hair bundles and vestibular defects, and were unresponsive to startle stimuli (Seiler et al., 2004).
Cadherin 23 (CDH23)
Mutations in CDH23 cause the DFNB12 form of hearing loss and are also involved in Usher syndrome type 1D (Bork et al., 2001; Hilgert et al., 2009). In zebrafish, circler mutant sputnik was linked to mutations in the cdh23 gene (Nicolson et al., 1998; Söllner et al., 2004). cdh23 mRNA is expressed in the developing inner ear, and later in the neuroepithelium of the inner ear and lateral line neuromasts. The Cdh23 protein localizes near the tip of hair bundles and the tip links required for sound detection (which are absent in the mutants). cdh23 mutants do not respond to startle stimuli and have abnormal swimming behavior.
Protocadherin Related 15 (PCDH15)
Mutations in the PCDH15 gene cause Usher syndrome type 1F and the DFNB23 form of non-syndromic hearing loss (Ahmed et al., 2008a; Ahmed et al., 2003a). In zebrafish, the orbiter mutant from the forward genetic screen showed vestibular dysfunction, failed to respond to startle stimuli and lacked hair cell microphonic potentials (Seiler et al., 2005). The gene responsible for the orbiter phenotype was later mapped to the pcdh15 gene. There are two paralogs of pcdh15 in zebrafish, pcdh15a, and pcdh15b. The hearing phenotypes were determined to be caused by mutations in the pcdh15a gene.
Otoferlin (OTOF)
Mutations in the human OTOF gene cause the DFNB9 form of non-syndromic hearing loss. There are two copies of the otof gene (otofa, and otofb) in zebrafish. mRNA from both otofa and otofb is expressed in the otic placodes within 3 dpf, and by 5 dpf only otofb mRNA was detected in the lateral line hair cells (Chatterjee et al., 2015). Knockdown of otoferlin genes causes hearing and balance dysfunction, and otoferlin morphant animals did not have inflated swim bladders. Otof mRNA from mouse was able to rescue the morphant phenotype in zebrafish, showing the conserved role of OTOF across the species (Chatterjee et al., 2015).
Deafness Autosomal 5 (DFNA5)
The autosomal dominant form of hearing loss DFNA5 is caused by mutations in the GSDME gene in humans (Van Laer et al., 1998). In zebrafish, the dfna5 gene is duplicated with two paralogs: dfna5a and dfna5b. dfna5b mRNA is expressed ubiquitously in early-stage embryos, but is later restricted to inner ear tissues (Busch-Nentwich et al., 2004). MO-mediated knockdown of dfna5b mRNA causes abnormal semicircular canals, and a higher dose of MO also causes malformed ventral jaw development due to impaired pharyngeal cartilage development. The malformed jaw phenotype was not recapitulated in the genetic mutant generated by CRISPR/Cas9 (Varshney et al., 2015b). Dfna5 KO mouse does not display impaired facial cartilage differentiation as well (Op de Beeck et al., 2011). Absence of cartilage differentiation phenotypes both in zebrafish genetic mutant and mouse KO could be due to genetic compensation.
Grainyhead Like Transcription Factor 2 (GRHL2)
Mutations in the GRHL2 gene are responsible for the DFNA28 form of non-syndromic hearing loss in humans (Vona et al., 2013). Due to duplication of the zebrafish genome, grhl2 has two paralogs: grhl2a and grhl2b. grhl2b mutants have smaller or missing otoliths (Han et al., 2011). Human mRNA overexpression in the mutants rescues the phenotypes, indicating a conserved role of grhl2 in inner ear development in vertebrates. However, the phenotype failed to rescue when human mRNA with a pathogenic variant was injected suggesting a pathogenic nature of the variant (Han et al., 2011).
Adenylate Cyclase 1 (ADCY1)
ADCY1 is a member of the adenylate cyclase group of enzymes that catalyze the conversion of ATP to cyclic AMP (cAMP). Production of cAMP is important for mechanotransduction in the inner ear (Santos-Cortez et al., 2014). Mutations in ADCY1 cause autosomal recessive non-syndromic hearing loss DFNB44. A nonsense mutation in ADCY1 was first identified in a consanguineous family from Pakistan. ADCY1 is duplicated in zebrafish with two copies adcy1a, and adcy1b. The function of adcy1 genes in zebrafish was investigated by MO-mediated mRNA knockdown. Morphant animals show smaller eyes, small brain, and curved bodies. Adcy1b morphant animals did not respond to acoustic startle stimuli, while adcy1a morphants have normal response similar to wild-type suggesting adcy1b is contributing to the hearing impairment (Santos-Cortez et al., 2014).
Immunoglobulin-Like Domain Containing Receptor 1 (ILDR1)
The Immunoglobulin-Like Domain Containing Receptor 1 (ILDR1) gene encodes an immunoglobulin-like domain-containing protein that functions as a multimeric receptor. Mutations in the ILDR1 gene cause non-syndromic DFNB42, and patients show bilateral non-progressive moderate-to-profound sensorineural hearing loss (Borck et al., 2011). Zebrafish has two copies of ildr1, ildr1a, and ildr1b. Based on in situ hybridization, only ildr1b has otic tissue-related expression. Ildr1b mRNA localizes to the otic vesicle, and the posterior lateral line primordium at 24–36 hpf, and the expression became more prominent in the anterior lateral line and inner ear (Borck et al., 2011; Sang et al., 2014). MO-mediated knockdown of ildr1b exerts delays in the development of the semicircular canals in the zebrafish inner ear, a reduced number of neuromasts and defective hearing and aberrant swimming pattern (Sang et al., 2014). The morphant embryos showed downregulation of the atp1b2b gene that encodes ATPase Na+/K+ transporting subunit beta 2b, and the overexpression of atp1b2b partially rescued the inner ear phenotypes. The disruption in posterior lateral line primordium migration is due to attenuated FGF signaling pathway, cxcr4b, and cxcr7b expression (Sang et al., 2014).
RHO family interacting cell polarization regulator 2 (RIPOR2)
RIPOR2 encodes an atypical inhibitor of the small G protein RhoA. RIPOR2 is a component of hair cell stereocilia, and mutations in this gene cause autosomal recessive deafness 104 (DFNB104). Diaz-Horta et al identified mutations in six members of a consanguineous Turkish family (Diaz-Horta et al., 2014). The mutation caused an in-frame skipping of exon 3. Zebrafish ortholog ripor2 mRNA expression was detected in the otic vesicle of 3-dpf zebrafish (Diaz-Horta et al., 2014). To investigate the function of ripor2 in zebrafish, two different splice site blocking MOs were used to reduce the protein expression. Both morphant animals showed reduced numbers of saccular hair cells and fewer lateral line neuromasts, and did not show balance abnormalities. Furthermore, auditory function in morphants was detected by recording microphonic potentials from hair cells, and the morphants had weaker microphonic responses than the wild-type animals (Diaz-Horta et al., 2014).
Transmembrane Protein 132E (TMEM132E)
TMEM132E encodes for transmembrane protein 132e and mutations in this gene cause autosomal recessive deafness 99 (DFNB99) (Li et al., 2015). Homozygous missense mutations in TMEM132E were first identified in two siblings from consanguineous Chinese parents. Tmem132e is highly expressed in hair cells of the mouse cochlea. Functional studies were performed in zebrafish using MO-mediated knockdown of tmem132e mRNA. Hair cell development was monitored using cationic dye 4-Di-2-AS capable of penetrating into hair cells through mechanotransduction channels, therefore, serving as an indicator of mechanotransduction function of hair cells. Hair cells of the inner ear and the lateral line in morphant animals did not label with 4-Di-2-AS dye indicating non-functional hair cells. Co-injection of wild-type human TMEM132E mRNA together with MO rescued the phenotype. Scanning electron microscopy revealed fewer and shorter kinocilia and stereocilia in neuromasts in morphant animals. The morphant larvae showed delayed startle response, and significant reduction in microphonic potential. Together these data suggest non-functional mechanotransduction in hair cells (Li et al., 2015).
Transformation/Transcription domain-Associated Protein (TRRAP)
TRRAP encodes a multi-domain protein kinase that functions as a cofactor in the histone acetylation process. Whole-exome sequencing identified mutations in TRRAP genes with autosomal dominant deafness in a three generation Chinese family (Xia et al., 2019a). Later, three different variants were identified from molecular genetic testing of 66 sporadic cases of hearing loss. Functional studies for TRRAP genes were carried out in zebrafish using both MO-mediated knockdown and knockout animals. Zebrafish has a single trrap gene, and the loss of trrap function resulted in fewer and smaller neuromasts in the lateral line, and the number of hair cells and supporting cells were reduced in both knockdown and knockout animals. Scanning electron microscopy revealed that both kinocilia and stereocilia on hair cells per neuromast were shortened and were fewer in both mutants and morphants. Furthermore, hearing ability was measured by acoustic and c-shape startle response, and both knockdown and knockout animals have reduced responses compared to the wild-type animals, confirming the role of trrap in hearing (Xia et al., 2019a).
Sphingosine-1-phosphate Receptor 2 (S1PR2)
S1pr2 is a member of the sphingosine-1-phosphate receptor class of transmembrane G protein-coupled sphingolipid receptors. Mutations in the human S1PR2 gene cause non-syndromic autosomal recessive hearing loss DFNB68 (Santos-Cortez et al., 2016). The expression of s1pr2 mRNA in zebrafish was investigated by whole-mount in situ hybridization. s1pr2 mRNA expresses in somites, in the encephalic region at 24 hpf, and later in the lateral line and midbrain/hindbrain boundary at 48 hpf stage (Hu et al., 2013). MO-mediated knockdown of s1rp2 led to abnormal semicircular canals and otoliths, abnormal deposition of neuromasts in the lateral line, and fewer hair cells. Morphant animals also showed downregulation of many hearing-related genes. Morphant animals showed an increased number of apoptotic hair cells and dead hair cells as stained by apoptotic marker Hoechst 33324 (Hu et al., 2013).
Cell Division-Cycle 14A (CDC14A)
CDC14A encodes a dual specificity protein tyrosine phosphatase. Pathogenic mutations in CDC14A are associated with autosomal recessive hearing loss (DFNB32). Biallelic nonsense mutations in CDC14A were identified in consanguineous Iranian families affected by severe or profound congenital deafness (Delmaghani et al., 2006; Imtiaz et al., 2018). In zebrafish, the cdc14a gene has two paralogs, cdc14aa, and cdc14ab. It has been shown that cdc14a plays an important role in ciliogenesis in Kupffer’s vesicle, and other ciliated organs (Clement et al., 2012). Based on this observation, Delmaghani et al performed MO-mediated knockdown of cdc14aa gene in zebrafish to investigate its role in hearing, the morphant animals showed shortening of kinocilia in inner ear hair cells (Delmaghani et al., 2006). Subsequently, the shortening of kinocilia phenotype was not recapitulated in genetic mutants generated by CRISPR/Cas9 in another independent study (Imtiaz et al., 2018). The cdc14aa mutant is predicted to ablate phosphatase activity, and did not show any difference in startle response, or mechanotransduction by uptake of Yo-Pro1 live dye compared to control animals. It is possible that discrepancy in the phenotypes between MO-mediated knockdown and genetic mutant could be due to genetic compensation (Rossi et al., 2015).
Phosphoribosyl Pyrophosphate Synethetase-1 (PRPS1)
PRPS1 encodes for phosphoribosyl pyrophosphate synethetase-1, an important enzyme in nucleotide biosynthesis, and pathogenic mutations in PRPS1 have been implicated in X-linked non-syndromic sensorineural hearing loss (DFNX1/DFN2), as well as other diseases such Charcot-Marie-Tooth disease-5, and ARTS syndrome. Presently, more than 25 pathogenic mutations have been identified (Liu et al., 2010). The function of prps1 was studied in zebrafish by two different groups using genetic mutants and MO-mediated knockdown (DeSmidt et al., 2019; Pei et al., 2016). Zebrafish has two paralogs, prps1a, and prps1b. The prps1a mutants showed pigmentation defects, small eyes, and a reduced number of neuromast hair cells, whereas prps1b mutants showed no overt phenotype. However, prps1a;prps1b double mutants displayed more severe defects in the eye, pigmentation, and the neuromast hair cells (Pei et al., 2016). MO-mediated knockdown of either prps1 a or prps1b led to small otic vesicles, fewer hair cells in the inner ear, and reduced microphonic potential than the control animals. Taken together, loss of prps1a and prps1b functions lead to sensorineural hearing loss in zebrafish (DeSmidt et al., 2019; Pei et al., 2016).
Methionine Sulfoxide Reductase B3 (MSRB3)
MSRB3 encodes for methionine sulfoxide reductase that catalyzes the reduction of oxidized methionine in damaged proteins (Ahmed et al., 2011). Mutations in MSRB3 cause autosomal recessive hearing loss, DFNB74, two homozygous mutations were reported in 8 consanguineous families from Pakistan, these mutations led to truncation of MSRB3 gene suggesting its essential role in hearing. The function of msrb3 was also investigated in zebrafish using MO-mediated knockdown of msrb3 mRNA (Shen et al., 2015). Msrb3 mRNA expressed broadly at 12 hpf, and by 26 hpf, it was highly expressed in the inner ear. MO-mediated knockdown animals displayed abnormal otoliths, semicircular canals, as well as fewer and disordered neuromasts in the lateral line. Phalloidin staining showed shorter and thinner stereocilia in the inner ear, and scanning electron microscopy revealed damaged kinocilia in lateral line. Morphant embryos showed either reduced or no startle response than the control zebrafish (Shen et al., 2015).
While there are only a limited number of zebrafish models that exist for non-syndromic hearing loss, the number of disease models are expected to rise with the advancement of CRISPR/Cas9-targeted mutagenesis approaches. This is expected to subsequently accelerate our ability to unravel the functional mechanisms for genes involved in hearing loss.
Conclusions
With the major advancements in sequencing technologies made in the past decade, there is an unprecedented opportunity to exploit animal models to explore gene-disease associations and uncover the molecular mechanisms involved in human deafness. Over the past two decades, zebrafish has become a popular vertebrate model organism for human disease. Development of new genetic tools and techniques is fueling the growth of disease models, including those for hearing and balance disorders. Until 2013, the majority of the mutants available for studies have been generated by random mutagenesis because systematic targeted mutagenesis was not possible; this limited functional genomic studies of hearing loss genes. The development of targeted mutagenesis methods such as TALENs and CRISPR/Cas9 made it possible to generate gene knockouts at will. CRISPR/Cas9 technology was quickly and widely adopted due to its simplicity and cost-effective nature. It also allows for simultaneous targeting of more than one gene, making it possible to target two paralogs simultaneously in zebrafish. With the widespread availability of these newer genetic tools, it is now possible to validate human hearing loss genes in zebrafish in a targeted manner in order to understand their functional mechanisms a vertebrate organism.
In spite of exciting developments in the genome editing field, it remains to be seen how CRISPR-based base editing technologies will accelerate the generation of pathogenic variants in zebrafish, and how effectively they can be applied to recapitulate the variants that have been identified in humans with hereditary hearing loss. There is also a need to develop more innovative approaches to understand the role of multiple genes, epigenetics, and the environment in disease pathology. Zebrafish will be important in overcoming these challenges because of its ability to generate a large number of animals and facilitate observation of minor effects and low penetrance phenotypes. Although zebrafish are phylogenetically distant from mammals, its distinct features as a vertebrate model organism and the fact that more than 90% of human hearing-related genes have orthologs in zebrafish make it an ideal model in which to study hearing loss. While zebrafish offers many advantages over other model organisms such as mouse to model hearing loss, lack of auditory organ such as cochlea makes it harder to recapitulate all hearing functions from humans.
Highlights.
Advances in high-throughput sequencing have greatly enhanced our knowledge of the mutational signatures responsible for hereditary hearing loss
The greatest hurdles in the genetics of hereditary hearing loss concern functional validation of novel genes and uncharacterized variants
The zebrafish is an ideal model for rapid validation and determination of pathological mechanisms of novel hearing loss genes
Genome editing technologies may provide a promising avenue to address one of the greatest bottlenecks in the field
Acknowledgements and author contributions
We thank Sheng-Jia Lin, Cassidy Petree, and Rachel Smith for their help in generating Figure 3. BV and GKV reviewed the literature, wrote the manuscript and generated figures. JD and MAHH generated a table and figure, reviewed and edited the manuscript. TH provided supervision, reviewed and edited the manuscript. All authors have read and approve of the final article.
Funding: This work was supported by intramural funding from the University of Tübingen (2545-1-0) to BV, as well as NIH/COBRE GM103636 (Project 3), and the Presbyterian Health Foundation (PHF) Grant to GKV.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abe S, Katagiri T, Saito-Hisaminato A, Usami S-I, Inoue Y, Tsunoda T, Nakamura Y 2003. Identification of CRYM as a candidate responsible for nonsyndromic deafness, through cDNA microarray analysis of human cochlear and vestibular tissues. Am. J. Hum. Genet. 72, 73–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed ZM, Riazuddin S, Aye S, Ali RA, Venselaar H, Anwar S, Belyantseva PP, Qasim M, Riazuddin S, Friedman TB 2008a. Gene structure and mutant alleles of PCDH15: nonsyndromic deafness DFNB23 and type 1 Usher syndrome. Hum Genet 124, 215–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed ZM, Smith TN, Riazuddin S, Makishima T, Ghosh M, Bokhari S, Menon PSN, Deshmukh D, Griffith AJ, Riazuddin S, Friedman TB, Wilcox ER 2002. Nonsyndromic recessive deafness DFNB18 and Usher syndrome type IC are allelic mutations of USHIC. Hum. Genet. 110, 527–531. [DOI] [PubMed] [Google Scholar]
- Ahmed ZM, Riazuddin S, Ahmad J, Bernstein SL, Guo Y, Sabar MF, Sieving P, Riazuddin S, Griffith AJ, Friedman TB, Belyantseva IA, Wilcox ER 2003a. PCDH15 is expressed in the neurosensory epithelium of the eye and ear and mutant alleles are responsible for both USH1F and DFNB23. Hum. Mol. Genet. 12, 3215–3223. [DOI] [PubMed] [Google Scholar]
- Ahmed ZM, Morell RJ, Riazuddin S, Gropman A, Shaukat S, Ahmad MM, Mohiddin SA, Fananapazir L, Caruso RC, Husnain T, Khan SN, Griffith J, Friedman TB, Wilcox ER 2003b. Mutations of MYO6 are associated with recessive deafness, DFNB37. Am J Hum Genet 72, 1315–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed ZM, Morell RJ, Riazuddin S, Gropman A, Shaukat S, Ahmad MM, Mohiddin SA, Fananapazir L, Caruso RC, Husnain T, Khan SN, Riazuddin S, Griffith AJ, Friedman TB, Wilcox ER 2003c. Mutations of MYO6 are associated with recessive deafness, DFNB37. Am. J. Hum. Genet. 72, 1315–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed ZM, Yousaf R, Lee BC, Khan SN, Lee S, Lee K, Husnain T, Rehman AU, Bonneux S, Ansar M, Ahmad W, Leal SM, Gladyshev VN, Belyantseva IA, Van Camp G, Riazuddin S, Friedman TB 2011. Functional null mutations of MSRB3 encoding methionine sulfoxide reductase are associated with human deafness DFNB74. Am J Hum Genet 88, 19–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed ZM, Masmoudi S, Kalay E, Belyantseva IA, Mosrati MA, Collin RW, Riazuddin S, Hmani-Aifa M, Venselaar H, Kawar MN, Tlili A, van der Zwaag B, Khan SY, Ayadi L, Riazuddin SA, Morell RJ, Griffith AJ, Charfedine I, Caylan R, Oostrik J, Karaguzel A, Ghorbel A, Riazuddin S, Friedman TB, Ayadi H, Kremer H 2008b. Mutations of LRTOMT, a fusion gene with alternative reading frames, cause nonsyndromic deafness in humans. Nat Genet 40, 1335–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ain Q, Nazli S, Riazuddin S, Jaleel A-U, Riazuddin SA, Zafar AU, Khan SN, Husnain T, Griffith AJ, Ahmed ZM, Friedman TB, Riazuddin S 2007. The autosomal recessive nonsyndromic deafness locus DFNB72 is located on chromosome 19p13.3. Hum. Genet. 122, 445–450. [DOI] [PubMed] [Google Scholar]
- Alvarez A, del Castillo I, Villamar M, Aguirre LA, Gonzalez-Neira A, Lopez-Nevot A, Moreno-Pelayo MA, Moreno F 2005. High prevalence of the W24X mutation in the gene encoding connexin-26 (GJB2) in Spanish Romani (gypsies) with autosomal recessive non-syndromic hearing loss. Am J Med Genet A 137A, 255–8. [DOI] [PubMed] [Google Scholar]
- Amsterdam A, Yoon C, Allende M, Becker T, Kawakami K, Burgess S, Gaiano N, Hopkins N 1997. Retrovirus-mediated insertional mutagenesis in zebrafish and identification of a molecular marker for embryonic germ cells. Cold Spring Harb Symp Quant Biol 62, 437–50. [PubMed] [Google Scholar]
- Anderson JL, Mulligan TS, Shen M-C, Wang H, Scahill CM, Du SJ, Busch- Nentwich EM, Farber SA 2017. mRNA processing in mutant zebrafish lines generated by chemical and CRISPR-mediated mutagenesis produces potentially functional transcripts. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong GA, Liao M, You Z, Lissouba A, Chen BE, Drapeau P 2016. Homology Directed Knockin of Point Mutations in the Zebrafish tardbp and fus Genes in ALS Using the CRISPR/Cas9 System. PLoS One 11, e0150188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azaiez H, Booth KT, Bu F, Huygen P, Shibata SB, Shearer AE, Kolbe D, Meyer N, Black-Ziegelbein EA, Smith RJH 2014. TBC1D24 mutation causes autosomal-dominant nonsyndromic hearing loss. Hum. Mutat. 35, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azaiez H, Booth KT, Ephraim SS, Crone B, Black-Ziegelbein EA, Marini RJ, Shearer AE, Sloan-Heggen CM, Kolbe D, Casavant T, Schnieders MJ, Nishimura C, Braun T, Smith RJH 2018. Genomic Landscape and Mutational Signatures of Deafness-Associated Genes. Am. J. Hum. Genet. 103, 484–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azaiez H, Decker AR, Booth KT, Simpson AC, Shearer AE, Huygen PL, Bu F, Hildebrand MS, Ranum PT, Shibata SB, Turner A, Zhang Y, Kimberling WJ, Cornell RA, Smith RJ 2015. HOMER2, a stereociliary scaffolding protein, is essential for normal hearing in humans and mice. PLoS Genet 11, e1005137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bademci G, Abad C, Incesulu A, Elian F, Reyahi A, Diaz-Horta O, Cengiz FB, Sineni CJ, Seyhan S, Atli EI, Basmak H, Demir S, Nik AM, Footz T, Guo S, Duman D, Fitoz S, Gurkan H, Blanton SH, Walter MA, Carlsson P, Walz K, Tekin M 2019. FOXF2 is required for cochlear development in humans and mice. Hum. Mol. Genet. 28, 1286–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bademci G, Abad C, Incesulu A, Rad A, Alper O, Kolb SM, Cengiz FB, Diaz- Horta O, Silan F, Mihci E, Ocak E, Najafi M, Maroofian R, Yilmaz E, Nur BG, Duman D, Guo S, Sant DW, Wang G, Monje PV, Haaf T, Blanton SH, Vona B, Walz K, Tekin M 2018. MPZL2 is a novel gene associated with autosomal recessive nonsyndromic moderate hearing loss. Hum. Genet. 137, 479–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bang PI, Yelick PC, Malicki JJ, Sewell WF 2002. High-throughput behavioral screening method for detecting auditory response defects in zebrafish. J Neurosci Methods 118, 177–87. [DOI] [PubMed] [Google Scholar]
- Barashkov NA, Dzhemileva LU, Fedorova SA, Teryutin FM, Posukh OL, Fedotova EE, Lobov SL, Khusnutdinova EK 2011. Autosomal recessive deafness 1A (DFNB1A) in Yakut population isolate in Eastern Siberia: extensive accumulation of the splice site mutation IVS1+1G>A in GJB2 gene as a result of founder effect. J. Hum. Genet. 56, 631–639. [DOI] [PubMed] [Google Scholar]
- Barta CL, Liu H, Chen L, Giffen KP, Li Y, Kramer KL, Beisel KW, He DZ 2018. RNA-seq transcriptomic analysis of adult zebrafish inner ear hair cells. Sci Data 5, 180005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baxendale S, Whitfield TT 2016. Methods to study the development, anatomy, and function of the zebrafish inner ear across the life course. Methods Cell Biol 134, 165–209. [DOI] [PubMed] [Google Scholar]
- Behlouli A, Bonnet C, Abdi S, Bouaita A, Lelli A, Hardelin J-P, Schietroma C, Rous Y, Louha M, Cheknane A, Lebdi H, Boudjelida K, Makrelouf M, Zenati A, Petit C 2014. EPS8, encoding an actin-binding protein of cochlear hair cell stereocilia, is a new causal gene for autosomal recessive profound deafness. Orphanet J. Rare Dis. 9, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Said M, Grati M.h., Ishimoto T, Zou B, Chakchouk I, Ma Q, Yao Q, Hammami B, Yan D, Mittal R, Nakamichi N, Ghorbel A, Neng L, Tekin M, Shi XR, Kato Y, Masmoudi S, Lu Z, Hmani M, Liu X 2016. A mutation in SLC22A4 encoding an organic cation transporter expressed in the cochlea strial endothelium causes human recessive non-syndromic hearing loss DFNB60. Hum. Genet. 135, 513–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bespalova IN, Van Camp G, Bom SJ, Brown DJ, Cryns K, DeWan AT, Erson AE, Flothmann K, Kunst HP, Kurnool P, Sivakumaran TA, Cremers CW, Leal SM, Burmeister M, Lesperance MM 2001. Mutations in the Wolfram syndrome 1 gene (WFS1) are a common cause of low frequency sensorineural hearing loss. Hum. Mol. Genet. 10, 2501–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bharadwaj AK, Kasztejna JP, Huq S, Berson EL, Dryja TP 2000. Evaluation of the myosin VIIA gene and visual function in patients with Usher syndrome type I. Exp Eye Res 71, 173–81. [DOI] [PubMed] [Google Scholar]
- Blanco-Sanchez B, Clement A, Phillips JB, Westerfield M 2017. Zebrafish models of human eye and inner ear diseases. Methods Cell Biol 138, 415–467. [DOI] [PubMed] [Google Scholar]
- Blanco-Sanchez B, Clément A, Fierro J Jr., Stednitz S, Phillips JB, Wegner J, Panlilio JM, Peirce JL, Washbourne P, Westerfield M 2018. Grxcr1 Promotes Hair Bundle Development by Destabilizing the Physical Interaction between Harmonin and Sans Usher Syndrome Proteins. Cell Rep. 25, 1281–1291.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth KT, Kahrizi K, Najmabadi H, Azaiez H, Smith RJ 2018. Old gene, new phenotype: splice-altering variants in cause recessive non-syndromic hearing impairment. J. Med. Genet. 55, 555–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth KT, Askew JW, Talebizadeh Z, Huygen PLM, Eudy J, Kenyon J, Hoover D, Hildebrand MS, Smith KR, Bahlo M, Kimberling WJ, Smith RJH, Azaiez H, Smith SD 2019. Splice-altering variant in COL11A1 as a cause of nonsyndromic hearing loss DFNA37. Genet. Med. 21, 948–954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borck G, Ur Rehman A, Lee K, Pogoda HM, Kakar N, von Ameln S, Grillet N, Hildebrand MS, Ahmed ZM, Nurnberg G, Ansar M, Basit S, Javed Q, Morell RJ, Nasreen N, Shearer AE, Ahmad A, Kahrizi K, Shaikh RS, Ali RA, Khan SN, Goebel I, Meyer NC, Kimberling WJ, Webster JA, Stephan DA, Schiller MR, Bahlo M, Najmabadi H, Gillespie PG, Nurnberg P, Wollnik B, Riazuddin S, Smith RJ, Ahmad W, Muller U, Hammerschmidt M, Friedman TB, Leal SM, Ahmad J, Kubisch C 2011. Loss-of-function mutations of ILDR1 cause autosomal-recessive hearing impairment DFNB42. Am J Hum Genet 88, 127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bork JM, Peters LM, Riazuddin S, Bernstein SL, Ahmed ZM, Ness SL, Polomeno R, Ramesh A, Schloss M, Srisailpathy CR, Wayne S, Bellman S, Desmukh D, Ahmed Z, Khan SN, Kaloustian VM, Li XC, Lalwani A, Riazuddin S, Bitner-Glindzicz M, Nance WE, Liu XZ, Wistow G, Smith RJ, Griffith AJ, Wilcox ER, Friedman TB, Morell RJ 2001. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 68, 26–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buniello A, Ingham NJ, Lewis MA, Huma AC, Martinez-Vega R, Varela-Nieto I, Vizcay-Barrena G, Fleck RA, Houston O, Bardhan T, Johnson SL, White JK, Yuan H, Marcotti W, Steel KP 2016. Wbp2 is required for normal glutamatergic synapses in the cochlea and is crucial for hearing. EMBO Mol. Med. 8, 191–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busch-Nentwich E, Sollner C, Roehl H, Nicolson T 2004. The deafness gene dfna5 is crucial for ugdh expression and HA production in the developing ear in zebrafish. Development 131, 943–51. [DOI] [PubMed] [Google Scholar]
- Chakraborty AA, Nakamura E, Qi J, Creech A, Jaffe JD, Paulk J, Novak JS, Nagulapalli K, McBrayer SK, Cowley GS, Pineda J, Song J, Wang YE, Carr SA, Root DE, Signoretti S, Bradner JE, Kaelin WG Jr. 2017. HIF activation causes synthetic lethality between the VHL tumor suppressor and the EZH1 histone methyltransferase. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan DK, Chang KW 2014. GJB2-associated hearing loss: systematic review of worldwide prevalence, genotype, and auditory phenotype. Laryngoscope 124, E34–53. [DOI] [PubMed] [Google Scholar]
- Chang N, Sun C, Gao L, Zhu D, Xu X, Zhu X, Xiong JW, Xi JJ 2013. Genome editing with RNA-guided Cas9 nuclease in zebrafish embryos. Cell Res 23, 465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang-Chien J, Yen YC, Chien KH, Li SY, Hsu TC, Yang JJ 2014. The connexin 30.3 of zebrafish homologue of human connexin 26 may play similar role in the inner ear. Hearing research 313, 55–66. [DOI] [PubMed] [Google Scholar]
- Charizopoulou N, Lelli A, Schraders M, Ray K, Hildebrand MS, Ramesh A, Srisailapathy CR, Oostrik J, Admiraal RJ, Neely HR, Latoche JR, Smith RJ, Northup JK, Kremer H, Holt JR, Noben-Trauth K 2011. Gipc3 mutations associated with audiogenic seizures and sensorineural hearing loss in mouse and human. Nat Commun 2, 201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chatterjee P, Padmanarayana M, Abdullah N, Holman CL, LaDu J, Tanguay RL, Johnson CP 2015. Otoferlin deficiency in zebrafish results in defects in balance and hearing: rescue of the balance and hearing phenotype with full-length and truncated forms of mouse otoferlin. Mol Cell Biol 35, 1043–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D-Y, Liu X-F, Lin X-J, Zhang D, Chai Y-C, Yu D-H, Sun C-L, Wang X-L, Zhu W-D, Chen Y, Sun L-H, Wang X-W, Shi F-X, Huang Z-W, Yang T, Wu H 2017. A dominant variant in DMXL2 is linked to nonsyndromic hearing loss. Genet. Med. 19, 553–558. [DOI] [PubMed] [Google Scholar]
- Chen W, Kahrizi K, Meyer NC, Riazalhosseini Y, Van Camp G, Najmabadi H, Smith RJH 2005. Mutation of COL11A2 causes autosomal recessive non-syndromic hearing loss at the DFNB53 locus. J. Med. Genet. 42, e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Zhu Y, He S, Lu Y, Chen J, Han B, Petrillo M, Wrzeszczynski KO, Yang S, Dai P, Zhai S, Han D, Zhang MQ, Li W, Liu X, Li H, Chen Z-Y, Yuan H 2011. Functional mutation of SMAC/DIABLO, encoding a mitochondrial proapoptotic protein, causes human progressive hearing loss DFNA64. Am. J. Hum. Genet. 89, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement A, Solnica-Krezel L, Gould KL 2012. Functional redundancy between Cdc14 phosphatases in zebrafish ciliogenesis. Dev Dyn 241, 1911–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin RW, Kalay E, Tariq M, Peters T, van der Zwaag B, Venselaar H, Oostrik J, Lee K, Ahmed ZM, Caylan R, Li Y, Spierenburg HA, Eyupoglu E, Heister A, Riazuddin S, Bahat E, Ansar M, Arslan S, Wollnik B, Brunner HG, Cremers CW, Karaguzel A, Ahmad W, Cremers FP, Vriend G, Friedman TB, Riazuddin S, Leal SM, Kremer H 2008. Mutations of ESRRB encoding estrogen-related receptor beta cause autosomal-recessive nonsyndromic hearing impairment DFNB35. Am J Hum Genet 82, 125–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmani M, Ammar-Khodja F, Bonnet C, Lefèvre GM, Hardelin J-P, Ibrahim H, Mallek Z, Petit C 2015. EPS8L2 is a new causal gene for childhood onset autosomal recessive progressive hearing loss. Orphanet J. Rare Dis. 10, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AC 1989. The prevalence of hearing impairment and reported hearing disability among adults in Great Britain. Int. J. Epidemiol. 18, 911–917. [DOI] [PubMed] [Google Scholar]
- de Kok YJ, van der Maarel SM, Bitner-Glindzicz M, Huber I, Monaco AP, Malcolm S, Pembrey ME, Ropers HH, Cremers FP 1995. Association between X-linked mixed deafness and mutations in the POU domain gene POU3F4. Science 267, 685–688. [DOI] [PubMed] [Google Scholar]
- del Castillo I, Villamar M, Moreno-Pelayo MA, del Castillo FJ, Alvarez A, Telleria D, Menendez I, Moreno F 2002. A deletion involving the connexin 30 gene in nonsyndromic hearing impairment. N Engl J Med 346, 243–9. [DOI] [PubMed] [Google Scholar]
- Delmaghani S, Aghaie A, Michalski N, Bonnet C, Weil D, Petit C 2012. Defect in the gene encoding the EAR/EPTP domain-containing protein TSPEAR causes DFNB98 profound deafness. Hum Mol Genet 21, 3835–44. [DOI] [PubMed] [Google Scholar]
- Delmaghani S, Aghaie A, Bouyacoub Y, El Hachmi H, Bonnet C, Riahi Z, Chardenoux S, Perfettini I, Hardelin J-P, Houmeida A, Herbomel P, Petit C 2016. Mutations in CDC14A, Encoding a Protein Phosphatase Involved in Hair Cell Ciliogenesis, Cause Autosomal-Recessive Severe to Profound Deafness. Am. J. Hum. Genet. 98, 1266–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmaghani S, del Castillo FJ, Michel V, Leibovici M, Aghaie A, Ron U, Van Laer L, Ben-Tal N, Van Camp G, Weil D, Langa F, Lathrop M, Avan P, Petit C 2006. Mutations in the gene encoding pejvakin, a newly identified protein of the afferent auditory pathway, cause DFNB59 auditory neuropathy. Nat. Genet. 38, 770–778. [DOI] [PubMed] [Google Scholar]
- DeSmidt AA, Zou B, Grati M.h., Yan D, Mittal R, Yao Q, Richmond MT, Denyer S, Liu X-Z, Lu Z 2019. Zebrafish Model for Nonsyndromic X-Linked Sensorineural Deafness, DFNX1. Anat. Rec. [DOI] [PubMed] [Google Scholar]
- Diaz-Horta O, Subasioglu-Uzak A, Grati M, DeSmidt A, Foster J 2nd, Cao L, Bademci G, Tokgoz-Yilmaz S, Duman D, Cengiz FB, Abad C, Mittal R, Blanton S, Liu XZ, Farooq A, Walz K, Lu Z, Tekin M 2014. FAM65B is a membrane-associated protein of hair cell stereocilia required for hearing. Proc Natl Acad Sci U S A 111, 9864–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Horta O, Abad C, Sennaroglu L, Foster J 2nd, DeSmidt A, Bademci G, Tokgoz-Yilmaz S, Duman D, Cengiz FB, Grati M.h., Fitoz S, Liu XZ, Farooq A, Imtiaz F, Currall BB, Morton CC, Nishita M, Minami Y, Lu Z, Walz K, Tekin M 2016. ROR1 is essential for proper innervation of auditory hair cells and hearing in humans and mice. Proc. Natl. Acad. Sci. U. S. A. 113, 5993–5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donaudy F, Snoeckx R, Pfister M, Zenner H-P, Blin N, Di Stazio M, Ferrara A, Lanzara C, Ficarella R, Declau F, Pusch CM, Nürnberg P, Melchionda S, Zelante L, Ballana E, Estivill X, Van Camp G, Gasparini P, Savoia A 2004. Nonmuscle myosin heavy-chain gene MYH14 is expressed in cochlea and mutated in patients affected by autosomal dominant hearing impairment (DFNA4). Am. J. Hum. Genet. 74, 770–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driever W, Solnica-Krezel L, Schier AF, Neuhauss SC, Malicki J, Stemple DL, Stainier DY, Zwartkruis F, Abdelilah S, Rangini Z, Belak J, Boggs C 1996. A genetic screen for mutations affecting embryogenesis in zebrafish. Development 123, 37–46. [DOI] [PubMed] [Google Scholar]
- Du X, Schwander M, Moresco EM, Viviani P, Haller C, Hildebrand MS, Pak K, Tarantino L, Roberts A, Richardson H, Koob G, Najmabadi H, Ryan AF, Smith RJ, Muller U, Beutler B 2008. A catechol-O-methyltransferase that is essential for auditory function in mice and humans. Proc Natl Acad Sci U S A 105, 14609–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebermann I, Phillips JB, Liebau MC, Koenekoop RK, Schermer B, Lopez I, Schafer E, Roux AF, Dafinger C, Bernd A, Zrenner E, Claustres M, Blanco B, Nurnberg G, Nurnberg P, Ruland R, Westerfield M, Benzing T, Bolz HJ 2010. PDZD7 is a modifier of retinal disease and a contributor to digenic Usher syndrome. J Clin Invest 120, 1812–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenberger T, Di Donato N, Decker C, Delle Vedove A, Neuhaus C, Nürnberg G, Toliat M, Nürnberg P, Mürbe D, Bolz HJ 2018. A C-terminal nonsense mutation links PTPRQ with autosomal-dominant hearing loss, DFNA73. Genet. Med. 20, 614–621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Brolosy MA, Kontarakis Z, Rossi A, Kuenne C, Gunther S, Fukuda N, Kikhi K, Boezio GLM, Takacs CM, Lai SL, Fukuda R, Gerri C, Giraldez AJ, Stainier DYR 2019. Genetic compensation triggered by mutant mRNA degradation. Nature 568, 193–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elepfandt A 1988. Processing of wave patterns in the lateral line system parallels to auditory processing. Acta Biol Hung 39, 251–65. [PubMed] [Google Scholar]
- Erickson T, Nicolson T 2015. Identification of sensory hair-cell transcripts by thiouracil-tagging in zebrafish. BMC Genomics 16, 842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson T, Morgan CP, Olt J, Hardy K, Busch-Nentwich E, Maeda R, Clemens R, Krey JF, Nechiporuk A, Barr-Gillespie PG, Marcotti W, Nicolson T 2017. Integration of Tmc1/2 into the mechanotransduction complex in zebrafish hair cells is regulated by Transmembrane O-methyltransferase (Tomt). Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernest S, Rauch GJ, Haffter P, Geisler R, Petit C, Nicolson T 2000. Mariner is defective in myosin VIIA: a zebrafish model for human hereditary deafness. Hum Mol Genet 9, 2189–96. [DOI] [PubMed] [Google Scholar]
- Feng Y, Chen C, Han Y, Chen Z, Lu X, Liang F, Li S, Qin W, Lin S 2016. Expanding CRISPR/Cas9 Genome Editing Capacity in Zebrafish Using SaCas9. G3 (Bethesda) 6, 2517–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaiano N, Amsterdam A, Kawakami K, Allende M, Becker T, Hopkins N 1996. Insertional mutagenesis and rapid cloning of essential genes in zebrafish. Nature 383, 829–32. [DOI] [PubMed] [Google Scholar]
- Gao J, Wang Q, Dong C, Chen S, Qi Y, Liu Y 2015. Whole Exome Sequencing Identified MCM2 as a Novel Causative Gene for Autosomal Dominant Nonsyndromic Deafness in a Chinese Family. PLoS One 10, e0133522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Yuan Y-Y, Lin Q-F, Xu J-C, Wang W-Q, Qiao Y-H, Kang D-Y, Bai D, Xin F, Huang S-S, Qiu S-W, Guan L-P, Su Y, Wang G-J, Han M-Y, Jiang Y, Liu H-K, Dai P 2018. Mutation of, an interferon lambda receptor 1, is associated with autosomal-dominant non-syndromic hearing loss. J. Med. Genet. 55, 298–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, Liu DR 2017. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghysen A, Dambly-Chaudiere C 2004. Development of the zebrafish lateral line. Curr Opin Neurobiol 14, 67–73. [DOI] [PubMed] [Google Scholar]
- Gleason MR, Nagiel A, Jamet S, Vologodskaia M, Lopez-Schier H, Hudspeth AJ 2009. The transmembrane inner ear (Tmie) protein is essential for normal hearing and balance in the zebrafish. Proc Natl Acad Sci U S A 106, 21347–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golling G, Amsterdam A, Sun Z, Antonelli M, Maldonado E, Chen W, Burgess S, Haldi M, Artzt K, Farrington S, Lin SY, Nissen RM, Hopkins N 2002. Insertional mutagenesis in zebrafish rapidly identifies genes essential for early vertebrate development. Nat Genet 31, 135–40. [DOI] [PubMed] [Google Scholar]
- Granato M, van Eeden FJ, Schach U, Trowe T, Brand M, Furutani-Seiki M, Haffter P, Hammerschmidt M, Heisenberg CP, Jiang YJ, Kane DA, Kelsh RN, Mullins MC, Odenthal J, Nusslein-Volhard C 1996. Genes controlling and mediating locomotion behavior of the zebrafish embryo and larva. Development 123, 399–413. [DOI] [PubMed] [Google Scholar]
- Grati M, Chakchouk I, Ma Q, Bensaid M, Desmidt A, Turki N, Yan D, Baanannou A, Mittal R, Driss N, Blanton S, Farooq A, Lu Z, Liu XZ, Masmoudi S 2015. A missense mutation in DCDC2 causes human recessive deafness DFNB66, likely by interfering with sensory hair cell and supporting cell cilia length regulation. Hum Mol Genet 24, 2482–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grati M.h., Yan D, Raval MH, Walsh T, Ma Q, Chakchouk I, Kannan-Sundhari A, Mittal R, Masmoudi S, Blanton SH, Tekin M, King M-C, Yengo CM, Liu XZ 2016. MYO3A Causes Human Dominant Deafness and Interacts with Protocadherin 15-CD2 Isoform. Hum. Mutat. 37, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grifa A, Wagner CA, D’Ambrosio L, Melchionda S, Bernardi F, Lopez-Bigas N, Rabionet R, Arbones M, Monica MD, Estivill X, Zelante L, Lang F, Gasparini P 1999. Mutations in GJB6 cause nonsyndromic autosomal dominant deafness at DFNA3 locus. Nat. Genet. 23, 16–18. [DOI] [PubMed] [Google Scholar]
- Grillet N, Schwander M, Hildebrand MS, Sczaniecka A, Kolatkar A, Velasco J, Webster JA, Kahrizi K, Najmabadi H, Kimberling WJ, Stephan D, Bahlo M, Wiltshire T, Tarantino LM, Kuhn P, Smith RJH, Müller U 2009. Mutations in LOXHD1, an evolutionarily conserved stereociliary protein, disrupt hair cell function in mice and cause progressive hearing loss in humans. Am. J. Hum. Genet. 85, 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haffter P, Granato M, Brand M, Mullins MC, Hammerschmidt M, Kane DA, Odenthal J, van Eeden FJ, Jiang YJ, Heisenberg CP, Kelsh RN, Furutani-Seiki M, Vogelsang E, Beuchle D, Schach U, Fabian C, Nusslein-Volhard C 1996. The identification of genes with unique and essential functions in the development of the zebrafish, Danio rerio. Development 123, 1–36. [DOI] [PubMed] [Google Scholar]
- Han Y, Mu Y, Li X, Xu P, Tong J, Liu Z, Ma T, Zeng G, Yang S, Du J, Meng A 2011. Grhl2 deficiency impairs otic development and hearing ability in a zebrafish model of the progressive dominant hearing loss DFNA28. Human molecular genetics 20, 3213–26. [DOI] [PubMed] [Google Scholar]
- Hardison AL, Lichten L, Banerjee-Basu S, Becker TS, Burgess SM 2005. The zebrafish gene claudinj is essential for normal ear function and important for the formation of the otoliths. Mech Dev 122, 949–58. [DOI] [PubMed] [Google Scholar]
- Hildebrand MS, Thorne NP, Bromhead CJ, Kahrizi K, Webster JA, Fattahi Z, Bataejad M, Kimberling WJ, Stephan D, Najmabadi H, Bahlo M, Smith RJ 2010. Variable hearing impairment in a DFNB2 family with a novel MYO7A missense mutation. Clin Genet 77, 563–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgert N, Smith RJ, Van Camp G 2009. Forty-six genes causing nonsyndromic hearing impairment: which ones should be analyzed in DNA diagnostics? Mutat Res 681, 189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn HF, Brownstein Z, Lenz DR, Shivatzki S, Dror AA, Dagan-Rosenfeld O, Friedman LM, Roux KJ, Kozlov S, Jeang K-T, Frydman M, Burke B, Stewart CL, Avraham KB 2013. The LINC complex is essential for hearing. J. Clin. Invest. 123, 740–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshijima K, Jurynec MJ, Klatt Shaw D, Jacobi AM, Behlke MA, Grunwald DJ 2019. Highly Efficient CRISPR-Cas9-Based Methods for Generating Deletion Mutations and F0 Embryos that Lack Gene Function in Zebrafish. Dev Cell 51, 645–657 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K, Clark MD, Torroja CF, Torrance J, Berthelot C, Muffato M, Collins JE, Humphray S, McLaren K, Matthews L, McLaren S, Sealy I, Caccamo M, Churcher C, Scott C, Barrett JC, Koch R, Rauch GJ, White S, Chow W, Kilian B, Quintais LT, Guerra-Assuncao JA, Zhou Y, Gu Y, Yen J, Vogel JH, Eyre T, Redmond S, Banerjee R, Chi J, Fu B, Langley E, Maguire SF, Laird GK, Lloyd D, Kenyon E, Donaldson S, Sehra H, Almeida-King J, Loveland J, Trevanion S, Jones M, Quail M, Willey D, Hunt A, Burton J, Sims S, McLay K, Plumb B, Davis J, Clee C, Oliver K, Clark R, Riddle C, Elliot D, Threadgold G, Harden G, Ware D, Begum S, Mortimore B, Kerry G, Heath P, Phillimore B, Tracey A, Corby N, Dunn M, Johnson C, Wood J, Clark S, Pelan S, Griffiths G, Smith M, Glithero R, Howden P, Barker N, Lloyd C, Stevens C, Harley J, Holt K, Panagiotidis G, Lovell J, Beasley H, Henderson C, Gordon D, Auger K, Wright D, Collins J, Raisen C, Dyer L, Leung K, Robertson L, Ambridge K, Leongamornlert D, McGuire S, Gilderthorp R, Griffiths C, Manthravadi D, Nichol S, Barker G, et al. 2013. The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hruscha A, Krawitz P, Rechenberg A, Heinrich V, Hecht J, Haass C, Schmid B 2013. Efficient CRISPR/Cas9 genome editing with low off-target effects in zebrafish. Development 140, 4982–7. [DOI] [PubMed] [Google Scholar]
- Hu ZY, Zhang QY, Qin W, Tong JW, Zhao Q, Han Y, Meng J, Zhang JP 2013. Gene miles-apart is required for formation of otic vesicle and hair cells in zebrafish. Cell Death Dis 4, e900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huebner AK, Gandia M, Frommolt P, Maak A, Wicklein EM, Thiele H, Altmüller J, Wagner F, Viñuela A, Aguirre LA, Moreno F, Maier H, Rau I, Giesselmann S, Nürnberg G, Gal A, Nürnberg P, Hübner CA, del Castillo I, Kurth I 2011. Nonsense mutations in SMPX, encoding a protein responsive to physical force, result in X-chromosomal hearing loss. Am. J. Hum. Genet. 88, 621–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imtiaz A, Kohrman DC, Naz S 2014. A frameshift mutation in GRXCR2 causes recessively inherited hearing loss. Hum. Mutat. 35, 618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imtiaz A, Belyantseva IA, Beirl AJ, Fenollar-Ferrer C, Bashir R, Bukhari I, Bouzid A, Shaukat U, Azaiez H, Booth KT, Kahrizi K, Najmabadi H, Maqsood A, Wilson EA, Fitzgerald TS, Tlili A, Olszewski R, Lund M, Chaudhry T, Rehman AU, Starost MF, Waryah AM, Hoa M, Dong L, Morell RJ, Smith RJH, Riazuddin S, Masmoudi S, Kindt KS, Naz S, Friedman TB 2018. CDC14A phosphatase is essential for hearing and male fertility in mouse and human. Hum. Mol. Genet. 27, 780–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingham NJ, Pearson SA, Vancollie VE, Rook V, Lewis MA, Chen J, Buniello A, Martelletti E, Preite L, Lam CC, Weiss FD, Powis Z, Suwannarat P, Lelliott CJ, Dawson SJ, White JK, Steel KP 2019. Mouse screen reveals multiple new genes underlying mouse and human hearing loss. PLoS Biol. 17, e3000194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irion U, Krauss J, Nusslein-Volhard C 2014. Precise and efficient genome editing in zebrafish using the CRISPR/Cas9 system. Development 141, 4827–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jao LE, Wente SR, Chen W 2013. Efficient multiplex biallelic zebrafish genome editing using a CRISPR nuclease system. Proc Natl Acad Sci U S A 110, 13904–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaworek TJ, Richard EM, Ivanova AA, Giese AP, Choo DI, Khan SN, Riazuddin S, Kahn RA 2013. An alteration in ELMOD3, an Arl2 GTPase-activating protein, is associated with hearing impairment in humans. PLoS genetics 9, e1003774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalay E, Li Y, Uzumcu A, Uyguner O, Collin RW, Caylan R, Ulubil-Emiroglu M, Kersten FFJ, Hafiz G, van Wijk E, Kayserili H, Rohmann E, Wagenstaller J, Hoefsloot LH, Strom TM, Nürnberg G, Baserer N, den Hollander AI, Cremers FPM, Cremers CWRJ, Becker C, Brunner HG, Nürnberg P, Karaguzel A, Basaran S, Kubisch C, Kremer H, Wollnik B 2006. Mutations in the lipoma HMGIC fusion partner-like 5 (LHFPL5) gene cause autosomal recessive nonsyndromic hearing loss. Hum. Mutat. 27, 633–639. [DOI] [PubMed] [Google Scholar]
- Kelsell DP, Dunlop J, Stevens HP, Lench NJ, Liang JN, Parry G, Mueller RF, Leigh IM 1997. Connexin 26 mutations in hereditary non-syndromic sensorineural deafness. Nature 387, 80–83. [DOI] [PubMed] [Google Scholar]
- Kettleborough RN, Busch-Nentwich EM, Harvey SA, Dooley CM, de Bruijn E, van Eeden F, Sealy I, White RJ, Herd C, Nijman IJ, Fenyes F, Mehroke S, Scahill C, Gibbons R, Wali N, Carruthers S, Hall A, Yen J, Cuppen E, Stemple DL 2013. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature 496, 494–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SY, Ahmed ZM, Shabbir MI, Kitajiri S, Kalsoom S, Tasneem S, Shayiq S, Ramesh A, Srisailpathy S, Khan SN, Smith RJ, Riazuddin S, Friedman TB 2007. Mutations of the RDX gene cause nonsyndromic hearing loss at the DFNB24 locus. Hum Mutat 28, 417–23. [DOI] [PubMed] [Google Scholar]
- Kindt KS, Sheets L 2018. Transmission Disrupted: Modeling Auditory Synaptopathy in Zebrafish. Front Cell Dev Biol 6, 114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kok FO, Shin M, Ni C, Gupta A, Grosse AS, van Impel A, Kirchmaier BC, Peterson-Maduro J, Kourkoulis G, Male I, DeSantis DF, Sheppard-Tindell S, Ebarasi L, Betsholtz C, Schulte-Merker S, Wolfe SA, Lawson ND 2014. Reverse Genetic Screening Reveals Poor Correlation between Morpholino-Induced and Mutant Phenotypes in Zebrafish. Developmental cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR 2016. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubisch C, Schroeder BC, Friedrich T, Lutjohann B, El-Amraoui A, Marlin S, Petit C, Jentsch TJ 1999. KCNQ4, a novel potassium channel expressed in sensory outer hair cells, is mutated in dominant deafness. Cell 96, 437–46. [DOI] [PubMed] [Google Scholar]
- Kurima K, Peters LM, Yang Y, Riazuddin S, Ahmed ZM, Naz S, Arnaud D, Drury S, Mo J, Makishima T, Ghosh M, Menon PS, Deshmukh D, Oddoux C, Ostrer H, Khan S, Deininger PL, Hampton LL, Sullivan SL, Battey JF Jr., Keats BJ, Wilcox ER, Friedman TB, Griffith AJ 2002. Dominant and recessive deafness caused by mutations of a novel gene, TMC1, required for cochlear hair-cell function. Nature genetics 30, 277–84. [DOI] [PubMed] [Google Scholar]
- Lalwani AK, Goldstein JA, Kelley MJ, Luxford W, Castelein CM, Mhatre AN 2000. Human nonsyndromic hereditary deafness DFNA17 is due to a mutation in nonmuscle myosin MYH9. Am. J. Hum. Genet. 67, 1121–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law SH, Sargent TD 2014. The serine-threonine protein kinase PAK4 is dispensable in zebrafish: identification of a morpholino-generated pseudophenotype. PLoS One 9, e100268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Bademci G, Subasioglu A, Diaz-Horta O, Zhu Y, Liu J, Mitchell TG, Abad C, Seyhan S, Duman D, Cengiz FB, Tokgoz-Yilmaz S, Blanton SH, Farooq A, Walz K, Zhai RG, Tekin M 2019. Dysfunction of, encoding the GRB2-related adaptor protein, is linked to sensorineural hearing loss. Proc. Natl. Acad. Sci. U. S. A. 116, 1347–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Zhao X, Xin Q, Shan S, Jiang B, Jin Y, Yuan H, Dai P, Xiao R, Zhang Q, Xiao J, Shao C, Gong Y, Liu Q 2015. Whole-exome sequencing identifies a variant in TMEM132E causing autosomal-recessive nonsyndromic hearing loss DFNB99. Hum. Mutat. 36, 98–105. [DOI] [PubMed] [Google Scholar]
- Li W, Sun J, Ling J, Li J, He C, Liu Y, Chen H, Men M, Niu Z, Deng Y, Li M, Li T, Wen J, Sang S, Li H, Wan Z, Richard EM, Chapagain P, Yan D, Liu XZ, Mei L, Feng Y 2018. ELMOD3, a novel causative gene, associated with human autosomal dominant nonsyndromic and progressive hearing loss. Hum. Genet. 137, 329–342. [DOI] [PubMed] [Google Scholar]
- Li XC, Everett LA, Lalwani AK, Desmukh D, Friedman TB, Green ED, Wilcox ER 1998. A mutation in PDS causes non-syndromic recessive deafness. Nat. Genet. 18, 215–217. [DOI] [PubMed] [Google Scholar]
- Li Y, Pohl E, Boulouiz R, Schraders M, Nurnberg G, Charif M, Admiraal RJ, von Ameln S, Baessmann I, Kandil M, Veltman JA, Nurnberg P, Kubisch C, Barakat A, Kremer H, Wollnik B 2010. Mutations in TPRN cause a progressive form of autosomal-recessive nonsyndromic hearing loss. Am J Hum Genet 86, 479–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Petree C, Requena T, Varshney P, Varshney GK 2019. Expanding the CRISPR Toolbox in Zebrafish for Studying Development and Disease. Front Cell Dev Biol 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Han D, Li J, Han B, Ouyang X, Cheng J, Li X, Jin Z, Wang Y, Bitner-Glindzicz M, Kong X, Xu H, Kantardzhieva A, Eavey RD, Seidman CE, Seidman JG, Du LL, Chen ZY, Dai P, Teng M, Yan D, Yuan H 2010. Loss-of-function mutations in the PRPS1 gene cause a type of nonsyndromic X-linked sensorineural deafness, DFN2. Am J Hum Genet 86, 65–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XZ, Walsh J, Tamagawa Y, Kitamura K, Nishizawa M, Steel KP, Brown SD 1997a. Autosomal dominant non-syndromic deafness caused by a mutation in the myosin VIIA gene. Nat. Genet. 17, 268–269. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Walsh J, Mburu P, Kendrick-Jones J, Cope MJ, Steel KP, Brown SD 1997b. Mutations in the myosin VIIA gene cause non-syndromic recessive deafness. Nat. Genet. 16, 188–190. [DOI] [PubMed] [Google Scholar]
- Liu XZ, Ouyang XM, Xia XJ, Zheng J, Pandya A, Li F, Du LL, Welch KO, Petit C, Smith RJ, Webb BT, Yan D, Arnos KS, Corey D, Dallos P, Nance WE, Chen ZY 2003. Prestin, a cochlear motor protein, is defective in non-syndromic hearing loss. Hum Mol Genet 12, 1155–62. [DOI] [PubMed] [Google Scholar]
- Livingston G, Sommerlad A, Orgeta V, Costafreda SG, Huntley J, Ames D, Ballard C, Banerjee S, Burns A, Cohen-Mansfield J, Cooper C, Fox N, Gitlin LN, Howard R, Kales HC, Larson EB, Ritchie K, Rockwood K, Sampson EL, Samus Q, Schneider LS, Selbæk G, Teri L, Mukadam N 2017. Dementia prevention, intervention, and care. Lancet 390, 2673–2734. [DOI] [PubMed] [Google Scholar]
- Lu Z, DeSmidt AA 2013. Early development of hearing in zebrafish. J Assoc Res Otolaryngol 14, 509–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukasz D, Kindt KS 2018. In Vivo Calcium Imaging of Lateral-line Hair Cells in Larval Zebrafish. J Vis Exp. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundberg YW, Xu Y, Thiessen KD, Kramer KL 2015. Mechanisms of otoconia and otolith development. Dev Dyn 244, 239–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch ED, Lee MK, Morrow JE, Welcsh PL, Leon PE, King MC 1997. Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science 278, 1315–8. [PubMed] [Google Scholar]
- Ma Z, Zhu P, Shi H, Guo L, Zhang Q, Chen Y, Chen S, Zhang Z, Peng J, Chen J 2019. PTC-bearing mRNA elicits a genetic compensation response via Upf3a and COMPASS components. Nature 568, 259–263. [DOI] [PubMed] [Google Scholar]
- Malicki J, Schier AF, Solnica-Krezel L, Stemple DL, Neuhauss SC, Stainier DY, Abdelilah S, Rangini Z, Zwartkruis F, Driever W 1996. Mutations affecting development of the zebrafish ear. Development 123, 275–83. [DOI] [PubMed] [Google Scholar]
- Matern MS, Beirl A, Ogawa Y, Song Y, Paladugu N, Kindt KS, Hertzano R 2018. Transcriptomic Profiling of Zebrafish Hair Cells Using RiboTag. Front Cell Dev Biol 6, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mburu P, Mustapha M, Varela A, Weil D, El-Amraoui A, Holme RH, Rump A, Hardisty RE, Blanchard S, Coimbra RS, Perfettini I, Parkinson N, Mallon A-M, Glenister P, Rogers MJ, Paige AJ, Moir L, Clay J, Rosenthal A, Liu XZ, Blanco G, Steel KP, Petit C, Brown SDM 2003. Defects in whirlin, a PDZ domain molecule involved in stereocilia elongation, cause deafness in the whirler mouse and families with DFNB31. Nat. Genet. 34, 421–428. [DOI] [PubMed] [Google Scholar]
- McGuirt WT, Prasad SD, Griffith AJ, Kunst HP, Green GE, Shpargel KB, Runge C, Huybrechts C, Mueller RF, Lynch E, King MC, Brunner HG, Cremers CW, Takanosu M, Li SW, Arita M, Mayne R, Prockop DJ, Van Camp G, Smith RJ 1999. Mutations in COL11A2 cause non-syndromic hearing loss (DFNA13). Nat. Genet. 23, 413–419. [DOI] [PubMed] [Google Scholar]
- Melchionda S, Ahituv N, Bisceglia L, Sobe T, Glaser F, Rabionet R, Arbones ML, Notarangelo A, Di Iorio E, Carella M, Zelante L, Estivill X, Avraham KB, Gasparini P 2001. MYO6, the human homologue of the gene responsible for deafness in Snell’s waltzer mice, is mutated in autosomal dominant nonsyndromic hearing loss. Am. J. Hum. Genet. 69, 635–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mencia A, Modamio-Hoybjor S, Redshaw N, Morin M, Mayo-Merino F, Olavarrieta L, Aguirre LA, del Castillo I, Steel KP, Dalmay T, Moreno F, Moreno-Pelayo MA 2009. Mutations in the seed region of human miR-96 are responsible for nonsyndromic progressive hearing loss. Nat Genet 41, 609–13. [DOI] [PubMed] [Google Scholar]
- Metcalfe WK, Kimmel CB, Schabtach E 1985. Anatomy of the posterior lateral line system in young larvae of the zebrafish. J Comp Neurol 233, 377–89. [DOI] [PubMed] [Google Scholar]
- Modamio-Hoybjor S, Mencia A, Goodyear R, del Castillo I, Richardson G, Moreno F, Moreno-Pelayo MA 2007. A mutation in CCDC50, a gene encoding an effector of epidermal growth factor-mediated cell signaling, causes progressive hearing loss. Am. J. Hum. Genet. 80, 1076–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno-Mateos MA, Fernandez JP, Rouet R, Vejnar CE, Lane MA, Mis E, Khokha MK, Doudna JA, Giraldez AJ 2017. CRISPR-Cpf1 mediates efficient homology-directed repair and temperature-controlled genome editing. Nature Communications 8, 2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan A, Vuckovic D, Krishnamoorthy N, Rubinato E, Ambrosetti U, Castorina P, Franzè A, Vozzi D, La Bianca M, Cappellani S, Di Stazio M, Gasparini P, Girotto G 2019. Next-generation sequencing identified SPATC1L as a possible candidate gene for both early-onset and age-related hearing loss. Eur. J. Hum. Genet. 27, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton CC, Nance WE 2006. Newborn Hearing Screening — A Silent Revolution. N Engl J Med 354, 2151–2164. [DOI] [PubMed] [Google Scholar]
- Mosrati MA, Hammami B, Rebeh IB, Ayadi L, Dhouib L, Ben Mahfoudh K, Hakim B, Charfeddine I, Mnif J, Ghorbel A, Masmoudi S 2011. A novel dominant mutation in SIX1, affecting a highly conserved residue, result in only auditory defects in humans. Eur. J. Med. Genet. 54, e484–8. [DOI] [PubMed] [Google Scholar]
- Mujtaba G, Schultz JM, Imtiaz A, Morell RJ, Friedman TB, Naz S 2015. A mutation of MET, encoding hepatocyte growth factor receptor, is associated with human DFNB97 hearing loss. J. Med. Genet. 52, 548–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mustapha M, Weil D, Chardenoux S, Elias S, El-Zir E, Beckmann JS, Loiselet J, Petit C 1999. An alpha-tectorin gene defect causes a newly identified autosomal recessive form of sensorineural pre-lingual non-syndromic deafness, DFNB21. Hum. Mol. Genet. 8, 409–412. [DOI] [PubMed] [Google Scholar]
- Nakanishi H, Kawashima Y, Kurima K, Chae JJ, Ross AM, Pinto-Patarroyo G, Patel SK, Muskett JA, Ratay JS, Chattaraj P, Park YH, Grevich S, Brewer CC, Hoa M, Kim HJ, Butman JA, Broderick L, Hoffman HM, Aksentijevich I, Kastner DL, Goldbach-Mansky R, Griffith AJ 2017. mutation and cochlear autoinflammation cause syndromic and nonsyndromic hearing loss DFNA34 responsive to anakinra therapy. Proc. Natl. Acad. Sci. U. S. A. 114, E7766–E7775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano Y, Kelly MC, Rehman AU, Boger ET, Morell RJ, Kelley MW, Friedman TB, Bánfi B 2018. Defects in the Alternative Splicing-Dependent Regulation of REST Cause Deafness. Cell 174, 536–548.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasevicius A, Ekker SC 2000. Effective targeted gene ‘knockdown’ in zebrafish. Nat Genet 26, 216–20. [DOI] [PubMed] [Google Scholar]
- Naz S, Griffith AJ, Riazuddin S, Hampton LL, Battey JF Jr., Khan SN, Riazuddin S, Wilcox ER, Friedman TB 2004. Mutations of ESPN cause autosomal recessive deafness and vestibular dysfunction. J. Med. Genet. 41, 591–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naz S, Giguere CM, Kohrman DC, Mitchem KL, Riazuddin S, Morell RJ, Ramesh A, Srisailpathy S, Deshmukh D, Griffith AJ, Friedman TB, Smith RJ, Wilcox ER 2002. Mutations in a novel gene, TMIE, are associated with hearing loss linked to the DFNB6 locus. Am J Hum Genet 71, 632–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolson T 2005. The genetics of hearing and balance in zebrafish. Annu Rev Genet 39, 9–22. [DOI] [PubMed] [Google Scholar]
- Nicolson T 2017. The genetics of hair-cell function in zebrafish. J Neurogenet 31, 102–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicolson T, Rusch A, Friedrich RW, Granato M, Ruppersberg JP, Nusslein-Volhard C 1998. Genetic analysis of vertebrate sensory hair cell mechanosensation: the zebrafish circler mutants. Neuron 20, 271–83. [DOI] [PubMed] [Google Scholar]
- Nissen RM, Yan J, Amsterdam A, Hopkins N, Burgess SM 2003. Zebrafish foxi one modulates cellular responses to Fgf signaling required for the integrity of ear and jaw patterning. Development 130, 2543–54. [DOI] [PubMed] [Google Scholar]
- Nyegaard M, Rendtorff ND, Nielsen MS, Corydon TJ, Demontis D, Starnawska A, Hedemand A, Buniello A, Niola F, Overgaard MT, Leal SM, Ahmad W, Wikman FP, Petersen KB, Crüger DG, Oostrik J, Kremer H, Tommerup N, Frödin M, Steel KP, Tranebjærg L, Børglum AD 2015. A Novel Locus Harbouring a Functional CD164 Nonsense Mutation Identified in a Large Danish Family with Nonsyndromic Hearing Impairment. PLoS Genet. 11, e1005386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obholzer N, Swinburne IA, Schwab E, Nechiporuk AV, Nicolson T, Megason SG 2012. Rapid positional cloning of zebrafish mutations by linkage and homozygosity mapping using whole-genome sequencing. Development 139, 4280–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obholzer N, Wolfson S, Trapani JG, Mo W, Nechiporuk A, Busch-Nentwich E, Seiler C, Sidi S, Sollner C, Duncan RN, Boehland A, Nicolson T 2008. Vesicular glutamate transporter 3 is required for synaptic transmission in zebrafish hair cells. J Neurosci 28, 2110–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Op de Beeck K, Van Camp G, Thys S, Cools N, Callebaut I, Vrijens K, Van Nassauw L, Van Tendeloo VF, Timmermans JP, Van Laer L 2011. The DFNA5 gene, responsible for hearing loss and involved in cancer, encodes a novel apoptosis-inducing protein. Eur J Hum Genet 19, 965–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang XM, Xia XJ, Verpy E, Du LL, Pandya A, Petit C, Balkany T, Nance WE, Liu XZ 2002. Mutations in the alternatively spliced exons of USH1C cause non-syndromic recessive deafness. Hum. Genet. 111, 26–30. [DOI] [PubMed] [Google Scholar]
- Pei W, Xu L, Varshney GK, Carrington B, Bishop K, Jones M, Huang SC, Idol J, Pretorius PR, Beirl A, Schimmenti LA, Kindt KS, Sood R, Burgess SM 2016. Additive reductions in zebrafish PRPS1 activity result in a spectrum of deficiencies modeling several human PRPS1-associated diseases. Sci Rep 6, 29946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips JB, Blanco-Sanchez B, Lentz JJ, Tallafuss A, Khanobdee K, Sampath S, Jacobs ZG, Han PF, Mishra M, Titus TA, Williams DS, Keats BJ, Washbourne P, Westerfield M 2011. Harmonin (Ush1c) is required in zebrafish Muller glial cells for photoreceptor synaptic development and function. Dis Model Mech 4, 786–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prykhozhij SV, Fuller C, Steele SL, Veinotte CJ, Razaghi B, Robitaille JM, McMaster CR, Shlien A, Malkin D, Berman JN 2018. Optimized knock-in of point mutations in zebrafish using CRISPR/Cas9. Nucleic Acids Res 46, e102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raible DW, Kruse GJ 2000. Organization of the lateral line system in embryonic zebrafish. J Comp Neurol 421, 189–98. [PubMed] [Google Scholar]
- Ramanagoudr-Bhojappa R, Carrington B, Ramaswami M, Bishop K, Robbins GM, Jones M, Harper U, Frederickson SC, Kimble DC, Sood R, Chandrasekharappa SC 2018. Multiplexed CRISPR/Cas9-mediated knockout of 19 Fanconi anemia pathway genes in zebrafish revealed their roles in growth, sexual development and fertility. PLoS Genet 14, e1007821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman AU, Morell RJ, Belyantseva IA, Khan SY, Boger ET, Shahzad M, Ahmed ZM, Riazuddin S, Khan SN, Friedman TB 2010. Targeted capture and next-generation sequencing identifies C9orf75, encoding taperin, as the mutated gene in nonsyndromic deafness DFNB79. Am J Hum Genet 86, 378–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman AU, Gul K, Morell RJ, Lee K, Ahmed ZM, Riazuddin S, Ali RA, Shahzad M, Jaleel A-U, Andrade PB, Khan SN, Khan S, Brewer CC, Ahmad W, Leal SM, Riazuddin S, Friedman TB 2011. Mutations of GIPC3 cause nonsyndromic hearing loss DFNB72 but not DFNB81 that also maps to chromosome 19p. Hum. Genet. 130, 759–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehman AU, Santos-Cortez RL, Morell RJ, Drummond MC, Ito T, Lee K, Khan AA, Basra MA, Wasif N, Ayub M, Ali RA, Raza SI, University of Washington Center for Mendelian, G., Nickerson DA, Shendure J, Bamshad M, Riazuddin S, Billington N, Khan SN, Friedman PL, Griffith AJ, Ahmad W, Riazuddin S, Leal SM, Friedman TB 2014. Mutations in TBC1D24, a gene associated with epilepsy, also cause nonsyndromic deafness DFNB86. Am J Hum Genet 94, 144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazuddin S, Ahmed ZM, Fanning AS, Lagziel A, Kitajiri S, Ramzan K, Khan SN, Chattaraj P, Friedman PL, Anderson JM, Belyantseva IA, Forge A, Riazuddin S, Friedman TB 2006a. Tricellulin is a tight-junction protein necessary for hearing. Am J Hum Genet 79, 1040–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazuddin S, Nazli S, Ahmed ZM, Yang Y, Zulfiqar F, Shaikh RS, Zafar AU, Khan SN, Sabar F, Javid FT, Wilcox ER, Tsilou E, Boger ET, Sellers JR, Belyantseva IA, Friedman TB 2008. Mutation spectrum of MYO7A and evaluation of a novel nonsyndromic deafness DFNB2 allele with residual function. Hum Mutat 29, 502–11. [DOI] [PubMed] [Google Scholar]
- Riazuddin S, Khan SN, Ahmed ZM, Ghosh M, Caution K, Nazli S, Kabra M, Zafar AU, Chen K, Naz S, Antonellis A, Pavan WJ, Green ED, Wilcox ER, Friedman PL, Morell RJ, Friedman TB 2006b. Mutations in TRIOBP, which encodes a putative cytoskeletal-organizing protein, are associated with nonsyndromic recessive deafness. Am J Hum Genet 78, 137–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riazuddin S, Belyantseva IA, Giese AP, Lee K, Indzhykulian AA, Nandamuri SP, Yousaf R, Sinha GP, Lee S, Terrell D, Hegde RS, Ali RA, Anwar S, Andrade-Elizondo PB, Sirmaci A, Parise LV, Basit S, Wali A, Ayub M, Ansar M, Ahmad W, Khan SN, Akram J, Tekin M, Riazuddin S, Cook T, Buschbeck EK, Frolenkov GI, Leal SM, Friedman TB, Ahmed ZM 2012. Alterations of the CIB2 calcium- and integrin-binding protein cause Usher syndrome type 1J and nonsyndromic deafness DFNB48. Nat Genet 44, 1265–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson NG, Lu L, Heller S, Merchant SN, Eavey RD, McKenna M, Nadol JB Jr., Miyamoto RT, Linthicum FH Jr., Lubianca Neto JF, Hudspeth AJ, Seidman CE, Morton CC, Seidman JG 1998. Mutations in a novel cochlear gene cause DFNA9, a human nonsyndromic deafness with vestibular dysfunction. Nat. Genet. 20, 299–303. [DOI] [PubMed] [Google Scholar]
- Rohacek AM, Bebee TW, Tilton RK, Radens CM, McDermott-Roe C, Peart N, Kaur M, Zaykaner M, Cieply B, Musunuru K, Barash Y, Germiller JA, Krantz ID, Carstens RP, Epstein DJ 2017. ESRP1 Mutations Cause Hearing Loss due to Defects in Alternative Splicing that Disrupt Cochlear Development. Dev. Cell 43, 318–331.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossi A, Kontarakis Z, Gerri C, Nolte H, Holper S, Kruger M, Stainier DY 2015. Genetic compensation induced by deleterious mutations but not gene knockdowns. Nature 524, 230–3. [DOI] [PubMed] [Google Scholar]
- Rost S, Bach E, Neuner C, Nanda I, Dysek S, Bittner RE, Keller A, Bartsch O, Mlynski R, Haaf T, Müller CR, Kunstmann E 2014. Novel form of X-linked nonsyndromic hearing loss with cochlear malformation caused by a mutation in the type IV collagen gene COL4A6. Eur. J. Hum. Genet. 22, 208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruel J, Emery S, Nouvian R, Bersot T, Amilhon B, Van Rybroek JM, Rebillard G, Lenoir M, Eybalin M, Delprat B, Sivakumaran TA, Giros B, El Mestikawy S, Moser T, Smith RJ, Lesperance MM, Puel JL 2008. Impairment of SLC17A8 encoding vesicular glutamate transporter-3, VGLUT3, underlies nonsyndromic deafness DFNA25 and inner hair cell dysfunction in null mice. Am J Hum Genet 83, 278–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Runge CL, Indap A, Zhou Y, Kent JW Jr., King E, Erbe CB, Cole R, Littrell J, Merath K, James R, Rüschendorf F, Kerschner JE, Marth G, Hübner N, Göring HHH, Friedland DR, Kwok W-M, Olivier M 2016. Association of TMTC2 With Human Nonsyndromic Sensorineural Hearing Loss. JAMA Otolaryngol. Head Neck Surg. 142, 866–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San B, Rougeot J, Voeltzke K, van Vegchel G, Aben M, Andralojc KM, Flik G, Kamminga LM 2019. The ezh2(sa1199) mutant zebrafish display no distinct phenotype. PLoS One 14, e0210217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sang Q, Zhang J, Feng R, Wang X, Li Q, Zhao X, Xing Q, Chen W, Du J, Sun S, Chai R, Liu D, Jin L, He L, Li H, Wang L 2014. Ildr1b is essential for semicircular canal development, migration of the posterior lateral line primordium and hearing ability in zebrafish: implications for a role in the recessive hearing impairment DFNB42. Hum. Mol. Genet. 23, 6201–6211. [DOI] [PubMed] [Google Scholar]
- Santos-Cortez RL, Lee K, Azeem Z, Antonellis PJ, Pollock LM, Khan S, Irfanullah, Andrade-Elizondo PB, Chiu I, Adams MD, Basit S, Smith JD, University of Washington Center for Mendelian, G., Nickerson DA, McDermott BM Jr., Ahmad W, Leal SM 2013. Mutations in KARS, encoding lysyl-tRNA synthetase, cause autosomal-recessive nonsyndromic hearing impairment DFNB89. Am J Hum Genet 93, 132–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Cortez RL, Lee K, Giese AP, Ansar M, Amin-Ud-Din M, Rehn K, Wang X, Aziz A, Chiu I, Hussain Ali R, Smith JD, University of Washington Center for Mendelian, G., Shendure J, Bamshad M, Nickerson DA, Ahmed ZM, Ahmad W, Riazuddin S, Leal SM 2014. Adenylate cyclase 1 (ADCY1) mutations cause recessive hearing impairment in humans and defects in hair cell function and hearing in zebrafish. Hum Mol Genet 23, 3289–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos-Cortez RLP, Faridi R, Rehman AU, Lee K, Ansar M, Wang X, Morell RJ, Isaacson R, Belyantseva IA, Dai H, Acharya A, Qaiser TA, Muhammad D, Ali RA, Shams S, Hassan MJ, Shahzad S, Raza SI, Bashir Z-EH, Smith JD, Nickerson DA, Bamshad MJ, University of Washington Center for Mendelian, G., Riazuddin S, Ahmad W, Friedman TB, Leal SM 2016. Autosomal-Recessive Hearing Impairment Due to Rare Missense Variants within S1PR2. Am. J. Hum. Genet. 98, 331–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schibler A, Malicki J 2007. A screen for genetic defects of the zebrafish ear. Mech Dev 124, 592–604. [DOI] [PubMed] [Google Scholar]
- Schneider E, Märker T, Daser A, Frey-Mahn G, Beyer V, Farcas R, Schneider-Rätzke B, Kohlschmidt N, Grossmann B, Bauss K, Napiontek U, Keilmann A, Bartsch O, Zechner U, Wolfrum U, Haaf T 2009. Homozygous disruption of PDZD7 by reciprocal translocation in a consanguineous family: a new member of the Usher syndrome protein interactome causing congenital hearing impairment. Hum. Mol. Genet. 18, 655–666. [DOI] [PubMed] [Google Scholar]
- Schonberger J, Wang L, Shin JT, Kim SD, Depreux FF, Zhu H, Zon L, Pizard A, Kim JB, Macrae CA, Mungall AJ, Seidman JG, Seidman CE 2005. Mutation in the transcriptional coactivator EYA4 causes dilated cardiomyopathy and sensorineural hearing loss. Nat Genet 37, 418–22. [DOI] [PubMed] [Google Scholar]
- Schraders M, Oostrik J, Huygen PL, Strom TM, van Wijk E, Kunst HP, Hoefsloot LH, Cremers CW, Admiraal RJ, Kremer H 2010a. Mutations in PTPRQ are a cause of autosomal-recessive nonsyndromic hearing impairment DFNB84 and associated with vestibular dysfunction. Am J Hum Genet 86, 604–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraders M, Haas SA, Weegerink NJ, Oostrik J, Hu H, Hoefsloot LH, Kannan S, Huygen PL, Pennings RJ, Admiraal RJ, Kalscheuer VM, Kunst HP, Kremer H 2011. Next-generation sequencing identifies mutations of SMPX, which encodes the small muscle protein, X-linked, as a cause of progressive hearing impairment. Am J Hum Genet 88, 628–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraders M, Lee K, Oostrik J, Huygen PL, Ali G, Hoefsloot LH, Veltman JA, Cremers FP, Basit S, Ansar M, Cremers CW, Kunst HP, Ahmad W, Admiraal RJ, Leal SM, Kremer H 2010b. Homozygosity mapping reveals mutations of GRXCR1 as a cause of autosomal-recessive nonsyndromic hearing impairment. Am J Hum Genet 86, 138–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraders M, Ruiz-Palmero L, Kalay E, Oostrik J, del Castillo FJ, Sezgin O, Beynon AJ, Strom TM, Pennings RJ, Seco CZ, Oonk AM, Kunst HP, Dominguez-Ruiz M, Garcia-Arumi AM, del Campo M, Villamar M, Hoefsloot LH, Moreno F, Admiraal RJ, del Castillo I, Kremer H 2012. Mutations of the gene encoding otogelin are a cause of autosomal-recessive nonsyndromic moderate hearing impairment. Am J Hum Genet 91, 883–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrauwen I, Melegh BI, Chakchouk I, Acharya A, Nasir A, Poston A, Cornejo-Sanchez DM, Szabo Z, Karosi T, Bene J, Melegh B, Leal SM 2019. Hearing impairment locus heterogeneity and identification of PLS1 as a new autosomal dominant gene in Hungarian Roma. Eur. J. Hum. Genet. 27, 869–878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schrauwen I, Helfmann S, Inagaki A, Predoehl F, Tabatabaiefar MA, Picher MM, Sommen M, Seco CZ, Oostrik J, Kremer H, Dheedene A, Claes C, Fransen E, Chaleshtori MH, Coucke P, Lee A, Moser T, Van Camp G 2012. A mutation in CABP2, expressed in cochlear hair cells, causes autosomal-recessive hearing impairment. Am J Hum Genet 91, 636–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz JM, Khan SN, Ahmed ZM, Riazuddin S, Waryah AM, Chhatre D, Starost MF, Ploplis B, Buckley S, Velasquez D, Kabra M, Lee K, Hassan MJ, Ali G, Ansar M, Ghosh M, Wilcox ER, Ahmad W, Merlino G, Leal SM, Riazuddin S, Friedman TB, Morell RJ 2009. Noncoding mutations of HGF are associated with nonsyndromic hearing loss, DFNB39. Am J Hum Genet 85, 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott HS, Kudoh J, Wattenhofer M, Shibuya K, Berry A, Chrast R, Guipponi M, Wang J, Kawasaki K, Asakawa S, Minoshima S, Younus F, Mehdi SQ, Radhakrishna U, Papasavvas MP, Gehrig C, Rossier C, Korostishevsky M, Gal A, Shimizu N, Bonne-Tamir B, Antonarakis SE 2001. Insertion of beta-satellite repeats identifies a transmembrane protease causing both congenital and childhood onset autosomal recessive deafness. Nat. Genet. 27, 59–63. [DOI] [PubMed] [Google Scholar]
- Seco CZ, Oonk AM, Dominguez-Ruiz M, Draaisma JM, Gandia M, Oostrik J, Neveling K, Kunst HP, Hoefsloot LH, del Castillo I, Pennings RJ, Kremer H, Admiraal RJ, Schraders M 2015. Progressive hearing loss and vestibular dysfunction caused by a homozygous nonsense mutation in CLIC5. Eur J Hum Genet 23, 189–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler C, Finger-Baier KC, Rinner O, Makhankov YV, Schwarz H, Neuhauss SC, Nicolson T 2005. Duplicated genes with split functions: independent roles of protocadherin15 orthologues in zebrafish hearing and vision. Development 132, 615–23. [DOI] [PubMed] [Google Scholar]
- Seiler C, Ben-David O, Sidi S, Hendrich O, Rusch A, Burnside B, Avraham KB, Nicolson T 2004. Myosin VI is required for structural integrity of the apical surface of sensory hair cells in zebrafish. Dev Biol 272, 328–38. [DOI] [PubMed] [Google Scholar]
- Shabbir MI, Ahmed ZM, Khan SY, Riazuddin S, Waryah AM, Khan SN, Camps RD, Ghosh M, Kabra M, Belyantseva IA, Friedman TB 2006. Mutations of human TMHS cause recessively inherited non-syndromic hearing loss. Journal of medical genetics 43, 634–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahin H, Walsh T, Sobe T, Abu Sa’ed J, Abu Rayan A, Lynch ED, Lee MK, Avraham KB, King MC, Kanaan M 2006. Mutations in a novel isoform of TRIOBP that encodes a filamentous-actin binding protein are responsible for DFNB28 recessive nonsyndromic hearing loss. Am J Hum Genet 78, 144–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shearer AE, Hildebrand MS, Smith RJH 2017. Hereditary Hearing Loss and Deafness Overview. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, (Eds.), GeneReviews. [Google Scholar]
- Shearer AE, Kolbe DL, Azaiez H, Sloan CM, Frees KL, Weaver AE, Clark ET, Nishimura CJ, Black-Ziegelbein EA, Smith RJH 2014. Copy number variants are a common cause of non-syndromic hearing loss. Genome Med. 6, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield AM, Smith RJH 2019. The Epidemiology of Deafness. Cold Spring Harb. Perspect. Med. 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen X, Liu F, Wang Y, Wang H, Ma J, Xia W, Zhang J, Jiang N, Sun S, Wang X, Ma D 2015. Down-regulation of msrb3 and destruction of normal auditory system development through hair cell apoptosis in zebrafish. Int. J. Dev. Biol. 59, 195–203. [DOI] [PubMed] [Google Scholar]
- Simon M, Richard EM, Wang X, Shahzad M, Huang VH, Qaiser TA, Potluri P, Mahl SE, Davila A, Nazli S, Hancock S, Yu M, Gargus J, Chang R, Al-Sheqaih N, Newman WG, Abdenur J, Starr A, Hegde R, Dorn T, Busch A, Park E, Wu J, Schwenzer H, Flierl A, Florentz C, Sissler M, Khan SN, Li R, Guan M-X, Friedman TB, Wu DK, Procaccio V, Riazuddin S, Wallace DC, Ahmed ZM, Huang T, Riazuddin S 2015. Mutations of human NARS2, encoding the mitochondrial asparaginyl-tRNA synthetase, cause nonsyndromic deafness and Leigh syndrome. PLoS Genet. 11, e1005097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sineni CJ, Yildirim-Baylan M, Guo S, Camarena V, Wang G, Tokgoz-Yilmaz S, Duman D, Bademci G, Tekin M 2019. A truncating CLDN9 variant is associated with autosomal recessive nonsyndromic hearing loss. Hum. Genet. 138, 1071–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirmaci A, Erbek S, Price J, Huang M, Duman D, Cengiz FB, Bademci G, Tokgöz-Yilmaz S, Hişmi B, Ozdağ H, Oztürk B, Kulaksizoğlu S, Yildirim E, Kokotas H, Grigoriadou M, Petersen MB, Shahin H, Kanaan M, King M-C, Chen Z-Y, Blanton SH, Liu XZ, Zuchner S, Akar N, Tekin M 2010. A truncating mutation in SERPINB6 is associated with autosomal-recessive nonsyndromic sensorineural hearing loss. Am. J. Hum. Genet. 86, 797–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slijkerman R, Goloborodko A, Broekman S, de Vrieze E, Hetterschijt L, Peters T, Gerits M, Kremer H, van Wijk E 2018. Poor Splice-Site Recognition in a Humanized Zebrafish Knockin Model for the Recurrent Deep-Intronic c.7595–2144A>G Mutation in USH2A. Zebrafish 15, 597–609. [DOI] [PubMed] [Google Scholar]
- Sloan-Heggen CM, Bierer AO, Shearer AE, Kolbe DL, Nishimura CJ, Frees KL, Ephraim SS, Shibata SB, Booth KT, Campbell CA, Ranum PT, Weaver AE, Black-Ziegelbein EA, Wang D, Azaiez H, Smith RJH 2016. Comprehensive genetic testing in the clinical evaluation of 1119 patients with hearing loss. Hum. Genet. 135, 441–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits JJ, Oostrik J, Beynon AJ, Kant SG, de Koning Gans PAM, Rotteveel LJC, Klein Wassink-Ruiter JS, Free RH, Maas SM, van de Kamp J, Merkus P, Consortium D, Koole W, Feenstra I, Admiraal RJC, Lanting CP, Schraders M, Yntema HG, Pennings RJE, Kremer H 2019. De novo and inherited loss-of-function variants of ATP2B2 are associated with rapidly progressive hearing impairment. Hum. Genet. 138, 61–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollner C, Rauch GJ, Siemens J, Geisler R, Schuster SC, Muller U, Nicolson T, Tubingen Screen C 2004. Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature 428, 955–9. [DOI] [PubMed] [Google Scholar]
- Söllner C, Rauch G-J, Siemens J, Geisler R, Schuster SC, Müller U, Nicolson T, Tübingen Screen C 2004. Mutations in cadherin 23 affect tip links in zebrafish sensory hair cells. Nature 428, 955–959. [DOI] [PubMed] [Google Scholar]
- Solnica-Krezel L, Schier AF, Driever W 1994. Efficient recovery of ENU-induced mutations from the zebrafish germline. Genetics 136, 1401–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stainier DYR, Raz E, Lawson ND, Ekker SC, Burdine RD, Eisen JS, Ingham PW, Schulte-Merker S, Yelon D, Weinstein BM, Mullins MC, Wilson SW, Ramakrishnan L, Amacher SL, Neuhauss SCF, Meng A, Mochizuki N, Panula P, Moens CB 2017. Guidelines for morpholino use in zebrafish. PLoS Genet 13, e1007000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stooke-Vaughan GA, Obholzer ND, Baxendale S, Megason SG, Whitfield TT 2015. Otolith tethering in the zebrafish otic vesicle requires Otogelin and α-Tectorin. Development 142, 1137–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thoenes M, Zimmermann U, Ebermann I, Ptok M, Lewis MA, Thiele H, Morlot S, Hess MM, Gal A, Eisenberger T, Bergmann C, Nürnberg G, Nürnberg P, Steel KP, Knipper M, Bolz HJ 2015. OSBPL2 encodes a protein of inner and outer hair cell stereocilia and is mutated in autosomal dominant hearing loss (DFNA67). Orphanet J. Rare Dis. 10, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tlili A, Mannikko M, Charfedine I, Lahmar I, Benzina Z, Ben Amor M, Driss N, Ala-Kokko L, Drira M, Masmoudi S, Ayadi H 2005. A novel autosomal recessive non-syndromic deafness locus, DFNB66, maps to chromosome 6p21.2–22.3 in a large Tunisian consanguineous family. Hum Hered 60, 123–8. [DOI] [PubMed] [Google Scholar]
- Vahava O, Morell R, Lynch ED, Weiss S, Kagan ME, Ahituv N, Morrow JE, Lee MK, Skvorak AB, Morton CC, Blumenfeld A, Frydman M, Friedman TB, King MC, Avraham KB 1998. Mutation in transcription factor POU4F3 associated with inherited progressive hearing loss in humans. Science 279, 1950–1954. [DOI] [PubMed] [Google Scholar]
- Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt PJ, Van de Heyning P, McGuirt WT, Smith RJ, Willems PJ, Legan PK, Richardson GP, Van Camp G 1998. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat. Genet. 20, 194–197. [DOI] [PubMed] [Google Scholar]
- van Wijk E, Krieger E, Kemperman MH, De Leenheer EM, Huygen PL, Cremers CW, Cremers FP, Kremer H 2003. A mutation in the gamma actin 1 (ACTG1) gene causes autosomal dominant hearing loss (DFNA20/26). J Med Genet 40, 879–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney GK, Sood R, Burgess SM 2015a. Understanding and Editing the Zebrafish Genome. Adv Genet 92, 1–52. [DOI] [PubMed] [Google Scholar]
- Varshney GK, Huang H, Zhang S, Lu J, Gildea DE, Yang Z, Wolfsberg TG, Lin S, Burgess SM 2013a. The Zebrafish Insertion Collection (ZInC): a web based, searchable collection of zebrafish mutations generated by DNA insertion. Nucleic Acids Res 41, D861–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney GK, Pei W, LaFave MC, Idol J, Xu L, Gallardo V, Carrington B, Bishop K, Jones M, Li M, Harper U, Huang SC, Prakash A, Chen W, Sood R, Ledin J, Burgess SM 2015b. High-throughput gene targeting and phenotyping in zebrafish using CRISPR/Cas9. Genome Res 25, 1030–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney GK, Lu J, Gildea DE, Huang H, Pei W, Yang Z, Huang SC, Schoenfeld D, Pho NH, Casero D, Hirase T, Mosbrook-Davis D, Zhang Jao, L.E., Zhang B, Woods IG, Zimmerman S, Schier AF, Wolfsberg TG, Pellegrini M, Burgess SM, Lin S 2013b. A large-scale zebrafish gene knockout resource for the genome-wide study of gene function. Genome Res 23, 727–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeven K, Van Laer L, Kirschhofer K, Legan PK, Hughes DC, Schatteman I, Verstreken M, Van Hauwe P, Coucke P, Chen A, Smith RJ, Somers T, Offeciers FE, Van de Heyning P, Richardson GP, Wachtler F, Kimberling WJ, Willems PJ, Govaerts PJ, Van Camp G 1998. Mutations in the human alpha-tectorin gene cause autosomal dominant non-syndromic hearing impairment. Nat. Genet. 19, 60–62. [DOI] [PubMed] [Google Scholar]
- Verpy E, Masmoudi S, Zwaenepoel I, Leibovici M, Hutchin TP, Del Castillo I, Nouaille S, Blanchard S, Laine S, Popot JL, Moreno F, Mueller RF, Petit C 2001. Mutations in a new gene encoding a protein of the hair bundle cause non-syndromic deafness at the DFNB16 locus. Nat Genet 29, 345–9. [DOI] [PubMed] [Google Scholar]
- von Ameln S, Wang G, Boulouiz R, Rutherford MA, Smith GM, Li Y, Pogoda H-M, Nürnberg G, Stiller B, Volk AE, Borck G, Hong JS, Goodyear RJ, Abidi O, Nürnberg P, Hofmann K, Richardson GP, Hammerschmidt M, Moser T, Wollnik B, Koehler CM, Teitell MA, Barakat A, Kubisch C 2012. A mutation in PNPT1, encoding mitochondrial-RNA-import protein PNPase, causes hereditary hearing loss. Am. J. Hum. Genet. 91, 919–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vona B, Nanda I, Neuner C, Muller T, Haaf T 2013. Confirmation of GRHL2 as the gene for the DFNA28 locus. Am J Med Genet A 161A, 2060–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vona B, Nanda I, Hofrichter MA, Shehata-Dieler W, Haaf T 2015. Non-syndromic hearing loss gene identification: A brief history and glimpse into the future. Mol Cell Probes 29, 260–70. [DOI] [PubMed] [Google Scholar]
- Vona B, Hofrichter MAH, Chioza BA, Crosby AH, Nanda I, Haaf T Genetic Elucidation of Nonsyndromic Hearing Loss in the High-Throughput Sequencing Era Vona B, Haaf T (eds): Genetics of Deafness. Monogr Hum Genet; Basel, Karger, 2016, vol 20, pp 56–72. doi: 10.1159/000444599 [DOI] [Google Scholar]
- Walsh T, Walsh V, Vreugde S, Hertzano R, Shahin H, Haika S, Lee MK, Kanaan M, King M-C, Avraham KB 2002. From flies’ eyes to our ears: mutations in a human class III myosin cause progressive nonsyndromic hearing loss DFNB30. Proc. Natl. Acad. Sci. U. S. A. 99, 7518–7523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh T, Pierce SB, Lenz DR, Brownstein Z, Dagan-Rosenfeld O, Shahin H, Roeb W, McCarthy S, Nord AS, Gordon CR, Ben-Neriah Z, Sebat J, Kanaan M, Lee MK, Frydman M, King M-C, Avraham KB 2010. Genomic duplication and overexpression of TJP2/ZO-2 leads to altered expression of apoptosis genes in progressive nonsyndromic hearing loss DFNA51. Am. J. Hum. Genet. 87, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Liang Y, Fridell RA, Probst FJ, Wilcox ER, Touchman JW, Morton CC, Morell RJ, Noben-Trauth K, Camper SA, Friedman TB 1998. Association of unconventional myosin MYO15 mutations with human nonsyndromic deafness DFNB3. Science 280, 1447–1451. [DOI] [PubMed] [Google Scholar]
- Wang H, Lin C, Yao J, Shi H, Zhang C, Wei Q, Lu Y, Chen Z, Xing G, Cao X 2019. Deletion of OSBPL2 in auditory cells increases cholesterol biosynthesis and drives reactive oxygen species production by inhibiting AMPK activity. Cell Death Dis 10, 627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Feng Y, Yan D, Qin L, Grati M.h., Mittal R, Li T, Sundhari AK, Liu Y, Chapagain P, Blanton SH, Liao S, Liu X 2018. A dominant variant in the PDE1C gene is associated with nonsyndromic hearing loss. Hum. Genet. 137, 437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasfy MM, Matsui JI, Miller J, Dowling JE, Perkins BD 2014. myosin 7aa(−/−) mutant zebrafish show mild photoreceptor degeneration and reduced electroretinographic responses. Exp. Eye Res. 122, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayne S, Robertson NG, DeClau F, Chen N, Verhoeven K, Prasad S, Tranebjarg L, Morton CC, Ryan AF, Van Camp G, Smith RJ 2001. Mutations in the transcriptional activator EYA4 cause late-onset deafness at the DFNA10 locus. Hum Mol Genet 10, 195–200. [DOI] [PubMed] [Google Scholar]
- Weil D, Kussel P, Blanchard S, Levy G, Levi-Acobas F, Drira M, Ayadi H, Petit C 1997. The autosomal recessive isolated deafness, DFNB2, and the Usher 1B syndrome are allelic defects of the myosin-VIIA gene. Nat Genet 16, 191–3. [DOI] [PubMed] [Google Scholar]
- Wesdorp M, de Koning Gans PAM, Schraders M, Oostrik J, Huynen MA, Venselaar H, Beynon AJ, van Gaalen J, Piai V, Voermans N, van Rossum MM, Hartel BP, Lelieveld SH, Wiel L, Verbist B, Rotteveel LJ, van Dooren MF, Lichtner P, Kunst HPM, Feenstra I, Admiraal RJC, Consortium D, Yntema HG, Hoefsloot LH, Pennings RJE, Kremer H 2018a. Heterozygous missense variants of LMX1A lead to nonsyndromic hearing impairment and vestibular dysfunction. Hum. Genet. 137, 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wesdorp M, Murillo-Cuesta S, Peters T, Celaya AM, Oonk A, Schraders M, Oostrik J, Gomez-Rosas E, Beynon AJ, Hartel BP, Okkersen K, Koenen HJPM, Weeda J, Lelieveld S, Voermans NC, Joosten I, Hoyng CB, Lichtner P, Kunst HPM, Feenstra I, de Bruijn SE, Consortium D, Admiraal RJC, Yntema HG, van Wijk E, Del Castillo I, Serra P, Varela-Nieto I, Pennings RJE, Kremer H 2018b. MPZL2, Encoding the Epithelial Junctional Protein Myelin Protein Zero-like 2, Is Essential for Hearing in Man and Mouse. Am. J. Hum. Genet. 103, 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitfield TT 2002. Zebrafish as a model for hearing and deafness. J Neurobiol 53, 157–71. [DOI] [PubMed] [Google Scholar]
- Whitfield TT, Riley BB, Chiang MY, Phillips B 2002. Development of the zebrafish inner ear. Dev Dyn 223, 427–58. [DOI] [PubMed] [Google Scholar]
- Whitfield TT, Granato M, van Eeden FJ, Schach U, Brand M, Furutani-Seiki M, Haffter P, Hammerschmidt M, Heisenberg CP, Jiang YJ, Kane DA, Kelsh RN, Mullins MC, Odenthal J, Nusslein-Volhard C 1996. Mutations affecting development of the zebrafish inner ear and lateral line. Development 123, 241–54. [DOI] [PubMed] [Google Scholar]
- Wibowo I, Pinto-Teixeira F, Satou C, Higashijima S, Lopez-Schier H 2011. Compartmentalized Notch signaling sustains epithelial mirror symmetry. Development 138, 1143–52. [DOI] [PubMed] [Google Scholar]
- Wierson WA, Welker JM, Almeida MP, Mann CM, Webster DA, Weiss TJ, Torrie ME, Vollbrecht MK, Lan M, McKeighan KC, Ming Z, Wehmeier A, Mikelson CS, Haltom JA, Kwan KM, Chien C-B, Balciunas D, Ekker SC, Clark KJ, Webber BR, Moriarity B, Solin SL, Carlson DF, Dobbs DL, McGrail M, Essner JJ 2018. GeneWeld: a method for efficient targeted integration directed by short homology. bioRxiv, 431627. [Google Scholar]
- Wilcox ER, Burton QL, Naz S, Riazuddin S, Smith TN, Ploplis B, Belyantseva I, Ben-Yosef T, Liburd NA, Morell RJ, Kachar B, Wu DK, Griffith AJ, Riazuddin S, Friedman TB 2001. Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell 104, 165–172. [DOI] [PubMed] [Google Scholar]
- Wu RS, Lam II, Clay H, Duong DN, Deo RC, Coughlin SR 2018. A Rapid Method for Directed Gene Knockout for Screening in G0 Zebrafish. Dev Cell 46, 112–125 e4. [DOI] [PubMed] [Google Scholar]
- Xia JH, Liu CY, Tang BS, Pan Q, Huang L, Dai HP, Zhang BR, Xie W, Hu DX, Zheng D, Shi XL, Wang DA, Xia K, Yu KP, Liao XD, Feng Y, Yang YF, Xiao JY, Xie DH, Huang JZ 1998. Mutations in the gene encoding gap junction protein beta-3 associated with autosomal dominant hearing impairment. Nat. Genet. 20, 370–373. [DOI] [PubMed] [Google Scholar]
- Xia W, Hu J, Ma J, Huang J, Wang X, Jiang N, Zhang J, Ma Z, Ma D 2019a. Novel TRRAP mutation causes autosomal dominant non-syndromic hearing loss. Clin. Genet. 96, 300–308. [DOI] [PubMed] [Google Scholar]
- Xia W, Hu J, Ma J, Huang J, Jing T, Deng L, Zhang J, Jiang N, Ma D, Ma Z 2019b. Mutations in TOP2B cause autosomal-dominant hereditary hearing loss via inhibition of the PI3K-Akt signalling pathway. FEBS Lett. 593, 2008–2018. [DOI] [PubMed] [Google Scholar]
- Xia W, Hu J, Liu F, Ma J, Sun S, Zhang J, Jin K, Huang J, Jiang N, Wang X, Li W, Ma Z, Ma D 2017. New role of LRP5, associated with nonsyndromic autosomal-recessive hereditary hearing loss. Hum. Mutat. 38, 1421–1431. [DOI] [PubMed] [Google Scholar]
- Xiao T, Roeser T, Staub W, Baier H 2005. A GFP-based genetic screen reveals mutations that disrupt the architecture of the zebrafish retinotectal projection. Development 132, 2955–67. [DOI] [PubMed] [Google Scholar]
- Xing G, Yao J, Wu B, Liu T, Wei Q, Liu C, Lu Y, Chen Z, Zheng H, Yang X, Cao X 2015. Identification of OSBPL2 as a novel candidate gene for progressive nonsyndromic hearing loss by whole-exome sequencing. Genet. Med. 17, 210–218. [DOI] [PubMed] [Google Scholar]
- Yan D, Zhu Y, Walsh T, Xie D, Yuan H, Sirmaci A, Fujikawa T, Wong AC, Loh TL, Du L, Grati M, Vlajkovic SM, Blanton S, Ryan AF, Chen ZY, Thorne PR, Kachar B, Tekin M, Zhao HB, Housley GD, King MC, Liu XZ 2013. Mutation of the ATP-gated P2X(2) receptor leads to progressive hearing loss and increased susceptibility to noise. Proc Natl Acad Sci U S A 110, 2228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Q, DeSmidt AA, Tekin M, Liu X, Lu Z 2016. Hearing Assessment in Zebrafish During the First Week Postfertilization. Zebrafish 13, 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Q, Wang L, Mittal R, Yan D, Richmond M, Denyer S, Requena T, Liu K, Varshney GK, Lu Z, Liu X 2019. Transcriptomic Analyses of Inner Ear Sensory Epithelia in Zebrafish. Anat Rec (Hoboken), 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yariz KO, Duman D, Seco CZ, Dallman J, Huang M, Peters TA, Sirmaci A, Lu N, Schraders M, Skromne I, Oostrik J, Diaz-Horta O, Young JI, Tokgoz-Yilmaz S, Konukseven O, Shahin H, Hetterschijt L, Kanaan M, Oonk AM, Edwards YJ, Li H, Atalay S, Blanton S, Desmidt AA, Liu XZ, Pennings RJ, Lu Z, Chen ZY, Kremer H, Tekin M 2012. Mutations in OTOGL, encoding the inner ear protein otogelin-like, cause moderate sensorineural hearing loss. Am J Hum Genet 91, 872–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasunaga S, Grati M, Cohen-Salmon M, El-Amraoui A, Mustapha M, Salem N, El-Zir E, Loiselet J, Petit C 1999. A mutation in OTOF, encoding otoferlin, a FER-1-like protein, causes DFNB9, a nonsyndromic form of deafness. Nat Genet 21, 363–9. [DOI] [PubMed] [Google Scholar]
- Young TL, Ives E, Lynch E, Person R, Snook S, MacLaren L, Cater T, Griffin A, Fernandez B, Lee MK, King MC 2001. Non-syndromic progressive hearing loss DFNA38 is caused by heterozygous missense mutation in the Wolfram syndrome gene WFS1. Hum. Mol. Genet. 10, 2509–2514. [DOI] [PubMed] [Google Scholar]
- Yousaf R, Gu C, Ahmed ZM, Khan SN, Friedman TB, Riazuddin S, Shears SB, Riazuddin S 2018a. Mutations in Diphosphoinositol-Pentakisphosphate Kinase PPIP5K2 are associated with hearing loss in human and mouse. PLoS Genet. 14, e1007297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousaf R, Ahmed ZM, Giese AP, Morell RJ, Lagziel A, Dabdoub A, Wilcox ER, Riazuddin S, Friedman TB, Riazuddin S 2018b. Modifier variant of METTL13 suppresses human GAB1-associated profound deafness. J. Clin. Invest. 128, 1509–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zazo Seco C, Serrão de Castro L, van Nierop JW, Morin M, Jhangiani S, Verver EJJ, Schraders M, Maiwald N, Wesdorp M, Venselaar H, Spruijt L, Oostrik J, Schoots J, Baylor-Hopkins Center for Mendelian, G., van Reeuwijk J, Lelieveld SH, Huygen PLM, Insenser M, Admiraal RJC, Pennings RJE, Hoefsloot LH, Arias-Vásquez A, de Ligt J, Yntema HG, Jansen JH, Muzny DM, Huls G, van Rossum MM, Lupski JR, Moreno-Pelayo MA, Kunst HPM, Kremer H 2015. Allelic Mutations of KITLG, Encoding KIT Ligand, Cause Asymmetric and Unilateral Hearing Loss and Waardenburg Syndrome Type 2. Am. J. Hum. Genet. 97, 647–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Zhao F, Zong L, Zhang P, Guan L, Zhang J, Wang D, Wang J, Chai W, Lan L, Li Q, Han B, Yang L, Jin X, Yang W, Hu X, Wang X, Li N, Li Y, Petit C, Wang HY, Wang Q 2013. Exome sequencing and linkage analysis identified tenascin-C (TNC) as a novel causative gene in nonsyndromic hearing loss. PLoS One 8, e69549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Miller KK, Yang T, Hildebrand MS, Shearer AE, DeLuca AP, Scheetz TE, Drummond J, Scherer SE, Legan PK, Goodyear RJ, Richardson GP, Cheatham MA, Smith RJ, Dallos P 2011. Carcinoembryonic antigen-related cell adhesion molecule 16 interacts with alpha-tectorin and is mutated in autosomal dominant hearing loss (DFNA4). Proc. Natl. Acad. Sci. U. S. A. 108, 4218–4223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu M, Yang T, Wei S, DeWan AT, Morell RJ, Elfenbein JL, Fisher RA, Leal SM, Smith RJ, Friderici KH 2003. Mutations in the gamma-actin gene (ACTG1) are associated with dominant progressive deafness (DFNA20/26). Am J Hum Genet 73, 1082–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong L, Guan J, Ealy M, Zhang Q, Wang D, Wang H, Zhao Y, Shen Z, Campbell CA, Wang F, Yang J, Sun W, Lan L, Ding D, Xie L, Qi Y, Lou X, Huang X, Shi Q, Chang S, Xiong W, Yin Z, Yu N, Zhao H, Wang J, Wang J, Salvi RJ, Petit C, Smith RJH, Wang Q 2015. Mutations in apoptosis-inducing factor cause X-linked recessive auditory neuropathy spectrum disorder. J. Med. Genet. 52, 523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou B, Desmidt AA, Mittal R, Yan D, Richmond M, Tekin M, Liu X, Lu Z 2019. The Generation of Zebrafish Mariner Model Using the CRISPR/Cas9 System. Anat Rec (Hoboken). [DOI] [PubMed] [Google Scholar]
- Zu Y, Tong X, Wang Z, Liu D, Pan R, Li Z, Hu Y, Luo Z, Huang P, Wu Q, Zhu Z, Zhang B, Lin S 2013. TALEN-mediated precise genome modification by homologous recombination in zebrafish. Nat Methods 10, 329–31. [DOI] [PubMed] [Google Scholar]
- Zwaenepoel I, Mustapha M, Leibovici M, Verpy E, Goodyear R, Liu XZ, Nouaille S, Nance WE, Kanaan M, Avraham KB, Tekaia F, Loiselet J, Lathrop M, Richardson G, Petit C 2002. Otoancorin, an inner ear protein restricted to the interface between the apical surface of sensory epithelia and their overlying acellular gels, is defective in autosomal recessive deafness DFNB22. Proc. Natl. Acad. Sci. U. S. A. 99, 6240–6245. [DOI] [PMC free article] [PubMed] [Google Scholar]